

Abstract
Chimeric antigen receptor (CAR) T cells targeting CD123, an acute myeloid leukemia (AML) antigen, hold the promise of improving outcomes for patients with refractory/recurrent disease. We generated five lentiviral vectors encoding CD20, which may serve as a target for CAR T cell depletion, and 2nd or 3rd generation CD123-CARs since the benefit of two costimulatory domains is model dependent. Four CARs were based on the CD123-specific single-chain variable fragment (scFv) 26292 (292) and one CAR on the CD123-specific scFv 26716 (716), respectively. We designed CARs with different hinge/transmembrane (H/TM) domains and costimulatory domains, in combination with the zeta (z) signaling domain: 292.CD8aH/TM.41BBz (8.41BBz), 292.CD8aH/TM.CD28z (8.28z), 716.CD8aH/TM.CD28z (716.8.28z), 292.CD28H/TM. CD28z (28.28z), and 292.CD28H/TM.CD28.41BBz (28.28.41BBz). Transduction efficiency, expansion, phenotype, and target cell recognition of the generated CD123-CAR T cells did not significantly differ. CAR constructs were eliminated for the following reasons: (1) 8.41BBz CARs induced significant baseline signaling, (2) 716.8.28z CAR T cells had decreased anti-AML activity, and (3) CD28.41BBz CAR T cells had no improved effector function in comparison to CD28z CAR T cells. We selected the 28.28z CAR since CAR expression on the cell surface of transduced T cells was higher in comparison to 8.28z CARs. The clinical study (NCT04318678) evaluating 28.28z CAR T cells is now open for patient accrual.
Keywords: immunotherapy, CAR, T cell therapy, AML, CD123, leukemia, chimeric antigen receptor
Graphical Abstract
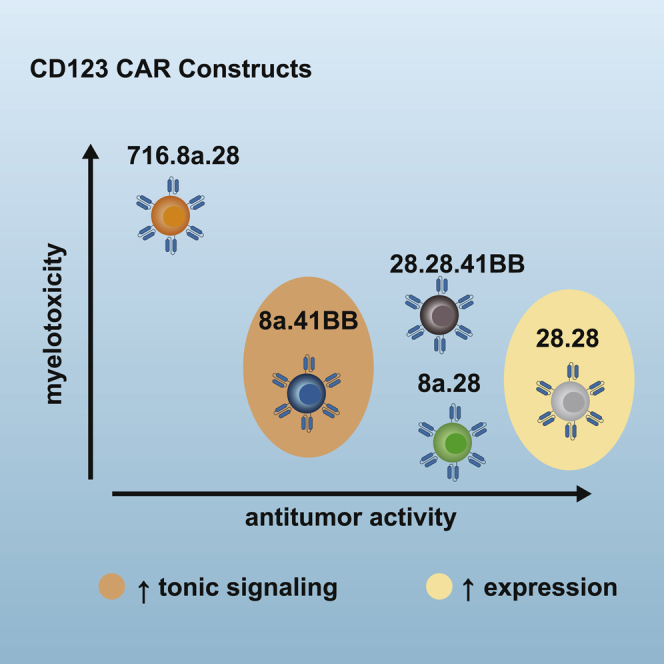
Acute myeloid leukemia (AML) remains a challenging disease to treat. Chimeric antigen receptor (CAR) T cells specific for CD123, an antigen expressed on AML blasts and to a lesser degree on normal hematopoietic progenitor cells (HPCs), have the potential to improve outcomes for patients with relapsed or refractory disease. Here we describe the generation of a panel of 5 lentiviral constructs encoding a CD20 safety switch and a 2nd or 3rd generation CD123-CAR. We perform a comprehensive analysis of T cells expressing this panel of CD123-CAR constructs including transgene expression, expansion kinetics, immunophenotype, and antitumor activity in vitro and in vivo. In addition, we demonstrate the functionality of the CD20 safety switch, which would allow for rapid elimination of T cells in the event of toxicity. This careful analysis allowed us to elucidate subtle differences between constructs that could be linked to differences in the antigen recognition, hinge/transmembrane (H/TM), and/or signaling domain of the designed CARs. As a result, we were able to select a CD123-CAR construct for our upcoming phase I trial (NCT04318678).
Introduction
Acute myeloid leukemia (AML) remains a clinically challenging disease because of its high morbidity, mortality, and relapse rates.1 Changes in supportive care have contributed to decreased treatment-related mortality in recent years. However, increased toxicities have dampened the benefit of intensive chemotherapy regimens on overall survival, and novel therapies are needed.
Chimeric antigen receptor (CAR) T cells combine the specificity of the single-chain variable fragment (scFv) of an antibody with the hinge/transmembrane (H/TM), costimulatory, and activation domains of T cells to specifically bind a tumor antigen, leading to T cell activation and tumor cell lysis. The CAR T cells specific for CD19 have proven successful in patients with CD19+ B cell acute lymphoblastic leukemia and lymphoma.2, 3, 4, 5, 6, 7, 8 However, identifying a targetable antigen for AML remains challenging because of a marked overlap between the antigens expressed on leukemic blasts and healthy tissues.9,10 The currently pursued CAR target antigens for AML include CD33, CLL-1, and CD123.11, 12, 13 Although these are expressed on a high percentage of AML blasts, they are also present at varying levels on mature neutrophils (CLL-1) and hematopoietic progenitor cells (HPCs; CD33 and CD123), raising on target/off cancer toxicity concerns.11, 12, 13
Several investigators have generated CD123-CARs with different endodomains, with a specific focus on CARs with a CD28 or 4-1BB endodomain for early phase clinical testing.12,14,15 Both CD28z- and 41BBz-CAR T cells targeting CD19 demonstrated potent antileukemia and lymphoma activity in humans, leading to their FDA approval.16,17 However, CD28z-CAR T cells persist for a shorter period of time in humans than do 41BBz-CAR T cells. Therefore, CD28z-CAR T cells are preferred for targeting CD123+ AML to limit HPC toxicity. CD28-CAR T cells, however, can recognize lower levels of antigens expressed on the cell surface of target cells than can 41BBz-CAR T cells, suggesting that CARs with a 41BBz endodomain are preferred.18 In addition, the nonsignaling components of CARs, including the H/TM domain, influence CAR expression and CAR T cell function.19, 20, 21
Although clinical studies are needed to assess the role of CAR design on the efficacy and safety of CD123-CAR T cell therapy, careful comparison of CAR T cells in preclinical studies is also needed to select a CAR construct for clinical testing. Here, we generated and compared five lentiviral vectors (LVs) encoding CD20, which may serve as a target for T cell product depletion, and CARs that differed in H/TM and signaling domains, with the goal of selecting a construct for future clinical testing. Although we only observed minor differences between constructs, we selected a CAR with a CD28 H/TM and CD28z signaling domain. This CAR will be evaluated in an FDA-approved bridge-to-transplant phase 1 clinical study designed to test CD123-CAR T cell safety and efficacy.
Results
Generation of CD123-CARCD20 T Cells
We designed five bicistronic LVs encoding CD20, a 2A peptide sequence, and different CD123-CARs (Figure 1A). For four CARs, we used the CD123-specific scFv 26292 (292), and for one CAR the CD123-specific scFv 26716 (716), respectively. We generated CARs with different H/TM domains and costimulatory domains, in combination with the zeta (z) signaling domain. We used either CD28 or 41BB as costimulatory domains. In addition, we explored a 3rd generation CAR design since at present the benefit of adding 41BB to CD28 costimulation is model dependent.22,23 Based on this, we constructed the following CARs: (1) CD20-2A-292.CD8aH/TM.41BBz (8.41BBz), (2) CD20-2A-292.CD8a H/TM.CD28z (8.28z), (3) CD20-2A-716.CD8aH/TM.CD28z (716.8.28z), (4) CD20-2A-292.CD28H/TM.CD28z (28.28z), and (5) 292.CD28H/TM.CD28.41BBz (28.28.41BBz). The amino acid sequence of the five CAR constructs is provided in the Supplemental Information (Figure S1). We generated T cells expressing CD123-CARs and CD20 (CD123-CARCD20) after lentiviral transduction, which mirrors our clinical-grade CAR T cell production process (Figure 1B). Transduction efficiency was determined by vector copy number (VCN) per cell and flow cytometry analysis. The mean VCN ranged from 1.31 to 2.25 (±0.40). CD123-CAR expression ranged from 71% to 95% (±3.2%), and CD20 expression ranged from 83.2% to 97.7% (±1.4%; Figures 1C–1E). CD123-CAR and CD20 expression followed a linear relationship for all designed CARs (Figure 1F), and the mean fluorescence intensity (MFI) levels of CD123-CAR and CD20 expression were not different (Figure S2). When we compared all five CAR constructs, we found no significant differences in VCN and transgene expression (p > 0.05). Comparison of CD8a and CD28 H/TM domain-containing CARs with the same signaling domain revealed a mean 2.3-fold (range, 1.9–2.6) higher MFI of CD28 H/TM CARs than that of CD8a H/TM CARs on the cell surface of T cells (Figure S3). In contrast, the mean fold changes for all other transduction parameters (i.e., percent CAR+, VCN, percent CD20+, and CD20 MFI) between T cells transduced with the CD20-2A-CD8a or CD28 H/TM CAR LVs were between 0.8 and 1.2 (Figure S3).
Figure 1.

Generation of CD123-Specific CAR T Cells
(A) Scheme of lentiviral vectors (LVs). Data throughout the figures are represented by the color code of the circles to the left of each construct. (B) Schematic for lentiviral transduction process. (C) Vector copy number (VCN) was determined by digital drop PCR analysis with primers within the lentiviral backbone (n = 5; p > 0.05 for all CAR T cell construct comparisons). (D–F) Flow cytometry evaluation was performed on day 8 after initial T cell activation. (D) CD123-CARCD20 expression (n = 5; p > 0.05). (E) CD20 transgene expression (n = 5; p > 0.05). (F) Coexpression of CD123-CAR and CD20. Representative histograms and dot plots are shown. The inset bar graph shows the mean and standard deviation values for cells expressing CD123-CAR and CD20 (n = 5; p > 0.05).
CAR Design Does Not Influence Expansion, Viability, or Phenotype of CD123-CARCD20 T Cell Products
CD123-CARCD20 T cells expanded more than 50-fold after 7 days in culture and exhibited viability of greater than 80% (Figures 2A and 2B; n = 5, p > 0.05). Determination of immunophenotype subsets (naive, CCR7+CD45RO–; central memory [CM], CCR7+CD45RO+; terminally differentiated [TD], CCR7–CD45RO–; and effector memory [EM], CCR7–CD45RO+) on day 8 revealed an approximate CD4:CD8 ratio of 1:1 for all constructs (Figure S4A; n = 5, p > 0.05). Most T cells demonstrated an EM or CM phenotype (Figure 2C). Only non-transduced (NT) T cells contained a significant percentage of naive T cells when compared with CD123-CARCD20 T cells (percentage of CD4+ cells in NT versus CD123-CARCD20 T cells: n = 5, p < 0.01; percentage of CD8+ cells in NT versus CD123-CARCD20 T cells: p < 0.0001). Flow cytometry analysis of the activation markers CD27, Tim3, and PD1 revealed no significant differences of single-positive (Figure 2B–2D; Figures S4B–S4D) or double-positive (Tim3+/PD1+) populations between CD123-CARCD20 T cells for either the CD4+ or CD8+ subsets (Figure 2D; n = 5, p > 0.05).
Figure 2.

CD123-CARCD20 T Cells Have Similar Kinetics and Immunophenotypes
(A) Fold expansion of the CD123-CARCD20 and non-transduced (NT) T cells (n = 5; p ≥ 0.05). (B) Viability of the indicated populations was determined by acridine orange/propidium iodide (AO/PI) exclusion (n = 5; p ≥ 0.05). (C and D) Immunophenotype and exhaustion phenotype was determined by flow cytometry on day 8. (C) T cell immunophenotypes (T cell subsets: naive, CCR7+CD45RO–; central memory (CM), CCR7+CD45RO+; terminally differentiated (TD), CCR7–CD45RO–; and effector memory (EM), CCR7–CD45RO+; n = 5. (D) CD4, CD8, Tim3, and PD1 expression (n = 5; p > 0.05 among CD123-CAR T cell groups).
T Cells Expressing CD123-CARCD20 Recognize and Kill CD123+ Targets
To test the functional activity of CD123-CARCD20 T cells, we maintained effector cells in media alone or in co-cultures with either CD123– (K562) or CD123+ (Molm13) cells and assayed for interferon-γ (IFN-γ) production. All CD123-CARCD20 T cell populations exhibited potent cytokine secretion in response to CD123+ target cells, as compared with NT T cells (Figure 3A; n = 5, IFN-γ secreted by CD123-CARCD20 T cells: range = 6,176–39,000 pg/mL; p < 0.0001). At baseline (media or K562 conditions), T cells expressing the 8.41BBz-CAR produced significantly higher IFN-γ levels (Figure 3A; p < 0.0001) than did the other CAR constructs. CD123-CARCD20 T cells exhibited significant in vitro antitumor activity against CD123+ target cells (Figure 3B; n = 5; p < 0.0001) but not against CD123– cells (K562). In contrast, NT T cells did not secrete IFN-γ or kill CD123+ target cells (Figure 3). Thus, all CD123-CARCD20 T cell products had the desired specificity, and only the 8.41BBz-CAR induced significant IFN-γ production and thereby baseline T cell activation. In addition, all CD123-CARCD20 T cell populations were efficiently eliminated (Figure S4E; n = 15, p = 0.0007) in the presence of rituximab and complement, with no differences between constructs.
Figure 3.

CD123-CARCD20 T Cells Recognize and Kill CD123+ Targets in an Antigen-Specific Manner
(A) Effector cells were grown in cocultures with media, K562 (CD123–), or Molm13 (CD123+) at an E:T ratio of 2:1 for 24 h. Supernatants were collected and evaluated for IFN-γ content by ELISA (n = 5; p < 0.0001 for NT versus CD123-CARCD20 T cell groups, and p > 0.05 for comparison among CD123-CARCD20 T cell groups). Scale magnification of data in (A; n = 5; p < 0.01 for comparison of 8.41BBz versus all other CD123-CARCD20 T cell groups). (B) Target cell populations were labeled with CFSE, incubated with effector T cells at the indicated ratios overnight and analyzed by flow cytometry by using absolute counting beads to determine cytotoxicity. n = 5; p > 0.05 for comparison on K562 targets and p < 0.0001 for CD123-CAR CD20, as compared with NT on Molm13.
CD34+ HPCs Are Recognized to a Greater Extent by 716 Than by 292 scFv-Based CARs
Because we observed no difference in AML target recognition among the constructs, we next compared the potential on target/off cancer toxicity of CD123-CARCD20 T cells against CD34+ HPCs in a standard colony-forming unit (CFU) assay at two effector to target ratios (E:T; 1:1 and 5:1). At an E:T ratio of 1:1, three (716.8.28z, 28.28z, and 28.28.41BBz) of the five evaluated CD123-CARCD20 T cell populations were cytotoxic to CD34+ target cells (Figure 4; n = 6 biological replicates). At an E:T ratio of 5:1, all CD123-CARCD20 T cells significantly reduced the number of CFUs formed (p < 0.05). At this higher E:T ratio, 716.8.28z CAR T cells induced a greater reduction in CFUs (Figure 4) than did the other CD123-CARCD20 T cells.
Figure 4.

Recognition of CD123+ Hematopoietic Precursor Cells by CD123-CARCD20 T Cells
Effector cells were incubated with CD34+ HPCs for 4 h at E:T ratios of 5:1 and 1:1, plated on semisolid media, and evaluated 12–14 days later (n = 6 biological replicates; ∗p < 0.05; black asterisk: comparison to NT T cells; red asterisk: comparison among CAR constructs).
T Cells Expressing 292 scFv-Based CARs Have Superior Antitumor In Vivo Activity
We used a xenograft mouse model to assess each CD123-CARCD20 T cell population for in vivo anti-AML activity. Molm13.ffluc cells were intravenously injected into the tail veins of non-obese diabetic severe combined immunodeficiency (NOD-SCID) gamma (NSG) mice, followed by tail vein injection of 1 × 107 or 3 × 106 effector cells on day 7 (Figure 5A). AML burden was longitudinally followed by bioluminescence imaging. At a cell dose of 1 × 107, CD123-CARCD20 T cells had potent antitumor activity regardless of evaluated CAR construct in comparison to control mice (n = 5 mice per group; p < 0.05; Figure 5B; Figure S5A). This resulted in a marked survival advantage (Figure 5C). At the end of the experiment (day 80 post AML injection), all 28.28z CAR T cell treated mice remained disease free in contrast to other treatment groups. At a cell dose of 3 × 106 CAR T cells, all CAR constructs had significant antitumor activity as judged by a significant survival advantage in comparison to untreated controls (n = 5 mice per group; p < 0.05; Figure 5D; Figure S5B). Mice treated with 716.8.28z CAR T cells had a significant lower overall survival than mice that had received 28.28z or 8.28z CAR T cells (p < 0.05; Figure 5E). Tumor-free mice treated at both cell doses experienced no weight loss, indicating that the infusion of CD123-CARCD20 T cells is well tolerated (Figure S5C).
Figure 5.

CD123-CARCD20 T Cells Have Potent Antitumor Activity In Vivo
(A) Schematic of experimental design. (B–E) Animals were intravenously injected with Molm13-expressing luciferase, followed by infusion of either 3 × 106 or 1 × 107 effector T cells and in vivo imaging for evaluation of tumor burden. (B) Bioluminescence signal over time (total flux in photons/s) of group receiving 1 × 107 T cells. (C) Kaplan-Meier survival curves for animal groups receiving 1 × 107 T cells. Statistical significance was determined with a Wilcoxon rank-sum test (p < 0.05; blue shading in table). (D) Bioluminescence signal over time (total flux in photons/s) of group receiving 3 × 106 T cells. (E) Kaplan-Meier survival curves for animal groups receiving 3 × 106 T cells. Statistical significance was determined by Wilcoxon rank-sum test (p < 0.05; blue shading in table).
Discussion
In this study, we generated five different LV constructs encoding CD20 and CD123-CARs that differed in their antigen binding, H/TM, and/or signaling domains. The CD123 CARCD20 T cell populations shared similar immunophenotypes and effector functions in vitro and in vivo, except for differences in baseline signaling and recognition of HPCs. On the basis of our comprehensive analysis, we selected one construct for clinical testing.
The effector function of CAR T cells directly results from the interplay between the antigen recognition domain (e.g., scFv affinity and antigen density),18 length and flexibility of the H/TM domain,19, 20, 21 and the costimulatory domain selected.22,24,25 More recently, small amino acid variations in and around the H/TM domain were found to affect the activity of CD19-CAR T cells in patients.26 However, at present, no general rules of CAR design have emerged, and CAR design for a particular antigen remains largely empiric. We therefore tested two different scFvs (26292 and 32716) with CD8a or CD28 H/TM domains and CD28 and/or 4-1BB as costimulatory domains in our study.
Because CAR T cell function is influenced by the manufacturing process27, 28, 29, 30 and our intent was to select a construct for clinical testing, we used a CAR T cell generation method that is established in our Current Good Manufacturing Practice facility. This method relies on CD4/CD8 selection, followed by activation, transduction, and expansion of CAR T cells in the presence of interleukin-7 (IL-7) and IL-15 in G-Rex culture devices. The resulting CAR T cell products exhibited a CD4 to CD8 ratio that approached 1:1, which is deemed favorable by other investigators.29,30 Of note, this was achieved without manufacturing CD4+ and CD8+ CAR T cells separately, simplifying the manufacturing process. The constructs expressing CD28 H/TM domains that also contained the same signaling domain resulted in higher levels of CAR expression than did their counterparts expressing CD8a H/TM. Although the influence of the H/TM domain on CAR cell surface expression has been reported by others,19, 20, 21,31 our study demonstrated that this is not due to differences in the transduction efficiencies of the CAR-encoding vectors.
The resulting CD123-CARCD20 T cells exhibited similar immunophenotypes, with a predominance of central and effector memory T cell subsets. The role that different T cell subsets play in determining CAR T cell in vivo efficacy is now emerging. For example, a recent study showed that CD19.28z-CAR T cell products containing a decreased amount of naive T cells correlate with increased progression-free survival (PFS) for poor risk and relapsed and refractory B cell non-Hodgkin’s lymphoma.32 In the same study, the presence of CD8+ CAR central memory T cells marginally improved PFS.32 Others have also reported that CD19-CAR T cell populations that are CD27+/PD-1–/CD8+ are a predictor for sustained remission.33
Of the CD123-CARs we generated, only the construct encoding a 4-1BB costimulatory domain induced baseline (tonic) signaling, which was evidenced by marked IFN-γ production without antigen-specific stimulation. However, this did not translate to increased expression of the markers associated with exhaustion, such as Tim3 and PD1, or decreased effector function. Nevertheless, we excluded this CAR as a potential candidate from clinical testing because of reports by others that tonic signaling can negatively affect CAR T cell function.22 While studies have indicated expression of exhaustion markers such as Tim3, PD1, and LAG3 correlate with CAR T cell effector function,22 we have observed no or only transient differences in several models.34, 35, 36 In addition, a recent publication suggests that silencing of PD-1 has the potential to impair CAR T cell function.37 Since at present there is no clear definition of T cell exhaustion,38 investigators have focused on defining epigenetic programs that define T cell plasticity.39 Indeed our recent studies indicate that deletion of the de novo DNA methyltransferase (DNMT3A) in multiple human CAR T cell systems resulted in a preservation of the CAR T cell’s ability to proliferate and mount an effector response during chronic antigen exposure.40 Thus, future studies are needed to define the epigenetic programs of CD123-CAR T cell populations.
We found that T cells expressing the 716 scFv-based CAR recognized HPCs to a greater extent than did 292 scFv-based CARs. Both scFvs bind to different epitopes; however, they have similar affinities for CD123 that are in the nanomolar range.41 Despite these similar affinities, only the 292 scFv-based immunotoxin imparted notable cytotoxic activity, indicating that the different binding sites within the extracellular domain of the antigen can affect the activity of immune-based approaches.42 This was also reported for CAR T cells.43,44 One study compared both scFvs in the context of CD28z-CARs and found no HPC toxicity for either CAR;14 however, these CARs contained a longer H domain (immunoglobulin G4 [IgG4]-Fc) than did ours. Recognition of HPCs by T cells expressing a 716 scFv-based 41BBz-CAR has been reported, and several safety switches are being actively explored to mitigate on target/off cancer toxicity. 292 and 716 scFv-based CAR T cells have the potential to recognize and kill HPCs in humans. Thus, having a suitable hematopoietic stem cell donor is one of the eligibility criteria of most current clinical studies evaluating the safety and efficacy of CD123-CAR T cells. Lastly, while CD123 splice variants have been described,45 a recent study suggest that there is no evidence that normal HPCs express different splice variants than AML blasts.46 We and others have previously demonstrated that transgenic expression of CD20 in T cells is a promising strategy to eliminate T cells with the clinical grade CD20 antibody rituximab, and we therefore included CD20 in our CD123-CAR encoding LVs as a safety switch.12,47,48
We evaluated the antitumor activity of CD123-CARCD20 T cells at two dose levels. Although we did not observe significant differences between the CAR T cell groups at higher doses, the lower dose 716 scFv-based CD123-CARCD20 T cells significantly decreased antitumor activity. No difference in in vivo antitumor activity of 292 scFv- and 716 scFv-based CD28z-CD123-CAR T cells was reported, but only a single T cell dose was evaluated.14 The addition of a 4-1BB signaling domain to CD28.z-CAR T cells did not improve antitumor activity in our study, which is consistent with reports by others that the benefit of incorporating a 4-1BB signaling domain into CD28.z-CAR T cells is model dependent.22,23 One study compared the expansion of CD28.z- and CD28.41BBz-CAR T cells targeting CD19 in individual patients with lymphoma and observed increased CD28.41BBz-CAR T cell expansion with low disease burden.49 We selected the CD28.CD28z CAR for clinical development since it was expressed at higher levels on T cells in comparison to CD8a.CD28z CARs. Higher cell surface expression of CD28 H/TM CARs in comparison to CD8a H/TMs was not observed in a recent study.50 However, investigators found that CD28 H/TM CARs formed more stable immunological synapses than CD8a H/TMs,50 supporting the selection of our CD28.CD28z CAR for clinical testing.
Our study has several limitations; first, we did not determine the in vivo expansion and persistence of infused CD123-CAR T cells. These studies should ideally be performed in syngeneic mouse models to exclude xenogeneic CAR T cell stimulation as a confounding factor. In this regard, we are currently developing an immune competent model to evaluate CD123-CAR T cells. In addition, we did not compare 716-based (28.28z) CARs to 292-based CARs (292.28.z) directly in our initial studies. In subsequent studies we have now shown that there are no significant differences in transduction efficiency, phenotype, expansion, and effector function between both CAR constructs in vitro studies (Figures S6–S8).
In conclusion, no clear winner emerged from our evaluation of the five CAR constructs. However, subtle differences emerged, leading us to make an informed decision: one CAR demonstrated tonic signaling; one CAR had limited antitumor activity; and one CAR did not endow T cells with improved effector function despite having a more complex design (i.e., two costimulatory endodomains). Of the remaining two CARs, we selected the CAR with a CD28 H/TM domain for clinical testing because it resulted in higher levels of CAR expression. The safety and efficacy of CD123-CARCD20 T cells generated with the selected CAR will be evaluated in an approved clinical study (NCT04318678) that is now open for patient accrual.
Materials and Methods
Cells and Culture Conditions
Deidentified apheresis products from healthy donors were purchased from Key Biologics (Memphis, TN, USA; 16761) and were used in accordance with the Helsinki Declaration. Authenticated K562 and Molm13 cell lines were obtained from ATCC. Molm13 cells expressing a GFP firefly luciferase (Molm13.ffluc) fusion molecule was previously described.48 All cell lines were maintained in RPMI culture media (GE Healthcare Life Sciences, Logan, UT, USA; SH30096.01) supplemented with 10% fetal bovine serum (FBS; GIBCO/Thermo Fisher Scientific, Waltham, MA, USA; 10082-147) and L-glutamine (GlutaMAX; GIBCO/Thermo Fisher Scientific; 35050-061).
LVs
The LV backbone used for this study has been previously described,51 except that the insulators were removed from the self-inactivating 3′ partially deleted viral long terminal repeats, according to the safety records of LVs in clinical trials.52,53 The expression cassette of the LV is under the control of the MND promoter (myeloproliferative sarcoma virus enhancer, negative control region deleted, dl587rev primer-binding site substituted).51 Mini genes encoding CD20, 2A, and the CD123-specific CARs were synthesized by GeneArt (Thermo Fisher, Waltham, MA, USA) and subcloned by standard techniques. All cloned CD20-2A-CD123-CAR constructs were verified by sequencing at the Hartwell Center at St. Jude Children’s Research Hospital (St. Jude). Purified lentiviral particles were produced by the St. Jude Vector Core Laboratory by using transient transfection, followed by fast protein liquid chromatography purification.54
CD4 and CD8 T Cell Isolation
CD4+/CD8+ T cells were magnetically isolated on a CliniMACS Plus instrument (Miltenyi Biotec; Bergisch Gladsbach, Germany) with CD4 (Miltenyi Biotec; 130-030-401) and CD8 (Miltenyi Biotec; 130-030-801) microbeads and the enrichment program 1.1, per the manufacturer instructions. Aliquots of enriched CD4+/CD8+ T cells were cryopreserved and thawed before use for CAR T cell generation.
CD123-CARCD20 T Cell Generation
Enriched CD4+/CD8+ T cells were resuspended at 1 × 106 per mL in X-VIVO 15 (Lonza, Walkersville, MD, USA; 04-744Q) supplemented with 5% human AB serum (Corning, Corning, NY, USA; 35-060-CI) and 10 ng/mL each IL-7 and IL-15 (Miltenyi Biotec; 170-076-111 and 170-076-114, respectively). Cells were activated by plating overnight with T cell TransAct (Miltenyi Biotec; 130-019-011). On day 1, 2 × 106 T cells were plated with LVs and transduced overnight at a multiplicity of infection of 10 to 12. After transduction, the cells were transferred to 6-well G-Rex plates (Wilson Wolf, New Brighton, MN, USA; 180102-1) and expanded for 7–10 days. On day 6, half of the media was removed and replaced with complete media with cytokines.
VCN
Transduced T cells were harvested, and total genomic DNA was isolated with the Zymo Research Quick-DNA 96-well kit (Zymo Research, Irvine, CA, USA; D3012). To determine the VCN per cell, we digested the genomic DNA with MspI and used as a template in PCR by using a digital droplet PCR instrument (QX200 Bio-Rad, Carlsbad, CA, USA). The following primer-probe sets were used to amplify the HIV psi sequence located on the vector genome and the endogenous control gene RPP30: 5′-ACTTGAAAGCGAAAGGGAAAC-3′, 5′-CACCCATCTCTCTCCTTCTAGCC-3′, and probe 5′-FAM-AGCTCTCTCGACGCAGGACTCGGC-3′ and 5′-GCGGCTGTCTCCACAAGT-3′, 5′-GATTTGGACCTGCGAGCG-3′, and probe 5′-HEX-CTGACCTGAAGGCTCT-3′, respectively. The reaction mixture contained ddPCR Supermix for probes without UTP (BioRad; 64180520). The cycled droplets were read with a QX200 droplet reader (Bio-Rad). The ratio of the numbers of molecules of these two genes was determined by the sample’s gene of interest relative copy number analyzed with QuantaSoft droplet reader software, version 1.7.4.0917 (Bio-Rad).
Flow Cytometry
Cells were stained with fluorochrome-conjugated primary antibodies for 30 min at 4°C and washed with fluorescence-activated cell sorting (FACS) buffer (2% FBS in 1 × PBS) before analysis. For CAR staining, cells were washed with 1 × PBS twice and then incubated with a recombinant CD123-Fc fusion protein (Abcam; ab88358) in PBS for 30 min at 4°C. The cells were then washed, incubated with the secondary antibody in FACS buffer for 30 min at 4°C, and washed with FACS buffer before analysis. Stained cells were analyzed with a CytoFLEX instrument (Beckman Coulter, Indianapolis, IN, USA) and FlowJo software. The following antibodies were used: CD4 (Clone OKT4, BV785, BioLegend; 317442), CD8 (Clone SK1, APC-Cy7, BD PharMingen; 557834), CCR7 (Clone REA546, PE, Miltenyi Biotech; 130-108-285), CD45RO (Clone UCHL1, APC, Tonbo; 20-0457-T100), Tim3 (Clone F38-2E2, PE-Cy7, Biolegend; 345014), PD1 (Clone EH12.2H7, BV421, Biolegend; 329920), CD20 (Clone 2H7, FITC, Tonbo; 35-0209-T100), and goat anti-human Fc-IgG (pooled goat antisera, PE, Southern Biotech; 2048-09).
Cytotoxicity Assay
Cytotoxic activity was evaluated with a flow-cytometry-based assay. Target cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) (Cayman Chemical, Ann Arbor, MI, USA; 600120) for 20 min at 37°C. We incubated 50,000 CFSE-labeled target cells overnight either alone or with effector T cells in round bottom 96-well plates (Corning, Corning, NY, USA; 353077) at an E:T ratio of 3:1. The cells were washed and resuspended in PBS containing Count Bright Absolute Counting Beads (Life Technologies, Eugene, OR, USA; C36950). CFSE was measured by flow cytometry with a BD FACSLyric instrument (Becton Dickinson, Franklin Lakes, NJ, USA) and analyzed with FlowJo software (Becton Dickinson). Lysis was calculated with the following formula: % lysis = 100 – (average CFSE+ events per 100 beads/average of CFSE+ events per 100 beads in wells with target alone) × 100.
Cytokine Production
Effector cells were grown in culture at a 2:1 ratio with target cells or in the presence of media alone for 24 h in a 24-well plate (Corning; 353047). Supernatants were collected, and IFN-γ was determined with a Quantikine ELISA kit (R&D, Minneapolis, MN, USA; SIF50), according to the manufacturer’s instructions.
CFU Assay
An apheresis product of mobilized peripheral blood was purchased from Key Biologics (Memphis, TN, USA), and CD34+ cells were isolated by the Human Applications Laboratory at St. Jude with a CliniMACS device, per the manufacturer instructions (Miltenyi Biotec). A modified CFU assay was performed. In brief, CD34+ cells (5 × 104) were incubated with CD123-CARCD20 T cells at ratios of 5:1 and 1:1 (T:CD34) for 4 h in 96-well round bottom plates. For each co-culture, three replicates (input equivalent of 2,000 CD34+ cells) were plated into 1 mL MethoCult H4434 media (Stem Cell Technologies, Vancouver, BC, Canada; 04434), following the manufacturer’s instructions. Colonies were counted 12–14 days later.
Safety Switch Activation Assay
T cells were resuspended in RPMI/1% human serum (Corning; 35-060-CI; heat inactivated) and incubated for 1 h at 37°C with 10 μg of rituximab (Biogen, Cambridge, MA; 502-051021) and 10% baby rabbit complement (Cedarlane, Burlington, NC; CL3441) in a 96-well round bottom plate, as previously published.48 The cells were washed and analyzed by flow cytometry for CD20 expression. Percent transgene expression was determined by the following formula: (%CD20+before – %CD20+after)/%CD20+before × (1–%CD20+after).
Xenograft Model
In vivo experiments were performed under a protocol approved by the St. Jude Institutional Animal Care and Use Committee. Animals were housed in specific pathogen-free rooms for the duration of the experiments. Female NSG mice (NOD-SCID IL-2Rgammanull, NOD-SCID IL-2Rgnull, NSG, NOD-SCID gamma) were obtained from the St. Jude breeding colony at 8–10 weeks of age. The mice received 5 × 103 Molm13 tumor cells modified to express a GFP.ffluc fusion gene (Molm13ffluc) via tail vein injection. 7 days later, mice in the treatment groups were infused with effector cells. Animals receiving tumor only samples were used as controls. Serial imaging was subsequently performed in the St. Jude Center for In Vivo Imaging and Therapeutics with a Xenogen IVIS-200 imaging system (IVIS, Xenogen, Alameda, CA, USA), as previously described.55 The mice were euthanized at predefined endpoints or when they met euthanasia criteria in accordance with St. Jude Animal Resource Center policy.
Statistics
Data were summarized using descriptive statistics. A Friedman or permutation test was used to examine overall differences in continuous variables between lentiviral CD123-CARCD20 constructs. The overall test was followed by pairwise comparisons with Wilcoxon signed-rank or paired-permutation tests, when appropriate (i.e., overall test p < 0.05). A two-way repeated-measures analysis of variance (ANOVA) with a rank transformation was used to examine overall differences in continuous variables. A Friedman test was used to examine the overall differences in CD4:CD8 ratios between constructs. The overall test was followed by pairwise comparisons with Wilcoxon signed-rank tests, when appropriate. The constructs were then compared to the control sample with Wilcoxon signed-rank tests. The CD4:CD8 ratio for each construct was compared to a value of 1 with Wilcoxon signed-rank tests (i.e., H0: ratio = 1; H1: ratio ≠ 1). The bias-corrected and accelerated bootstrap confidence intervals are reported for the median ratio for each construct. The Kruskal-Wallis test was used to examine overall differences in bioluminescence on day 12 between constructs. The overall test was followed by pairwise comparisons with Wilcoxon rank-sum tests. A two-way repeated-measures ANOVA with a rank transformation was used to examine overall differences in bioluminescence over time and between constructs. Time, construct, and their interaction were considered in the model. Survival was compared among constructs with Wilcoxon rank-sum tests. Statistical analyses were conducted with R software, version 3.6.0 (Lucent Technologies, Murray Hill, New Providence, NJ, USA).
Author Contributions
Conceptualization, J.M.R., C.L.B., S.G., and M.P.V.; Data Analysis, A.S., N.S., J.M.R., S.G., and M.P.V.; Investigation, J.M.R., S.Z., F.Z., Y.-I.K., J.M., and A.V.; Resources, R.E.T. and B.R.; Writing – Original Draft, J.M.R., J.M., R.E.T., S.G., and M.P.V.; Writing – Review & Editing, J.M.R., S.Z., F.Z., Y.-I.K., J.M., A.V., R.E.T., A.S., N.S., C.L.B., B.R., S.G., and M.P.V.; Funding Acquisition, S.G., and M.P.V.; Supervision, S.G. and M.P.V.
Conflicts of Interest
J.M.R., S.Z., F.Z., Y.-I.K., A.V., R.E.T., C.L.B., B.R., S.G., and M.P.V. have patent applications in the field of Cell and Gene Therapy.
Acknowledgments
We would like to acknowledge the St. Jude Children’s Research Hospital Scientific Writing Department for their help with manuscript editing. Preclinical imaging was performed by the Center for In Vivo Imaging and Therapeutics, which is supported in part by NIH grants P01CA096832 and R50CA211481. Preclinical procedures were performed in collaboration with the St. Jude Animal Resource Center. This project was supported in part by The Leukemia Lymphoma Society, St. Baldrick's Foundation, Assisi Foundation of Memphis, Cancer Prevention & Research Institute of Texas (RP101335), and American Lebanese Syrian Associated Charities. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.06.024.
Supplemental Information
References
- 1.Zwaan C.M., Kolb E.A., Reinhardt D., Abrahamsson J., Adachi S., Aplenc R., De Bont E.S., De Moerloose B., Dworzak M., Gibson B.E. Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. J. Clin. Oncol. 2015;33:2949–2962. doi: 10.1200/JCO.2015.62.8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude S.L. Tisagenlecleucel in pediatric patients with acute lymphoblastic leukemia. Clin. Adv. Hematol. Oncol. 2018;16:664–666. [PubMed] [Google Scholar]
- 4.Locke F.L., Neelapu S.S., Bartlett N.L., Siddiqi T., Chavez J.C., Hosing C.M., Ghobadi A., Budde L.E., Bot A., Rossi J.M. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol. Ther. 2017;25:285–295. doi: 10.1016/j.ymthe.2016.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee D.W., Kochenderfer J.N., Stetler-Stevenson M., Cui Y.K., Delbrook C., Feldman S.A., Fry T.J., Orentas R., Sabatino M., Shah N.N. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kochenderfer J.N., Dudley M.E., Kassim S.H., Somerville R.P., Carpenter R.O., Stetler-Stevenson M., Yang J.C., Phan G.Q., Hughes M.S., Sherry R.M. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015;33:540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davila M.L., Riviere I., Wang X., Bartido S., Park J., Curran K., Chung S.S., Stefanski J., Borquez-Ojeda O., Olszewska M. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardner R.A., Finney O., Annesley C., Brakke H., Summers C., Leger K., Bleakley M., Brown C., Mgebroff S., Kelly-Spratt K.S. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322–3331. doi: 10.1182/blood-2017-02-769208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perna F., Berman S.H., Soni R.K., Mansilla-Soto J., Eyquem J., Hamieh M., Hendrickson R.C., Brennan C.W., Sadelain M. Integrating Proteomics and Transcriptomics for Systematic Combinatorial Chimeric Antigen Receptor Therapy of AML. Cancer Cell. 2017;32:506–519. doi: 10.1016/j.ccell.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ehninger A., Kramer M., Röllig C., Thiede C., Bornhäuser M., von Bonin M., Wermke M., Feldmann A., Bachmann M., Ehninger G., Oelschlägel U. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014;4:e218. doi: 10.1038/bcj.2014.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gill S., Tasian S.K., Ruella M., Shestova O., Li Y., Porter D.L., Carroll M., Danet-Desnoyers G., Scholler J., Grupp S.A. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123:2343–2354. doi: 10.1182/blood-2013-09-529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tasian S.K., Kenderian S.S., Shen F., Ruella M., Shestova O., Kozlowski M., Li Y., Schrank-Hacker A., Morrissette J.J.D., Carroll M. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood. 2017;129:2395–2407. doi: 10.1182/blood-2016-08-736041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tashiro H., Sauer T., Shum T., Parikh K., Mamonkin M., Omer B., Rouce R.H., Lulla P., Rooney C.M., Gottschalk S., Brenner M.K. Treatment of Acute Myeloid Leukemia with T Cells Expressing Chimeric Antigen Receptors Directed to C-type Lectin-like Molecule 1. Mol. Ther. 2017;25:2202–2213. doi: 10.1016/j.ymthe.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mardiros A., Dos Santos C., McDonald T., Brown C.E., Wang X., Budde L.E., Hoffman L., Aguilar B., Chang W.C., Bretzlaff W. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122:3138–3148. doi: 10.1182/blood-2012-12-474056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Budde L., Song J., Kim Y., Blanchard S., Wagner J., Stein A., Weng L., Del Real M., Hernandez R., Marcucci E. Remissions of Acute Myeloid Leukemia and Blastic Plasmacytoid Dendritic Cell Neoplasm Following Treatment with CD123-Specific CAR T Cells: A First-in-Human Clinical Trial. Blood. 2017;130:811. [Google Scholar]
- 16.Bouchkouj N., Kasamon Y.L., de Claro R.A., George B., Lin X., Lee S., Blumenthal G.M., Bryan W., McKee A.E., Pazdur R. FDA Approval Summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-cell Lymphoma. Clin. Cancer Res. 2019;25:1702–1708. doi: 10.1158/1078-0432.CCR-18-2743. [DOI] [PubMed] [Google Scholar]
- 17.O’Leary M.C., Lu X., Huang Y., Lin X., Mahmood I., Przepiorka D., Gavin D., Lee S., Liu K., George B. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients with Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2019;25:1142–1146. doi: 10.1158/1078-0432.CCR-18-2035. [DOI] [PubMed] [Google Scholar]
- 18.Ramakrishna S., Highfill S.L., Walsh Z., Nguyen S.M., Lei H., Shern J.F., Qin H., Kraft I.L., Stetler-Stevenson M., Yuan C.M. Modulation of Target Antigen Density Improves CAR T-cell Functionality and Persistence. Clin. Cancer Res. 2019;25:5329–5341. doi: 10.1158/1078-0432.CCR-18-3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hudecek M., Lupo-Stanghellini M.T., Kosasih P.L., Sommermeyer D., Jensen M.C., Rader C., Riddell S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013;19:3153–3164. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watanabe N., Bajgain P., Sukumaran S., Ansari S., Heslop H.E., Rooney C.M., Brenner M.K., Leen A.M., Vera J.F. Fine-tuning the CAR spacer improves T-cell potency. OncoImmunology. 2016;5:e1253656. doi: 10.1080/2162402X.2016.1253656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guest R.D., Hawkins R.E., Kirillova N., Cheadle E.J., Arnold J., O’Neill A., Irlam J., Chester K.A., Kemshead J.T., Shaw D.M. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J. Immunother. 2005;28:203–211. doi: 10.1097/01.cji.0000161397.96582.59. [DOI] [PubMed] [Google Scholar]
- 22.Long A.H., Haso W.M., Shern J.F., Wanhainen K.M., Murgai M., Ingaramo M., Smith J.P., Walker A.J., Kohler M.E., Venkateshwara V.R. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drent E., Poels R., Ruiter R., van de Donk N.W.C.J., Zweegman S., Yuan H., de Bruijn J., Sadelain M., Lokhorst H.M., Groen R.W.J. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor-engineered T Cells. Clin. Cancer Res. 2019;25:4014–4025. doi: 10.1158/1078-0432.CCR-18-2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawalekar O.U., O’Connor R.S., Fraietta J.A., Guo L., McGettigan S.E., Posey A.D., Jr., Patel P.R., Guedan S., Scholler J., Keith B. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44:380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 25.Gomes-Silva D., Mukherjee M., Srinivasan M., Krenciute G., Dakhova O., Zheng Y., Cabral J.M.S., Rooney C.M., Orange J.S., Brenner M.K., Mamonkin M. Tonic 4-1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell Rep. 2017;21:17–26. doi: 10.1016/j.celrep.2017.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ying Z., Huang X.F., Xiang X., Liu Y., Kang X., Song Y., Guo X., Liu H., Ding N., Zhang T. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. 2019;25:947–953. doi: 10.1038/s41591-019-0421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berger C., Jensen M.C., Lansdorp P.M., Gough M., Elliott C., Riddell S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Invest. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alizadeh D., Wong R.A., Yang X., Wang D., Pecoraro J.R., Kuo C.F., Aguilar B., Qi Y., Ann D.K., Starr R. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol. Res. 2019;7:759–772. doi: 10.1158/2326-6066.CIR-18-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turtle C.J., Hanafi L.A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sommermeyer D., Hudecek M., Kosasih P.L., Gogishvili T., Maloney D.G., Turtle C.J., Riddell S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Künkele A., Johnson A.J., Rolczynski L.S., Chang C.A., Hoglund V., Kelly-Spratt K.S., Jensen M.C. Functional Tuning of CARs Reveals Signaling Threshold above Which CD8+ CTL Antitumor Potency Is Attenuated due to Cell Fas-FasL-Dependent AICD. Cancer Immunol. Res. 2015;3:368–379. doi: 10.1158/2326-6066.CIR-14-0200. [DOI] [PubMed] [Google Scholar]
- 32.Sauter C.S., Senechal B., Riviere I., Ni A., Bernal Y., Wang X., Purdon T., Hall M., Singh A.N., Szenes V.Z. CD19 CAR T Cells Following Autologous Transplantation in Poor Risk Relapsed and Refractory B cell non-Hodgkin Lymphoma. Blood. 2019;134:626–635. doi: 10.1182/blood.2018883421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fraietta J.A., Lacey S.F., Orlando E.J., Pruteanu-Malinici I., Gohil M., Lundh S., Boesteanu A.C., Wang Y., O’Connor R.S., Hwang W.T. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018;24:563–571. doi: 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Velasquez M.P., Szoor A., Vaidya A., Thakkar A., Nguyen P., Wu M.F., Liu H., Gottschalk S. CD28 and 41BB Costimulation Enhances the Effector Function of CD19-Specific Engager T Cells. Cancer Immunol. Res. 2017;5:860–870. doi: 10.1158/2326-6066.CIR-17-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mata M., Gerken C., Nguyen P., Krenciute G., Spencer D.M., Gottschalk S. Inducible Activation of MyD88 and CD40 in CAR T Cells Results in Controllable and Potent Antitumor Activity in Preclinical Solid Tumor Models. Cancer Discov. 2017;7:1306–1319. doi: 10.1158/2159-8290.CD-17-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prinzing B., Bell M., Schreiner P., Fan Y., Krenciute G., Gottschalk S. Chimeric Antigen Receptors with MyD88 and CD40 endodomain Endow T cell with superior antitumor activity. Mol. Ther. 2019;27:431. [Google Scholar]
- 37.Wei J., Luo C., Wang Y., Guo Y., Dai H., Tong C., Ti D., Wu Z., Han W. PD-1 silencing impairs the anti-tumor function of chimeric antigen receptor modified T cells by inhibiting proliferation activity. J. Immunother. Cancer. 2019;7:209. doi: 10.1186/s40425-019-0685-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blank C.U., Haining W.N., Held W., Hogan P.G., Kallies A., Lugli E., Lynn R.C., Philip M., Rao A., Restifo N.P. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019;19:665–674. doi: 10.1038/s41577-019-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghoneim H.E., Fan Y., Moustaki A., Abdelsamed H.A., Dash P., Dogra P., Carter R., Awad W., Neale G., Thomas P.G. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell. 2017;170:142–157. doi: 10.1016/j.cell.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zebley C.C., Peterson T.C., Prinzing B., Bell M., Fan Y., Crawford J.C., Houke H., Haydar D., Yi Z., Nguyen P. De novo DNA methylation programs regulate CAR T-cell exhaustion. The Journal of Immunology. 2020;204 246.5. [Google Scholar]
- 41.Du X., Ho M., Pastan I. New immunotoxins targeting CD123, a stem cell antigen on acute myeloid leukemia cells. J. Immunother. 2007;30:607–613. doi: 10.1097/CJI.0b013e318053ed8e. [DOI] [PubMed] [Google Scholar]
- 42.Kunik V., Peters B., Ofran Y. Structural consensus among antibodies defines the antigen binding site. PLoS Comput. Biol. 2012;8:e1002388. doi: 10.1371/journal.pcbi.1002388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haso W., Lee D.W., Shah N.N., Stetler-Stevenson M., Yuan C.M., Pastan I.H., Dimitrov D.S., Morgan R.A., FitzGerald D.J., Barrett D.M. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Z., Jiang D., Yang H., He Z., Liu X., Qin W., Li L., Wang C., Li Y., Li H. Modified CAR T cells targeting membrane-proximal epitope of mesothelin enhances the antitumor function against large solid tumor. Cell Death Dis. 2019;10:476. doi: 10.1038/s41419-019-1711-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kovtun Y., Jones G.E., Adams S., Harvey L., Audette C.A., Wilhelm A., Bai C., Rui L., Laleau R., Liu F. A CD123-targeting antibody-drug conjugate, IMGN632, designed to eradicate AML while sparing normal bone marrow cells. Blood Adv. 2018;2:848–858. doi: 10.1182/bloodadvances.2018017517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Underwood J.G., Smith J.L., Call L.F., Tseng E., Hylkema T.A., Ries R.E., Leonti A.R., Farrar J.E., Triche T.J., Meshinchi S. Discovery of Novel IL3RA (CD123) Isoforms By Long Read Transcriptomics, Heterogeneous Expression Among AML Patient Cohorts and the Implications for Anti-CD123 Therapeutics. Blood. 2019;134:4658. [Google Scholar]
- 47.Griffioen M., van Egmond E.H., Kester M.G., Willemze R., Falkenburg J.H., Heemskerk M.H. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica. 2009;94:1316–1320. doi: 10.3324/haematol.2008.001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonifant C.L., Szoor A., Torres D., Joseph N., Velasquez M.P., Iwahori K., Gaikwad A., Nguyen P., Arber C., Song X.T. CD123-Engager T Cells as a Novel Immunotherapeutic for Acute Myeloid Leukemia. Mol. Ther. 2016;24:1615–1626. doi: 10.1038/mt.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramos C.A., Rouce R., Robertson C.S., Reyna A., Narala N., Vyas G., Mehta B., Zhang H., Dakhova O., Carrum G. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018;26:2727–2737. doi: 10.1016/j.ymthe.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Majzner R.G., Rietberg S.P., Sotillo E., Dong R., Vachharajani V.T., Labanieh L., Myklebust J.H., Kadapakkam M., Weber E.W., Tousley A.M. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov. 2020;10:702–723. doi: 10.1158/2159-8290.CD-19-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chan W.K., Suwannasaen D., Throm R.E., Li Y., Eldridge P.W., Houston J., Gray J.T., Pui C.H., Leung W. Chimeric antigen receptor-redirected CD45RA-negative T cells have potent antileukemia and pathogen memory response without graft-versus-host activity. Leukemia. 2015;29:387–395. doi: 10.1038/leu.2014.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McGarrity G.J., Hoyah G., Winemiller A., Andre K., Stein D., Blick G., Greenberg R.N., Kinder C., Zolopa A., Binder-Scholl G. Patient monitoring and follow-up in lentiviral clinical trials. J. Gene Med. 2013;15:78–82. doi: 10.1002/jgm.2691. [DOI] [PubMed] [Google Scholar]
- 53.Cornetta K., Duffy L., Turtle C.J., Jensen M., Forman S., Binder-Scholl G., Fry T., Chew A., Maloney D.G., June C.H. Absence of Replication-Competent Lentivirus in the Clinic: Analysis of Infused T Cell Products. Mol. Ther. 2018;26:280–288. doi: 10.1016/j.ymthe.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Throm R.E., Bauler M., Wu C.C., Roberts J.K., Fan B., Ferrara F., Wiegolsz M., Ryu B. Production of Lentiviral Vectors Using 293T Cells Adapted to Grow in Suspension with Serum-Free Media. Mol. Ther. Methods Clin. Dev. 2018;26:58–68. [Google Scholar]
- 55.Shaffer D.R., Savoldo B., Yi Z., Chow K.K., Kakarla S., Spencer D.M., Dotti G., Wu M.F., Liu H., Kenney S., Gottschalk S. T cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. Blood. 2011;117:4304–4314. doi: 10.1182/blood-2010-04-278218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.