

Abstract
Background
The delta opioid receptor (DOR) has been identified as a therapeutic target for migraine, with DOR agonists exhibiting low abuse potential compared with conventional µ-opioid agonists. TRV250 is a novel small molecule agonist of the DOR that is preferentially selective for G-protein signaling, with relatively little activation of the β-arrestin2 post-receptor signaling pathway. This selectivity provides reduced susceptibility to proconvulsant activity seen with non-selective DOR agonists. TRV250 significantly reduced nitroglycerin-evoked hyperalgesia in rodents, indicating a potential utility in acute migraine without the risk of seizure activity or abuse potential.
Objective
This trial evaluated the safety, tolerability, and pharmacokinetics of ascending dose levels of TRV250 administered subcutaneously (SC) and the relative bioavailability of TRV250 administered orally compared with SC administration.
Methods
This was a two-part, single ascending dose study. Part A included four cohorts of healthy adults (N = 38). Each cohort was dosed on three occasions (placebo and two different dose levels of TRV250, allocated in randomized order and administered by SC route). In Part B, a single cohort of nine subjects received an oral dose of either TRV250 (n = 7) or placebo (n = 2) in a fed or fasted state. Serial blood samples were obtained for pharmacokinetic determination across a 24-h post-dose period. Safety assessments included clinical laboratory measures, vital signs, 12-lead electrocardiogram (ECG), and electroencephalogram (EEG) pre- and post-dosing.
Results
TRV250 was well tolerated. There were no serious adverse events (SAEs), and all AEs were mild in severity. Injection-site reactions and headache were the most common AEs. One subject was withdrawn from the study due to a TRV250-related AE of postural orthostatic tachycardia. There were no clinically relevant changes in physical examination, hematology, clinical chemistry, urinalysis, suicidal ideation, or vital signs, with the exception of orthostatic changes in some subjects. No subject experienced abnormalities in EEGs or experienced a change from baseline in heart-rate-corrected QT interval (QTcF) > 60 ms, or an absolute QTcF interval > 480 ms at any post-dosing observation. Peak and total plasma exposure to TRV250 increased in a dose-proportional manner following 0.1–30 mg SC doses, with the mean half-life ranging from 2.39 to 3.76 h. Oral bioavailability of TRV250 ranged from 14% (fasting) to 19% (fed) relative to SC dosing, while administration with food increased the AUC but decreased the rate of absorption as reflected by a modest delay in median time to maximum concentration and a slight reduction in maximum concentration.
Conclusion
The findings from the first-in-human study support further evaluation of TRV250, a G-protein selective DOR agonist, in the treatment of acute migraine.
Key Points
| This trial is a first‐in‐human trial conducted in healthy volunteers to explore the safety, tolerability, and pharmacokinetics of TRV250. |
| TRV250 was well tolerated, with the most common AEs of injection-site reaction (including erythema and pain) and headaches, both of which were mild in most subjects and not dose related. |
| There were no clinically relevant changes in physical exams, ECGs, EEGs, suicidal ideation, hematology, clinical chemistries, urinalysis, or vital signs observed after TRV250 administration, with the exception that there were some TRV250-related orthostatic blood pressure and heart rate changes in some subjects. |
| Peak and total plasma exposure to TRV250 increased in a dose-proportional manner following 0.1–30 mg SC doses, with the mean half-life ranging from 2.39 to 3.76 h. Oral bioavailability of TRV250 was acceptable and ranged from 14% (fasting) to 19% (fed) relative to SC dosing. |
Introduction
Migraine headache is a debilitating disorder that affects daily functioning, and is the sixth most prevalent disease worldwide, with an estimated three billion people reporting a migraine and tension headache [1, 2]. Migraine is a chronic, recurrent neurologic disease characterized by episodes of severe headache pain, typically lasting from 4 to 72 h [3]. Headaches are usually unilateral, with a pulsating quality, can be aggravated by routine physical activity, and are often accompanied by nausea, photophobia, and phonophobia [2]. Migraine attacks can occur with or without an aura (a temporary neurologic disturbance of the sensory or motor functions) [2]. Functional imaging studies during a migraine demonstrate changes in blood flow along with changes in neuronal activity in various cortical locations, suggesting involvement of both vascular and neuronal components in the pathophysiology of the disorder [4]. Recent data point to a central role for calcitonin gene-related peptide (CGRP) in the development of migraine. Concentrations of CGRP in the CNS appear to be temporally related to the development of migraines. Chronic migraine patients have elevated CGRP levels in the CNS, and anti-CGRP therapies have been shown to be effective treatments in the clinic [5]. Delta receptors may modulate the release of CGRP, as these receptors are co-located in some of the dorsal root ganglia, which also produce CGRP [6, 7].
Conventionally, the use of triptans and nonsteroidal anti-inflammatory drugs (NSAIDs) represent therapy of choice for patients with acute migraine headache [8]. Although μ-opioid agonists are typically avoided, in patients with contraindications to triptans and who do not respond to NSAIDs, the use of μ-opioid medications is a treatment option [9]. Recently, the delta opioid receptor (DOR) has been identified as a potential therapeutic target for migraine [10]. In addition to producing analgesia, DOR agonists are associated with low abuse potential, display properties suggesting anxiolytic and antidepressant activity in animals, and no evidence of the development of hyperalgesia with chronic dosing.
Agonists acting at the DOR appear to be promising development candidates for acute migraine; however, clinical development of the earlier DOR agonist (SNC80) was halted due to its untoward convulsant effects [11, 12]. The second-generation DOR agonists (ARM-390 and ADL-5859) are reported to be devoid of convulsant properties [12]; however, their development was terminated due to failure to show pain reduction in early phase II trials [13].
Data suggest that β-arrestin2 recruitment plays a critical role in DOR-mediated convulsions [14, 15]. TRV250 is a novel small molecule agonist of the DOR that acts in a manner preferentially selective for G-protein signaling, with relatively little activation of the β-arrestin2 post-receptor signaling pathway. Findings from nonclinical studies have shown a substantial reduction in proconvulsant activity with TRV250 compared with other, non G-protein selective DOR agonists, with an established 50- to 80-fold margin between therapeutic effect and seizures in rodents and monkeys (Data on File). In the nitroglycerin-evoked hyperalgesia model in rodents used to screen candidates for potential utility in acute migraine [16], TRV250 significantly reduced hyperalgesia (Data on File). Thus, TRV250 could be an effective therapeutic candidate for the acute treatment of migraine without the risk of seizure that has been observed with previous candidates in the DOR agonist class. Here we report the findings from a first-in-human study evaluating the safety, tolerability, and pharmacokinetics (PK) of ascending single dose levels of TRV250 administered subcutaneously (SC), as well as the relative bioavailability of TRV250 administered orally compared with SC administration.
Methods
Compliance with Ethical Standards
The study was performed in accordance with International Conference on Harmonization Good Clinical Practice guidelines and ethical principles that have their origin in the Declaration of Helsinki. Protocols were approved by an Independent Ethics Committee before eligibility screening. Each subject provided written informed consent before any study-related procedures were performed.
Subjects
Healthy adult male or female subjects between 18 and 50 years of age with a body weight ≥ 50 kg and body mass index between 18 and 32.0 kg/m2, and who were capable of giving informed consent were enrolled. Subjects were determined as healthy by a responsible physician, based on a medical evaluation including history, physical examination, vital signs, electrocardiogram (ECG), electroencephalogram (EEG), and laboratory tests. Female subjects of child-bearing age were required to use an effective contraceptive method. Male subjects were required to use barrier methods of birth control for 90 days after the last dosing period.
Subjects were excluded if they had (a) any significant central nervous system, cardiac, pulmonary, metabolic, renal, hepatic, gastrointestinal, or psychiatric conditions or history of fainting or syncope that would interfere with the interpretation of safety or tolerability data obtained in this trial, (b) abnormal ECG, heart-rate-corrected QT interval (QTcF) > 450 ms, or abnormal EEG at screening, (c) aspartate aminotransferase (AST) or alanine aminotransferase (ALT) > upper limit of normal (ULN) at screening, (d) positive urine test for drugs of abuse, or alcohol, (e) positive for Human Immunodeficiency Virus (HIV) antibody, hepatitis B virus surface antigen, or hepatitis C virus antibody at screening, (f) history of smoking, (g) previously participated in a clinical trial within 30 days or 5 half-lives of the previous medication, (h) used any prescription or non-prescription medications (except acetaminophen ≤ 2 g/day), including herbal and dietary supplements (including St. John’s wort) within 14 days prior to the first dose of study medication, or (i) consumed grapefruit or grapefruit juice within 14 days prior to dosing. In addition, female subjects who had a positive pregnancy test or were pregnant at screening or prior to dosing in any period, as well as female subjects who were breastfeeding or planned to initiate breastfeeding within 90 days after their final dose of study medication, were also excluded.
Overall Study Design
The trial was conducted between 3 April 2017 (first subject dosed) and 13 February 2018 (last patient last visit) in a single center in the United Kingdom. This study was a randomized, single-blind, placebo-controlled study conducted in two parts. During Part A, subjects received single ascending doses of TRV250, or placebo, administered SC in order to evaluate the safety, tolerability, and single-dose PK of subcutaneously administered TRV250. During Part B, subjects received either single oral doses of TRV250 (as powder-in-capsule in the fed or fasted state) or placebo in order to determine the safety, PK, and relative oral bioavailability compared with the SC route of administration (Fig. 1). All subjects and the central EEG raters were blinded to treatment allocation, while the Investigator and the investigational pharmacist responsible for preparing the doses for administration were not. All subjects returned for a follow-up visit 7–10 days after their last dose of study medication for safety evaluation and lab measures.
Fig. 1.

Study design. During Part A, subjects received single ascending doses of TRV250, or placebo, administered subcutaneously (SC). Cohort 4 in Part A had 10 subjects dosed with TRV250 due to inclusion of a backfill subject. During Part B, subjects received either single oral doses of TRV250 (as powder-in-capsule in the fed state or fasted state) or placebo
Subcutaneous Dosing (Part A)
A total of four cohorts, each consisting of nine or ten subjects, were studied in part A. Eight sequences of allocation were prepared, and each sequence allocated nine subjects to two active doses of TRV250 and one placebo dose. The sequence allowed for sentinel dosing in two subjects (one on TRV250 and one on placebo) with no subject receiving a sentinel dose more than once. The overall allocation within a dose schedule was in the ratio of two subjects receiving TRV250 to every one receiving placebo. Cohorts were randomly assigned to one of the sequences, and subjects within the cohort were randomly assigned to one of the allocations.
Each subject received two SC doses of TRV250 and one placebo SC dose, with each dose separated by a minimum of 5 days, for a total of up to three dosing periods. Once a cohort completed three study periods, a new cohort of approximately nine subjects was initiated to receive ascending doses in the same fashion. Subjects who had been dosed in a previous cohort were not permitted to participate in subsequent cohorts. The starting dose of TRV250 was 0.1 mg SC; subsequent dosing levels were escalated in a stepwise fashion up to a dose of 30 mg, with each escalation only conducted following evaluation of the available safety, tolerability, and PK results from the previous dosing groups. The doses studied among the four cohorts were as follows: Cohort 1: 0.1 mg, 0.3 mg, 0.9 mg; Cohort 2: 2.5 mg, 4 mg, 6 mg; Cohort 3: 9 mg, 13.5 mg, 20 mg; and Cohort 4: 30 mg on two occasions. A matched volume (0.5–1.0 mL) of placebo solution (containing the same excipients in comparable amounts as in the TRV250 formulation) for injection was administered for each dose level. The site of injection was the abdomen and when multiple injections were required (for doses > 10 mg), different sites in the abdomen, each 2 cm away from any other injection site, were used. Subjects were fasted (with water permitted ad libitum during the fast until 1 h before dosing) from midnight the evening prior to each dose administration and for 4 h after dosing (with water permitted ad libitum after 1-h post-dose).
Oral Dosing (Part B)
In Part B, only a single cohort of nine subjects was enrolled. Seven subjects were allocated at random to receive two doses of TRV250 with one dose being received in the fed state and one in the fasted state. Two subjects were allocated at random to receive two doses of placebo with one dose being received in the fed state and one in the fasted state.
Subjects were randomized to receive either two active oral doses of TRV250 6 mg (as powder-in-capsule; n = 7), or two placebo doses (microcrystalline cellulose as a capsule; n = 2), in both the fed and fasted state.
All subjects began fasting at midnight, with water permitted ad libitum during the fast until 1 h before dosing. The fast ended approximately 30 min prior to dosing in the fed state, and 4 h after dosing in the fasted state. Water was permitted ad libitum starting 1 h after dosing. During the fed state, subjects received a standardized high fat meal (with approximately 250, 210, and 550 cal from protein, carbohydrate, and fat, respectively, for a total of approximately 1000 cal) and the study drug was administered within 30 min after the start of the meal with 240 mL of water. Subjects were instructed to complete the full meal within 30 min or less.
Sample Size
The sample size was selected to provide sufficient descriptive information on the safety, tolerability, and PK of TRV250. There were no prospective calculations of statistical power.
Plasma Assay
Plasma samples were analyzed using a validated LC–MS/MS assay with tandem mass spectrometric detection with an established limit of quantification of 0.1 ng/mL. Intra-day precision in the validation ranged from 0.8% to 4.8% and accuracy (bias) ranged from − 10.9% to − 0.8%. Inter-day precision and accuracy ranged from 1.7 to 5.8% and − 5.5 to − 2.3%, respectively. Plasma samples were stable at − 60 °C to − 80 °C for at least 535 days and were stable for at least 24 h at room temperature. Freeze–thaw stability was demonstrated across at least five cycles. Under refrigerated conditions (2–8 °C), processed samples were stable for at least 119 h.
Incurred sample reanalysis (ISR) was conducted on study samples to confirm the reproducibility of the bioanalytic method. It was observed that 77% of the repeat and original results were within 20% of the mean of the two values, meeting the pre-established acceptance criteria of 67%, confirming reproducibility of the assay.
Pharmacokinetic Assessment
Blood samples for pharmacokinetic (PK) assessment were collected pre-dose; and 0.25 h, 0.5 h, 0.75 h, 1 h, 1.5 h, 2 h, 4 h, 6 h, 9 h, 12 h, 16 h, and 24 h after dosing. PK parameters were calculated following SC and oral dosing, including area under the concentration–time curve to the time of the last quantifiable concentration (AUCt), area under the concentration–time curve extrapolated to infinity (AUCinf), maximum observed concentration (Cmax), time to maximum observed concentration (tmax), half-life (t½), and relative bioavailability (Frel).
Safety Assessments
The safety and tolerability of TRV250 were evaluated by recording the incidence and severity of adverse events (AEs) throughout the study, and from review of clinical laboratory assessments, vital signs, ECGs, physical examination, and EEG assessments. AE verbatim terms were coded according to the terminology of the Medical Dictionary for Regulatory Activities (MedDRA), Version 20.0.
EEG Evaluation
At screening, EEGs were performed in order to exclude subjects with spike/sharp wave (SSW) abnormalities suggesting a risk for seizure. During the study, spontaneous EEGs (Grass Technologies, West Warwick, RI, USA) were obtained over 10-min periods pre-dose and at approximately 15 and 30 min, and 1 and 4 h following dose administration. An EEG headcap was worn starting pre-dose and for up to 8 h post-dose for scheduled recordings and for any unscheduled recording, requested by the study physician, in association with any clinical phenomena considered to suggest the presence of seizure activity. Each EEG was independently scored as normal or abnormal by two central EEG raters prior to treatment condition and prior to each dose escalation.
Safety Stopping Criteria (ECG Evaluation)
Any subject observed to show a QTcF > 500 ms or uncorrected QT > 600 ms or change from baseline in QTcF > 60 ms at any timepoint was withdrawn from the study. In addition, if these changes occurred in two or more subjects at a given dose level, dosing at that level was ceased and no further dose escalation was carried forward.
Safety Stopping Criteria (Additional)
Any subject who experienced symptomatic hypotension, bradycardia, or tachycardia necessitating medical intervention was withdrawn from the study. If two or more subjects administered TRV250 experienced changes in blood pressure or heart rate in a dosing period necessitating withdrawal from the study, dosing in that period was ceased and no further dose escalation occurred. If any subject administered TRV250 in a cohort experienced increases in AST or ALT > 5-fold ULN, or > 3-fold ULN with concomitant serum total bilirubin > 2-fold ULN (including > 35% direct bilirubin), dose escalation was ceased, and no further dose escalation occurred.
Additional criteria necessitating withdrawal from the study were (a) a serious AE (SAE) observed within a cohort that was possibly related or probably related to study drug, (b) a severe AE occurring in two or more subjects administered TRV250 at a given dose level or within a cohort, or (c) significant adverse events or significant findings from safety assessments (e.g., vital signs, laboratory tests).
Pharmacokinetic Stopping Criteria
The study also included PK stopping criteria, where the dose was not to be further escalated if, at the next projected dose, there was a > 5% probability (based on simulations from data obtained from previous doses) that any individual subject was predicted to meet or exceed an AUC of 1128 ng*h/mL or a Cmax of 482 ng/mL. These limits correspond to total concentrations (after accounting for differences in protein binding between monkeys and humans) in monkeys receiving TRV250 2.5 mg/kg who had transient decreases in heart rate, mild to moderate decreases in arterial blood pressure (≥ 15%), and increases in the QTcF (all monitorable tolerability parameters that are reversible).
Statistical Analysis
The safety population included all subjects who received at least one dose of TRV250 or placebo. Descriptive summaries of safety and tolerability data were performed. Vital signs, clinical laboratory tests, ECG, and oxygen saturation were listed and summarized descriptively by treatment group and also reported as the change from baseline. EEG findings, the number and percentage of subjects with physical examination findings for each body system, and the number and percentage of subjects with an injection-site reaction were summarized.
The PK population included all subjects who received at least one dose of TRV250 and who had sufficient data to compute AUCt and other PK parameters. Plasma TRV250 concentration–time data were analyzed by non-compartmental methods using Phoenix® WinNonlin® 6.3 (Certara, Princeton, NJ, USA) for the interim analyses and PKPlus™ 2 (Simulations Plus, Lancaster, CA, USA) for the final analysis. Plasma concentration–time data and PK parameters were listed by treatment and subjects and summarized by treatment. To determine PK stopping criteria, simulations were conducted to predict the Cmax and AUCinf for projected doses following each dosing period using ModelRisk® Software (Vose Software).
For Part B, the relative bioavailability (%Frel) was estimated by comparison of AUCinf and Cmax for each 6-mg oral dose (capsule in fed state and fasted state) with SC and by comparison of the capsule in the fed state with the capsule in the fasted state. Geometric least squared means (GLSM), ratio of GLSM, and 90% confidence intervals were obtained from mixed-effect models for each comparison and parameter, fitted to the natural log of the PK parameter (with subsequent back transformation), with method of administration as a fixed effect and subject as a random effect.
Results
Baseline Characteristics
Forty-seven subjects were enrolled; 38 participated in Part A and nine participated in Part B (Fig. 2). Of the 38 subjects enrolled in Part A, 32 completed the study. Among the six subjects who discontinued early, two subjects voluntarily withdrew from the study, one subject was removed from participation due to a positive test for drugs of abuse (cocaine, opioids, benzodiazepines, cannabinoids, amphetamine, barbiturates) following the first dosing period, one subject was removed due to Investigator decision, one subject was withdrawn due to an adverse event, and one subject, who was a replacement subject, completed only a single dosing period. All nine subjects enrolled in Part B completed the study. In general, study subjects were predominantly young and White (Table 1). The demographics were similar among subjects participating in Part A and Part B, with the exceptions that the subjects in Part B were approximately 6 years younger and weighed an average of 7 kg less than subjects in Part A (Table 1).
Fig. 2.

Study disposition. D/C discontinued
Table 1.
Baseline demographics of subjects in Part A and Part B of the study
| Characteristic | Part A (n = 38) | Part B (n = 9) |
|---|---|---|
| Male, n (%) | 21 (55.3) | 5 (55.6) |
| Female, n (%) | 17 (44.7) | 4 (44.4) |
| Age, years, mean ± SD | 31.8 ± 9.7 | 26.0 ± 8.6 |
| Race, n (%) | ||
| White | 36 (94.7) | 9 (100) |
| Asian | 1 (2.6) | |
| African American | 1 (2.6) | |
| Weight, kg, mean ± SD | 76.3 ± 14.4 | 68.9 ± 5.8 |
| BMI, kg/m2, mean ± SD | 25.2 ± 3.2 | 24.1 ± 2.9 |
SD standard deviation, BMI body mass index
Pharmacokinetics
Pharmacokinetics after TRV250 Subcutaneous Administration
Absorption of TRV250 after SC administration was rapid, with average peak concentrations reached about 1.5 h after administration. Following Cmax, TRV250 concentrations decreased in a monophasic manner over 24 h (Fig. 3).
Fig. 3.
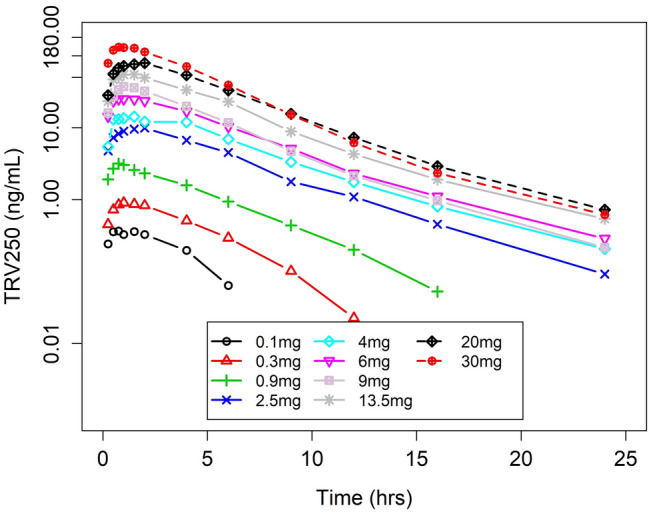
Mean plasma concentrations of TRV250 after subcutaneous (SC) dosing over time (Part A)
PK parameters by dose are summarized in Table 2. Geometric mean Cmax and AUCinf values following a 30-mg SC dose of TRV250 were 151 ng/mL and 652 ng*h/mL, respectively. The median tmax ranged from 0.75 to 2 h, and the t½ remained relatively constant across all doses, with the geometric mean ranging from 2.39 to 3.76 h (Table 2).
Table 2.
TRV250 pharmacokinetic parameters by dose after SC administration (Part A)
| Dose | Cmaxa (ng/mL) | tmaxb (h) | AUCinfa (ng*h/mL) | t½a (h) |
|---|---|---|---|---|
| 0.1 mg (n = 6) | 0.388 (34.0%) [0.240–0.568] | 1 [0.5–1.5] | 2.81 (15.9%) [2.27–3.46] | 2.80 (34.7%) [2.06–5.14] |
| 0.3 mg (n = 5) | 0.950 (13.6%) [0.774–1.10] | 1 [0.5–1.5] | 5.47 (11.3%) [5.04–5.69] | 2.39 (17.9%) [1.94–3.00] |
| 0.9 mg (n = 6) | 3.18 (33.7%) [2.27–5.15] | 0.75 [0.5–1.03] | 15.7 (14.6%) [13.2–19.3] | 2.64 (27.7%) [1.82–3.52] |
| 2.5 mg (n = 7) | 10.5 (11.7%) [9.26–12.5] | 1.5 [0.5–2] | 60.8 (30.7%) [43.6–96.8] | 2.72 (26.1%) [1.93–3.97] |
| 4.0 mg (n = 6) | 16.8 (36.6%) [11.0–25.8] | 1.53 [0.5–4.35] | 97.7 (23.1%) [77.9–141] | 3.49 (15.8%) [3.05–4.61] |
| 6.0 mg (n = 6) | 30.2 (26.5%) [20.4–42.2] | 1.13 [0.5–2.07] | 157 (21.8%) [120–217] | 3.64 (28.4%) [2.33–5.00] |
| 9.0 mg (n = 6) | 38.7 (32.0%) [27.8–60.2] | 1.26 [0.75–1.5] | 187 (18.0%) [160–244] | 3.37 (27.7%) [2.13–4.37] |
| 13.5 mg (n = 6) | 56.5 (20.2%) [38.7–69.1] | 1.25 [0.75–2] | 318 (19.0%) [256–412] | 3.76 (21.6%) [2.58–4.63] |
| 20 mg (n = 5) | 79.6 (24.7%) [58.2–102] | 2 [1–2] | 474 (27.5%) [377–698] | 3.39 (21.8%) [2.78–4.87] |
| 30 mgc (n = 16) | 151 (20.9%) [97.8–230] | 1 [0.5–2] | 652 (17.9%) [522–939] | 3.17 (21.6%) [2.29–4.53] |
AUCinf area under the concentration–time curve extrapolated to infinity, Cmax maximum observed concentration, CV coefficient of variance, SC subcutaneous, tmax time to maximum observed concentration, t½ half-life
aGeometric mean (geometric CV%) [min–max]
bMedian [min–max]
cData from DP1–3 combined (n = 16)
Pharmacokinetics After TRV250 Oral Administration
The mean concentration–time data for a TRV250 6-mg oral dose administered in the fed and fasted state are represented in Fig. 4. The plasma concentration–time profiles following oral administration of TRV250 6 mg in the fed and fasted state are overlaid with the SC administration of TRV250 6 mg in Fig. 4. The tmax following oral administration was later than after SC administration in the fed state, but similar to SC administration in the fasted state. Concentrations of TRV250 were much lower after oral administration than after the same dose given SC. Following the peak, TRV250 concentrations decreased in an approximate monophasic manner over 16 h.
Fig. 4.
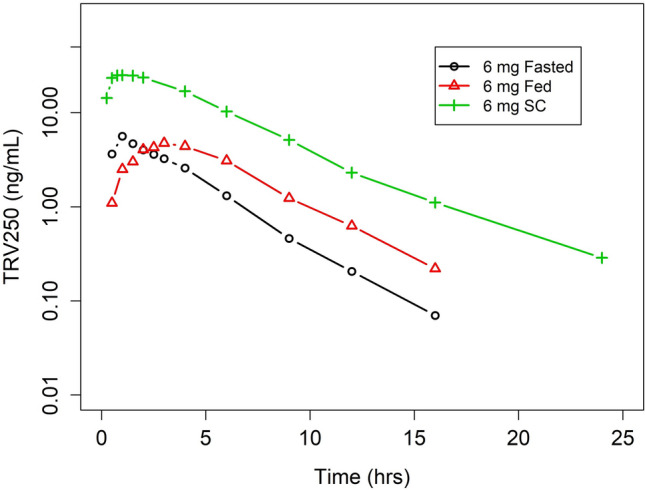
Mean plasma concentrations of TRV250 by fed status and dosage (oral vs SC) form. SC subcutaneous
PK parameters following TRV250 6 mg as an oral dose administered in the fed and fasted states are summarized in Table 3. Geometric mean Cmax values following TRV250 6 mg orally in the fed and fasted states were 5.28 ng/mL and 6.06 ng/mL, respectively. Geometric mean AUCinf values following TRV250 6 mg orally in the fed and fasted states were 31.6 and 22.8 ng*h/mL, respectively. Relative to TRV250 administered orally in the fasted state, the AUC was higher based on geometric least square mean ratio, expressed as a percentage (138%; 90% CI 123–156) and the Cmax was lower (87%; 90% CI 73.0–104) when TRV250 was administered in the fed state.
Table 3.
TRV250 pharmacokinetic parameters after oral administration in the fed and fasted state (Part B)
| Treatment | Cmaxa (ng/mL) | tmaxb (h) | AUCinfa (ng*h/mL) | t½a (h) |
|---|---|---|---|---|
| Fed (6 mg) | 5.28 (40.1%) [2.61–7.68] | 3.22 [3–6] | 31.6 (34.2%) [17.0–50.3] | 2.60 (21.6%) [1.69–3.32] |
| Fasted (6 mg) | 6.06 (24.3%) [4.82–8.59] | 1 [1–3] | 22.8 (25.3%) [16.2–30.8] | 2.52 (31.1%) [1.60–4.12] |
AUCinf area under the concentration–time curve extrapolated to infinity, Cmax maximum observed concentration, CV coefficient of variance, tmax time to maximum observed concentration, t½ half-life
aGeometric mean (geometric CV%) [min–max]
bMedian [min–max]
Compared with SC, the oral bioavailability of TRV250 was relatively lower regardless of whether it was administered in the fasted or fed state. Based on an assessment using total exposure (AUCinf), the relative bioavailability of oral compared with SC administration was higher for TRV250 administered in the fed state (19.1%; 90% CI 15.6–23.3) than in the fasted state (13.8%; 90% CI 11.3–16.8).
Safety and Tolerability of TRV250
Of the 38 subjects in Part A, 29 experienced at least one adverse event (AE). All events were considered mild with the exception of four AEs (in three subjects) that were of moderate intensity and included pain at injection site (n = 1, subject receiving placebo), headache (n = 1, subject received TRV250 0.9-mg dose), postural orthostatic tachycardia and increased heart rate (both in the same patient receiving TRV250 0.1-mg dose) occurring 4.5 h after dosing. The patient with the postural orthostatic tachycardia was withdrawn from the study. In Part B, among the seven subjects who received oral TRV250 6 mg, four subjects in the fasted state and two subjects in the fed state experienced mild AEs, with only one subject experiencing muscle fasciculation and lethargy possibly related to the study drug.
A summary of AEs by preferred term for both Part A and Part B is listed in Tables 4 and 5, respectively. The most common AEs after SC dosing were injection-site reaction, including pain and erythema reported at TRV250 dose levels of 0.1 mg (n = 2, Cohort 1), 2.5 mg (n = 1, Cohort 2), 9 mg (n = 2, Cohort 3), 30 mg (n = 3, Cohort 4), and placebo (n = 7); and headache reported at TRV250 dose levels of 0.3 mg (n = 2, Cohort 1), 0.9 mg (n = 2, Cohort 1), 2.5 mg (n = 1, Cohort 2), 20 mg (n = 1, Cohort 3), 30 mg (n = 4, Cohort 4), and placebo (n = 3).
Table 4.
Treatment-emergent AEs by MedDRA preferred term with TRV250 SC administration (Part A)
| AE | Part A TRV250 | |||||||
|---|---|---|---|---|---|---|---|---|
| Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | |||||
| TRV250 N = 9 |
Placebo N = 8 |
TRV250 | Placebo | TRV250 | Placebo | TRV250 | Placebo | |
| Subjects with at least one AE | 8 (88.9) | 3 (37.5) | 6 (60.0) | 0 | 6 (60.0) | 5 (62.5) | 6 (75.0) | 7 (77.8) |
| Number of events | 19 | 5 | 10 | 0 | 9 | 6 | 24 | 11 |
| Injection-site reaction (including pain and erythema) | 2 (22.2) | 1 (12.5) | 1 (10.0) | 0 | 2 (20.0) | 3 (37.5) | 3 (37.5) | 3 (33.3) |
| Catheter-site pain | 0 | 0 | 0 | 0 | 0 | 0 | 2 (25.0) | 0 |
| Headache | 4 (44.4) | 1 (12.5) | 1 (10.0) | 0 | 1 (10.0) | 0 | 4 (50.0) | 2 (22.2) |
| Nausea | 2 (22.2) | 1 (12.5) | 1 (10.0) | 0 | 0 | 1 (11.1) | ||
| Procedural nausea | 0 | 0 | 0 | 0 | 0 | 0 | 1 (12.5) | 0 |
| Dizziness | 3 (33.3) | 0 | 0 | 0 | 0 | 0 | 1 (12.5) | 0 |
| Back pain | 0 | 0 | 0 | 0 | 1 (10.0) | 2 (25.0) | 1 (12.5) | 1 (11.1) |
| Oropharyngeal pain | 0 | 0 | 2 (20.0) | 0 | 0 | 0 | 3 (37.5) | 0 |
| Arthralgia | 0 | 0 | 1 (10.0) | 0 | 0 | 0 | 0 | 0 |
| Amylase increased | 1 (11.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dysmenorrhea | 2 (22.2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Heart rate increased | 1 (11.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Lipase increased | 1 (11.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Nasal congestion | 1 (11.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Postural orthostatic tachycardia | 1 (11.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Rash | 0 | 1 (12.5) | 0 | 0 | 0 | 0 | 0 | 0 |
| Swelling face | 0 | 1 (12.5) | 0 | 0 | 0 | 0 | 0 | 0 |
| Vomiting | 1 (11.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Medical device site pruritis | 0 | 0 | 1 (10.0) | 0 | 0 | 0 | 0 | 0 |
| Musculoskeletal chest pain | 0 | 0 | 1 (10.0) | 0 | 0 | 0 | 0 | 0 |
| Musculoskeletal discomfort | 0 | 0 | 1 (10.0) | 0 | 0 | 0 | 0 | 0 |
| Red blood cell count decreased | 0 | 0 | 0 | 1 (10.0) | 0 | 0 | 0 | 0 |
| Abdominal pain lower | 0 | 0 | 0 | 0 | 1 (10.0) | 0 | 0 | 0 |
| Dysgeusia | 0 | 0 | 0 | 0 | 0 | 1 (12.5) | 0 | 0 |
| Epistaxis | 0 | 0 | 0 | 0 | 1 (10.0) | 0 | 0 | 0 |
| Menorrhagia | 0 | 0 | 0 | 0 | 1 (10.0) | 0 | 0 | 0 |
| Traumatic hematoma | 0 | 0 | 0 | 0 | 1 (10.0) | 0 | 0 | 0 |
| Viral upper respiratory tract infection | 0 | 0 | 0 | 0 | 0 | 0 | 3 (37.5) | 0 |
| Presyncope | 0 | 0 | 0 | 0 | 0 | 0 | 2 (25.0) | 0 |
| Head injury | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (11.1) |
| Influenza-like illness | 0 | 0 | 0 | 0 | 0 | 0 | 1 (12.5) | 0 |
| Lymph gland infection | 0 | 0 | 0 | 0 | 0 | 0 | 1 (12.5) | 0 |
| Rhinalgia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (12.5) |
| Supraventricular extrasystole | 0 | 0 | 0 | 0 | 0 | 0 | 1 (12.5) | 0 |
Percentages are based on the number of subjects in each treatment. Subjects summarized by actual treatment received. Adverse events were coded using the MedDRA Dictionary, version 20.0
TRV250 dosing Cohort 1: 0.1 mg, 0.3 mg, 0.9 mg; Cohort 2: 2.5 mg, 4 mg, 6 mg; Cohort 3: 9 mg, 13.5 mg, 20 mg; and Cohort 4: all patients received 30 mg
AE adverse event, MedDRA Medical Dictionary for Regulatory Activities, SC subcutaneous
Table 5.
Treatment-emergent AEs by MedDRA preferred term with TRV250 oral administration (Part B)
| AE | 6 mg Fed N = 7 |
6 mg Fasted N = 7 |
Placebo N = 2 |
|---|---|---|---|
| Subjects with at least one AE | 2 (28.6) | 4 (57.1) | 1 (50.0) |
| Number of events | 2 | 5 | 2 |
| Cystitis | 1 (14.3) | 0 | 0 |
| Dysmenorrhea | 0 | 1 (14.3) | 0 |
| Lethargy | 0 | 1 (14.3) | 0 |
| Medical device site irritation | 0 | 1 (14.3) | 0 |
| Medical device site pruritis | 0 | 0 | 1 (50.0) |
| Muscle contractions (involuntary) | 0 | 1 (14.3) | 0 |
| Peripheral swelling | 0 | 1 (14.3) | 0 |
| Postural orthostatic tachycardia | 0 | 0 | 1 (50.0) |
| Presyncope | 1 (14.3) | 0 | 0 |
Percentages are based on the number of subjects in each treatment. Subjects summarized by actual treatment received. Adverse events were coded using the MedDRA Dictionary, version 20.0
The AEs in the placebo group were in the fed state
AE adverse event, MedDRA Medical Dictionary for Regulatory Activities
There were 23 AEs in Part A and two AEs in Part B that were considered possibly or probably related to TRV250 (Tables 6, 7). Of these, only one AE (in a subject receiving TRV250 0.1 mg) led to withdrawal due to postural orthostatic tachycardia (as mentioned above). There were no deaths or serious AEs reported in the study.
Table 6.
Summary of AEs possibly or probably related to study treatment by cohort in Part A
| AE | Cohort 1 (0.1–0.9 mg) | Cohort 2 (2.5–6 mg) | Cohort 3 (9–20 mg) | Cohort 4 (30 mg) | Placebo |
|---|---|---|---|---|---|
| Dizziness | 3 | 0 | 0 | 1 | 0 |
| Injection-site reaction (including pain and erythema) | 2 | 1 | 2 | 3 | 7 |
| Headache | 1 | 0 | 1 | 3 | 1 |
| Postural orthostatic tachycardia | 1 | 0 | 0 | 0 | 0 |
| Epistaxis | 0 | 0 | 1 | 0 | 0 |
| Vasovagal episodes | 0 | 0 | 0 | 2 | 0 |
| Nausea | 0 | 0 | 0 | 0 | 1 |
| Supraventricular extrasystoles | 0 | 0 | 0 | 1 | 0 |
| Presyncope | 0 | 0 | 0 | 3 | 0 |
AE adverse event
Table 7.
Summary of AEs possibly or probably related to study treatment by cohort in Part B
| AE | TRV250 | Placebo | ||
|---|---|---|---|---|
| 6 mg Fed | 6 mg Fasted | Fed | Fasted | |
| Lethargy | 0 | 1 | 0 | 0 |
| Muscle contractions | 0 | 1 | 0 | 0 |
Both AEs occurred in the same subject
AE adverse event
There were no clinically relevant changes in physical examination, hematology, clinical chemistry, urinalysis, suicidal ideation, or vital signs, with the exception of orthostatic changes in few subjects (Table 8).
Table 8.
Orthostatic changes
| N | Dose of TRV250 | Description |
|---|---|---|
| Symptomatic changes | ||
| 1 | 0.9 mg | Dizziness, BP of ~ 75/40 at 4-h time point |
| 1 | 30 mg | Pre-syncopal episode at 4 h on standing; associated non-sustained sinus tachycardia of 146 beats/min |
| 1 | 30 mg (at the third dosing period) | Low baseline BP pre-dose that dipped below normal for 4 h post-dose and three pre-syncopal episodes on standing at 4 h |
| Asymptomatic changesa | ||
| 2 | 0.1 mg | Drop in SBP or DBP of > 5 mmHg, with an increase in HR of > 20 beats/min |
| 2 | 0.3 mg | |
| 2 | 0.9 mg | |
| 1 | 2.5 mg | |
| 1 | 6 mg | |
| 1 | 20 mg | |
| 2 | 30 mg | |
BP blood pressure, DBP diastolic blood pressure, HR heart rate, SBP systolic blood pressure
aAsymptomatic changes also observed in three placebo subjects
There were no clinically significant changes from baseline observed upon review of EEGs in individual subjects. No individual subject responded in the affirmative to any question posed in the Columbia-Suicide Severity Rating Scale (C-SSRS) during the study and follow-up assessment throughout the study, including follow-up.
There were no dose-dependent, drug-related changes in categorical QTcF interval measures among individual subjects (Table 9). In particular, there were no subjects with a > 60 ms increase in QTcF or with QTcF values > 500 ms.
Table 9.
QTcF changes related to study treatment by cohort
| Study parts | ΔQTcF > 30 but < 60 ms |
ΔQTcF > 60 s |
QTcF > 450 ms |
QTcF > 500 ms |
||
|---|---|---|---|---|---|---|
| TRV250 dose, n | Placebo, n | TRV250 dose, n | Placebo, n | |||
| Part A: Cohort 1 | 0.3 mg, n = 1 | n = 1 | 0 |
0.3 mg, n = 1 0.9 mg, n = 2 |
n = 2 | 0 |
| Part A: Cohort 2 |
4 mg, n = 2 6 mg, n = 1 |
n = 2 | 0 | 4 mg, n = 1 | n = 2 | 0 |
| Part A: Cohort 3 |
9 mg, n = 2 13.5 mg, n = 1 20 mg, n = 2 |
n = 3 | 0 |
13.5 mg, n = 2 20 mg, n = 1 |
n = 2 | 0 |
| Part A: Cohort 4 |
30 mg, n = 1(DP1) n = 2 (DP2) n = 3 (DP3) |
n = 1 | 0 |
30 mg n = 1 (DP2) n = 1 (DP3) |
n = 1 | 0 |
| Part B | 0 | 0 | 0 | 6 mg fasted, n = 1 | 0 | 0 |
DP dosing period, QTcF heart-rate-corrected QT interval
Discussion
Pharmacokinetics
This trial was a first‐in-human trial conducted in healthy volunteers to explore the safety, tolerability, and PK of TRV250. TRV250 showed predictable PK after SC administration, with exposures increasing proportionally with doses between 0.1 mg and 30 mg SC. The observed half-life was consistent across all doses, ranging between 2.39 and 3.76 h. Oral bioavailability of TRV250 was lower (14–19%) when compared with SC administration, regardless of whether TRV250 was administered in a fasted or fed state. When administered after a meal, a food effect was observed that was characterized by an increase in bioavailability but a decrease in the rate of absorption, with a slightly lower Cmax (87%), a modest delay in the median tmax, and a higher AUC (138%). This was consistent with the delayed gastric emptying usually observed after a high-fat meal. These PK data suggest that oral dosing of TRV250 is feasible. The oral bioavailability of TRV250 at 14–19% relative to SC is also similar to those values reported for sumatriptan [17].
Safety
TRV250 was well tolerated; the most common AEs were injection-site reactions and headache, both of which were mild in most subjects and were not dose related. There were no serious AEs reported or deaths in this study. One subject receiving TRV250 (0.1 mg) discontinued due to experiencing postural orthostatic tachycardia 4.5 h after dosing. There were no clinically relevant changes in hematology, clinical chemistries, or urinalysis observed after TRV250 administration. According to the C-SSRS responses, no subjects reported any suicide-related thoughts or behaviors.
Of note, there were no clinically relevant changes in EEG. Absence of abnormal EEG findings support the lack of proconvulsant activity with TRV250 as seen in animal studies [18].
There were no clinically significant findings in vital signs, with the exception of orthostatic changes (symptomatic, including blood pressure changes or presyncopal episodes; or asymptomatic, defined as a drop in systolic or diastolic blood pressure of ≥ 5 mmHg, with an increase in HR of > 20 beats/min) in some subjects. These findings were confounded by the fact that subjects were in the supine position for 4 h and may not have been well hydrated during this time. Nevertheless, an effect of TRV250 on the orthostatic vital signs cannot be ruled out.
There were no dose-dependent changes in QTcF interval, and no excess of categorical outliers across the dose range studied. Few subjects both in the active phase or placebo phase experienced changes in QTcF between 30 and 60 ms or QTcF > 450 ms. There were no subjects with a > 60-ms increase in QTcF or with QTcF values ≥ 500 ms. There were no clinically relevant changes in ECG.
Limitations
There are a few limitations to the conclusions that can be made from this study. Because subjects were healthy, the pharmacokinetic data collected represents the best case and does not include variability due to patient covariates. As the study was performed in a small number of subjects, conclusions related to safety can only be made for common adverse events, but not rare adverse events.
Conclusions
TRV250 SC doses of 0.6–30 mg and oral doses of 6 mg in the fed and fasted state were generally well tolerated. Based on the available data from this first-in-human study demonstrating safety, tolerability, and an attractive PK profile, further development of TRV250, a G protein-selective DOR agonist, for the treatment of acute migraine is warranted.
Acknowledgements
The authors would like to thank the volunteers who took part in the trial, as well as the staff who assisted with the trial. They would also like to thank Kanaka Sridharan, (Scientific Communications, Trevena, Inc.) for providing editorial support and Linda Wase (VP Medical Affairs, Trevena, Inc.) for her review of the manuscript.
Data Availability
The datasets generated and/or analyzed during the current trial are not yet publicly available.
Compliance with Ethical Standards
Funding
This trial was funded by Trevena, Inc. Nuventra Pharma Sciences was the consulting firm that analyzed the data. Funding for open access was provided by Trevena, Inc.
Conflict of interest
Michael J. Fossler, Michael S. Kramer, Kelly Arscott and Mark A. Demitrack are employees of Trevena, Inc. and own stocks/stock options in the company. At the time of completion of the study, Ian E. James was an employee at Trevena, Inc. Virginia Schmith is a paid consultant of Trevena, Inc. At the time of conduct of the study, Stephen Greene was a fellow at the University of North Carolina at Chapel Hill and worked on the project for Nuventra Pharma Sciences. Lauren Lohmer is an employee at Nuventra Pharma Sciences.
Research involving human participants
This trial was conducted in accordance with International Conference on Harmonisation Good Clinical Practice guidelines and ethical principles that have their origin in the Declaration of Helsinki. Protocols were approved by an Independent Ethics Committee before eligibility screening.
Informed consent
Written informed consent was obtained from each subject before any trial-related procedures were performed.
References
- 1.GBDH Collaborators Global, regional, and national burden of migraine and tension-type headache, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018;17(11):954–976. doi: 10.1016/S1474-4422(18)30322-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen D, Willis-Parker M, Lundberg GP. Migraine headache: Is it only a neurological disorder? Links between migraine and cardiovascular disorders. Trends Cardiovasc Med. 2019 doi: 10.1016/j.tcm.2019.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Migraine Facts. National Headache Foundation; 2007; https://headaches.org/2007/11/20/migraine-facts/. Accessed Feb 7, 2020.
- 4.Charles A, Brennan KC. The neurobiology of migraine. Handb Clin Neurol. 2010;97:99–108. doi: 10.1016/S0072-9752(10)97007-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charles A. The pathophysiology of migraine: implications for clinical management. Lancet Neurol. 2018;17(2):174–182. doi: 10.1016/S1474-4422(17)30435-0. [DOI] [PubMed] [Google Scholar]
- 6.Bertels Z, Pradhan AAA. Emerging treatment targets for migraine and other headaches. Headache. 2019;59(Suppl 2):50–65. doi: 10.1111/head.13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edvinsson L, Haanes KA, Warfvinge K, Krause DN. CGRP as the target of new migraine therapies—successful translation from bench to clinic. Nat Rev Neurol. 2018;14(6):338–350. doi: 10.1038/s41582-018-0003-1. [DOI] [PubMed] [Google Scholar]
- 8.Pomes LM, Guglielmetti M, Bertamino E, Simmaco M, Borro M, Martelletti P. Optimising migraine treatment: from drug-drug interactions to personalized medicine. J Headache Pain. 2019;20(1):56. doi: 10.1186/s10194-019-1010-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ong JJY, De Felice M. Migraine treatment: current acute medications and their potential mechanisms of action. Neurotherapeutics. 2018;15(2):274–290. doi: 10.1007/s13311-017-0592-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moye LS, Tipton AF, Dripps I, Sheets Z, Crombie A, Violin JD, et al. Delta opioid receptor agonists are effective for multiple types of headache disorders. Neuropharmacology. 2019;148:77–86. doi: 10.1016/j.neuropharm.2018.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Danielsson I, Gasior M, Stevenson GW, Folk JE, Rice KC, Negus SS. Electroencephalographic and convulsant effects of the delta opioid agonist SNC80 in rhesus monkeys. Pharmacol Biochem Behav. 2006;85(2):428–434. doi: 10.1016/j.pbb.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung PCS, Boehrer A, Stephan A, Matifas A, Scherrer G, Darcq E, et al. Delta opioid receptors expressed in forebrain GABAergic neurons are responsible for SNC80-induced seizures. Behav Brain Res. 2015;278:429–434. doi: 10.1016/j.bbr.2014.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spahn V, Stein C. Targeting delta opioid receptors for pain treatment: drugs in phase I and II clinical development. Expert Opin Investig Drugs. 2017;26(2):155–160. doi: 10.1080/13543784.2017.1275562. [DOI] [PubMed] [Google Scholar]
- 14.Dripps IJ, Boyer BT, Neubig RR, Rice KC, Traynor JR, Jutkiewicz EM. Role of signalling molecules in behaviours mediated by the δ opioid receptor agonist SNC80. Br J Pharmacol. 2018;175(6):891–901. doi: 10.1111/bph.14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vicente-Sanchez A, Dripps IJ, Tipton AF, Akbari H, Akbari A, Jutkiewicz EM, et al. Tolerance to high-internalizing δ opioid receptor agonist is critically mediated by arrestin 2. Br J Pharmacol. 2018;175(14):3050–3059. doi: 10.1111/bph.14353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moye LS, Pradhan AAA. Animal model of chronic migraine-associated pain. Curr Protoc Neurosci. 2017;80:9.60.1–9.60.9. doi: 10.1002/cpns.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferrari A, Tiraferri I, Neri L, Sternieri E. Why pharmacokinetic differences among oral triptans have little clinical importance: a comment. J Headache Pain. 2011;12(1):5–12. doi: 10.1007/s10194-010-0258-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crombie A, Arezzo J, Cowan C, Dewire S, Gowen-MacDonald W, Hawkins M, et al. TRV250: a novel biased ligand at the delta receptor for the potential treatment of migraine. Cephalalgia. 2015;35:1225–1226. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analyzed during the current trial are not yet publicly available.