

Abstract
Background and Purpose
ACE inhibitors (ACEIs) and AT1 receptor antagonists (ARBs) are first‐line drugs that are believed to reduce the progression of end‐stage renal disease in diabetic patients. Differences in the effects of ACEIs and ARBs are not well studied and the mechanisms responsible are not well understood.
Experimental Approach
Male diabetic CD‐1 mice were treated with ACEI, ARB, N‐acetyl‐seryl‐aspartyl‐lysyl‐proline (AcSDKP), ACEI + AcSDKP, ARB + AcSDKP, glycolysis inhibitors or non‐treatment. Moreover, prolyl oligopeptidase inhibitor (POPi)‐injected male diabetic C57Bl6 mice were treated with ACEI, AcSDKP and ARB or non‐treatment. Western blot and immunofluorescent staining were used to examine key enzymes and regulators of central metabolism.
Key Results
The antifibrotic action of ACEI imidapril is due to an AcSDKP‐mediated antifibrotic mechanism, which reprograms the central metabolism including restoring SIRT3 protein and mitochondrial fatty acid oxidation and suppression of abnormal glucose metabolism in the diabetic kidney. Moreover, the POPi S17092 significantly blocked the AcSDKP synthesis, accelerated kidney fibrosis and disrupted the central metabolism. ACEI partly restored the kidney fibrosis and elevated the AcSDKP level, whereas the ARB (TA‐606) did not show such effects in the POPi‐injected mice. ACE inhibition and AcSDKP suppressed defective metabolism‐linked mesenchymal transformations and reduced collagen‐I and fibronectin accumulation in the diabetic kidneys.
Conclusion and Implications
The study envisages that AcSDKP is the endogenous antifibrotic mediator that controls the metabolic switch between glucose and fatty acid metabolism and that suppression of AcSDKP leads to disruption of kidney cell metabolism and activates mesenchymal transformations leading to severe fibrosis in the diabetic kidney.
Abbreviations
- 2‐DG
2‐deoxyglucose
- ACEI
angiotensin converting enzyme inhibitor
- ACEi
angiotensin converting enzyme inhibition
- AcSDKP
N‐acetyl‐seryl‐aspartyl‐lysyl‐proline
- ARB
AT1 antagonist/blocker
- CPT1a
carnitine palmitoyltransferase 1a
- DCA
dichloroacetate
- GLUT1
glucose transporter protein 1
- HIF1α
hypoxia‐inducible factor‐1α
- HK2
hexokinase 2
- PDK4
pyruvate dehydrogenase kinase 4
- PGC1α
PPARγ coactivator 1α
- PKM2
pyruvate kinase M2 type
- POP
prolyl oligopeptidase
- RAAS
renin angiotensin aldosterone system
- SIRT3
NAD‐dependent deacetylase sirtuin‐3
- STZ
streptozotocin
- αSMA
α‐smooth muscle actin
What is already known
ACE inhibition is protective against diabetic kidney disease.
AT1 antagonists have minimal reno‐protective action in the mouse model of diabetic kidney disease.
What this study adds
Protective nature of ACE inhibition is due to the induction of AcSDKP‐mediated antifibrotic action.
ACE inhibition and AcSDKP disrupt the defective metabolism‐linked mesenchymal transformations in the diabetic kidney.
What is the clinical significance
AcSDKP alone or in combination with ACEIs can be directly used in combating diabetic kidney disease.
Glycolysis inhibitors are antifibrotic agents which elevate AcSDKP and could be used in kidney fibrosis.
1. INTRODUCTION
Diabetic kidney disease is the leading cause of end‐stage renal disease (ESRD) worldwide (Held et al., 1991; Koye, Magliano, Nelson, & Pavkov, 2018). Kidney fibrosis is the final consequence of diabetic kidney disease (Held et al., 1991; Lovisa et al., 2015; Srivastava et al., 2019). Kidney fibroblasts play an important role in the progression of kidney fibrosis (LeBleu et al., 2013; Srivastava et al., 2019; Zeisberg, Potenta, Sugimoto, Zeisberg, & Kalluri, 2008). BP control is the key to minimizing the progression from diabetes to diabetic nephropathy (Ganesh & Viswanathan, 2011; Hsu, Lin, Ou, Huang, & Wang, 2017). Conventional therapy of diabetic nephropathy includes renin angiotensin aldosterone system (RAAS) inhibitors, such as ACE inhibitors (ACEI) and angiotensin II receptor antagonists (AT1 antagonists/ARBs), which are first‐line drugs that can effectively reduce the incidence of end‐stage renal disease (Gu et al., 2016; Hsu et al., 2017; Palmer et al., 2015). However, the effects of ACE inhibition and AT1 antagonism in mouse models of diabetic nephropathy have not been well analysed, as well as having diverse clinical effects (Mauer et al., 2009). We have demonstrated that AT1 antagonism was unable to restore kidney structure and did not suppress renal fibrosis in the streptozotocin (STZ)‐induced diabetic CD‐1 mice, whereas ACE inhibition significantly suppressed the renal fibrosis and restored the kidney structure in these diabetic mice (Nagai et al., 2014). In line with this study, we recently demonstrated that ACE regulates antifibrotic microRNAs crosstalk level and dipeptidyl peptidase 4 (DPP‐4) associated fibrogenic processes in the kidneys of diabetic mice (Srivastava, Goodwin, Kanasaki, & Koya, 2020). Inhibiting ACE increases the plasma level of endogenous peptide N‐acetyl‐seryl‐aspartyl‐lysyl‐proline (AcSDKP) (Kanasaki, Nagai, Kitada, Koya, & Kanasaki, 2011). AcSDKP is a tetrapeptide that is normally present in human plasma and is exclusively hydrolysed by ACE (Kanasaki et al., 2011). AcSDKP is released from its precursor thymosin β4 (Tβ4) by two enzymatic steps mediated by meprin‐α and prolyl oligopeptidase (POP) (Kanasaki et al., 2011). Renal release of AcSDKP has been studied (Romero et al., 2019). Blockade of AcSDKP synthesis by inhibiting POP enzyme using small chemicals has been investigated in various in vivo and in vitro models (Macconi et al., 2012; Romero et al., 2019). We reported that AcSDKP alone or in combination with ACE inhibition can prevent renal fibrosis by inhibiting the endothelial‐to‐mesenchymal transition program in the kidneys of diabetic mice (Nagai et al., 2014; Srivastava et al., 2016). AcSDKP has demonstrated protective effects on organ fibrosis in several experimental animal models of fibrosis (Nitta et al., 2016; Omata et al., 2006; Shibuya et al., 2005).
Increased mesenchymal activation in the diabetic kidney has been identified as one of the mechanisms causing fibrosis (Srivastava, Koya, & Kanasaki, 2013; Srivastava, Shi, Koya, & Kanasaki, 2014). Snail1 is the zinc‐finger transcription factor which is involved in cell differentiation and survival, two of the processes focused on in fibroblast research in kidneys. Snail1 has a pivotal role in the regulation of epithelial‐to‐mesenchymal transition, the process by which epithelial cells acquire a migratory, mesenchymal phenotype, as a result of its repression of E‐cadherin (Grande et al., 2015; Lovisa et al., 2015). Alteration in fuel‐source preferences (glucose, fatty acids, glutamine or ketone bodies) has emerged as an important mechanism of cell differentiation (DeBerardinis & Thompson, 2012).
Metabolic reprogramming is a crucial constituent of malignant transformation (Oldfield et al., 2001). However, little is known about the metabolism of renal epithelial cells (Rowe et al., 2013). TGFβ1 is a well‐known mesenchymal inducer (Grande et al., 2015), suppresses fatty acid oxidation (Kang et al., 2015) and induces glucose metabolism in high‐glucose‐treated cultured renal tubular epithelial cells (TECs) (Srivastava et al., 2018). Renal tubular epithelial cells require high levels of baseline energy consumption and are highly dependent on fatty acid oxidation (Kang et al., 2015).
Kidney fibrosis is associated with an increased rate of sirtuin 3 (SIRT3) deficiency‐linked abnormal glucose metabolism and mesenchymal activation (Srivastava et al., 2018). SIRT3 is a major mitochondrial deacetylase that targets several diverse enzymes involved in central metabolism resulting in the activation of many oxidative pathways (Kim et al., 2010; Yin & Cadenas, 2015). SIRT3 blocks organ fibrosis by controlling TGFβ/smad3 signalling (Bindu et al., 2017; Chen et al., 2015; Sosulski, Gongora, Feghali‐Bostwick, Lasky, & Sanchez, 2017). Moreover, disruption in central metabolism leads to kidney injury (Kang et al., 2015; Poyan Mehr et al., 2018; Srivastava et al., 2018; Tran et al., 2016; Zhou et al., 2019). We have observed that SIRT3 deficiency leads to induction of abnormal glucose metabolism through higher pyruvate kinase M2 type (PKM2) dimer formation and hypoxia‐inducible factor‐1α (HIF1α) accumulation (Srivastava et al., 2018). This is similar to that observed in diabetic subjects with chronic kidney disease, in that oxygen consumption remains elevated with higher lactate levels in the kidney and there are also increased rates of glycolysis (Blantz, 2014). Glycolysis inhibitors or PKM2 activators disrupt such metabolic reprogramming resulting in significant suppression of fibrosis, indicating that they can be utilized as a new therapeutic approach to combate diabetic kidney complications (Qi et al., 2017; Srivastava et al., 2018). A recent preclinical study suggests that sodium glucose transporter 2 inhibition abolished the defective glucose metabolism and associated epithelial‐to‐mesenchymal transitions in the diabetic kidneys, resulting in remarkable improvements in the kidney's structure, functions and fibrosis (Li et al., 2020).
Some of the alterations of energy metabolism reported so far in mouse models of ischaemic acute kidney injury (AKI) include increased lactate release into the interstitium (Eklund, Wahlberg, Ungerstedt, & Hillered, 1991), elevated pyruvate kinase in kidney homogenates after ischaemia reperfusion injury (Fukuhara et al., 1991), increased glycolysis after mercuric chloride‐induced acute kidney injury (Ash & Cuppage, 1970) and reduced mitochondrial number in atrophic tubular cells in rats (Lan et al., 2016). Glycolysis‐derived methylglyoxal causes changes in kidney function among individuals with type 2 diabetes mellitus (Jensen et al., 2016). Aberrant glycolysis in autosomal dominant polycystic kidney disease shares similar features with aerobic glycolysis; treatment with glycolysis inhibitor 2‐deoxyglucose (2‐DG) suppressed the disease phenotype (Rowe et al., 2013).
Herein, we hypothesized that AcSDKP disrupts metabolic reprogramming in fibrotic kidneys associated with diabetes. This could provide a new insight into combating diabetic kidney disease.
2. METHODS
2.1. Reagents and antibodies
AcSDKP was a gift from Dr. Omata from Asabio Bio Technology (Osaka, Japan). Imidapril (ACE‐I) and TA‐606 (ARB) were provided by Mitsubishi Tanabe Pharma (Osaka Japan) through an MTA. For rabbit polyclonal anti‐pyruvate kinase (PK) isozyme M2 (Cat# 4053, RRID:AB_1904096), rabbit anti‐HK‐2 (Cat# 2867, RRID:AB_2232946), carnitine palmitoyltransferase 1a (CPT1a) (Cat# 12252, RRID:AB_2797857) and PPARγ coactivator 1α (PGC1α) (Cat# 2178, RRID:AB_823600) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). A mouse monoclonal pyruvate dehydrogenase kinase 4 (PDK4) (Cat# ab71240, RRID:AB_1269709), rabbit polyclonal Tβ4 (Cat# ab14335, RRID:AB_301115) and a rabbit polyclonal ACE (Cat# ab28311, RRID:AB_726126) were purchased from Abcam (Cambridge, UK). The goat anti‐Sirt3 antibody (Cat# sc‐365175, RRID:AB_10710522) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). The mouse monoclonal anti‐β‐actin (AC‐74) (Cat# A2228, RRID:AB_476697) antibody was obtained from Sigma (St. Louis, MO, USA). For immunofluorescence analysis, a mouse anti‐HIF1α (Cat# ab51608, RRID:AB_880418) antibody was purchased from Abcam (Cambridge, UK). A rabbit polyclonal anti‐Ki67 (Cat# sc‐15402, RRID:AB_2250495) was purchased from Santa Cruz Biotechnology. A goat polyclonal anti‐Snail antibody (Cat# ab53519, RRID:AB_881666), a rabbit polyclonal anti‐GLUT1 (Cat# ab153309, RRID:AB_301844) antibody and a rabbit anti‐αSMA (Abcam Cat# ab5694, RRID:AB_2223021) and a mouse anti‐αSMA (Abcam Cat# ab7817, RRID:AB_262054) antibodies were purchased from Abcam (Cambridge, UK). Fluorescence‐, Alexa Fluor 647‐ and rhodamine‐conjugated secondary antibodies were obtained from Jackson Immuno Research (West Grove, PA, USA).
2.2. Animal experimentations and development of mouse model of diabetic kidney disease
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology. The experiments in the methods sections are carried out in accordance with Kanazawa Medical University animal protocols (protocol numbers 2014‐89, 2013‐114 and 2014‐101) and protocol approved by the Institutional Care and use Committee at Yale University School of Medicine, New Haven, CT, USA, and were consistent with the National Institutes of Health Guidelines for the Care of Laboratory Animals. Authors confirm that all the experiments are performed in accordance with guidelines and regulations for scientific and ethical experimentation.
We purchased 7‐week‐old male CD‐1 (MGI Cat# 5659424, RRID:MGI:5659424) and male C57Bl6 (MGI Cat# 5655052, RRID:MGI:5655052) mice from Jackson laboratory. The induction of diabetes in the CD‐1 model was performed according to the previously established experimental protocol (Kanasaki et al., 2014; Li et al., 2017; Shi et al., 2015; Srivastava et al., 2016; Srivastava et al., 2018). In brief, diabetes was induced in male 8‐week‐old CD‐1 mice with a single intraperitoneal injection of STZ at 200 mg·kg−1 in 10 mmol·L−1 citrate buffer (pH 4.5). The induction of diabetes was confirmed as a blood glucose level > 16 mmol·L−1 2 weeks after STZ injection. After randomization, we utilized a fibrotic diabetic kidney disease model (STZ‐treated CD‐1 mice) for the interventional study. Sixteen weeks after the induction of diabetes, the randomized male diabetic CD‐1 mice were divided into the following six groups:‐ the ACE inhibitor (ACEI) imidapril (2.5 mg·kg BW−1·day−1); the AT1 receptor antagonist (ARB) TA‐606 (Hashimoto et al., 1998) (3 mg·kg BW−1·day−1); AcSDKP (500 μg·kg BW−1·day−1 using an osmotic mini‐pump); AcSDKP (500 μg·kg BW−1·day−1 using an osmotic mini‐pump) + imidapril; AcSDKP (500 μg·kg BW−1·day−1 using an osmotic mini‐pump) + TA‐606 (3 mg·kg BW−1·day−1) and non‐treatment (control). Imidapril or TA‐606 was provided in the drinking water. In the second set of experiments, five consecutive low‐dosed STZ (50 mg·kg−1·day−1) was injected in the 8‐week‐old male C57Bl6 mice. The diabetic C57Bl6 mice were randomized and divided into following five groups:‐ POP inhibitor (POPI; S17092), i.p. at the dose of 40 mg·kg−1 three times in a week for 8 weeks); imidapril + S17092; TA‐606 + S17092; AcSDKP + S17092 and vehicle‐treated diabetic control mice. In the third set of experiments, the male randomized diabetic CD‐1 mice were divided into the following three groups:‐ a control group; dichloroacetate (DCA) treatment group (1 g·L body weight−1·day−1 in drinking water) and 2‐DG treatment group for 500 μg·kg−1 i.p. twice a week for 4 weeks. In the fourth set of experiments, using an osmotic mini‐pump we administered AcSDKP at a low dose ( 200 μg·kg BW−1·day−1), at a medium dose (500 μg·kg BW−1·day−1) and at a high dose (1,000 μg·kg BW−1·day−1) in randomized male diabetic CD‐1 mice for 8 weeks. All of these mice had free access of food and water during experiments. The mice were killed 24 weeks after the induction of diabetes. Blood was withdrawn from the retro‐orbital plexus of each mouse and plasma was separated. The blood glucose was measured before sacrifice of each mouse using glucose strips. The urine was collected using metabolic cages. After the sacrifice of each mouse, we excised the kidneys from each mouse. Kidneys were cut into pieces and put at −80°C for gene expression and protein level analysis. However, immediately, we put one halves of kidney into optimal cutting temperature compound for frozen sections and the other halves into the 4% paraformaldehyde for the Masson's trichrome‐stained sections and for the paraffin‐embedded kidney sections. The BP of each mouse was monitored using the tail‐cuff method with a BP‐98A instrument (Softron Co. Beijing, China) within the week prior to euthanasia.
2.3. Metabolic reprogramming in the kidney
For analysis of metabolic reprogramming in the kidneys, we studied expression level of the critical regulators (SIRT3, HIF1α and PGC1α) and key enzymes of central metabolism (hexokinase 2 [HK2], PKM2 and PDK4), glucose transporter 1 (GLUT1) and CPT1a, an enzyme that is critical in fatty acid oxidation into the mitochondria. We analysed the expression analysis through immunohistochemical, immunofluorescence methods in the kidney sections and western blot analysis in the kidney homogenate. For the mesenchymal and fibrogenic analysis, we analysed α‐smooth muscle actin (αSMA), fibronectin and fibroblast specific protein 1 (FSP‐1). To evaluate, the specific contribution of metabolic defects with the mesenchymal inductions, we evaluated co‐labelling of key enzyme and regulators with αSMA. For our study, we characterized metabolic reprogramming by analysis of key regulators and enzymes of glucose metabolism and fatty acid metabolism.
2.4. In vivo silencing studies by using SIRT3 siRNA
For the SIRT3 in vivo knockdown study, we utilized diabetic CD‐1 mice which had experienced 8 weeks of diabetes. They were divided into two groups: scramble group and SIRT3 siRNA group. A chemically modified HPLC purified SIRT3 siRNA duplex (sense strand 5′GUCUGAAGCAGUACAGAAAtt and antisense strand 5′UUUCUGUACUGCUUCAGACaa) and scramble siRNA duplex were purchased from Invitrogen in vivo ready siRNAs. All of the oligos were dissolved in buffer (Atelo gene, Koken Co., Ltd., Japan) and injected into the tail vein (100 μl) twice weekly for 3 weeks at the dose of 5 mg·kg−1 body weight. We selected the way of administration and dose selection of SIRT3 siRNA and control siRNA from our previous published results (Srivastava et al., 2018).
2.5. AcSDKP measurements
Blood of each mouse was harvested into a heparinized tube containing captopril (final concentration of 10 μmol·L−1) and centrifuged at 3,000× g for 15 min at 4°C. We obtained estimated plasma and urine AcSDKP concentrations using a competitive enzyme immunoassay kit (SPI‐BIO, Massy, France) according to the manufacturer's instruction. Urine AcSDKP was normalized at the urine creatinine level.
2.6. ACE and prolyl oligopeptidase enzyme activity assay
ACE and POP activity were measured using commercially available kits from BioVision, CA, USA, and BPS Biosciences, CA, USA.
2.7. Morphological evaluation
For the fibrosis measurements, Masson's trichrome‐stained sections were evaluated by ImageJ (RRID:SCR_003070) software and the fibrotic areas were estimated. For the Sirius red staining, deparaffinized sections were incubated with picrosirius red solution for 1 h at room temperature. The slides were washed twice with acetic acid solution for 30 s per wash. Then the slides were dehydrated in absolute alcohol three times. The slides were cleared in xylene and mounted with a synthetic resin. Sirius red staining was imaged and analysed using the ImageJ software, and the fibrotic areas were quantified. For each mouse, images of six different fields of view were evaluated at 40× magnification.
2.8. Immunohistochemistry
The Immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018). Paraffin‐embedded kidney sections (5 μm thick) were deparaffinized and rehydrated (2 min in xylene, four times; 1 min in 100% ethanol, twice; 1 min in 95% ethanol; 45 s in 70% ethanol; and 1 min in distilled water), and the antigen was retrieved in a 10‐mM citrate buffer pH 6 at 98°C for 60 min. To block the endogenous peroxidase, all sections were incubated in 0.3% hydrogen peroxide for 10 min. The immunohistochemistry was performed using a Vectastain ABC Kit (Vector Laboratories, Burlingame, CA, USA). Rabbit polyclonal PKM2 (Cell Signaling Technology Cat# 4053, RRID:AB_1904096; 1:100), PDK4 (Abcam Cat# ab71240, RRID:AB_1269709; 1:100), PGC1α (Cell Signaling Technology Cat# 2178, RRID:AB_823600; 1:100) and CPT1a (Cell Signaling Technology Cat# 12252, RRID:AB_2797857; 1:100) antibodies were purchased from Cell Signaling Technology. Goat polyclonal anti‐SIRT3 antibody (Santa Cruz Biotechnology Cat# sc‐365175, RRID:AB_10710522; 1:200) and anti‐Ki67 (Santa Cruz Biotechnology Cat# sc‐15402, RRID:AB_2250495; 1:100) were purchased from Santa Cruz Biotechnology. A goat polyclonal anti‐Snail antibody (Abcam Cat# ab53519, RRID:AB_881666; 1:100) and a rabbit polyclonal anti‐GLUT1 (Abcam Cat# ab15309, RRID:AB_301844; 1:100) antibody were purchased from Abcam (Cambridge, MA, USA). In the negative controls, the primary antibody was omitted and replaced with the blocking solution.
2.9. Immunofluorescence
Frozen kidney sections (5 μm) were used for immunofluorescence; double positive labelling with SIRT3/αSMA, HK2/αSMA, PKM2/αSMA, PDK4/αSMA, Ki67/αSMA and HIF1α/αSMA was measured. Briefly, frozen sections were dried and placed in acetone for 10 min at −30°C. Once the sections were dried, they were washed twice in PBS for 5 min and then blocked in 2% BSA/PBS for 30 min at room temperature. Thereafter, the sections were incubated in primary antibody of SIRT3 (Santa Cruz Biotechnology Cat# sc‐365175, RRID:AB_10710522; 1:100), HK‐2 (Cell Signaling Technology Cat# 2867, RRID:AB_2232946; 1:100), HIF1α (Cat# ab51608, RRID:AB_880418; 1:100) and PKM2 (Cell Signaling Technology Cat# 4053, RRID:AB_1904096; 1:100) for 1 h and washed in PBS (5 min) three times. Next, the sections were incubated with the secondary antibodies for 30 min, washed with PBS three times (5 min each) and mounted with mounting medium with DAPI (Vector Laboratories). The immune‐labelled sections were analysed by fluorescence microscopy (Axio Vert.A1, Carl Zeiss Microscopy GmbH, Jena, Germany). For each mouse, original magnification of 400× pictures was obtained from six different areas, and quantification was performed.
2.10. In vitro experiment in renal tubular epithelial cells
Human renal tubular epithelial (HK‐2, ATCC Cat# CRL‐22190, RRID:CVCL_0302) cells were cultured in DMEM and Keratinocyte‐SFM (1×) medium (Life Technologies, Green Island, NY, USA), respectively. When the cells on the adhesion reagent reached 70% confluence, 25 ng·ml−1 recombinant human TGFβ1 for 48 h was placed in the serum diluted medium with or without ACEi (100 nM), AcSDKP (100 nM) and ARB (100 nM). AcSDKP, ACEi and ARB were pre‐incubated 2 h before TGFβ1 stimulation. Protein samples were harvested using RIPA buffer.
2.11. Extracellular acidification rate (ECAR) measurements by Seahorse Technology
High‐glucose (30 mM)‐pretreated renal tubular epithelial HK‐2 cells were plated in XF 96 plates (Agilent Technologies, Santa Clara, CA, USA) until confluent. AcSDKP was stimulated before 2 h in the TGFβ1‐treated and ‐untreated cells. Basal extracellular acidification rate measurement for 2 h at 37°C incubator without CO2 supplementation was performed as per the manufacturer's instructions. The data of basal extracellular acidification rate were normalized by μg protein in each well.
2.12. Transfection
The human renal tubular HK‐2 cells were used for the transfection studies. The HK‐2 cells were pretreated with glucose (30 mM) for 48 h and transfected with 100 nM of specifically designed siRNA for Tβ4 and ACE using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. Cells were harvested for qPCR and for western blot analysis.
2.13. Western blotting
Protein lysates were denatured in an SDS sample buffer at 100°C for 5 min. After centrifugation (17,000× g for 10 min at 4°C), supernatants were separated on SDS‐PAGE and blotted onto PVDF membranes (Pall Corporation, Pensacola, FL, USA) using the semidry method. PKM2 (Cell Signaling Technology Cat# 4053, RRID:AB_1904096; 1:1,000), PDK4 (Abcam Cat# ab71240, RRID:AB_1269709; 1:1,000), PGC1α (Cell Signaling Technology Cat# 2178, RRID:AB_823600; 1:1,000) and CPT1a (Cell Signaling Technology Cat# 12252, RRID:AB_2797857; 1:1,000), anti‐SIRT3 antibody (Santa Cruz Biotechnology Cat# sc‐365175, RRID:AB_10710522; 1:250) and GLUT1 (Abcam Cat# ab15309, RRID:AB_301844; 1:500) were used. The immunoreactive bands were developed using an enhanced chemiluminescence detection system (Pierce Biotechnology, Rockford, IL, USA) and detected using an ImageQuant LAS 400 digital biomolecular imaging system (GE Healthcare Life Sciences, Uppsala, Sweden).
2.14. RNA isolation and qPCR
Frozen kidney tissues were first placed on the RNAlater®‐I (Life Technologies) for 16 h at −20°C before the subsequent homogenization process. Total RNA was isolated using the RNeasy mini kit (Qiagen) following the manufacturer's instructions and was quantified with a NanoDrop spectrophotometer (ND‐1000, NanoDrop Technologies, DE, USA). cDNA was generated by RT kit (Takara Bio Inc.) using the concentration of 500 ng·μl−1 mRNA. mRNA gene expression was quantified by using SYBR green PCR kit (Takara Bio Inc.). qPCRs were performed in a 7900HT Fast Real‐Time PCR System (Life Technologies) and quantified using the δ–δ‐cycle threshold (Ct) method (ΔΔCt). All experiments were performed in triplicate, and 18S was utilized as an internal control. The mature sequences of specific primers were designed by Hokkaido System Science Co. (Hokkaido, Japan). The sequence of forward primer for TGFβ1 is 5′‐AAAACCAAAGACATCTCACAC and reverse primer 5′‐GAATCGAAAGCCCTGTATTCC; for Snail1 forward primer 5′‐CCGGAAGCCCAACTATAGCGA and reverse primer 5′‐TTCAGAGCGCCCAGGCTGAGGTACT; for Twist1 forward primer 5′‐GCAAGATCATCCCCACGCTG and reverse primer 5′‐GCAGGACCTGGTAGAGGAAG; for αSMA forward primer 5′‐CTGACAGAGGCACCACTGAA and reverse primer 5′‐GAAATAGCCAAGCTCAG; for FSP‐1 forward primer 5′‐TTCCAGAAGGTGATGAG and reverse primer 5′‐TCATGGCAATGCAGGACAGGAAGA; whereas for 18S forward primer 5′‐CGAAAGCATTTGCCAAGAAT and reverse primer 5′‐AGTCGGCATCGTTTATGGTC.
2.15. Statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). All values are expressed as means ± SEM and analysed using the statistical package for the GraphPad Prism 7 (GraphPad Software, Inc., La Jolla, CA, USA, RRID:SCR_002798). No data points were excluded from the analysis in any experiment. The one‐way ANOVA, followed by Tukey's test, was employed to analyse the significance when comparing multiple independent groups. The post hoc tests were run only if F achieved P < 0.05 and there was no significant variance inhomogeneity. Previous published studies and power analysis suggested that when using ANOVA, the sample sizes used would yield sufficient power to reliably detect differences in the mean among the group with sufficient power (i.e. >90%). The declared group size is the number of independent value, and the statistical analysis was performed using these independent values. In each experiment, N represents the number of separate experiments (in vitro) and the number of mice (in vivo). Technical replicates were used to ensure the reliability of single values. Data analyses were blinded. The data were considered statistically significant at P < 0.05.
2.16. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018) and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. AcSDKP is the key peptide for antifibrotic actions of ACE inhibition and protects against diabetic kidney disease
STZ‐induced diabetic CD‐1 mice is the established mouse model for the study of diabetic kidney disease (Srivastava et al., 2018). Here, we analysed the comparative effect of ACE inhibition, AT1 antagonism and AcSDKP on renal protection in the diabetic CD‐1 mice. During analysis of Sirius red staining, we found that the kidneys of vehicle‐treated diabetic CD‐1 mice showed profound fibrosis. ACE inhibition (ACEi) alone with imidapril and AcSDKP administration alone significantly suppressed the renal fibrosis, while AT1 antagonism with TA‐606 alone did not (Figure 1a). ACE inhibition in combination with AcSDKP displayed an additive effect in preventing kidney fibrosis, when compared to either ACE inhibition or AcSDKP alone. However, AT1 antagonism in combination with AcSDKP did not change the ability of AcSDKP to suppress kidney fibrosis (Figure 1a). AcSDKP also showed a dose‐dependent effect on renal fibrosis (Figure S1). Immunofluorescence analysis revealed higher deposition of collagen‐I and fibronectin in the kidneys of diabetic CD‐1 mice. ACE inhibition and AcSDKP caused significant suppression in the collagen‐I and fibronectin deposition, however AT1 antagonism did not (Figure 1b). ACE inhibition in combination with AcSDKP caused additive effect in suppressing collagen‐I and fibronectin level when compared to either ACE inhibition or AcSDKP. Again, AT1 antagonism in combination with AcSDKP did not affect AcSDKP ability to suppress kidney fibrosis (Figure 1b). The kidneys of diabetic mice showed up‐regulated gene expression level of mesenchymal marker (αSMA) and FSP‐1. ACE inhibition alone and AcSDKP alone down‐regulated the expression level of αSMA and FSP‐1, whereas AT1 antagonism did not (Figure 1c). ACE inhibition in combination with AcSDKP was more effective in down‐regulating αSMA and FSP‐1 expression level. Again, AT1 antagonism in combination with AcSDKP did not affect AcSDKP ability down‐regulated αSMA and FSP‐1 expression level (Figure 1c). Plasma and urine AcSDKP level was found lower in the diabetic group. ACE inhibition caused significant elevation in the AcSDKP level, while AT1 antagonism had little effect (Figure 1d). ACE inhibition in combination with AcSDKP was more effective in elevating AcSDKP level. Again, AT1 antagonism in combination with AcSDKP did not affect the AcSDKP level when compared to AcSDKP alone (Figure 1d). Moreover, ACE inhibition caused significant suppression of ACE activity, while AT1 antagonism suppressed the level of AT1 receptor protein expression (Figures 1e and S2A). However, ACE inhibition and AT1 antagonism did not cause any rovert difference in the level of prolyl oligopeptidase (POP) enzyme activity in the kidneys of diabetic mice (Figure 1f). The kidneys of diabetic mice showed higher deposition of angiotensin II. ACE inhibition and AcSDKP diminished protein expression level of angiotensin II, whereas AT1 antagonism did not suppress (Figure S2B). ACE inhibition in combination with AcSDKP showed additive effect in reducing angiotensin II protein expression. However, AT1 antagonism in combination with AcSDKP did not affect AcSDKP ability to reduce angiotensin II protein expression (Figure S2B). Moreover, angiotensin II stimulation enhanced gene expression level of αSMA and FSP‐1 high‐glucose‐treated renal tubular HK‐2 epithelial cells (Figure S2C).
FIGURE 1.

AcSDKP is key molecule for antifibrotic actions of ACE inhibitors (ACEIs) in the kidneys of diabetic mice. (a) Sirius red staining in the kidneys of non‐diabetic control, diabetic (DM) and imidapril (ACE inhibition; ACEi), TA‐606 (AT1 receptor blockade; ARB), AcSDKP, ACEi + AcSDKP and ARB + AcSDKP‐treated diabetic CD‐1 mice. Representative pictures are shown. Relative fibrosis is calculated by ImageJ program. Scale bar: 50 μm. Six mice were evaluated in each group. (b) Immunofluorescence analysis of collagen‐I/DAPI and fibronectin/DAPI in the kidney of control, DM, ACEi, ARB, AcSDKP, ACEi + AcSDKP and ARB + AcSDKP‐treated diabetic CD‐1 mice. The representative pictures are shown. Scale bar: 50 μm. Collagen‐I and fibronectin rhodamine labelled and DAPI blue. Six mice were evaluated in each group. (c) Gene expression analysis of αSMA and FSP‐1 by qPCR in the kidneys of control, DM, ACEi, ARB, AcSDKP, ACEi + AcSDKP and ARB + AcSDKP‐treated diabetic CD‐1 mice. 18S was used as internal control to normalize the expression level. Six mice were analysed in each group. (d) Plasma and urine concentration of AcSDKP in the indicated group of mice. The plasma of six mice in the non‐diabetic control and seven mice in the diabetic and ACEi, ARB, AcSDKP, ACEi + AcSDKP and ARB + AcSDKP‐treated diabetic group were analysed. However, urine of six mice were analysed in each group. Urine AcSDKP concentration was normalized by urine creatinine level and is shown in the graph. (e) ACE and (f) POP enzyme activity analysis by fluorimeter in kidney homogenates of indicated groups. Six mice were analysed in each group. Data in the graph are presented as mean ± SEM. Statistical significance: * P < 0.05. DM represents diabetic group
3.2. AcSDKP disrupts the metabolic reprogramming in diabetic kidney disease
To test the contribution of ACE inhibition and AT1 antagonism on central metabolism, we performed western blot analysis in the lysates from kidneys of diabetic CD‐1 mice subjected to ACE inhibition alone or with AcSDKP (combination treatment) or AT1 antagonism alone treatment. Our results showed significant suppression in the protein levels of SIRT3, CPT1a and PGC1α and significant induction of the protein levels of GLUT1, PKM2 and PDK4 in the kidneys of diabetic mice when compared to kidneys of control CD‐1 mice (Figure 2a). ACE inhibition and combination treatments (ACE inhibition + AcSDKP) restored the protein levels of SIRT3, CPT1a and PGC1α and suppressed the levels of GLUT1, PKM2 and PDK4 in these diabetic kidneys. However, these effects were more prominent with combination treatment when compared to ACE inhibition treatment alone (Figure 2a). AT1 antagonist treatment with TA‐606 in diabetic mice could neither restore the protein levels of SIRT3, CPT1a and PGC1α nor suppress the levels of GLUT1, PKM2 and PDK4 in the kidneys (Figure 2a). Furthermore, AcSDKP treatment alone in the diabetic mice caused significant elevation in the protein level of SIRT3, CPT1a and PGC1α and significant reduction in the induced levels of GLUT1, PKM2 and PDK4 in the kidneys when compared to the kidneys of vehicle‐treated diabetic CD‐1 mice (Figure 2b). To localize these metabolic changes, we performed immunohistochemistry for key regulators which play a crucial role in central metabolism, they are SIRT3, PKM2, PDK4, PGC1α, GLUT1 and CPT1a in the kidneys (Figures 2c and S3). We found remarkable suppression of the protein level of SIRT3, PGC1α and CPT1a in the tubulo‐interstitial compartment in kidneys of diabetic mice when compared to non‐diabetic control. ACE inhibition alone and AcSDKP alone or ACE inhibition in combination with AcSDKP significantly restored SIRT3, PGC1α and CPT1a, whereas AT1 antagonism failed to restore these markers (Figures 2c and S3). The kidneys of the diabetic mice had significantly higher protein expression of PKM2, PDK4 and GLUT1 in the damaged tubular area when compared to the tubular area in the kidneys of non‐diabetic control. ACE inhibition alone and AcSDKP alone or ACE inhibition in combination with AcSDKP significantly diminished the induced level of PKM2, PDK4 and GLUT1, whereas AT1 antagonism did not suppress (Figures 2c and S3). ACE inhibition + AcSDKP was more effective in elevating SIRT3, PGC1α and CPT1a and in suppressing PKM2, PDK4 and GLUT1 level, whereas AT1 antagonism + AcSDKP did not show any additive effects (Figures 2c and S3).
FIGURE 2.
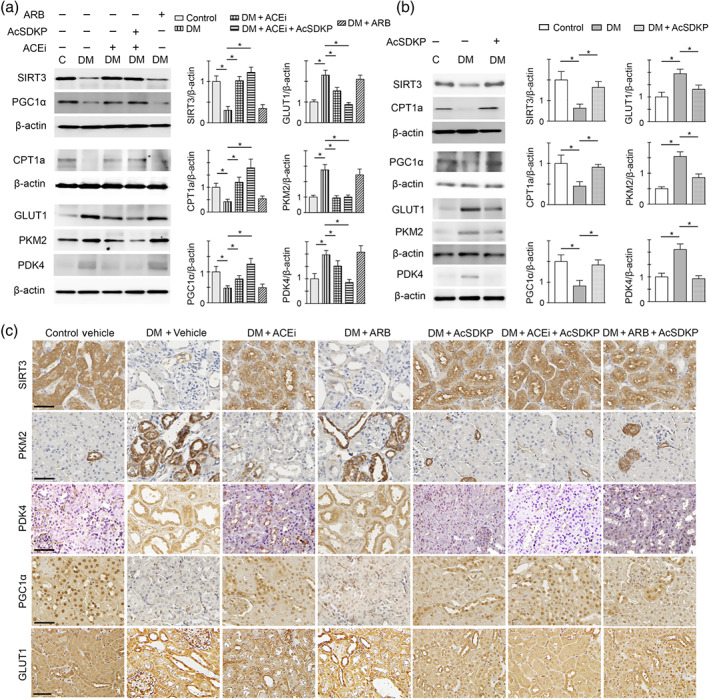
AcSDKP restores central metabolism in diabetic kidney disease. (a) Western blot analysis of SIRT3, PGC1α, CPT1a, GLUT1, PKM2 and PDK4 in the kidney of control, diabetic, ACE inhibited with imidapril (ACEi), combination (ACEi + AcSDKP) and TA‐606 (AT1 receptor blockade; ARB)‐treated diabetic mice. Representative pictures from five blots are shown. Densitometry quantification of SIRT3, PGC1α, CPT1a, GLUT1, PKM2 and PDK4 was performed on ImageJ. The data were normalized to β‐actin. The kidneys of five mice were analysed. (b) Western blot analysis of SIRT3, PGC1α, CPT1a, GLUT1, PKM2 and PDK4 in the kidneys of the control, diabetic and AcSDKP‐treated diabetic mice. Representative pictures from five blots are shown. Densitometry quantification of SIRT3, PGC1α, CPT1a, GLUT1, PKM2 and PDK4 was performed on ImageJ. The data were normalized to β‐actin. The kidneys of five mice were analysed. The data are expressed as the means ± SEM and are included in the graph. (c) Immunohistochemistry analysis of SIRT3, PKM2, PDK4, PGC1α and GLUT1 in the kidneys of the control, diabetic and ACEi, ARB, AcSDKP, ACEi + AcSDKP and ARB + AcSDKP‐treated diabetic CD‐1 mice. Five mice were analysed in each group. Representative pictures in each panel are shown. Scale bar: 50 μm. Data in the graph are presented as mean ± SEM. Statistical significance: * P < 0.05. DM represents diabetic group
3.3. AcSDKP is a critical endogenous anti‐mesenchymal peptide and inhibits defective central metabolism of myofibroblasts
To study the effects of ACE inhibition, AT1 antagonism, AcSDKP, ACE inhibition + AcSDKP and AT1 antagonism + AcSDKP on the mesenchymal transcriptional induction in the kidneys, we analysed the immunohistochemical analysis of Snail1 protein expression (Figure 3a). The data revealed the induction of Snail1 protein expression in the kidney of diabetic mice when compared to control (Figure 3a). The kidneys from imidapril (ACE inhibitor), AcSDKP and imidapril+ AcSDKP‐treated diabetic mice exhibited significant suppression in the Snail1 protein expression in the tubular area when compared to the kidneys of vehicle‐treated diabetic mice. However, the kidneys of TA‐6006 (AT1 receptor antagonist)‐treated diabetic mice did not show any remarkable changes (Figure 3a). The kidneys of ACE inhibited + AcSDKP showed additive effect in suppressing Snail1 protein expression, whereas AT1 antagonist + AcSDKP did not (Figure 3a). Moreover, the kidney of diabetic mice showed up‐regulated gene expression level of Snail1, Twist1 and TGFβ1 when compared to control. ACE inhibition alone, AcSDKP alone and ACE inhibition + AcSDKP caused significant down‐regulation, but AT1 antagonism did not alter the expression level (Figure 3b). To test the contribution of HIF1α‐associated abnormal glucose metabolism in the induction of α‐SMA, we performed co‐immunolabelling of the SIRT3 with αSMA, HK2 with αSMA and HIF1α with αSMA (Figure 3c). We observed that the kidneys of diabetic CD‐1 mice displayed higher levels of αSMA deposition with significantly reduced levels of SIRT3 protein expression and higher levels of HK2 and HIF1α when compared to kidneys of control CD‐1 mice. The kidneys of imidapril (ACEI) alone, AcSDKP alone and imidapril + AcSDKP‐treated diabetic mice restored SIRT3 and suppressed HK2 and HIF1α protein level, whereas TA‐606 (AT1 antagonist) did not (Figure 3c).
FIGURE 3.
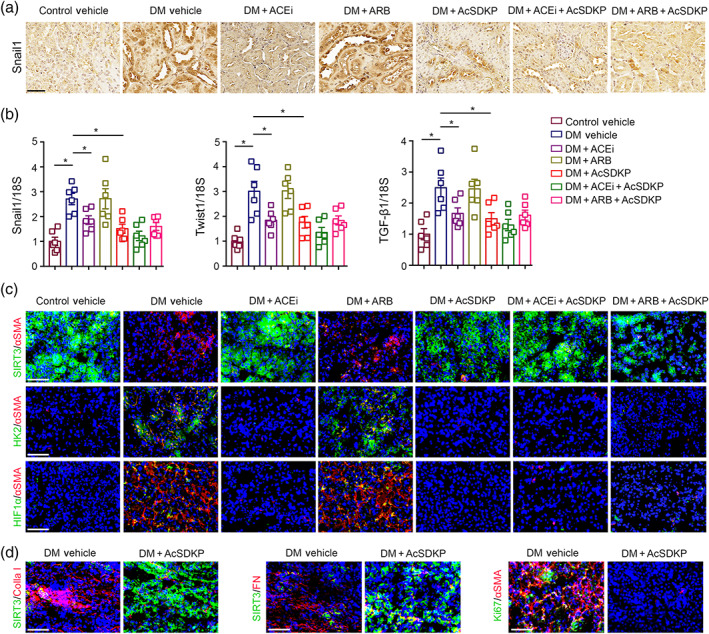
AcSDKP disrupts the metabolic reprogramming of myofibroblasts in diabetic kidneys. (a) Immunohistochemistry analysis of Snail1 in the kidney of non‐diabetic control, diabetic (DM) and imidapril (ACE inhibition; ACEi), TA‐606 (AT1 receptor blockade; ARB), AcSDKP, ACEi + AcSDKP and ARB + AcSDKP‐treated diabetic CD‐1 mice. Representative pictures are shown. Scale bar: 50 μm. Five mice were analysed in each group. (b) Gene expression analysis of Snail1, Twist1 and TGFβ1 by qPCR in the kidneys of indicated groups. The kidneys of six mice were analysed in each group. (c) Co‐immunofluorescence analysis of SIRT3/αSMA/DAPI, HK2/αSMA/DAPI and HIF1α/αSMA/DAPI in the kidneys of indicated groups. Representative pictures are shown. Scale bar: 50 μm. In the first set of experiments, SIRT3 FITC labelled green, αSMA rhodamine labelled and DAPI blue; in the second set of experiments, HK2 FITC (green) labelled, αSMA rhodamine labelled and DAPI blue. In the third set of experiments, HIF1α FITC labelled green, αSMA rhodamine labelled and DAPI blue. The kidneys of five mice were analysed in each group. (d) Co‐immunofluorescence analysis of SIRT3/collagen‐I, SIRT3/fibronectin and proliferation marker ki67 with αSMA was analysed. The kidneys of five mice were analysed in each group. Scale bar: 50 μm. Representative pictures in each panel are shown. Data in the graph are presented as mean ± SEM. Statistical significance: * P < 0.05. DM represents diabetic group
During co‐immunofluorescence analysis of SIRT3/collagen‐I and SIRT3/fibronectin in the kidneys of AcSDKP‐treated diabetic mice, we observed significant increase in the SIRT3 protein expression (Figure 3d). However there was a remarkable reduction in the collagen‐I and fibronectin expression in the kidneys of AcSDKP‐treated diabetic mice when compared to kidneys of vehicle‐treated diabetic mice (Figure 3d). The kidneys of AcSDKP‐treated diabetic mice showed significant suppression in the expression level of Ki67/αSMA when compared to the kidneys of diabetic mice (Figure 3d).
3.4. Blockade of AcSDKP synthesis by inhibiting prolyl oligopeptidase enzyme causes diabetic kidney disease
To understand further the contribution of AcSDKP in the regulation of renal fibrosis, we used the competitive inhibitor (S17092; POPi) of POP enzyme and thus partly blocked AcSDKP synthesis. Initially we carried out our experiment in the partial fibrotic C57Bl6 mouse strain (Figure 4a). The kidney from diabetic C57Bl6 mouse showed minimal fibrotic alteration when compared to the kidney from diabetic CD‐1 mouse (Srivastava et al., 2016; Srivastava et al., 2018). We treated with imidapril, TA‐606 or AcSDKP in the POPi (S17092)‐injected diabetic C57Bl6 mice (Figure 4a). At the time of sacrifice, the POPi‐treated diabetic mice and vehicle‐treated diabetic mice had similar blood glucose, body weight and BP. However, POPi‐injected diabetic mice had higher kidney weight and albumin‐to‐creatinine ratio when compared to vehicle‐treated diabetic mice (Figure 4b). Imidapril treatment (ACE inhibition) did not cause any remarkable changes in body weight and blood glucose, only a slight lowering in kidney weight and BP, although there was a remarkable suppression in albumin‐to‐creatinine ratio when compared to POPi‐injected diabetic mice (Figure 4b). TA‐606 treatment (AT1 antagonism) did not alter body weight, blood glucose or kidney weight in the POPi‐injected diabetic mice (Figure S4). AcSDKP treatment also did not alter body weight and blood glucose but there was a remarkably lowering of kidney in the POPi‐injected diabetic mice (Figure S5). POPi significantly suppressed both plasma and urine AcSDKP level. ACE inhibition and AcSDKP elevated the level of plasma and urine AcSDKP, whereas AT1 antagonism did not elevate in the POPi‐injected diabetic mice (Figures 4c, S4 and S5). POPi did not cause remarkable alteration in angiotensin converting enzyme (ACE) activity in the kidneys. However, ACE inhibition treatment in POPi‐injected mice caused significant lowering in ACE activity (Figure 4d). The kidneys of POPi‐injected diabetic mice had reduced POP enzyme activity. ACE inhibition, AT1 antagonism and AcSDKP did not alter the POP enzyme activity (Figures 4e, S4 and S5).
FIGURE 4.

Blockade of AcSDKP synthesis by inhibiting prolyl oligopeptidase (POP) enzyme exacerbates renal fibrosis in mouse model of diabetic kidney disease. (a) The schematic charts depicting the treatment protocol of POP inhibitor (Pi) S17092 in the reduced fibrotic diabetic C57Bl6 mice. Imidapril (ACE inhibition, ACEi) and TA‐606 (AT1 receptor antagonist, ARB) were given for 8 weeks in the POP inhibitor (POPi)‐treated diabetic C57Bl6 mice. (b) Physiological characteristics (body weight, blood glucose, kidney weight, albumin‐to‐creatinine ratio and BP) of diabetic control, Pi and ACEi treatment in POPi‐injected diabetic C57Bl6 mice. After 24 weeks, six mice were analysed in each group. (c) The level of AcSDKP in the plasma and in urine was analysed by elisa. Urine AcSDKP levels were normalized by the urine creatinine level. Six mice were analysed in each group. (d) ACE activity analysis by fluorimeter in kidney homogenates of indicated groups. Six mice were analysed in each group. (e) POP enzyme activity analysis by fluorimeter in kidney homogenate of indicated groups. The kidneys of six mice were analysed in each group. (f) Masson's trichrome staining (MTS) in the kidneys of indicated groups and the quantification of the relative area fibrosis (RAF) by ImageJ. Scale bar: 50 μM. The kidneys of six mice were analysed in each group. (g) Sirius red staining in the kidneys of indicated groups and quantification by ImageJ. Scale bar: 50 μM. The kidneys of six mice were analysed in each group. (h) Immunofluorescence analysis of collagen‐I/DAPI in the kidneys of indicated groups. Relative collagen deposition (RCD) quantification by ImageJ. Representative pictures are shown. Scale bar: 50 μM in each panel. Merged pictures are shown. The kidneys of six mice were analysed in each group. (i) Gene expression analysis of αSMA and FSP‐1 by qPCR analysis in the kidneys of indicated groups. 18S was used as internal control to normalize the expression level. The kidneys of six mice were analysed in each group. Data are expressed as the mean ± SEM and are shown in the graph. Statistical significance: * P < 0.05. DC represents diabetic control; Pi represents POP inhibitor treatment group
During histological analysis, we found that the kidneys of POPi‐injected diabetic mice had higher deposition of extracellular matrix, higher area fibrosis and collagen deposition when compared to kidney of vehicle‐treated diabetic mice (Figure 4f–h). ACE inhibition partly, whereas AcSDKP completely restored the kidney structure, suppressed the renal fibrosis and collagen deposition in the POPi‐injected diabetic mice, whereas AT1 antagonism did not suppress the renal fibrogenic phenotype (Figures 4f–h, S4 and S5).
3.5. Prolyl oligopeptidase inhibition disrupts the kidney homeostasis and metabolism
POPi elevated Snail1 protein/mRNA expression in the kidney of diabetic mice when compared to vehicle‐treated diabetic mice (Figure 5a,b). Imidapril (ACEI) treatment and AcSDKP treatment in POPi‐injected diabetic mice exhibited suppression in the Snail1 protein/mRNA expression. However, TA‐606 (AT1 antagonist) treatment in POPi‐injected diabetic mice did not show remarkable changes in the Snail1 protein expression (Figures 5a,b and S6A,B). Moreover, to test the contribution of POPi in the central metabolism, we analysed SIRT3/αSMA, PKM2/αSMA and HIF1α/αSMA co‐staining in the kidneys of POPi‐treated diabetic mice and compared with the kidneys of vehicle‐treated diabetic mice (Figure 5c). We found that the kidneys of POPi‐injected diabetic mice displayed higher αSMA positive cells, with significant suppression in the SIRT3 protein expression and induced protein expression of PKM2 and HIF1α when compared to the kidneys of vehicle‐treated diabetic mice (Figure 5c). The kidneys from the POPi‐injected diabetic mice that had received imidapril had partly restored SIRT3 protein expression and showed suppressed αSMA level, whereas blockage of AT1 receptors had no effect on these changes. ACE inhibition diminished the level of PKM2/αSMA and HIF1α/αSMA, whereas AT1 antagonism was ineffective (Figure 5c). AcSDKP completely restored the SIRT3 protein expression and showed diminished αSMA level in the kidneys from POPi‐injected diabetic mice. AcSDKP treatment also reduced the level of PKM2/αSMA and HIF1α/αSMA in the kidneys from POPi‐injected diabetic mice (Figure S6C). Immunohistochemical analysis in these kidneys from POPi‐injected diabetic mice revealed the suppression of the protein expression level of SIRT3, PGC1α and CPT1a and induction in the level PKM2, GLUT1 and PDK4 in the tubular area when compared to that kidneys of vehicle‐treated diabetic mice (Figure 5d). ACE inhibition partly restored the SIRT3, PGC1α and CPT1a protein expression and partly suppressed the level of PDK4, PKM2 and GLUT1 in the kidneys from POPi‐injected diabetic mice, whereas AT1 antagonism did not show such effect (Figure 5d). AcSDKP treatment restored the SIRT3, PGC1α and CPT1a protein expression and reduced the level of PDK4 and PKM2 in the kidneys from POPi‐injected diabetic mice (Figure S6D).
FIGURE 5.

Blockade of AcSDKP synthesis disrupts the metabolic homeostasis in kidney. (a) Immunohistochemistry analysis of Snail1 in the kidney of diabetic (vehicle treated), S17092 treated (prolyl oligopeptidase inhibited; POPi) and imidapril (ACE inhibited; ACEi) and TA‐606 (AT1 receptor blockade; ARB) intervention in POPi‐treated diabetic C57Bl6 mice. Representative pictures are shown. Scale bar: 50 μm. Five mice were analysed in each group. (b) Gene expression analysis of Snail1 and TGFβ1 by qPCR in the kidneys of indicated groups. The kidneys of five mice were analysed in each group. Gene expression data were normalized by 18S. (c) Co‐immunofluorescence analysis of SIRT3, PKM2 and HIF1α, with α‐SMA, was analysed by using fluorescence microscope. The kidneys of five mice were analysed in each group. Representative pictures in each panel are shown. (d) Immunohistochemistry analysis of SIRT3, PKM2, PDK4, CPT1a, PGC1α and GLUT1, in the kidneys of the vehicle‐treated diabetic, POPi and ACEi and ARB intervened POPi‐treated diabetic mice. Five mice were analysed in each group. Representative pictures in each panel are shown. Scale bar: 50 μm. Data in the graph are presented as mean ± SEM. Statistical significance: * P < 0.05. Vehicle represents diabetic group
3.6. AcSDKP treatment inhibits TGFβ1‐associated defective central metabolism in vitro
To define the changes in the central metabolism pathways specific to TGFβ1, we examined protein expression analysis of regulatory enzymes in the human proximal tubular epithelial cells (HK‐2). We found that TGFβ1 stimulation caused significant suppression in the protein levels of SIRT3, PGC1α and CPT1a and concomitant induction in the protein levels of HK2, PKM2 and PDK4 in high‐glucose‐treated HK‐2 cells (Figure 6a). Both ACE inhibition and AcSDKP restore the protein levels of SIRT3, PGC1α and CPT1a, while significantly reducing the elevated protein level of HK2, PKM2 and PDK4 in the TGFβ1‐stimulated high‐glucose‐treated HK‐2 cells. AT1 antagonism could neither restore the TGFβ1‐stimulated suppressed protein levels of SIRT3, PGC1α and CPT1A nor suppress the TGFβ1‐stimulated elevation of HK2, PKM2 and PDK4 (Figure 6a,b). While analysing the extracellular acidification rate by seahorse flux analyser, we found that TGFβ1 stimulation reduced the extracellular acidification rate level in HK‐2 cells treated with basal glucose (5 mM), whereas cells treated with high glucose (25 mM) had higher extracellular acidification rate level (Figure 7a). The HK‐2 cells treated with high glucose had higher lactate release and suppressed intracellular ATP (Figure 7b,c). AcSDKP intervention significantly suppressed the higher level of basal extracellular acidification rate and lactate release and induced the intracellular ATP level in the high‐glucose‐treated HK‐2 cells (Figure 7a–c).
FIGURE 6.
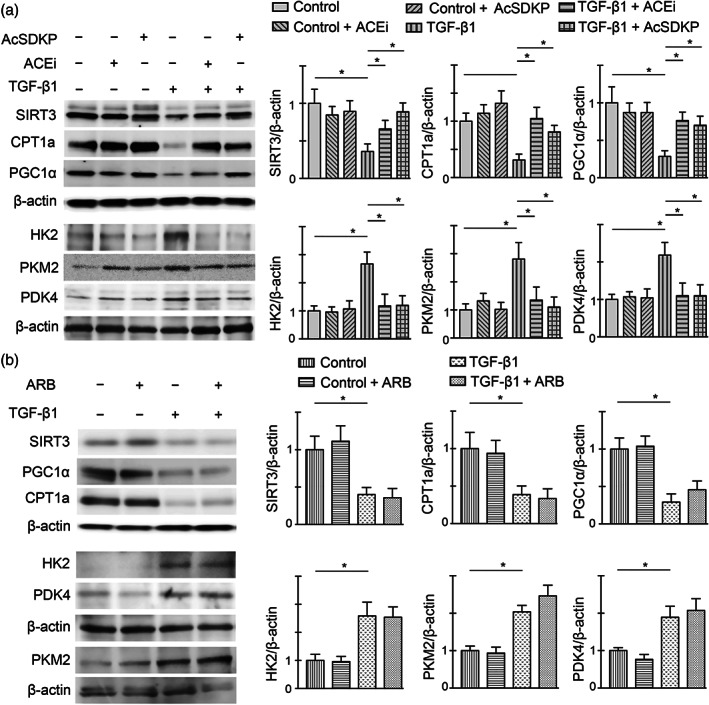
ACE inhibition and AcSDKP stimulation cancel the TGFβ1‐driven abnormal metabolism in vitro. (a) Western blot analysis of SIRT3, CPT1a, PGC1α, HK2, PKM2 and PDK4 in the imidapril (ACE inhibition; ACEi) and AcSDKP‐treated tubular epithelial cells (HK‐2 cells) in the presence and absence of TGFβ1 (25 ng·ml−1) in high glucose condition. Representative blots are shown. Quantification of SIRT3, CPT1a, PGC1α, HK2, PKM2 and PDK4, respectively, by densitometry. The data were normalized to β‐actin. Five independent sets of experiments were analysed. (b) Western blot analysis of SIRT3, CPT1a, PGC1α, HK2, PKM2 and PDK4 in the ARB‐treated HK‐2 cells in the presence and absence of TGFβ1 in high glucose condition. Representative blots are shown. Quantification of SIRT3, CPT1a, PGC1α, HK2, PKM2 and PDK4, respectively, by densitometry. The data were normalized by β‐actin. Five independent sets of experiments were analysed. Data in the graph are presented as mean ± SEM. Statistical significance: * P < 0.05
FIGURE 7.

AcSDKP stimulation cancel the TGFβ1‐driven extracellular acidification in high glucose condition. (a) Measurement of basal extracellular acidification rate (ECAR) in the AcSDKP‐intervened TGFβ1‐treated HK‐2 cells with basal glucose (5 mM) and high glucose (25 mM) was measured with an XF96 Extracellular Flux Analyser (Seahorse Bioscience). Seven wells in each group were analysed. Results were normalized by μg protein in each well. (b) Lactate release. (c) Intracellular ATP level in the AcSDKP‐intervened TGFβ1‐treated HK‐2 cells with high glucose (25 mM) was measured using commercial kit. Seven wells in each group were analysed. Data in the graph are presented as mean ± SEM. Statistical significance: * P < 0.05
To further analyse the effects of AcSDKP in the metabolic homeostasis, we knockdown the Tβ4 and angiotensin converting enzyme (ACE) by using specific siRNAs in high‐glucose‐treated HK‐2 cells. Tβ4 and ACE silencing resulted into significant suppression in the mRNA and protein level of Tβ4 and ACE (Figure 8a,b). Tβ4 knockdown caused up‐regulation in gene expression level of FSP‐1, PKM2 and PDK4 and down‐regulation in PPARα and PGC1α expression levels, whereas ACE knockdown caused down‐regulation in gene expression level of FSP‐1, PKM2 and PDK4 and up‐regulation in PPARα and PGC1α expression levels (Figure 8a). In addition, Tβ4 knockdown caused elevation in the fibronectin, PKM2 and PDK4 and reduction in CPT1a protein levels, whereas ACE knockdown caused reduction in FSP‐1, PKM2 and PDK4 and induction in CPT1a protein levels (Figure 8c). Tβ4 silencing suppressed whereas ACE silencing elevated the AcSDKP level in the cell medium (Figure 8d).
FIGURE 8.
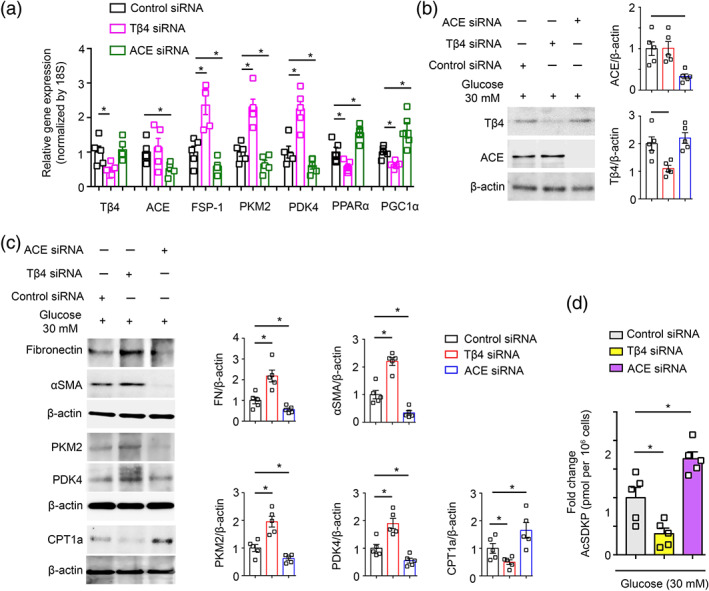
Thymosin β4 knockdown disrupts whereas ACE knockdown improves the central metabolism. (a) Gene expression analysis of Tβ4, ACE, FSP‐1, PKM2, PDK4, PPARα and PGC1α by qPCR in the Tβ4 siRNA, and ACE siRNA transfected high glucose (30 mM 48 h pretreated)‐treated HK‐2 cells. Five independent sets of experiments were analysed. Gene expression data were normalized by 18S. (b) Western blot analysis of Tβ4 and ACE in the Tβ4 siRNA and ACE siRNA transfected high‐glucose‐treated HK‐2 cells. Five independent sets of experiments were analysed. Densitometry calculations were normalized to β‐actin. Representative blots are shown. (c) Western blot analysis of fibronectin, αSMA, PKM2, PDK4 and CPT1a in the Tβ4 siRNA and ACE siRNA transfected HK‐2 cells. Five independent sets of experiments were analysed. Densitometry calculations were normalized to β‐actin. Representative blots are shown. (d) AcSDKP levels were measured in the cell medium of Tβ4 siRNA and ACE siRNA transfected HK‐2 cells. Five independent sets of experiments were analysed. Data in the graph are presented as mean ± SEM. Statistical significance: * P < 0.05
3.7. Glycolysis inhibition suppresses renal fibrosis via elevating AcSDKP level
Furthermore, we analysed the comparative effects of glycolysis inhibitors on collagen deposition in diabetic kidney (Figure 9a). Immunofluorescence data revealed that dichloroacetate (DCA) and 2‐deoxyglucose (2‐DG) significantly suppressed collagen‐I in the kidneys of diabetic mice (Figure 9b). DCA and 2‐DG restored the level of plasma and urine AcSDKP (Figure 9c,d). SIRT3 silencing in the mice caused remarkable reduction in the plasma and urine AcSDKP level (Figure 9e,f). SIRT3 knockdown in diabetic mice showed down‐regulated gene expression level of SIRT3 and CPT1a whereas up‐regulated the gene expression level of HIF1α and FSP‐1 in the kidneys (Figure S7A). SIRT3 knockdown did not cause any alteration in the expression and activity level of POP and ACE in the kidneys of diabetic mice (Figure S7A,B). DCA and 2‐DG treatment in the diabetic mice did not alter the POP activity, however showed suppressed trends of ACE activity in the kidneys (Figure S7C).
FIGURE 9.
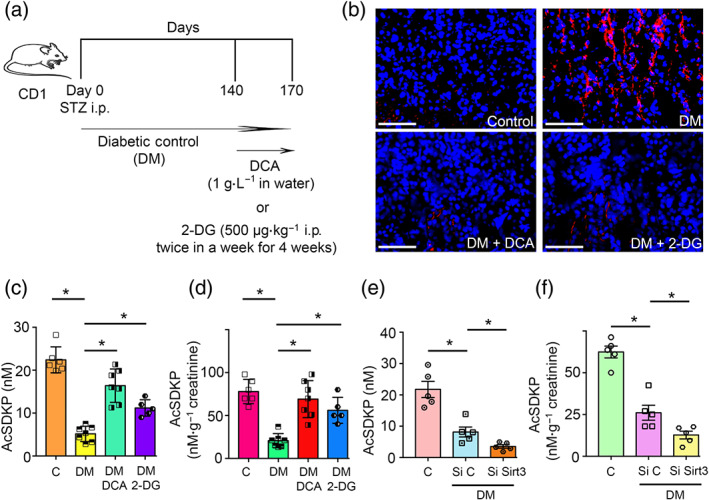
Glycolysis inhibitions cancel renal fibrosis by elevating the AcSDKP level. (a) Schematic charts depicting the treatment protocol of glycolysis inhibitors, dichloroacetate (DCA) and 2‐deoxyglucose (2‐DG) in the fibrotic diabetic CD‐1 mice. (b) Immunofluorescence analysis of collagen‐I/DAPI in the kidneys of control, DM and DM mice treated with either DCA or 2‐DG. Representative pictures are shown. Scale bar: 50 μM in each panel. Merged pictures are shown. The kidneys of six mice were analysed in each group. (c) The level of AcSDKP in the plasma of control, DM and DM mice treated with either DCA or 2‐DG was analysed by ELISA . The plasma of six mice were analysed in control group, seven mice in DM and DCA‐treated diabetic mice and five mice in 2‐DG‐treated diabetic mice (d) Urine AcSDKP levels were measured by ELISA in the control, DM and DM mice treated with either DCA or 2‐DG group. Urine AcSDKP levels were normalized to the urine creatinine level. The urine of six mice were analysed in control group, seven mice in DM and DCA‐treated diabetic mice and five mice in 2‐DG‐treated diabetic mice. (e)The level of AcSDKP in the plasma of tail vein‐injected scramble and SIRT3 siRNA in diabetic mice. The plasma of five mice were analysed in each group. (f) Urine AcSDKP levels were measured by ELISA in the tail vein‐injected scramble and SIRT3 siRNA in diabetic mice, and urine AcSDKP was normalized by the urine creatinine level. The urine of five mice were analysed in each group. The data are expressed as the means ± SEM and are included in the graph. Statistical significance: * P < 0.05. DM represents diabetic group, Si C represents scramble siRNA injected, while Si Sirt3 represents SIRT3 siRNA injected group
4. DISCUSSION
Activation of matrix‐associated fibroblasts plays a major role during fibrosis (LeBleu et al., 2013; Lovisa et al., 2015; Zeisberg et al., 2008). In the present study, we proposed that AcSDKP is one of the essential antifibrotic peptides that regulates central metabolism and mesenchymal transformations in the kidney. From the results, it is clear that ACE inhibition mediated renal protection is due to its ability to activate the AcSDKP‐associated renal protections. This study also established the differential effects of ACE inhibitors and AT1 antagonists (RAAS inhibitors) on the defective metabolism in the diabetic kidney. AT1 antagonists alone or combination with ACE inhibitors are used to reduce the risk of end‐stage renal disease (Jacobsen, Andersen, Jensen, & Parving, 2003; Kalender et al., 2002; Palmer et al., 2015; Scheen, 2004; Wang et al., 2012). However, the safe use and the renal protection action of these RAAS inhibitors are still a matter of ongoing research. AT1 antagonists alone or in combination with ACE inhibitors have been shown to have a surprising variety of effects (Mauer et al., 2009; Mauer & Fioretto, 2005). ACE inhibition causes suppression of angiotensin II formation, resulting in the less angiotensin II binding to the AT1 receptor as well as AT2 receptor (Wolf & Ritz, 2005). However, when AT1 antagonist is used receptor signalling is reduced/blocked increasing the availability angiotensin II to act on other pathways, such as increasing AT2 receptor activation (Naito et al., 2010). The biological consequence of increased AT2 receptor activation in such condition could be beneficial (Carey, Wang, & Siragy, 2000; Danyel, Schmerler, Paulis, Unger, & Steckelings, 2013; Matavelli, Huang, & Siragy, 2011; Padia & Carey, 2013; Naito et al., 2010), but some others have reported a detrimental effect on organ protection (Waseda et al., 2008; Mifune et al., 2000; Tejera et al., 2004; Cao et al., 2002). In our analysis, in any case, such increased activation AT2 receptors would likely not have any major beneficial role. However, the role of increased AT2 receptor activation in organ protection needs further investigation.
There is not sufficient data from clinical trials that clearly demonstrate any significant differences between ACE inhibitors and AT1 antagonists in renal outcome in diabetes. However even with this limitation, Mauer et al. (2009) analysed over 5 years the effect of the AT1 antagonist losartan and the ACEI enalapril (compared to placebo) used in normotensive type I diabetic patient without albuminuria. They found that losartan was associated with significant increase in the onset of microalbuminuria when compared to placebo, while enalapril was associated with an insignificant suppression in the onset of microalbuminuria when compared to placebo. Furthermore, Mauer et al. (2009) performed renal biopsies before and after the drug intervention and found that only the losartan group tended to have an increase in glomerular mesangial matrix fractions in post‐intervention biopsy samples but not in the pre‐intervention samples (n = 50 in each group, P = 0.07). Although this not as expected it may suggest an important difference between AT1 antagonists (ARBs) and ACEI superiority suggested by meta‐analysis (Wu et al., 2013). However, some studies have shown that combination therapies may not be renal‐protective, despite a remarkable reduction in the albuminuria (Kunz, Friedrich, Wolbers, & Mann, 2008; Mann et al., 2008; Mogensen et al., 2000). The data from the previous studies in diabetic and non‐diabetic subjects have shown differential effects in terms of renal function and protection (Hsu et al., 2017; Laverman, Remuzzi, & Ruggenenti, 2004; Wu et al., 2017). However, some studies have shown better effects for ACE inhibition over AT1 antagonism on renal protections in diabetic patients who have features of chronic kidney disease (Baltatzi, Savopoulos, & Hatzitolios, 2011; Hsu et al., 2017; Laverman et al., 2004; Mavridis, Palmer, & Strippoli, 2016; Wu et al., 2017). In some studies, AT1 antagonist treatment is not protective for glomerular health (Fernandez‐Fernandez et al., 2012; Nagai et al., 2020; Nakanishi et al., 2011). The discrepancies between these trials could be due to differences in experimental design and medicine used. In the present study, the combination treatment (ACEI + AT1 antagonist) was not the focus of the study, however we analysed the differential renal protection abilities of ACE inhibition and AT1 receptor block. From the data, it is clear that the renal protection ability of ACE inhibition is associated with an elevated level of AcSDKP, while the AT1 antagonist TA‐606 did not show such protective effects. ACE inhibition in combination with AcSDKP is more effective in reducing the renal fibrogenic phenotypes, suggesting that antifibrotic mechanisms of AcSDKP are partly involved for renal protection ability of ACEIs. AcSDKP is an alternative substrate for the ACE (Kanasaki et al., 2011). One study that shows that ACE inhibition elevates the AcSDKP level by fourfold to fivefold (Kanasaki et al., 2011) which confirms our results. From the results of POP inhibition with S17092 in the reduced fibrotic diabetic mice model, which was found to partially block AcSDKP synthesis and accelerates the renal fibrosis, it is imperative to understand the mechanism behind the clear antifibrotic action of AcSDKP. AcSDKP clearly abolished whereas ACE inhibition only partly abolished the renal fibrogenic processes in the POPi‐injected mice, which have a lower level of AcSDKP. AT1 antagonism did not show any renal protective effects, suggesting that AcSDKP is one of the many antifibrotic molecules by which ACE inhibitors mediate their antifibrotic actions.
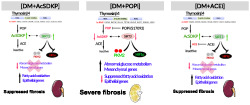
To investigate further the antifibrotic mechanism of AcSDKP, we analysed the keys enzymes of central metabolism in the kidneys. In the diabetic kidneys, the disruption in central metabolism can accelerate the epithelial‐to‐mesenchymal transition and endothelial‐to‐mesenchymal transition derived‐fibroblasts formation and proliferation. However, disruption in central metabolism can also lead to the development of intermediated cell types which accelerate in the fibrogenic phenotypes in diabetic kidneys (Kang et al., 2015; Srivastava et al., 2018,2019). Defective fatty acid oxidation and concomitant induction in the abnormal glucose metabolism contributes in the activation of mesenchymal mechanisms in the kidney (Kang et al., 2015; Srivastava et al., 2018). Altered metabolic shifts, which produce the required primary metabolic precursors for survival of the matrix associated fibroblasts, are a key mechanism for myofibroblast formation in the diabetic kidneys (Srivastava et al., 2018). We observed that SIRT3 regulates the metabolic shift in the kidney and its deficiency causes metabolic insults in diabetic kidney (Srivastava et al., 2018). SIRT3 is key intermediate molecule to regulate glucose metabolism in cytosol and fatty acid oxidation in mitochondria in diabetic kidneys (Srivastava et al., 2018). It is evident from our data that the protective nature of ACE inhibition is due to its ability to restore SIRT3 protein level, restore normal mitochondrial metabolism, fatty acid oxidation and suppress the HIF1α‐inducible proteins (GLUT1, HK2, PKM2 and PDK4)‐linked defective glucose metabolism. AT1 receptor blockade failed to restore the SIRT3 protein level and, hence, was unable to restore the normal kidney metabolism in diabetes. Similarly, our results from cultured‐epithelial cells demonstrated that Tβ4 silencing suppressed AcSDKP level in the cell medium was associated with disruption of central metabolism and gain of fibrogenic features. Whereas, ACE silencing elevated the AcSDKP level and improved the metabolic homeostasis, further validating that AcSDKP is an important protein required for epithelial cell homeostasis. But how do ACE inhibition and AcSDKP restore the normal glucose and fatty acid metabolism? To understand the association between AcSDKP and SIRT3, we analysed the AcSDKP level in the plasma and urine of SIRT3 silenced diabetic mice and the data suggest that SIRT3 suppression is associated with a lower AcSDKP level and did not affect ACE and POP enzyme activities in diabetic kidneys. It is evident from these results that lower level of AcSDKP in the SIRT3 silenced mice could be due to a secondary effect, not a direct effect. It is possible that lower level of AcSDKP could be due to severe fibrosis and inflammation by the systemic loss of SIRT3. Interestingly, glycolysis inhibitors have shown to cause elevated levels of AcSDKP in the kidney and plasma. However, glycolysis inhibitors have shown tendency to lower ACE activity. Figure 10 depicts schematic diagrams showing the regulation of AcSDKP in central metabolism. AcSDKP is produced by Tβ4 by catalysis of POP enzyme. ACE degrades AcSDKP into the inactive form. POPi partly blocks the AcSDKP synthesis and thus shows reduced level of AcSDKP and hence fibrosis in the diabetic kidneys. Reduction ACE activity by ACEIs therefore elevates the level of AcSDKP, whereas AT1 antagonists does not alter the AcSDKP level. The kidney protective mechanism of ACE inhibition is partly due to its ability to elevate the AcSDKP level, which is associated with SIRT3‐mediated restoration of fatty acid oxidation and reduction in abnormal glucose metabolism, and hence, AcSDKP is the key endogenous peptide for energy homeostasis in kidney cells.
FIGURE 10.
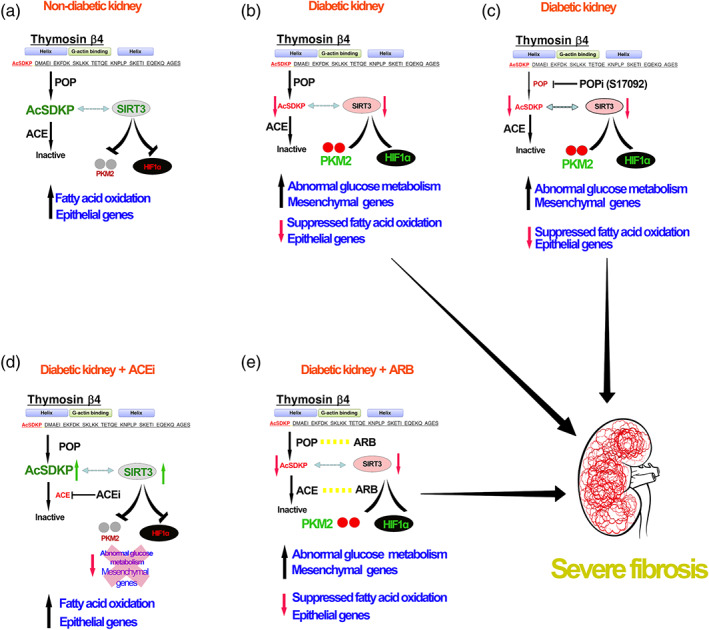
Working hypothesis depicts the critical role of AcSDKP in the kidney metabolism. Suppressed level of AcSDKP in diabetes is linked with SIRT3‐deficiency associated disruption in central metabolism and hence, fibrosis in kidney. POP inhibitor (S17092), partially blocks the AcSDKP level and hence further enhanced the severity of defective metabolism‐associated kidney fibrosis. ACE inhibition elevates the AcSDKP level, improve the renal metabolic health whereas, ARB does not alter the AcSDKP level , and hence, unable to improve the renal metabolic health in diabetic kidney, suggests that AcSDKP is essential peptide for renal metabolic health and protects from fibrosis
In conclusion, AcSDKP is a vital peptide which is associated with normal kidney metabolism. Administration of AcSDKP leads to disruption of defective cell metabolism in diabetic kidneys. The present study adds to the understanding of the antifibrotic mechanisms of ACE inhibition and the biology of AcSDKP.
AUTHOR CONTRIBUTIONS
S.P.S. performed the experiments, proposed the original idea, generated the figures and wrote the manuscript. J.G. provided intellectual input. K.K. supervised the experiments, provided intellectual input and performed final editing of the manuscript. D.K. provided intellectual input.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry and Animal Experimentation, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S1 Dose dependent antifibrotic response of AcSDKP in the kidneys of diabetic mice
Figure S2 Angiotensin II level in the kidneys
Figure S3 CPT1a level in kidneys
Figure S4 Effect of ARB in the POP inhibitor treated diabetic mice
Figure S5 Effect of AcSDKP in the POP inhibitor treated diabetic mice
Figure S6 Effect of AcSDKP on the renal metabolism in the POP inhibitor treated diabetic mice
Figure S7 Effect of SIRT3 knockdown in the ACE and POP activity in the kidneys of diabetic mice
Data S1. Supporting information
ACKNOWLEDGEMENTS
We thank Dr. Omata from Asabio Bio Technology for providing us AcSDKP. Also, we thank Mitsubishi Tanabe Pharma for providing ACEI (Imidapril) and AT1 receptor antagonist (TA‐606) through an MTA agreement. This work was partially supported by grants from the Japan Society for the Promotion of Science for K.K. (23790381) and D.K. (25282028 and 25670414), and research grants from the Japan Research Foundation for Clinical Pharmacology to K.K. (2011) and Takeda Visionally Research Grant to K.K. (2013). This work was also partially supported by a Grant for Collaborative Research awarded to D.K. (C2011‐4 and C2012‐1) and a Grant for Promoted Research awarded to K.K. (S2011‐1 and S2012‐5) from Kanazawa Medical University. In addition, this work was also partially supported by a grant from the Foundation for the National Institutes of Health (USA) (R01HL131952) to J.G. K.K. who was also supported by several foundational grants, including grants from the Japan Research Foundation for Clinical Pharmacology, the Daiichi‐Sankyo Foundation of Life Science, the Ono Medical Research Foundation, the NOVARTIS Foundation (Japan) for the Promotion of Science, the Takeda Science Foundation and the Banyu Foundation. K.K. and D.K. received lecture fees from Boehringer Ingelheim, Eli Lilly and Mitsubishi Tanabe Pharma. Boehringer Ingelheim, Eli Lilly and Mitsubishi Tanabe Pharma donated to Kanazawa Medical University and were not directly associated with this project.
Srivastava SP, Goodwin JE, Kanasaki K, Koya D. Metabolic reprogramming by N‐acetyl‐seryl‐aspartyl‐lysyl‐proline protects against diabetic kidney disease. Br J Pharmacol. 2020;177:3691–3711. 10.1111/bph.15087
Contributor Information
Keizo Kanasaki, Email: kkanasak@med.shimane-u.ac.jp.
Daisuke Koya, Email: koya0516@kanazawa-med.ac.jp.
REFERENCES
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, S. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Mathie, A. , Peter, J. A. , … CGTP Collaborators . (2019). The concise guide to pharmacology 2019/2020: G protein‐coupled receptors. British Journal of Pharmacology, 176, S21–S141. 10.1111/bph.14748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash, S. R. , & Cuppage, F. E. (1970). Shift toward anaerobic glycolysis in the regenerating rat kidney. The American Journal of Pathology, 60, 385–402. [PMC free article] [PubMed] [Google Scholar]
- Baltatzi, M. , Savopoulos, C. , & Hatzitolios, A. (2011). Role of angiotensin converting enzyme inhibitors and angiotensin receptor blockers in hypertension of chronic kidney disease and renoprotection. Study results. Hippokratia, 15, 27–32. [PMC free article] [PubMed] [Google Scholar]
- Bindu, S. , Pillai, V. B. , Kanwal, A. , Samant, S. , Mutlu, G. M. , Verdin, E. , … Gupta, M. P. (2017). SIRT3 blocks myofibroblast differentiation and pulmonary fibrosis by preventing mitochondrial DNA damage. American Journal of Physiology. Lung Cellular and Molecular Physiology, 312, L68–L78. 10.1152/ajplung.00188.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blantz, R. C. (2014). Phenotypic characteristics of diabetic kidney involvement. Kidney International, 86, 7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey, R. M. , Wang, Z. Q. , & Siragy, H. M. (2000). Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension, 35, 155–163. 10.1161/01.hyp.35.1.155 [DOI] [PubMed] [Google Scholar]
- Cao, Z. (2002). Angiotensin Type 2 Receptor Antagonism Confers Renal Protection in a Rat Model of Progressive Renal Injury. Journal of the American Society of Nephrology, 13, (7), 1773–1787. 10.1097/01.asn.0000019409.17099.33 [DOI] [PubMed] [Google Scholar]
- Chen, T. , Li, J. , Liu, J. , Li, N. , Wang, S. , Liu, H. , … Bu, P. (2015). Activation of SIRT3 by resveratrol ameliorates cardiac fibrosis and improves cardiac function via the TGF‐beta/Smad3 pathway. American Journal of Physiology. Heart and Circulatory Physiology, 308, H424–H434. 10.1152/ajpheart.00454.2014 [DOI] [PubMed] [Google Scholar]
- Curtis M.J., Alexander S., Cirino G., Docherty J.R., George G.H., Giembycz M.A.,… Ahluwali, A. (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology 175, 987‐993. https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danyel, L. A. , Schmerler, P. , Paulis, L. , Unger, T. , & Steckelings, U. M. (2013). Impact of AT2‐receptor stimulation on vascular biology, kidney function, and blood pressure. Integr Blood Press Control, 6, 153–161. 10.2147/IBPC.S34425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis, R. J. , & Thompson, C. B. (2012). Cellular metabolism and disease: what do metabolic outliers teach us? Cell, 148, 1132–1144. 10.1016/j.cell.2012.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund, T. , Wahlberg, J. , Ungerstedt, U. , & Hillered, L. (1991). Interstitial lactate, inosine and hypoxanthine in rat kidney during normothermic ischaemia and recirculation. Acta Physiologica Scandinavica, 143, 279–286. 10.1111/j.1748-1716.1991.tb09233.x [DOI] [PubMed] [Google Scholar]
- Fernandez‐Fernandez, B. , Ortiz, A. , Gomez‐Guerrero, C. , Barat, A. , Martin‐Cleary, C. , & Egido, J. (2012). Juxtaglomerular apparatus hyperplasia under dual angiotensin blockade. A footprint of adequate RAS inhibition or a concern for renal fibrosis? BMC Nephrology, 13, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara, Y. , Yamamoto, S. , Yano, F. , Orita, Y. , Fujiwara, Y. , Ueda, N. , … Tanaka, T. (1991). Changes in activities and mRNA levels of glycolytic enzymes of ischemia‐reperfused rat kidney. Contributions to Nephrology, 95, 222–228. 10.1159/000420663 [DOI] [PubMed] [Google Scholar]
- Ganesh, J. , & Viswanathan, V. (2011). Management of diabetic hypertensives. Indian J Endocrinol Metab, 15(Suppl 4), S374–S379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande, M. T. , Sanchez‐Laorden, B. , Lopez‐Blau, C. , De Frutos, C. A. , Boutet, A. , Arevalo, M. , … Nieto, A. (2015). Snail1‐induced partial epithelial‐to‐mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nature Medicine, 21, 989–997. 10.1038/nm.3901 [DOI] [PubMed] [Google Scholar]
- Gu, J. , Yang, M. , Qi, N. , Mei, S. , Chen, J. , Song, S. , … Mei, C. (2016). Olmesartan Prevents Microalbuminuria in db/db Diabetic Mice Through Inhibition of Angiotensin II/p38/SIRT1‐Induced Podocyte Apoptosis. Kidney & Blood Pressure Research, 41, 848–864. 10.1159/000452588 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto, Y. , Ohashi, R. , Kurosawa, Y. , Minami, K. , Kaji, H. , … Murata.S. (1998). Pharmacologic Profile of TA‐606, a Novel Angiotensin II‐receptor Antagonist in the Rat. Journal of Cardiovascular Pharmacology, 31, 568–575. 10.1097/00005344-199804000-00015 [DOI] [PubMed] [Google Scholar]
- Held, P. J. , Port, F. K. , Webb, R. L. , Wolfe, R. A. , Garcia, J. R. , Blagg, C. R. , & Agodoa, L. Y. (1991). The United States Renal Data System's 1991 annual data report: an introduction. American Journal of Kidney Diseases, 18, 1–16. [PubMed] [Google Scholar]
- Hsu, F. Y. , Lin, F. J. , Ou, H. T. , Huang, S. H. , & Wang, C. C. (2017). Renoprotective Effect of angiotensin‐converting enzyme inhibitors and angiotensin ii receptor blockers in diabetic patients with proteinuria. Kidney & Blood Pressure Research, 42, 358–368. 10.1159/000477946 [DOI] [PubMed] [Google Scholar]
- Jacobsen, P. , Andersen, S. , Jensen, B. R. , & Parving, H. H. (2003). Additive effect of ACE inhibition and angiotensin II receptor blockade in type I diabetic patients with diabetic nephropathy. J Am Soc Nephrol, 14, 992–999. 10.1097/01.asn.0000054495.96193.bf [DOI] [PubMed] [Google Scholar]
- Jensen, T. M. , Vistisen, D. , Fleming, T. , Nawroth, P. P. , Rossing, P. , Jorgensen, M. E. , … Witte, D. R. (2016). Methylglyoxal is associated with changes in kidney function among individuals with screen‐detected Type 2 diabetes mellitus. Diabetic Medicine, 33, 1625–1631. 10.1111/dme.13201 [DOI] [PubMed] [Google Scholar]
- Kalender, B. , Ozturk, M. , Tuncdemir, M. , Uysal, O. , Dagistanli, F. K. , Yegenaga, I. , … Erek, E. (2002). Renoprotective effects of valsartan and enalapril in STZ‐induced diabetes in rats. Acta Histochemica, 104, 123–130. 10.1078/0065-1281-00643 [DOI] [PubMed] [Google Scholar]
- Kanasaki, K. , Shi, S. , Kanasaki, M. , He, J. , Nagai, T. , Nakamura, Y. , … Koya, D. (2014). Linagliptin‐mediated DPP‐4 inhibition ameliorates kidney fibrosis in streptozotocin‐induced diabetic mice by inhibiting endothelial‐to‐mesenchymal transition in a therapeutic regimen. Diabetes, 63, 2120–2131. 10.2337/db13-1029 [DOI] [PubMed] [Google Scholar]
- Kanasaki, M. , Nagai, T. , Kitada, M. , Koya, D. , & Kanasaki, K. (2011). Elevation of the anti‐fibrotic peptide N‐acetyl‐seryl‐aspartyl‐lysyl‐proline: A blood pressure‐independent beneficial effect of angiotensin I‐converting enzyme inhibitors. Fibrogenesis & Tissue Repair, 4, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, H. M. , Ahn, S. H. , Choi, P. , Ko, Y. A. , Han, S. H. , Chinga, F. , … Susztak, K. (2015). Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nature Medicine, 21, 37–46. 10.1038/nm.3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H. S. , Patel, K. , Muldoon‐Jacobs, K. , Bisht, K. S. , Aykin‐Burns, N. , Pennington, J. D. , … Gius, D. (2010). SIRT3 is a mitochondria‐localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell, 17, 41–52. 10.1016/j.ccr.2009.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koye, D. N. , Magliano, D. J. , Nelson, R. G. , & Pavkov, M. E. (2018). The Global Epidemiology of Diabetes and Kidney Disease. Advances in Chronic Kidney Disease, 25, 121–132. 10.1053/j.ackd.2017.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz, R. , Friedrich, C. , Wolbers, M. , & Mann, J. F. (2008). Meta‐analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Annals of Internal Medicine, 148, 30–48. [DOI] [PubMed] [Google Scholar]
- Lan, R. , Geng, H. , Singha, P. K. , Saikumar, P. , Bottinger, E. P. , Weinberg, J. M. , & Venkatachalam, M. A. (2016). Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J Am Soc Nephrol, 27, 3356–3367. 10.1681/ASN.2015020177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laverman, G. D. , Remuzzi, G. , & Ruggenenti, P. (2004). ACE inhibition versus angiotensin receptor blockade: which is better for renal and cardiovascular protection? J am Soc Nephrol, 15(Suppl 1), S64–S70. [DOI] [PubMed] [Google Scholar]
- LeBleu, V. S. , Taduri, G. , O'Connell, J. , Teng, Y. , Cooke, V. G. , Woda, C. , … Kalluri, R. (2013). Origin and function of myofibroblasts in kidney fibrosis. Nature Medicine, 19, 1047–1053. 10.1038/nm.3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Liu, H. , Takagi, S. , Nitta, K. , Kitada, M. , Srivastava, S. P. , … Koya, D. (2020). Renal protective effects of empagliflozin via inhibition of EMT and aberrant glycolysis in proximal tubules. JCI Insight, 5 10.1172/jci.insight.129034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Shi, S. , Srivastava, S. P. , Kitada, M. , Nagai, T. , Nitta, K. , … Koya, D. (2017). FGFR1 is critical for the anti‐endothelial mesenchymal transition effect of N‐acetyl‐seryl‐aspartyl‐lysyl‐proline via induction of the MAP 4K4 pathway. Cell Death & Disease, 8, e2965 10.1038/cddis.2017.353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovisa, S. , LeBleu, V. S. , Tampe, B. , Sugimoto, H. , Vadnagara, K. , Carstens, J. L. , … Kalluri, R. (2015). Epithelial‐to‐mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nature Medicine, 21, 998–1009. 10.1038/nm.3902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macconi, D. , Tomasoni, S. , Romagnani, P. , Trionfini, P. , Sangalli, F. , Mazzinghi, B. , … Benigni, A. (2012). MicroRNA‐324‐3p promotes renal fibrosis and is a target of ACE inhibition. J Am Soc Nephrol, 23, 1496–1505. 10.1681/ASN.2011121144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann, J. F. , Schmieder, R. E. , McQueen, M. , Dyal, L. , Schumacher, H. , Pogue, J. , … ONTARGET investigators . (2008). Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double‐blind, controlled trial. Lancet, 372, 547–553. 10.1016/S0140-6736(08)61236-2 [DOI] [PubMed] [Google Scholar]
- Matavelli, L. C. , Huang, J. , & Siragy, H. M. (2011). Angiotensin AT(2) receptor stimulation inhibits early renal inflammation in renovascular hypertension. Hypertension, 57, 308–313. 10.1161/HYPERTENSIONAHA.110.164202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauer, M. , & Fioretto, P. (2005). Preventing microalbuminuria in type 2 diabetes. The New England Journal of Medicine, 352, 833–834. author reply 833‐834 [PubMed] [Google Scholar]
- Mauer, M. , Zinman, B. , Gardiner, R. , Suissa, S. , Sinaiko, A. , Strand, T. , … Klein, R. (2009). Renal and retinal effects of enalapril and losartan in type 1 diabetes. The New England Journal of Medicine, 361, 40–51. 10.1056/NEJMoa0808400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavridis, D. , Palmer, S. C. , & Strippoli, G. F. (2016). Comparative Superiority of ACE Inhibitors Over Angiotensin Receptor Blockers for People With CKD: Does It Matter? American Journal of Kidney Diseases, 67, 713–715. 10.1053/j.ajkd.2016.02.031 [DOI] [PubMed] [Google Scholar]
- McGrath, John C , & Lilley Elliot (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. British Journal of Pharmacology, 172(13), 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mifune, M. , Sasamura, H. , Shimizu‐Hirota, R. , Miyazaki, H. , & Saruta, T. (2000). Angiotensin II Type 2 Receptors Stimulate Collagen Synthesis in Cultured Vascular Smooth Muscle Cells. Hypertension, 36(5), 845–850. 10.1161/01.hyp.36.5.845 [DOI] [PubMed] [Google Scholar]
- Mogensen, C. E. , Neldam, S. , Tikkanen, I. , Oren, S. , Viskoper, R. , Watts, R. W. , & Cooper, M. E. (2000). Randomised controlled trial of dual blockade of renin‐angiotensin system in patients with hypertension, microalbuminuria, and non‐insulin dependent diabetes: the candesartan and lisinopril microalbuminuria (CALM) study. BMJ, 321, 1440–1444. 10.1136/bmj.321.7274.1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai, T. , Kanasaki, M. , Srivastava, S. P. , Nakamura, Y. , Ishigaki, Y. , Kitada, M. , … Koya, D. (2014). N‐acetyl‐seryl‐aspartyl‐lysyl‐proline inhibits diabetes‐associated kidney fibrosis and endothelial‐mesenchymal transition. BioMed Research International, 2014, 696475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai, Y. , Yamabe, F. , Sasaki, Y. , Ishii, T. , Nakanishi, K. , Nakajima, K. , … Yamanaka, N. (2020). A Study of Morphological Changes in Renal Afferent Arterioles Induced by Angiotensin II Type 1 Receptor Blockers in Hypertensive Patients. Kidney & Blood Pressure Research, 45, 194–208. 10.1159/000505025 [DOI] [PubMed] [Google Scholar]
- Naito, T. , Ma, L. J. , Yang, H. , Zuo, Y. , Tang, Y. , Han, J. Y. , … Fogo, A. B. (2010). Angiotensin type 2 receptor actions contribute to angiotensin type 1 receptor blocker effects on kidney fibrosis. American Journal of Physiology. Renal Physiology, 298, F683–F691. 10.1152/ajprenal.00503.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi, K. , Nagai, Y. , Piao, H. , Akimoto, T. , Kato, H. , Yanakieva‐Georgieva, N. , … Oite, T. (2011). Changes in renal vessels following the long‐term administration of an angiotensin II receptor blocker in Zucker fatty rats. Journal of the Renin‐Angiotensin‐Aldosterone System, 12, 65–74. 10.1177/1470320310387844 [DOI] [PubMed] [Google Scholar]
- Nitta, K. , Shi, S. , Nagai, T. , Kanasaki, M. , Kitada, M. , Srivastava, S. P. , … Koya, D. (2016). Oral administration of N‐acetyl‐seryl‐aspartyl‐lysyl‐proline ameliorates kidney disease in both type 1 and type 2 diabetic mice via a therapeutic regimen. BioMed Research International, 2016, 9172157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield, M. D. , Bach, L. A. , Forbes, J. M. , Nikolic‐Paterson, D. , McRobert, A. , Thallas, V. , … Cooper, M. E. (2001). Advanced glycation end products cause epithelial‐myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). The Journal of Clinical Investigation, 108, 1853–1863. 10.1172/JCI11951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omata, M. , Taniguchi, H. , Koya, D. , Kanasaki, K. , Sho, R. , Kato, Y. , … Inomata, N. (2006). N‐acetyl‐seryl‐aspartyl‐lysyl‐proline ameliorates the progression of renal dysfunction and fibrosis in WKY rats with established anti‐glomerular basement membrane nephritis. Journal of the American Society of Nephrology: JASN, 17, 674–685. 10.1681/ASN.2005040385 [DOI] [PubMed] [Google Scholar]
- Padia, S. H. , & Carey, R. M. (2013). AT2 receptors: beneficial counter‐regulatory role in cardiovascular and renal function. Pflügers Archiv, 465, 99–110. 10.1007/s00424-012-1146-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, S. C. , Mavridis, D. , Navarese, E. , Craig, J. C. , Tonelli, M. , Salanti, G. , … Strippoli, G. F. (2015). Comparative efficacy and safety of blood pressure‐lowering agents in adults with diabetes and kidney disease: A network meta‐analysis. Lancet, 385, 2047–2056. 10.1016/S0140-6736(14)62459-4 [DOI] [PubMed] [Google Scholar]
- Poyan Mehr, A. , Tran, M. T. , Ralto, K. M. , Leaf, D. E. , Washco, V. , Messmer, J. , … Parikh, S. M. (2018). De novo NAD(+) biosynthetic impairment in acute kidney injury in humans. Nature Medicine, 24, 1351–1359. 10.1038/s41591-018-0138-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, W. , Keenan, H. A. , Li, Q. , Ishikado, A. , Kannt, A. , Sadowski, T. , … King, G. L. (2017). Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nature Medicine, 23, 753–762. 10.1038/nm.4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero, C. A. , Kumar, N. , Nakagawa, P. , Worou, M. E. , Liao, T. D. , Peterson, E. L. , & Carretero, O. A. (2019). Renal release of N‐acetyl‐seryl‐aspartyl‐lysyl‐proline is part of an antifibrotic peptidergic system in the kidney. American Journal of Physiology. Renal Physiology, 316, F195–F203. 10.1152/ajprenal.00270.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe, I. , Chiaravalli, M. , Mannella, V. , Ulisse, V. , Quilici, G. , Pema, M. , … Boletta, A. (2013). Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nature Medicine, 19, 488–493. 10.1038/nm.3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheen, A. J. (2004). Renin‐angiotensin system inhibition prevents type 2 diabetes mellitus. Part 1. A meta‐analysis of randomised clinical trials. Diabetes & Metabolism, 30, 487–496. 10.1016/s1262-3636(07)70146-5 [DOI] [PubMed] [Google Scholar]
- Shi, S. , Srivastava, S. P. , Kanasaki, M. , He, J. , Kitada, M. , Nagai, T. , … Koya, D. (2015). Interactions of DPP‐4 and integrin beta1 influences endothelial‐to‐mesenchymal transition. Kidney International, 88, 479–489. 10.1038/ki.2015.103 [DOI] [PubMed] [Google Scholar]
- Shibuya, K. , Kanasaki, K. , Isono, M. , Sato, H. , Omata, M. , Sugimoto, T. , … Koya, D. (2005). N‐acetyl‐seryl‐aspartyl‐lysyl‐proline prevents renal insufficiency and mesangial matrix expansion in diabetic db/db mice. Diabetes, 54, 838–845. 10.2337/diabetes.54.3.838 [DOI] [PubMed] [Google Scholar]
- Sosulski, M. L. , Gongora, R. , Feghali‐Bostwick, C. , Lasky, J. A. , & Sanchez, C. G. (2017). Sirtuin 3 Deregulation Promotes Pulmonary Fibrosis. The Journals of Gerontology. Series a, Biological Sciences and Medical Sciences, 72, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, S. P. , Goodwin, J. E. , Kanasaki, K. , & Koya, D. (2020). Inhibition of angiotensin‐converting enzyme ameliorates renal fibrosis by mitigating DPP‐4 level and restoring antifibrotic microRNAs. Genes (Basel), 11 10.3390/genes11020211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, S. P. , Hedayat, F. A. , Kanasaki, K. , & Goodwin, J. E. (2019). microRNA Crosstalk Influences Epithelial‐to‐Mesenchymal, Endothelial‐to‐Mesenchymal, and Macrophage‐to‐Mesenchymal Transitions in the Kidney. Frontiers in Pharmacology, 10, 904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, S. P. , Koya, D. , & Kanasaki, K. (2013). MicroRNAs in Kidney Fibrosis and Diabetic Nephropathy: Roles on EMT and EndMT. BioMed Research International, 2013, 125469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, S. P. , Li, J. , Kitada, M. , Fujita, H. , Yamada, Y. , Goodwin, J. E. , … Koya, D. (2018). SIRT3 deficiency leads to induction of abnormal glycolysis in diabetic kidney with fibrosis. Cell Death & Disease, 9, 997 10.1038/s41419-018-1057-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, S. P. , Shi, S. , Kanasaki, M. , Nagai, T. , Kitada, M. , He, J. , … Koya, D. (2016). Effect of antifibrotic microRNAs crosstalk on the action of N‐acetyl‐seryl‐aspartyl‐lysyl‐proline in diabetes‐related kidney fibrosis. Scientific Reports, 6, 29884 10.1038/srep29884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, S. P. , Shi, S. , Koya, D. , & Kanasaki, K. (2014). Lipid mediators in diabetic nephropathy. Fibrogenesis & Tissue Repair, 7, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejera, N. , Go´mez‐Garre, D. , La´zaro, A. , Gallego‐Delgado, J. , Alonso, C. , Blanco, J. , … Egido, J. (2004). Persistent Proteinuria Up‐Regulates Angiotensin II Type 2 Receptor and Induces Apoptosis in Proximal Tubular Cells. American J Pathol, 164(5), 1817–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, M. T. , Zsengeller, Z. K. , Berg, A. H. , Khankin, E. V. , Bhasin, M. K. , Kim, W. , … Parikh, S. M. (2016). PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature, 531, 528–532. 10.1038/nature17184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B. , Komers, R. , Carew, R. , Winbanks, C. E. , Xu, B. , Herman‐Edelstein, M. , … Kantharidis, P. (2012). Suppression of microRNA‐29 expression by TGF‐beta1 promotes collagen expression and renal fibrosis. J am Soc Nephrol, 23, 252–265. 10.1681/ASN.2011010055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waseda, Y. , Yasui, M. , Nishizawa, Y. , Inuzuka, K. , Takato, H. , Ichikawa, Y. , … Nakao, S. (2008). Angiotensin II type 2 receptor antagonist reduces bleomycin‐induced pulmonary fibrosis in mice. Respiratory Research, 9(1), 10.1186/1465-9921-9-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, G. , & Ritz, E. (2005). Combination therapy with ACE inhibitors and angiotensin II receptor blockers to halt progression of chronic renal disease: pathophysiology and indications. Kidney International, 67, 799–812. 10.1111/j.1523-1755.2005.00145.x [DOI] [PubMed] [Google Scholar]
- Wu, H. Y. , Huang, J. W. , Lin, H. J. , Liao, W. C. , Peng, Y. S. , Hung, K. Y. , … Chien, K. L. (2013). Comparative effectiveness of renin‐angiotensin system blockers and other antihypertensive drugs in patients with diabetes: systematic review and bayesian network meta‐analysis. BMJ, 347, f6008 10.1136/bmj.f6008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, H. Y. , Peng, C. L. , Chen, P. C. , Chiang, C. K. , Chang, C. J. , Huang, J. W. , … Chien, K. L. (2017). Comparative effectiveness of angiotensin‐converting enzyme inhibitors versus angiotensin II receptor blockers for major renal outcomes in patients with diabetes: A 15‐year cohort study. PLoS ONE, 12, e0177654 10.1371/journal.pone.0177654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, F. , & Cadenas, E. (2015). Mitochondria: the cellular hub of the dynamic coordinated network. Antioxidants & Redox Signaling, 22, 961–964. 10.1089/ars.2015.6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg, E. M. , Potenta, S. E. , Sugimoto, H. , Zeisberg, M. , & Kalluri, R. (2008). Fibroblasts in kidney fibrosis emerge via endothelial‐to‐mesenchymal transition. J Am Soc Nephrol, 19, 2282–2287. 10.1681/ASN.2008050513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H. L. , Zhang, R. , Anand, P. , Stomberski, C. T. , Qian, Z. , Hausladen, A. , … Stamler, J. S. (2019). Metabolic reprogramming by the S‐nitroso‐CoA reductase system protects against kidney injury. Nature, 565, 96–100. 10.1038/s41586-018-0749-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Dose dependent antifibrotic response of AcSDKP in the kidneys of diabetic mice
Figure S2 Angiotensin II level in the kidneys
Figure S3 CPT1a level in kidneys
Figure S4 Effect of ARB in the POP inhibitor treated diabetic mice
Figure S5 Effect of AcSDKP in the POP inhibitor treated diabetic mice
Figure S6 Effect of AcSDKP on the renal metabolism in the POP inhibitor treated diabetic mice
Figure S7 Effect of SIRT3 knockdown in the ACE and POP activity in the kidneys of diabetic mice
Data S1. Supporting information