

Abstract
Osimertinib, a mutant-specific third generation EGFR TKI, is emerging as the preferred first-line therapy for EGFR mutant lung cancer, yet resistance inevitably develops in patients. We modeled acquired resistance to osimertinib in transgenic mouse models of EGFRL858R-induced lung adenocarcinoma and found that it is mediated largely through secondary mutations in EGFR – either C797S or L718V/Q. Analysis of circulating free DNA data from patients revealed that L718Q/V mutations almost always occur in the context of an L858R driver mutation. Therapeutic testing in mice revealed that both erlotinib and afatinib caused regression of osimertinib-resistant C797S-containing tumors, whereas only afatinib was effective on L718Q mutant tumors. Combination first-line osimertinib plus erlotinib treatment prevented the emergence of secondary mutations in EGFR. These findings highlight how knowledge of the specific characteristics of resistance mutations are important for determining potential subsequent treatment approaches and suggest strategies to overcome or prevent osimertinib resistance in vivo.
Graphical abstract
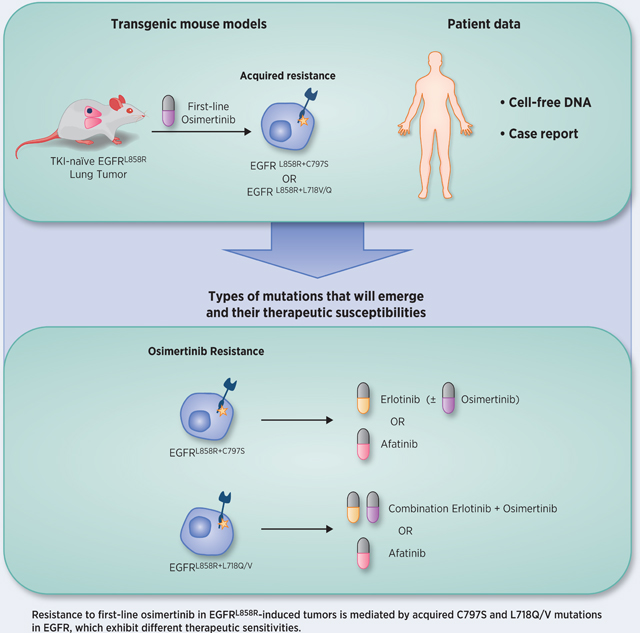
INTRODUCTION
Lung adenocarcinoma (LUAD)-associated mutations in exons encoding the tyrosine kinase domain (TKD) of EGFR confer sensitivity to 1st and 2nd generation tyrosine kinase inhibitors (TKIs) such as erlotinib, gefitinib, afatinib and dacomitinib (1); however, almost all patients develop acquired resistance to these drugs about one year after starting treatment. About 60% of these resistant tumors have a secondary mutation in exon 20 (T790M), which increases the ATP-binding affinity of the mutated protein and renders the tumor resistant to 1st and 2nd generation TKIs (2). Several other mechanisms of acquired resistance to TKIs have been identified, such as mutations in genes encoding proteins that signal downstream of EGFR, histological transformation, and amplification of other receptor tyrosine kinases (3).
Third generation inhibitors (e.g. osimertinib) were developed that target mutated EGFR (including the T790M mutation) over wild-type, presumably resulting in decreased toxicity (4). Based on its clinical activity, osimertinib was first approved for the second-line treatment of T790M-positive EGFR mutant lung cancer (5). However, resistance to second-line osimertinib has also been described in patients – and in ~30% of cases is due to the emergence of additional mutations in EGFR such as C797S/G, G796S/R, L792F/H, L718Q/V, and G724S (6–16). The C797S mutation removes the cysteine side-chain with which osimertinib reacts covalently, thus preventing drug binding to EGFR. Mutations at L718 and G796 have been predicted to prevent drug binding by sterically altering the drug-binding pocket (7, 17).
Osimertinib has also shown promising activity in the first-line (18, 19), leading to its recent approval for use in this setting. This raises important questions concerning likely mechanisms of resistance to first-line osimertinib, about which very little is known. Analysis of circulating tumor DNA (ctDNA) in patients whose tumors progressed on first-line osimertinib (20, 21) has shown that MET amplification was the most common mechanism of resistance (14%), followed by the C797S (7%) and L718Q EGFR mutations (2% of cases). Given this limited information, it remains unclear which therapeutic strategies might be effective at overcoming resistance to first-line osimertinib. One possible strategy is to sequentially apply, or combine, different generations of EGFR TKIs. Several studies have demonstrated that a 1st generation TKI is effective in cell line models with C797S (22, 23), and this is supported by case reports of several patients who responded to treatment with a 1st generation EGFR TKI (24, 25) or the combination of osimertinib plus a 1st generation EGFR TKI (26) after developing resistance to osimertinib mediated by C797S. Indeed, a clinical trial is currently underway to evaluate the combination of osimertinib and gefitinib in TKI-naïve EGFR mutant lung cancer patients [NCT03122717]. Similarly, several in vitro studies and one case report have suggested that 2nd generation TKIs can be effective in cells that have acquired osimertinib resistance through acquisition of an L718 mutation (9, 15, 23, 27).
To investigate mechanisms of resistance to first-line osimertinib more exhaustively, we used a genetically-engineered mouse model (GEMM) of EGFR mutant lung cancer to generate osimertinib-resistant tumors and evaluate the efficacy of sequential or combination EGFR TKIs to overcome or prevent resistance. Further, we analyzed mutational data obtained from a large cohort of lung cancer patients and uncovered allele specificity of osimertinib resistance EGFR mutations. Finally, we report a case of a patient with a tumor harboring both the L718V and L718Q mutations at resistance to first-line osimertinib who benefited from afatinib treatment.
MATERIALS AND METHODS
Mouse husbandry
All animals were kept in pathogen-free micro-isolator housing under BSL2 guidelines approved by the Yale University IACUC and in agreement with the NIH Guide for the Care and Use of Laboratory Animals. The generation of doxycycline-inducible EGFRL858R, EGFRL858R+T790M, and CCSP-rtTA mice has been previously described (28–30).
Cell culture
HEK 293T cells (purchased from ATCC) were maintained at 5% CO2 and cultured in DMEM (Gibco) with 10% FBS (Gibco) and 1% penicillin/streptomycin (Gibco) and kept in a 37˚C incubator. PC9 and PC9-VanR cells (purchased from ECACC-Sigma-Aldrich) and II-18 cells (obtained the global cell bank of AstraZeneca) were maintained at 5% CO2 in RPMI with 10% FCS and 1X GlutaMAX (Thermo Fisher Scientific). H1975 (NCI-H1975) cells (purchased from ATCC) were cultured at 5% CO2 in RPMI-1640 (Thermo Fisher) supplemented with 10% FBS (Sigma), 1% Glutamax (Thermo Fisher) and 1% penicillin/streptomycin (Thermo Fisher). Cell lines were periodically checked for mycoplasma contamination and found to be negative, and were authenticated by STR profiling.
Patient cell-free DNA analysis
We performed a retrospective review of EGFR mutation positive cases with advanced NSCLC from the Guardant Health de-identified database of patients who underwent cfDNA sequencing as part of standard clinical care between September 2017 and March 2019 (Guardant Health, Redwood City, CA). Testing was performed in a Clinical Laboratory Improvement Amendments (CLIA)-certified, College of American Pathologists (CAP)-accredited, New York State Department of Health-approved clinical laboratory at Guardant Health, Inc. Analysis was completed under a Quorum Review Institution Review Board protocol for deidentified and limited datasets and did not require specific written informed consent. During this study period the Guardant360 ctDNA assay included complete exon sequencing of critical exons in 73 genes with reporting of single nucleotide variants in all 73 genes and indels, fusions and copy number variation in a subset of genes. Plasma was analyzed per methods previously described (31). From this cohort, we specifically analyzed cases with the common EGFR driver mutations L858R and E746_A750. We looked at unique patient cases and counted the number of reported C797S, L718Q, or L718V mutations co-occurring with each driver mutation. For patients who had serial testing, each resistance mutation was counted only once per patient – the first time it was reported (unless noted in the Figure legend). For the analysis of T790M-positivity, the status of T790M was considered only within the same blood draw as the osimertinib-resistance mutation. Two cases of T790M-negative/L718Q-positive, noted by a ‘&’ in Figure 2, later gained a T790M mutation and maintained the L718Q mutations, but were counted as T790M-negative. In cases resistant to second-line osimertinib with “T790M loss,” the resistance mutation was present only without T790M, so they were counted as T790M-negative. The complete dataset is available in the Supplementary Materials.
Figure 2. Allele-specific pattern of osimertinib resistance mutations in mice and patients.
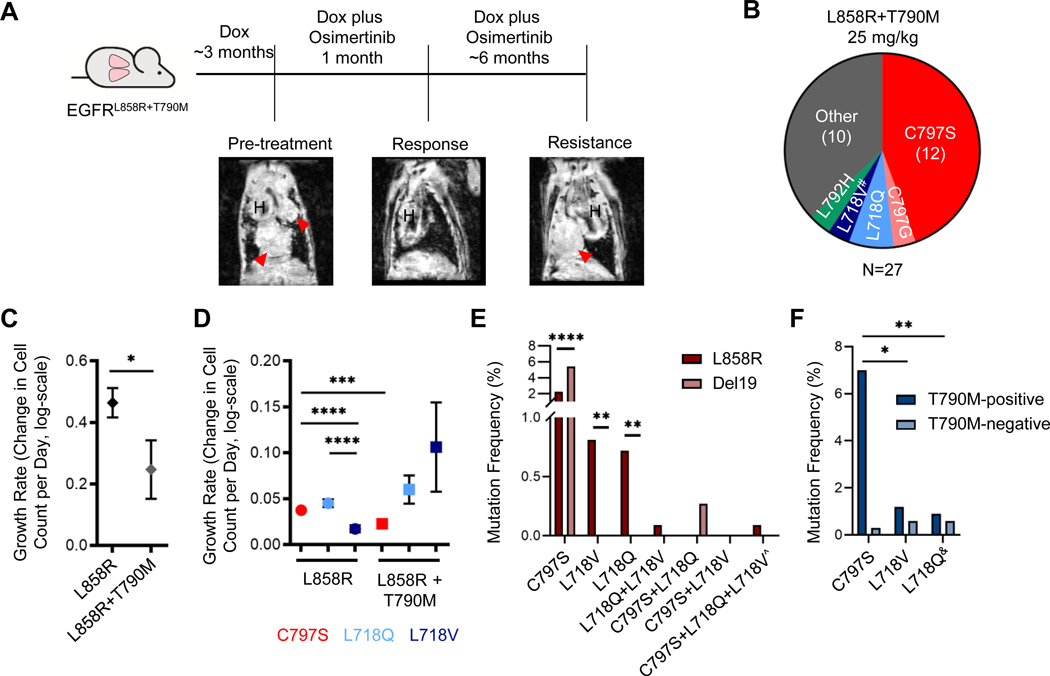
A. Schema of the experiment. CCSP-rtTA;TetO-EGFRL858R+T790M mice were administered doxycycline for the duration of the experiment and developed tumors after ~3 months on doxycycline. When tumors were detected by MRI (see pre-treatment image), mice were put on continuous osimertinib treatment (QD M-F) which elicited a response (see representative response MRI) and treated until the emergence of resistant tumors. Coronal MR images are shown, in which H indicates heart and red arrows indicate tumor. The osimertinib-resistant tumors were then collected and analyzed to identify the resistance mechanism. B. Pie-chart illustrating the resistance mechanisms found in osimertinib-resistant tumors from EGFRL858R+T790M mice. #, tumor that lost T790M. C. Estimated growth rates of TKI-naïve tumors for both the EGFRL858R and EGFRL858R+T790M models. D. Estimated growth rates of the osimertinib-resistant tumors treated with 25 mg/kg osimertinib by resistance mutation for both the EGFRL858R and EGFRL858R+T790M models. Growth rates are depicted as the relative change in cell count per day (log-scale). Error bars represent SEM. P-values were obtained by performing two-sided t tests. E and F. Graphs showing the frequency of the indicated osimertinib resistance mutations in cfDNA from E. patients with tumors harboring the indicated baseline EGFR mutation or F. L858R-positive cases with or without a T790M mutation. ^, the mutations in this case were called in 2 separate blood draws. &, 2 of the T790M-negative L718Q cases listed here later gained a T790M mutation and maintained L718Q in a later blood draw. Two-sided Fisher’s exact test; *, p<0.05; **, p<0.005; ****, p<0.0001.
Patient and clinical sample collection
The patient underwent treatment with EGFR targeted therapies based on the treating oncologist’s recommendations. The patient’s response to therapy was monitored by CT or MRI and clinical examinations as per standard practice. Response assessment was determined by the treating clinician. Formalin-fixed paraffin-embedded sections were prepared from tumor biopsies for histological assessment and genetic testing at various time points. For the pre-treatment biopsy, the sequencing performed was a targeted NGS Illumina MiSeqDx platform, and the liver biopsy at progression was analyzed using Foundation One CDx sequencing. The case report was included with written informed consent from the patient, approved by the Institutional Review Board (IRB #2055–13) at Lifespan Cancer Institute at the Rhode Island Hospital and in accordance with the Declaration of Helsinki.
RESULTS
Acquired resistance to first-line osimertinib is mediated by secondary EGFR mutations in vivo
To identify mechanisms of resistance to first-line osimertinib in EGFR mutant LUAD, we modeled acquired resistance to this TKI using the CCSP-rtTA;TetO-EGFRL858R GEMM (28). In this model, lung-specific expression of the human EGFR transgene is induced by doxycycline administration and the mice develop multifocal peripheral LUADs that closely mirror the human disease. Mice were treated continuously 5 days/week with osimertinib once a day (QD) and monitored by magnetic resonance imaging (MRI) for the emergence of osimertinib-resistant tumors (Figure 1A). The 5 mg/kg dose was used initially (4), but in subsequent studies it was determined that 25 mg/kg QD was approximately equivalent to the 80 mg QD human clinical dose (32). Therefore, once tumors resistant to 5 mg/kg appeared, mice were switched to 25 mg/kg to confirm resistance. We also treated an independent cohort of mice with 25 mg/kg osimertinib QD from the start. MRIs from a representative subset of 16 mice revealed that 15 (94%) exhibited complete responses to 25 mg/kg osimertinib after one month of treatment (Figure 1B), and all (100%) exhibited complete responses by 2 months.
Figure 1. Acquired resistance to first-line osimertinib arises primarily due to the emergence of secondary mutations in EGFR.
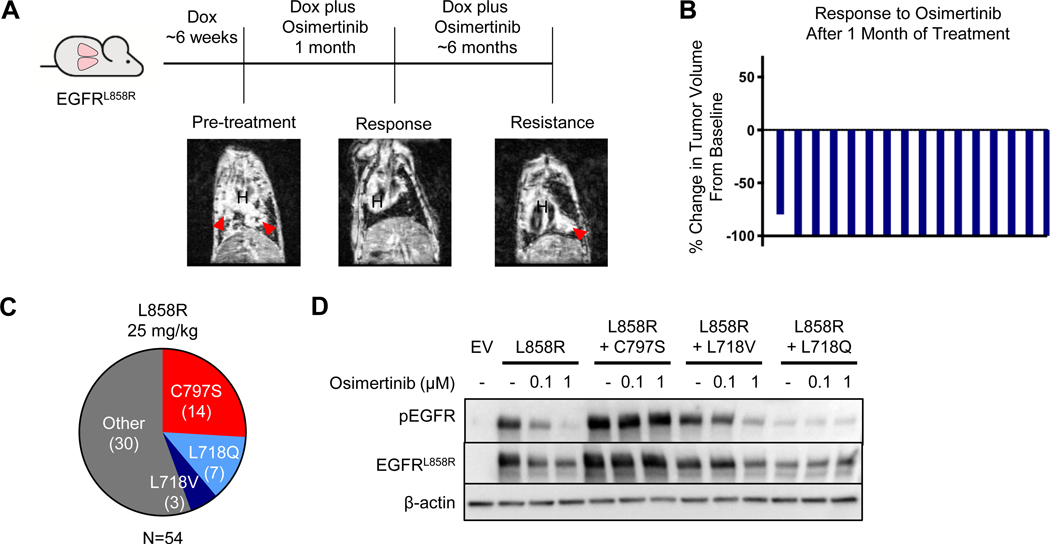
A. Schema of the experiment. CCSP-rtTA;TetO-EGFRL858R mice were administered doxycycline for the duration of the experiment and developed tumors after ~6 weeks on doxycycline. When tumors were detected by MRI (see pre-treatment image), osimertinib treatment was initiated (QD M-F) which elicited a response (see representative response MRI) and treated until the emergence of resistant tumors by MRI. Coronal MR images are shown, in which ‘H’ indicates heart and red arrows indicate tumor. The osimertinib-resistant tumors were then collected and analyzed to determine the resistance mechanisms present. B. Waterfall plot showing tumor volume changes after 1 month of osimertinib treatment in individual CCSP-rtTA;TetO-EGFRL858R mice, normalized to baseline tumor. C. Pie-charts illustrating the resistance mechanisms found in osimertinib-resistant tumors treated with 25 mg/kg of osimertinib. D. Western blot analysis of 293T cells transiently transfected with pcDNA3.1(–) plasmids encoding the indicated EGFR alleles, treated for 1 hour with varying concentrations of osimertinib. Blots are representative of n=2 biological replicates. EV, empty vector.
In the 5➔25 mg/kg cohort, the first osimertinib-resistant tumor, defined by confirmed growth in subsequent MRIs and a size of at least 30 mm3, emerged after 4.4 months of treatment (average 5.3 months). Twelve of the 14 mice (86%) developed one or more resistant tumors. To ensure availability of sufficient material for analysis, resistant tumors were allowed to grow to 200–300 mm3 before mice were sacrificed and tumors collected. To establish the mechanism of resistance to osimertinib in the tumors, we used targeted Sanger sequencing of cDNA extracted from the tumors to identify secondary mutations in the EGFR transgene. A mutation encoding the C797S substitution was seen in 5 of 17 (29.4%) osimertinib-resistant tumors (Supplementary Figure S1). An additional 5 tumors (29.4%) harbored L718V mutations, and the remaining 7 (41.2%) had no acquired EGFR mutation. None of the tumors had the T790M mutation commonly seen at resistance to 1st generation TKIs, consistent with clinical observations (20, 21), and all tumors retained the activating L858R mutation. Sequencing of cDNA from a subset of these tumors confirmed that the resistance mutations emerged in cis with L858R (Supplementary Table S1). All resistance mechanisms found were mutually exclusive within each resistant tumor and distinct resistance mechanisms were uncovered in different tumors from within the same mouse.
In the cohort of mice treated with 25 mg/kg osimertinib QD only, 26 (93%) developed osimertinib-resistant tumors – on average after 7.8 months. Of the 54 tumors collected, there were 14 (25.9%) C797S, 7 (13%) L718Q, and 3 (5.6%) L718V mutant tumors, whereas the remaining 30 (55.6%) had no secondary EGFR mutations (Figure 1C). Therefore, the two dosing regimens resulted in different frequencies of L718V-mutated tumors (p=0.0162). Histological analysis of the resistant tumors confirmed that all tumors were lung adenocarcinomas consistent with treatment-naïve tumors from this model. No resistant tumors exhibited cellular state or mesenchymal changes.
To examine the effects of these secondary mutations on EGFR phosphorylation in Western blots, we engineered the C797S, L718V and L718Q mutations individually into EGFRL858R cDNAs and transiently transfected the resulting plasmids into 293T cells. Whereas osimertinib efficiently suppresses EGFR phosphorylation in EGFRL858R mutant cells, it fails to block phosphorylation of the EGFRL858R+C797S or EGFRL858R+L718Q variants even at 1μM (Figure 1D). The EGFRL858R+L718V variant exhibits an intermediate phenotype. These data suggest that, although all three mutations confer osimertinib resistance, the L718V mutation does so less effectively than the C797S and L718Q mutations. This is consistent with L718V mutations arising more frequently in tumors that originally received lower osimertinib doses in the mice. Interestingly, EGFRL858R+L718Q mutant cells also exhibit reduced levels of EGFR protein and of basal EGFR phosphorylation, suggesting reduced stability and/or kinase activity of this variant.
Until recently, osimertinib was predominantly used as a second-line EGFR-targeted therapy for T790M-positive tumors. To compare osimertinib resistance in T790M-negative tumors to that observed in T790M-positive tumors directly, we utilized the CCSP-rtTA;TetO-EGFRL858R+T790M GEMM (29) and treated mice continuously with osimertinib (Figure 2A). Twenty out of the 49 treated mice (41%) developed osimertinib-resistant tumors, on average after 8.0 months. Of the 27 tumors collected, we found 12 (44.4%) C797S, 1 (3.7%) C797G, 2 (7.4%) L718Q, 1 (3.7%) L718V mutant tumor with concomitant loss of T790M, 1 (3.7%) L792H, and 10 tumors (37.0%) with no acquired EGFR mutation (Figure 2B). Interestingly, the C797G and L792H mutations were never observed in the EGFRL858R model, only in the context of T790M. These data suggest that C797X mutations may be more frequent in the EGFRL858R+T790M than EGFRL858R setting, although the difference is not significant (p=0.0787) due to low numbers in the EGFRL858R+T790M model.
The reversion to wild-type T790 seen with gain of L718V in one tumor (Supplementary Figure S2) was surprising and mirrors two clinical case reports (9, 33). We subcloned the cDNA extracted from this tumor and confirmed that loss of T790M had occurred on the same allele as the L718V mutation. Since the T790M mutation is in the EGFR transgene in the mice, this finding suggests that it has reverted back to wild-type. To confirm this hypothesis, we performed targeted deep sequencing of osimertinib-resistant tumors and untreated control tumors. We did not detect wild-type T790 alleles in any of 8 untreated control tumors. Further, all other osimertinib-resistant tumors retained T790M, indicating that the loss of T790M was unique to the L718V mutant tumor.
Motivated by previous findings that the T790M mutation confers a growth disadvantage (34), we next investigated whether the osimertinib resistance mutations alter the growth dynamics of EGFR mutant tumors. We estimated the growth rates by applying a linear mixed effects model to our MRI tumor volume measurements. The growth rates are estimated as the relative change in number of tumor cells per day (see Supplementary Methods). We found that tumors in the EGFRL858R+T790M model do indeed exhibit a slower growth rate than tumors in the EGFRL858R model (p=0.007; Figure 2C). Within the EGFRL858R model, L718V mutant tumors grew slower than both the C797S and L718Q mutant tumors (both p<0.0001). Within the EGFRL858R+T790M model, L718Q mutant tumors exhibited faster growth than C797S mutant tumors (p=0.0205; Figure 2D). In addition, C797S mutant tumors grew faster in the EGFRL858R than EGFRL858R+T790M model (p=0.0002). These results indicate that the resistance mutations can indeed differentially alter the growth dynamics of resistant tumors, which would impact the spectrum of resistance mutations that emerges.
L718V/Q and C797S mutations are observed at different frequencies in different allelic contexts in patient cfDNA
To explore the prevalence of mutations that confer osimertinib resistance in patients, we investigated a large cohort of EGFR-mutated cases – 1117 L858R and 1123 E746_A750 (“Del19”) – from the Guardant Health de-identified database of patients with advanced non-small cell lung cancer (NSCLC) undergoing routine clinical genomic testing by plasma next generation sequencing (31). Within each driver mutation group, we examined the frequency of C797S, L718V, L718Q and any combination of these specific mutations (Figure 2E) – without considering the treatment for patients, since this was not available for all cases. Collectively, these mutations were observed in 3.9% of L858R and 5.7% of Del19 tumors (p=0.0606). The C797S mutation was present in 25 L858R (2.2%) and 61 Del19 cases (5.4%), whereas L718V was found in 9 L858R (0.8%) and L718Q was observed in 8 L858R cases (0.7%). We found that while the C797S mutation was observed more frequently in Del19 than L858R mutant tumors (p<0.0001), L718V and L718Q were more frequent in L858R cases (L718V, p=0.0019; L718Q, p=0.0038). In fact, the L718Q mutation was only observed with a Del19 mutation when C797S was also present (n=3), and the L718V mutation was never observed with a Del19 mutation.
We further separated the data into T790M-positive and -negative subsets. The C797S, L718V and L718Q mutations occurred more frequently with T790M than without it (Figure 2F and Supplementary Figure S3). This could potentially be due to the fact that a higher proportion of patients who have received osimertinib to date are within the T790M-positive subset and might be represented in the database. However, within the L858R subset, C797S mutations occurred more frequently together with T790M (28 times more T790M-positive than –negative) than L718V (1.9 times) or L718Q (1.4 times) mutations (L718V, p=0.0074; L718Q, p=0.0041). The ratio of C797S T790M-positive to –negative cases was similar in the Del19 subset (Supplementary Figure S3). These data further indicate that the relative frequency of L718Q/V mutations compared to C797S may be higher in T790M-negative compared to T790M-positive tumors.
CRISPR-edited human LUAD cell lines harboring L718Q/V mutations are selected for in the presence of osimertinib
To test whether the L718Q and L718V substitutions in EGFR confer resistance to osimertinib in human lung cancer cells, we utilized CRISPR/Cas9 technology to model the presence of subclonal cell populations containing these mutations in an endogenous gene context. We designed a specific CRISPR RNA targeting EGFR proximal to L718 as well as donor single-strand DNA (ssDNA) containing either the L718Q or L718V mutations (see Supplementary Methods and Supplementary Table S2). Three human LUAD cell lines, PC9 (EGFRDel19), PC9-VanR (EGFRDel19+T790M; vandetinib-resistant), and II-18 (EGFRL858R), were transfected with ribonucleoprotein complexes and the specific ssDNA and seeded into two wells (Figure 3A). Three days after transfection, cells from one well were collected as the ‘pre-osimertinib’ sample. In parallel, the other well was treated with osimertinib for 3–4 weeks then collected (‘post-osimertinib’). This timepoint was chosen to ensure that drug-resistant growth would be due to the specific CRISPR-edited mutations, and not to other resistance mechanisms that might emerge in the cells under long-term osimertinib treatment.
Figure 3. EGFR L718Q and L718V mutations confer resistance to osimertinib in human lung cancer cells.
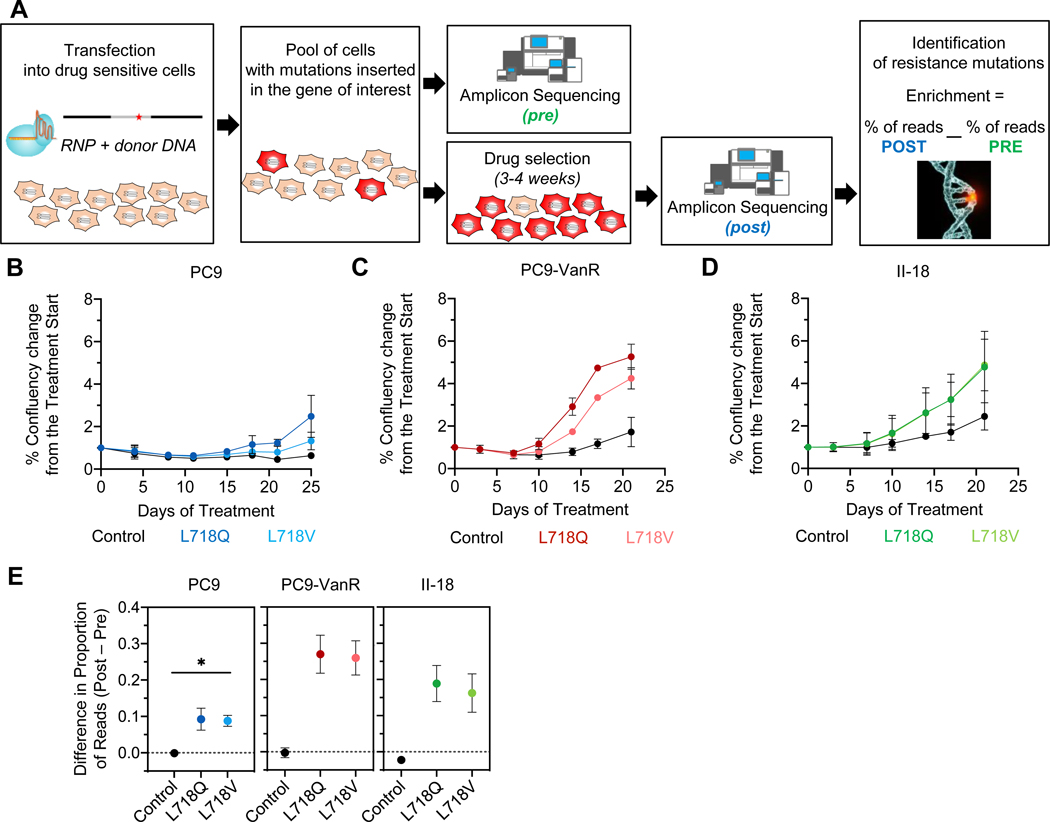
A. Schematic workflow of the CRISPR experiment (see Supplementary Methods). B-D. Cell growth of CRISPR-edited-PC9 (blue), PC9-VanR (red), and II-18 (green, lines superimposed) cells during osimertinib treatment as measured by Cell Metric. E. Histograms showing the difference in the proportion of reads for the indicated codon in the corresponding sample before (pre) and after osimertinib selection (post). As a reference, the mean of the top 5 indels found in the control sample is shown. The dashed line is the background threshold. Error bars show SEM for three biological replicates for PC9 and two for PC9-VanR and II-18 for B-D and E. Two-sided t test, * p<0.05.
PC9, PC9-VanR, and II-18 populations that contained cells with the L718Q or L718V mutations showed more proliferation in the presence of osimertinib than control cells (Figures 3B-D and Supplementary Figure S4). Amplicon-sequencing analysis confirmed that the L718Q/V mutations were enriched in all cell lines (Figure 3E and Supplementary Figure S5A). The T790M mutation was absent from PC9-VanR cells that grew out with the L718Q/V mutations (Supplementary Figure S5B) – resembling the GEMM tumor and patient case described above that both gained L718V and lost T790M, further suggesting that L718 mutations may be more likely to confer resistance in the absence of T790M.
Erlotinib is effective in cells with C797S but not L718Q/V mutations, whereas afatinib is effective in both
Having developed models of resistance to first-line osimertinib treatment, we next sought to investigate therapeutic strategies to treat these tumors. Previous studies have suggested that a 1st generation TKI is effective against lung cancer cells harboring C797S (22, 24, 25, 27). In transiently-transfected cells, erlotinib inhibited pEGFR in both EGFRL858R+C797S and EGFRL858R+L718V mutated variants but did not decrease phosphorylation in the EGFRL858R+L718V mutant to the same extent as in EGFRL858R cells (Figure 4A and Supplementary Figure S6A). The EGFRL858R+L718Q mutant was not inhibited substantially by erlotinib, although the low basal level of pEGFR makes it difficult to interpret any differences (Figure 4A and Supplementary Figure S6A). Conversely, afatinib caused complete inhibition of EGFRL858R+L718V/Q phosphorylation (Figure 4B and Supplementary Figure S6B). Afatinib also inhibited the EGFRL858R+C797S variant, but required 10-fold more drug for complete inhibition of C797S than L718Q/V mutant cells – consistent with previous data (23). Most importantly, complete inhibition of the EGFRL858R+L718V/Q variants by afatinib was achieved at concentrations ~10-fold lower than those needed to inhibit EGFRT790M (Figure 4C and 4D). This is highly significant since it is well-established that, although afatinib can suppress EGFRL858R+T790M activity in vitro, it cannot do this at clinically achievable doses. We extended these studies to the Del19 setting and saw similar changes in EGFR phosphorylation (Figure 4 E-H). These data are consistent with a report from Nishino et al., in which they perform cell inhibition assays with Ba/F3 cells stably expressing either L858R or Del19 with or without the osimertinib-resistance mutations. They demonstrated that L718Q and L718V confer an >70-fold increase in the IC50 for osimertinib compared with the L858R parental cells. They also showed that the IC50 for erlotinib on L718Q and L718V mutant cells was >20-fold that for the parental cells. In contrast, cells harboring the L718Q and L718V mutations had only slightly different IC50s for afatinib (5.5-fold and 1.1-fold, respectively) (27).
Figure 4. Afatinib suppresses phosphorylation of EGFR mutants containing the L718V/Q mutations.
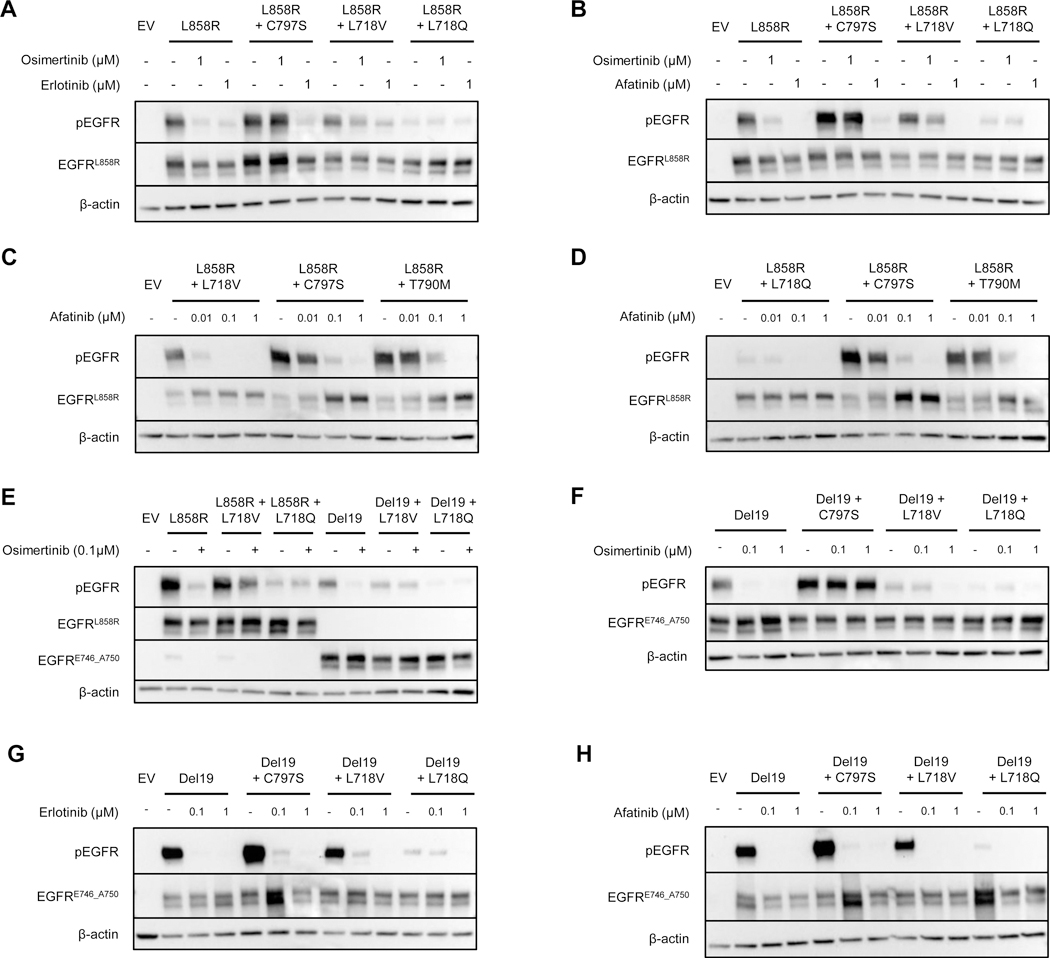
Western blots of 293T cells transiently transfected with pcDNA3.1(–) containing EGFR with the indicated mutations, treated for one hour with varying concentrations of the indicated TKIs. A and B show a direct comparison of EGFRL858R, EGFRL858R+C797S, EGFRL858R+L718V and EGFRL858R+L718Q treated with 100 nM osimertinib, erlotinib, or afatinib. C and D show dose-dependent changes in EGFR phosphorylation of EGFRL858R+L718V and EGFRL858R+L718Q compared with EGFRL858R+C797S and EGFRL858R+T790M treated with increasing doses of afatinib. E. Comparison of the effect of 100 nM osimertinib on L858R and exon 19 deletion mutants. Sensitivity of the exon 19 deletion mutants with or without the indicated mutations to varying concentrations of osimertinib (F), erlotinib (G) or afatinib (H). All blots are representative of n=2 biological replicates. EV, empty vector.
To determine whether the observed effects of the TKIs on the different osimertinib-resistant mutations in vitro correlated with in vivo drug sensitivity, we treated mice with EGFRL858R osimertinib-resistant tumors with erlotinib or afatinib for 3 weeks (Figure 5A). Tumors were considered ‘resistant’ when their volume had increased by ≥20% from the point at which the TKI treatment was switched, and ‘sensitive’ when their volume had decreased by ≥30%. We followed 24 osimertinib-resistant tumors that were switched to erlotinib and found that 7 (29.2%) responded to the TKI. Five of these harbored the C797S mutation (Figure 5B and Supplementary Figure S7A), whereas no acquired EGFR mutation could be identified in the other two. By contrast, 16 tumors (66.7%) became resistant and one (4.2%) remained stable. Six of the erlotinib-resistant tumors harbored mutations at L718 (3 L718V and 3 L718Q). While L718Q mutant tumors never decreased in size following erlotinib treatment and grew rapidly, tumors with the L718V mutation exhibited a transient response to erlotinib before becoming truly resistant (Figure 5B and Supplementary Figure S7A). No acquired EGFR mutations could be detected in the remaining 10 resistant tumors or the 1 stable tumor.
Figure 5. The sensitivity of osimertinib-resistant tumors to erlotinib or afatinib treatment depends on the specific osimertinib resistance mutation present.
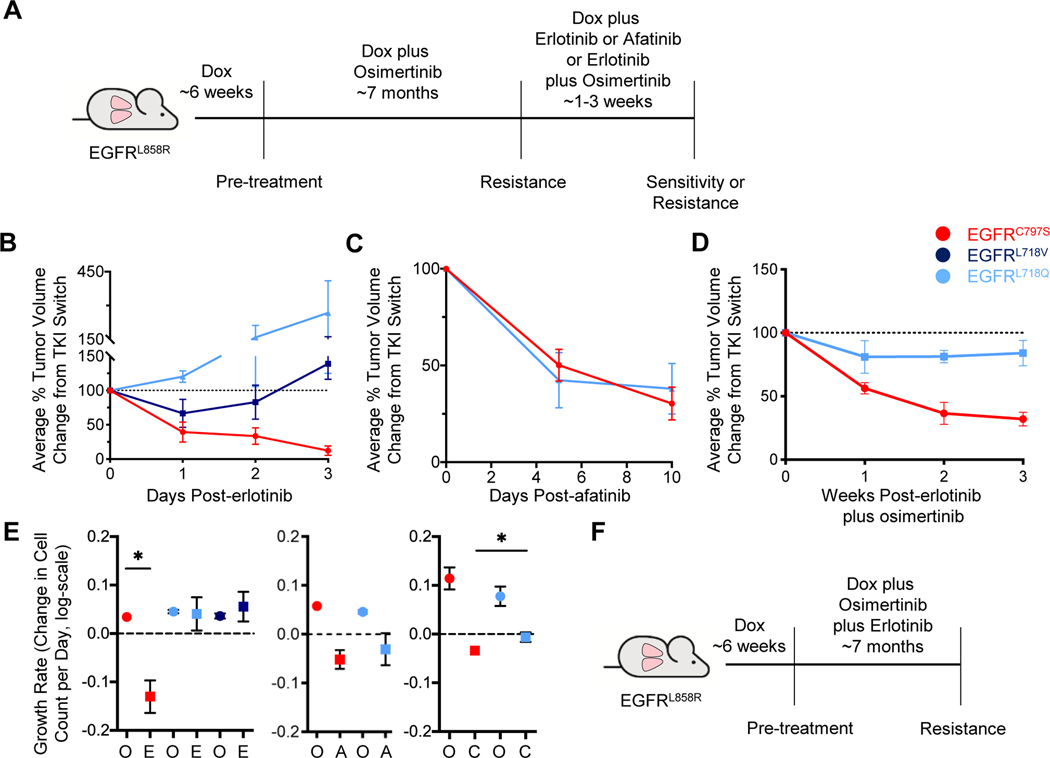
A. Schema of the experiment. CCSP-rtTA;TetO-EGFRL858R mice were treated with 25 mg/kg osimertinib until the emergence of resistant tumors, as in Figure 1. Mice were then switched to either erlotinib, afatinib, or the combination of erlotinib plus osimertinib for 1–3 weeks. B, C, and D. Average tumor volume changes for the osimertinib-resistant tumors switched to 25 mg/kg erlotinib for 3 weeks (B), 25 mg/kg afatinib for 10 days (C), and erlotinib plus osimertinib (D, 25 mg/kg each) as determined by MRI. Tumor volume is normalized to the point of TKI switch. Error bars represent SEM. For B, curves are the average of n=11 total tumors (C797S n=5; L718V n=3; L718Q n=3). For C, n=12 total tumors (C797S n=7; L718Q n=5). For D, n=10 total tumors (C797S n=7; L718Q n=3). E. Graphs of tumor growth rates for the osimertinib-resistant tumors before and after they were switched to erlotinib (left), afatinib (center), or erlotinib plus osimertinib (right). Growth rates are depicted as the relative change in cell count per day (log-scale). F. Schema of the experiment. TKI-naïve CCSP-rtTA;TetO-EGFRL858R mice were treated with the combination of osimertinib plus erlotinib (25 mg/kg each) until the emergence of resistant tumors. Error bars represent SEM. (* indicates p<0.05). O, osimertinib; E, erlotinib; A, afatinib; C, erlotinib plus osimertinib.
We next assessed the effect of afatinib treatment on 24 osimertinib-resistant EGFRL858R tumors (Figure 5A). Fourteen tumors (58.3%) regressed after this switch, whereas 7 (29.2%) grew by ≥20% on afatinib within 1–3 weeks of treatment, and 3 (12.5%) remained stable. The disease control rate (sensitive and stabilized tumors) on afatinib (70.8%) was greater than on erlotinib (33.3%, p=0.0199). Mice were treated with afatinib for 10 days because in the first set of mice, 2 responding tumors responded completely, leaving no tumor cells for analysis of the resistance mechanisms (Supplementary Figure S7B). Of the 12 tumors responding to afatinib that could be sequenced, all but one had secondary mutations in EGFR: 7 C797S and 4 L718Q. An additional tumor with stable disease also harbored the L718Q mutation (Figure 5C and Supplementary Figure S7C). This demonstrates in vivo that tumors with these mutations respond to a 2nd generation EGFR TKI. It is important to note that the dose of afatinib used in these experiments is not effective against tumors with the T790M mutation in vivo (35).
We treated a cohort of 7 osimertinib-resistant EGFRL858R tumors with 7.5 mg/kg QD afatinib and monitored their growth for 3 weeks. One tumor (14%) responded, 1 (14%) exhibited stable disease, and 5 (71%) were resistant. Three of these tumors harbored the C797S mutations and exhibited mixed responses, whereas 2 tumors harboring the L718Q mutation were resistant to this dose of afatinib (Supplementary Figure S7D and E). Two additional resistant tumors did not have a secondary mutation in EGFR. This result demonstrates that the response to afatinib of C797S and L718Q mutant tumors is dose-dependent.
We also treated a set of osimertinib-resistant EGFRL858R tumors with the combination of 25 mg/kg erlotinib plus 25 mg/kg osimertinib to assess whether using these TKIs together could overcome resistance to first-line osimertinib. Intriguingly, this strategy has been shown to be effective on cells harboring the L718Q mutation (23), despite the mutation conferring resistance to each TKI individually. Eight out of 16 tumors (50%) responded, 2 (13%) exhibited stable disease, and 6 (38%) were resistant. Seven sensitive tumors harbored the C797S mutation (Figure 5D and Supplementary Figure S7F). The remaining responding tumor – as well as the 2 tumors with stable disease – harbored L718Q mutations (Figure 5D and Supplementary Figure S7F). None of the 6 resistant tumors had secondary EGFR mutations.
We compared the growth rates of tumors with specific mutations before and after they were switched to erlotinib, afatinib, or the combination of erlotinib plus osimertinib (Figure 5E). The positive or negative growth rates correlated with resistance or sensitivity to the TKI, respectively. We observed that only the C797S mutant tumors exhibited a negative growth rate on erlotinib treatment compared with positive growth on osimertinib (p=0.0115, Figure 5E, left). Both the C797S and L718Q mutant tumors exhibited negative growth rates on afatinib treatment (Figure 5E, center). Both C797S and L718Q mutant tumors exhibited negative growth rates under the combination of erlotinib plus osimertinib, though this was lower for C797S (p=0.0392; Figure 5E, right). These data suggest that L718Q mutant tumors may respond (or at least stabilize) when treated with the combination of 1st and 3rd generation EGFR TKIs.
Combination first-line erlotinib plus osimertinib prevents secondary mutations in EGFR
Given that L718V/Q mutations are not observed at resistance to first-line erlotinib, and the combination of erlotinib and osimertinib was effective against L718Q mutant tumors, we wondered whether this combination as initial therapy could prevent the emergence of L718 mutations. To test this hypothesis, we treated 17 tumor-bearing CCSP-rtTA;TetO-EGFRL858R mice with a combination of osimertinib and erlotinib (25 mg/kg QD continuously M-F each; Figure 5F). Six out of 17 mice (35.3%) developed resistant tumors, on average after 6.9 months. This time to resistance is slightly shorter than it was for the EGFRL858R mice treated with 25 mg/kg osimertinib only, although the difference is not significant (6.9 versus 7.8 months, p=0.2767). Of 10 resistant tumors collected, none harbored secondary mutations in EGFR. This result suggests that the combination of 1st and 3rd generation TKIs may indeed be effective at preventing secondary EGFR mutations from arising as a resistance mechanism, although it did not necessarily delay the emergence of resistance overall. Ten mice (58.8%) were sacrificed before the end of the study without developing resistant tumors due to declining health, possibly due to kidney toxicity, as morphologic abnormalities were found in the kidneys including fibrosis and some atrophy. Therefore, it is possible that dosing will need to be optimized when using this combination of TKIs.
Mutations in Kras confer resistance to osimertinib in vivo
In efforts to identify mechanisms of osimertinib resistance in tumors without EGFR secondary mutations we first surveyed Kras. Alterations in this gene have been implicated in resistance to osimertinib and previously found at resistance to other EGFR-directed therapies in these models (10–12, 20, 36, 37). In the EGFRL858R tumors with acquired resistance to first-line osimertinib described in Figure 1C, 28 of 54 tumors (51.9%) resistant to 25 mg/kg osimertinib harbored Kras mutations at codon 12, 13 or 61, as did 4 of 17 (23.5%) tumors resistant to the 5➔25 mg/kg osimertinib regimen and 7 out of 27 (25.9%) osimertinib-resistant EGFRL858R+T790M tumors (Supplementary Figure S8). Finally, 9 of 10 tumors (90%) resistant to combination first-line erlotinib plus osimertinib harbored mutations in Kras (Supplementary Figure S8). The Kras mutations (Supplementary Table S3) were all mutually exclusive with acquired EGFR mutations, yet the engineered EGFR L858R or L858R+T790M mutations were always maintained in the Kras mutant tumors.
We also did not observe any significant amplification of Met – an established EGFR TKI resistance mechanism (1–3, 10, 11, 13, 20, 21) – in the tumors that did not acquire either an EGFR or Kras mutation (Supplementary Figure S9). Therefore, acquired EGFR and Kras mutations account for almost all cases of osimertinib-resistant tumors in these GEMMs (Supplementary Figure S8).
Benefit from afatinib treatment in a patient with an L858R mutant tumor harboring L718Q and L718V mutations
To investigate the potential benefit of 2nd generation EGFR TKIs in overcoming resistance due to L718 mutations in patients, we studied a patient who had developed resistance to first-line osimertinib and was subsequently treated with afatinib. A 62-year-old woman with a 4 pack-year smoking history presented with back and flank pain. CT and MRI scans showed a 3.7 cm mass in the right upper lobe of the lung, multiple sub-centimeter lung nodules, 3 liver lesions, an adrenal lesion, multiple lesions in the thoracic spine, a compression fracture at T9 with evidence of spinal cord compression, and punctate lesions in the left parietal lobe and cerebellum. Biopsy of a liver lesion demonstrated adenocarcinoma positive for TTF-1 and Napsin-A by immunohistochemistry. An EGFRL858R mutation was detected in the tumor with targeted NGS using the Illumina MiSeqDx platform (Figure 6A, Supplementary Table S4). The patient underwent a T9 corpectomy and spine fusion followed by consolidative radiation after which she was started on 80 mg osimertinib QD. She had an excellent response, with shrinkage of all sites of disease and resolution of brain metastases. Imaging performed every ~2 months showed a continued response, until 8.5 months after initiation of osimertinib when the patient was found to have growth of a liver lesion. A repeat biopsy was performed on the growing lesion and showed adenocarcinoma consistent with the original pathology. This tissue was sent for NGS using Foundation One CDx testing. The original EGFRL858R mutation was detected (allelic frequency of 84%), along with EGFRL718Q (73%) and EGFRL718V (3%) mutations. The patient underwent thermal ablation to the enlarging liver lesion, however 2 months later she was found to have progression in the liver and lungs. At that time, she was started on 40 mg afatinib QD given the acquired L718V and L718Q mutations. Imaging performed ~2 months later showed shrinkage in a previously-growing lung lesion (Figure 6B) – although this site may not necessarily harbor the L718Q/V mutations – and stable disease at all other sites. RECIST1.1 measurements confirmed that afatinib led to a partial response (Supplementary Table S5).
Figure 6. Benefit from afatinib in a patient who developed resistance to first-line osimertinib.
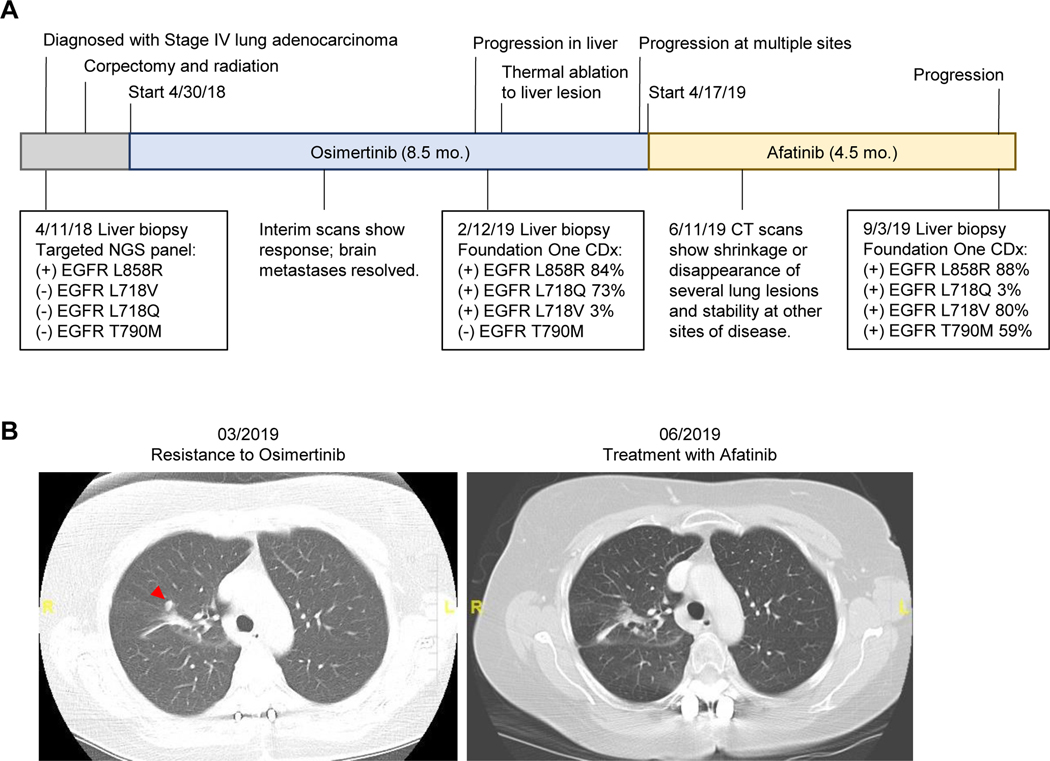
A. Timeline of the patient’s treatment history and clinical testing results. Osimertinib was given at 80 mg QD and afatinib was given at 40 mg QD. Timeline not to scale. B. CT scans of a lung lesion (indicated with red arrow) that was new prior to starting afatinib, and then shrunk after treatment with afatinib.
After ~4.5 months on afatinib treatment, the patient developed disease progression. Another biopsy was performed on a progressing liver lesion and the tissue was sent for NGS using Foundation One CDx testing. This biopsy revealed the emergence of the EGFRT790M mutation (allelic frequency of 59%). Interestingly, this was accompanied by an increase in the allelic frequency of the L718V mutation (80%), and a decrease in the frequency of the L718Q mutation (3%). This report suggests that while L718Q/V mutant tumors could benefit from transient responses to afatinib treatment, they can acquire the T790M mutation and eventually become resistant to all available EGFR TKIs.
DISCUSSION
The mean progression-free survival for osimertinib in the first-line is 19 months, with an overall survival of 39 months, which is superior to that observed with other TKIs (18, 19). Patients are not cured, however, and a better understanding of acquired resistance to osimertinib is needed. Here, using GEMMs of EGFR mutant LUAD, we show that resistance to first-line osimertinib can be due to acquired C797S, L718Q and L718V mutations in EGFR. This is the first report of an in vivo first-line osimertinib resistance model that acquires EGFR mutations. Data emerging from the FLAURA trial of front-line osimertinib indicate that, at acquired resistance, the C797S and L718Q mutations are detected in 7% and 2% of cases from ctDNA (21). The higher frequency of C797S mutations versus L718Q mutations observed in these patient samples (with a variety of driver mutations) compared with the more equal distribution of C797S and L718Q mutations in our mouse model may reflect the allele specificity of the resistance mutations. Indeed, our analysis of cfDNA from patients with EGFR mutant lung cancer clearly show that C797S mutations predominate in the Del19 subset of tumors, and that L718Q and L718V mutations generally only arise in the context of L858R, consistent with the few published reports of L718Q/V mutations (8–10, 15, 16, 33). Why L718Q/V mutations are found more readily in the context of L858R mutations in patients remains to be determined. It is possible that it is due to differential stability of the different mutation combinations or differences in other biochemical properties such as their kinase activity or dimerization properties. Interestingly, in vitro data in our study and others’ (38) show that Del19 cells harboring a L718Q/V mutation can exist and are resistant to osimertinib suggesting the intriguing possibility that resistance mechanisms that emerge spontaneously in vivo may be somewhat different than those observed in vitro.
The mechanism by which the C797S substitution causes osimertinib resistance is straightforward as it removes the side-chain thiol with which osimertinib reacts covalently. Models obtained using molecular dynamics-based energy minimization provide some insight into how L718V/Q mutations might affect drug-binding. The L718 side-chain contacts the phenyl ring of the drug in a crystal structure of osimertinib-bound wild-type EGFR TKD (PDB ID 4ZAU) (39). Substituting L718 with V or Q is therefore highly likely to alter the mode or orientation of osimertinib binding, as others have noted (8, 17, 23). We confirmed this by modeling non-covalent complexes of L858R-mutated EGFR TKD with osimertinib, erlotinib, and afatinib (Figure 7). While the pose of osimertinib in the energy-minimized L858R TKD complex (with leucine at position 718) is maintained, loss of van der Waals interactions is seen for the L718V mutation, and a steric clash with the glutamine side-chain is seen for the L718Q mutation (upper panels, Figure 7). These effects are compensated for in the models by reorientation of osimertinib in the drug-binding site, which may make its orientation sub-optimal for covalent reaction with C797 – as argued previously for L718Q/T790M EGFR (17). There is also some reduction in osimertinib binding energy suggesting that the affinity of the ‘encounter complex’ prior to covalent reaction may be reduced – which would also reduce efficacy. The same mutations are predicted to have similar effects on erlotinib binding energy and orientation (middle panel, Figure 7). In this case, the lack of any covalent aspect suggests that observed resistance must arise from weakened binding. Indeed, L718 contacts erlotinib directly in EGFR/erlotinib crystal structures (40). Finally, the effects of the L718V/Q mutations on afatinib binding energy are only ~80% of those calculated for osimertinib, suggesting that they will have less severe effects on residence time of afatinib for its covalent reaction. Moreover, the orientation of afatinib appears less affected by the mutations – in Figure 7, there is no clash between afatinib in its L858R-bound orientation and the Q718 side-chain, although interactions with L718 are lost in the L718V variant.
Figure 7. Possible structural consequences of L718V/Q-mutations for EGFR binding to TKIs.
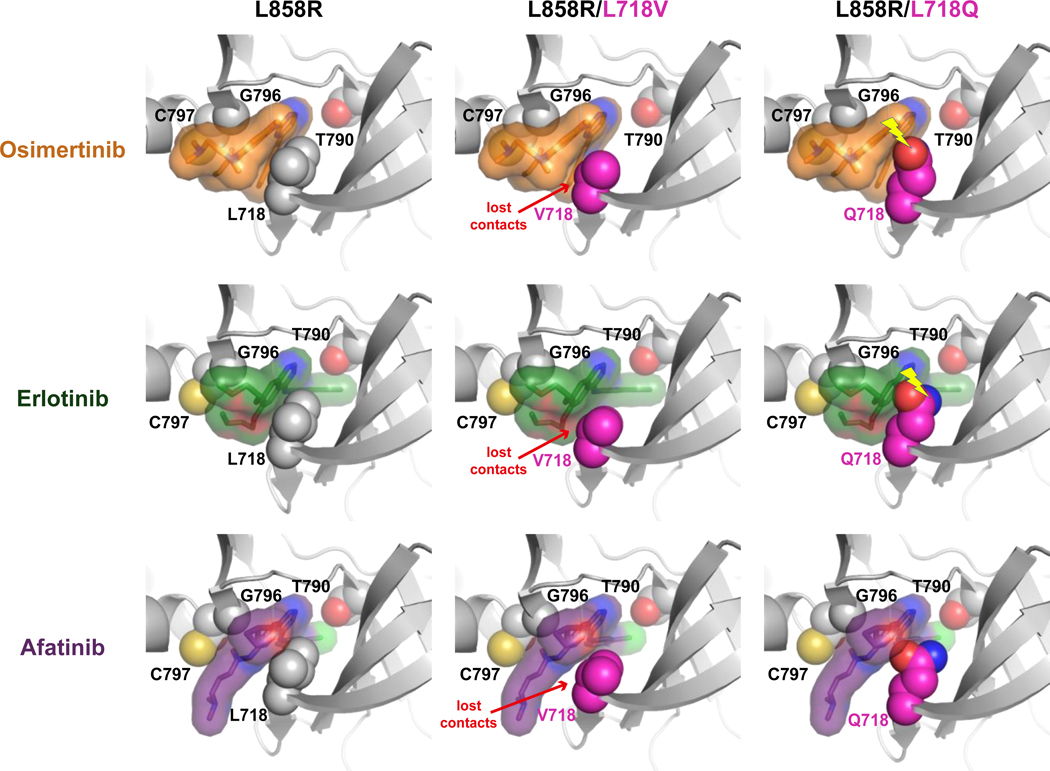
Structural models for the L858R-mutated EGFR kinase domain bound to osimertinib, erlotinib, or afatinib are shown, with (or without) the incorporation of L718V or L718Q mutations as described in the Supplementary Methods. In each case, the bound drug molecule of the energy minimized model has been replaced with that bound to the L858R TKD lacking an L718 mutation in order to show how drug binding is impacted. Interactions with drug are lost in all L718V variants (and drug is reoriented slightly to compensate). Clashes between the Q718 side-chain and drug (denoted by yellow lightning bolt) are seen for the osimertinib and erlotinib complexes (with resulting drug reorientation), but not for afatinib.
Through experiments in CRISPR-edited human EGFR mutant LUAD cell lines – to mimic the acquisition of resistance when low frequency sub-clonal mutations are present – we show that sub-clonal L718V/Q mutations confer resistance to osimertinib. However, the L718V mutation retained some sensitivity to osimertinib at higher doses, and its frequency was reduced in vivo with higher osimertinib doses. The only three reports of patient tumor biopsies revealing an L718V mutation also coincided with loss of T790M (9, 10, 33), which was replicated in one of our mice and the CRISPR-edited PC9-VanR cells that gained L718V/Q mutations. It is possible that destabilization of EGFR by L718 mutations prevents them from occurring in the presence of T790M.
With osimertinib now as the preferred first-line therapy for advanced EGFR mutant tumors, it is critical to develop strategies to overcome resistance. Our findings confirm the erlotinib-sensitivity of C797S mutant tumors in vivo, suggested by several in vitro studies and case reports (22, 24, 25), but L718V/Q mutant tumors were not sensitive to erlotinib. Afatinib caused tumor regression in all tumors harboring C797S or L718Q mutations, as seen in previous in vitro studies (9, 23). These data are consistent with our report of a patient who developed resistance to first-line osimertinib through the acquisition of both L718Q and L718V mutations in the tumor and whose disease was stabilized by afatinib treatment, similarly to two recent second-line osimertinib case reports with L858R+L718Q (15) and L858R+L718V mutant tumors (33). Afatinib is currently FDA-approved for the treatment of tumors harboring G719X mutations based on its clinical efficacy (41), suggesting a similar mechanism could underlie the sensitivity of L718 and G719 mutants to afatinib.
It is unclear why L718 mutations have never been observed in patient tumors resistant to 1st generation EGFR TKIs, given that they confer resistance to these inhibitors in vitro and in vivo based on our work and others’ (9, 23). It is possible that this could in part be due to the specific sensitivity of different alleles depending on the baseline mutation present. Moreover, whether a mutation emerges may depend on several factors including the ease with which a mutation occurs (i.e. base change requirements), the strength of TKI-binding and the extent of the selective pressure exerted by the TKI. Thus, a combination of EGFR TKIs may be able to prevent secondary EGFR mutations from emerging or delay the emergence of resistance. We tested the approach of combining a 1st and 3rd generation TKI in vivo and, importantly, none of the ten resistant tumors that developed had acquired mutations in EGFR. However, resistance did emerge. Therefore, it is possible that by effectively suppressing EGFR-dependent mechanisms of resistance with the combination therapy, EGFR-independent mechanisms will become more prominent. How this will affect clinical outcomes for patients remains to be determined. Indeed, clinical studies to evaluate the safety and efficacy of combination EGFR TKIs are ongoing (e.g. NCT03122717). Given that we observed toxicities of the drug combination in vivo, future experiments with optimized doses of the agents will be important to conclusively determine whether this approach can be used to prevent EGFR-dependent resistance mechanisms. This also highlights the importance of dose optimization to minimize toxicities and maximize efficacy in clinical trials. Nevertheless, our data present an encouraging new therapeutic approach to treat EGFR mutant tumors upfront when tumors are most likely to be maximally addicted to mutant EGFR signaling.
Not all EGFR TKI-resistant tumors acquire secondary mutations in EGFR, and we identified Kras mutations in osimertinib-resistant tumors. This is not surprising given that we previously observed these mutations arising in TKI-naïve tumors and at resistance to other EGFR inhibitors in these models (36, 37, 42), and activation of the MAPK/ERK pathway has been described as a mechanism of resistance to 3rd generation EGFR TKIs (43, 44). Whether the relatively high abundance of Kras mutations in our mouse model is due to spontaneous tumor development in aging mice (45) or whether these are enriched with osimertinib requires additional investigation. Nevertheless, KRAS mutations are certainly of clinical relevance given that they are being observed at osimertinib resistance in patients (10–12, 20, 46).
Acquired EGFR and Kras mutations account for almost all cases of osimertinib-resistant GEMM tumors described here. However, other mechanisms of resistance have been observed in patients (3, 10, 21, 47) and overall, secondary mutations in EGFR are found more frequently in our mouse models than in osimertinib-resistant human tumors. This could reflect the reduced genomic complexity of the GEMM tumors (42), and the fact that expression of the mutant EGFR transgene is constantly being driven by the presence of doxycycline, which may confer a greater propensity for on-target resistance. Therefore, other models should be used for further discovery of EGFR-independent resistance mechanisms and evaluation of therapeutic approaches to overcome them (44, 48, 49). Nonetheless, our study provides important insight into EGFR-dependent (and proximal) resistance mechanisms to osimertinib and highlights the complexity and heterogeneity of osimertinib resistance. Two key avenues for clinical investigation suggested by our findings are: i). Evaluation of sequential TKI treatment that includes osimertinib followed by a 2nd generation TKI such as afatinib or dacomitinib, and ii). Evaluation of TKI combinations in the first-line.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
This study provides insight into the biological and molecular properties of first-line osimertinib resistance EGFR mutations and evaluates therapeutic strategies to overcome resistance.
ACKNOWLEDGEMENTS
The authors would like to thank all members of the Politi Laboratory for helpful discussions and support. We also thank Emily Forrest for critical reading and insights into the manuscript. We thank Kaya Bilguvar, Christopher Castaldi, James Knight, and The Yale Center for Genome Analysis for assistance with the NGS and analysis of mouse tumors. We also thank Damla Etal, Julia Lindgren, Ben Taylor and Mike Firth for the CRISPR NGS run and analysis. We thank Fangyong Li and The Yale Center for Analytical Sciences for assistance with statistical analysis. We also thank Daniel O’Neill for assistance with data analysis.
This work was supported by Yale’s Specialized Program of Research Excellence in Lung Cancer grant (to K. Politi, S.B. Goldberg and M.A. Lemmon, P50 CA196530) and funding from AstraZeneca (to K. Politi). Additional support came from the NIH/NCI-funded Yale Cancer Biology Training Program T32 CA193200-01A1 and F31 CA228268-01A1 (to J.H. Starrett), R01 CA198164 (M.A. Lemmon), the Ginny and Kenneth Grunley Fund for Lung Cancer Research, and the Canadian Institutes of Health Research Project Grant PJT-148725 (to W.W. Lockwood). W.W. Lockwood is supported by a Michael Smith Foundation for Health Research Scholar and NIHR New Investigator Awards, A. Guernet is a fellow funded by the IMED AstraZeneca postdoc program, A. Nagelberg is supported by a scholarship from the CIHR, and K.D. Ashtekar is an Arnold and Mabel Beckman Foundation Postdoctoral Fellow. Yale Cancer Center Shared Resources used in this article were in part supported by NIH/NCI Cancer Center Support Grant P30 CA016359.
AG, MEC, PS and DC are employees and shareholders at AstraZeneca PLC. K.Price is an employee with stock ownership in Guardant Health, Inc. K.Politi is co-inventor on a patent licensed to Molecular MD for EGFR T790M mutation testing (through MSKCC). K.Politi has received Honoraria/Consulting fees from Takeda, NCCN, Novartis, Merck, AstraZeneca, Tocagen, Maverick Therapeutics and Dynamo Therapeutics and research support from AstraZeneca, Kolltan, Roche and Symphogen. SMK has received research support from Roche, AstraZeneca and Tempest Therapeutics. SMK is on the scientific advisory board of Celsius Therapeutics and a co-founder of Jetix. SBG has received research support from AstraZeneca and honoraria/consulting feeds from AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Genentech, Eli Lilly, Amgen and Spectrum.
Footnotes
Conflict of Interest Statement
JHS, DA, MG, HK, TFS, AK, RH, FM, K.Poels, IKvR, MAL, AN, DF, KDA, WWL and AU do not declare any conflicts of interest.
REFERENCES
- 1.Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11(8):473–81. [DOI] [PubMed] [Google Scholar]
- 2.Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10(11):760–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol. 2013;31(8):1070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer discovery. 2014;4(9):1046–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372(18):1689–99. [DOI] [PubMed] [Google Scholar]
- 6.Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21(6):560–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ou SI, Cui J, Schrock AB, Goldberg ME, Zhu VW, Albacker L, et al. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/R and L792F/H mutations in one EGFR (L858R/T790M) NSCLC patient who progressed on osimertinib. Lung Cancer. 2017;108:228–31. [DOI] [PubMed] [Google Scholar]
- 8.Bersanelli M, Minari R, Bordi P, Gnetti L, Bozzetti C, Squadrilli A, et al. L718Q Mutation as New Mechanism of Acquired Resistance to AZD9291 in EGFR-Mutated NSCLC. J Thorac Oncol. 2016;11(10):e121–3. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Li Y, Ou Q, Wu X, Wang X, Shao YW, et al. Acquired EGFR L718V mutation mediates resistance to osimertinib in non-small cell lung cancer but retains sensitivity to afatinib. Lung Cancer. 2018;118:1–5. [DOI] [PubMed] [Google Scholar]
- 10.Yang Z, Yang N, Ou Q, Xiang Y, Jiang T, Wu X, et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients. Clin Cancer Res. 2018;24(13):3097–107. [DOI] [PubMed] [Google Scholar]
- 11.Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018;4(11):1527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le X, Puri S, Negrao MV, Nilsson MB, Robichaux J, Boyle T, et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin Cancer Res. 2018;24(24):6195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov. 2018;8(12):1529–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown BP, Zhang YK, Westover D, Yan Y, Qiao H, Huang V, et al. On-target resistance to the mutant-selective EGFR inhibitor osimertinib can develop in an allele specific manner dependent on the original EGFR activating mutation. Clin Cancer Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J, Jin B, Su H, Qu X, Liu Y. Afatinib helped overcome subsequent resistance to osimertinib in a patient with NSCLC having leptomeningeal metastasis baring acquired EGFR L718Q mutation: a case report. BMC Cancer. 2019;19(1):702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Z, Yang J, Chen Y, Shao YW, Wang X. Acquired EGFR L718V Mutation as the Mechanism for Osimertinib Resistance in a T790 M-Negative Non-Small-Cell Lung Cancer Patient. Target Oncol 2019. [DOI] [PubMed] [Google Scholar]
- 17.Callegari D, Ranaghan KE, Woods CJ, Minari R, Tiseo M, Mor M, et al. L718Q mutant EGFR escapes covalent inhibition by stabilizing a non-reactive conformation of the lung cancer drug osimertinib. Chem Sci. 2018;9(10):2740–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med. 2018;378(2):113–25. [DOI] [PubMed] [Google Scholar]
- 19.Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N Engl J Med. 2019. [DOI] [PubMed] [Google Scholar]
- 20.Ramalingam SS, Yang JC, Lee CK, Kurata T, Kim DW, John T, et al. Osimertinib As First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2018;36(9):841–9. [DOI] [PubMed] [Google Scholar]
- 21.Ramalingam SS, Rukazenkov Y, Todd A, Markovets A, Chmielecki J, Barrett JC, et al. LBA50Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Annals of Oncology. 2018;29(suppl_8). [Google Scholar]
- 22.Niederst MJ, Hu H, Mulvey HE, Lockerman EL, Garcia AR, Piotrowska Z, et al. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin Cancer Res. 2015;21(17):3924–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, et al. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin Cancer Res. 2015;21(17):3913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chic N, Mayo-de-Las-Casas C, Reguart N. Successful Treatment with Gefitinib in Advanced Non-Small Cell Lung Cancer after Acquired Resistance to Osimertinib. J Thorac Oncol. 2017;12(6):e78–e80. [DOI] [PubMed] [Google Scholar]
- 25.Rangachari D, To C, Shpilsky JE, VanderLaan PA, Kobayashi SS, Mushajiang M, et al. Brief Report: EGFR-mutated lung cancers resistant to osimertinib through EGFR-C797S respond to 1(st) generation reversible EGFR inhibitors but eventually acquire EGFR-T790M/C797S in preclinical models and clinical samples. J Thorac Oncol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Z, Yang JJ, Huang J, Ye JY, Zhang XC, Tu HY, et al. Lung Adenocarcinoma Harboring EGFR T790M and In Trans C797S Responds to Combination Therapy of First- and Third-Generation EGFR TKIs and Shifts Allelic Configuration at Resistance. J Thorac Oncol. 2017;12(11):1723–7. [DOI] [PubMed] [Google Scholar]
- 27.Nishino M, Suda K, Kobayashi Y, Ohara S, Fujino T, Koga T, et al. Effects of secondary EGFR mutations on resistance against upfront osimertinib in cells with EGFR-activating mutations in vitro. Lung Cancer. 2018;126:149–55. [DOI] [PubMed] [Google Scholar]
- 28.Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20(11):1496–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Regales L, Balak MN, Gong Y, Politi K, Sawai A, Le C, et al. Development of new mouse lung tumor models expressing EGFR T790M mutants associated with clinical resistance to kinase inhibitors. PLoS One. 2007;2(8):e810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tichelaar JW, Lu W, Whitsett JA. Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J Biol Chem. 2000;275(16):11858–64. [DOI] [PubMed] [Google Scholar]
- 31.Odegaard JI, Vincent JJ, Mortimer S, Vowles JV, Ulrich BC, Banks KC, et al. Validation of a Plasma-Based Comprehensive Cancer Genotyping Assay Utilizing Orthogonal Tissue- and Plasma-Based Methodologies. Clin Cancer Res. 2018;24(15):3539–49. [DOI] [PubMed] [Google Scholar]
- 32.Ballard P, Yates JW, Yang Z, Kim DW, Yang JC, Cantarini M, et al. Preclinical Comparison of Osimertinib with Other EGFR-TKIs in EGFR-Mutant NSCLC Brain Metastases Models, and Early Evidence of Clinical Brain Metastases Activity. Clin Cancer Res. 2016;22(20):5130–40. [DOI] [PubMed] [Google Scholar]
- 33.Fang W, Gan J, Huang Y, Zhou H, Zhang L. Acquired EGFR L718V Mutation and Loss of T790M-Mediated Resistance to Osimertinib in a Patient With NSCLC Who Responded to Afatinib. Journal of Thoracic Oncology. 2019;14(12):e274–e5. [DOI] [PubMed] [Google Scholar]
- 34.Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med. 2011;3(90):90ra59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest. 2009;119(10):3000–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Politi K, Fan PD, Shen R, Zakowski M, Varmus H. Erlotinib resistance in mouse models of epidermal growth factor receptor-induced lung adenocarcinoma. Dis Model Mech. 2010;3(1–2):111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pirazzoli V, Ayeni D, Meador CB, Sanganahalli BG, Hyder F, de Stanchina E, et al. Afatinib plus Cetuximab Delays Resistance Compared to Single-Agent Erlotinib or Afatinib in Mouse Models of TKI-Naive EGFR L858R-Induced Lung Adenocarcinoma. Clin Cancer Res. 2016;22(2):426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, et al. EGFR mutations and resistance to Irreversible pyrimidine based EGFR inhibitors. Clin Cancer Res. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yosaatmadja Y, Silva S, Dickson JM, Patterson AV, Smaill JB, Flanagan JU, et al. Binding mode of the breakthrough inhibitor AZD9291 to epidermal growth factor receptor revealed. J Struct Biol. 2015;192(3):539–44. [DOI] [PubMed] [Google Scholar]
- 40.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277(48):46265–72. [DOI] [PubMed] [Google Scholar]
- 41.Yang JC, Sequist LV, Geater SL, Tsai CM, Mok TS, Schuler M, et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015;16(7):830–8. [DOI] [PubMed] [Google Scholar]
- 42.McFadden DG, Politi K, Bhutkar A, Chen FK, Song X, Pirun M, et al. Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc Natl Acad Sci U S A. 2016;113(42):E6409–E17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A, et al. Combined EGFR/MEK Inhibition Prevents the Emergence of Resistance in EGFR-Mutant Lung Cancer. Cancer Discov. 2015;5(9):960–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eberlein CA, Stetson D, Markovets AA, Al-Kadhimi KJ, Lai Z, Fisher PR, et al. Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015;75(12):2489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dragani TA, Manenti G, Pierotti MA. Genetics of murine lung tumors. Adv Cancer Res. 1995;67:83–112. [DOI] [PubMed] [Google Scholar]
- 46.Del Re M, Tiseo M, Bordi P, D’Incecco A, Camerini A, Petrini I, et al. Contribution of KRAS mutations and c.2369C > T (p.T790M) EGFR to acquired resistance to EGFR-TKIs in EGFR mutant NSCLC: a study on circulating tumor DNA. Oncotarget. 2017;8(8):13611–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schoenfeld AJ, Chan JM, Kubota D, Sato H, Rizvi H, Daneshbod Y, et al. Tumor Analyses Reveal Squamous Transformation and Off-Target Alterations As Early Resistance Mechanisms to First-line Osimertinib in EGFR-Mutant Lung Cancer. Clin Cancer Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ichihara E, Westover D, Meador CB, Yan Y, Bauer JA, Lu P, et al. SFK/FAK Signaling Attenuates Osimertinib Efficacy in Both Drug-Sensitive and Drug-Resistant Models of EGFR-Mutant Lung Cancer. Cancer Res. 2017;77(11):2990–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Galvani E, Sun J, Leon LG, Sciarrillo R, Narayan RS, Sjin RT, et al. NF-kappaB drives acquired resistance to a novel mutant-selective EGFR inhibitor. Oncotarget. 2015;6(40):42717–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.