

Abstract
The fraction of administered antibiotics that reach the cecum and colon causes dysbiosis of the gut microbiome, resulting in various diseases. Protection of the gut microbiome from antibiotics using antibiotic adsorbents in the cecum and colon is a promising method to overcome this issue. Previously, activated charcoal (AC) has been reported to protect the gut microbiome of host animals. AC is an adsorbent that is widely used to capture toxic compounds and overdosed drugs in the gastrointestinal tract. The specificity of adsorbents for antibiotics is critical to avoid the risk of unexpected side effects caused by nonspecific adsorption of biological compounds in the intestinal fluid, such as bile acids and essential micronutrients. Here, we have developed specific adsorbents for vancomycin (VCM), which is known to cause gut dysbiosis. The adsorbents were composed of polyethyleneglycol-based microparticles (MPs) in which a specific ligand for VCM, D-Ala-D-Ala-OH, was attached via dendrons of D-lysine to raise the content of the ligand in the MPs. The MPs successfully protected Staphylococcus lentus from VCM in vitro because of the adsorption of VCM in the culture media. Pre-administration of MPs to mice reduced the amount of free VCM in the feces to an undetectable level. This treatment minimized the effect of VCM on gut microbiota and provided protection against Clostridioides difficile infection after oral challenge with spores.
Keywords: adsorbent, antibiotic, dysbiosis, microparticle, microbiome
INTRODUCTION
Antibiotics are used extensively for the treatment of infectious diseases. However, during antibiotic treatment, the non-absorbed fraction of orally administered antibiotics, along with the fraction excreted into the upper intestine via bile of both orally and parenterally administered antibiotics, reaches the large intestine, where it affects the gut microbiome and induces dysbiosis [1]. Dysbiosis in the gut microbiome results in various consequences in the short and long term, including Clostridioides difficile infection (CDI) [2], the generation of antibiotic-resistant bacteria [3], allergies, and metabolic syndromes [4].
To avoid gut dysbiosis induced by antibiotics, strategies to protect the gut microbiome during antibiotic treatment need to be developed. Two strategies have been reported to date. One of these strategies is the oral administration of β-lactamase to degrade residual β-lactam antibiotics that reach the large intestine [5,6,7,8]. It has been reported recently that a clinical trial of this therapy for the suppression of antibiotic-induced CDI was successful [9]. However, this therapy is only applicable to β-lactam antibiotics. The other reported strategy to protect the gut microbiome is the use of activated charcoal (AC) as an adsorbent of antibiotics in the large intestine [10, 11]. Because AC is a nonspecific adsorbent, this method is potentially applicable to a variety of antibiotics. However, the nonspecific characteristics of AC pose the risk of side effects; they may affect the metabolism of host animals by the nonspecific adsorption of biological compounds in the intestinal fluid, such as bile acids and essential micronutrients. In the treatment of patients with renal failure, orally administered AC nonspecifically adsorbed bile acids [12], which is suspected to be a risk factor for cardiovascular disease [13]. To avoid the nonspecific adsorption of biological compounds in the upper intestine, a colon-specific AC has been developed in which the AC was coated with pectin, which is degradable by colonic microflora [14]. A phase I clinical trial of this AC formulation showed that the fecal free-antibiotic level was reduced to a negligible level, without affecting the blood concentration of some biological compounds [10].
MATERIALS AND METHODS
Preparation and analysis of ligand-modified microparticles (MPs)
Preparation of MPs
D-lysine dendrons terminated with the ligand were synthesized on NovaPEG amino resin (0.2 mmol of amine group) by Fmoc solid-phase peptide synthesis (SPPS) and tert-butyl SPPS for the D-lysine dendron and ligand, respectively. First, the D-lysine dendron was synthesized using Fmoc-D-Lys(Fmoc)-OH and N-[1-(cyano-2-ethoxy-2-oxoethylideneaminooxy) dimethylamino(morpholino)] uronium hexafluorophosphate (COMU) as the carboxyl-group activating agent. Then, succinic anhydride was attached to the lysine α- and ɛ-amine groups using DMAP followed by addition of Fmoc-L-Lys-OH•HCl using COMU. After removing the Fmoc group, the α-amine group of L-lysine was acetylated with acetic anhydride. The amounts of cleaved Fmoc groups were used to quantify the number of modified L-lysine resides. H-D-Ala-OtBu was attached to the α-carboxyl group of lysine using COMU. After removing the tert-butyl group using trifluoroacetic acid/dichloromethane (9:1), addition of H-D-Ala-OtBu and removal of the tert-butyl group were repeated. After completion of the reaction, the resin was washed with tetrahydrofuran MeOH, water, and citrate buffer (pH 3.0) and dried in a desiccator.
Field emission scanning electron microscope observations
Dried MPs were placed on carbon sheets, and their morphology was observed with a field emission scanning electron microscope (SEM; Hitachi SU8000, Japan) operated at 10 kV.
Adsorption of vancomycin (VCM) into MPs
MPs (2.0 mg) swelled in distilled water for 24 hr at 37°C were mixed with VCM (1.0–50 µg/mL) in 500 µL of phosphate buffered saline (PBS; pH 7.4) and shaken (200 rpm) in a shaker for 24 hr at room temperature (r.t.). Then the concentration of VCM in the aqueous phase was determined by high-performance liquid chromatography (HPLC). VCM was quantified by a validated HPLC method using a LaChrom Elite L2455 diode array detector (Hitachi, Japan) on a COSMOSIL 5C4 column (5 µm, 10 × 150 mm). An isocratic mobile phase consisting of 10% methanol and 90% Milli-Q water was used with a flow rate of 1.0 mL/min and a 50 µL injection volume. VCM was detected at a wavelength of 225 nm. The adsorption isotherm was fitted based on the Langmuir equation as follows using Prism 8 (GraphPad Software):
| Ce/Qe = Kd/Qm + Ce/Qm, |
where Ce (M) is the equilibrium concentration of VCM in solution, Qe (mol-VCM/g-MPs) is the amount of VCM adsorbed at equilibrium, Qm (mol-VCM/g-MPs) is the adsorption capacity of the MPs, and Kd (M) is the dissociation constant.
Evaluation of the minimal inhibitory concentration 50 value
Swelled MPs in MD12 medium (500 µL) were added into the top compartments of transwells (3 µm porosity; BD Biosciences) in 6-well plates (Corning). Staphylococcus lentus isolated in the logarithmic growth period was added to MD12 medium and incubated at 37°C for 24 hr. Then, aliquots (50 µL) were taken from the cell suspensions and mixed with the VCM-containing MD12 medium, and the mixtures (1.5 mL) were applied gently into the bottom compartments of the transwells. The plates were then incubated at 37°C for 24 hr in a constant temperature incubator. The minimal inhibitory concentration (MIC) value was determined as the lowest VCM concentration to show no growth of turbidity with the plate reader at a wavelength of 600 nm. The MIC50 value was defined as the lowest concentration of VCM at which 50% of the bacteria were inhibited.
Determination of the dose of MPs in vivo
C5/BL/6 J mice (male; 8 weeks old) were purchased from Kyudo Co., Ltd. (Saga, Japan). Animal experiments were performed according to the guidelines of the animal care and use committee, Kyushu University. Mice were fed 30 kGy γ-irradiated CE-2 (CLEA Japan Inc.) and had access to bedding and tap water. They also had a light cycle comprised of 12 hr of light and 12 hr of darkness. For in vivo capacity testing, mice (n=3) were administered MP-G4 (3.0 or 10.0 mg/100 µL Dulbecco’s phosphate-buffered saline [D-PBS]), and after 1 hr, VCM (300 µg/100 µL sterile water) was administered by oral gavage. Feces were collected over 23 hr after administration. Distilled water was then added to 1.0 g of each feces sample, and the dispersion was vortex mixed. It was then centrifuged at 8,000 rpm for 10 min at 4°C. The supernatant was then filtered through a 0.45 µm filter and diluted with mobile phase before being analyzed using HPLC. The HPLC analysis procedure was the same as that described in Section “Adsorption of vancomycin (VCM) into MPs”. To examine the effect of repeated daily treatments, mice were divided into four groups (n=3 per group) and administered either D-PBS or MP-G4 (10.0 mg/100 µL D-PBS), and after 1 hr, either sterile water or VCM (300 µg/100 µL sterile water) was administered by oral gavage. The treatment was conducted once a day for 7 days, and body weight was measured daily.
Preservation of the microbiome and protection from CDI
C. difficile spore preparation
C. difficile VPI10463 (ATCC 43255) spores were prepared as follows. C. difficile VPI10463 was grown overnight in 5 mL of Columbia Broth at 37°C under anaerobic conditions. The next day, 40 mL of Clospore medium was added to the inoculum. The culture medium was anaerobically incubated at 37°C for 7 days. Spores were harvested by centrifugation and washed with cold water three times. Spore stocks were stored at 4°C in sterile water. Viable spores were enumerated by plating for colony-forming units (CFU)/mL on taurocholate, cefoxitin, cycloserine, fructose agar (TCCFA) to determine the challenge dose.
VCM and MP administration and challenge with C. difficile
C57BL/6 J mice (female; 5 weeks old) were purchased from CLEA Japan Inc. Mice were fed 30 kGy γ-irradiated CE-2 (CLEA Japan Inc.) and had access to bedding and tap water. They also had a light cycle comprised of 12 hr of light and 12 hr of darkness. The procedure for the animal experiment is shown in Supplementary Fig. 1. The C57BL/6 J mice (female; 5 weeks old) were divided into four groups (n=6 per group) and administered D-PBS or MP-G4 (10.0 mg/100 µL D-PBS), and after 1 hr, either sterile water or vancomycin (300 µg/100 µL sterile water) was administered by oral gavage for 5 days (= treatment period). Following the treatment period, mice were allowed to drink normal water for 2 days, and the next day, they were challenged with 1 × 104 CFU C. difficile spores by oral gavage. These animal experiments were conducted using protocols approved by the Keio University Ethics Committee for Animal Experiments.
16S ribosomal DNA analysis
Mouse fecal samples were collected and preserved at −80°C. Bacterial DNA was extracted in accordance with the E.Z.N.A. Stool DNA Kit pathogen detection protocol (OMEGA). The V3–V4 region of the 16S rDNA gene was amplified following DNA extraction using universal primers (Supplementary Table 1). The PCR reaction mixture consisted of 5 ng/µL of DNA extraction mix (2.5 µL), 2 × CAPA HiFi mix (Illumina; 12.5 µL), and 1 µM of each primer (5 µL). The cycling conditions were as follows: 95°C for 3 min; 25 cycles of 95°C for 30 sec, 55°C for 30 sec, and 72°C for 30 sec; and then a final elongation step at 72°C for 5 min. The amplicon DNA was purified using AMPure XP beads (Beckman Coulter) followed by a second PCR reaction with a mixture consisting of purified DNA (5 µL), 2 × CAPA HiFi mix (Illumina; 25 µL), distilled water (10 µL), and 1 µM of each primer (Nextera XT Index Kit; Illumina; 5 µL). The cycling conditions were as follows: 95°C for 3 min; 8 cycles of 95°C for 30 sec, 55°C for 30 sec, and 72°C for 30 sec, and then a final elongation step at 72°C for 5 min. Likewise, tagged DNA was purified with AMPure XP beads and diluted in 10 mM Tris-HCl buffer (pH 8.5) to 12 pM; and then all samples were pooled. The completed library was sequenced with an Illumina MiSeq 600-cycle v3-v4 kit (Illumina).
Evaluation of fecal CFUs
C. difficile CFUs were determined using fecal samples collected on day 1 after infection. Fecal samples were weighed and suspended in 1 mL of D-PBS and homogenized in an anaerobic chamber. An aliquot (50 µL) of the suspensions was plated on TCCFA and incubated at 37°C under anaerobic conditions for 3 days. Then, the number of colonies was enumerated.
ELISA of fecal lipocalin-2
Feces collected on day 1 after infection were weighed and stored at −80°C. The fecal samples were suspended in D-PBS containing 0.1% Tween-20 (100 mg/mL) and vortexed for 20 min. The suspension was centrifuged at 4°C at 12,000 rpm for 10 min. ELISA was conducted according to the protocol of the ELISA kit (DY1857, R&D Systems). Samples were diluted in the kit-recommended reagent diluent (1% BSA in D-PBS).
Statistical analysis
Statistical analyses were performed using R studio (Version 1.1.456). The survival rate and significance between the four groups were tested by the Kaplan-Meier test using the R Survival package. Bartlett’s test was used to determine the significance of variances between the four groups. If the differences were significant (p<0.05), Scheffe’s test was used for comparisons of discontinuous variables between the four groups. The Tukey HSD test was used for comparisons between the four groups with similar variances. The diversity of the gut microbiota was analyzed by QIIME 13 and R using the R vegan and multcomp packages. Intra- and intergroup β-diversity analyses were analyzed by R using the R multcomp package.
RESULTS
Strategy and design of adsorbents
Here, we sought to develop a specific adsorbent for antibiotics, free from the risks associated with nonspecific adsorbents (Fig. 1). As a target antibiotic, we selected VCM, which is used widely against Gram-positive bacteria. VCM treatment via both oral and parenteral administration causes gut dysbiosis [15, 16], as well as the generation of VCM-resistant bacteria colonies in the rectum [17]. VCM specifically binds to the carboxyl-terminus of peptidoglycan precursors terminating in the sequence D-Ala-D-Ala-OH to block the cross-linking reaction in the growing bacterial cell wall [18]. We used this peptide as a ligand to adsorb VCM. It has been reported that cell-wall mimetic peptides, such as N,N’-diacetyl-L-Lys-D-Ala-D-Ala-OH, have relatively high binding affinity for VCM (dissociation constant Kd = 106 M−1) [19]. To immobilize the peptide ligand, we used a cross-linked polymeric support for the solid phase peptide synthesis because it enables high loading of the ligand peptide. A high ligand content is important to achieve adsorption of VCM in the large intestine using a practical dosage. To raise the loading of the ligand in the MPs, the ligand was attached to the MPs via a linker of dendritic D-lysine [20] (Fig. 1b). Using the obtained ligand-modified MPs, we investigated the adsorption of VCM in vivo and the suppression of CDI in a mouse model.
Fig. 1.
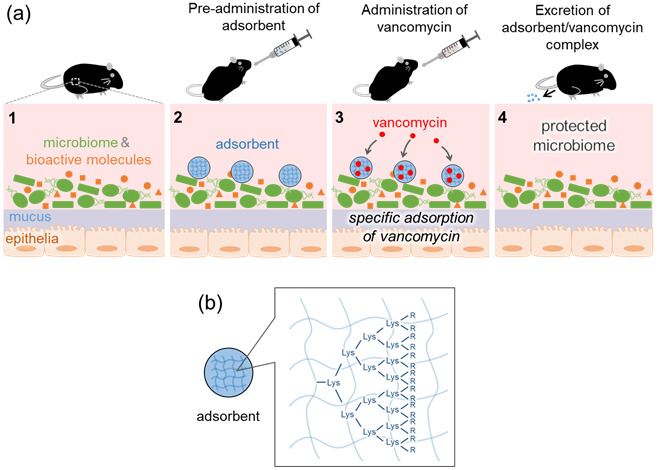
(a) Mechanism of action of the vancomycin (VCM)-specific adsorbent microparticles (MPs) in the large intestine. Pre-administered adsorbent specifically captures VCM, protects the gut microbiome, and minimizes the effect on the concentrations of bioactive molecules in the intestinal fluid. (b) Schematic drawing of an MP. The MP is a spherical hydrogel of cross-linked polyethyleneglycol with a VCM-specific ligand (D-Ala-D-Ala-OH) immobilized via D-lysine dendrons. R indicates the ligand.
Preparation of ligand-modified MPs
A dendritic ligand unit was synthesized by SPPS on a commercially available resin (Fig. 2). Here, we used a water-swellable NovaPEG amino resin composed of cross-linked PEG [21], as water-swellable properties are a prerequisite for the adsorption of VCM in the intestinal fluid. First, 3rd or 4th generation D-lysine dendron units were synthesized on the resin using Fmoc-D-Lys(Fmoc)-OH as the monomer. D-lysine was employed here for its resistance against peptidases in the gastrointestinal fluid. Then, the α- and ɛ-amine groups of terminal D-lysine residues were converted to carboxyl groups using succinic anhydride (Supplementary Scheme 1). On the carboxyl groups, ɛ-L-lysine was introduced, and then the ligand (D-Ala-D-Ala-OH) was synthesized using H-D-Ala-OtBu as the monomer via tert-butyl SPPS [22]. Before modification of H-D-Ala-OtBu, the number of terminal L-lysine residues was quantified from the amount of Fmoc groups released from the terminal L-lysine, and the yield for this step was calculated based on the number of initial amine groups in the resin (yield in Table 1). The yield became lower with increasing generations, probably because of the steric repulsion in the crowded termini of the dendrons of higher generations. Indeed, the 4th generation is reported to be the maximum for quantitative preparation [23]. The amount of loaded ligand per weight of MP was not very different between MP-G3 and MP-G4 (Table 1) because the weights of the dendritic ligand units (68% for MP-G3 and 83% for MP-G4) comprised most of the total weights of these MPs.
Fig. 2.
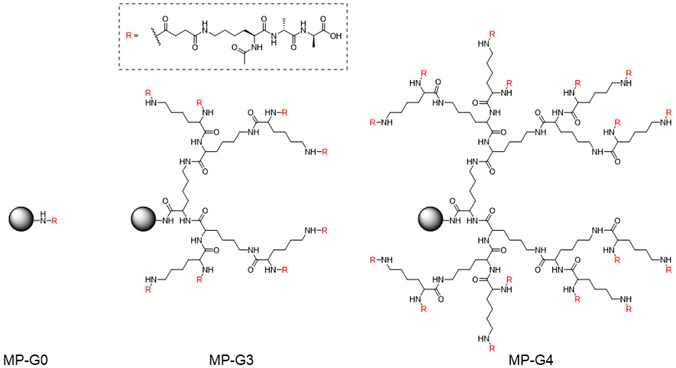
Chemical structures of the ligand-modified microparticles (MPs) with three levels of dendritic ligand generations. Spheres indicate the resin matrix.
Table 1. Characteristics of microparticles (MPs).
| MP-G0 | MP-G3 | MP-G4 | |
|---|---|---|---|
| Yield (%) a | 100 | 58 | 42 |
| Loaded ligand (µmol/mg) | 0.44 | 1.1 | 1.2 |
| Diameter (µm)b | 111 ± 20 | 139 ± 24 | 150 ± 31 |
| Qmax (µmol/mg) | 0.24 | 0.35 | 0.35 |
| Qmax,g (g/g) | 0.36 | 0.51 | 0.51 |
| Qmax,mol (mol/mol) | 0.86 | 0.46 | 0.41 |
| Kd (µM) | 14 | 29 | 20 |
aOverall yield until the modification of l-lysine residue.
bDetermined from SEM images.
SEM images of each dry MP showed that a spherical shape was maintained even for the high-weight fraction of the dendritic ligand unit (Fig. 3). MPs with higher generation dendrons were larger in size, reflecting the increase in the weight fraction of the dendritic ligand unit with each generation. The size of the MP (>100 µm) was sufficient to avoid penetration of the intestinal epithelium [24].
Fig. 3.

SEM images of ligand-modified MPs. (a) MP-G0, (b) MP-G3, and (c) MP-G4. Scale bars indicate 500 µm.
Adsorption of VCM into MPs
The adsorption characteristics of the ligand-modified MPs with respect to VCM were evaluated. After incubating a mixture of each MP and VCM for 24 hr at 37°C, unbound VCM was quantified using HPLC. The adsorption isotherms of each MP are summarized in Fig. 4. Intact NovaPEG amino resin (naked MP; NP in Fig. 4) showed negligible adsorption of VCM. In contrast, the ligand-modified MPs showed adsorption of VCM depending on the generation level, indicating the specific interaction of the ligand of the MPs with VCM. The maximum adsorption amount (Qmax) and dissociation constant (Kd) values were calculated using the Langmuir equation and are summarized in Table 1. The Kd values were not affected by the generation level of the dendron unit. The Qmax values of the MPs with dendritic ligands were higher than that of MP-G0, but the increments were smaller than the values expected from the loaded ligand. The Qmax,mol value, which is a stoichiometric expression of Qmax, showed that only half of the ligands in MP-G3 and MP-G4 functioned to capture VCM, indicating that the sterically crowded state of the ligands on the dendrons will impede the recognition of VCM. The typical Qmax values of AC for various antibiotics are reported to be 10−1 to 10° g/g [25]. Thus, the Qmax values of the MPs were equivalent to those of AC, a representative adsorbent with a high capacity. Compared with the reported Kd value of a complex between a free ligand and VCM (up to 1 µM) [26], the Kd values of the MPs were one order of magnitude higher. The lower affinity of the ligands in the MPs compared with the free ligand can be explained by the aforementioned result showing that half of the ligands cannot bind VCM because of steric hindrance.
Fig. 4.
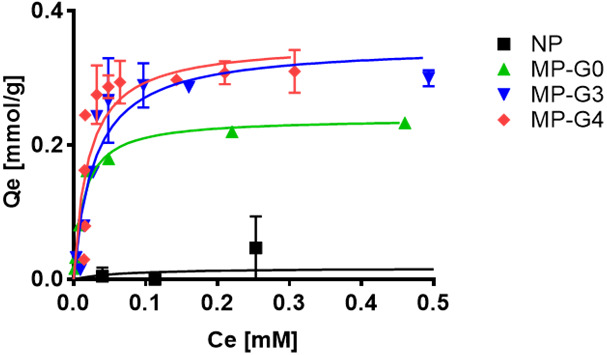
Adsorption isotherms of vancomycin (VCM) for the microparticles (MPs). VCM and MPs (4.0 mg/mL) were mixed in PBS (pH 7.4) and incubated for 24 hr at r.t. The concentration of free VCM was determined by HPLC.
We also confirmed the specificity of MP-G4 for VCM over other antibiotics (cefoperazone, clarithromycin) (Supplementary Fig. 2). We found that MP-G4 specifically adsorbed VCM, while AC, as a control, adsorbed cefoperazone and clarithromycin by nonspecific hydrophobic interaction. Thus, MP-G4 is specific adsorbent for VCM.
The ability of the MPs to protect bacteria from VCM was determined from the MIC50 value. The Gram-positive bacterium S. lentus was cultured in the presence of VCM and each MP for 24 hr. As shown in Fig. 5a, the MIC50 value of VCM toward S. lentus was 6.0 µg/mL in the absence of MPs, which is consistent with the reported value [21]. Naked MPs (NPs) showed almost no effect on the MIC50 value, while the ligand-modified MPs clearly raised the MIC50 values; the MIC50 values in the presence of MP-G3 and MP-G4 were 33 and 63 µg/mL, respectively. MP-G4 showed a slightly higher protective effect than MP-G3. Figure 5b shows the concentration dependence of the protective effect of MP-G4. The MIC50 value correlated with the concentration of MP-G4. These results showed the ability of the MPs to protect bacteria from VCM.
Fig. 5.
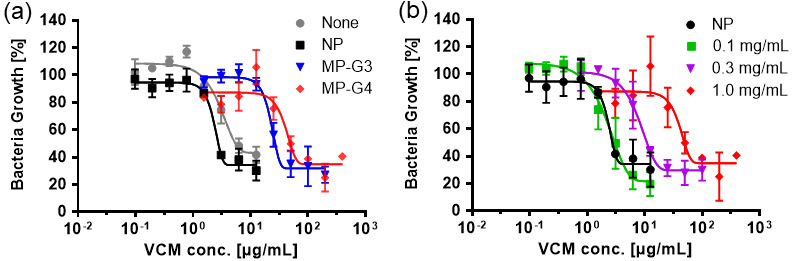
Protection of S. lentus from vancomycin (VCM) by different microparticles (MPs) (a) and by MP-G4 at different concentrations (b). S. lentus was incubated in MD12 medium containing VCM with or without MPs (1.0 mg/mL in panel a) at 37°C for 24 hr. The MIC50 value was determined from the turbidity at 600 nm. Data are presented as the mean ± SD (n=3).
Adsorption of VCM into MPs in vivo to protect the microbiome
The required dosage of MP-G4 to capture VCM in the gastrointestinal tract was determined. First, MP-G4 was administered by oral gavage. After 1 hr (during this period, microparticles start to reach the cecum [27, 28]), VCM (300 µg) was administered orally. After VCM administration, feces were collected over 23 hr, and the amount of free VCM in the feces was quantified by HPLC analysis. As shown in Fig. 6a, without the pre-administration of MP-G4, approximately 70% of orally administered VCM was detected in the feces. In the presence of MP-G4, the amount of VCM in the feces was reduced in a dose-dependent manner. A dosage of 10.0 mg of MP-G4 was found to be sufficient to capture all the VCM; thus, hereafter, we used a 10.0 mg dosage of MP-G4. The binding capacity of MP-G4 calculated from 3.0 mg of MP-G4 was 0.023 mg/mg. Though this value is one order of magnitude lower than the binding capacity determined in PBS shown in Fig. 4 (0.51 mg/mg), the VCM adsorption efficacy of MP-G4 appears to be high. This is desirable considering the presence of a large amount of biological compounds in the gastrointestinal fluid that will compete with VCM in binding with the ligand. According to the dose conversion equation based on the surface area and body weight [28], the calculated dosage of MP-G4 for human application would be 40.5 mg/kg, which is comparable with the dosage of pectin-coated AC in a clinical trial (7.5 g/human; 125 mg/kg) [10]. When this administration regimen was conducted once a day for 7 days in mice, no detectable loss of body weight was observed (Fig. 6b).
Fig. 6.
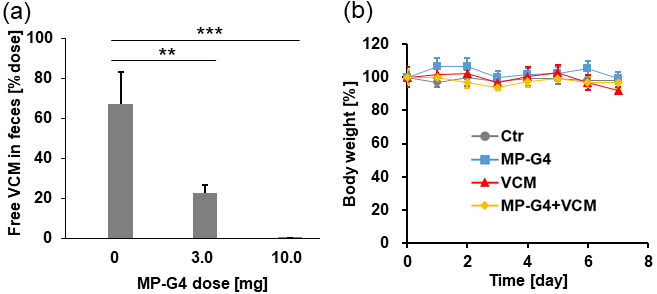
(a) Amount of free vancomycin (VCM) in the feces of mice orally treated with VCM and MP-G4 (n=3 per group). **p<0.01; ***p<0.001. VCM (300 µg/100 µL) was administered 1 hr after MP-G4 (0–10.0 mg/100 µL). (b) The body weight changes in the four groups of mice (n=3 per group) treated once a day for 7 days.
To examine whether pre-administration of MP-G4 could prevent VCM-induced gut dysbiosis in vivo, we analyzed the gut microbiota composition before and after VCM (300 µg) treatment by 16S rDNA sequencing. The composition of the gut microbiota did not differ between any of the groups before treatment but changed significantly after VCM treatment. The administration of MP-G4 did not affect the gut microbial community. Pre-administration of MP-G4 prevented the changes in gut microbial composition induced by VCM treatment (Fig. 7a and Supplementary Fig. 3). Specifically, the abundance of Enterobacteriales and Erysipelotrichales was significantly increased in the VCM-treated group compared with the control group. In contrast, this increase in the abundance of these bacteria was not observed in the VCM-treated group pre-administered MP-G4 (Fig. 7b and Supplementary Fig. 4).
Fig. 7.
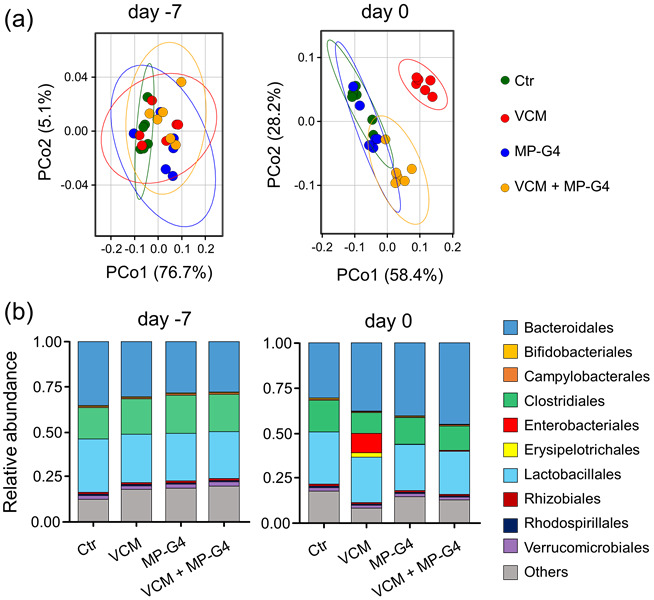
Pre-administration of MP-G4 prevented vancomycin (VCM)-induced perturbation in the gut microbiota. (a) Principal coordinates analysis plot of weighted-UniFrac distances by water (Ctr), VCM, MP-G4, or VCM with MP-G4-treated groups (n=6 per group). (b) Fecal microbiota composition of mice orally treated with water (Ctr), VCM, MP-G4, or VCM with MP-G4 on day −7 (just before treatment) and day 0 (after treatment; n=6 per group). Each bar shows the mean for individual mice from each group. Data are representative of two independent experiments.
VCM is active against most Gram-positive bacteria and causes the overgrowth of Enterobacteriales in the gut [16, 29]. In general, a bloom of Enterobacteriales is a hallmark of gut dysbiosis [30]. Therefore, our results indicated that the pre-administration of MP-G4 protected the gut microbiota from VCM-induced dysbiosis by capturing VCM in the gastrointestinal tract.
Protection from C. difficile infection
Disruption of the gut microbiota induced by the use of antibiotics enables C. difficile colonization in the gut [31]. In particular, an increase in the abundance of Enterobacteriales is associated with higher susceptibility to C. difficile colonization [32, 33]. Therefore, we next investigated whether the pre-administration of MP-G4 protected VCM-treated mice against CDI. While all the control mice survived, all VCM-treated mice succumbed to CDI on the second day after infection. However, both the VCM-treated or -untreated mice, pre-administered with MP-G4, all survived post CDI (Fig. 8a). C. difficile reached up to 108 CFU/g feces 1 day after infection in the VCM-treated mice. In contrast, the burden of C. difficile in the feces of both the control and VCM-treated mice pre-administered MP-G4, as well as that of the control mice not pre-administered MP-G4, was not detectable (< 102) or under 105 CFU/g feces (Fig. 8b). Furthermore, the level of lipocalin-2, an antimicrobial protein released from the epithelia during inflammation [34], increased in the VCM-treated mice but not in the control mice nor in the control and VCM-treated mice pre-administered MP-G4 (Fig. 8c). These results indicated that the pre-administration of MP-G4 protected VCM-treated mice against CDI.
Fig. 8.
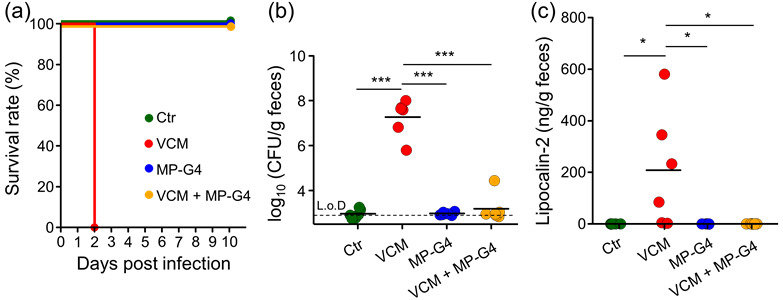
Pre-administration of MP-G4 protected vancomycin (VCM)-treated mice against CDI. (a) Mouse survival over time after CDI in C57BL/6J female mice treated with water (Ctr), MP-G4, VCM, or VCM with MP-G4 (n=6 per group). (b) Pathogen loads [CFU/g] in feces were detected 1 day after infection. Each dot represents an individual mouse. L.o.D.: limit of detection. (c) ELISA measurement of lipocalin-2 in feces from C. difficile-infected mice from each group (n=6 per group). *p<0.05; ***p<0.001. Data are representative of two independent experiments.
DISCUSSION
We proposed the use of antibiotic-specific adsorbents to protect the gut microbiome from VCM-induced dysbiosis. We prepared adsorbent MPs composed of hydrogels of cross-linked PEG with a peptide ligand specific to VCM immobilized via dendritic D-lysine as a linker to achieve a high content of the ligand in the MPs. The prepared MPs showed a relatively high capacity for VCM adsorption in aqueous media (up to 0.5 g/g). This is equivalent to the reported capacity of activated charcoal, a representative nonspecific adsorbent, toward various antibiotics. In vitro tests showed that the MPs improved the resistance of S. lentus toward VCM. After oral administration at a practical dosage, MP-G4 adsorbed VCM in the intestine almost completely and suppressed dysbiosis of the gut microbiome in mice. As a consequence of the protection from gut dysbiosis, the mice were resistant against an oral challenge with the spores of C. difficile. The composition of fecal metabolites, such as the levels of carbohydrates, amino acids, and bile acids, have been shown to be different between CDI patients and healthy individuals [35, 36]. Therefore, it would be predicted that treatment with MPs would also protect against metabolic changes induced by VCM. Further studies will reveal whether the MPs can also prevent metabolic fitness for C. difficile induced by VCM treatment.
We have shown that MPs prevented gut dysbiosis induced by VCM and protected mice from CDI. Because VCM is not normally absorbed into the body, oral treatment with VCM works only in the intestine. Currently, oral VCM treatment is applied for patients with CDI and Staphylococcus aureus enteritis. It could be speculated that the MPs prevent not only gut dysbiosis but also the elimination of such enteric pathogens without any systemic effect after oral VCM treatment. Therefore, it is possible that the MPs reduce the efficacy of VCM against C. difficile or enteric S. aureus infection. For this reason, the MPs cannot be applied for patients who are infected with enteric pathogens, including C. difficile, and will be received with VCM. Although clinical application of the MPs is difficult at the moment, further development of absorbents for specific antibiotics that have systemic effects when orally treated will provide good strategies to protect the host from both pathogens and gut dysbiosis.
Supplementary
Acknowledgments
This work was supported in part by a Grant-in-Aid for Young Scientists A (17H05068) and the Yakult Bio-Science Foundation (YK).
REFERENCES
- 1.Dethlefsen L, Relman DA. 2011. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA 108 Suppl 1: 4554–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Theriot CM, Young VB. 2015. Interactions between the gastrointestinal microbiome and Clostridium difficile. Annu Rev Microbiol 69: 445–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burton S, Washington N, Steele RJC, Musson R, Feely L. 1995. Intragastric distribution of ion-exchange resins: a drug delivery system for the topical treatment of the gastric mucosa. J Pharm Pharmacol 47: 901–906. [DOI] [PubMed] [Google Scholar]
- 4.Belkaid Y, Hand TW. 2014. Role of the microbiota in immunity and inflammation. Cell 157: 121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harmoinen J, Vaali K, Koski P, Syrjänen K, Laitinen O, Lindevall K, Westermarck E. 2003. Enzymic degradation of a β-lactam antibiotic, ampicillin, in the gut: a novel treatment modality. J Antimicrob Chemother 51: 361–365. [DOI] [PubMed] [Google Scholar]
- 6.Harmoinen J, Mentula S, Heikkilä M, van der Rest M, Rajala-Schultz PJ, Donskey CJ, Frias R, Koski P, Wickstrand N, Jousimies-Somer H, Westermarck E, Lindevall K. 2004. Orally administered targeted recombinant Beta-lactamase prevents ampicillin-induced selective pressure on the gut microbiota: a novel approach to reducing antimicrobial resistance. Antimicrob Agents Chemother 48: 75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stiefel U, Harmoinen J, Koski P, Kääriäinen S, Wickstrand N, Lindevall K, Pultz NJ, Bonomo RA, Helfand MS, Donskey CJ. 2005. Orally administered recombinant metallo-β-lactamase preserves colonization resistance of piperacillin-tazobactam-treated mice. Antimicrob Agents Chemother 49: 5190–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffman A, Horwitz E, Hess S, Cohen-Poradosu R, Kleinberg L, Edelberg A, Shapiro M. 2008. Implications on emergence of antimicrobial resistance as a critical aspect in the design of oral sustained release delivery systems of antimicrobials. Pharm Res 25: 667–671. [DOI] [PubMed] [Google Scholar]
- 9.Kokai-Kun JF, Roberts T, Coughlin O, Le C, Whalen H, Stevenson R, Wacher VJ, Sliman J. 2019. Use of ribaxamase (SYN-004), a β-lactamase, to prevent Clostridium difficile infection in β-lactam-treated patients: a double-blind, phase 2b, randomised placebo-controlled trial. Lancet Infect Dis 19: 487–496. [DOI] [PubMed] [Google Scholar]
- 10.de Gunzburg J, Ghozlane A, Ducher A, Le Chatelier E, Duval X, Ruppé E, Armand-Lefevre L, Sablier-Gallis F, Burdet C, Alavoine L, Chachaty E, Augustin V, Varastet M, Levenez F, Kennedy S, Pons N, Mentré F, Andremont A. 2018. Protection of the human gut microbiome from antibiotics. J Infect Dis 217: 628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Gunzburg J, Ducher A, Modess C, Wegner D, Oswald S, Dressman J, Augustin V, Feger C, Andremont A, Weitschies W, Siegmund W. 2015. Targeted adsorption of molecules in the colon with the novel adsorbent-based medicinal product, DAV132: a proof of concept study in healthy subjects. J Clin Pharmacol 55: 10–16. [DOI] [PubMed] [Google Scholar]
- 12.Araki Y, Tsujikawa T, Andoh A, Sasaki M, Fujiyama Y, Bamba T. 2000. Therapeutic effects of an oral adsorbent on acute dextran sulphate sodium-induced colitis and its recovery phase in rats, especially effects of elimination of bile acids in gut lumen. Dig Liver Dis 32: 691–698. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Coresh J, Eustace JA, Longenecker JC, Jaar B, Fink NE, Tracy RP, Powe NR, Klag MJ. 2004. Association between cholesterol level and mortality in dialysis patients: role of inflammation and malnutrition. JAMA 291: 451–459. [DOI] [PubMed] [Google Scholar]
- 14.Khoder M, Tsapis N, Domergue-Dupont V, Gueutin C, Fattal E. 2010. Removal of residual colonic ciprofloxacin in the rat by activated charcoal entrapped within zinc-pectinate beads. Eur J Pharm Sci 41: 281–288. [DOI] [PubMed] [Google Scholar]
- 15.Halpin AL, de Man TJB, Kraft CS, Perry KA, Chan AW, Lieu S, Mikell J, Limbago BM, McDonald LC. 2016. Intestinal microbiome disruption in patients in a long-term acute care hospital: A case for development of microbiome disruption indices to improve infection prevention. Am J Infect Control 44: 830–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun L, Zhang X, Zhang Y, Zheng K, Xiang Q, Chen N, Chen Z, Zhang N, Zhu J, He Q. 2019. Antibiotic-induced disruption of gut microbiota alters local metabolomes and immune responses. Front Cell Infect Microbiol 9: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donskey CJ, Helfand MS, Pultz NJ, Rice LB. 2004. Effect of parenteral fluoroquinolone administration on persistence of vancomycin-resistant Enterococcus faecium in the mouse gastrointestinal tract. Antimicrob Agents Chemother 48: 326–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheldrick GM, Jones PG, Kennard O, Williams DH, Smith GA. 1978. Structure of vancomycin and its complex with acetyl-D-alanyl-D-alanine. Nature 271: 223–225. [DOI] [PubMed] [Google Scholar]
- 19.Kannan R, Harris CM, Harris TM, Waltho JP, Skelton NJ, Williams DH. 1988. Function of the amino sugar and N-terminal amino acid of the antibiotic vancomycin in its complexation with cell wall peptides. J Am Chem Soc 110: 2946–2953. [Google Scholar]
- 20.Swali V, Wells NJ, Langley GJ, Bradley M. 1997. Solid-phase dendrimer synthesis and the generation of super-high-loading resin beads for combinatorial chemistry. J Org Chem 62: 4902–4903. [Google Scholar]
- 21.Cho WM, Joshi BP, Cho H, Lee KH. 2007. Design and synthesis of novel antibacterial peptide-resin conjugates. Bioorg Med Chem Lett 17: 5772–5776. [DOI] [PubMed] [Google Scholar]
- 22.Gutheil WG, Xu Q. 2002. N-to-C solid-phase peptide and peptide trifluoromethylketone synthesis using amino acid tert-butyl esters. Chem Pharm Bull (Tokyo) 50: 688–691. [DOI] [PubMed] [Google Scholar]
- 23.Fischer G, Wängler B, Wängler C. 2014. Optimized solid phase-assisted synthesis of dendrons applicable as scaffolds for radiolabeled bioactive multivalent compounds intended for molecular imaging. Molecules 19: 6952–6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt C, Lautenschlaeger C, Collnot EM, Schumann M, Bojarski C, Schulzke JD, Lehr CM, Stallmach A. 2013. Nano- and microscaled particles for drug targeting to inflamed intestinal mucosa: a first in vivo study in human patients. J Control Release 165: 139–145. [DOI] [PubMed] [Google Scholar]
- 25.Ahmed MJ. 2017. Adsorption of quinolone, tetracycline, and penicillin antibiotics from aqueous solution using activated carbons: review. Environ Toxicol Pharmacol 50: 1–10. [DOI] [PubMed] [Google Scholar]
- 26.Rao J, Yan L, Lahiri J, Whitesides GM, Weis RM, Warren HS. 1999. Binding of a dimeric derivative of vancomycin to L-Lys-D-Ala-D-lactate in solution and at a surface. Chem Biol 6: 353–359. [DOI] [PubMed] [Google Scholar]
- 27.Hodges GM, Carr EA, Hazzard RA, Carr KE. 1995. Uptake and translocation of microparticles in small intestine. Morphology and quantification of particle distribution. Dig Dis Sci 40: 967–975. [DOI] [PubMed] [Google Scholar]
- 28.Padmanabhan P, Grosse J, Asad ABMA, Radda GK, Golay X. 2013. Gastrointestinal transit measurements in mice with 99mTc-DTPA-labeled activated charcoal using NanoSPECT-CT. EJNMMI Res 3: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walsh C. 2000. Molecular mechanisms that confer antibacterial drug resistance. Nature 406: 775–781. [DOI] [PubMed] [Google Scholar]
- 30.Zeng MY, Inohara N, Nuñez G. 2017. Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunol 10: 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seekatz AM, Young VB. 2014. Clostridium difficile and the microbiota. J Clin Invest 124: 4182–4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freedberg DE, Toussaint NC, Chen SP, Ratner AJ, Whittier S, Wang TC, Wang HH, Abrams JA. 2015. Proton pump inhibitors alter specific taxa in the human gastrointestinal microbiome: a crossover trial. Gastroenterology 149: 883–5.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang L, Dong D, Jiang C, Li Z, Wang X, Peng Y. 2015. Insight into alteration of gut microbiota in Clostridium difficile infection and asymptomatic C. difficile colonization. Anaerobe 34: 1–7. [DOI] [PubMed] [Google Scholar]
- 34.Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, Vijay-Kumar M. 2012. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS One 7: e44328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Theriot CM, Koenigsknecht MJ, Carlson PE, Jr, Hatton GE, Nelson AM, Li B, Huffnagle GB, Z Li J, Young VB. 2014. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun 5: 3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Battaglioli EJ, Hale VL, Chen J, Jeraldo P, Ruiz-Mojica C, Schmidt BA, Rekdal VM, Till LM, Huq L, Smits SA, Moor WJ, Jones-Hall Y, Smyrk T, Khanna S, Pardi DS, Grover M, Patel R, Chia N, Nelson H, Sonnenburg JL, Farrugia G, Kashyap PC. 2018. Clostridioides difficile uses amino acids associated with gut microbial dysbiosis in a subset of patients with diarrhea. Sci Transl Med 10: eaam7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.