

Abstract
Clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 loaded by vectors could induce high rates of specific site genome editing and correct disease-causing mutations. However, most monogenic genetic diseases such as hemophilia are caused by different mutations dispersed in one gene, instead of an accordant mutation. Vectors developed for correcting specific mutations may not be suited to different mutations at other positions. Site-specific gene addition provides an ideal solution for long-term, stable gene therapy. We have demonstrated SaCas9-mediated homology-directed factor IX (FIX) in situ targeting for sustained treatment of hemophilia B. In this study, we tested a more efficient dual adeno-associated virus (AAV) strategy with lower vector dose for liver-directed genome editing that enables CRISPR-Cas9-mediated site-specific integration of therapeutic transgene within the albumin gene, and we aimed to develop a more universal gene-targeting approach. We successfully achieved coagulation function in newborn and adult hemophilia B mice by a single injection of dual AAV vectors. FIX levels in treated mice persisted even after a two-thirds partial hepatectomy, indicating stable gene integration. Our results suggest that this CRISPR-Cas9-mediated site-specific gene integration in hepatocytes could transform into a common clinical therapeutic method for hemophilia B and other genetic diseases.
Keywords: AAV, CRISPR-Cas9, hemophilia B, targeted integration, albumin
Graphical Abstract
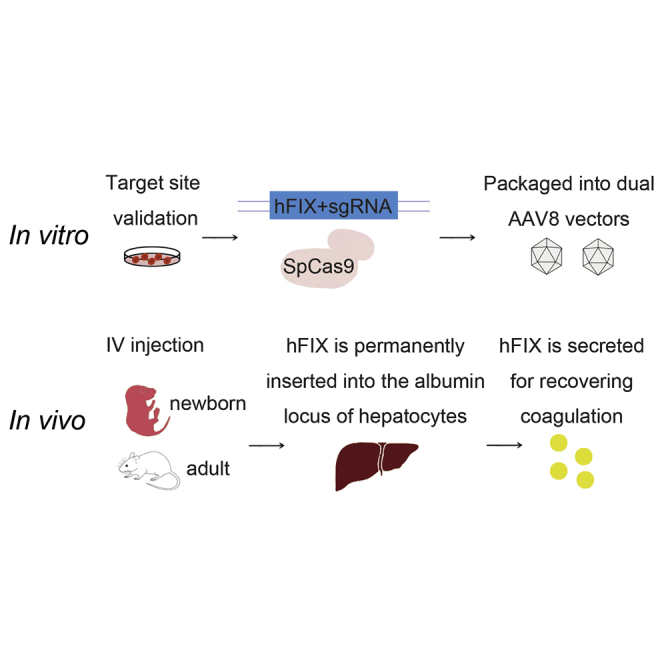
Wang et al. provide a method of CRISPR-Cas9-mediated gene site-specific integration for hemophilia B gene therapy. Through a dual AAV8 delivery system targeting mice hepatocytes, hFIX could be efficiently integrated into mAlb intron 1 locus. Treated adult and newborn hemophilia B mice could efficiently express hFIX with high clotting activity.
Introduction
Hemophilia B is an X-linked congenital hemorrhagic disease that is caused by a deficiency or dysfunction of coagulation factor IX (FIX). Disabled FIX results in bleeding manifestations, and severe hemophilia B-induced recurrent spontaneous hemarthrosis could result in deformity. In addition, intracranial hemorrhage would threaten patient life.1,2 Mainstream treatment relies on an additional plasma-derived or recombinant FIX protein injection for on-demand or prophylaxis administration. However, this strategy is expensive and hard to sustain because of the cost of recombinant proteins and frequent infusion.3 More importantly, inhibitors might be induced by recombinant FIX protein,4 and it would make the treatment of such patients more complicated and difficult. Adeno-associated virus (AAV)-mediated gene therapy provides a promising treatment approach for remedial expression of FIX. It has been confirmed that applying AAV, delivering a transgene expressing hyperactive FIX variant, could achieve sustained expression of FIX.2 Nevertheless, the virus genome could be lost during cell turnover due to the non-integrating character of AAV. When applied in liver-directed gene therapy, it might lead to a decline in therapeutic effects along with hepatocyte division.5 Loading AAV with genome-editing elements for targeted gene modification of host DNA might be a promising solution. With the aid of genome-editing tools, especially the emergence of CRISPR-Cas9, this targeted site genome-editing strategy has been tested in hemophilia B,6 mucopolysaccharidosis,7 Duchenne muscular dystrophy (DMD),8 and tyrosinemia.9
In our previous work, we developed a dual AAV system for transferring the CRISPR-Cas9 system-mediated homology-directed repair (HDR) to correct an ornithine transcarbamylase (OTC) point mutation in the liver of neonatal OTC-deficient mice. Mutations could be corrected in nearly 10% of hepatocytes, and survival increased in mice challenged with a high-protein diet.10 Based on these targeted-site gene modifications mediated by AAV, we hoped to develop a more universal liver-directed therapeutic technology compatible for other genetic mutations. Replacement of the entire gene with an episomal expression from the normal gene could reverse the clinical picture for some monogenetic diseases. However, most monogenic genetic diseases such as hemophilia B are induced by different mutations scattered in one gene, and a genome-editing strategy tackling multiple defects at different positions in the same gene would make a genome modification approach more complex. Targeting and integrating the remedial gene at a specific and safe locus might overcome the obstacle. Recently, we successfully recovered FIX expression through in situ integration.6 However, the FIX integration strategy in situ is limited by insufficient expression of the endogenic FIX promoter. In our previous research, we increased vector dosage for AAV8.SaCas9 by 5-fold in combination with increased donor vector doses. The increments of FIX protein activity levels were insignificant, and the expression level of FIX was nearly 10%.6 Although this approach could recover FIX expression to the therapeutic level, the limitation of this approach is that a vector-targeted FIX locus cannot be flexibly applied to other genetic diseases. Albumin (Alb) is a liver-directed protein that has high transcriptional activity.11 It provides an ideal versatile platform for multifarious therapeutic transgene integration. Sharma et al.11 applied zinc finger nuclease (ZFN)- mediated targeting the Alb locus to correct disease phenotype in mouse models of hemophilia A and B. However, this approach of ZFNs-mediated in vivo gene therapy requires co-transduction of three AAV vectors in the same hepatocyte, which would limit the therapy efficiency in clinical application. In addition, more vectors might trigger innate immunity response and influence the outcome of clinical gene transfer.
In this study, we developed a simpler and more universal liver-targeted dual AAV system loaded with CRISPR-Cas9 and verified the treatment effect on hemophilia B mice. By integrating codon-optimized partial human FIX (hFIX) in the mouse Alb (mAlb) locus, we successfully improved the coagulation function in both neonatal and adult hemophilia B mice. The treated mice survived a two-thirds partial hepatectomy challenge. hFIX expression from hepatocytes was efficient and sustained. Hence, we established a practical therapeutic strategy with high HDR frequency, more efficient FIX expression, and lower vectors dose, which would be well suited for many other genetic diseases, including those that are induced by dysfunctional proteins.
Results
Development of a Dual AAV Vector System for Cas9-Mediated hFIX Gene Integration
Based on our previous work,6 we aimed to develop a high-efficiency CRISPR-mediated gene integration vector for hemophilia B treatment. Following Cas9-induced double-strand breaks (DSBs), the codon-optimized, promoterless hFIX cDNA sequence spanning exon 2 to exon 8 and carrying hyperactive Padua mutation (hFIXco-Padua)12 would be integrated into the first mAlb intron. This would lead to the transcription of a chimeric m-hFIX mRNA by the strong mAlb promoter. In addition, the first mAlb exon would encode a universal secretory peptide, so that protein hFIX would be expressed and secreted after the specific integration (Figure 1A). We designed and engineered four single guide RNA (sgRNAs) (Table S1) targeting mAlb intron 1, and confirmed their activities in vitro in the murine cell line, H2.35, via Surveyor nuclease assays. We observed cleavage at frequencies ranging from 14.1% to 50.3% (Figure 1B), and sgRNA4 had a higher on-target editing efficiency than did the other three sgRNAs; hence, it was selected for subsequent studies. Next, we constructed AAV vectors for in vivo liver-directed hFIX integration. One vector expressed the SpCas9 gene from an engineered truncated liver-specific promoter, a shortened version of the liver-specific thyroxine binding globulin promoter (TBG-S1)13 (referred to as AAV8.SpCas9). The targeting donor vector encoded sgRNA4 and hFIXco-Padua, followed by bovine growth hormone (bGH) poly(A) and was flanked by length-optimized homology arms 0.5-kb on the 5′ side and 0.7-kb on the 3′ side (referred to as AAV8.sgRNA.donor) (Figure 1C). The untargeted donor vector contains all elements except for the 20-nt target sequence of the sgRNA (referred to as AAV8.control.donor). These vectors were produced for treatment of hemophilia B mice with the dual AAV system.
Figure 1.

In Vivo Genome Editing of Albumin Locus in Mouse Liver by AAV.CRISPR-Cas9
(A) Schematic illustrating albumin targeting strategy. (B) In vitro validation of sgRNAs targeted to mAlb1 in the H2.35 mouse cell line by transient transfection and Surveyor nuclease assays. sgRNA4 showed the highest efficiency in inducing indels in the targeted loci and was therefore chosen for subsequent studies. Arrows denote Surveyor nuclease cleaved fragments of the Alb PCR products. Results were replicated in two independent experiments. (C) Dual AAV vector system for liver-directed and SpCas9-mediated gene insertion. The AAV8.sgRNA.donor vector encoded a promoterless hFIX cassette containing exons 2–8 of the hFIX gene flanked by a SA signal and a poly(A) (PA) sequence.
Efficacy of In Vivo Gene Integration in Adult and Neonatal Hemophilia B Mouse Liver via Dual AAV Strategy
To determine the optimal donor vector dose for in vivo gene integration, we performed tail vein injection with 9 × 1010 genome copies (GC) of AAV8.SpCas9 and either 9 × 1010, 2.7 × 1011, or 4.5 × 1011 GC of the AAV8.sgRNA.donor vector. After 4 weeks of injection, we detected hFIX expression in the liver via immunofluorescence (Figures S1A and S1B) and measured plasma hFIX protein levels by an enzyme-linked immunosorbent assay (ELISA) (Figure S1C). Increasing the donor vector dose resulted in a significant increase in hFIX expression in the liver and plasma. We selected the highest donor dose (4.5 × 1011 GC/mouse) for subsequent experiments to obtain a better therapeutic effect. In adult mouse experiments, we treated 8-week-old male hemophilia B mice with 9 × 1010 GC of AAV8.SpCas9 and 4.5 × 1011 GC of AAV8.sgRNA.donor. Untargeted hemophilia B mice were injected with 9 × 1010 GC of AAV8.SpCas9 and 4.5 × 1011 GC of AAV8.control.donor. Plasma hFIX was measured via ELISA, starting at the week of injection (Figure 2A). Plasma hFIX levels plateaued at 40% of normal levels in targeted mice. For the untargeted and untreated groups, hFIX plasma levels were hardly detectable, suggesting that in the targeted group, hFIX expression was a result of on-target editing. The activity of hFIX, as determined by activated partial thromboplastin time (aPTT) in targeted mice, showed above-normal levels (162% ± 29% of normal) of activity owing to the hFIXco-Padua incorporated into the donor vector (Figure 2B). We sacrificed mice at 16 weeks post-injection and harvested liver samples. Liver tissue sections were analyzed by immunofluorescence for hFIX expression. No signal (<1%) was detected in untreated and untargeted mice. Targeted mice showed significantly higher numbers of hFIX-expressing cells (Figure 2C). Next, for evaluating the treatment effect at the neonatal stage, we performed temporal vein injection with AAV8.SpCas9 (3 × 1010 GC/mouse) and AAV8.sgRNA.donor (1.5 × 1011 GC/mouse) in newborn male hemophilia B mice. Plasma hFIX was measured by ELISA, beginning at 4 weeks post-injection. For the targeted group, plasma hFIX levels plateaued at 120% of the normal levels (Figure 2C), and hFIX activity increased more than 500% (590% ± 61%) of normal (Figure 2D). We performed two-third partial hepatectomy on the targeted mice at 14 weeks post-injection and harvested the liver samples. All five mice survived the procedure without complications or interventions. hFIX expression and activity levels remained stable until the end of the study (week 48) (Figures 2C and 2D). For the untargeted and untreated groups, hFIX protein and activity levels were hardly detectable (Figures 2C and 2D). We sacrificed a subset of control vector-treated mice (n = 3) at 14 weeks post-injection and harvested liver samples to serve as controls. Excisional liver tissue sections were analyzed by immunofluorescence for hFIX expression. No obvious signal was observed in the untreated and untargeted groups. Targeted animals showed obvious patches of hFIX-expressing cells in the liver. Morphometry indicated that an average of 15% of the hepatocytes from targeted mice 14 weeks post-injection expressed hFIX. Liver samples of the targeted group collected at 48 weeks post-injection showed a similar hFIX expression level (14.8% ± 2%) (Figure 2F), which indicated the persisted hFIX expression even after a two-thirds partial hepatectomy. These results demonstrated that this dual AAV system could recover the coagulation function in both adult and newborn hemophilia B mice.
Figure 2.

Efficient Restoration of hFIX Expression in Hemophilia B Mice Treated by Dual AAV8 Strategy
(A) Plasma hFIX was measured by ELISA following tail vein injections of 8-week-old male hemophilia B mice with the AAV8.SpCas9 (9 × 1010 GC/mouse) and AAV8.sgRNA.donor (4.5 × 1011 GC/mouse) (n = 5). Untargeted hemophilia B mice received AAV8.SpCas9 (9 × 1010 GC/mouse) and AAV8.control.donor (4.5 × 1011 GC/mouse) (n = 5). Untreated hemophilia B mice (n = 5) were included as control. (B) hFIX activity in adult-treated hemophilia B mouse plasma. (C) Plasma hFIX was measured by ELISA following temporal vein injections of newborn male hemophilia B mice with the AAV8.SpCas9 (3 × 1010 GC/mouse) and AAV8.sgRNA.donor (1.5 × 1011 GC/mouse) (n = 5). All targeted mice were subjected to two-thirds partial hepatectomy (PH) 14 weeks after vector treatment. Untargeted hemophilia B mice received AAV8.SpCas9 (3 × 1010 GC/mouse) and AAV8.control.donor (1.5 × 1011 GC/mouse) (n = 6). Three untargeted mice were sacrificed at week 14 for analyses. Untreated hemophilia B mice (n = 3) were included as control. (D) hFIX activity in hemophilia B mouse plasma after neonatal vector treatment. (E) Immunofluorescence staining with antibodies against hFIX (red) with 4′,6-diamidino-2-phenylindole (DAPI) nuclear counterstain (blue) on liver sections, which were treated as adults collected at 16 weeks after injection (left). Scale bars, 100 μm. Quantification of gene expression was based on the percentage of area on liver sections expressing hFIX by immunostaining (right). (F) Immunofluorescence staining with antibodies against hFIX with DAPI nuclear counterstain on liver sections, which were treated as newborns collected at 14 and 48 weeks after injection (left). Scale bars, 100 μm. Quantification of gene expression was based on the percentage of area on liver sections expressing hFIX by immunostaining (right). Error bars represent mean ± SEM. Dunnett’s test. n.s., not significant.
On-target Indel Frequency and HDR-Mediated Gene Integration Efficiency
For analyzing the editing efficiency, we extracted liver tissue DNA from adult hemophilia B mice at 16 weeks after vector treatment. Then, we analyzed the on-target indel effect via next-generation sequencing (NGS) of PCR amplicons of the targeted mAlb locus. We observed evidence of indel at the desired site in all targeted mice. The indel frequency was 21%–34%, whereas untreated or untargeted mice had background levels of 0.9% and 1% (Figure 3A). Neonates treated with the targeting vector had a mean indel frequency of 32.8% (26.1%–40.1%, n = 5) at week 14 and 33.4% (28.2%–41%, n = 5) at week 48 (Figure 3B). Untargeted mice had background levels of 1% and 1.1% at 14 and 48 weeks, respectively, similar to untreated mice (Figure 3B).
Figure 3.

Indel and HDR-Mediated Gene Integration Efficiency and Transgene Expression Analyses
Liver mRNA and DNA were isolated from hemophilia B mice at 14, 16, and 48 weeks after treatment with dual gene integration vectors or untargeted vectors. DNA and mRNA from untreated hemophilia B mice served as control. Indel analysis on the mAlb locus was analyzed by NGS on liver DNA. HDR-mediated gene integration was analyzed by LM-PCR following digestion with XbaI on liver DNA. The pooled PCR amplicons were performed by NGS. Chimeric m-hFIX mRNA levels in liver were measured by qPCR using primers spanning the junction of mAlb and hFIX cDNA. (A) Indel frequency in mice treated as adults. (B) Indel frequency in mice treated as neonates. (C) HDR frequency in mice treated as adults. (D) HDR frequency in mice treated as neonates. (E) Quantification of chimeric m-hFIX mRNA levels in the liver treated as adults and neonates. Symbols represent individual mice. Means ± SEM are shown. Dunnett’s test. n.s., not significant.
To characterize the editing results in the targeted mAlb locus and calculate the gene integration efficiency at the DNA level, we applied a method called ligation-mediated PCR (LM-PCR), and coupled it with NGS (Figure S2). We first digested the genomic DNA isolated from adult and newborn groups after vector injection using XbaI, which had recognition sites in the homology arms and in the donor vector. After digested DNA was ligated to an XbaI-compatible linker, we proceeded with LM-PCR to generate similar sizes of PCR amplicons from untreated, untargeted, and targeted mAlb loci. After the third round of amplification, PCR amplicons were followed by NGS. We analyzed the mapped reads to calculate the ratio of reads containing the expected hFIXco-Padua sequence and total mapped reads, and defined it as HDR-mediated gene targeting efficiency. The targeted mice (n = 5) treated as adults showed a targeting efficiency of 3.8% ± 0.8% (Figure 3C). The untreated (n = 3) and untargeted mice (n = 5) showed background levels of ~0.1% (Figure 3C). The targeted mice treated as neonates showed similar targeting efficiency at 14 weeks (8.2%–16.1%) and 48 weeks (8.9%–15.9%) after vector injection liver samples (Figure 3D). The untargeted and untreated mice showed background levels of ~0.1% (Figure 3D). These observations confirmed that hFIX integration occurred at the expected mAlb genome locus.
Expression of hFIX after Treatment with the Targeting Vector
We measured chimeric m-hFIX mRNA copies in liver harvested at 16 weeks post-injection for adult animals, 14 and 48 weeks post-injection for neonatal mice. Via reverse transcription, followed by quantitative PCR (qPCR) using primers spanning the junction of mAlb and hFIX, we calculated hFIX expression. We detected high levels of the chimeric mRNA in both adult and neonatal targeted groups but did not observe differences in these levels between weeks 14 and 48. Untreated and untargeted mice showed baseline levels (Figure 3E). These data confirmed the expected expression of chimeric m-hFIX mRNA.
Off-target Efficiency and Toxicity Evaluation of the Dual AAV System Treatment
For evaluating the off-target activity of sgRNA4 in vivo, we designed primers encompassing 10 of the most probable off-target loci for sgRNA4. The top 10 sites were predicted by the online tool Benchling (https://www.benchling.com; Tables S1 and S3). Amplicons of these sequences were generated by PCR on liver genomic DNA extracted at 16 weeks after vector treatment of adult mice and 48 weeks after vector treatment of newborn mice. Then, we quantified the indel by NGS in the predicted off-target regions. We observed similar indel percentages in these loci from mice treated with targeted vectors compared to untreated mice (Figures 4A and 4B), suggesting that sgRNA4 is highly specific for its intended target sequence.
Figure 4.

In Vivo Off-target Validation on Liver DNA Samples
(A) NGS data of off-target sites for liver DNA samples isolated from adult mice 16 weeks after vector treatment. Cumulative indel mutations within the protospacer region were plotted for each off-target locus in the targeted group (n = 5) compared to untreated mice (n = 3). (B) NGS data of off-target sites for liver DNA samples isolated from newborn mice 14 weeks after vector treatment. Cumulative indel mutations within the protospacer region were plotted for each off-target locus in the targeted group (n = 5) compared to untreated mice (n = 3). Means ± SEM are shown.
Meanwhile, AAV, which we used to deliver gene integration elements for hepatocyte stable hFIX expression, might contribute to immune and genome toxicity. Hence, we evaluated liver toxicity in animals treated with the dual AAV system. Histological analysis of adult livers harvested 16 weeks following the dual AAV system revealed no change in pathology (Figure 5A). Detailed analysis of transaminase levels (both alanine [ALT] and aspartate [AST] aminotransferases) did not show a toxicity effect (Figures 5B and 5C). The differences of SpCas9 mRNA and DNA levels were insignificant in targeted and untargeted groups (Figures 5D and 5E). Western blot and qPCR analysis showed similar Alb protein and mRNA levels in untreated, untargeted, and targeted mice (Figures 5F and 5G). Similar studies were conducted on treated newborn mice (Figures 5H–5N). SpCas9 mRNA/vector copy decreased to very low but still detectable levels over time (Figures 5K and 5L). In conclusion, our hFIX specific integration approach for hemophilia B treatment had therapeutic benefits for both adult and neonatal mice, and without significant off-target and toxicity effects.
Figure 5.

Liver Function Tests and Toxicity Examination in Adult and Newborn Animals Treated with Dual AAV8 Vectors
(A) Histological analysis by hematoxylin and eosin stain on adult livers harvested 16 weeks following dual vector treatment. Scale bar, 100 μm. (B and C) Liver transaminase (B, ALT; C, AST) levels in untreated hemophilia B mice (n = 3) or 16 weeks following targeted vector (n = 5) and untargeted vector (n = 5) treatment. (D) Quantification of SpCas9 mRNA levels in liver isolated from adult mice 16 weeks after vector treatment by qPCR. (E) Quantification of SpCas9 vector genome in liver from adult treatment group by qPCR. (F) Western blot analysis. Liver lysates were prepared from adult untreated or hemophilia B mice treated with the dual AAV vectors for detection of Alb protein. (G) Quantification of Alb mRNA in the liver by qPCR. (H) Histological analysis by hematoxylin and eosin stain on newborn livers harvested 14 weeks following dual vector treatment. Scale bar, 100 μm. (I and J) ALT and AST levels in untreated hemophilia B mice (n = 5) or newbron mice 14 weeks after targeted vector (n = 5) and untargeted vector (n = 5) treatment. (K) Quantification of SpCas9 mRNA levels in liver isolated from newborn mice 14 and 48 weeks after vector treatment by qPCR. (L) Quantification of SpCas9 vector genome in liver from newborn treatment group by qPCR. (M) Western blot analysis. Liver lysates were prepared from untreated or newborn hemophilia B mice treated with the dual AAV vectors for detection of Alb protein. (N) Quantification of Alb mRNA in the liver by qPCR in newborn treatment group. Means ± SEM are shown. Dunnett’s test. ∗p < 0.05, ∗∗p < 0.01. n.s., not significant.
Discussion
In our previous work, we had successfully corrected the OTC mutation in newborn livers by CRISPR-Cas9-mediated HDR.10 This strategy is appropriate for diseases induced by single nucleotide mutation; however, for scattered genetic mutations, it seems unpractical to apply one vector to all abnormal genomic phenomena; hence, we aimed to develop a more general approach for genetic deficiencies. In this study, we applied a dual AAV system-mediated gene integration strategy to stably express hFIX in adult and neonatal hemophilia B mice. In order to make SpCas9 compatible with the capacity of AAV, we applied an engineered truncated liver-specific promoter so that AAV8.SpCas9 was limited to 4.98 kb without influencing efficiency of genome editing. In addition, we also optimized the sequences of splice acceptor (SA) for efficient gene expression. According to the results of targeted mice, hFIX was efficiently integrated in mAlb intron 1 through Cas9-induced genome editing. We found that hFIX could be efficiently expressed in the plasma along with the recovery of coagulation function. In addition, treated mice could survive after two-thirds partial hepatectomy without influencing hFIX expression.
In recent decades, AAV-mediated gene therapies have shown promise in curing hemophilia B.14, 15, 16, 17 A single dose of AAV8-mediated FIX expression could restore 2%–11% of normal levels18 and sustained for more than 3 years.19 In addition, preclinical and clinical trials showed a sustained expression of hFIX of 6.7 ± 1.0 years, which supposed a part of integration.20 By delivering the hyperactive FIX variant, FIX activity could achieve in the range of 30%–40% of normal.2 However, due to the non-integrated character, in most cases the expression of FIX levels in humans would reduce over time.18,19,21 For liver-targeted gene therapy, the vector genome could be lost over time during hepatocyte turnover, which might lead to a gradual decline of therapeutic effects. Hence, the strategy of site-specific integration was developed. Targeting intron 1 of FIX through AAV-loaded genome-editing tool-mediated HDR could yield therapeutic levels of gene expression in hemophilia B mice.22,23 Another approach without nuclease aimed at integrating a 2A-peptide sequence in the last exon of Alb before the stop codon.24 Recently, we incorporated the hFIX partial cDNA with a hyperactive Padua mutation into the 5′ end of mouse FIX exon 2 to apply the endogenous FIX promoter permanently expressing the FIX protein.6 The “promoterless” method without nuclease or with the endogenous promoter in situ ensured a favorable safety profile to a certain degree. However, the efficiency of HDR is usually quite low, resulting in a limited number of modified cells.25 Utilizing the strong Alb promoter, one study focused on ZFN-mediated integration in intron 1 of the Alb gene.11 However, the mechanism of ZFN is DNA-protein binding; carrier construction is complex and time-consuming.26 In addition, the efficiency of this ZFN-mediated gene integration was no more than 2% on the mRNA level in Sharma et al.’s11 work. For a simpler and more effective treatment strategy, we applied CRISPR-Cas9-induced hFIX integration targeting mAlb. In treated newborn mice, circulating hFIX protein levels could plateau at 120% of the normal level, which was 10-fold higher than our previous work (Figure 2C). The hyperactive Padua mutation increased the hFIX protein activity. In our results by applying a lower dose, hFIX activity was above the normal level in both adult and newborn groups (Figures 2B and 2D), which was also higher than found in our previous work.6 In addition, the targeting efficiencies in adult (~4%) and newborn (~11%) treated mice were much higher than those in studies by Ohmori et al.23 and Barzel et al. (~0.5%). Because of the high transcription activity of Alb, this “safe harbor” versatile platform could express various proteins and substituted the donor for each respective therapeutic transgene. Thus, our strategy could be applied for nearly all hemophilia B patients, including those with mutations in the FIX promoter or in the 5′ end of the FIX coding sequence.27 Additionally, our therapeutic strategy could be widely used in other genetic diseases through specific integration sites of the remedial gene. While intron 1 of the Alb locus is an attractive locus for gene integration, this locus might not be applied to non-secreted therapeutic proteins. We noticed that our method may have the risk to change Alb gene expression after successful targeted gene integration. Thus, we detected the expression of wild-type Alb protein and mRNA levels. Perhaps because of the Alb homeostasis,28 we found that the total expression level of Alb did not change significantly after genome editing (Figures 5F, 5G, 5M, and 5N).
For genome editing-mediated gene therapy, the off-target effect must be concerned for avoiding unwanted mutation-induced limitation of clinical application. In this study, we estimated the in vivo treatment specificity of sgRNA4 via NGS validation of the top 10 potential off-target sites identified by a computer algorithm. Besides efficient on-target editing, we did not detect any off-target effect, confirming the high specificity of sgRNA4. As the CRISPR system shows substantial promise for human gene therapy, Akcakaya et al.29 described a highly sensitive strategy that could robustly identify genome-wide CRISPR-Cas nuclease off-target effects in vivo. The detection of a genome-wide off-target effect is necessary for clinical application. In our preceding research, we increased donor AAV vector dose up to 1.5 × 1013 GC/mouse with the result of elevated hFIX level no more than ~20% of normal.6 Such a high dose of the vector might increase the risk of immune responses in a clinical application. Note that host immune responses induced by editing nucleases and viral vectors might attenuate the therapeutic effect and lead to unexpected adverse results.30 In an in vitro model of canine hemophilia B, an all-in-one single adenoviral system containing all the functional editing elements was confirmed to achieve higher efficiency than a two-vector system.31 However, the adenovirus was associated with strong immunity and fatal systemic inflammation.32 Although AAV has low immunogenicity, researchers had found that high-dose intravenous AAV gene therapy administration would lead to severe toxicity affecting liver in nonhuman primates (NHPs) and piglets.33 Our targeted gene integration strategy could achieve stable and complete normalization of hemostasis in both adult and newborn hemophilia B mice while the vector doses used in this study were lower than those used in Sharma et al.’s11 gene integration therapy study and in our previous work.6 Our efficient therapy strategy with lower vector dose might be more applicable in the clinical state. In addition, without genome-editing nucleases, AAV-mediated NHP fetal gene therapy also showed long-term treatment effects.34, 35, 36 The expression of FIX was sustained in the absence of toxicity.35,36 These NHP studies by fetal organ-targeted gene therapy provided another strategy for hemophilia B long-term therapeutic effects without immune response. Although AAV has the character of non-integration, genome toxicity is still a concern. AAV could be integrated into the host genome, albeit at a very low frequency compared to that of lentivirus.37,38 Some murine studies have documented that AAV-mediated gene therapy could result in insertional mutagenesis.39,40 Hanlon et al.41 found high levels of AAV vector integration into CRISPR-induced DSBs. Deep sequencing could be a powerful method for validated measurement of AAV integration frequencies.42,43 We found different parts of the AAV vector sequences inserted into DSBs in our previous work.6 Due to the limitation of amplicon sequencing method in this study, we could not determine the extent of vector sequence integration around target region. There is the possibility that some hFIX expression is derived from the donor vector inserted by non-homologous end joining (NHEJ) rather than HDR. Further studies are necessary to detect the NHEJ-mediated integration of the gene-targeting vector. Moreover, site-specific gene integration might influence the protein expression of the targeted locus. In this context, animals treated with dual AAV did not show abnormal liver function, and Alb expression interference was not observed (Figure 5), implying the safety of our dual AAV system for clinical use. We advocate that further studies are necessary to ensure the safety of our therapeutic strategy in the clinical transformation.
In summary, based on our previous work, we have extended the use of the dual AAV system. Through the liver-targeted genome-editing strategy of gene integration at the mAlb locus, we achieved the recovery of hFIX expression levels and improvement in the coagulation function in newborn and adult hemophilia B mice models through lower vector dose. We should recognize that the efficacy of genome editing is still lower than the efficacy of vector transduction in gene addition therapy. Nevertheless, our strategy could permanently recover FIX expression, and this strategy is potentially useful for mutation position-specific and other genetic diseases. The robust and sustained gene expression was clinically beneficial and could be developed into a universal genome-editing tool for life-saving therapy.
Materials and Methods
Plasmid Construction and AAV Vector Production
Four 20-nt target sequences preceding a 5′-NGG protospacer-adjacent motif (PAM) sequence located in the mAlb intron 1 were selected. These sequences were cloned into pX330.hSpCas9 plasmid (Addgene plasmid #42230). The sequences of sgRNAs and primers are described in Tables S2 and S3. The AAV mAlb gene-targeting vector contains (1) the U6-mAlb sgRNA4 that specifically targets a region in intron 1 of mAlb, (2) an optimized SA sequence derived from the hFIX gene followed by hFIXco-Padua,12 and (3) the bGH poly(A) flanked by 0.5 kb on left side and 0.7 kb on right side. The untargeted vector lacks the protospacer sequence from the U6-mAlb sgRNA4 element. The SpCas9 expression AAV vector system consists of TBG-S1,13 an engineered truncated liver-specific promoter, and SpCas9 cDNA. Final AAV production plasmids were generated using an EndoFree Plasmid Megaprep kit (QIAGEN, Hilden, Germany).
AAV vectors used in Figure 1C were generated by triple plasmid transfection of human embryonic kidney 293 cells (ATCC, Manassas, VA, USA). All AAV8 vectors were produced as previously described.44 The genome titer (GC/mL) of AAV vectors was carried out by qPCR (Roche, catalog no. 06402682001). qPCR was performed to measure AAV8.SpCas9 genome titer using forward primer 5′-GGACCAGGAACTGGACATCA-3′, reverse primer 5′-TCGATGGAGTCGTCCTTCAG-3′, and probe 5′-Fam-CCACATCGTAGTCGGACAGCCGGT-Tamra-3′. The genome titer of AAV8.sgRNA.donor and AAV8.control.donor was measured using forward primer 5′-GCCAGCCATCTGTTGT-3′, reverse primer 5′-GGAGTGGCACCTTCCA-3′, and probe 5′-Fam-TCCCCCGTGCCTTCCTTGACC-Tamra-3′.
Cell Culture and Transfection
H2.35 cells (ATCC) were maintained in DMEM medium supplemented with 10% fetal bovine serum (FBS) cultured at 37°C with 5% CO2. For target validation, plasmid containing SpCas9 and sgRNA was cotransfected with plasmid expressing GFP and puromycin resistance into H2.35 cells using the TranslT-X2 system (Mirus, Marietta, GA, USA) per the manufacturer’s recommendations. Transfected cells were under puromycin (1 μg/mL) selection for 24 h to enrich transfected cells.
Animal Experiments
Hemophilia B mice (B6.129P2-F9tm1Dws/J) were obtained from The Jackson Laboratory (Sacramento, CA, USA). Mating cages were monitored daily for births. Newborn postnatal day 2 (P2) male mice received a temporal vein injection of a mixture of two AAV vectors at a volume of 50 μL. Plasma samples for hFIX assays were obtained by retro-orbital bleeding 4 weeks after vector treatment, and every 2–4 weeks afterward. A two-thirds partial hepatectomy was performed on a subset of targeting vector-treated hemophilia B mice 14 weeks after AAV injection. A subset of mice treated with control vector was sacrificed at 14 weeks after AAV injection and liver tissues were harvested as controls. The rest of mice were sacrificed 48 weeks post-injection. In the adult mouse study, 8- to 10-week-old hemophilia B male mice received a mixture of two AAV vectors via the tail vein. Mice were bled at various time points starting 1 week post-injection. Mice were sacrificed 16 weeks post-injection. All animal protocols were approved by the Institutional Animal Care and Concern Committee at Sichuan University, and animal care was in accordance with the committee’s guidelines.
On- and Off-Target Mutagenesis Analysis
Genomic mutations at the Alb locus in vitro were detected using the Surveyor nuclease assay (Integrated DNA Technologies, Coralville, IA, USA). Genomic DNA from the H2.35 mouse cell line by transient transfection was isolated by QuickExtract solution (Epicentre Biotechnologies, Madison, WI, USA) for in vitro validation of each sgRNA. Loci were amplified through nested PCR amplifications using Q5 high-fidelity DNA polymerase (NEB, Ipswich, MA, USA). Equal amounts of test and reference PCR products were denatured and re-annealed using a thermal cycler, and then treated with Surveyor nuclease. DNA fragments were analyzed by 4%–20% gradient polyacrylamide TBE gel (Thermo Fisher Scientific, Invitrogen, catalog no. EC62255BOX) electrophoresis.
On- and off-target mutagenesis analysis in vivo was detected by NGS. In vivo genome-editing efficiency was measured on mice liver tissues 14, 16, and 48 weeks following vector administration. Genomic DNA was extracted by a genome extraction kit (TIANGEN Biotech (Beijing), catalog no. DP304-02). After nest PCR amplification with primers flanking the on- and off-target loci, PCR products were used for an NGS assay. Indel analyses were performed on NGS of the PCR amplicons. The most likely off-target sites were determined using the algorithm described in the Benchling website (https://www.benchling.com), referred to as OT1 through OT10. Purified PCR fragments were subjected to NGS. Libraries were made from the second PCR products and sequenced on Illumina MiSeq (2 × 300-bp paired end, Personal Biotechnology, Shanghai, China). Data were processed according to standard Illumina sequencing analysis procedures. Processed reads were mapped to the expected PCR amplicons as reference sequences using custom scripts. Reads that did not map to the reference were discarded. Indels were determined by comparison of reads against reference using custom scripts. All candidate indels for OT1 through OT10 sites were manually curated for confirmation. The oligonucleotide primer sequences used to detect mutations are presented in Tables S2 and S3.
hFIX Antigen and Activity
hFIX protein levels in the plasma were quantified by an ELISA kit (Affinity Biologicals, Ancaster, ON, Canada) and are shown as a percentage of normal level according to the manufacturer’s protocol. FIX activity was measured by one-step aPTT using a SCA2000 veterinary coagulation analyzer (Diagnostica Stago, Parsippany, NJ, USA) according to the instructions of the manufacturer. A standard curve was generated using factor assay control plasma (FACT, George King Bio-Medical) in FIX-deficient plasma (Instrumentation Laboratory, catalog no. 0020011900). FACT was calibrated using a reference standard (i.e., the fourth World Health Organization [WHO] International Standard NIBSC code 07/182). Samples were compared to the standard curve to obtain the relative activity of hFIX. Samples were diluted 2- to 50-fold in FIX-deficient plasma to obtain levels within the interpretable range.
Western Blot
Western blot analyses were performed on liver lysates. Alb protein was detected by rabbit polyclonal antibody (1:10,000 dilution, Proteintech, catalog no. 16475-1-AP). Rabbit anti-GAPDH antibody (1:100,000 dilution, ABclonal, catalog no. AC001) was used to detect GAPDH. Blots were imaged and analyzed by an iBright CL1000 imaging systems (Thermo Fisher Scientific, Invitrogen).
Gene Expression Analysis and qPCR
RNA was isolated from liver using TRIzol (Thermo Fisher Scientific, Invitrogen, catalog no. 15596018). High-capacity cDNA produced from a PrimeScript RT reagent kit (Takara, catalog no. RR047A) served as a template for qPCR assays (Roche, catalog no. 06402712001). We measured SpCas9, GAPDH, wild-type mAlb, and chimeric m-hFIX using gene-specific forward and reverse primers. Gene expression levels were normalized to GAPDH levels. The percentage of chimeric m-hFIX mRNA on-target was calculated as the ratio between the abundance of template for chimeric m-hFIX to the abundance of template for wild-type mAlb. For untreated controls, no qPCR signal was detected with primers for chimeric m-hFIX. DNA purified from untargeted and targeted mice livers served as templates for SpCas9 copy number assay. We quantified the abundance of mouse genomes using SpCas9 primers and was calculated using its standard curve, which was performed by the packing plasmid of AAV8.SpCas9. The sequences of primers are presented in Table S2.
Gene Targeting Efficiency Analysis
HDR-mediated integration of the hFIX into the mAlb locus was quantified by LM-PCR coupled with NGS (Figure S2). 1 μg of genomic DNA was digested with 20 U of XbaI for 4 h at 37°C to produce fragments of similar sizes and the same sticky ends from both targeted and wild-type alleles. Digested DNA was purified with Agencourt AMPure XP beads (Beckman Coulter, catalog no. A63880) at a 2× ratio and eluted in 20 μL of elution buffer (QIAGEN, Hilden, Germany). Purified DNA was quantified using the Quant-It PicoGreen double-stranded DNA (dsDNA) assay (Thermo Fisher Scientific, Invitrogen). In all, a total of 180 ng of purified DNA was ligated to an XbaI-compatible linker. Ligated DNA was purified with AMPure XP beads at a 0.9× ratio and eluted in 15 μL of elution buffer. Linker ligation was followed by three-step nested PCR. DNA was amplified by touchdown PCR using Q5 high-fidelity DNA polymerase. The PCR program consisted of one cycle of 95°C for 5 min; 15 cycles of 95°C for 30 s, 70°C for 2 min (−1°C per cycle), 72°C for 1.5 min; and 15 cycles of 95°C for 30 s, 55°C for 1 min, and 72°C for 1.5 min. PCR product was purified with AMPure XP beads at a 0.9× ratio and eluted in 15 μL of elution buffer. The third PCR product was purified and analyzed by NGS. We analyzed the mapped reads to calculate the ratio of reads containing the expected hFIXco sequence and total mapped reads, and defined it as HDR-mediated gene-targeting efficiency. The sequences of primers and oligonucleotides are presented in Table S2.
Liver Immunofluorescence, Histopathology
Liver lobes were collected from mice, rinsed in ice-cold phosphate-buffered saline (PBS), blotted dry, mounted in OCT, and frozen in a liquid nitrogen-cooled isopentane bath. Samples were placed at −80°C until sectioning. In all samples, a minimum of two liver sections from varying depths were cut at 5 μm and mounted onto positively charged glass slides. Frozen sections were thawed to room temperature and fixed in 4% paraformaldehyde (all solutions in PBS) for 10 min. Slides were then washed three times in PBS for 5 min. Sections were then blocked with 1% donkey serum containing 0.2% Triton X-100 for 30 min at room temperature in a humidified chamber. Serum was removed and slides were incubated with 100 μL of goat anti-human FIX immunoglobulin G (IgG) primary antibody (Affinity Biologicals, catalog no. GAFIX-AP) at 1:100 in PBS containing 1% donkey serum for 1.5 h and then washed three times in PBS for 5 min. Slides were then incubated with 100 μL of donkey anti-goat IgG secondary antibody (Abcam, catalog no. ab150132) at 1:100 in PBS containing 1% donkey serum for 40 min and then washed three times in PBS for 5 min. Slides were mounted with 100 μL of DAPI nuclear counterstain and covered with a coverslip. Images were overlaid by Adobe Photoshop CS6.
To quantify percentages of hFIX-expressing hepatocytes, five random images were taken with a 10× objective from each liver section stained for hFIX expression. Using ImageJ software (W.S. Rasband, National Institutes of Health, USA; https://imagej.nih.gov/ij/), images were thresholded for hFIX-positive area (i.e., the hFIX-positive area was selected), and the percentage of the hFIX-positive area was determined for each image. In a second measurement the images were for “empty” area (e.g., veins and sinusoids) to determine the percentage of the area not occupied by liver tissue. This was possible as a result of the presence of weak background fluorescence of the liver tissue. The final percentage of hFIX-positive liver tissue (i.e., hFIX+ hepatocytes) was then calculated per adjusted area (total area minus empty area), and the values were averaged for each liver.
For histologic examination, liver lobes were collected from mice, rinsed in ice-cold PBS, blotted dry, and fixed in 4% paraformaldehyde. These fixed liver samples were embedded by paraffin and performed on sections. Hematoxylin and eosin (H&E) staining was performed on these processed sections according to standard protocols. Sections were analyzed for any abnormalities compared to livers from untreated animals.
Assessment of Plasma Liver Enzymes
Serum ALT and AST measurements were performed on mouse serum obtained via retro-orbital bleeding and analyzed by WestChina-Frontier PharmaTech (Chengdu, China).
Statistical Analysis
Graphpad Prism was used to perform all statistical tests. Values express mean ± SEM. Statistical analysis was by a Dunnett’s test, as indicated in the figure legends. In all tests, p <0.05 was considered significant.
Author Contributions
Y.Y. conceived this study and designed the experiments. L.M. constructed the donor vectors. Q.W. and X.Z. performed mouse studies. Q.W. performed indel and targeting efficiency analyses. Q.L. performed the hF9 ELISA and aPTT assay. Y.L. performed qPCR analysis. J.S. and X.Z. performed immunofluorescence assays. Q.W. wrote the manuscript. Y.Y. and H.D. edited the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the Joint Funds of the National Natural Science Foundation of China (Grant no. U19A2002), the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (no. 2018ZX09733001-005-002), and by the Science and Technology Major Project of Sichuan Province (no. 2017SZDZX0011).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.06.025.
Supplemental Information
References
- 1.Mannucci P.M., Tuddenham E.G. The hemophilias—from royal genes to gene therapy. N. Engl. J. Med. 2001;344:1773–1779. doi: 10.1056/NEJM200106073442307. [DOI] [PubMed] [Google Scholar]
- 2.George L.A., Sullivan S.K., Giermasz A., Rasko J.E.J., Samelson-Jones B.J., Ducore J., Cuker A., Sullivan L.M., Majumdar S., Teitel J. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med. 2017;377:2215–2227. doi: 10.1056/NEJMoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Young G. New challenges in hemophilia: long-term outcomes and complications. Hematology (Am. Soc. Hematol. Educ. Program) 2012;2012:362–368. doi: 10.1182/asheducation-2012.1.362. [DOI] [PubMed] [Google Scholar]
- 4.DiMichele D. Inhibitor development in haemophilia B: an orphan disease in need of attention. Br. J. Haematol. 2007;138:305–315. doi: 10.1111/j.1365-2141.2007.06657.x. [DOI] [PubMed] [Google Scholar]
- 5.Wang L., Wang H., Bell P., McMenamin D., Wilson J.M. Hepatic gene transfer in neonatal mice by adeno-associated virus serotype 8 vector. Hum. Gene Ther. 2012;23:533–539. doi: 10.1089/hum.2011.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L., Yang Y., Breton C.A., White J., Zhang J., Che Y., Saveliev A., McMenamin D., He Z., Latshaw C. CRISPR/Cas9-mediated in vivo gene targeting corrects hemostasis in newborn and adult factor IX-knockout mice. Blood. 2019;133:2745–2752. doi: 10.1182/blood.2019000790. [DOI] [PubMed] [Google Scholar]
- 7.Gomez-Ospina N., Scharenberg S.G., Mostrel N., Bak R.O., Mantri S., Quadros R.M., Gurumurthy C.B., Lee C., Bao G., Suarez C.J. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019;10:4045. doi: 10.1038/s41467-019-11962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amoasii L., Hildyard J.C.W., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Harron R., Stathopoulou T.R., Massey C., Shelton J.M. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin H., Song C.Q., Dorkin J.R., Zhu L.J., Li Y., Wu Q., Park A., Yang J., Suresh S., Bizhanova A. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 2016;34:328–333. doi: 10.1038/nbt.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y., Wang L., Bell P., McMenamin D., He Z., White J., Yu H., Xu C., Morizono H., Musunuru K. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol. 2016;34:334–338. doi: 10.1038/nbt.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma R., Anguela X.M., Doyon Y., Wechsler T., DeKelver R.C., Sproul S., Paschon D.E., Miller J.C., Davidson R.J., Shivak D. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood. 2015;126:1777–1784. doi: 10.1182/blood-2014-12-615492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cantore A., Nair N., Della Valle P., Di Matteo M., Màtrai J., Sanvito F., Brombin C., Di Serio C., D’Angelo A., Chuah M. Hyperfunctional coagulation factor IX improves the efficacy of gene therapy in hemophilic mice. Blood. 2012;120:4517–4520. doi: 10.1182/blood-2012-05-432591. [DOI] [PubMed] [Google Scholar]
- 13.Greig J.A., Wang Q., Reicherter A.L., Chen S.J., Hanlon A.L., Tipper C.H., Clark K.R., Wadsworth S., Wang L., Wilson J.M. Characterization of adeno-associated viral vector-mediated human factor VIII gene therapy in hemophilia A mice. Hum. Gene Ther. 2017;28:392–402. doi: 10.1089/hum.2016.128. [DOI] [PubMed] [Google Scholar]
- 14.Anguela X.M., Sharma R., Doyon Y., Miller J.C., Li H., Haurigot V., Rohde M.E., Wong S.Y., Davidson R.J., Zhou S. Robust ZFN-mediated genome editing in adult hemophilic mice. Blood. 2013;122:3283–3287. doi: 10.1182/blood-2013-04-497354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arruda V.R., Stedman H.H., Haurigot V., Buchlis G., Baila S., Favaro P., Chen Y., Franck H.G., Zhou S., Wright J.F. Peripheral transvenular delivery of adeno-associated viral vectors to skeletal muscle as a novel therapy for hemophilia B. Blood. 2010;115:4678–4688. doi: 10.1182/blood-2009-12-261156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miesbach W., Meijer K., Coppens M., Kampmann P., Klamroth R., Schutgens R., Tangelder M., Castaman G., Schwäble J., Bonig H. Gene therapy with adeno-associated virus vector 5-human factor IX in adults with hemophilia B. Blood. 2018;131:1022–1031. doi: 10.1182/blood-2017-09-804419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spronck E.A., Liu Y.P., Lubelski J., Ehlert E., Gielen S., Montenegro-Miranda P., de Haan M., Nijmeijer B., Ferreira V., Petry H., van Deventer S.J. Enhanced factor IX activity following administration of AAV5-R338L “Padua” factor IX versus AAV5 WT human factor IX in NHPs. Mol. Ther. Methods Clin. Dev. 2019;15:221–231. doi: 10.1016/j.omtm.2019.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nathwani A.C., Tuddenham E.G., Rangarajan S., Rosales C., McIntosh J., Linch D.C., Chowdary P., Riddell A., Pie A.J., Harrington C. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nathwani A.C., Reiss U.M., Tuddenham E.G., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nathwani A.C., Reiss U., Tuddenham E., Chowdary P., McIntosh J., Riddell A., Pie, J, Mahlangu J.N., Recht M., Shen Y.-M. Adeno-associated mediated gene transfer for hemophilia B: 8 year follow up and impact of removing” empty viral particles” on safety and efficacy of gene transfer. Blood. 2018;132(Suppl 1):491. [Google Scholar]
- 21.Pipe S., Stine K., Rajasekhar A., Everington T., Poma A., Crombez E., Hay C.R.M. 101HEMB01 is a phase 1/2 open-label, single ascending dose-finding trial of DTX101 (AAVrh10FIX) in patients with moderate/severe hemophilia B that demonstrated meaningful but transient expression of human factor IX (hFIX) Blood. 2017;130(Suppl 1):3331. [Google Scholar]
- 22.Li H., Haurigot V., Doyon Y., Li T., Wong S.Y., Bhagwat A.S., Malani N., Anguela X.M., Sharma R., Ivanciu L. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–221. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohmori T., Nagao Y., Mizukami H., Sakata A., Muramatsu S.I., Ozawa K., Tominaga S.I., Hanazono Y., Nishimura S., Nureki O., Sakata Y. CRISPR/Cas9-mediated genome editing via postnatal administration of AAV vector cures haemophilia B mice. Sci. Rep. 2017;7:4159. doi: 10.1038/s41598-017-04625-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barzel A., Paulk N.K., Shi Y., Huang Y., Chu K., Zhang F., Valdmanis P.N., Spector L.P., Porteus M.H., Gaensler K.M., Kay M.A. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature. 2015;517:360–364. doi: 10.1038/nature13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cox D.B., Platt R.J., Zhang F. Therapeutic genome editing: prospects and challenges. Nat. Med. 2015;21:121–131. doi: 10.1038/nm.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li T., Miller C.H., Payne A.B., Craig Hooper W. The CDC Hemophilia B mutation project mutation list: a new online resource. Mol. Genet. Genomic Med. 2013;1:238–245. doi: 10.1002/mgg3.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bern M., Sand K.M., Nilsen J., Sandlie I., Andersen J.T. The role of albumin receptors in regulation of albumin homeostasis: implications for drug delivery. J. Control. Release. 2015;211:144–162. doi: 10.1016/j.jconrel.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 29.Akcakaya P., Bobbin M.L., Guo J.A., Malagon-Lopez J., Clement K., Garcia S.P., Fellows M.D., Porritt M.J., Firth M.A., Carreras A. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature. 2018;561:416–419. doi: 10.1038/s41586-018-0500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.High K.A., Roncarolo M.G. Gene therapy. N. Engl. J. Med. 2019;381:455–464. doi: 10.1056/NEJMra1706910. [DOI] [PubMed] [Google Scholar]
- 31.Gao J., Bergmann T., Zhang W., Schiwon M., Ehrke-Schulz E., Ehrhardt A. Viral vector-based delivery of CRISPR/Cas9 and donor DNA for homology-directed repair in an in vitro model for canine hemophilia B. Mol. Ther. Nucleic Acids. 2019;14:364–376. doi: 10.1016/j.omtn.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raper S.E., Chirmule N., Lee F.S., Wivel N.A., Bagg A., Gao G.P., Wilson J.M., Batshaw M.L. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003;80:148–158. doi: 10.1016/j.ymgme.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 33.Hinderer C., Katz N., Buza E.L., Dyer C., Goode T., Bell P., Richman L.K., Wilson J.M. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum. Gene Ther. 2018;29:285–298. doi: 10.1089/hum.2018.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rahim A.A., Wong A.M., Hoefer K., Buckley S.M., Mattar C.N., Cheng S.H., Chan J.K., Cooper J.D., Waddington S.N. Intravenous administration of AAV2/9 to the fetal and neonatal mouse leads to differential targeting of CNS cell types and extensive transduction of the nervous system. FASEB J. 2011;25:3505–3518. doi: 10.1096/fj.11-182311. [DOI] [PubMed] [Google Scholar]
- 35.Mattar C.N.Z., Gil-Farina I., Rosales C., Johana N., Tan Y.Y.W., McIntosh J., Kaeppel C., Waddington S.N., Biswas, A, Choolani, M In utero transfer of adeno-associated viral vectors produces long-term factor IX levels in a cynomolgus macaque model. Mol. Ther. 2017;25:1843–1853. doi: 10.1016/j.ymthe.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan J.K.Y., Gil-Farina I., Johana N., Rosales C., Tan Y.W., Ceiler J., Mcintosh J., Ogden B., Waddington S.N., Schmidt M. Therapeutic expression of human clotting factors IX and X following adeno-associated viral vector-mediated intrauterine gene transfer in early-gestation fetal macaques. FASEB J. 2019;33:3954–3967. doi: 10.1096/fj.201801391R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakai H., Yant S.R., Storm T.A., Fuess S., Meuse L., Kay M.A. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J. Virol. 2001;75:6969–6976. doi: 10.1128/JVI.75.15.6969-6976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller D.G., Petek L.M., Russell D.W. Adeno-associated virus vectors integrate at chromosome breakage sites. Nat. Genet. 2004;36:767–773. doi: 10.1038/ng1380. [DOI] [PubMed] [Google Scholar]
- 39.Deyle D.R., Russell D.W. Adeno-associated virus vector integration. Curr. Opin. Mol. Ther. 2009;11:442–447. [PMC free article] [PubMed] [Google Scholar]
- 40.Chandler R.J., Sands M.S., Venditti C.P. Recombinant adeno-associated viral integration and genotoxicity: insights from animal models. Hum. Gene Ther. 2017;28:314–322. doi: 10.1089/hum.2017.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanlon K.S., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Volak A., Spirig S.E., Muller A., Sousa A.A., Tsai S.Q., Bengtsson N.E. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019;10:4439. doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nowrouzi A., Penaud-Budloo M., Kaeppel C., Appelt U., Le Guiner C., Moullier P., von Kalle C., Snyder R.O., Schmidt M. Integration frequency and intermolecular recombination of rAAV vectors in non-human primate skeletal muscle and liver. Mol. Ther. 2012;20:1177–1186. doi: 10.1038/mt.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Breton C., Clark P.M., Wang L., Greig J.A., Wilson J.M. ITR-Seq, a next-generation sequencing assay, identifies genome-wide DNA editing sites in vivo following adeno-associated viral vector-mediated genome editing. BMC Genomics. 2020;21:239. doi: 10.1186/s12864-020-6655-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lock M., Alvira M., Vandenberghe L.H., Samanta A., Toelen J., Debyser Z., Wilson J.M. Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum. Gene Ther. 2010;21:1259–1271. doi: 10.1089/hum.2010.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.