

Abstract
The synergism between cardiomyogenesis and angiogenesis is essential for cardiac regeneration. Circular RNAs (circRNAs) play pivotal roles in cell growth and angiogenesis, but their functions in cardiac regeneration are not yet known. In this study, we investigated the role and underlying mechanisms of circRNA Hipk3 (circHipk3) in both cardiomyogenesis and angiogenesis during cardiac regeneration. We found that circHipk3 was overexpressed in the fetal or neonatal heart of mice. The transcription factor Gata4 bound to the circHipk3 promoter and increased circHipk3 expression. Cardiomyocyte (CM) proliferation in vitro and in vivo was inhibited by circHipk3 knockdown and increased by circHipk3 overexpression. Moreover, circHipk3 overexpression promoted coronary vessel endothelial cell proliferation, migration, and tube-forming capacity and subsequent angiogenesis. More importantly, circHipk3 overexpression attenuated cardiac dysfunction and decreased fibrotic area after myocardial infarction (MI). Mechanistically, circHipk3 promoted CM proliferation by increasing Notch1 intracellular domain (N1ICD) acetylation, thereby increasing N1ICD stability and preventing its degradation. In addition, circHipk3 acted as a sponge for microRNA (miR)-133a to promote connective tissue growth factor (CTGF) expression, which activated endothelial cells. Our findings suggested that circHipk3 might be a novel therapeutic target for preventing heart failure post-MI.
Keywords: circRNA, angiogenesis, cardiac regeneration
Graphical Abstract
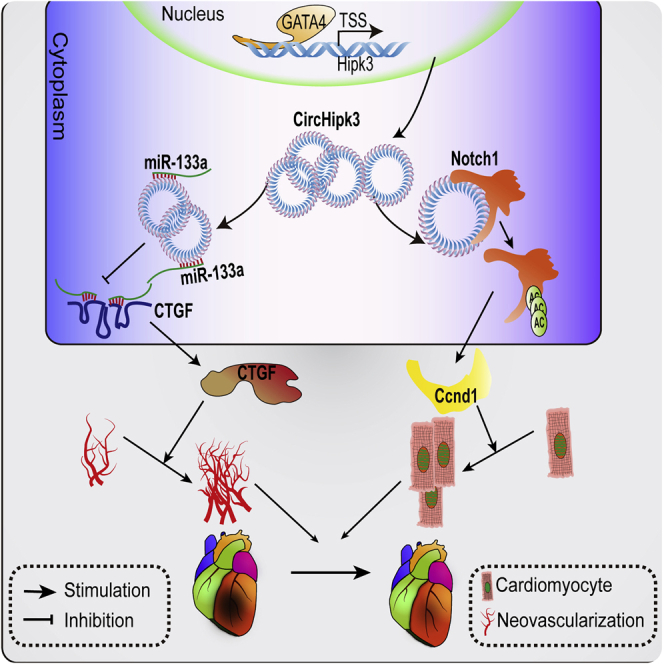
The function of circRNA in cardiac regeneration is largely known. In this study, circHipk3 was identified as a novel regulator of the synergism between cardiomyogenesis and angiogenesis during cardiac regeneration. Our findings suggested that circHipk3 might be a novel therapeutic target for preventing heart failure after myocardial infarction.
Introduction
The stimulation of endogenous cardiac regeneration is a promising approach to replace the lost myocardium after injury or diseases, such as myocardial infarction (MI), myocarditis, and primary cardiomyopathy.1 Recently, therapeutic strategies involving the transfer of cardiogenic factors, including transcription factors,2 growth factors,3 extracellular matrix,4 microRNAs (miRNAs), and long noncoding RNAs (lncRNAs),5,6 to the ischemic myocardium have been found to induce the dedifferentiation and proliferation of pre-existing cardiomyocytes (CMs), thereby reducing the scar area and promoting the functional recovery of the heart after MI. The involvement of CM proliferation in endogenous cardiac regeneration has been extensively investigated, but the role of angiogenesis in mediating functional recovery after MI has been largely neglected. Recent experiments confirmed that the synergy between CM proliferation and angiogenesis is required and essential for structural and functional repair during cardiac regeneration.7,8 Several regulators, including the Erbb2 and Azin2 splice variants, have been shown to induce both myogenesis and angiogenesis after myocardial injury.3,7 Interestingly, these regulators significantly improved the functional recovery and outcome after MI, suggesting that they are promising candidates for regenerative medicine in the heart. Despite their clinical significance, these regulators are easily degraded and have relatively poor tissue specificity, which might limit therapeutic efficacy and future clinical application.
Noncoding RNAs (ncRNAs) are showing increasing potential to induce CM proliferation and angiogenesis during cardiac regeneration due to their important roles in regulating cardiac development, endothelial activation, and cell proliferation.9, 10, 11 Circular RNAs (circRNAs) are a new class of ncRNAs that adopt a covalently closed loop structure, lack a 3′ terminus, and are resistant to RNA exonucleases. Thus, compared to linear RNAs, circRNAs have extraordinary stability and longer durations of action.12,13 Moreover, circRNAs are more likely than their parent linear transcripts to be expressed in a tissue-specific manner, indicating their promise as therapeutic targets for regenerative medicine.14 Of all of the identified circRNAs, circRNA Hipk3 (circHipk3) is a highly abundant, conserved mammalian circRNA that plays important roles in regulating cell growth and viability. Emerging evidence shows that circHipk3 regulates the proliferation of multiple cancerous and noncancerous cells by sponging miRNAs.15, 16, 17, 18 circHipk3 is involved in endothelium activation and angiogenesis through the inhibition of microRNA (miR)-30a-3p activity.19 Importantly, high-throughput RNA sequencing recently revealed that circHipk3 is highly expressed in the neonatal heart, suggesting that circHipk3 likely participates in heart regeneration.20 Therefore, circHipk3 might induce myogenesis and angiogenesis after MI via sponging relevant miRNAs.
In line with our hypotheses, this study was designed to investigate the potential role of circHipk3 on CM proliferation and angiogenesis both in vitro and in vivo. The underlying mechanism of circHipk3 regulating cardiac regeneration was also explored.
Results
circHipk3 Is Highly Expressed in Fetal and Neonatal Hearts
Previous reports characterize one abundant circRNA derived from exon2 of the Hipk3 host gene, named circHipk3.15 The circRNA ID numbers of circHipk3 from human and mouse genome in circBase are hsa_circ_0000284 and mmu_circ_0001052, respectively. The sequence of circHipk3 was highly conservative among human, rat, and mouse (Figure S1). With the use of a transcriptomic dataset reported in a previous study,20 we found that the circHipk3 expression level was significantly higher in the neonatal (postnatal day 1 [P1]) rat heart than in the adult rat heart (Figure 1A). qRT-PCR assays further confirmed that circHipk3 expression in the mouse heart and in CMs was significantly decreased during cardiac development (Figures 1B and 1C). Moreover, we detected circHipk3 expression in different tissues of neonatal mice and found that circHipk3 was highly expressed in the fetal heart (Figure 1D). We also ascertained the circHipk3 expression profile in the main cell types of the neonatal and adult heart and found that circHipk3 was highly expressed in CMs in myocardial tissue (Figures 1E and 1F). We then investigated the stability of this circRNA by RNase R digestion and actinomycin D (an inhibitor of transcription) treatment. It was shown that circHipk3 was resistant to RNase R digestion, whereas the associated linear transcript was easily degraded (Figure 1G). A qRT-PCR analysis revealed that circHipk3 was highly stable, with a transcript half-life exceeding 24 h (Figure 1H). By contrast, the linear Hipk3 (linearHipk3) mRNA exhibited a half-life of <12 h. To knock down circHipk3 expression in CMs, we designed three small interfering (si)RNAs: one siRNA targeting the backsplice sequence, another siRNA targeting a sequence only in the linear transcript, and a third siRNA targeting a sequence in a circularized exon shared by both linear and circular species (Figure S2). As expected, si-circHipk3 and si-both, but not si-Hipk3, decreased the expression of a circular transcript (Figure 1I). These results further confirmed the circular form of circHipk3. Then, we performed in situ hybridization (ISH) for circHipk3 expression in mouse myocardial tissue. In accordance with our expectations, circHipk3 expression was significantly higher in the fetal heart than that in the adult heart (Figure 1J). Additionally, fluorescence ISH (FISH) and qRT-PCR assays showed that circHipk3 was enriched in the cytoplasm of 7-day-old (P7) CMs (Figures 1K and 1L).
Figure 1.

circHipk3 Is Highly Expressed in the Neonatal Heart
(A) Conserved circRNAs were differentially expressed in the adult and neonatal rodent heart. The expression level of circRNA is shown in RPM (reads per million mapped reads). Dashed lines indicate the interval of 2-fold change, n = 3. (B) circHipk3 expression in the heart of P1, P7, and adult mice; ∗p < 0.05 versus adult mice, n = 6. (C) circHipk3 expression in CMs isolated from P1, P7, and adult mice; ∗p < 0.05 versus adult mice, n = 6. (D) circHipk3 expression in different tissues of P1 mice; ∗p < 0.05 versus heart, n = 6. (E) circHipk3 expression in different cell types within the P1 neonatal heart; ∗p < 0.05 versus CM, n = 6. CM, cardiomyocyte; EC, endothelial cell; FC, fibrocyte. (F) circHipk3 expression in different cell types within the adult heart; ∗p < 0.05 versus CM. (G) circHipk3 and Hipk3 mRNA expression levels in P0 CMs after RNase R treatment; ∗p < 0.05 versus RNase− treatment, n = 6. (H) circHipk3 and Hipk3 mRNA expression levels in P0 CMs after actinomycin D treatment; ∗p < 0.05 versus Hipk3 mRNA at each time point, n = 5. (I) The effect of different siRNAs on circHipk3 expression; ∗p < 0.05 versus control treatment, #p < 0.05 versus si-Hipk3 treatment, n = 6. (J) Detection of circHipk3 abundance in the mouse hearts by ISH; ∗p < 0.05, n = 6. (K and L) qRT-PCR (K) and RNA-FISH assays (L) for circHipk3 distribution in the cytoplasm and nucleus of P7 CMs. (M) ChIP analysis for H3K27ac modifications and Gata4 occupancy at the circHipk3 promoter, which was defined as −1,000b to +1,000b relative to the transcription start site of the Hipk3 gene. The y axis represents the intensity of ChIP-seq reads. (N) EMSA assays detecting the direct binding of Gata4 to the circHipk3 promoter. (O) The effect of Gata4 knockdown on circHipk3 and linearHipk3 expression in P0 CMs; ∗p < 0.05, n = 6.
Next, we explored the upstream mechanism regulating circHipk3 expression in CMs. Histone modification is known to be associated with chromatin-dependent processes, including transcription. Previous studies revealed that H3K27ac is one of histone modifications that is necessary to predict gene expression accurately.21,22 In accordance with the expression profile, chromatin immunoprecipitation sequencing (ChIP-seq) showed that the levels of H3K27ac at the circHipk3 promoter were markedly reduced in the adult heart compared to the embryonic heart (Figure 1M). Previous studies have reported that circRNA expression is regulated by the binding of transcription factors to their promoters.19,23 Thus, we utilized ChIP-seq to map the genome-wide occupancy of Gata4, a transcription factor responsible for CM proliferation and cardiac regeneration. Interestingly, we found a notable peak overlapping with the circHipk3 promoter, indicating the important role of Gata4 in regulating circHipk3 expression (Figure 1M). Furthermore, electrophoretic mobility shift assays (EMSAs) confirmed the direct interaction between Gata4 and the circHipk3 promoter (Figure 1N). qRT-PCR assays further confirmed the Gata4-mediated regulation of circHipk3 expression (Figure 1O). We also explored the effect of Gata4 knockdown linearHipk3 mRNA expression, which shared the same promoter with circular transcript. It was observed that Gata4 knockdown had no significant effect on linearHipk3 mRNA expression (Figure 1O). Our results demonstrated the regulatory role of Gata4 on circHipk3 expression.
circHipk3 Overexpression Promotes CM Proliferation In Vitro
We assessed the effect of circHipk3 downregulation on P0 CM proliferation using short hairpin RNA (shRNA) targeting the backsplice of circHipk3 and showed that circHipk3 knockdown significantly decreased the percentage of P0 CMs expressing EdU, Ki67, and pH3 (Figures 2A–2C). To determine the function of circHipk3 in CM proliferation, we then increased circHipk3 expression in P7 CMs by transfection with adenovirus (ADV) carrying vectors expressing circHipk3 (ADV-circHipk3). As expected, circHipk3 overexpression significantly increased the ratio of P7 CMs expressing EdU, Ki67, pH3, and Aurora B (Figures 2D–2G). Moreover, we found that P7 CMs underwent cell division after circHipk3 overexpression, whereas almost none of these cells divided after control treatment (Figure 2H, Video S1). In addition, flow cytometry assays further showed that circHipk3 overexpression increased the percentage of P7 CMs accumulating in the S and G2/M phase and reduced the percentage in G0/G1 (Figure 2I). To explore the role of circHipk3 on CM apoptosis, we detected a TUNEL+ signal in ADV-circHipk3- and ADV-vector-transfected CM after H2O2 treatment. Expectedly, our results showed that circHipk3 overexpression led to the decreased number of TUNEL+ CM after H2O2 treatment (Figure S3), suggesting that circHipk3 overexpression could repress CM apoptosis.
Figure 2.

circHipk3 Regulates CM Proliferation In Vitro
(A) Detection of Ki67+ P0 CMs after transfection with si-circHipk3 or si-NC. Ki67+ CMs are indicated by arrows; n = 500 CMs, ∗p < 0.05, scale bars, 50 μm. (B) Detection of EdU+ P0 CMs after transfection with si-circHipk3 or si-NC. EdU+ CMs are indicated by arrows; n = 500 CMs, ∗p < 0.05, scale bars, 50 μm. (C) Detection of pH3+ P0 CMs after transfection with si-circHipk3 or si-NC. pH3+ CMs are indicated by arrows; n = 500 CMs, ∗p < 0.05, scale bars, 50 μm. (D) Detection of Aurora B+ P7 CMs after ADV-circHipk3 or ADV-vector infection; n = 500 CMs, ∗p < 0.05, scale bars, 50 μm. (E) Detection of Ki67+ P7 CMs after ADV-circHipk3 or ADV-vector infection. Ki67+ CMs are indicated by arrows; n = 500 CMs, ∗p < 0.05 versus, scale bars, 50 μm. (F) Detection of EdU+ P7 CMs after ADV-circHipk3 or ADV-vector infection. EdU+ CMs are indicated by arrows; n = 500 CMs, ∗p < 0.05, scale bars, 50 μm. (G) Detection of pH3+ P7 CMs after ADV-circHipk3 or ADV-vector infection. pH3+ CMs are indicated by arrows; n = 500 CMs, ∗p < 0.05, scale bars, 50 μm. (H) Quantification and representative images of 24 h time-lapse videos of P7 CMs transfected with ADV-circHipk3. ∗p < 0.05 versus; n = 250 CMs, scale bars, 50 μm. The arrows indicate the dividing CM. (I) Flow cytometry analysis of P7 CMs 48 h after ADV-circHipk3 or ADV-vector transfection; ∗p < 0.05 versus vector group in each stage.
circHipk3 Maintains Coronary Artery Endothelial Cell (CAEC) Function
Next, we assessed the role of circHipk3 in CAECs using human CAECs (HCAECs). We used shRNAs targeting the backsplice of circHipk3 to decrease circHipk3 expression without affecting the linear transcript in HCAECs. HCAEC proliferation, as evidenced using EdU incorporation assays, increased upon circHipk3 overexpression but decreased after circHipk3 knockdown (Figures S4A and S4B). Matrigel tube formation and cell scratch assays showed that HCAEC tube formation and migration were promoted by circHipk3 overexpression (Figures S4C and S4E) but inhibited by circHipk3 knockdown (Figures S4D and S4F). Additionally, coculture of CMs and HCAECs was performed to explore whether circHipk3 will be transferred from CMs to cardiac ECs. We found that coculture of HCAECs and ADV-circHipk3-transfected CMs has no effect on the HCAEC proliferation, suggesting there might be no transfer of circHipk3 between mouse CMs and ECs (Figures S5A–S5C). Moreover, we found that when cocultured with mouse, CM/EC has no effect on the circHipk3 expression level of EC/CM, respectively (Figures S6A and S6B). We further extracted exosome, an important secretion factor of cell-to-cell communication, from the supernatant of CM and EC. The PCR assays showed that circHipk3 was hardly expressed in exosome isolated from CM or EC (Figure S6C). Additional cell functional studies showed that coculture with the exosome from circHipk3 overexpressing CM/EC had no effect on EC/CM proliferation, respectively (Figures S6D and S6E). These results further indicated that circHipk3 may not be involved in CM/EC crosstalk by modulating the secretion factors.
To establish the specificity of the function of circHipk3, we investigated the association between EC and CM proliferation and the other three circRNAs, which are also highly expressed in neonatal hearts (Figure S7A). We used siRNAs targeting the backsplicing site of these circRNAs to decrease their expression specifically in HCAECs and CMs (Figure S7B). We found that silencing the circRNA expression has no significant effect on HCAEC or CM proliferation (Figures S7C and S7D), suggesting the specificity of circHipk3 function.
To exclude the effect from Hipk3 mRNA, we detected Hipk3 mRNA expression after circHipk3 knockdown using shRNAs targeting the circHipk3 backsplicing site. Expectedly, we found that circHipk3 knockdown has no effect on Hipk3 mRNA expression in CMs or cardiac ECs (Figure S8A). Moreover, we found that only Hipk3 mRNA knockdown did not significantly increase the percent of proliferative CM or cardiac EC compared to the control (Figures S8B and S8C). These results suggested that the Hipk3 linear transcript was not associated with CM or cardiac EC proliferation. Furthermore, we constructed the vector to overexpress the circHipk3 sequence without circularization and investigated its effect on CM and EC proliferation (Figures S9A and S9B). It was found that the overexpressed circHipk3 sequence without circularization did not significantly increase the rate of CM or EC proliferation, demonstrating that the circularized form is essential for circHipk3 (Figures S9C and S9D).
circHipk3 Overexpression Induces Cardiac Regeneration in Adult Mice
To determine the function of circHipk3 overexpression in vivo, adeno-associated virus 9 (AAV9)-circHipk3 was delivered to adult mice, and proliferating CMs were detected. Green fluorescent protein (GFP)-based assays and qRT-PCR assays indicated effective vector transduction (Figures S10A and 10B). Consistent with the in vitro findings, circHipk3 overexpression significantly increased the ratio of CMs expressing Ki67, pH3, and Aurora B (Figures 3A–3C). Moreover, we performed ex vivo studies by isolating CMs from adult mice to further determine the effect of circHipk3 on CM proliferation. The ratio of Ki67+ CMs was significantly higher in the AAV9-circHipk3 group than in the AAV9-vector group (Figure 3D). Wheat germ agglutinin (WGA) staining showed that circHipk3 overexpression had no effect on CM hypertrophy (Figure 3E).
Figure 3.

circHipk3 Overexpression Promotes CM Proliferation In Vivo
(A) Detection of Ki67+ adult CMs, 2 weeks after AAV9-circHipk3 or AAV9-vector injection. Ki67+ CMs are indicated by arrows; n = 4,000 CMs, ∗p < 0.05, scale bars, 50 μm. (B) Detection of pH3+ adult CMs, 2 weeks after AAV9-circHipk3 or AAV9-vector injection. pH3+ CMs are indicated by arrows; n = 4,000 CMs, ∗p < 0.05, scale bars, 50 μm. (C) Detection of Aurora B+ adult CMs, 2 weeks after AAV9-circHipk3 or AAV9-vector injection. Aurora B+ CMs are indicated by arrows; n = 3,200 CMs, ∗p < 0.05, scale bars, 50 μm. (D) Detection of Ki67+ ex vivo CMs isolated from adult mouse hearts, 2 weeks after AAV9-circHipk3 or AAV9-vector injection; n = 300 CMs, ∗p < 0.05, scale bars, 50 μm. (E) WGA staining of the adult mouse heart, 2 weeks after AAV9-circHipk3 or AAV9-vector injection; n = 400 CMs, scale bars, 50 μm. (F) Detection of Ki67+ adult CMs, 2 weeks after MI. Ki67+ CMs are indicated by arrows; n = 4,000 CMs, ∗p < 0.05, scale bars, 50 μm. (G) Detection of pH3+ adult CMs, 2 weeks after MI. pH3+ CMs are indicated by arrows; n = 4,000 CMs, ∗p < 0.05, scale bars, 50 μm. (H) Detection of TUNEL+ adult CMs, 2 weeks after MI. TUNEL+ CMs are indicated by arrows; n = 4,500 CMs, ∗p < 0.05, scale bars, 50 μm.
Furthermore, using an MI mouse model, we investigated whether circHipk3 overexpression might be a therapeutic strategy for MI by inducing regenerative repair. We injected AAV9-circHipk3 and AAV9-vector into the peri-infarcted area of mice with MI. The results showed that compared to control, circHipk3 overexpression significantly increased the population of Ki67+ and pH3+ CMs and decreased that of TUNEL+ CMs in the peri-infarcted area at 14 days post MI (Figures 3F–3H).
We also measured effective vector transduction for EC in mouse hearts after injection of AAV9-circHipk3-GFP by immunofluorescence assay. It was found that GFP could be expressed in cardiac ECs (Figure S10C). We also isolated ECs from hearts, 4 weeks after injection of AAV9-circHipk3-GFP. By detection of GFP signals, we found that the transduction efficiency of ECs for AAV9-circHipk3-GFP reached about 70% (Figure S10D). Moreover, qRT-PCR assays revealed that injection of AAV9-circHipk3 significantly increased circHipk3 expression in cardiac ECs compared to the controls (Figure S10E). We subsequently investigated the effect of circHipk3 on angiogenesis post-MI. Antibodies against von Willebrand factor (vWF) and integrin β2 (IB4) were used to label vessels in the peri-infarcted and infarcted zone. As expected, the vessel density was markedly higher in the infarcted and border zone in the AAV9-circHipk3 group than in the AAV9-vector group at 14 days post-MI in adult mice (Figures 4A–4D and S11). Moreover, we used alpha-smooth muscle actin (α-SMA) and cluster of differentiation 31 (CD31) double-staining to detect arterioles. Consistently, we found that circHipk3 overexpression significantly increased arteriole density in the infarcted and border zone (Figures 4E and 4F).
Figure 4.

circHipk3 Overexpression Promotes Angiogenesis in Adult Mice after MI
(A) vWF staining in the peri-infarcted zone in the shcircHipk3 and shNC groups, 14 days after MI, ∗p < 0.05, n = 6, scale bars, 50 μm. (B) vWF staining in the infarcted zone in the shcircHipk3 and shNC groups, 14 days after MI, ∗p < 0.05, n = 6, scale bars, 50 μm. (C) IB4 staining in the peri-infarcted zone in the shcircHipk3 and shNC groups, 14 days after MI, ∗p < 0.05, n = 6, scale bars, 50 μm. (D) IB4 staining in the infarcted zone in the shcircHipk3 and shNC groups, 14 days after MI, ∗p < 0.05, n = 6, scale bars, 50 μm. (E) α-SMA and CD31 costaining in the peri-infarcted zone in the shcircHipk3 and shNC groups, 14 days after MI, ∗p < 0.05, n = 6, scale bars, 50 μm. (F) α-SMA and CD31 costaining in the infarcted zone in the shcircHipk3 and shNC groups, 14 days after MI, ∗p < 0.05, n = 6, scale bars, 50 μm.
Next, we investigated whether circHipk3 overexpression improves cardiac function after MI. Echocardiography showed that the left ventricular ejection fraction (LVEF) and fractional shortening (FS) values were significantly higher in mice injected with AAV9-circHipk3 than in mice injected with AAV9-vector after MI (Figure 5A). Additionally, positron emission tomography (PET)/computed tomography (CT) was used to detect myocardial perfusion after MI. circHipk3 overexpression markedly increased the myocardial perfusion score compared to control (Figure 5B). Moreover, Masson trichrome and triphenyltetrazolium chloride (TTC) staining showed that circHipk3 overexpression after MI significantly decreased the scar size compared to control (Figures 5C and 5D). Additionally, we investigated the effect of circHipk3 on fibrocyte (FC) in vitro. The result of Ki67 immunostaining showed that circHipk3 overexpression had no significant effect on FC proliferation in vitro (Figure S12).
Figure 5.

circHipk3 Overexpression Improves Cardiac Function after MI
(A) Cardiac function measured by echocardiography after MI. Time interval, 33 ms. Quantitative analysis of left ventricular ejection fraction (LVEF) and fractional shortening (FS). ∗p < 0.05 versus sham group, δp < 0.05 versus vector group, n = 8. (B) PET-CT of the mouse heart at 2 and 4 weeks after MI. ∗p < 0.05 versus sham group, δp < 0.05 versus vector group, n = 6, scale bars, 0.8 mm. (C) Masson trichrome staining of mouse heart cross-sections at 2 and 4 weeks after MI. ∗p < 0.05 versus vector group, n = 8, scale bars, 1 mm. (D) TTC staining of mouse heart cross-sections at 2 and 4 weeks after MI. ∗p < 0.05 versus vector group, n = 6, scale bars, 0.8 mm.
circHipk3 Is Required for Neonatal CM Proliferation In Vivo
To investigate whether circHipk3 is essential for endogenous cardiac regeneration, we treated P0 mice with ADV expressing shRNA targeting circHipk3 (ADV-shcircHipk3) or negative control (NC) shRNA (ADV-shNC). qRT-PCR assays demonstrated that ADV-shcircHipk3 significantly decreased circHipk3 expression in P0 neonatal mouse hearts compared to ADV-shNC (Figure S10F). We found that circHipk3 knockdown significantly reduced the number of CMs expressing Ki67, pH3, and Aurora B in P4 neonatal mouse hearts (Figures 6A, 6B, and 6E). When ADV-shcircHipk3 or ADV-shNC was injected into the infarcted border zone of neonatal MI mice, the ratio of Ki67+ and pH3+ CMs was much lower in the peri-infarcted zone of the ADV-shcircHipk3 group than that of the ADV-shNC group (Figures 6C and 6D). Additionally, circHipk3 downregulation significantly increased the number of TUNEL+ CMs in the peri-infarcted zone (Figure 6F). WGA staining indicated that circHipk3 knockdown had no effect on CM size in P0 neonatal mouse hearts (Figure 6G). We also investigated the effect of circHipk3 downregulation on cardiac function of P0 neonatal mouse hearts after MI. As expected, circHipk3 knockdown significantly reduced the LVEF of P0 neonatal mouse hearts after MI (Figures 6H and 6I). Masson staining showed that circHipk3 knockdown increased the scar area in P0 neonatal mouse hearts after MI (Figures 6J and 6K).
Figure 6.

circHipk3 Knockdown Impairs Regenerative Repair in the Neonatal Mouse Heart after MI
(A) Detection of Ki67+ CMs, 4 days after ADV-shcircHipk3 or ADV-shNC injection. Ki67+ CMs are indicated by arrows; n = 3,600 CMs, ∗p < 0.05, scale bars, 50 μm. (B) Detection of pH3+ CMs, 4 days after ADV-shcircHipk3 or ADV-shNC injection. pH3+ CMs are indicated by arrows; n = 3,600 CMs, ∗p < 0.05, scale bars, 50 μm. (C) Detection of Ki67+ CMs, 1 week after MI. Ki67+ CMs are indicated by arrows; n = 3,000 CMs, ∗p < 0.05, scale bars, 50 μm. (D) Detection of pH3+ CMs, 1 week after MI. pH3+ CMs are indicated by arrows; n = 3,000 CMs, ∗p < 0.05 versus the ADV-shNC group, scale bars, 50 μm. (E) Detection of Aurora B+ CMs, 4 days after ADV-shcircHipk3 or ADV-shNC injection. Aurora B+ CMs are indicated by arrows; n = 3,000 CMs, ∗p < 0.05, scale bars, 50 μm. (F) Detection of TUNEL+ CMs, 1 week after MI. TUNEL+ CMs are indicated by arrows; n = 2,800 CMs, ∗p < 0.05, scale bar, 50 μm. (G) WGA staining of the neonatal mouse heart, 1 week after ADV-shcircHipk3 or ADV-shNC injection; n = 400 CMs, scale bars, 50 μm. (H and I) Cardiac function measured by echocardiography, 1 week post MI (H). Time interval, 33 ms. Quantitative analysis of LVEF (I); n = 8, ∗p < 0.05 versus sham group, δp < 0.05 versus shNC group. (J and K) Masson trichrome staining of the neonatal mouse heart at 1 week after MI (J). Quantitative analysis of fibrosis area (K); n = 6, ∗p < 0.05 versus shNC group, scale bars, 0.2 mm.
The circHipk3-miR-133a-Connective Tissue Growth Factor (CTGF) Axis Regulates HCAEC Function and Angiogenesis after MI
circHipk3 was reported to regulate retinal EC function by miRNA sponging.19 Herein, we investigated whether circHipk3 regulates coronary vessel ECs by acting as a miRNA sponge. The Segal Lab bioinformatic tools predicted that several miRNAs may bind to circHipk3 (Table S3). We investigated the effect of circHipk3 overexpression on miRNAs that have more than one binding site on the circHipk3 sequence and are associated with cardiac protection. It was found that circHipk3 overexpression significantly decreased the expression of miR-133a, suggesting that miR-133a is the target of circHipk3 (Figure 7A). miR-133a, which has been found to be essential for heart development and protection, has two conserved binding sites in circHipk3 (Figure S13A). Next, we constructed pGL3 luciferase reporters harboring the entire circHipk3 sequence (LUC-circHipk3) or a mutant circHipk3 sequence without the miR-133a binding sites (LUC-circHipk3 mutant). miR-133a mimics significantly reduced LUC-circHipk3 activity but did not affect LUC-circHipk3 mutant activity (Figure 7B). Moreover, RNA FISH experiments in HCAECs showed overlap between circHipk3 and miR-133a (Figure 7C). To further determine the interaction between circHipk3 and miR-133a, RNA pulldown assay was performed using the probe targeting circHipk3 backsplice. The following qRT-PCR assay demonstrated the direct interaction between circHipk3 and miR-133a (Figure S13B). Moreover, we also measured miR-133a expression after transfection of vectors overexpressing linearHipk3 in ECs. It was shown that linearHipk3 overexpression had no significant effect on miR-133a expression (Figure S13C). miR-133a has been reported to regulate EC proliferation and migration.19 We found that miR-133a mimic treatment significantly decreased the ratio of EdU+ HCAEC, which could reverse the effect of circHipk3 on HCAEC proliferation (Figure 7H). The results suggest that miR-133a is the main downstream effector of circHipk3 in HCAEC.
Figure 7.

circHipk3 Regulates Coronary Angiogenesis by Sponging miR-133a
(A) Changes in miRNA expression after circHipk3 overexpression in HCAECs. ∗p < 0.05 versus si-NC in each group, n = 4. (B) Luciferase activity in HCAECs transfected with luciferase-circHipk3-wild type (WT) or luciferase-circHipk3-mutant (Mut). ∗p < 0.05 versus mimic-NC treatment, n = 6. (C) RNA-FISH detection of circHipk3 and miR-133a colocalization, scale bars, 50 μm. (D) Luciferase activity in HCAECs transfected with luciferase-CTGF 3′ UTR-WT or luciferase-CTGF 3′ UTR-Mut. ∗p < 0.05 versus psi-CTGF, #p < 0.05 versus psi-CTGF + circHipk3, &p < 0.05 versus psi-CTGF + circHipk3 + miR-133a, n = 4. (E) CTGF protein expression levels in ADV-circHipk3 and ADV-vector group. ∗p < 0.05, n = 5. (F) CTGF protein expression levels in HCAECs after miR-133a mimic or miR-133a inhibitor treatment. ∗p < 0.05 versus mimic-NC, #p < 0.05 versus antagomir (anta)-NC, n = 4. (G) CTGF protein expression levels in HCAECs after circHipk3 and miR-133a interference. ∗p < 0.05 versus ADV-vector + mimic-NC, #p < 0.05 versus ADV-circHipk3 + mimic-NC, &p < 0.05 versus ADV-vector + miR-133a mimics, n = 4. (H) EdU staining in HCAECs after circHipk3 and miR-133a interference. ∗p < 0.05 versus ADV-vector + mimic-NC, #p < 0.05 versus ADV-circHipk3 + mimic-NC, &p < 0.05 versus ADV-vector + miR-133a mimics, n = 4, scale bars, 50 μm. (I) CD34 staining in the peri-infarcted zone of the corresponding groups, 14 days after MI. ∗p < 0.05 versus AAV9-vector, #p < 0.05 versus AAV9-circHipk3, &p < 0.05 versus AAV9-miR-133a, n = 6, scale bars, 50 μm. (J) pH3 staining in the peri-infarcted zone of the corresponding groups, 14 days after MI. ∗p < 0.05 versus AAV9-vector, #p < 0.05 versus AAV9-circHipk3, &p < 0.05 versus AAV9-miR-133a, n = 6, scale bars, 50 μm. (K) Cardiac function measured by echocardiography, 14 days after MI. ∗p < 0.05 versus AAV9-vector, #p < 0.05 versus AAV9-circHipk3, &p < 0.05 versus AAV9-miR-133a, n = 6. Time interval, 33 ms. (L) Masson trichrome staining of the corresponding groups, 14 days after MI. ∗p < 0.05 versus AAV9-vector, #p < 0.05 versus AAV9-circHipk3, &p < 0.05 versus AAV9-miR-133a, n = 6, scale bars, 6 mm.
It was reported that CTGF, a growth factor closely associated with angiogenesis, is the target gene of miR-133.24,25 We found that the miR-133a binding site in the CTGF 3′ UTR is highly conserved across multiple species (Figure S13D). In our study, luciferase assays showed that miR-133a directly bound to the CTGF 3′ UTR in HCAECs (Figure 7D). Furthermore, we found that circHipk3 overexpression could significantly increase CTGF expression (Figure 7E), and CTGF expression was reduced by miR-133a mimics and increased by miR-133a inhibitors (Figure 7F). Moreover, the circHipk3-induced effect on CTGF expression could be abolished by miR-133a mimic transfection (Figure 7G). Thus, CTGF might be the common target of miR-133a in coronary vessel ECs.
To determine the association between circHipk3 and miR-133a, we coinjected AAV9-circHipk3 and AAV9-miR-133a to mouse heart after MI. We found that AAV9-miR-133a effectively diminished circHipk3-induced miR-133a downregulation (Figure S13E). As expected, AAV9-miR-133a significantly reversed circHipk3-induced angiogenesis and CM proliferation (Figures 7I and 7J). More importantly, AAV9-miR-133a diminished the effect of circHipk3 overexpression on fibrosis reduction and cardiac function improvement after MI (Figures 7K and 7L). These results suggested that miR-133a-induced angiogenesis is essential for the function of circHipk3 during cardiac regeneration.
circHipk3 Induces CM Proliferation by Increasing Notch1 Intracellular Domain (N1ICD) Stability
Next, we explored the mechanism by which circHipk3 induces CM proliferation. Unexpectedly, the expression level of miR-133a was not changed in circHipk3-overexpressing CMs, and the miR-133a mimic has no effect on circHipk3-induced CM proliferation (Figure S13F and S13G). The relative low abundance of miR-133a in CMs might result into the irrelevant circHipk3/miR-133a interaction in CMs (Figure S13H). Moreover, RNA immunoprecipitation (RIP) assays showed that circHipk3 did not bind to Ago2, which mediates miRNA-dependent RNA silencing in CMs (data not shown). These results suggest that circHipk3 acts independent of miRNA sponging in CMs.
Because several circRNAs have a protein-binding capacity, we subsequently performed RNA pulldown assays to identify potential circHipk3-binding proteins in CMs. Biotinylated probes against the circHipk3 backsplice sequence were used, and the precipitants were analyzed by mass spectrometry, which led to the identification of an important regulator of CM proliferation, Notch1 (Figures 8A and S14A; Table S4). The RNA-Protein Interaction Prediction (RPISeq) program predicted that circHipk3 and N1ICD are likely to interact (Figure S14B). This interaction between N1ICD and circHipk3 was confirmed by western blotting after RNA pulldown (Figure 8A).
Figure 8.

circHipk3 Regulates CM Proliferation by Promoting Notch1 Stability
(A) Protein immunoprecipitated using the probe targeting the circHipk3 backsplice site. (Bottom) Notch1 intracellular domain (N1ICD) protein detected by western blotting. (B) circHipk3 knockdown decreased the expression of N1ICD, which could be reversed by MG132 treatment. ∗p < 0.05 versus si-NC, n = 4. (C) Western blotting analysis of N1ICD protein levels in P0 CMs at the indicated periods of time after 20 μg/mL cycloheximide treatment. ∗p < 0.05 circHipk3 versus si-circHipk3, #p < 0.05 si-circHipk3 + MG132 versus si-circHipk3, δp < 0.05 si-NC versus si-circHipk3, n = 4. (D) circHipk3 promotes N1ICD nuclear translocation after MG132 or DMSO treatment. ∗p < 0.05, n = 4. (E) Protein immunoprecipitated by an acetylation-specific antibody and detected by western blotting using the anti-N1ICD antibody. (F) Western blotting analysis of Ccnd1 protein levels in P0 CMs after circHipk3 and Notch1 interference. ∗p < 0.05 versus circHipk3 + si-Notch1, n = 4. (G) Detection of EdU+ P7 CMs after circHipk3 and Notch1 interference. ∗p < 0.05 versus circHipk3 + si-Notch1, n = 4, scale bars, 50 μm.
Then, we investigated the effect of the interaction between circHipk3 and N1ICD. We found that N1ICD protein expression was significantly decreased by circHipk3 knockdown, which could be reversed by the MG132 treatment (Figure 8B). However, circHipk3 had no effect on Notch1 mRNA expression (Figure S14C). We next explored whether circHipk3 regulates N1ICD protein expression by affecting its stability and half-life. We found that circHipk3 overexpression increased, whereas circHipk3 knockdown decreased the half-life of N1ICD (Figure 8C). MG132 treatment reversed the effect of circHipk3 knockdown on the N1ICD half-life (Figure 8C). Furthermore, we found that circHipk3 promoted N1ICD nuclear translocation, suggesting that circHipk3 affects the transcriptional activity of N1ICD (Figure 8D). A previous study indicated that N1ICD acetylation is associated with its stability and function during CM proliferation.26 Consistently, we also found that circHipk3 knockdown decreased N1ICD acetylation using immunoprecipitation (IP) assays (Figures 8E and S14D). We further found that Notch1 knockdown blocked circHipk3-induced Ccnd1 overexpression and CM proliferation (Figures 8F and 8G). Collectively, these results indicated that the effect of circHipk3 on CM proliferation might be achieved by modulating N1ICD stability.
In addition, we performed RIP assay to detected the interaction between circHipk3 and N1ICD protein in ECs. We found that circHipk3 was likely not to interact with N1ICD in HCAEC (Figure S14E). Moreover, we detected N1ICD expression after circHipk3 knockdown in HCAECs. Western blotting assays showed that circHipk3 knockdown had no effect on N1ICD expression in HCAECs (Figure S14F). These results suggest that circHipk3 and N1ICD interaction is not relevant in cardiac ECs.
Discussion
In the current study, circHipk3 was identified as a novel regulator of the synergism between cardiomyogenesis and angiogenesis during cardiac regeneration, resulting in reduced scar size and improved cardiac function after MI. Moreover, we found that circHipk3 promoted CM proliferation by increasing Notch1 acetylation and induced endothelial activation by inhibiting miR-133a. The cardiac transcription factor Gata4 bound to the circHipk3 promoter and regulated its expression through an upstream mechanism. Our findings suggest that circHipk3 might be a valuable therapeutic target for inducing cardiac regeneration and improving prognosis after MI.
In this study, we demonstrated that circRNAs control the synergism between cardiomyogenesis and angiogenesis, as circHipk3 promoted both CM proliferation and endothelial activation during cardiac regeneration. Our study confirms a crucial role of circHipk3 in coronary angiogenesis, since circHipk3 promoted HCAEC proliferation, migration, and tube-forming capacity and increased the capillary density in the peri-infarcted area. These results are consistent with those of a previous study that reported that circHipk3 is involved in diabetes-induced retinal microvascular dysfunction.19 In addition to its role in coronary angiogenesis, the important role of circHipk3 in endogenous cardiac regeneration was also shown in this study. We found that circHipk3 overexpression increased the ratio of CMs expressing proliferation-related markers and undergoing cell division both in vitro and in vivo. Our data indicated that circHipk3 also directly controls CM proliferation by regulating cell-cycle re-entry. We further demonstrated that the regulation of both cardiomyogenesis and angiogenesis by circHipk3 is valuable for facilitating cardiac repair after MI. In our study, miR-133a overexpression acted as an angiogenesis inhibitor and decreased the positive effects of circHipk3 in hearts with MI, indicating that circHipk3-induced revascularization is required and essential for the induction of CM proliferation. Moreover, the circHipk3-induced improvement in cardiac function post-MI suggests that modulating the synergism between CM proliferation and coronary angiogenesis by targeting circHipk3 might have a therapeutic benefit. In addition, the RNase digestion experiments revealed that circHipk3 is more stable than its corresponding linear transcripts, suggesting that circHipk3 might have a longer duration of function. We also found that circHipk3 is highly expressed in neonatal mouse hearts. These excellent characteristics of circHipk3 make it a potentially ideal candidate for improving prognosis post-MI.
circHipk3 has been shown in previous studies to exert its function by sponging relevant miRNAs.17,19 Herein, we extended knowledge of the mechanism of action of circHipk3 by showing that circHipk3 also functions by regulating protein modification; specifically, circHipk3 promoted Notch1 acetylation and stability in CMs. The bioinformatic tools showed that circHipk3 was enriched for miRNA seed sequences. However, the expression levels of miRNAs with the most predicted circHipk3 binding sites were not altered in circHipk3-overexpressing CMs, indicating that circHipk3 acts independently of miRNA sponging. Next, we performed RNA affinity purification assays and identified Notch1 as an RNA-binding protein that binds circHipk3 and potentially participates in cardiac regeneration. Notch1 signaling plays an essential role in maintaining CM proliferation and differentiation and chamber formation during embryonic heart development.26, 27, 28 Our study showed that the interaction between circHipk3 and N1ICD increased the acetylation and protein stability of the N1ICD. Consistent with our results, previous studies showed that N1ICD is acetylated by (Sirt1) in CMs and thus, has increased stability, and these processes are involved in CM proliferation and cardiac regeneration.26 We speculate that the interaction of N1ICD with circHipk3 might change the conformation of N1ICD and thus, prevent exposure of its concealed acetylation sites to Sirt1. Moreover, Notch1 expression was found to be associated with Ccnd1s, further indicating its role in regulating CM cell-cycle progression. Therefore, Notch1 might be downstream of circHipk3 and involved in the regulation of CM proliferation.
This study also showed that circHipk3 expression was regulated by the transcription factor Gata4 in CMs. circRNA expression is controlled by interactions between transcription factors and promoters. With the use of EMSAs and ChIP-seq assays, we confirmed that Gata4 bound to specific sites in the circHipk3 promoter and positively regulated circHipk3 expression levels. Previous studies have demonstrated that Gata4 is a core transcription factor in the cardiac regulatory network.29 Recently, Gata4 was found to participate in embryonic CM proliferation and activation of the endogenous cardiac-regenerative response after injury.30,31 In this study, we further revealed that circHipk3 downregulation could repress the effect of Gata4, suggesting that circHipk3 might be an important downstream effector of Gata4 during CM proliferation. Elucidation of the Gata4/circHipk3/Notch1 regulatory axis provides novel insight into CM proliferation and endogenous cardiac regeneration.
Cell-to-cell communication is believed to be critical on the synergism between cardiomyogenesis and angiogenesis.32 In our study, we also investigated the role of circHipk3 on regulating CM/EC crosstalk. Unexpectedly, the CM/EC coculture system failed to find out the role of circHipk3 on cell-to-cell communication between CM and EC in vitro. The study on exosome further indicated that circHipk3 may not be involved in CM/EC crosstalk by modulating the secretion factors. Actually, there are multiple means by which cells communicate to each other.33 Herein, we prostrated that circHipk3 expressed in EC regulated CM proliferation in vivo by inducing revascularization and then providing nutrient and oxygen to regenerative tissue. circHipk3 might also be involved in CM/EC communication by additional means in vivo, either by direct cell interactions through membrane receptors and ligands or by releasing soluble molecules, such as growth factors, cytokines, and chemokines.33 These ways of CM/EC communication should be explored by future studies.
There are some limitations of the current study. First, our study demonstrated that circHipk3 overexpression triggered both CM proliferation and angiogenesis during cardiac regeneration and that circHipk3 expression in CMs was regulated by binding of the transcription factor Gata4 to the promoter. To better understand the roles of circHipk3 in ECs, the upstreaming mechanisms regulating circHipk3 expression in ECs need to be defined in the future. Second, we confirmed that circHipk3 was involved in CM proliferation and endothelial activation both in vivo and in vitro, and the related mechanisms were also investigated in vitro. CM- and EC-specific knockout mice might be helpful in dissecting the underlying molecular mechanisms by which circHipk3 overexpression reduces infarct size and improves survival rates.
In conclusion, circHipk3, which is regulated by the transcription factor Gata4, triggers CM proliferation and angiogenesis by activating Notch1 signaling and inhibiting miR-133a activity, respectively. Overexpression of circHipk3 induced cardiac regeneration and angiogenesis and reduced the infarct size after MI, which significantly restored cardiac function. These findings suggest that circHipk3 might be an effective therapeutic target for preventing heart failure after MI.
Materials and Methods
MI Model Establishment
C57BL/6 mice were obtained from Guangdong Medical Laboratory Animal Center. All animal experiments were approved by the Animal Research Committee of Southern Medical University and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Briefly, adult male mice (56–70 days old) were anesthetized by an intraperitoneal injection of 3% pentobarbital sodium (40 mg/kg). The left anterior descending (LAD) coronary artery was ligated with a silk suture, 1 mm from the ascending aorta. In sham-operated animals, an analogous surgical operation was performed without occlusion of the LAD. All mice were sacrificed by cervical dislocation at the indicated time points.
Cardiac function was detected by transthoracic echocardiography using a Vevo 2100 high-resolution imaging system equipped with a 30-MHz transducer (RMV-707B; VisualSonics, Toronto, ON, Canada).
Myocardial perfusion was detected by PET-CT using a NanoScan PET/CT system. Fluorodeoxyglucose (18F-FDG; 15 MBq) in 100 mL 0.9% saline was injected intravenously, 1 h before each scan. List-mode data were detected for 15 min and subsequently reconstructed into a single image with a volume of 110 × 60 × 60 mm3 and a voxel size of 0.4 × 0.4 × 0.4 mm3.
Cell Isolation and Culture
Neonatal CMs were isolated from P1 and P7 C57BL/6 mice, as previously described.34 Briefly, after the heart was removed from a neonatal mouse, it was split and digested with 0.25% trypsin (Sigma) at 4°C overnight. Then, tissue fragments were incubated in a mixture of type II collagenase (Roche), bovine serum albumin (BSA; Sigma), and phosphate-buffered saline (PBS) at 37°C for 15 min. The cell suspensions were collected after centrifugation and removal of the supernatant and resuspended in DMEM/F12 medium (Life Technologies), supplemented with 10% fetal bovine serum (FBS). Then, the cells were placed for 2 h in cell-culture incubators at 37°C in 5% CO2 and a humidified atmosphere to separate CMs and fibroblasts based on their different adhesion properties. The supernatant containing nonadherent CMs was then collected, and the cells were pelleted, resuspended in the aforementioned culture medium, and plated at the appropriate density. The neonatal CMs prepared using this procedure consistently had a purity of approximately 90%.
Adult mouse CMs were isolated from 10-week-old adult mice using a modified protocol as described.35 Briefly, when the adult mouse was anesthetized, the heart was rapidly excised and mounted on a Langendorff apparatus. Then, the heart was perfused with calcium-free perfusion buffer (113 mM NaCl, 4.7 mM KCl, 0.6 mM KH2PO4, 0.6 mM Na2HPO4, 1.2 mM MgSO4, 10 mM Na-HEPES, 12 mM NaHCO3, 10 mM KHCO3, 0.032 mM phenol red, 30 mM taurine, 10 mM 2,3-butanedione monoxime [BDM], and 5.5 mM glucose) for 5 min, followed by the addition of 50 mL digestion buffer composed of 15,000 U of type II collagenase (Roche) and 50 μM CaCl2 for 10 min. The heart was subsequently cut into small pieces, which were vigorously disrupted with a Pasteur pipette to obtain individual cells. Then, the cells were centrifuged at low speed, and the supernatant was removed. The adult mouse CMs were plated onto culture slides coated with laminin (10 μg/mL−1; Life Technologies; 23017015) and incubated in F12 medium containing 10% FBS.
ECs and FCs of mouse hearts were isolated according to previous studies.36, 37, 38, 39 Briefly, mouse hearts were dissociated into suspension with a heart dissociation MACS kit (Miltenyi Biotec). Then, the suspension was filtered with MACS SmartStrainer (Miltenyi Biotec) and added with anti-CD31 Microbeads (Miltenyi Biotec) for EC or anti-CD90 Microbeads (Miltenyi Biotec) for FC. Afterward, ECs were isolated by separation over a LS MACS Column. Purity of the ECs and FC was tested by immunofluorescence assay using the CD31 antibody and CD90 antibody, respectively.
siRNA, ADV, AAV9, and Vector Transfection
siRNAs and miR-133a mimics and inhibitors were obtained from RiboBio (Guangzhou, China). A scrambled siRNA was used as a NC. Transfections were performed with the LipoFilter kit (MicroAire) according to the manufacturer’s instructions. siRNAs and miR-133a mimics and inhibitors were transfected at a final concentration of 50 nM. Subsequent experiments were performed 48 h after transfection.
The ADV and AAV9 vectors harboring GFP for circHipk3 depletion or overexpression, linearHipk3 overexpression, and miR-133a overexpression were synthesized by Vigene (Shandong, China). Neonatal CMs or ECs were transfected with ADV-circHipk3 for 48 h in order to overexpress circHipk3. The multiplicity of infection (MOI) ranged from 10 to 20. qRT-PCR assays were used to determine that circHipk3 expression was successfully upregulated.
The animal studies were conducted as previously described.35,40 Briefly, adult mice were intramyocardially injected at 3–5 sites with AAV9 vectors expressing circHipk3 or empty vectors (AAV9-circHipk3 or AAV9-vector) at a dose of 1,011 viral genome particles per mouse (approximately 30 μL) using an insulin syringe with a 30-gauge needle. For the infarcted mice, AAV9 was injected into myocardial tissue close to the infarct zone immediately after the left anterior descending coronary artery ligation. Myocardial tissues were collected 28 days after injection. In P0 neonatal mice, ADV vectors expressing shRNA targeting circHipk3 or empty vectors (ADV-shcircHipk3 or ADV-NC) were injected into 3 sites in the left ventricle at a dose of 5 × 109 viral genome particles per animal (approximately 5 μL) by using an insulin syringe with a 30-gauge needle. The ADV was also injected in peri-infarcted myocardium immediately after LAD ligation. The hearts of the injected mice were isolated 1 week after ADV injection.
Ad-GFP was used to investigate the distribution of ADV or AAV9 vector in the mouse heart. A Bruker In Vivo FX Pro system (Billerica, MA, USA) was used to detect GFP in the mouse heart. Quantitative real-time PCR assays were used to examine circHipk3 expression after transfection.
Immunofluorescence Analysis
Myocardial tissues were placed a in tissue-freezing medium (Sakura Finetek USA) and frozen at −20°C. After fixation in 4% polyformaldehyde for 30 min, tissue sections and cultured cells were permeabilized with 1% Triton X-100 in PBS for 5 min and then incubated in 3% BSA for 1 h at room temperature. The samples were subsequently incubated at 4°C overnight with the following primary antibodies diluted in 3% BSA: anti-Ki67 (1:200, 275R; Cell Marque), anti-pH3 (ab170904; Abcam), anti-cardiac troponin T (cTnT; ab33589; Abcam), anti-Aurora B (ab2254; Abcam), anti-α-SMA (ab7817; Abcam), anti-vWF (ab11713; Abcam), and anti-IB4 (ab181548; Abcam). A Click-it EdU Imaging Kit (Life Technologies; #C10638) was used to detect EdU incorporation according to the manufacturer’s instruction. An In Situ Cell Death Detection Kit (Roche, Shanghai, China) was used to detect apoptotic cells in samples. After three washes with PBS, the samples were stained with fluorescent secondary antibodies (Alexa Fluor 488 and/or 594; Abcam) for 1 h at room temperature, followed by 20 min of 4′,6-diamidino-2-phenylindole (DAPI) staining (ab104139; Abcam). Images were captured using confocal laser-scanning microscopy (Carl Zeiss).
Histological Examinations
Myocardial tissues were fixed with 4% polyformaldehyde for 24 h, embedded in wax, and sectioned. The sections were subsequently deparaffinized through a graded alcohol series, boiled in a pressure cooker, cooled for 3 h, and processed as indicated.
To evaluate capillary density, specimens were blocked with 4% goat serum and incubated with anti-vWF antibodies (ab11713; Abcam). The number of capillaries per unit area in imaged fields at 200× magnification was determined using an Olympus BX51 microscope.
Masson trichrome (MST 8004; MST Biotechnology) staining was used to detect the fibrotic area according to the manufacturer’s instructions. The area of fibrosis in the infarcted heart was determined by detecting collagen deposition (blue) using ImageJ software.
The expression levels of circHipk3 in human and mouse heart tissue were detected by ISH, as described previously.41 Sections were incubated in 3% pepsin, diluted in fresh citrate buffer at 37°C for 30 min, and then prehybridized with prehybridization solution for 2 h at 37°C. Subsequently, sections were incubated with digoxigenin (DIG)-labeled RNA probes (Biosense Bioscience, Guangzhou, China) overnight at 37°C. After blocking with 3% BSA for 30 min at 37°C, the sections were incubated with alkaline phosphatase-conjugated sheep anti-DIG Fab fragments for 1 h at room temperature.
Real-Time PCR
TRIzol reagent (R6830-01, E.Z.N.A.; Omega Bio-Tek) was used to extract total RNA from cell or tissue lysates. The nuclear and cytoplasmic fractions were isolated by using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific) following the manufacturer’s instructions. Reverse transcription was performed by using PrimeScript RT Master Mix (Takara, Dalian, China), and quantitative PCR (qPCR) was performed by using SYBR Green PCR Master Mix (Takara, Dalian, China) in a LightCycler 480 System (Roche, Germany). The primers are shown in Table S1.
RNase R Treatment
RNase R treatment was performed according to a previously described protocol. Briefly, 2 μg total RNA was treated with or without 3 U μg−1 RNase R (Sigma) at 37°C for 30 min. The treated RNA was subsequently purified with an RNeasy MinElute Cleanup Kit (QIAGEN).
Exosome Isolation
Exosome extraction was performed according to previous studies.42,43 Briefly, 30 mL of cell-culture supernatant was centrifuged at 1,000 × g for 5 min to remove cell pellets. The supernatant was transferred to a sterile vessel, centrifuged at 3,000 × g for 15 min and then filtered using a 0.20-μm syringe filter to remove remaining cells and cell debris. The resultant supernatant was ultracentrifuged at 200,000 × g for 90 min at 4 °C (SW 41 Ti Rotor). The ultracentrifuged supernatant was transferred to a fresh tube, and the pellet was suspended using PBS. This eluate containing exosomes was stored at 4°C overnight or used immediately for experiments.
Western Blot Analysis
Total protein from cells or tissue lysates was prepared using radioimmunoprecipitation assay (RIPA) lysis buffer (boster biological technology [BestBio]) containing Protease Inhibitor Cocktail Set I (BestBio). Proteins were separated in 10% SDS-PAGE gels and transferred to nitrocellulose membranes. The following primary antibodies were used: anti-Ccnd1 (ab16663; Abcam), anti-N1ICD (#4147; Cell Signaling Technology [CST]), and anti-CTGF (ab6992; Abcam). As a control, we used the mouse anti-Gapdh antibody (1:1,000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Alexa Fluor 680-conjugated anti-rabbit immunoglobulin G (IgG; 1:10,000 dilution; Abcam, USA) was used as the secondary antibody. Signals were captured by an Odyssey detection system (LI-COR Biosciences, Lincoln, NE, USA).
RNA-FISH
Cy3-labeled RNA probes targeting the circHipk3 backsplice sequence and miR-133a were purchased from RiboBio (Guangzhou, China) and used in RNA-FISH assays, performed as described previously.44 Briefly, cultured cells were washed with PBS, permeabilized with 0.5% Triton X-100, and incubated with RNA probes in hybridization buffer (RiboBio, Guangzhou, China). Then, cells were incubated with anti-cTnT (ab33589; Abcam), followed by fluorescent secondary antibodies (Alexa Fluor 594; Abcam) and DAPI staining (ab104139; Abcam). Images were captured using confocal laser-scanning microscopy (Carl Zeiss).
RNA Pulldown Assays
RNA pulldown assays were performed as described previously with minor modifications. Briefly, cells were washed in ice-cold PBS, lysed in coimmunoprecipitation buffer, and incubated at room temperature for 4 h with 3 μg biotinylated DNA oligo probes targeting the circHipk3 backsplice sequence. Then, then cells were treated with 50 μL washed streptavidin-coated magnetic beads (SA1004; Invitrogen) at room temperature for another hour. RNase-free BSA and yeast tRNA (Sigma, Shanghai, China) were used to prevent nonspecific binding. RNA bound to beads was isolated by the TRIzol kit, and bound protein was detected by western blotting. The specific bands were extracted and analyzed by mass spectrometry.
TTC Staining
TTC assays were performed as previously described.6 Briefly, the mouse heart was dissected and sectioned using a metal slicer into 3 mm-thick slices. The slices were incubated in 1% TTC (Sigma-Aldrich) in PBS for 15 min at room temperature. The specimens were washed with PBS to stop the staining process and then photographed.
Luciferase Reporter Assay
The circHipk3 sequence and the 3′ UTR of CTGF were cloned into the psiCHECK-2 luciferase vector, which was purchased from Saicheng Bio (Guangzhou, China). All constructs were confirmed by sequencing. For luciferase reporter assays, ADV-circHipk3 or miR-133a mimics were cotransfected into CMs with the indicated luciferase reporters using Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific), 24 h before luciferase reporter transfection. Luciferase activity was detected using the Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA).
EMSAs
EMSAs were performed using an EMSA Kit (Biosense, Guangzhou) according to previous descriptions.41 Labeled and unlabeled wild-type Gata4 probes were mixed for 10 min for competition experiments. The BioSens Gel Imaging System (BIOTOP, China) was used to detect bands. The sequences of the wild-type and mutated probes are 5′-GTAGATTTCTATCTCTCTCTCT-3′ and 5′-GTAGATCGAACGAGACGATTCT-3′, respectively.
Time-Lapse Videos
Primary P7 CMs were isolated and cultured in confocal dishes with culture medium as described above Video S1. Time-lapse videos were acquired at 40× magnification for 24 h at 10-min intervals by using a Leica (TCS Sp8) confocal microscope. Then, immunostaining with an anti-cTnT antibody was used to identify CMs.
CM/HCAEC Coculture
Coculture of CM/HCAEC was performed according to a previous study’s description.45 We used a double-chamber coculture system in which CMs were cultured in the upper chamber, and HCAECs were cultured in the lower chamber.
RIP
RIP experiments were conducted as described previously.35,40 The N1ICD antibody (#4147; CST) was used to precipitate RNA, and then PCR assays were used to present the binding.
Data Availability
circRNA expression profiles in adult and neonatal (P1) rat hearts and conserved sequence identification from datasets were obtained from a previous study.20 The threshold for up- and downregulated circRNAs was fold change ≥ 2 and false discovery rate (FDR) <0.05.
The ChIP-seq data are available through the Gene Expression Omnibus (GEO) using the following accession numbers: E14.5 mouse heart H3K27ac (GEO: GSM1264374), P0 mouse heart H3K27ac (GEO: GSM1264378), P7 mouse heart H3K27ac (GEO: GSM1264380), P56 mouse heart H3K27ac (GEO: GSM1264384), adult heart Gata4 (GEO: GSM1260008), and fetal heart Gata4 (GEO: GSM1260024). ChIP data were analyzed as previously described.46
Statistical Analysis
The results were statistically analyzed with SPSS 18.0 software. All data are presented in graphs as the mean ± SD. For the statistical comparison of 2 groups, an unpaired, 2-tailed Student’s t test was used. One-way ANOVA, followed by the least significant difference post hoc test, was used for the comparison of ≥3 groups. The p value was calculated to determine the significance of each difference, and p <0.05 was considered to indicate statistical significance.
Author Contributions
X.S., H.Z., G.W., S.H., and J.B. designed the study. X.S., H.Z., G.W., C.L., and S.Z. performed the experiments. M.L., Wei Li, H.W., H.G., and J.S. contributed to the acquisition, analysis, and interpretation of data. Wangjun Liao, Y.L., S.H., and J.B. wrote the manuscript. All authors read and approved the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (nos. 81771857, 81571698, 81271640, and 81960047); Guangdong Basic and Applied Basic Research Fund (no. 2019A1515110687); Guangzhou Basic and Applied Basic Research Fund (no. 202002030198); and Outstanding Youths Development Scheme of Nanfang Hospital, Southern Medical University (no. 2019J003).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.06.024.
Contributor Information
Senlin Huang, Email: huangsenlin11@qq.com.
Jianping Bin, Email: jianpingbin@126.com.
Supplemental Information
References
- 1.van Berlo J.H., Molkentin J.D. An emerging consensus on cardiac regeneration. Nat. Med. 2014;20:1386–1393. doi: 10.1038/nm.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tao G., Kahr P.C., Morikawa Y., Zhang M., Rahmani M., Heallen T.R., Li L., Sun Z., Olson E.N., Amendt B.A., Martin J.F. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature. 2016;534:119–123. doi: 10.1038/nature17959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.D’Uva G., Aharonov A., Lauriola M., Kain D., Yahalom-Ronen Y., Carvalho S., Weisinger K., Bassat E., Rajchman D., Yifa O. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat. Cell Biol. 2015;17:627–638. doi: 10.1038/ncb3149. [DOI] [PubMed] [Google Scholar]
- 4.Bassat E., Mutlak Y.E., Genzelinakh A., Shadrin I.Y., Baruch Umansky K., Yifa O., Kain D., Rajchman D., Leach J., Riabov Bassat D. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature. 2017;547:179–184. doi: 10.1038/nature22978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eulalio A., Mano M., Dal Ferro M., Zentilin L., Sinagra G., Zacchigna S., Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376–381. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- 6.Li X., He X., Wang H., Li M., Huang S., Chen G., Jing Y., Wang S., Chen Y., Liao W. Loss of AZIN2 splice variant facilitates endogenous cardiac regeneration. Cardiovasc. Res. 2018;114:1642–1655. doi: 10.1093/cvr/cvy075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X., Sun Y., Huang S., Chen Y., Chen X., Li M., Si X., He X., Zheng H., Zhong L. Inhibition of AZIN2-sv induces neovascularization and improves prognosis after myocardial infarction by blocking ubiquitin-dependent talin1 degradation and activating the Akt pathway. EBioMedicine. 2019;39:69–82. doi: 10.1016/j.ebiom.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karra R., Foglia M.J., Choi W.Y., Belliveau C., DeBenedittis P., Poss K.D. Vegfaa instructs cardiac muscle hyperplasia in adult zebrafish. Proc. Natl. Acad. Sci. USA. 2018;115:8805–8810. doi: 10.1073/pnas.1722594115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Devaux Y., Zangrando J., Schroen B., Creemers E.E., Pedrazzini T., Chang C.P., Dorn G.W., 2nd, Thum T., Heymans S., Cardiolinc network Long noncoding RNAs in cardiac development and ageing. Nat. Rev. Cardiol. 2015;12:415–425. doi: 10.1038/nrcardio.2015.55. [DOI] [PubMed] [Google Scholar]
- 10.Miao Y., Ajami N.E., Huang T.S., Lin F.M., Lou C.H., Wang Y.T., Li S., Kang J., Munkacsi H., Maurya M.R. Enhancer-associated long non-coding RNA LEENE regulates endothelial nitric oxide synthase and endothelial function. Nat. Commun. 2018;9:292. doi: 10.1038/s41467-017-02113-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmitt A.M., Chang H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell. 2016;29:452–463. doi: 10.1016/j.ccell.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li M., Ding W., Sun T., Tariq M.A., Xu T., Li P., Wang J. Biogenesis of circular RNAs and their roles in cardiovascular development and pathology. FEBS J. 2018;285:220–232. doi: 10.1111/febs.14191. [DOI] [PubMed] [Google Scholar]
- 13.Qu S., Yang X., Li X., Wang J., Gao Y., Shang R., Sun W., Dou K., Li H. Circular RNA: A new star of noncoding RNAs. Cancer Lett. 2015;365:141–148. doi: 10.1016/j.canlet.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Xia S., Feng J., Lei L., Hu J., Xia L., Wang J., Xiang Y., Liu L., Zhong S., Han L., He C. Comprehensive characterization of tissue-specific circular RNAs in the human and mouse genomes. Brief. Bioinform. 2017;18:984–992. doi: 10.1093/bib/bbw081. [DOI] [PubMed] [Google Scholar]
- 15.Zheng Q., Bao C., Guo W., Li S., Chen J., Chen B., Luo Y., Lyu D., Li Y., Shi G. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016;7:11215. doi: 10.1038/ncomms11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen B., Yu J., Guo L., Byers M.S., Wang Z., Chen X., Xu H., Nie Q. Circular RNA circHIPK3 Promotes the Proliferation and Differentiation of Chicken Myoblast Cells by Sponging miR-30a-3p. Cells. 2019;8:177. doi: 10.3390/cells8020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen G., Shi Y., Liu M., Sun J. circHIPK3 regulates cell proliferation and migration by sponging miR-124 and regulating AQP3 expression in hepatocellular carcinoma. Cell Death Dis. 2018;9:175. doi: 10.1038/s41419-017-0204-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X., Liu B., Zhou M., Fan F., Yu M., Gao C., Lu Y., Luo Y. Circular RNA HIPK3 regulates human lens epithelial cells proliferation and apoptosis by targeting the miR-193a/CRYAA axis. Biochem. Biophys. Res. Commun. 2018;503:2277–2285. doi: 10.1016/j.bbrc.2018.06.149. [DOI] [PubMed] [Google Scholar]
- 19.Shan K., Liu C., Liu B.H., Chen X., Dong R., Liu X., Zhang Y.Y., Liu B., Zhang S.J., Wang J.J. Circular Noncoding RNA HIPK3 Mediates Retinal Vascular Dysfunction in Diabetes Mellitus. Circulation. 2017;136:1629–1642. doi: 10.1161/CIRCULATIONAHA.117.029004. [DOI] [PubMed] [Google Scholar]
- 20.Werfel S., Nothjunge S., Schwarzmayr T., Strom T.M., Meitinger T., Engelhardt S. Characterization of circular RNAs in human, mouse and rat hearts. J. Mol. Cell. Cardiol. 2016;98:103–107. doi: 10.1016/j.yjmcc.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Karlić R., Chung H.R., Lasserre J., Vlahoviček K., Vingron M. Histone modification levels are predictive for gene expression. Proc. Natl. Acad. Sci. USA. 2010;107:2926–2931. doi: 10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S., Zang C., Xiao T., Fan J., Mei S., Qin Q., Wu Q., Li X., Xu K., He H.H. Modeling cis-regulation with a compendium of genome-wide histone H3K27ac profiles. Genome Res. 2016;26:1417–1429. doi: 10.1101/gr.201574.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Z., Huang S., Cao Y., Yao Y., Li J., Chen J., Jiang B., Yuan X., Xiang X., Xiong J., Deng J. YAP1 inhibits circRNA-000425 expression and thus promotes oncogenic activities of miR-17 and miR-106. Biochem. Biophys. Res. Commun. 2018;503:2370–2375. doi: 10.1016/j.bbrc.2018.06.163. [DOI] [PubMed] [Google Scholar]
- 24.Duan L.J., Qi J., Kong X.J., Huang T., Qian X.Q., Xu D., Liang J.H., Kang J. MiR-133 modulates TGF-β1-induced bladder smooth muscle cell hypertrophic and fibrotic response: implication for a role of microRNA in bladder wall remodeling caused by bladder outlet obstruction. Cell. Signal. 2015;27:215–227. doi: 10.1016/j.cellsig.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 25.Yang L., Hou J., Cui X.H., Suo L.N., Lv Y.W. MiR-133b regulates the expression of CTGF in epithelial-mesenchymal transition of ovarian cancer. Eur. Rev. Med. Pharmacol. Sci. 2017;21:5602–5609. doi: 10.26355/eurrev_201712_14001. [DOI] [PubMed] [Google Scholar]
- 26.Collesi C., Felician G., Secco I., Gutierrez M.I., Martelletti E., Ali H., Zentilin L., Myers M.P., Giacca M. Reversible Notch1 acetylation tunes proliferative signalling in cardiomyocytes. Cardiovasc. Res. 2018;114:103–122. doi: 10.1093/cvr/cvx228. [DOI] [PubMed] [Google Scholar]
- 27.Koenig S.N., Bosse K., Majumdar U., Bonachea E.M., Radtke F., Garg V. Endothelial Notch1 Is Required for Proper Development of the Semilunar Valves and Cardiac Outflow Tract. J. Am. Heart Assoc. 2016;5:e3075. doi: 10.1161/JAHA.115.003075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Metrich M., Bezdek Pomey A., Berthonneche C., Sarre A., Nemir M., Pedrazzini T. Jagged1 intracellular domain-mediated inhibition of Notch1 signalling regulates cardiac homeostasis in the postnatal heart. Cardiovasc. Res. 2015;108:74–86. doi: 10.1093/cvr/cvv209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bisping E., Ikeda S., Kong S.W., Tarnavski O., Bodyak N., McMullen J.R., Rajagopal S., Son J.K., Ma Q., Springer Z. Gata4 is required for maintenance of postnatal cardiac function and protection from pressure overload-induced heart failure. Proc. Natl. Acad. Sci. USA. 2006;103:14471–14476. doi: 10.1073/pnas.0602543103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu W., Huang X., Tian X., Zhang H., He L., Wang Y., Nie Y., Hu S., Lin Z., Zhou B. GATA4 regulates Fgf16 to promote heart repair after injury. Development. 2016;143:936–949. doi: 10.1242/dev.130971. [DOI] [PubMed] [Google Scholar]
- 31.Zhou L., Liu J., Xiang M., Olson P., Guzzetta A., Zhang K., Moskowitz I.P., Xie L. Gata4 potentiates second heart field proliferation and Hedgehog signaling for cardiac septation. Proc. Natl. Acad. Sci. USA. 2017;114:E1422–E1431. doi: 10.1073/pnas.1605137114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Talman V., Kivelä R. Cardiomyocyte—endothelial cell interactions in cardiac remodeling and regeneration. Front. Cardiovasc. Med. 2018;5:101. doi: 10.3389/fcvm.2018.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiodoni C., Di Martino M.T., Zazzeroni F., Caraglia M., Donadelli M., Meschini S., Leonetti C., Scotlandi K. BioMed Central; 2019. Cell Communication and Signaling: How to Turn Bad Language into Positive One. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li B., Hu Y., Li X., Jin G., Chen X., Chen G., Chen Y., Huang S., Liao W., Liao Y. Sirt1 Antisense Long Noncoding RNA Promotes Cardiomyocyte Proliferation by Enhancing the Stability of Sirt1. J. Am. Heart Assoc. 2018;7:e009700. doi: 10.1161/JAHA.118.009700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Y., Li X., Li B., Wang H., Li M., Huang S., Sun Y., Chen G., Si X., Huang C. Long Non-coding RNA ECRAR Triggers Post-natal Myocardial Regeneration by Activating ERK1/2 Signaling. Mol. Ther. 2019;27:29–45. doi: 10.1016/j.ymthe.2018.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song J., Chen X., Wang M., Xing Y., Zheng Z., Hu S. Cardiac endothelial cell-derived exosomes induce specific regulatory B cells. Sci. Rep. 2014;4:7583. doi: 10.1038/srep07583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jelonek K., Walaszczyk A., Gabryś D., Pietrowska M., Kanthou C., Widłak P. Cardiac endothelial cells isolated from mouse heart - a novel model for radiobiology. Acta Biochim. Pol. 2011;58:397–404. [PubMed] [Google Scholar]
- 38.Weber S.C., Gratopp A., Akanbi S., Rheinlaender C., Sallmon H., Barikbin P., Koehne P.S. Isolation and culture of fibroblasts, vascular smooth muscle, and endothelial cells from the fetal rat ductus arteriosus. Pediatr. Res. 2011;70:236–241. doi: 10.1203/PDR.0b013e318225f748. [DOI] [PubMed] [Google Scholar]
- 39.Guo J., Zheng L., Chen L., Luo N., Yang W., Qu X., Liu M., Cheng Z. Lipopolysaccharide activated TLR4/NF-κB signaling pathway of fibroblasts from uterine fibroids. Int. J. Clin. Exp. Pathol. 2015;8:10014–10025. [PMC free article] [PubMed] [Google Scholar]
- 40.Huang S., Li X., Zheng H., Si X., Li B., Wei G., Li C., Chen Y., Chen Y., Liao W. Loss of Super-Enhancer-Regulated circRNA Nfix Induces Cardiac Regeneration After Myocardial Infarction in Adult Mice. Circulation. 2019;139:2857–2876. doi: 10.1161/CIRCULATIONAHA.118.038361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao M., Luo R., Liu Y., Gao L., Fu Z., Fu Q., Luo X., Chen Y., Deng X., Liang Z. miR-3188 regulates nasopharyngeal carcinoma proliferation and chemosensitivity through a FOXO1-modulated positive feedback loop with mTOR-p-PI3K/AKT-c-JUN. Nat. Commun. 2016;7:11309. doi: 10.1038/ncomms11309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.La Shu S., Yang Y., Allen C.L., Maguire O., Minderman H., Sen A., Ciesielski M.J., Collins K.A., Bush P.J., Singh P. Metabolic reprogramming of stromal fibroblasts by melanoma exosome microRNA favours a pre-metastatic microenvironment. Sci. Rep. 2018;8:12905. doi: 10.1038/s41598-018-31323-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Z., Wang C., Li T., Liu Z., Li L. Comparison of ultracentrifugation and density gradient separation methods for isolating Tca8113 human tongue cancer cell line-derived exosomes. Oncol. Lett. 2014;8:1701–1706. doi: 10.3892/ol.2014.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du W.W., Yang W., Chen Y., Wu Z.K., Foster F.S., Yang Z., Li X., Yang B.B. Foxo3 circular RNA promotes cardiac senescence by modulating multiple factors associated with stress and senescence responses. Eur. Heart J. 2017;38:1402–1412. doi: 10.1093/eurheartj/ehw001. [DOI] [PubMed] [Google Scholar]
- 45.Wang X., Huang W., Liu G., Cai W., Millard R.W., Wang Y., Chang J., Peng T., Fan G.C. Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J. Mol. Cell. Cardiol. 2014;74:139–150. doi: 10.1016/j.yjmcc.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
circRNA expression profiles in adult and neonatal (P1) rat hearts and conserved sequence identification from datasets were obtained from a previous study.20 The threshold for up- and downregulated circRNAs was fold change ≥ 2 and false discovery rate (FDR) <0.05.
The ChIP-seq data are available through the Gene Expression Omnibus (GEO) using the following accession numbers: E14.5 mouse heart H3K27ac (GEO: GSM1264374), P0 mouse heart H3K27ac (GEO: GSM1264378), P7 mouse heart H3K27ac (GEO: GSM1264380), P56 mouse heart H3K27ac (GEO: GSM1264384), adult heart Gata4 (GEO: GSM1260008), and fetal heart Gata4 (GEO: GSM1260024). ChIP data were analyzed as previously described.46