

SUMMARY
We address whether combinations with a pan-RAF inhibitor (RAFi) would be effective in KRAS mutant pancreatic ductal adenocarcinoma (PDAC). Chemical library and CRISPR genetic screens identify combinations causing apoptotic anti-tumor activity. The most potent combination, concurrent inhibition of RAF (RAFi) and ERK (ERKi), is highly synergistic at low doses in cell line, organoid, and rat models of PDAC, whereas each inhibitor alone is only cytostatic. Comprehensive mechanistic signaling studies using reverse phase protein array (RPPA) pathway mapping and RNA sequencing (RNA-seq) show that RAFi/ERKi induced insensitivity to loss of negative feedback and system failures including loss of ERK signaling, FOSL1, and MYC; shutdown of the MYC transcriptome; and induction of mesenchymal-to-epithelial transition. We conclude that low-dose vertical inhibition of the RAF-MEK-ERK cascade is an effective therapeutic strategy for KRAS mutant PDAC.
Graphical Abstract
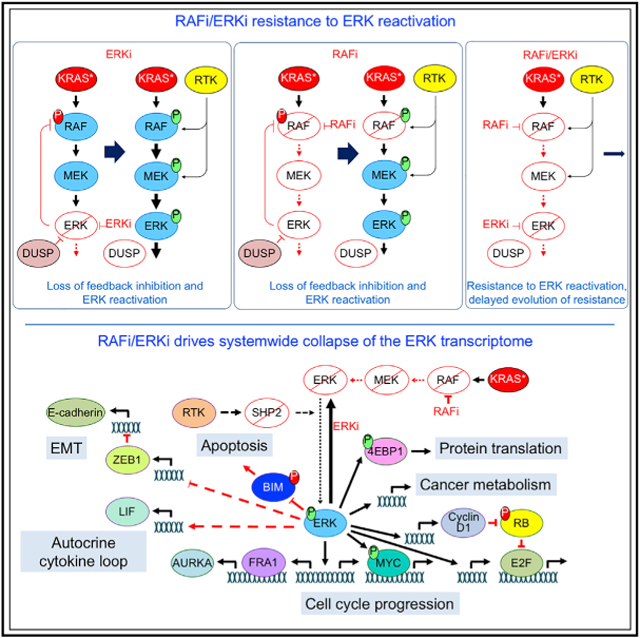
In Brief
Ozkan-Dagliyan et al. apply a chemical screen and identify concurrent inhibition of RAF and ERK as the most potent combination that causes low-dose apoptotic death of KRAS mutant pancreatic cancer. This vertical inhibition combination is resistant to loss of negative feedback mechanisms that reactivate ERK and potently suppresses diverse ERK-dependent processes.
INTRODUCTION
The genetic basis of pancreatic ductal adenocarcinoma (PDAC) is well established (Ryan et al., 2014), yet current standards of care comprise conventional cytotoxic drugs rather than targeted therapies. Among the four major genetic alterations in PDAC, only KRAS functions as an oncogene. Given the >95% KRAS mutation frequency in PDAC and substantial experimental evidence that KRAS is essential for PDAC maintenance (Collins et al., 2012; Ying et al., 2012), KRAS is the most attractive target for therapeutic intervention in this disease (Waters and Der, 2018).
Despite significant recent progress in developing direct inhibitors of mutant KRAS (Janes et al., 2018; Ostrem and Shokat, 2016), with two now under clinical evaluation, these are selective for KRASG12C, a mutant that is found infrequently (only ~2%) in PDAC (Cox et al., 2014). Inhibitors of KRAS effector signaling remain promising KRAS-targeted therapies (Papke and Der, 2017; Ryan and Corcoran, 2018). Of the multitude of effectors, substantial experimental studies and PDAC patient data support the key role of the RAF-MEK-ERK mitogen-activated protein kinase (MAPK) cascade in driving KRAS-dependent PDAC growth. Mutationally activated BRAFV600E can phenocopy mutant KRAS and drive the development of invasive and metastatic PDAC (Collisson et al., 2012), and BRAF mutations are found in ~50% of the rare PDAC that are KRAS wild type (WT) (TCGA, 2017). Further, an effector small interfering RNA (siRNA) screen demonstrated that KRAS-dependent cancers are driven largely by RAF (Yuan et al., 2018). These observations support the RAF-MEK-ERK cascade as the key effector pathway driving KRAS-dependent PDAC. However, to date, therapeutic targeting of MEK in KRAS mutant lung cancer demonstrated limited to no efficacy in patients (Blumenschein et al., 2015; Jänne et al., 2017). Challenges to the effective use of inhibitors of ERK MAPK signaling include toxicity in normal cells (Blasco et al., 2011) and adaptive responses to inhibitor treatment, resulting in ERK reactivation and bypass of inhibitor action (Duncan et al., 2012).
Another challenge in targeting the ERK MAPK cascade is determining which level of the three-tiered kinase cascade will provide the most effective and long-term therapeutic response. At the top of the pathway are the three highly related RAF isoforms—ARAF, BRAF, and CRAF/RAF1—that exhibit distinct roles in RAS-driven cancers (Desideri et al., 2015). BRAF-selective inhibitors caused paradoxical activation of ERK signaling in RAS mutant cancers (Hatzivassiliou et al., 2010; Poulikakos et al., 2010). Pan-RAF inhibitors (RAFis) overcome paradoxical activation and showed greater activity in KRAS mutant cancers (Peng et al., 2015; Yen et al., 2018). However, genetic deletion studies in Kras-driven mouse models argue that pan-RAF inhibition may be limited by normal cell toxicity and that a CRAF-selective strategy may provide a tumor-selective therapy (Blasco et al., 2011; Karreth et al., 2011). In contrast, Craf deficiency in a KrasG12D-driven PDAC model had no inhibitory effect on tumor development or progression (Eser et al., 2013). Thus, the role of specific RAF isoforms in KRAS-driven oncogenesis may be highly tissue selective.
Recently, we addressed the limitations of RAFis and MEK inhibitors (MEKis) by using ERK1/2-selective inhibitors in KRAS mutant PDAC (Hayes et al., 2016). We found that ERK1/2 inhibition suppressed PDAC growth and that sensitivity correlated with ERK inhibition-mediated loss of MYC protein. In search of combinations to enhance the anti-tumor efficacy of inhibitors of the RAF-MEK-ERK cascade, we identified inhibitors that target a spectrum of functionally diverse proteins that synergized with a pan-RAFi to cause apoptotic death. Surprisingly, the strongest synergistic and apoptotic activity resulted from concurrent inhibition of RAF and ERK, which was more effective than concurrent inhibition of either RAF and MEK or MEK and ERK and was particularly striking at low doses. We have delineated a multi-faceted mechanistic basis for the anti-tumor potency of this vertical inhibition of the ERK MAPK cascade.
RESULTS
ARAF, BRAF, and CRAF Contribute to Growth of KRAS Mutant Pancreatic Cancer
Previous studies evaluating the three RAF serine/threonine kinase isoforms suggested distinct tissue-specific dependencies in the development of KRAS-driven cancers, with Craf critical for Kras-driven lung cancer (Blasco et al., 2011; Karreth et al., 2011) but not pancreatic cancer (Eser et al., 2013). We previously determined that a panel of nine conventional KRAS mutant PDAC lines exhibited KRAS-dependent growth (Hayes et al., 2016). To further address the role of RAF isoforms in the growth of KRAS mutant PDAC, we additionally verified the KRAS-dependent growth of four KRAS mutant cell lines established from patient-derived xenograft (PDX) PDAC tumors (Figures 1A, S1A, and S1B). Applying previously characterized small hairpin RNA (shRNA) vectors for selective suppression of ARAF, BRAF, or CRAF (Freeman et al., 2013), we found that suppression of any RAF isoform alone was sufficient to partially impair growth of all six KRAS mutant PDX PDAC cell lines (Figures 1A, S1C, and S1D), demonstrating that each RAF gene contributes to KRAS-dependent PDAC growth, with the general hierarchy of significance CRAF>BRAF>ARAF. This finding is similar to that made by McCormick and colleagues, where concurrent siRNA suppression of all three RAF genes was required to cause an equivalent suppression of growth of KRAS mutant cell lines as seen with KRAS suppression (Yuan et al., 2018).
Figure 1. Concurrent Inhibition of All RAF Isoforms Diminishes PDAC Growth.

(A) PDAC cell lines were infected by lentivirus vectors encoding nonspecific (NS) control or distinct shRNAs targeting ARAF, BRAF, CRAF, or KRAS sequences. Colonies were stained by crystal violet ~10 days after plating. Data are presented as median. All p values shown are in comparison to the vehicle control for the individual graph. Adjusted p values are from Dunnett’s multiple comparison test. Adjusted p values: Pa01C (ARAF-sh1 = 0.6792, ARAF-sh2 = 0.6726, BRAF-sh1 = 0.8883, BRAF-sh2 = 0.0592, CRAF-sh1 = *, 0.0024, CRAF-sh2 = 0.6350), (KRAS-sh = ***, 0.0010). Pa02C (ARAF-sh1 = 0.7153, ARAF-sh2 = 0.0692, BRAF-sh1 = 0.8788, BRAF-sh2 = 0.1990, CRAF-sh1 = 0.0539, CRAF-sh2 = 0.2738), (KRAS-sh = **, 0.0068). Pa14C (ARAF-sh1 = ****, < 0.0001, ARAF-sh2 = ****, < 0.0001, BRAF-sh1 = *, 0.0103, BRAF-sh2 = ****, < 0.0001, CRAF-sh1 = ****, < 0.0001, CRAF-sh2 = ****, < 0.0001, KRAS-sh = ****, < 0.0001). Pa16C (ARAF-sh1 = 0.9995, ARAF-sh2 = 0.7500, BRAF-sh1 = 0.9977, BRAF-sh2 = ****, < 0.0001, CRAF-sh1 = ****, < 0.0001, CRAF-sh2 = ****, < 0.0001, KRAS-sh = ****, < 0.0001). MIA PaCa-2 (ARAF-sh1 = 0.5520, ARAF-sh2 = **, 0.0051, BRAF-sh1 = 0.2316, BRAF-sh2 = **, 0.0076, CRAF-sh1 = **, 0.0086, CRAF-sh2 = **, 0.0098). PANC-1 (ARAF-sh1 = 0.3213, ARAF-sh2 = ****, < 0.0001, BRAF-sh1 = 0.0586, BRAF-sh2 = **, 0.0025, CRAF-sh1 = ****, < 0.0001, CRAF-sh2 = ****, < 0.0001).
(B) PDAC cell lines were treated with RAFi (0.04–10 μM, 72 h). Cell lysates were immunoblotted to determine levels of the indicated proteins. Data are representative of three independent experiments.
(C) PDAC cell lines were treated with RAFi (0.01–2.5 μM, 72 h). Proliferation was measured by Calcein AM cell viability assay. Data are the mean average of three independent experiments. Error bars are shown as ± SEM.
(D) CRISPR screen. PDAC cell lines were infected with the CRISPR library and treated with vehicle control or RAFi (w2, 2 weeks; w4, 4 weeks; a and b indicate replicate samples). The enrichment score indicates either enrichment (red) or depletion (blue) of the indicated genes in cells treated with RAFi relative to vehicle control.
(E) Cell lines were infected by lentivirus vectors encoding NS or two distinct ARAF shRNAs (72 h) and treated with RAFi (0.01–2.5 μM, 120 h). Proliferation was measured by Calcein AM cell viability assay. Data are the mean average of three technical replicates. Error bars are shown as ± SEM. Summary of GI50 values.
This finding suggested that optimal inhibition of RAF in KRAS mutant PDAC will require a pan-RAFi. We utilized the pan-RAFi LY3009120, which displays potent nanomolar inhibition of all three RAF proteins in vitro (Peng et al., 2015). As observed previously (Hatzivassiliou et al., 2010; Poulikakos et al., 2010), the mutant BRAF-selective inhibitor vemurafenib caused dose-dependent paradoxical activation rather than inactivation of ERK in KRAS mutant PDAC cells (Figure S1E). In contrast, treatment with RAFi caused dose-dependent inhibition of ERK, with IC50 values ranging from 0.20 to 2.37 μM (Figures 1B and S1F; Table S1A).
We next determined the effects of RAFi treatment on the growth of a panel of KRAS mutant PDAC cell lines (Figure 1C; Tables S1A, S1C, and S1D). Defining sensitivity as a GI50 of <2.5 μM, a concentration where pERK is suppressed, we observed that four cell lines were sensitive (GI50 0.20–2.41 μM), and five lines were resistant (GI50 > 2.5 μM) after 3 days of treatment. However, after a 5-day treatment, only two lines remained resistant. Interestingly, sensitivity to RAFi did not correspond to sensitivity to either the MEK1/2-selective inhibitor selumetinib or the ERK1/2-selective inhibitor SCH772984 (Hayes et al., 2016) (Table S1A). These distinct sensitivities indicate that inhibition of the ERK MAPK cascade at different levels may not have equivalent consequences, although off-target activities of each inhibitor and other factors (e.g., mode of action, binding affinity) may contribute to these differences.
To identify a genetic basis for sensitivity and resistance to RAFi, we performed a CRISPR screen targeting the druggable genome in the presence of a sublethal (GI30) concentration of RAFi in a sensitive (Pa02C) and a resistant (Pa01C) PDAC line. We identified genes whose loss increased or decreased sensitivity to RAFi (Figures S1G–S1I). Shown in the heatmap are 45 genes where three or more short guide RNAs (sgRNAs) scored in the top 25% of ranked hits on average for each condition; for the majority of these, their loss increased RAFi sensitivity (Figure S1I). Among the 37 genes in that category are some identified previously to regulate sensitivity to MEKis/ERK inhibitors (ERKis) (e.g., HDAC7, PIK3CB, and PDGFRB) (Anderson et al., 2017; Sun et al., 2014). Surprisingly, ARAF was among the top 10% of hits (Figure 1D), with stable suppression of ARAF causing up to a 4-fold shift in GI50 in both RAFi-sensitive and RAFi-resistant PDAC cell lines (Figures 1E and S1J). Suppression of BRAF or CRAF also increased sensitivity to RAFi (Figure S1K). These results may reflect that non-kinase functions are not blocked by an ATP-competitive inhibitor of kinase activity.
Chemical Library Screen Identifies Synergistic ERK MAPK Vertical Inhibition Combinations
To determine if co-treatment with other signaling inhibitors could overcome RAFi de novo resistance, we utilized a 525-compound chemical library comprising approved or clinical candidate oncology inhibitors (Pemovska et al., 2013)(Table S1E) in a panel of 20 KRAS mutant human or mouse PDAC cell lines (Figure S2D). We applied both viability (CellTiter-Glo) and cytotoxicity (CellTox Green) assays to identify drug combinations that enhanced RAFi growth inhibitory or cytotoxic activity, respectively. Combinations were identified as synergistic when the delta drug sensitivity score (DSS) [DSS (drug+RAFi) – DSS (drug alone)] was >5 or antagonistic when deltaDSS was ≤5 Pemovska et al., 2013). Synergy or antagonism identified in two or more cell lines is shown for cytotoxicity and viability assays (Figures 2A and S2A). Reflecting the genetic heterogeneity of PDAC (Waters and Der, 2018), we observed significant cell line variability in drug sensitivity. The screens identified multiple chemically distinct inhibitors of the same functional class of proteins.
Figure 2. Identification of Synergistic Drug Combinations That Enhance ERK MAPK Inhibitor Cytotoxicity.

(A) Cell lines were treated with a 525-inhibitor library with or without RAFi (2 μM, 72 h). Cell death was measured by CellTox Green. Drug sensitivity score (δDSS) was used to quantify inhibitor responses and plotted as red (>additive), blue (<additive) or white (no effect).
(B–D) Pa02C and Pa16C were treated with RAFi (0.01–2.5 μM) alone or in combination with ERKi (0.08–1.25 μM) (B), MEKi (0.25–4 nM) (C), or EGFRi (0.08–1.25 μM) (D) for 120 h. Proliferation was measured by Calcein AM cell viability assay. Representative bliss synergy score heatmap for three independent experiments is shown (left). Red, synergy; green, antagonism; white, no effect. Averaged dose response curves of three independent experiments (with three technical replicates) are shown (right). Error bars are ± SEM. Synergyavg = average bliss synergy score.
We focused on the RAFi combinations identified in the cytotoxic assay (Figure 2A). As expected, we identified multiple inhibitors of PI3K-AKT-mTOR signaling in the viability screen and to a significantly lesser degree in the cytotoxicity screen (Figures 2A and S2A) (Engelman et al., 2008). Multiple inhibitors of EGFR/HER2 receptor tyrosine kinases, HSP90, histone deacetylases, and microtubule organization were also able to cause cell death when combined with RAFi.
Unexpectedly, we found that cytotoxic RAFi combinations included MEKis or ERKis, but not RAFis. Similarly, combinations with the MEKi trametinib or the ERKi SCH772984 were with EGFR/HER2, pan-RAF, and ERK, but not other MEKis (Figure S2B), or with EGFR/HER2 inhibitors, pan-RAFis, and MEKis, but not other ERKis (Figure S2C), respectively. These findings suggest that for a vertical combination to be synthetic lethal, each inhibitor must target a distinct node of the RAF-MEK-ERK pathway (Figure S2E).
We performed Bliss analyses to determine whether the inhibitory activities of each combination were additive or synergistic; Bliss scores greater than 1.0 indicate synergy. Concurrent inhibition of RAF and ERK (designated RAFi/ERKi hereafter) not only caused cytotoxicity in RAFi-sensitive KRAS mutant PDAC lines in a dose-dependent manner, but also sensitized RAFi-resistant cells (Figures 2B and S3A). This combination was highly synergistic, with average synergy scores of 12.5 and 15.5 for Pa02C and Pa16C cell lines, respectively (Table S1F), particularly at lower concentrations of each inhibitor. In ERKi-resistant Pa16C cells, the GI50 for RAFi alone was 839 nM, whereas it was only 5 nM when combined with a low dose of ERKi (80 nM) (Table S1D). Thus, 168-fold less RAFi was able to produce similar efficacy when used as a component of vertical pathway inhibition.
Interestingly, the ERKi screen did not identify BRAF-selective inhibitors (Figure S2C), and BRAF inihibitors (BRAFis) in combination with ERKis resulted in no significant enhancement in activity (Figure S3B; Table S1I). This result supports the requirement to inhibit all RAF isoforms to disrupt KRAS signaling to ERK (Figures 1A, S1C, and S1D).
Consistent with the requirement to target distinct nodes, ERKi did not synergize with the mechanistically distinct ERKi ulixertinib/BVD-523 (Figure S3C; Table S1J). On the other hand, a second pan-RAFi, lifirafenib/BGB-283, also synergized with ERKi (Figure S3D; Tables S1K and S1L). Similarly, RAFi also synergized with another ERKi, LY3214996 (Figure S3E; Tables S1M and S1N). Finally, leveraging additional combinations with RAFi targeting different nodes of the MAPK pathway, we found that RAFi, in combination with either erlotinib (EGFRi) or trametinib (MEKi), also synergistically suppressed growth (Figures 2C and 2D; Tables S1G and S1H). In summary, synergistic cytotoxic growth suppression was seen only when the combinations involve inhibitors of distinct nodes of the pathway.
Since a common mechanism for resistance to MEKis involves reactivation of ERK (Lake et al., 2016; Morrison, 2012; Samatar and Poulikakos, 2014), we speculated that the RAFi/ERKi combination is more effective than the RAFi/MEKi combination, in part due to resistance to ERK reactivation. To address this possibility, we applied MIB/MS (multiplexed kinase inhibitor beads and mass spectrometry) kinome profiling, a kinome-wide unbiased method that has been used to monitor drug-induced compensatory signaling activities (Duncan et al., 2012). The kinase activity/expression changes caused by 72-h RAFi/ERKi or RAFi/MEKi showed near-identical profiles (Figures S3F and S3G). However, RAFi/ERKi but not RAFi/MEKi showed strong suppression of ERK1/2 activities. Immunoblot analyses also showed that the RAFi/ERKi combination caused greater suppression of ERK signaling and phosphorylation of the ERK substrate RSK (Figure S3H). Thus, the RAFi/MEKi combination showed greater resistance to ERK reactivation.
RAFi/ERKi Treatment Suppresses ERK Signaling Despite Loss of Negative Feedback Inhibition
Immunoblot analyses of six PDAC cell lines verified that RAFi/ERKi synergistically reduced pERK levels (Figures 3C, S5C, and S5D) and that RAFi similarly synergized with the chemically distinct ERK1/2-selective inhibitor, LY3214996 (Figure S5E). To identify changes in signaling pathways and the resulting alterations in gene transcription that were quantitatively or qualitatively different upon combination treatment versus each inhibitor alone, we performed reverse phase protein array (RPPA) (Baldelli et al., 2017) pathway activation mapping (Figure 3A) and RNA sequencing (RNA-seq) analyses (Figure 3B). Together, these datasets show how RAFi/ERKi disrupted multiple signaling networks that in turn disrupted multiple transcription factor-driven transcriptomes (Figure S4).
Figure 3. Concurrent RAF and ERKi Inhibition Disrupts ERK-Dependent Signaling and Cellular Processes.

(A) RPPA analyses of PDAC cell lines treated with vehicle control, RAFi (0.3 μM), ERKi (0.04 μM), or the combination for multiple time points (0.25, 1, 8, 24 and 72 h). RAFi/ERKi-treated PDAC cells were normalized to their respective vehicle control. Proteins with significant phosphorylation or expression changes at 72-h time point are plotted as fold changes. Red, increased fold change; blue, decreased fold change; white, no change.
(B) GSEA of the cell lines shown in (A). Enriched or depleted gene sets treated with RAFi/ERKi compared to RAFi (upper graph) or ERKi (below graph) are shown (24 h).
(C) Pa16C cells were treated with vehicle control, RAFi (0.3 μM) and ERKi (0.04 μM) alone, or the combination (120 h). Cell lysates were immunoblotted to determine levels of pERK, total ERK, total MYC, and vinculin. Data are representative of three independent experiments.
RPPA-based pathway mapping revealed dynamic changes in protein phosphorylation and total protein components of signaling networks (Figure 3A; Tables S2A and S2B). These changes were much more robust upon concurrent treatment with RAFi/ERKi compared to the limited time-dependent alterations caused by either inhibitor alone. The combination suppressed ERK signaling (pRSK, MYC) more strongly than did single agents (Figure 3A). Additionally, RAFi/ERKi treatment strongly reduced SRC family kinase (SFK) phosphorylation and activation. SFK phosphorylation of ARAF and CRAF is a critical RAS-mediated step in the activation of RAF kinase activity (Morrison, 2012). Decision tree analysis confirmed the significant alterations of these proteins (Table S2C). Finally, consistent with the growth inhibitory consequences, the RAFi/ERKi combination also reduced phosphorylation and activation of mitotic kinases Aurora and PLK1 (Figure 3A).
We applied MIB/MS and confirmed that 3-day treatment with RAFi/ERKi caused additive or synergistic alterations in kinase activity and/or expression compared to RAFi or ERKi alone (Figure S6A). While RAFi alone reduced activity/expression of BRAF but not ERK1 or ERK2, and ERKi alone reduced ERK1 and ERK2 but not BRAF, the combination reduced both BRAF and ERK. Consistent with our previous observation that loss of MYC protein correlated with ERKi sensitivity, RPPA and MIB/MS revealed that the combination much more effectively reduced MYC protein levels (Figures 3A and S5C–S5E). Finally, MIB/MS also indicated that the combination reduced the activity/expression of Aurora and PLK1 (Figure S6A).
To compare the resulting gene transcription changes induced by RAFi/ERKi or each agent alone, we performed RNA-seq analyses after 4 or 24 h of treatment (Figure 3B). The strongest induction of gene transcription was of the interferon (IFN)-α/γ-related gene sets (Figure 3B; Tables S2D–S2I). This is consistent with a study of Kras-driven colon cancer, where Kras strongly downregulated IFN-α/γ gene sets, suppressing a T cell immune response (Liao et al., 2019). Consistent with the major role of the ERK MAPK effector pathway in driving KRAS-regulated gene expression, gene set enrichment analysis (GSEA) revealed that RAFi/ERKi suppressed genes upregulated by KRAS and increased expression of genes downregulated by KRAS (Figures 3B, S5A, and S5B). In particular, RAFi/ERKi strongly suppressed MYC transcription (Figure S6B) as well as potently silenced the expression of MYC-regulated genes (Figures 3B and S6C). RAFi/ERKi also robustly suppressed the expression of a second oncogenic transcription factor, FOSL1. Both MYC and FOSL1 are well-validated drivers of KRAS-dependent PDAC growth (Vallejo et al., 2017; Vaseva et al., 2018).
Previous studies showed that single-agent inhibition of any node of the RAF-MEK-ERK cascade is limited by the loss of negative feedback on the pathway, causing ERK reactivation (Lake et al., 2016; Morrison, 2012; Samatar and Poulikakos, 2014). Given its potent suppression of ERK signaling, RAFi/ERKi also robustly decreased inhibitory feedback on ERK. RPPA pathway mapping demonstrated that RAFi/ERKi increased phosphorylation at PAK phosphorylation sites on ARAF and CRAF that enhance RAF kinase activity (King et al., 1998) (Figure 3A). PAK signaling drives resistance to combined BRAF and MEK inhibition in BRAF mutant melanoma by causing ERK reactivation (Lu et al., 2017). Similarly, there was strong suppression of genes encoding diverse negative regulators of ERK signaling. These include proteins that directly dephosphorylate ERK (e.g., DUSP4–7) as well as proteins that inhibit RTK activation of RAS via its exchange factor SOS1 or that inactivate RAS via its negative regulator NF1 (e.g., SPRY2/4, SPRED1/2, respectively) (Figure S6B). To determine how RAFi/ERKi treatment was able to retain strong suppression of ERK signaling despite the extensive loss of negative feedback on ERK, we examined SHP2, essential for RTK activation of RAS and ERK (Ahmed et al., 2019). RAFi/ERKi treatment reduced SHP2 phosphorylation (Figure 3A) and suppressed transcription of the gene encoding SHP2 (PTPN11) (Figure S6D). RAFi/ERKi treatment also reduced phosphorylation of the SHP2 docking site on FRSα (Y436), an adaptor protein that links the fibroblast growth factor receptor (FGFR) RTK to SOS1 and RAS activation. FRSα is phosphorylated and inactivated by ERK feedback inhibition (Lax et al., 2002). Thus, downregulation of SHP2 contributes to the effectiveness of the RAFi/ERKi combination by creating insensitivity to the loss of ERK negative feedback.
RAFi/ERKi Treatment Decreases Cell Cycle Progression, Suppresses Protein Translation Signaling, and Increases Apoptosis
Pathway mapping at the levels of transcriptional control (RNA-seq), protein abundance (WB, RPPA, MIB/MS), and protein activity (RPPA, MIB/MS) all showed that the biological consequences of RAFi/ERKi treatment are due to multiple distinct mechanisms (Figure S4). RAFi/ERKi resulted in numerous changes that decreased the ERK-dependent events that facilitate G1 progression, including increased phosphorylation of p27KIP1, decreased expression of CCND1 (encoding cyclin D1) (Figure S6B), decreased phosphorylation of the RB tumor suppressor (Figure 3A), loss of E2F1 gene transcription (Figure S6D), and decreased transcription of E2F target genes (Figure S6E). RAFi/ERKi suppressed the genes normally upregulated during G2/M checkpoint progression more strongly than did either RAFi or ERKi alone (Figure S6E), and they suppressed the activities/expression of mitotic kinases that are critical regulators of the G2 to M transition (Figures 3A and S6E). Reflecting these activities was a time-dependent decrease in the proliferation marker Ki67 (Figure 3A).
Pathway activation mapping also indicated that RAFi/ERKi synergistically suppressed mTORC1 signaling—which promotes protein translation, as indicated by reduced phosphorylation of mTORC1 substrates 4EBP1 and S6K—and of the S6K substrate ribosomal protein S6 (Figure 3A). Strong suppression of mTORC1-stimulated gene expression was also seen with RAFi/ERKi (Figures 3B and S7F).
Further, RAFi/ERKi suppressed essentially all genes in the glycolytic pathway (Figure S6E), consistent with studies demonstrating that mutant KRAS drives increased transcription of glycolytic genes through ERK and MYC (Bryant et al., 2019; Ying et al., 2012). RAFi/ERKi also strongly suppressed genes involved in mitochondrial biogenesis, mitophagy, and impaired mitochondrial function (Figure S6E). Thus, RAFi/ERKi-mediated growth suppression involves the suppression of key metabolic processes.
Finally, pathway activation mapping also identified increases in markers of apoptosis caused by RAFi/ERKi; increased cleaved caspases 3, 6, and 7 and PARP; and increased expression of the pro-apoptotic proteins BIM and BAD (Figure 3A). RAFi/ERKi also more strongly increased the transcription of genes encoding pro-apoptotic proteins (e.g., HRK, BCL2L11 [encoding BIM], PUMA, CASP1) and reduced the transcription of genes encoding pro-survival proteins (BCL2L1 [encoding BCLXL], MCL1) than either single agent (Figures 4A, S7A, and S7B). RAFi/ERKi-treatment-mediated increases in BIM and BAD were further verified by immunoblotting (Figure 4B). Induction of apoptosis was verified by flow cytometry analysis. Whereas RAFi or ERKi treatment alone did not significantly increase apoptosis over vehicle control (<10%), the percentage of apoptotic cells was more than tripled with RAFi/ERKi (Figures 4C, 4D, and S7C–S7E). Thus, RAFi/ERKi treatment impairs processes that promote cell proliferation and enhances processes that lead to cell death.
Figure 4. Concurrent RAF and ERK Inhibition Causes Apoptosis.

(A) (left) Fold changes (log2) of RNA expression of pro-apoptosis and pro-survival genes as the averaged values of MIA PaCa-2, Pa02C, Pa14C, and Pa16C cell lines treated with RAFi/ERKi (0.3 μM and 0.04 μM, respectively) and compared to RAFi (0.3 μM) (24 h). Error bars are shown as standard error. All p values shown are in comparison to the vehicle control for individual graph; p values are from Wald test. Adjusted p values: BAX (0.2971), BAK1 (0.7226), BAD (*, 0.0332), BID (0.3363), BIK (0.2635), HRK (**, 0.003), BCL2L11 (****, 1.49E-5), PUMA/BBC3 (*, 0.0221), CASP1 (0.3642), CASP2 (**, 0.0034), CASP3 (0.6314), CASP4 (0.1526), CASP6 (0.5876), CASP7 (0.5114), CASP8 (0.9215), CASP9 (0.2887), BCL2 (***, 0.0014), BCL2L1 (0.1569), MCL1 (0.9338), BCL2L2 (0.9080). (Right) fold changes (log2) of the transcripts of the cells treated with RAFi/ERKi (0.3 μM and 0.04 μM, respectively) and compared to ERKi (0.04 μM) (24 h). Error bars are shown as standard error. All p values shown are in comparison to the vehicle control for individual graph; p values are determined from the Wald test. Adjusted p values: BAX (***, 0.0003), BAK1 (0.4834), BAD (0.6736), BID (0.1958), BIK (0.2171), HRK (***, 0.0011), BCL2L11 (****, 7.12E−6), PUMA/BBC3 (*, 0.0148), CASP1 (0.3406), CASP2 (*, 0.0250), CASP3 (0.7561), CASP4 (*, 0.0229), CASP6 (*, 0.0615), CASP7 (0.1599), CASP8 (0.4119), CASP9 (0.2582), BCL2 (****, 0.0001), BCL2L1 (**, 0.0032), MCL1 (0.7493), BCL2L2 (0.9250).
(B) Pa01C, Pa02C, or Pa14C cells were treated with vehicle, RAFi (0.3 μM), ERKi (0.04 μM), or the combination (72 h). Cell lysates were immunoblotted to determine levels of the indicated proteins.
(C) Percent apoptosis of Pa02C and Pa16C cells treated with the vehicle control, RAFi (0.3 or 0.6 μM), ERKi (0.04 or 0.08 μM), or the combinations (120 h). Error bars are shown as ± SEM.
(D) Representative images of percent apoptosis of the cell lines in (C). Fluorescence-activated cell sorting (FACS) analysis was used to measure apoptosis.
Combination RAFi/ERKi Treatment Stimulates Mesenchymal-to-Epithelial Transition
We noted from the RPPA analyses that combined RAFi/ERKi treatment increased E-cadherin protein levels (Figures 3A and S8A), suggesting induction of the mesenchymal-to-epithelial transition (MET) program. RAFi/ERKi treatment substantially increased E-cadherin expression (2.4- to 7.3-fold) in four of six cell lines evaluated (Figures 5A, 5B, S8A, and S8B; Tables S3A and S3B), whereas RAFi or ERKi treatment alone did so to a lesser degree (Figure 5B). RAFi/MEKi or MEKi/ERKi treatment also increased E-cadherin expression. In contrast, BRAFi in combination with either ERKi or MEKi did not significantly increase E-cadherin expression. The limited change in vimentin transcription and protein levels upon RAFi/ERKi treatment (Figures 3B, 5A, 5B, and S8B) is consistent with a partial MET program.
Figure 5. Concurrent RAF and ERK Inhibition Induces Mesenchymal-to-Epithelial Transition.

(A) Pa02C and Pa16C cells were treated with RAFi (0.3 μM), BRAFi (1 μM), MEKi (0.5 nM), or ERKi (0.04 μM) alone or in combination as indicated (120 h). Cell lysates were immunoblotted to determine the levels of the indicated proteins.
(B) Expression levels of the proteins in (A) are plotted as fold changes. All the proteins are normalized to loading control and their respective vehicle control. Error bars are shown as ± SEM.
(C) Representative immunofluorescence images of Pa02C cells treated with vehicle, RAFi (0.3 μM), ERKi (0.04 μM), or the combination to visualize E-cadherin expression and distribution (72 h). Scale bar, 20 μm.
(D) Fold changes in RNA expression of epithelial and mesenchymal markers are plotted for the mean average of MIA PaCa-2, Pa02C, Pa14C, and Pa16C cell lines. Error bars are shown as standard error. p values shown are from Wald test and are in comparison to the vehicle control. Adjusted p values: CDH1 (*, 0.0252), CLDN1 (0.8818), TJP1 (0.7301), VIM (0.8465), CDH2 (0.3881), CTNNB1 (0.2445), SNAI1 (0.2542), SNAI2 (0.5672), ZEB1 (*, 0.0262).
The degree of E-cadherin increase correlated with the degree of pERK reduction, which was caused most effectively by combination RAFi/ERKi compared to treatment with any single inhibitor or other combinations. Increases in E-cadherin also correlated with induction of apoptosis (Figures 4B–4D and S7C–S7E). In the lines in which E-cadherin was upregulated, RAFi/ERKi also synergistically induced apoptosis. In contrast, in Pa01C cells, RAFi/ERKi did not alter E-cadherin levels (Figure S8B) and caused only a weak induction of apoptosis (Figures 4B and S7C–S7E). Finally, immunofluorescence analyses determined that RAFi but not ERKi treatment alone enhanced E-cadherin staining at cell peripheries and at cell-cell junctions, and combined RAFi/ERKi treatment caused the strongest upregulation of E-cadherin (Figures 5C and S8C).
We used RNA-seq and GSEA to identify the mechanistic underpinnings of MET induced by RAFi/ERKi. Expression of CDH1, which encodes E-cadherin, was increased by the combination (Figure 5D), consistent with reduced levels of several transcription factors that suppress CDH1 transcription (SNAI1, SNAI2, and ZEB1) (Derynck and Weinberg, 2019) (Figure 5D). Conversely, RPPA analyses showed that TGFβ/SMAD2, a well-established signaling pathway that drives EMT (epithelial-mesenchymal transition), was also reduced by RAFi/ERKi (Figure 3A). RAFi/ERKi caused a significant reduction in PLK1 activity (Figures S6A and 3A), a driver of EMT through ERK and FRA1 (Wu et al., 2016), with FRA1 also significantly reduced transcriptionally (FOSL1) by this combination (Figure S6B). Thus, induction of MET-like reprogramming, synergistically induced by RAFi/ERKi treatment, correlated with induction of apoptosis.
Concurrent Inhibition of Compensatory Signaling Enhances RAFi/ERKi Cytotoxicity
We next addressed two additional potential improvements in response to the combination. First, we determined if sequential treatment could be more effective than concurrent treatment. We compared low-dose sequential inhibitor treatment to low-dose concurrent treatment. For sequential treatment, cells were treated with the initial RAFi or ERKi for 3 days, and then the other inhibitor ([RAFi + ERKi] or [ERKi + RAFi]) was added. In Pa02C cells, RAFi/ERKi resulted in 70% fewer cells than vehicle or either inhibitor alone (Figure 6A). In contrast, sequential treatment in either order caused a much more limited decrease (~20%–25%). In Pa14C cells, whereas treatment with either RAFi or ERKi alone caused a ~25% decrease, concurrent RAFi/ERKi treatment caused a near-complete suppression of proliferation (~95%). By comparison, RAFi followed by ERKi, or ERKi followed by RAFi, caused a ~75% or ~50% reduction, respectively. Thus, concurrent treatment was more effective than sequential inhibitor treatment.
Figure 6. Concurrent Inhibition of Compensatory Signaling Enhances RAFi/ERKi Growth Inhibition.

(A) Pa02C and Pa14C cells were treated with RAFi (0.3 μM), ERKi (0.04 μM), FRAX597 (PAKi, 1 μM), or MK2206 (AKTi, 0.6 μM) alone or in combination (slash indicates concurrent inhibition; plus sign indicates sequential inhibition, inhibitor addition after 72 h). Remaining cells were stained with crystal violet after a total of 5 days. Data are the mean average of two independent experiments. Error bars are shown as ± SEM.
(B) Pa02C and Pa14C cells were treated as in (A) for a total of 5 days. Cell lysates were immunoblotted to determine the levels of the indicated proteins. Data are representative of two independent experiments.
Second, we determined if further concurrent inhibition of additional feedback mechanisms can further enhance RAFi/ERKi treatment in Pa02C cells, where there was room for improvement over the RAFi/ERKi combination (Figure 6A). We observed that RAFi/ERKi was associated with increased PAK activity. For example, RPPA demonstrated increased phosphorylation at the PAK phosphorylation site in CRAF (S338) (King et al., 1998) (Figures 3A and S4), and this was verified by immunoblotting (Figure 6B). Increased LIMK phosphorylation and activation, supported by increased phosphorylation at S3 of the actin-severing LIMK substrate Cofilin (Figures 3A and 6B), was also consistent with upregulation of PAK activity. S3 phosphorylation inactivates Cofilin, stimulating actin polymerization (Kanellos and Frame, 2016), which is also consistent with the MET-associated changes (Derynck and Weinberg, 2019) that we observed. Immunofluorescence images revealed F-actin rearrangement upon RAFi and/or ERKi treatment (Figure S8C). Thus, RAFi/ERKi was associated with increased PAK activation, likely through upregulation of RTK signaling (Rane and Minden, 2019). Therefore, we determined if adding the PAK inhibitor (PAKi) FRAX597 would further enhance RAFi/ERKi growth inhibition. PAKi treatment alone reduced pCRAF without affecting proliferation. However, the triple combination of RAFi/ERKi/PAKi was able to nearly ablate proliferation (Figures 6A and S9A–S9C).
Another potential compensatory activity is PI3K-AKT activation, which has been described in response to MEKi treatment. This also explains how concurrent PI3Ki can enhance ERK MAPK inhibitory activity (Figure 2A). Accordingly, RAFi/ERKi treatment of Pa02C cells increased AKT pS473, which was blocked by the AKT inhibitor (AKTi) MK2206 (Figure 6B). Like PAKi, AKTi alone did not affect proliferation, but concurrent or sequential AKTi treatment caused more growth suppression than RAFi/ERKi treatment alone (Figures 6A and S9A–S9C). Thus, as with PAKi, the concurrent AKTi treatment blocked a compensatory activation mechanism and further enhanced RAFi/ERKi growth suppression.
Low-Dose RAFi/ERKi Vertical Inhibition Is Effective in KRAS Mutant Organoids and Tumor-Bearing Rats
We next evaluated the activity of the RAFi/ERKi combination in other KRAS mutant cancer cell lines and in PDAC organoids and tumor-bearing animals. RAFi/ERKi also caused synergistic growth suppression in KRAS mutant colon and lung cancer cell lines (Figures 7A and S10A). Extending these analyses to models that may better reflect patient tumor response, we observed strong synergistic growth inhibition due to the RAFi/ERKi combination in patient-derived KRAS mutant PDAC and CRC organoid models (Figures 7B, 7C, S10B, and S10C).
Figure 7. Vertical ERK MAPK Inhibition is Effective in Organoid and Rat Models of KRAS Mutant Cancers.

(A) KRAS mutant cancer cell lines were treated with RAFi (0.01–2.5 μM) and ERKi (0.08–1.25 μM) alone or in combination for 120 h. Proliferation was measured by Calcein AM cell viability assay. Representative bliss synergy score heatmaps for three independent experiments is shown (left). Red, synergy; green, antagonism; white, no effect. The averaged dose response curves of three independent experiments are shown (right). Error bars are ± SEM. Synergyavg = average bliss synergy score.
(B) KRAS mutant PDAC (10 days) and CRC organoids (5 days) were treated with RAFi (0.01–2.5 μM) and ERKi (0.04–0.63 μM) alone or in combination. Proliferation was measured by CellTiter-Glo 3D cell viability assay. Dose response curves and bliss synergy scores were calculated and represented as in (A).
(C) Representative images of PDAC organoid hM1A treated with the vehicle control DMSO or RAFi (0.16 μM) or ERKi (0.04 μM) alone or in combination (left). Scale bar, 200 μm. Representative images of CRC organoid OT238 treated with DMSO or RAFi (0.31 μM) or ERKi (0.04 μM) alone or in combination (right). Scale bar, 100 μm.
(D) Relative tumor volume of the NIH nude rats with implanted HPAF-II cells were treated with RAFi (20 mpk, BID) alone or in combination with the ERKi LY3214996 (LY ERKi, 10 mpk, QD) for 36 days (left). Body weight changes are shown (right). Error bars are shown as ± SEM.
(E) Quantitation of blot analysis to determine levels of pRSK of tumor lysates (n = 5 animals per group). Error bars are shown as ± SEM.
Finally, we extended the analyses of the RAFi/ERKi combination to evaluate anti-tumor activity. Since RAFi has poor pharmacokinetics in mice, our analyses were limited to analyses of PDAC-cell-line-induced tumors in immune-compromised rats. For these analyses, we utilized three PDAC lines that showed different degrees of response to vertical inhibition combinations. HPAF-II cells exhibit strong synergistic growth suppression with RAFi in combination with ERKi, MEKi, and EGFRi (Figures S10E–S10G; Tables S1D and S1F–S1H). CFPAC1 cells were also responsive to vertical inhibition combinations, but to a lesser degree, and SW1990 cells showed very limited increased growth suppression with the combinations.
Whereas ERKi and RAFi alone simply reduced the rate of tumor growth in HPAF-II PDAC xenografts, the RAFi/ERKi vertical inhibition combination was able to induce tumor regression even at low doses and did so without statistically significant toxicity (Figure 7D). The target-based mechanism of tumor regression is supported by immunoblot analyses of remaining tumor tissue isolated 4 h after the last treatment (day 21), where the combination reduced ERK signaling more potently than did either inhibitor alone (Figures 7E and S10D). Analyses of CFPAC-1 also determined that RAFi/ERKi showed greater activity than each inhibitor alone (Figures S10H and S10I). In contrast, RAFi alone did not significantly reduce SW1990 tumor growth, whereas ERKi alone or in combination with RAFi shows comparable limited tumor reduction (Figure S10J). No significant toxicity was observed for both models, as indicated by maintained body weight (Figures S10H and S10J). Thus, the in vivo responses closely mirrored the different sensitivities of each cell line when evaluated in cell culture.
DISCUSSION
Despite the essential role of the RAF-MEK-ERK cascade in KRAS-dependent PDAC growth, single-agent pharmacologic inhibition of this cascade has been ineffective due to the loss of ERK-dependent negative feedback inhibitory mechanisms that then cause ERK reactivation and drug resistance in cancer cells. Normal tissue toxicity has also been a limitation. To address these limitations, we applied a chemical screen to identify combinations that enhance the apoptotic activity of RAFi-MEKi-ER-Kis. While we identified multiple mechanistically distinct apoptotic combinations, surprisingly, the most potent combination was the concurrent inhibition of two distinct nodes of the three-tiered ERK MAPK cascade, where treatment with a pan-RAFi together with an ERK-selective inhibitor (RAFi/ERKi) exhibited the strongest synergistic activity.
Despite strong induction of compensatory signaling activities that can drive ERK reactivation, the RAFi/ERKi combination was able to cause synergistic suppression of ERK activation and system-wide disruption of ERK-dependent cellular processes, causing cell cycle arrest and apoptosis; loss of MYC-, E2F- and FRA1-dependent transcriptomes; impaired metabolism; and induction of MET. That the synergistic apoptotic growth suppression was strongest at lower inhibitor concentrations suggests that combined vertical inhibition of the ERK MAPK signaling circuitry can overcome the limitations seen with single-agent inhibition and lead to tumor regression rather than stasis at doses that can reduce normal tissue toxicity.
The concept of vertical inhibition of ERK MAPK was first demonstrated in BRAF mutant melanoma, where BRAFi/MEKi combinations are now approved. This combination strategy delays onset of resistance and reduces toxicity compared with BRAFi treatment alone, albeit without any reduction in the dosing compared with BRAFi treatment alone (Dummer et al., 2018; Flaherty et al., 2012; Larkin et al., 2014; Long et al., 2014). Extending this concept further, it was shown that a triple RAFi/MEKi/ERKi combination, with each used in the combination at the maximum tolerated dose, further delayed the onset of resistance, exhibited stronger suppression of BRAF mutant tumors, and further reduced toxicity compared with the double-combination MEKi/ERKi (Xue et al., 2017).
Similar to our RAFi/ERKi vertical inhibition strategy, two recent studies showed that concurrent RAFi/MEKi treatment blocked ERK reactivation caused by MEKi treatment alone in KRAS mutant or WT cancer cell lines (Lamba et al., 2014; Yen et al., 2018). A third study showed that concurrent MEKi/ERKi treatment exhibited stronger inhibition of ERK and increased anti-tumor activity than either inhibitor alone (Merchant et al., 2017). While our study found that combined inhibition of any two distinct nodes of the EGFR-RAF-MEK-ERK cascade is superior to any single-node treatment, our analyses support the RAFi/ERKi combination as the optimal combination. MEKis and ERKis are limited by CRAF reactivation, whereas RAFis and MEKis are limited by ERK reactivation; therefore, the RAFi/ERKi combination targets the two key reactivation nodes. We also determined that concurrent rather than sequential inhibitor treatment allowed more effective ERK inhibition and growth suppression. Finally, we demonstrated that triple combinations with a third inhibitor that targets additional compensatory mechanisms (PAK or AKT activation) further enhance RAFi/ERKi activity.
To address a mechanistic basis for the synergistic and apoptotic growth suppression of the RAFi/ERKi combination, we applied RPPA and RNA-seq analyses to identify activities seen with RAFi/ERKi but not with each inhibitor alone. Together, these analyses identified a spectrum of ERK-dependent signaling activities and cellular processes driven more potently by RAFi/ERKi than RAFi or ERKi alone, where any one perturbation alone would be expected to significantly impair cancer cell proliferation. Perhaps most significant is the synergistic loss of a key ERK substrate, MYC, and the suppression of MYC-regulated gene transcription. We showed recently that the loss of MYC is a key basis for ERKi sensitivity in PDAC and that MYC suppression alone impairs PDAC tumorigenic growth (Hayes et al., 2016; Vaseva et al., 2018).
RAFi/ERKi induced more robust suppression of ERK signaling than was achievable even by high-dose treatment with each inhibitor alone. Along with this, it also more robustly induced changes associated with a loss of ERK negative feedback mechanisms (Morrison, 2012; Samatar and Poulikakos, 2014), such as suppressing transcription of multiple DUSP family genes encoding protein phosphatases that dephosphorylate and inactivate ERK and activating RTKs that promote PAK-dependent phosphorylation and activation of CRAF. However, although these changes would be expected to reactivate ERK signaling, the combination nevertheless retained the ability to strongly suppress ERK signaling. Thus, another basis for the synergistic activity of RAFi/ERKi is insensitivity to the compensatory activation mechanisms that limit the effectiveness of RAFis, MEKis, or ERKis when used as single-agent therapies. An additional unexpected consequence of RAFi/ERKi was inhibition of SHP2, a key relay mechanism that connects RTK activation with downstream activation of RAS and ERK signaling. Recent studies showed that concurrent treatment with a SHP2 inhibitor can negate the RTK-mediated compensatory activities that are stimulated by ERK MAPK inhibition and can synergistically enhance MEKi activity (Ahmed et al., 2019; Fedele et al., 2018; Mainardi et al., 2018; Ruess et al., 2018; Wong et al., 2018).
Concurrent RAFi/MEKi treatment also caused apoptotic cell death not seen with each inhibitor alone. A mechanistic basis for this was identified, where the combination showed stronger promotion of pro-apoptotic and suppression of pro-survival activities. Similarly, RAFi/ERKi synergistically caused G1 arrest through RB activation of loss of transcription of E2F-mediated gene expression. Together, these activities provide a basis for the ability of RAFi/ERKi to suppress PDAC growth at lower concentrations of each inhibitor. Thus, vertical inhibition of the ERK MAPK cascade may reduce the normal tissue toxicity seen with single-agent therapy and promote cytotoxic rather than cytostatic inhibition of cancer cell proliferation.
We also observed that RAFi/ERKi induced a partial MET program by transcriptional suppression of SNAI1/2 and ZEB1, the latter of which encodes a transcription factor suppressor of CDH1 transcription (Derynck and Weinberg, 2019). With enhanced CDH1 expression promoting enhanced E-cadherin expression, a key driver of MET, RAFi/ERKi-treated PDAC cells exhibit a transition from a mesenchymal to an epithelial state. Further, we observed a trend in which MET was the strongest in the PDAC cell lines where RAFi/ERKi caused the strongest induction of apoptosis. This relationship mirrors the earlier findings, where KRAS mutant cancer cell lines with a mesenchymal phenotype were those that escaped KRAS addiction, whereas KRAS mutant cell lines with an epithelial phenotype were susceptible to KRAS suppression-induced apoptosis (Singh et al., 2009). Similarly, McCormick and colleagues found that KRAS mutant cancer cells with an epithelial but not a mesenchymal phenotype exhibited ERK dependency (Yuan et al., 2018).
Our determination that the RAFi/ERKi combination caused both G1 arrest and apoptosis in cancer cells shows the ability of this combination to block tumorigenic growth and cause tumor regression. Additionally, we found that RAFi/ERKi may enhance an anti-tumor immune response. Whereas EPHA2 has been shown to suppress anti-tumor T cell immunity (Markosyan et al., 2019), kinome profiling upon RAFi/ERKi treatment revealed a reduction of EPHA2 expression/activity. Further, one of the strongest consequences of RAFi/ERKi was stimulation of IFN-α/γ genes, which would also stimulate a T cell immune response (Liao et al., 2019). Thus, RAFi/ERKi may cause potent anti-tumor activity both by suppressing tumor cell growth and by stimulating a host immune response. Our analyses showed that RAFi/ERKi caused tumor regression in immune-suppressed rats. We speculate that the RAFi/ERKi combination will elicit robust tumor regression in syngeneic PDAC mouse models where there is an intact immune system.
During our studies, a clinical trial evaluating LY3009120 in patients with advanced or metastatic cancer was terminated early based on the lack of sufficient clinical efficacy observed (NCT02014116), emphasizing the need to consider vertical inhibition combination approaches. One ongoing clinical trial is evaluating the pan-RAFi LXH254 in combination with either an ERKi or a MEKi in patients with advanced or metastatic KRAS mutant lung cancer or NRAS mutant melanoma (NCT02974725). This study will provide a clinical comparison of a RAFi/ERKi versus RAFi/MEKi combination.
In summary, while we identified inhibitors of diverse cellular components that enhanced the anti-tumor activity of a pan-RAFi, the most potent combination involved vertical inhibition of the RAF-MEK-ERK cascade that was effective at low doses. Our evidence shows that RAFi/ERKi, despite stimulating robust compensatory mechanisms that can drive ERK reactivation, is refractory to these mechanisms and consequently achieves pathway suppression at a level not achievable with each inhibitor alone. This concept is further supported by our triple combinations with inhibitors of PAK- or AKT-dependent compensatory mechanisms. This causes the loss of a spectrum of ERK-dependent cellular processes driven by aberrant gene transcription (G1 progression, pro-survival, EMT) that then promotes cancer cell death and tumor regression. Finally, since we found that RAFi/ERKi was effective in KRAS mutant pancreatic, lung, and colorectal cancer cell lines, this combination may serve as a pan-KRAS mutant cancer therapy.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Requests for further information and reagents should be directed to and will be fulfilled by the Lead Contact, Channing J. Der (cjder@med.unc.edu).
Materials Availabilty
This study did not generate any new reagents.
Data and Code Availability
The accession number for the binary sequence alignment/map (BAM) files of RNA-seq data of cell lines MIA PaCA-2, Pa02C, Pa14C and Pa16C reported in this paper is [ENA] (the EMBL-EBI European Nucleotide Archive) PRJEB38063 (https://www.ebi.ac.uk/ena/, https://www.ebi.ac.uk/ena/data/view/PRJEB38063). The file names containing “R” indicate RAF inhibitor treated samples, “E” indicate ERK inhibitor treated samples, “C” indicate combination treated samples and “V” indicate vehicle control. The file names containing “4” and “24” indicate treatment time of 4 hours and 24 hours respectively.”
This study did not generate any unique codes.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
The patient-derived xenograft (PDX) human pancreatic cancer cell lines Pa01C, Pa02C, Pa03C, Pa04C, Pa14C and Pa16C were gifted by Dr. Anirban Maitra (MD Anderson Cancer Center). Conventional human pancreatic cancer cell lines (MIA PaCa-2, PANC-1, HPAF-II, CFPAC-1 and SW 1990), lung cancer cell lines (A549, NCI-H358 and SW900) and colorectal cancer cell line (SW620) were obtained from American Type Culture Collection (ATCC). PDX pancreatic and colorectal cancer cell lines were maintained in Dulbecco’s Modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). HPAF-II, CFPAC-1 and SW 1990 pancreatic cancer cell lines were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS). Lung cancer cell lines were maintained in RPMI medium supplemented with 10% FBS. Cell lines were tested for mycoplasma. Pancreatic cancer cell line identities were verified by Short Tandem Repeat (STR) analysis.
Patient-Derived Organoids
The human pancreatic cancer organoids were provided by Dr. David Tuveson (Cold Spring Harbor Laboratory). The patient-derived PDAC organoids hM1A KRASG12D and hT2 KRASG12R were cultured at 37°C in 5% CO2. Cells were seeded in growth factor reduced Matrigel (Corning) domes and fed with complete human feeding medium: advanced DMEM/F12 based WRN condition media (L-WRN (ATCC CRL-3276)), 1x B27 supplement, 10 mM HEPES, 0.01 μM GlutaMAX, 10 mM nicotinamide, 1.25 mM N-acetylcysteine, 50 ng/mL hEGF, 100 ng/mL hFGF10, 0.01 μM hGastrin I, 500 nM A83-01, 1 μM PGE2 and additionally 10.5 μM Y27632 (Boj et al., 2015). Organoids were tested for mycoplasma. The patient-derived colorectal organoids OT227 KRASG13D, OT238 KRASG12D and OT302 KRASG12D were previously fully characterized in terms of genomic alterations by Schütte et al. (2017). CRC organoids were cultured in crypt culture medium (CCM) containing advanced DMEM/F12 (GIBCO) supplemented with 1x GlutaMAX (GIBCO), 10 mM HEPES buffer (GIBCO), Penicillin/Streptomycin (100 U/ml/100 μg/ml), 1 mM N-acetylcysteine (Sigma), 1x N2 Supplement (GIBCO), 1x B27 Supplement (GIBCO) and prepared with freshly added hFGF basic/FGF2 (20 ng/ml) (Sigma) and hEGF (50 ng/ml) (Sigma).
Rats
All in vivo studies were performed in accordance with the American Association for Laboratory Animal Care institutional guidelines and approved by The Eli Lilly and Company Animal Care and Use Committee. HPAF-II, CFPAC-1 and SW1990 pancreatic cancer cells (ATCC cat#CRL-1997)) were cultured in MEM supplemented with 10% heat inactivated fetal bovine serum, sodium pyruvate, and nonessential amino acids. Cell lines were tested for mycoplasma and identity was confirmed by STR-based DNA finger printing and multiplex PCR (IDEXX-Radil). Logarithmically growing cells with < 7 passage from the thaw were used for implantation.
In all three studies (HPAF II, CFPAC1 and SW1990), 7-8 weeks old (120-145 g) female NIH Nude (NIHRNU-IVI) rats from Taconic Farms Inc were used.
Animal maintenance and care:
12 hr light/dark cycle, Temp: 68-75°C, Humidity: 30%–70% relative, 3-5 animals /cage, Bedding: Bed o cob, Filtered water, Feeding: Ad libitum, Feed: Standard chow
METHOD DETAILS
shRNA and Plasmid Transfections
The shRNAs targeting ARAF (TRCN0000000567, TRCN0000000568), BRAF (TRCN0000006290, TRCN0000006291), and CRAF (TRCN0000001065, TRCN0000001066) were provided Deborah K. Morrison (NCI), and shRNA targeting KRAS (TRCN0000010369) was provided by J. Settleman (Genentech). 0.9 × 106 HEK293T cells were seeded on T25 flasks and incubated overnight. To generate lentiviral particles, HEK293T cells were transfected with plasmids by using FuGENE6 (Roche) protocol. Vector (4 μg), pSPAX2 (3 μg) and pMD2.G (1 μg) plasmids were diluted in 400 μL Opti-MEM medium. Transfection reagent FuGENE6 (24 μl) was added into the diluted plasmid mixture and incubated for 15 min at room temperature. Transfection mixture was added onto HEK293T cells dropwise and incubated overnight. Transfection medium was replaced with DMEM with 20% FBS and incubated for 48 hr. Virus particles were collected. 106 cells were infected by 0.5 μL virus combined with polybrene (final concentration of 8 μg/μl) in 2 mL medium. The medium was replaced with complete medium (DMEM with 10% FBS) after 8 hr. Antibiotic selection was started after 12-16 hr of incubation in complete medium. Upon 72 or 120 hr of antibiotic selection, cells were collected for immunoblotting, anchorage-independent colony formation, or proliferation assays.
To quantify anchorage-independent colony formation, 400-1000 cells per well (depending on the cell line) were seeded on 6-well plates and incubated for ≥ 10 days at 37°C until colonies were formed. Colonies were stained by crystal violet and quantified by ImageJ (version 2.0.0). Briefly, the 6-well plate images were converted to 8-bit images. The same thresholding was applied to all the wells to subtract background. The mean intensities were normalized by dividing each value to the average intensity of its respective control well.
To quantify proliferation, 2×105 cells per well were seeded and incubated overnight at 37°C. The next day cells were treated with LY3009120 (0.3 μM), SCH772984 (0.04 μM), the PAK1 inhibitor FRAX-597 (0.5 μM), or the pan-AKT1/2/3 inhibitor MK-2206 (0.6 μM) alone or in various combinations as indicated in the figures for a total of 10 days. Cells were stained by crystal violet and quantified by ImageJ as described above.
Immunoblotting
The human pancreatic cancer cells were washed with ice-cold PBS, lysed with ice-cold 1% RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.01% SDS, 0.5% sodium deoxycholate, 1% NP-40) supplemented with protease (Roche) and phosphatase (Sigma) inhibitors, scraped and incubated in cold tubes for 10 min on ice. Cell lysates were centrifuged at 18,213 x g (12,700 rpm) at 4°C for 10 min, and the supernatant was used for determining protein concentration by Bradford assay (Bio-Rad). Standard immunoblotting procedures were followed. Membranes were blocked in 5% BSA diluted in TBST (TBS with 0.05% Tween 20) for 1 hr.
Proliferation Assays
The cancer cells (103 per well) were seeded in 96-well plates, incubated overnight, and treated with small molecule inhibitors. Cells were treated with a pan-RAF inhibitor LY3009120 (0.01-2.5 μM) alone or in combination with ERK1/2 inhibitor SCH772984 (0.04-1.25 μM, 72 or 120 hr), ERK1/2 inhibitor LY3214996 (0.04-1.25 μM, 120 hr), MEK1/2 inhibitor trametinib (Mekinist) (0.125-4.0 nM, 120 hr), or EGFR inhibitor erlotinib (Tarceva) (0.01-2.5 μM, 120 hr). Cells were treated with ERK1/2 inhibitor SCH772984 (0.04-0.16 μM) alone or in combination with mutant BRAF-selective inhibitor vemurafenib (Zelboraf) (0.01-2.5 μM), ERK1/2 inhibitor ulixertinib (BVD-523) (0.01-2.5 μM), or RAF family kinases and EGFR inhibitor BGB-283 (lifirafenib) (0.01-2.5 μM) for 120 hr. Vehicle control DMSO was kept ≤ 0.01%. Cells were incubated at 37°C 72 or 120 hr. Proliferation was measured by counting calcein AM (500 nM, 20 min) (Invitrogen) labeled live cells by using a SpectraMax i3x multimode detection platform (Molecular Devices). Bliss synergy scores were calculated using SynergyFinder (version 1.6.1) package of R (version 3.5.1) environment. An R script was used to convert the 96-well plate format data into SynergyFinder data table format. SynergyFinder data table includes the identifiers for inhbitiors, row and column numbers, the name of the inhibitors in a dose-response matrix, the concentration and its unit for each well, and the response, which is % inhibition in cell growth. SynergyFinder calculates a synergy score based on Bliss model (Bliss, 1939).
CRISPR/Cas9 Screen
Design and Cloning of the CRISPR Library
In order to assemble a library of druggable, cancer-relevant genes, we manually curated 2,240 genes from six broad domains of interest: chromatin modifiers (including epigenetic readers, writers, erasers), the full kinome, pathways responsible for mediating and repairing DNA damage, genes/proteins that represent the target of FDA-approved drugs for any indication, genes that are frequently mutated in cancer, and genes that comprise the pathways most frequently dysregulated in tumorigenesis, tumor maintenance, and drug resistance. Beyond these genes of interest, we also selected 150 genes, chosen for their demonstrated dispensability or non-dispensability across a series of essentiality studies (Hart et al., 2014), to be used as control genes. We selected five constructs to represent each of these 2,390 genes, producing a subtotal of 11,950 short guide RNA (sgRNA) constructs. Finally, we included 50 non-targeting control constructs for a total of 12,000 sgRNAs. All of the CRISPR constructs used in this library were selected from a previously characterized and published library (Wang et al., 2014). sgRNA inserts corresponding to the entire 12,000 sgRNA library were synthesized by CustomArray, Inc in the form:
GGAAAGGACGAAACACCGXXXXXXXXXXXXXXXXXXXXGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGC. Where “X” denotes variable 20-mer sgRNA sequence unique to each construct. The resulting library was cloned using previously published methods (Shalem et al., 2014). In brief, the oligo pool was diluted 1:100 in molecular biology grade water and amplified using NEB Phusion Hotstart Flex with the following primers and PCR protocol:
Array F: TAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCG
Array R: ACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC
PCR protocol: 98°C/30 s, 18 × [98°C/10 s, 63°C/10 s, 72°C/15 s], 72°C/3 min. Resulting inserts were processed with Axygen PCR clean-up beads (ratio of 1.8 × starting volume; Fisher Scientific) and reconstituted in half the volume of molecular biology grade water. In parallel, the lentiCRISPRv2 vector (Addgene ID 52961) was digested with BsmBI (Thermo Fisher) at 37°C for2 hr. The product was run out for size-selection on a 1% agarose gel and the ~13 kB band was gel-extracted. Using 100 ng of BsmBI-cleaved lentiCRISPRv2 and 40 ng of sgRNA oligo insert as substrates, a Gibson assembly reaction was performed (total volume of 20 μl, for 30 min, at 50°C). Following Gibson assembly, 1 μl of the product was electroporated into electrocompetent Lucigen 10G-elite cells, spread onto LB-ampicillin plates and incubated at 37°C for 16 hr. The efficiency of transformation was estimated by plating limiting dilutions on LB-ampicillin plates. Multiple electroporations were performed, producing an estimated 500,000 total colonies, sufficient to cover the entire library of 12,000 constructs more than 40-fold. The colonies were collected in LB and plasmid extra was performed using a plasmid maxiprep kit (QIAGEN). DNA was used to make lentivirus.
CRISPR/Cas9 Library Lentivirus Generation and Infection
Lentivirus generation was performed as described previously (Martz et al., 2014). HEK293T cells were seeded in 15 cm dishes and grown up to 50% confluency. Library plasmid (6.25 μg), psPAX2 (5.6 μg), pVSVg (0.625 μg) and transfection reagent FuGENE6 (Roche) was incubated for 30 min. The transfection mixture was added to the cells and incubated overnight. Harvest media (DMEM with 30% FBS) was added the next day and incubated for 48 hr. Virus particles were collected and filtered through a 0.45 μm filter. Virus tittering was performed as described previously (Martz et al., 2014).
Human pancreatic cancer cell lines (5×105 per well) were seeded in 6-well plates and incubated overnight. Next day, virus was added at an MOI of 0.3. Upon puromycin selection, a day 2 sample is taken to ensure library representation and the remaining cells were seeded in 500 cm dishes. The cells were maintained in puromycin media for 10 days to achieve 1,000x coverage of the library. The cells were either treated with vehicle control DMSO (≤0.01%) or LY3009120 RAF inhibitor (GI20-30). The cells were collected after 2 and 4 weeks, DNA was extracted with DNeasy Blood & Tissue Kit (QIAGEN) and prepared for sequencing.
Drug Sensitivity Resistance Testing (DSRT) Chemical Library Screen
The DSRT platform that has been described previously (Gautam et al., 2016; Pemovska et al., 2013) was adapted for the PDAC cell lines. 525 different oncological compounds were utilized in this study (Table S2J). The compounds were plated to white clear bottom 384-well plates (Corning #3712) in 5 concentrations in 10-fold dilution steps, thus covering an individually optimized 10,000-fold concentration range for each compound using an Echo 550 Liquid Handler (Labcyte). Cell killing 100 μM benzethonium chloride (BzCl2) and 0.1% dimethyl sulfoxide (DMSO) vehicle were used as positive and negative controls respectively. For the combination screenings, single concentration of RAFi (LY3009120), MEKi (trametinib), ERKi (SCH772984) in final concentrations of 2 μM, 25 nM, or 100 nM respectively, were added on top of 525 compound 5-concentrations plates using Echo 550. Pa01C, Pa02C, Pa03C, Pa04C, Pa14C, Pa18C and MDA-PATC53 were screened in combination with ravoxertinib whereas the remaining cell lines were screened in combination with SCH772984. All subsequent liquid handling was performed using a MultiDrop dispenser (Thermo Scientific). The pre-dispensed compounds were dissolved in 5 μL of culture media, containing viability and cytotoxicity measurement reagents, RealTime-Glo and CellTox Green (Promega), respectively and left on a plate shaker at room temperature for 30 min. Twenty μl cell suspension containing optimized number of cells per well were seeded in the drugged plates. After 72 hr incubation, the multiplexed cell viability (luminescence) and cytotoxicity (fluorescence) was recorded using a PheraStar plate reader (BMG Labtech). The raw luminescence and fluorescence data were analyzed in Breeze software, an in-house developed data analysis pipeline at Institute for molecular Medicine Finland (FIMM), to calculate the drug sensitivity scores (DSS) (Yadav et al., 2014). The drug combination selective effect was calculated as combination DSS minus single agent DSS, termed as “Delta DSS.”
Reverse Phase Protein Array (RPPA)
Human pancreatic cancer cell lines MIA PaCA-2, Pa02C, Pa14C and Pa16C were seeded (2×105 cells per well) onto 6-well plates. The next day cells were treated with vehicle control DMSO (≤0.01%), LY3009120 RAFi (0.3 μM), SCH772984 ERKi (0.04 μM), or the combination of the two inhibitors for 15 min, and 1, 8, 24 or 72 hr. Cells were lysed and processed as previously described (Baldelli et al., 2017). Coomassie Protein Assay Reagent kit (Thermo Fisher Scientific) was used to measure protein concentration, following the manufacturer’s instructions. 2X Tris-glycine SDS sample buffer (Life Technologies) with 5% β-mercaptoethanol was used to dilute cell lysates to 0.5 mg/ml. Samples were boiled for 8 min and stored at −20°C until arrayed. An Aushon 2470 automated system (Aushon BioSystems) (Pierobon et al., 2017) was used to immobilize cell lysates and the internal controls and print in technical replicates (n = 3) onto nitrocellulose-coated glass slides (Grace Bio-Labs). Sypro Ruby Protein Blot Stain (Molecular Probes) was used to quantify protein concentration in each sample, following the manufacturer’s instructions. Reblot Antibody Stripping solution (Chemicon) was used to pretreat the remaining arrays (15 min at room temperature). The arrays were washed with PBS and incubated in I-block (Tropix) for 5 hr before antibody staining (Baldelli et al., 2017). Arrays were incubated with 3% H2O2, Avidin, Biotin (DakoCytomation), and an additional serum-free protein block (DakoCytomation) to reduce nonspecific binding of endogenous proteins. Staining was performed using an automated system (DakoCytomation) was used. Each slide was probed for 30 min with one antibody targeting the protein of interest, with 157 antibodies that target proteins involved in signaling networks that regulate cell growth, survival and metabolism were used to probe arrays (Table S2J). All antibodies used were validated as described previously (Signore and Reeder, 2012). Signal amplification was determined by using biotinylated anti-rabbit (Vector Laboratories) or anti-mouse secondary antibody (DakoCytomation) and a commercially available tyramide-based avidin-biotin amplification system (Catalyzed Signal Amplification System, DakoCytomation). IRDye 680RD streptavidin (LI-COR Biosciences) fluorescent detection system was used. TECAN laser scanner was used to scan Sypro Ruby and antibody stained slides, and the images were analyzed using commercially available software (MicroVigene Version 5.1.0.0, Vigenetech) as previously described (Baldelli et al., 2015).
RNA Sequencing
The human pancreatic cancer cell lines MIA PaCA-2, Pa02C, Pa14C and Pa16C were seeded at ~50% confluency. The next day the cells were treated with the vehicle control DMSO (≤0.01%), LY3009120 (RAFi, 0.3 μM), SCH772984 (ERKi, 0.04 μM) or the combination of the two inhibitors for 4 or 24 hr. The cells were washed twice with ice-cold PBS and before scrape removal in ice-cold PBS. The cells were collected by centrifugation at 326 x g (500 rpm) at 4°C for 10 min, and the cell pellets were flash frozen by liquid nitrogen. Whole transcriptome libraries were generated from total RNA (50 ng) of the human pancreatic cancer cell lines by using Illumina’s Truseq RNA Sample Prep to perform RNA sequencing. Oligo(dT) magnetic beads were used to select Poly(A) mRNA, and TruSeq PCR Master Mix and primer cocktail were used to enrich the libraries. The Agilent Bioanalyzer and Invitrogen Qubit were used to clean and quantify the amplified products. The Illumina HiSeq 2500 was used to sequence the clustered flowcell for paired 100-bp reads by using Illumina’s TruSeq SBS Kit V3. Lane level fastq files were appended together if they were sequenced across multiple lanes. These fastq files were then aligned with STAR 2.3.1 to GRCh37.62 using ensembl.74.genes.gtf as GTF files. Transcript abundances were quantified by HTSeq in total read counts per transcript.
MIB/MS
The human pancreatic cancer cell line MIA PaCA-2 was treated with the vehicle control DMSO (≤0.01%), LY3009120 (RAFi; 0.3 μM), trametinib (MEKi; 0.5 nM), SCH772984 ERKi (0.04 μM), or the combinations of RAFi/MEKi and RAFi/ERKi for 72 hr. The samples were prepared as described previously (Duncan et al., 2012). Briefly, the cells were processed on ice by using MIB lysis buffer [50 mM HEPES (pH 7.5), 0.5% Triton X-100, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10 mM sodium fluoride, 2.5 mM sodium orthovanadate, 1X protease inhibitor cocktail (Roche), 1% phosphatase inhibitor cocktail 2 (Sigma-Aldrich), and 1% of phosphatase inhibitor cocktail 3 (Sigma-Aldrich)]. The cell lysates were sonicated (3 × 10 s) on ice and were collected by centrifugation (10,000 x g) at 4°C for 10 min. The supernatant was filtered through a 0.2 mm SFCA membrane. The lysates (~5 mg protein per experiment) were gravity-flowed over multiplexed kinase inhibitor beads (MIBs) (Sepharose conjugated to VI-16832, CTx-0294885, PP58, Purvalanol B, UNC8088A, UNC21474). MIBs were washed with high (1 M NaCl) and low salt (150 mM NaCl + 0.1% SDS) lysis buffers without the inhibitors. The samples were boiled with the elution buffer (100 mM Tris-HCl, 0.5% SDS, and 1% β-mercaptoethanol, pH 6.8) at 100°C for 5 min to elute the bound kinases from MIBs. The eluted kinases (proteins) were concentrated with Amicon Ultra-4 (10K cutoff) spin columns (Millipore), purified by removing the detergent using methanol/chloroform extraction, and digested by sequencing grade Trypsin (Promega) overnight at 37°C. Hydrated ethyl acetate extraction was used to remove triton, and PepClean C-18 spin columns (Pierce, Thermo Scientific) were used to de-salt the digested peptides.
Biological triplicates of the MIB samples were analyzed by LC-MS/MS as described previously (Vaseva et al., 2018). Briefly, each sample was injected onto an Easy Spray PepMap C18 column (75 μm id × 25 cm, 2 μm particle size) (Thermo Scientific) and separated over a 2 hr method. The gradient for separation consisted of 5%–32% mobile phase B at a 250 nl/min flow rate, where mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in ACN. The Thermo QExactive HF was operated in data-dependent mode where the 15 most intense precursors were selected for subsequent HCD fragmentation (set to 27%).
Flow Cytometry
TACS® Annexin V-FITC Kit (BD Biosciences) was used to measure apoptosis according to the manufacturer’s instructions. Detached cells in the culture medium and the trypsinized cells were collected, mixed and centrifuged at 300 x g for 5 min at room temperature. The cells were washed with ice-cold PBS and incubated in Annexin V Incubation Reagent (1% Annexin V-FITC, 1X propidium iodide solution, in 1X calcium-containing binding buffer) in the dark for 15 min at room temperature. Cell mixture was diluted 1:5 in 1X binding buffer. BD LSRFortessa flow cytometer was used to analyze the cells. FACSDiva v8.0.1 was used to collect and export 30,000 cells to be analyzed with Cytobank. A “cells” gate was generated to avoid small and large debris in the bottom right corner or off-scale on either axis by using a side scatter area (SSC-A) (y) versus forward scatter area (FSC-A) (x) dot plot. Propidium iodide area (PI-A) (y) versus fluorescein isothiocyanate area (FITC-A) (x) dot plot was used to measure apoptosis. Vehicle control DMSO (≤ 0.01%) treated cells were assigned as the healthy cell population by a quadrant gate (PI-A negative, FITC-A negative).
Immunofluorescence
Human pancreatic cancer cells Pa01C and Pa02C were plated on 10 μg/ml fibronectin-coated glass coverslips and treated with DMSO (≤0.01%), LY3009120 RAFi (0.3 μM), SCH772984 ERKi (0.04 μM), or the combination of the two inhibitors for 72 hr. The cells were washed twice with PBS, fixed in 3.7% formaldehyde (Fisher Scientific) for 10 min at room temperature, washed with PBS and permeabilized with 0.1% Triton X-100 (Sigma) for 5 min at room temperature. Non-specific signals were blocked using 2% BSA (Sigma) in PBS for 30 min at room temperature. Cells were incubated with the primary antibody (diluted in 2% BSA-PBS) for 40 min at room temperature, followed by three washes with PBS and incubation with the secondary antibody (diluted in 2% BSA-PBS) for 45 min at room temperature. After washing three times with PBS, cells were mounted with Prolong Diamond antifade mounting media (Invitrogen). E-cadherin was visualized by anti-rabbit E-cadherin (clone 24E10; Cell Signaling, 1:100) followed by a goat-anti-rabbit Alexa Fluor-568 conjugated secondary antibody (Invitrogen, 1:200). Phalloidin conjugated with an Alexa 488 fluorophore (Invitrogen, 1:200) was used to visualize F-actin and DAPI (Invitrogen, 1:10000) was used to label the nucleus. Images were acquired in five random fields on an Olympus FV1000 confocal microscope using a 40x objective (1.2 zoom) and a maximally opened pinhole.
Organoid Viability Assay
PDAC organoids were dissociated and 3000 single cells per well were seeded in 150 μl of 10% growth factor reduced Matrigel (Corning) and 90% human organoid feeding media + 10.5 μM Y27632 (Selleckchem) into Poly(2-hydroxyethyl methacrylate) (SIGMA) coated 96-well clear flat bottom plates (Corning Ref. 3903). On the second day after seeding organoids were drugged with SCH772984 (0.04 μM to 0.63 μM) and LY3009120 (0.01 μM to 2.5 μM). Ten days after drugging, organoids were imaged with the Molecular Devices SpectraMax i3x MiniMax 300 imaging cytometer. After image acquisition organoid viability was accessed with CellTiter-Glo® 3D Cell Viability Assay (Promega) according to manufactures protocol on the SpectraMax i3x plate reader. Colorectal organoid cultures were immersed in 90% Matrigel (BD, Cat# 356231), plated onto 24-well plates in 20 ml drops per well and upon solidification overlaid with 500 μl CCM. For inhibitor experiments the organoids were harvested and trypsinized until single cell suspension was available for plating onto flat bottom 96-well format (Corning, Cat. No. 3603) (outer wells filled with PBS only). Per well 1000-1500 single cell organoids were plated in a 6 μl Matrigel drop and 150 μl CCM was added to culture PD3D’s for two days prior inhibitor addition. The inhibitors were added to the CRC PD3D’s as specified using the Tecan 300D dispenser and incubated for 5 days prior monitoring of proliferation by microscopy (Exp1-3, Tecan Spark, Exp 4 and 5, Keyence BZ-X710) and CellTiter-Glo® 3D measurement.
Bliss synergy scores were calculated using SynergyFinder as described in “Proliferation assays.”
PDAC Xenograft Study
Five × 106 HPAF II (ATCC# CRL-1997), CFPAC-1 (ATCC# CRL-1918) or SW1990 (ATCC# CRL-2172) pancreatic cancer cells in a 1:1 Matrigel mix (HPAF-II and SW1990; 0.2 mL total volume) were injected subcutaneously into the right hind flank of 7- to 8-week old (125-150 g) female athymic NIH (NIHRNU-IVI) nude rats (Taconic Farms). After tumors reached 200-300 mm3, animals were randomized into groups of six. Both LY3214996/ERKi and LY3009120/RAFi were administered orally (gavage) in 0.2 mL volume of vehicle (1%HEC/0.25% Tween 80/0.05% Antifoam) for 21 days. Tumor volume and body weight are measured twice weekly. Tumor volume is estimated by using the formula: v = l x w2 × 0.536 where l = larger of measured diameter and w = smaller of perpendicular diameter.
QUANTIFICATION AND STATISTICAL ANALYSIS
Prism Software
GraphPad Prism (versions 7.0.4 and 8.0.2) built-in tests were used to analyze the data provided. For the boxplots and growth inhibition curves, data are the mean average of 3 independent experiments and error bars are represented as ± SEM (unless otherwise noted). p values are indicated on the graphs and the absolute values are denoted in the figure legends. All data presented are normalized to their respective vehicle control. Technical and biological replicates are as indicated in the figure legends.
ImageJ Software
ImageJ (version 2.0.0) software was used to analyze images (Schneider et al., 2012).
SynergyFinder
Bliss synergy scores were calculated using SynergyFinder (version 1.6.1) R (version 3.5.1) package (Ianevski et al., 2017) as described in “Proliferation assays.”
Decision Tree and Forest Analyses
Decision tree and forest analyses were performed by using Sci-Kit Learn (version 0.20.3) in Python (version 3.6.3). RPPA dataset was grouped into five categories: vehicle control versus RAFi, vehicle control versus ERKi, vehicle control versus RAFi + ERKi, RAFi versus RAFi + ERKi, and ERKi versus RAFi + ERKi. RPPA dataset was further grouped into two categories as training and test data. Training (70% of RPPA dataset) and test data (remining 30% of RPPA dataset) comprise equal distribution of samples from the five categories described above. Decision trees were generated by using these training and test data. Accuracy for decision trees was above 80%.
For decision forest analysis, 70% of the data from each of the five categories (described above) were fit to a decision forest by a Random Grid Search to optimize for the best fitting forest based on F1 macro score (number of estimators between 300 and 500 and max features between 30 and 150). Once a model was obtained, accuracy on the test set was accessed (greater than 90% for all categories) and feature importance’s from the model were used to rank the most critical proteins.
CRISPR Analysis
CRISPR sequencing was performed at UNC (High-Throughput Sequencing Facility, Chapel Hill, NC) and Hudson Alpha (HudsonAlpha Genomic Services Laboratory, Huntsville, AL). Single-end sequencing (75 bp) was performed by using Illumina NextSeq 500 with ~10% PhiX (varied depending on the run). Read counts for individual samples were quantified using the protocol outlined in Shalem et al. (2014). Briefly, unaligned reads in FASTQ format were used to generate a Burrows-Wheeler index using the Bowtie build-index function. Reads were then aligned to the index using the Bowtie aligner, followed by the quantification of the number of reads per sgRNA. For each pair of samples (vehicle control and drug treated), guide constructs were removed if they were not identified in the vehicle control. Missing values in the treated samples were imputed with the mean value of all guide constructs for that gene in the treated sample. Reads were then normalized between samples by converting them to a percentage of total reads per sample. The ratio of vehicle/treated for each construct yielded an “enrichment ratio” for the pair of samples per guide construct. To summarize results per gene, the average enrichment ratio (AER) was calculated for each gene among all constructs. Genes with a standard deviation in enrichment ratio greater than three were removed. Genes were rank ordered by their AER and assigned to bins according to their position in the ranked list. Genes were then given a value according to their bin assignment: top 10% sensitizers (bin value = 2), top 25% sensitizer, excluding top 10% (bin value = 1), top 10% antagonizer (bin value = −2), top 25% antagonizer, excluding top 10% (bin value = −1), neither sensitizer nor antagonizer (bin value = 0). Given the variability of genetic CRISPR-Cas9 screens (between samples and replicates), this bin placement method permitted greater comparability between sample pairs.
DSRT Chemical Library Screen Analysis
Inhibitors included in the screen were categorized by their known primary protein target (Gautam et al., 2016; Pemovskaet al., 2013; Yadavet al., 2014). Cell lines and drugs were hierarchically clustered using average linkage and Euclidean distance; δDSS values are displayed.
Reverse phase protein array (RPPA) analysis
Antibody intensity values were imported into R (version 3.5.2) and missing values were imputed with the median intensity value for the respective antibody. Data were log2 transformed and the fold change over the median vehicle value was calculated for each antibody, factored by cell line and time point. Euclidean distance and average linkage were utilized for semi-supervised hierarchical clustering of log2 fold-change values for antibodies.
RNA Sequencing Analysis
Counts data were imported into R (version 3.5.2) and differential expression analysis was performed with the DESeq2 package (version 1.22.2) (Liberzon et al., 2015, 2011; Love et al., 2014; Mootha et al., 2003; Subramanian et al., 2005). Significant hits were defined by an adjusted p value < 0.05. Gene set enrichment analysis (GSEA) was performed using the GSEA Desktop application (version 3.0, available at https://www.gsea-msigdb.org/gsea/index.jsp) and curated gene sets were obtained from MSigDB (v6.2, available at https://www.gsea-msigdb.org/gsea/msigdb/index.jsp). Specific gene sets are referenced in figure legends. GSEAPreranked was run using the DESeq2 test statistic as the weighted ranking factor for each comparison.
MIB/MS Analysis
Following MaxQuant processing of data and generation of Label-free quantification (LFQ) intensity values, data files were analyzed using R (version 3.5.2). A total of 207 kinases were identified with 191 present in > 50% of samples and containing two or more peptides. 27/191 kinases were missing one or more values after this filtering and these values were imputed. A normal distribution was modeled on the non-missing LFQ intensity values of the kinases containing missing intensity values. Imputed values were drawn randomly from this distribution. Following filtering and imputation, LFQ intensity values were log2 transformed and the fold change over the median vehicle value was calculated for each kinase. significant kinases between DMSO, ERKi, RAFi, and ERKi+RAFi treatments were determined using one-way ANOVA (Benjamini-Hochberg adjusted p value < 0.05). Euclidean distance and average linkage were utilized for unsupervised hierarchical clustering of log2 fold-change values for significant kinases.
Tumor Xenograft Statistical Analysis
The statistical analysis of the tumor volume data begins with a data transformation to a log scale to equalize variance across time and treatment groups. The log volume data are analyzed with a two-way repeated-measures analysis of variance by time and treatment using the MIXED procedures in SAS software (Version 9.3). The correlation model for the repeated-measures is Spatial Power. Treated groups are compared to the control group at each time point. The MIXED procedure is also used separately for each treatment group to calculate adjusted means and standard errors at each time point. Both analyses account for the autocorrelation within each animal and the loss of data that occurs when animals with large tumors are removed from the study early. The adjusted means and standard errors (s.e.) are plotted for each treatment group versus time. Analysis for tumor volume is based on log10 and spatial power covariance structure. P value is based on the comparison between two specific groups.
Tumor Xenograft Combination Analysis Method (Bliss independence)
First, the usual repeated-measures model is fit to log volume versus group and time. Then contrast statements are used to test for an interaction effect at each time point using the 2 specific treatments that are combined. This is equivalent to the Bliss Independence method and assumes that tumor volumes can, in theory, reach zero, i.e., complete regression. The expected additive response (EAR) for the combination is calculated on the tumor volume scale as: response (EAR) EAR volume = V1 * V2/V0, where V0, V1, and V2 are the estimated mean tumor volumes for the vehicle control, treatment 1 alone, and treatment 2 alone, respectively. If the interaction test is significant, the combination effect is declared statistically more than additive or less than additive depending on the observed combination mean volume being less than or more than the EAR volume, respectively. Otherwise, the statistical conclusion is additive. In addition, a biologically relevant range of additivity can be defined as X% above and below the EAR volume. Typically, X would be 25 to 40%. Then a biological conclusion can be made for the combination as synergistic (greater than additive), additive, or less than additive if the observed combination mean volume is below, in, or above the interval of additivity.
There may be situations were stasis is the best expected response. In those situations, the Bliss method can be applied directly to the % delta T/C values to obtain an EAR percent response: EAR % delta T/C = Y1 *Y2/100, where Y1 and Y2 are the percent delta T/C values for the single-agent treatments. Currently, there is no statistical test to compare the observed % delta T/C in the combination group versus the EAR, but the biological criterion described above can be applied.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) | Cell Signaling Technology | Cat# 4370; RRID:AB_2315112 |
| Rabbit polyclonal anti-p44/42 MAPK (ERK1/2) | Cell Signaling Technology | Cat# 9102; RRID:AB_330744 |
| Rabbit polyclonal anti-phospho-AKT (Ser473) | Cell Signaling Technology | Cat# 9271; RRID:AB_329825 |
| Rabbit polyclonal anti-Akt | Cell Signaling Technology | Cat# 9272; RRID:AB_329827 |
| Mouse monoclonal anti-Vinculin | Sigma-Aldrich | Cat# V9131; RRID:AB_477629 |
| Mouse monoclonal anti-c-K-Ras (Ab-1) (234-4.2) | Millipore | Cat# OP24; RRID:AB_2134115 |
| Mouse monoclonal anti-A-Raf (A-5) | Santa Cruz Biotechnology | Cat# sc-166771; Lot# A2617; RRID:AB_2060508 |
| Mouse monoclonal anti-Raf-B (F-7) | Santa Cruz Biotechnology | Cat# sc-5284; Lot# A0617; RRID:AB_626760 |
| Mouse monoclonal anti-Raf-1 (E-10) | Santa Cruz Biotechnology | Cat# sc-7267; Lot# I0117; RRID:AB_628196 |
| Rabbit polyclonal anti-phospho-MEK1 (Ser298) | Cell Signaling Technology | Cat# 9128; RRID:AB_330810 |
| Mouse monoclonal anti-MEK1/2 (L38C12) | Cell Signaling Technology | Cat# 4694; RRID:AB_10695868 |
| Rabbit monoclonal anti-c-Myc (D84C12) XP | Cell Signaling Technology | Cat# 5605; RRID:AB_1903938 |
| Rabbit polyclonal anti-phospho-p90RSK (Thr359/Ser363) | Cell Signaling Technology | Cat# 9344; RRID:AB_331650 |
| Rabbit monoclonal anti-RSK1/2/3 | Cell Signaling Technology | Cat# 9355; RRID:AB_659900 |
| Rabbit monoclonal anti-p27 kip1 | Cell Signaling Technology | Cat# 3688; RRID:AB_2077836 |
| Rabbit polyclonal anti-BAD | Cell Signaling Technology | Cat# 9292; RRID:AB_331419 |
| Rabbit monoclonal anti-Bim (C34C5) | Cell Signaling Technology | Cat# 2933; RRID:AB_1030947 |
| Rabbit monoclonal anti-E-Cadherin (24E10) | Cell Signaling Technology | Cat# 3195; RRID:AB_2291471 |
| Rabbit monoclonal anti-vimentin (D21H3) XP | Cell Signaling Technology | Cat# 5741; RRID:AB_10695459 |
| Rabbit polyclonal anti-phospho-FoxO1 (Thr24)/FoxO3a (Thr32) Antibody | Cell Signaling Technology | Cat# 9464; RRID:AB_329842 |
| Rabbit polyclonal anti-phospho-FoxO1 (Ser256) | Cell Signaling Technology | Cat# 9461; RRID:AB_329831 |
| Rabbit monoclonal anti-FoxO3a (D19A7) | Cell Signaling Technology | Cat# 12829; RRID:AB_2636990 |
| Rabbit monoclonal anti-phospho-c-Raf (Ser338) (56A6) | Cell Signaling Technology | Cat# 9427; RRID:AB_2067317 |
| Rabbit polyclonal anti-phospho-Cofilin (Ser3) | Cell Signaling Technology | Cat# 3311; RRID:AB_330238 |
| Rabbit polyclonal anti-Cofilin (D3F9) XP | Cell Signaling Technology | Cat# 5175; RRID:AB_10622000 |
| Rabbit monoclonal anti-phospho-RSK1 p90 (T359 + S363) antibody [E238] | Abcam | Cat# ab32413; RRID:AB_2181172 |
| Anti-RSK1 | Eli Lilly and Company | Cat# 334G |
| Mouse monoclonal anti-phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (E10) | Cell Signaling Technology | Cat# 9106; RRID:AB_331768 |
| Rabbit monoclonal anti-p44/42 MAPK (Erk1/2) (137F5) | Cell Signaling Technology | Cat# 4695; RRID:AB_390779 |
| Rabbit polyclonal anti-phospho-S6 Ribosomal Protein (Ser240/244) | Cell Signaling Technology | Cat# 2215; RRID:AB_331682 |
| Mouse monoclonal anti-S6 Ribosomal Protein (54D2) | Cell Signaling Technology | Cat# 2317; RRID:AB_2238583 |
| Mouse monoclonal anti-β-actin | Sigma-Aldrich | Cat# A5441; RRID:AB_476744 |
| Bacterial and Virus Strains | ||
| DH5α | Thermo Fisher | Cat# 18258012 |
| Biological Samples | ||
| HPAF-II KRAS mutant PDAC rat xenograft tumor tissue | Provided by Eli Lilly | Star methods section of this paper |
| CFPAC-1 KRAS mutant PDAC rat xenograft tumor tissue | Provided by Eli Lilly | Star methods section of this paper |
| SW1990 KRAS mutant PDAC rat xenograft tumor tissue | Provided by Eli Lilly | Star methods section of this paper |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BGB-283 (RAF family kinases and EGFR inhibitor) | Selleckchem | Cat# S7926 |
| BVD-523 (ERK1/2 inhibitor) | Provided by Biomed | Biomed |
| Valley Discoveries | Valley Discoveries | |
| Erlotinib (EGFR inhibitor) | Selleckchem | Cat# S7786 |
| FRAX-597 (PAK1/2/3 inhibitor) | Selleckchem | Cat# S7271 |
| LY3009120 (pan-RAF inhibitor) | Provided by Eli Lilly | Provided by Eli Lilly |
| LY3214996 (ERK1/2 inhibitor) | Provided by Eli Lilly | Provided by Eli Lilly |
| MK-2206 (AKT1/2/3 inhibitor) | Selleckchem | Cat# S1078 |
| SCH772984 (ERK1/2 inhibitor) | Selleckchem | Cat# S7101 |
| Trametinib (MEK1/2 inhibitor) | Selleckchem | Cat# S2673 |
| Vemurafenib (BRAF inhibitor) | Selleckchem | Cat# S1267 |
| Critical Commercial Assays | ||
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat# G7570 |
| RealTime-Glo MT Cell Viability Assay | Promega | Cat# G9711 |
| CellTox Green Cytotoxicity Assay | Promega | Cat# G8741 |
| TACS® Annexin V-FITC in situ apoptosis detection kit | Trevigen, Inc. | Cat# 4830 |
| Deposited Data | ||
| CRISPR | This paper | Star methods section of this paper |
| Drug sensitivity resistance testing (DSRT) | This paper | Star methods section of this paper |
| Reverse phase protein array (RPPA) | This paper | Star methods section of this paper |
| RNA sequencing | EMBL-EBI European Nucleotide Archive (ENA) database - https://www.ebi.ac.uk/ena/ | PRJEB38063 |
| Multiplexed kinase inhibitor beads and mass spectrometry (MIB/MS) | This paper | Star methods section of this paper |
| Experimental Models: Cell Lines | ||
| Human: HPAC (pancreatic adenocarcinoma) | ATCC | Cat# CRL-2119, RRID:CVCL_3517 |
| Human: MIA PaCa-2 (pancreatic ductal adenocarcinoma) | ATCC | CRM-CRL-1420, RRID:CVCL_0428 |
| Human: PANC-1 (pancreatic ductal adenocarcinoma) | ATCC | Cat# CRL-1469, RRID:CVCL_0480 |
| Human: Pa01C (pancreatic ductal adenocarcinoma) | (Jones et al., 2008) | (Jones et al., 2008) |
| Human: Pa02C (pancreatic ductal adenocarcinoma) | (Jones et al., 2008) | (Jones et al., 2008) |
| Human: Pa03C (pancreatic ductal adenocarcinoma) | (Jones et al., 2008) | (Jones et al., 2008) |
| Human: Pa04C (pancreatic ductal adenocarcinoma) | (Jones et al., 2008) | (Jones et al., 2008) |
| Human: Pa14C (pancreatic ductal adenocarcinoma) | (Jones et al., 2008) | (Jones et al., 2008) |
| Human: Pa16C (pancreatic ductal adenocarcinoma) | (Jones et al., 2008) | (Jones et al., 2008) |
| Human: A549 (lung adenocarcinoma) | ATCC | Cat# CCL-185, RRID:CVCL_0023 |
| Human: NCI-H358 (minimally invasive lung adenocarcinoma) | ATCC | Cat# CRL-5807, RRID:CVCL_1559 |
| Human: SW900 (squamous cell lung carcinoma) | ATCC | ATCC Cat# HTB-59, RRID:CVCL_1731 |
| Human: SW620 (Colon adenocarcinoma) | ATCC | Cat# CCL-227, RRID:CVCL_0547 |
| Human: HPAF-II (pancreatic adenocarcinoma) | ATCC | Cat# CRL-1997, RRID:CVCL_0313 |
| Human: CFPAC-1 (pancreatic ductal adenocarcinoma, cystic fibrosis) | ATCC | Cat# CRL-1918, RRID:CVCL_1119 |
| Human: SW1990 (pancreatic adenocarcinoma) | ATCC | Cat# CRL-2172, RRID:CVCL_1723 |
| Experimental Models: Organisms/Strains | ||
| Human: hM1A PDAC organoid | (Boj et al., 2015) | (Boj et al., 2015) |
| Human: hT2 PDAC organoid | (Boj et al., 2015) | (Boj et al., 2015) |
| Human: OT227 CRC organoid | (Schütte et al., 2017) | (Schütte et al., 2017) |
| Human: OT228 CRC organoid | (Schütte et al., 2017) | (Schütte et al., 2017) |
| Human: OT302 CRC organoid | (Schütte et al., 2017) | (Schütte et al., 2017) |
| Rat: HPAF II PDAC model | ATCC | ATCC# CRL-1997 |
| Rat: CFPAC-1 PDAC model | ATCC | ATCC# CRL-1918 |
| Rat: SW1990 PDAC model | ATCC | ATCC# CRL-2172 |
| Oligonucleotides | ||
| shRNA targeting sequence: ARAF #1 CCGGCCAGCCAATCAATGTTCGTCTCTCG AGAGACGAACATTGATTGGCTGGTTTTTT |
(Freeman et al., 2013) | TRCN0000000567 |
| shRNA targeting sequence: ARAF #2 CCGGGTAGAGGAGGTAGTGATGGAACTC GAGTTCCATCACTACCTCCTCTACTTTTT |
(Freeman et al., 2013) | TRCN0000000568 |
| shRNA targeting sequence: BRAF #1 CCGGCCGCTGTCAAACATGTGGTTACTCG AGTAACCACATGTTTGACAGCGGTTTTT |
(Freeman et al., 2013) | TRCN0000006290 |
| shRNA targeting sequence: BRAF #2 CCGGGCTGGTTTCCAAACAGAGGATCTCG AGATCCTCTGTTTGGAAACCAGCTTTTT |
(Freeman et al., 2013) | TRCN0000006291 |
| shRNA targeting sequence: CRAF #1 CCGGGCTTCCTTATTCTCACATCAACTC GAGTTGATGTGAGAATAAGGAAGCTTTTT |
(Freeman et al., 2013) | TRCN0000001065 |
| shRNA targeting sequence: CRAF #2 CCGGCGGAGATGTTGCAGTAAAGATCTC GAGATCTTTACTGCAACATCTCCGTTTTT |
(Freeman et al., 2013) | TRCN0000001066 |
| shRNA targeting sequence: KRAS #1 CAGTTGAGACCTTCTAATTGG | (Singh et al., 2009) | TRCN0000010369 |
| Software and Algorithms | ||
| ImageJ version 2.0.0-rc-69/1.52n software | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| DESeq2 package (version 1.22.2) | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GSEA (version 3.0) | (Subramanian et al., 2005) | http://www.gsea-msigdb.org/gsea/index.jsp |
| MSigDB (version 6.2) | (Liberzon et al., 2015) | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp |
| Prism software (version 8.0.2) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Python (version 3.6.3) | Python Software Foundation | https://www.python.org/ |
| R (version 3.5.1) | R Core Team (2013) | http://www.R-project.org/ |
| Sci-Kit Learn (version 0.20.3) | (Pedregosa et al., 2011) | https://scikit-learn.org |
| SynergyFinder (version 1.6.1) | (lanevski et al., 2017) | https://bioconductor.org/packages/release/bioc/html/synergyfinder.html |
Highlights.
Vertical inhibition of the RAF-MEK-ERK cascade drives apoptotic cell death rather than cytostasis
RAF+ERK inhibition induces powerful synergy, enabling effective treatment at low doses
Concurrent RAF+ERK inhibition renders treated cells insensitive to compensatory ERK reactivation
Concurrent RAF+ERK inhibition induces MET and silences the MYC-dependent transcriptome
ACKNOWLEDGMENTS
We thank Deborah Morrison (National Cancer Institute/NCI) for the RAF shRNA vectors and David Tuveson (Cold Spring Harbor Laboratory) for PDAC organoids. Support was provided by grants from the NCI to A.D.C. and/or C.J.D. from the NCI (R01CA42978, R01CA175747, R01CA223775, P50CA196510, U01CA199235, P01CA203657, and R35CA232113) and from the Pancreatic Cancer Action Network (15-90-25-DER), Department of Defense (W81XWH-15-1-0611), and the Lustgarten Foundation (388222) to C.J.D. I.O.-D. and J.N.D. were supported by fellowships from the Slomo and Cindy Silvian Foundation. J.N.D. was also supported by NCI T32CA071341 and F30CA243253. B.P. was supported by the Deutsche Forschungsgemeinschaft (DFG PA 3051/1-1). A.M.W. was supported a fellowship from the American Cancer Society (PF-18-061). C.M.G. was supported by NCI T32CA009156 and F32CA221005.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107764.
DECLARATION OF INTERESTS
C.J.D. is a consultant/advisory board member for Mirati Therapeutics and Deciphera Pharmaceuticals. C.J.D. has received research funding support from Mirati Therapeutics and Deciphera Pharmaceuticals and has consulted for Ribometrix, Jazz Therapeutics, SVB Leerink, Axon Advisors LLC, Third Bridge, SmartAnalyst, Turning Point Therapeutics, and Eli Lilly. A.D.C. has consulted for Eli Lilly and Mirati Therapeutics. E.F.P. and M.P. receive royalties from Avant Diagnostics. E.F.P. is a consultant to and shareholder of Avant Diagnostics, Inc. and Perthera, Inc. and received funding support from Mirati Therapeutics, Genentech, Inc., and Abbvie, Inc. R.V.T. was previously employed at Eli Lilly and Company and is now employed at Astellas Pharma US. S.V.B., S.-B.P., and R.V.T. are shareholders of Eli Lilly.
REFERENCES
- Ahmed TA, Adamopoulos C, Karoulia Z, Wu X, Sachidanandam R, Aaronson SA, and Poulikakos PI (2019). SHP2 Drives Adaptive Resistance to ERK Signaling Inhibition in Molecularly Defined Subsets of ERK-Dependent Tumors. Cell Rep. 26, 65–78.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GR,Winter PS, Lin KH, Nussbaum DP, Cakir M, Stein EM, Soderquist RS, Crawford L, Leeds JC, Newcomb R, et al. (2017). A Landscape of Therapeutic Cooperativity in KRAS Mutant Cancers Reveals Principles for Controlling Tumor Evolution. Cell Rep. 20, 999–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldelli E, Bellezza G, Haura EB, Crinó L, Cress WD, Deng J, Ludovini V, Sidoni A, Schabath MB, Puma F, et al. (2015). Functional signaling pathway analysis of lung adenocarcinomas identifies novel therapeutic targets for KRAS mutant tumors. Oncotarget 6, 32368–32379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldelli E, Calvert V, Hodge A, VanMeter A, Petricoin EF III, and Pierobon M (2017). Reverse Phase Protein Microarrays. Methods Mol. Biol 7606, 149–169. [DOI] [PubMed] [Google Scholar]
- Blasco RB, Francoz S, Santamaría D, Cañamero M, Dubus P, Charron J, Baccarini M, and Barbacid M (2011). c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell 79, 652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss CI (1939). The toxicity of poisons applied jointly. Ann. Appl. Biol 26, 585–615. [Google Scholar]
- Blumenschein GR Jr., Smit EF, Planchard D, Kim DW, Cadranel J, De Pas T, Dunphy F, Udud K, Ahn MJ, Hanna NH, et al. (2015). A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC). Ann. Oncol 26, 894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, et al. (2015). Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM, George SD, et al. (2019). Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med 25, 628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Bednar F, Zhang Y, Brisset JC, Galbán S, Galbán CJ, Rakshit S, Flannagan KS, Adsay NV, and Pasca di Magliano M (2012). Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Invest 722, 639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, Charles RP, Rabinovich BA, Hann B, Dankort D, et al. (2012). A central role for RAF→MEK→ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2, 685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AD, Fesik SW, Kimmelman AC, Luo J, and Der CJ (2014). Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov 13, 828–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, and Weinberg RA (2019). EMT and Cancer: More Than Meets the Eye. Dev. Cell 49, 313–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desideri E, Cavallo AL, and Baccarini M (2015). Alike but Different: RAF Paralogs and Their Signaling Outputs. Cell 161, 967–970. [DOI] [PubMed] [Google Scholar]
- Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, Garbe C, Schadendorf D, Krajsova I, Gutzmer R, et al. (2018). Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus bini-metinib versus vemurafenib or encorafenib (COLUMBUS): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 19, 1315–1327. [DOI] [PubMed] [Google Scholar]
- Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, et al. (2012). Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 149, 307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, et al. (2008). Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med 14, 1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, Hieber M, Arbeiter A, Klein S, Kong B, et al. (2013). Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 23, 406–420. [DOI] [PubMed] [Google Scholar]
- Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, Araki K, Ozerdem U, Simeone DM, Miller G, et al. (2018). SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov. 8, 1237–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, et al. (2012). Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med 367, 1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman AK, Ritt DA,and Morrison DK(2013).Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol. Cell 49, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam P, Karhinen L, Szwajda A, Jha SK, Yadav B, Aittokallio T, and Wennerberg K (2016). Identification of selective cytotoxic and synthetic lethal drug responses in triple negative breast cancer cells. Mol. Cancer 15, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Brown KR, Sircoulomb F, Rottapel R, and Moffat J (2014). Measuring error rates in genomic perturbation screens: gold standards for human functional genomics. Mol. Syst. Biol 10, 733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, et al. (2010). RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464, 431–435. [DOI] [PubMed] [Google Scholar]
- Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, Aziz M, Kassner M, Bryant KL, Pierobon M, et al. (2016). Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell 29, 75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianevski A, He L, Aittokallio T, and Tang J (2017). SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics 33, 2413–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, et al. (2018). Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 172, 578–589.e17. [DOI] [PubMed] [Google Scholar]
- Jänne PA, van den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crinò L, Orlov S, Blackhall F, Wolf J, Garrido P, et al. (2017). Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA 317, 1844–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. (2008). Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellos G, and Frame MC (2016). Cellular functions of the ADF/cofilin family at a glance. J. Cell Sci 129, 3211–3218. [DOI] [PubMed] [Google Scholar]
- Karreth FA, Frese KK, DeNicola GM, Baccarini M, and Tuveson DA (2011). C-Raf is required for the initiation of lung cancer by K-Ras(G12D). Cancer Discov. 1, 128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AJ, Sun H, Diaz B, Barnard D, Miao W, Bagrodia S, and Marshall MS (1998). The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature 396, 180–183. [DOI] [PubMed] [Google Scholar]
- Lake D, Corrêa SA, and Müller J (2016). Negative feedback regulation of the ERK1/2 MAPK pathway. Cell. Mol. Life Sci 73, 4397–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamba S, Russo M, Sun C, Lazzari L, Cancelliere C, Grernrum W, Lieftink C, Bernards R, Di Nicolantonio F, and Bardelli A (2014). RAF suppression synergizes with MEK inhibition in KRAS mutant cancer cells. Cell Rep. 8, 1475–1483. [DOI] [PubMed] [Google Scholar]
- Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, Mandalà M, Demidov L, Stroyakovskiy D, Thomas L, et al. (2014). Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med 371, 1867–1876. [DOI] [PubMed] [Google Scholar]
- Lax I, Wong A, Lamothe B, Lee A, Frost A, Hawes J, and Schlessinger J (2002). The docking protein FRS2alpha controls a MAP kinase-mediated negative feedback mechanism for signaling by FGF receptors. Mol. Cell 10, 709–719. [DOI] [PubMed] [Google Scholar]
- Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, Li J, Wang G, Lan Z, Li J, et al. (2019). KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 35, 559–572.e557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, et al. (2014). Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med 371, 1877–1888. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Liu S, Zhang G, Bin Wu, Zhu Y, Frederick DT, Hu Y, Zhong W, Randell S, Sadek N, et al. (2017). PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature 550, 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainardi S, Mulero-Sánchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, Lieftink C, Steinberg JD, de Wit N, Gonçalves-Ribeiro S, et al. (2018). SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med 24, 961–967. [DOI] [PubMed] [Google Scholar]
- Markosyan N, Li J, Sun YH, Richman LP, Lin JH, Yan F, Quinones L, Sela Y, Yamazoe T, Gordon N, et al. (2019). Tumor cell-intrinsic EPHA2 suppresses anti-tumor immunity by regulating PTGS2 (COX-2). J. Clin. Invest 130, 3594–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martz CA, Ottina KA, Singleton KR, Jasper JS, Wardell SE, Peraza-Penton A, Anderson GR, Winter PS, Wang T, Alley HM, et al. (2014). Systematic identification of signaling pathways with potential to confer anticancer drug resistance. Sci. Signal 7, ra121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant M, Moffat J, Schaefer G, Chan J, Wang X, Orr C, Cheng J, Hunsaker T, Shao L, Wang SJ, et al. (2017). Combined MEK and ERK inhibition overcomes therapy-mediated pathway reactivation in RAS mutant tumors. PLoS ONE 12, e0185862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003).PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Morrison DK (2012). MAP kinase pathways. Cold Spring Harb. Perspect. Biol 4, a011254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem JM, and Shokat KM (2016). Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat. Rev. Drug Discov 15, 771–785. [DOI] [PubMed] [Google Scholar]
- Papke B, and Der CJ (2017). Drugging RAS: Knowtheenemy. Science 355, 1158–1163. [DOI] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res 12, 2825–2830. [Google Scholar]
- Pemovska T, Kontro M, Yadav B, Edgren H, Eldfors S, Szwajda A, Almusa H, Bespalov MM, Ellonen P, Elonen E, et al. (2013). Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov. 3, 1416–1429. [DOI] [PubMed] [Google Scholar]
- Peng SB, Henry JR, Kaufman MD, Lu WP, Smith BD, Vogeti S, Rutkoski TJ, Wise S, Chun L, Zhang Y, et al. (2015). Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell 28, 384–398. [DOI] [PubMed] [Google Scholar]
- Pierobon M, Ramos C, Wong S, Hodge KA, Aldrich J, Byron S, Anthony SP, Robert NJ, Northfelt DW, Jahanzeb M, et al. (2017). Enrichment of PI3K-AKT-mTOR Pathway Activation in Hepatic Metastases from Breast Cancer. Clin. Cancer Res 23, 4919–4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, and Rosen N (2010). RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane CK, and Minden A (2019). P21 activated kinase signaling in cancer. Semin. Cancer Biol 54, 40–49. [DOI] [PubMed] [Google Scholar]
- Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu B , Görgülü K, Dantes Z, Wörmann SM, Diakopoulos KN, et al. (2018). Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med 24, 954–960. [DOI] [PubMed] [Google Scholar]
- Ryan MB, and Corcoran RB (2018). Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol 15, 709–720. [DOI] [PubMed] [Google Scholar]
- Ryan DP, Hong TS, and Bardeesy N (2014). Pancreatic adenocarcinoma. N. Engl. J. Med 371, 2140–2141. [DOI] [PubMed] [Google Scholar]
- Samatar AA, and Poulikakos PI (2014). Targeting RAS-ERK signalling in cancer: promises and challenges. Nat. Rev. Drug Discov 13, 928–942. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütte M, Risch T, Abdavi-Azar N, Boehnke K, Schumacher D, Keil M, Yildiriman R, Jandrasits C, Borodina T, Amstislavskiy V, et al. (2017). Molecular dissection of colorectal cancer in pre-clinical models identifies bio-markers predicting sensitivity to EGFR inhibitors. Nat. Commun 8, 14262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signore M, and Reeder KA (2012). Antibody validation by Western blotting. Methods Mol. Biol 823, 139–155. [DOI] [PubMed] [Google Scholar]
- Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, and Settleman J (2009). A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 15, 489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM, et al. (2014). Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508, 118–122. [DOI] [PubMed] [Google Scholar]
- TCGA (2017). Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 32, 185–203.e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo A, Perurena N, Guruceaga E, Mazur PK, Martinez-Canarias S, Zandueta C, Valencia K, Arricibita A, Gwinn D, Sayles LC, et al. (2017). An integrative approach unveils FOSL1 as an oncogene vulnerability in KRAS-driven lung and pancreatic cancer. Nat. Commun 8, 14294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaseva AV, Blake DR, Gilbert TSK, Ng S, Hostetter G, Azam SH, Ozkan-Dagliyan I, Gautam P, Bryant KL, Pearce KH, et al. (2018). KRAS Suppression-Induced Degradation of MYC Is Antagonized by a MEK5-ERK5 Compensatory Mechanism. Cancer Cell 34, 807–822.e807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, and Lander ES (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters AM, and Der CJ (2018). KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med 8, a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, Xu D, Schumacher SE, Puschhof J, McFarland J, et al. (2018). Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med 24, 968–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Ivanov AI, Fisher PB, and Fu Z (2016). Polo-like kinase 1 induces epithelial-to-mesenchymal transition and promotes epithelial cell motility by activating CRAF/ERK signaling. eLife 5, e10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Martelotto L, Baslan T, Vides A, Solomon M, Mai TT, Chaudhary N, Riely GJ, Li BT, Scott K, et al. (2017). An approach to suppress the evolution of resistance in BRAFV600E-mutant cancer. Nat. Med 23, 929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav B, Pemovska T, Szwajda A, Kulesskiy E, Kontro M, Karjalainen R, Majumder MM, Malani D, Murumägi A, Knowles J,et al. (2014). Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci. Rep 4, 5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen I, Shanahan F, Merchant M, Orr C, Hunsaker T, Durk M, La H, Zhang X, Martin SE, Lin E, et al. (2018). Pharmacological Induction of RAS-GTP Confers RAF Inhibitor Sensitivity in KRAS Mutant Tumors. Cancer Cell 34, 611–625.e617. [DOI] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan TL, Amzallag A, Bagni R, Yi M, Afghani S, Burgan W, Fer N, Strathern LA, Powell K, Smith B, et al. (2018). Differential Effector Engagement by Oncogenic KRAS. Cell Rep. 22, 1889–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the binary sequence alignment/map (BAM) files of RNA-seq data of cell lines MIA PaCA-2, Pa02C, Pa14C and Pa16C reported in this paper is [ENA] (the EMBL-EBI European Nucleotide Archive) PRJEB38063 (https://www.ebi.ac.uk/ena/, https://www.ebi.ac.uk/ena/data/view/PRJEB38063). The file names containing “R” indicate RAF inhibitor treated samples, “E” indicate ERK inhibitor treated samples, “C” indicate combination treated samples and “V” indicate vehicle control. The file names containing “4” and “24” indicate treatment time of 4 hours and 24 hours respectively.”
This study did not generate any unique codes.