

Summary
Glioblastoma is the most common and deadly primary brain malignancy. Despite advances in precision medicine oncology (PMO) allowing the identification of molecular vulnerabilities in glioblastoma, treatment options remain limited, and molecular assays guided by genomic and expression profiling to inform patient enrollment in life-saving trials are lacking. Here, we generate four-dimensional (4D) cell-culture arrays for rapid assessment of drug responses in glioblastoma patient-derived models. The arrays are 3D printed with thermo-responsive shape memory polymer (SMP). Upon heating, the SMP arrays self-transform in time from 3D cell-culture inserts into histological cassettes. We assess the utility of these arrays with glioblastoma cells, gliospheres, and patient derived organoid-like (PDO) models and demonstrate their use with glioblastoma PDOs for assessing drug sensitivity, on-target activity, and synergy in drug combinations. When including genomic and drug testing assays, this platform is poised to offer rapid functional drug assessments for future selection of therapies in PMO.
Subject Areas: Tissue Engineering, Medical Biotechnology, Cancer
Graphical Abstract
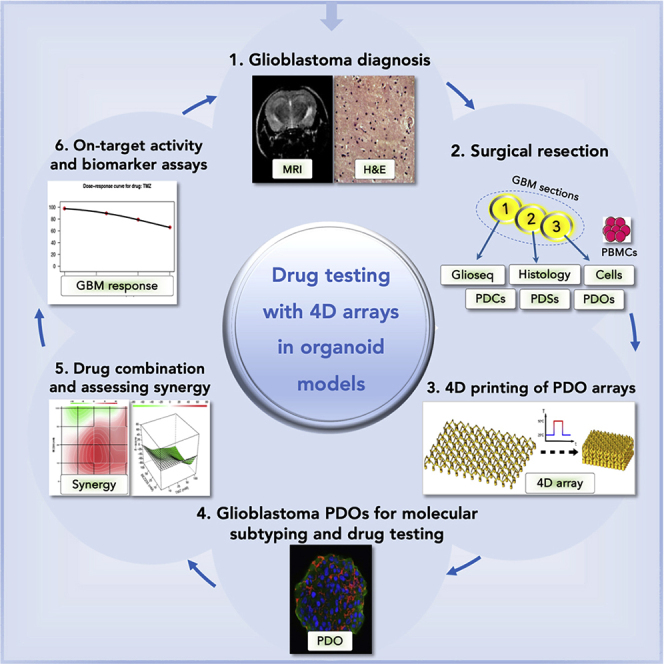
Highlights
-
•
4D printed thermo-responsive polymer arrays self-transform for 3D culture and histology
-
•
Patient-derived organoid models represent original tumors better than spheres in arrays
-
•
Cellular heterogeneity and drug responses are assessed in glioblastoma organoid models
-
•
Functional utility is shown by detecting drug sensitivity, on-target activity, and synergy
Tissue Engineering; Medical Biotechnology; Cancer
Introduction
Glioblastoma (GBM) is the most common and deadly primary brain malignancy (Siegel et al., 2020). The clinical suspicion of a brain tumor frequently follows a new onset of neurological deficits or seizures, with MRI being the imaging method of choice to detect brain tumors. However, a definite diagnosis of a GBM (World Health Organization grade IV brain tumor) can only be made histologically (Weller et al., 2015). Even when current therapies for GBM, including resection, irradiation, chemotherapy, and/or concurrent or adjuvant temozolomide (TMZ), are implemented shortly after diagnosis, tumors frequently recur, resulting in a median survival rate of patients with GBM averaging a dismal 15 months (Siegel et al., 2020). Extreme plasticity, the strong selective pressures occurring during early GBM development and with current therapies (Barthel et al., 2019), and intra- and inter-tumor heterogeneity in GBM, as revealed by single-cell RNA sequencing (scRNA-seq) studies (Neftel et al., 2019; Patel et al., 2014), impose the therapeutic failures. Moreover, the lack of patient-derived models of GBM that reflect these core elements of heterogeneity yet can be used to predict patient outcome; for instance predicting responses to therapy, resulted in high failure rates for many new drugs. In GBM, patient-derived models can be generated from bulk and/or GBM stem-like cells (GSCs). Although debated, the GSC phenotypic features (Gimple et al., 2019) and signaling pathways (Rajakulendran et al., 2019) could be employed to identify GBM molecular vulnerabilities in these models for examining targeted and personalized therapies. The Cancer Genome Atlas (TCGA) and other sequencing studies allowed the sub-classification of GBM into three molecular subtypes (Weller et al., 2015). However, scRNA-seq studies revealed that individual GBMs contain a spectrum of these subtypes (Patel et al., 2014), and are present in multiple cellular states within each tumor, with these states enriched by the genetic influence and/or the tumor microenvironment (TME) (Neftel et al., 2019).
Seminal studies established the ability of mammalian neural precursor cells, a presumed normal counterpart of GSCs, to respond to supplemented growth factors and to form either monolayer cultures of differentiated cells or free-floating spherical aggregates termed “neurospheres” (Reynolds and Weiss, 1992). Recent advances in three-dimensional (3D) culture allowed utilizing the remarkable self-organizing properties of embryonic stem cells (ESCs)/induced pluripotent stem cells (iPSCs) (Eiraku et al., 2008) to direct their neural developmental differentiation into human cerebral organoids, either in the presence (Lancaster et al., 2013) or absence (Pasca et al., 2015) of extracellular matrix (ECM) scaffolds. Notably, cerebral organoids recapitulated key neural cell growth, organization, and differentiation (Lancaster et al., 2013), thus substantiating their use in tissue regeneration and tumor modeling. When derived from GBM patient biopsies and/or surgical specimens, patient-derived cells (PDCs), patient-derived spheres (PDSs, also called gliospheres, analogous to glial neurospheres), and organoid-like cultures could be established. The development of 3D patient-derived organoid-like (PDO) culture of human primary tumors allowed for the initiation and long-term propagation of these PDO cultures from biopsies and/or surgical resections of multiple cancers (Tuveson and Clevers, 2019), including the derivation of GBM-PDOs from patients with recurrent and treated GBM (Hubert et al., 2016). These models could also be utilized for generating patient-derived xenografts (PDXs) at heterotopic (frequently subcutaneous) sites and/or in the brain (patient-derived orthotopic xenografts or PDOXs) for in vivo studies. Nevertheless, the xenografts remain expensive to generate, time consuming, and may become clonally distinct from the originating GBMs (Patrizii et al., 2018). Remarkably, the powerful potential of PDOs in modeling treatment responses and predicting clinical outcome of patients enrolled in clinical trials has been noted (Vlachogiannis et al., 2018). Moreover, a recent case report demonstrated the potential of using PDOs for tailoring treatment in GBM (Loong et al., 2020). Yet, the generation of PDOs for drug testing is still laborious and lengthy, requiring multiple steps including establishing and transporting of PDCs and ECM between thousands of wells for dissociation; making PDOs; allowing cellular growth for weeks to months; performing drug treatment with multiple compounds; assessing cell viability and tumorigenic assays; cell fixation and antibody staining; histologic processing; and final immunohistochemical (IHC) validations. These lengthy and laborious steps with manual transfers between each step prohibit the wider use of these assays in translational studies and make them unsuitable for integration into clinical diagnostic tests and/or large-scale drug screening. Targeted therapies could be designed to counter GBM heterogeneity (Prados et al., 2015), yet drug testing in PDOs for targeted therapy is a tedious process taking weeks to months to complete (Vlachogiannis et al., 2018). In addition, whereas using hydrogel-based carriers allowed simultaneous histological processing of spheroids and organoids (Parker et al., 2020), samples in these carriers still required time-consuming manual transfers between culture and histology vessels, also subjecting the delicate spheroids and organoids to the possibility of undesired distortions during processing.
We have previously utilized PDSs from primary tumors to model tumor heterogeneity and develop therapies to target the self-renewing stem-like cells (Bansal et al., 2016; Bartucci et al., 2017). In parallel, advances both in 3D and 4D printing (with 4D printing referring to 3D printing with smart materials that are responsive to stimuli, programming them to evolve from one 3D shape to another) allowed generating devices, implants, and scaffolds for tissue engineering. Here, we first generated expandable/collapsible smart material arrays by 3D printing. Upon heating, these 4D printed arrays self-transformed from cell-culture inserts into histological cassettes. Self-transformation occurred inclusive of their GBM-PDO contents, which remained in the same configuration throughout the entire assay, therefore allowing for rapid programmable drug testing and assessing indicators of effective and synergistic GBM combination therapy.
Results
Bioengineering of 4D Printed Cell-Culture Insert Arrays
As a first step toward streamlining pre-clinical studies of ex vivo models of GBM, we utilized 4D printing to fabricate the self-transformable cell-culture insert arrays. 4D printing refers to 3D prints with smart materials that change shape, properties, and/or functions in response to external stimuli, with the fourth dimension being time (Ge et al., 2016; Yang et al., 2019). The smart material utilized in this work was a shape memory polymer (SMP), and the high-precision 3D printing technique was projection micro-stereolithography (PμSL) (Zheng et al., 2012). Each cell-culture insert array consisted of interconnected wells and was capable of self-transforming, while maintaining the same layout, contents, and configurations, between the size of a standard 96-well plate and the size of a histology megacassette (3.6 times the overall dimensional change) (Figure 1 and Video S1). This process allowed completing the histological processing of the entire array in one sectioning step (see Transparent Methods). The SMPs could be fixed in a temporarily deformed shape (shape programming) and restored to the original shape (shape recovery) upon heating around glass-transition temperature (Tg) (Figures S1A and S1B). Each well of the cell-culture insert array consisted of a lower cell-culture compartment containing biological samples and four upper interconnecting helical bridges allowing self-transformation (Figures 1B and S1C). The self-transformable SMP cell-culture insert arrays were mounted on 96- or 6-well plates. The arrays were utilized for assessing the establishment of GBM PDCs in 3D PDSs and GBM-PDO cultures for 1 or 2 weeks or up to 12 weeks, respectively, after which cells were fixed and the entire cell-culture array self-transformed back to the size of a histology megacassette. Shape transformation was followed by microtome sectioning, staining, and imaging within the same platform, thus remarkably expediting the processing of these multiple histological assays. We first compared the growth conditions of GBM cells in our cell-culture arrays with the existing approaches using PDCs and GBM cells in 2D surface-coated adherence culture, 3D PDSs, and GBM-PDO cultures.
Figure 1.

Diagrams Representing Step-by-Step Operating Procedures of the 4D Cell-Culture Insert Array Shape Transformation
(A) At room temperature (RT), an insert in the cassette configuration was first mounted on a custom-built stretcher. The stretcher has eight rails that can simultaneously move all carriages sitting in rails between the dimension of a cassette and the dimension of a 96-well plate. The insert was stretched to a 96-well plate configuration by simply rotating the top and bottom plates against each other at RT. All helical bridges are unwound during the rotation of the stretcher. After rotation, both the insert and the stretcher were placed in an oven at 50°C for 10 min and then cooled down to RT to program the stretched shape. The insert array was then removed from the stretcher with the temporarily programmed shape. Cell-culture insert arrays were then mounted onto a fixture that has a perfect matching between edge units of the insert and a 96-well plate. The fixture with the insert was then placed on a 96-well plate for cell seeding. Cells, culture media, and treatment compounds were injected into insert wells using micropipettes (see Transparent Methods). After 3D cell culture, the insert was removed from the fixture and heated to 50°C to induce a shape recovery to the cassette configuration while maintaining the registry of the cultured cells. In the cassette configuration, the insert was ready for histological processing to obtain the histology of the entire cell-culture array.
(B) The diagram displays top and side views and the corresponding dimensions of an individual well component of the 4D printed cell-culture insert array. See also Figures S1 and S4 and Video S1 for demonstrations of the thermomechanical properties of the 4D printed cell-culture insert array and Figures S5–S7 for data determining the biocompatibility of these cell-culture arrays.
The printed insert that was stretched to the 96-well plate configuration transforms into the histological cassette configuration when heated to 50oC while maintaining the registry of the insert array. In the cassette configuration, the insert is ready for histological processing to obtain the histology of the entire 3D cell-culture insert array contents with a single histological processing and sectioning step. The movie was taken during the 20-min heat-responsive transformation and was set to 200X playback.
GBM-PDO Cultures Model Key GBM Features
Primary GBM were obtained from GBM patients undergoing craniotomy resection at Robert Wood Johnson University Hospital under an Institutional Review Board-approved protocol. Live cells were maintained in conditions to generate and maintain GBM PDSs (Mehta et al., 2015) or were used to generate organoid-like GBM-PDOs in conditions developed in our laboratory (see Transparent Methods), which were modified from those used to generate cerebral organoids (Lancaster et al., 2013) and GBM-PDOs (Hubert et al., 2016). Single cells were seeded at clonal densities in ultra-low attachment plates with culture inserts in serum-free growth factor-supplemented conditions for PDSs or in ECM droplets within inserts for GBM-PDOs (Figure 2A). To compare GBM-PDCs, GBM-PDSs, and GBM-PDOs in our system, we assessed if they resembled their corresponding originating tumors. We performed histological analyses and assessed variability in size, nuclear morphology, and cytologic features. Each PDS and PDO was independently confirmed by a neuropathologist to show key histological features of high-grade gliomas such as solid growth and diffuse invasive appearance using H&E staining. Although PDS cells multiplied significantly faster in the first 3–5 days when compared with PDO cells (Figures 2B and S2A), PDSs were smaller than PDOs at 2 weeks (PDSs: 100–300 μm, PDOs: 400–600 μm) (Figure S2B). GBM-PDOs grown in the 4D printed arrays were also larger than PDSs (Figure 2C). PDSs were still smaller than GBM-PDOs after 12 weeks of culture, where GBM-PDOs could be expanded to sizes reaching 2–3.5 mm (Figures S2B and S2C).
Figure 2.

Utilization of SMP Cell-Culture Insert Arrays for Histological Processing of GBM-PDOs
(A) Diagram displaying the cell-culture array when used in 6-well plates, and bright-field images of 14-day 3D PDO culture. Complexity of the organoid structures increased with time.
(B) Average cell doubling times during the first week of PDS and PDO culture from four patients with GBM.
(C) After 6–14 days of 3D PDS and/or PDO culture alone or in the array, PDSs and PDOs were fixed and stained with H&E. Images demonstrate a group of representative PDSs derived from GBM#50, with the lower image being a 4× magnification of a PDS in the top image. The lower right image demonstrates the phenotypes of PDOs from GBM#46 when cultured in the array.
(D) Sphere- and organoid-forming potentials were assessed using extreme limiting dilution analysis software at http://bioinf.wehi.edu.au/software/elda/. The frequencies based on log-fraction plot of the dilution model, log-active cell fractions, and 95% confidence interval are indicated from four GBM-PDSs and GBM-PDOs.
(E) Histological H&E analysis of original tissue (GBM#46), PDOXs generated from the same patient cells, PDSs, PDOs, and PDOs in the cell-culture insert array collected and fixed after 14 days of culture. Note that the cell density is different in patient and PDOX sections because the cell density depends on the number of engrafted or plated cells. See additional features of these PDSs and PDOs in Figure S2.
Comparison of doubling times and clonogenic potentials between PDSs versus PDOs in (B and D) are represented as mean ± SD of four replicates and were determined by two-way ANOVA with Bonferroni post-hoc test (∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05). Scale bars, 100 μm in (A, C, and E).
Quantitative analysis showed that PDSs had widely varying clonogenic sphere-forming potentials (Figure 2D). In contrast, clonogenic organoid-forming potential among GBM-PDOs were more uniform (Figure 2D). When compared morphologically with PDSs, GBM-PDOs showed notable pleomorphism in the central core and outside rim regions with significant variability in cell size, nuclear morphology, and cytologic features (Figure 2C). When examined histologically, GBM-PDOs showed histological features in H&E resembling the originating GBM patient tissue (Figure 2E). They also generated similar histological features when xenografted into the adult immune-deficient NSG mouse brain (Figure 2E), following procedures we described previously (Patrizii et al., 2018) (see Transparent Methods).
To characterize the cellular subset distribution within PDSs and GBM-PDOs from all four patient tumors, we examined neurodevelopmental markers associated with highly undifferentiated grade IV glioma and markers of self-renewal and differentiation by immunocytological assays. It was previously shown that the neurosphere assay enriches for neural stem cells (NSCs) and also likely GSCs, and when basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF) are removed from media or PDCs are cultured in the presence of serum and/or on polyornithine-coated surfaces, GBM cells undergo GSC loss (Mehta et al., 2015). Unlike PDSs that were enriched in NESTIN-expressing cells in the outer rim (Figure 3A), and PDCs that downregulated NESTIN but showed relatively more differentiation expression profiles toward mature astrocytic glial fibrillary acidic protein (GFAP)- and neuronal tubulin-beta-III (TUJ1)-expressing cells, we observed robust heterogeneity in size, cell identity, and morphology in GBM-PDOs between 2 and 12 weeks of culture (Figures 3B and S2). NESTIN expression was notably slightly denser near the outer rim of GBM-PDOs, perhaps due to the proximity to the stimulatory growth factors (EGF, bFGF). Yet, unlike PDSs, NESTIN was still present in cells within the GBM-PDO inner core. There was less dense overall staining in the GBM-PDO core, with the GBM-PDO centers containing multiple cellular areas in between the ECM (Figures 3B, 2E, and 4). Remarkably, 3D cultured genotypically matched GBM-PDOs remained heterogeneous when cultured in the 4D printed insert arrays for weeks and were capable of interconnecting (mimicking brain cells) and showing multiple cell phenotypes (Figures 3B and 4). Quantitative analysis showed that although PDSs are enriched in NESTIN-expressing neuroepithelial progenitors, they featured less GFAP-, galactosidase C (GALC)-, and TUJ1-expressing cell types (Figure 3C). These cellular subtype distributions were essentially reversed in PDCs cultured on polyornithine plates (or when bFGF and EGF were replaced with serum). PDCs downregulated NESTIN and featured GFAP- and TUJ1-expressing cells, but GALC was consistently less clearly detectable (Figure 3A). Therefore, PDSs and PDCs demonstrated distinct self-renewal and differentiation profile enrichment. On the other hand, GBM-PDOs demonstrated balanced NESTIN- GFAP-, GALC-, and TUJ1-expressing cell phenotypes (Figures 3B and 3C). NESTIN-expressing cells in the outer rim coexpressed other GSC markers like the orphan nuclear receptor tailless (TLX) (Figure 4A) and the iPSC and self-renewal protein SOX2, with TLX and SOX2 expressions being widely detected in PDSs, both at the outer and inner cores, albeit at lower levels in the inner core (Figures 4A and 4B).
Figure 3.

Comparing Cellular Components of Patient-Derived Models of GBM-PDCs, GBM-PDSs, and GBM-PDOs
(A) Immunofluorescence (IF) for NESTIN, which is used as a neuroepithelial progenitor marker, and glial fibrillary acidic protein (GFAP), galactosidase C (GALC), and tubulin-beta-III (TUJ1) (neuroepithelial differentiation markers). GFAP is used as a marker of CNS mature astrocytes and ependymal cells. GALC is a marker for early-stage oligodendroglia. TUJ1 is a neuron-specific early marker of signal commitment in primitive neuroepithelium. DAPI is used to label the nuclei, whereas phalloidin is used to mark the cell architectures within the PDCs, PDSs, and PDOs. Note that PDSs formed after GBM 3D culture for 1 week are enriched with NESTIN expression but not differentiation markers, but when the formed spheres were then cultured on polyornithine plates for an additional 10 days, GBM cells (PDCs) differentiated with partial expression of some differentiation markers detected.
(B) IF assays of GBM PDOs formed after 14 days of 3D PDO culture. PDOs readily express various progenitor (NESTIN in green) and/or differentiated neural markers (GFAP, GALC, or TUJ1 in green), representing GBM phenotypic cell heterogeneity. Images represent an overlay of green (NESTIN, GFAP, GALC, or TUJ1), red (phalloidin [ph]), and blue (DAPI [D] for nuclei) confocal images. Scale bars in (A) and (B) are 100 μm.
(C) Violin plot quantitation of phenotypic PDO expression profiles represented as mean ± SD from four representative GBMs in six independent experiments.
Figure 4.

Phenotypic Cellular Heterogeneity in GBM-PDOs
(A) IF mosaic imaging of NESTIN (green) and TLX (purple) proteins in GBM#76 PDSs.
(B) SOX2 (green) protein expression in nuclei (arrows) of GBM#76 PDSs.
(C and D) BMI1 IF from GBM#46 with PDSs (C) and PDOs (D). Images are representatives from three independent experiments. Scoring of IF level (0–3+) is demonstrated in (D) next to each corresponding representative cell. The PDS and PDO magnified regions of the mosaic span are indicated by dashed outlines. Scale bars in (A–D) are 100 μm.
(E) Images were quantified for the amount of BMI1 positive staining, as well as for intensity of the staining. For quantitative analyses of IF and IHC from patient tissues, H scores were calculated as (% at 0) × 0 + (% at 1+) × 1 + (% at 2+) × 2 + (% at 3+) × 3 = Range 0–300 based on analyses of at least 10 fields per slide averaged by two qualified examiners. Representative images and analyses of BMI1 expression in GBM tissues and PDOXs can be seen in Figure S3A. Comparison of H scores between patient tissues versus PDSs and PDOs are represented as mean ± SD of counts from two independent observers, and comparisons were determined by two-way ANOVA with Bonferroni post-hoc test (∗∗∗p < 0.001, ∗∗p < 0.01).
Transcriptional heterogeneity in GBM may reflect differences in chromatin compaction modified by the epigenetic polycomb repressive complex-1 (PRC1), which comprises the self-renewal protein BMI1; also GBM with mesenchymal transcriptional profiles showed enrichment of a BMI1 activation signature (Jin et al., 2017). Moreover, both BMI1 expression and activation signatures were associated with poor patient prognosis (Jin et al., 2017). Therefore, we compared BMI1 expression in PDSs and GBM-PDOs (Figures 4C and 4D) and correlated their levels to BMI1 levels in the originating GBMs (Figures 4C–4E and S3A). Similar to SOX2 and TLX expression profiles, BMI1 expression was enriched in PDSs when compared with the corresponding originating GBM from three patients, whereas BMI1 levels in GBM-PDOs were overall in line with those in the originating GBMs and their derived PDOX (Figure 4E). These data suggest that PDS profiles limit the ability to study heterogeneous GBM cell populations simultaneously, whereas the profiles of GBM-PDOs more relatively resemble GBM cellular subtype heterogeneity. We therefore utilized these GBM-PDOs to validate the use of the SMP cell-culture array in drug testing.
Biocompatibility of the Cell-Culture Insert Arrays
To examine the biocompatibility of the cell-culture arrays for generating GBM-PDSs and GBM-PDOs for histological processing and imaging, we first established optimum seeding densities for 3D cultures (see Transparent Methods) to test the culture inserts. We quantified PDS and PDO number, viability, and differentiation potential upon shape recovery of the inserts at 50°C. We were able to reduce the cell fixation time from an overnight fixation or fixation for several hours to fixation for 1 h. These data are in support of SMP compatibility with paraformaldehyde fixation, while maintaining cellular integrity either in individual inserts or complete arrays in the 12-step histological assays (Figure S4).
We examined the effects of SMP components on cell viability both in U87MG and primary GBM 3D cultures. U87MG cells and primary GBM#50 PDSs or PDOs grown with the arrays in the first prototype array initially failed to proliferate (Figure S5), even though the controls not included in the arrays, particularly GBM#50, exhibited normal growth, larger spheres, and diversified organoid-like growth with multicellular connection after 1 or 2 weeks (Figure S5). To assess any potential toxicity, we measured media levels in prolonged cultures and determined that the arrays were not absorbing media, thus limiting growth factor availability. In addition, 2-week culture media were slightly more alkaline compared with controls, suggesting that the arrays could be leaching a low-level chemical that may interfere with long-term GBM-PDO cultures. SMP components, including PEGDA 250, BPADMA, PI, and PA were each examined using the intracellular ATP cell viability assays. Only PEGDA250, when used at a three-log higher concentration of its median dose (1,000-fold in excess of EC50, which was 7.2 μM) significantly impacted cell viability (Figure S6), suggesting that low PEGDA 250 levels may be leaching from the inserts in prolonged GBM-PDO cultures. Preincubation of the insert array after 3D printing in culture media, PBS, 10% BSA, or β-mercaptoethanol (at 10 or 50 μM) did not reverse the cell loss phenotype, but pre-soaking in 100% acetone followed by PBS and ethanol washes did (Figure S7). Based on these data, we established the conditions for use of 4D printed SMP arrays for GBM-PDO generation and drug sensitivity assays.
Identifying GBM Molecular Subtypes for Examining Targeted Therapy
GBM patient tissues are routinely subjected to targeted exome sequencing (Glioseq) at our hospital. Glioseq analyzes 30 genes for single-nucleotide variants or indels, 24 genes for copy number variations (CNVs), and 14 types of structural alterations in BRAF, EGFR, and FGFR3 genes in a single workflow (Nikiforova et al., 2016). We analyzed Glioseq data to guide the selection of potential targeted therapies (Figure 5A). Glioseq findings revealed that GBM-PDOs from GBM#46, GBM#50, GBM#70, and GBM#76 had mutations in TERT, NF1 deletions, PTEN splice variants, ATRX mutations, and CDKN2A copy number loss. Further genomic and molecular analyses revealed that cells from these GBMs had WT exon 5 p53 without detectable hotspot mutations, but they showed no detectable expression of CDKN2a (Figure S3B), suggesting that CDKN2a copy number losses are homozygous in these cells, a common GBM feature (Weller et al., 2015).
Figure 5.

Targeted Exome Sequencing and Molecular Subtype Profiling of GBMs
(A) Summary of GBM patient profiles, molecular features, Glioseq profiles, and outcome based on targeted exome sequencing of originating tissues and corresponding GBM-PDOs.
(B) Heatmap of the molecular subtype and microenviromental marker expressions in four GBM samples displayed against a pool of five nonmalignant normal human brain samples, and the corresponding originating resected GBMs (labeled with a zero). Z scores in the heatmap are represented as mean expression levels and were calculated from qPCR ΔCt values from three independent experiments. See also Figures S3B and S3C for assessments of TP53 and CDKN2a expression in these GBMs.
Recent scRNA-seq-based models support that GBM cells remain heterogeneous, exhibit plasticity, and exist in four main cellular neurodevelopmental states: mesenchymal (MES), astrocytic, oligodendroglial precursor cell, and proneuronal progenitor cell states (Neftel et al., 2019). We performed GBM subtype gene signatures and gene expression profiling using a subset of markers based on TCGA database and signatures associated with tumor histology, grade, and defining molecular features (Jin et al., 2017). GBM46, GBM50, GBM70, and GBM76 displayed MES transcriptional gene expression profiles (Figure 5B). The frequency of cells in each GBM cellular state is influenced by CDK4, EGFR, and PDGFRA CNVs, as well as NF1 mutations (Neftel et al., 2019). Glioseq analyses revealed no CNVs in CDK4, EGFR, or PDGFR, but identified NF1 deletions or mutations in all four GBMs. As high expression of mesenchymal-related genes together with either high or low hypoxia-response genes defines either mesenchymal-like hypoxia-independent (MES1) or mesenchymal-like hypoxia-dependent (MES2) signatures, respectively (Neftel et al., 2019), we surmized that GBM46 and GBM50 PDOs have relatively matching MES1 signatures and were chosen for subsequent drug sensitivity assays.
Evaluation of Targeted and Combination Therapy
For GBM-PDO drug sensitivity assays, single cells were seeded at clonal densities in ultra-low attachment plates with culture inserts in serum-free growth factor supplemented conditions for PDS formation and within ECM droplets in inserts for GBM-PDO formation. After 2 weeks, clonal GBM-PDOs were treated for 72 h with either the standard chemotherapy TMZ and/or molecularly targeted agents, targeting mTOR, PI3K, or DNA damage response (DDR) among others. Following treatments, the entire 4D printed cell-culture arrays self-transformed to their original programmable cassette size upon heating for 20 min, for direct histological and IHC processing on the same day, while maintaining the starting tissue and array orientation and configuration. Interestingly, drugs tested had varying effects on different GBM-PDOs. TMZ was effective at reducing viability in GBM#46 more than GBM#50, whereas BEZ-235, a PI3K/mTOR dual inhibitor, was more effective in GBM#50, illuminating inter-tumor heterogeneity between GBM patient samples even with relatively similar molecular subtypes (Figure 6A). To test our hypothesis that combining compounds targeting GBM cell survival signaling in GBM cells with PTEN mutations (by targeting PI3K/mTOR signaling), NF1 and/or ATRX mutations (by using niraparib), and DDR (also by using niraparib), we assessed these combinations against TMZ in GBM-PDOs (Figure 6). Targeted therapies resulted in significantly higher antitumor activities relative to TMZ in GBM#50. To assess the synergy of two compounds in combination therapy, a Bliss independence model (Ianevski et al., 2017) for combination matrix surface evaluation was utilized for the combination of TMZ plus BEZ235 or niraparib plus BEZ235 treatment of GBM-PDOs. These studies revealed that whereas BEZ235 may protect from TMZ toxicities (Figure 6B), BEZ235 and niraparib could act synergistically (Figure 6C). We also assessed apoptosis induction, GBM cell migration, and ECM invasion using live cell imaging and fluorescent cell monitoring of treated GBM-PDOs by measuring the levels of activated caspase 3 (Figures 7A and 7B) and EGFP fluorescent migrating or invading GBM-PDO cells (Figures 7C and 7D). The results indicated that combination therapy increased apoptosis in GBM#46 PDOs that were relatively less sensitive to TMZ (Figures 7B–7D) and could significantly reduce migration and invasion of GBM-PDO cells compared with TMZ (Figures 7C and 7D), complementing the cell proliferation assays. Moreover, we detected on-target activities of these therapies by immunofluorescence against the GBM hallmark GFAP, GSC marker BMI1, and PI3K/mTOR marker pS6 (Figures 7E and 7F), providing rationale for further validating these combination therapies. In total, these data demonstrate that our developed system could be used for rapid histological processing and drug sensitivity testing in GBM-PDOs.
Figure 6.

Assessing Targeted and Combination Therapy in GBM-PDOs
(A) Treatment effects on GBM cell viability demonstrated in violin plots by measuring intracellular ATP levels of treated cells compared with untreated cells from the same patient GBM-PDOs. Dosage selection for each GBM-PDO was determined based on growth-inhibitory concentrations. Note the better antitumor effects for targeted therapies and combination (Comb.) therapies at equal concentration (1X of [growth inhibitory conc. at 50%] GI50 conc. for each drug), when compared with standard TMZ treatment. Data represent treatment of six independent experiments in GBM-PDOs from GBM#46 (left panels) and GBM#50 (right panels). Results are mean ± SD of eight replicates, and significance was determined using two-way ANOVA (∗∗∗p < 0.001).
(B) Isobologram for calculation and visualization of synergy scores from reduced GBM cell proliferation measured by ATP levels and synergistic response of the Bliss score surface assessed using the combination of TMZ plus BEZ235.
(C) Bliss score surface observed using the combination of niraparib plus BEZ235. Bliss plots were generated using the SynergyFinder application (Ianevski et al., 2017).
Figure 7.

Detecting Antitumor Activities of Targeted Therapy and On-Target Activity in Live GBM-PDOs
(A) Left, model depicting detection of activated caspase-3 (Casp-3) activity by DEVD-NucView488 and live GBM-PDO cell population-averaged apoptosis, migration, and invasion assays. Right, the diagram displays the PDO-activated caspase-3 assay. GFP-positive cells within the organoids indicate apoptotic cells. GBM cells that invade the semisolid extracellular Matrigel represent invading cells, whereas cells that migrate outside of the organoid represent migrated cells.
(B) Comparison of cell viability and activated caspase-3 between untreated, DMSO, TMZ, and targeted therapy groups was determined by two-way ANOVA with Bonferroni post-hoc test. Graph indicates significance versus TMZ with ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Violin plot on the top right represents real-time measurement of apoptotic cells in GBM-PDOs from GBM#46 and GBM#50 as untreated, DMSO-treated, or in response to treatment with TMZ, BEZ (targeting mTOR/PI3K), and combination therapy.
(C) GBM PDO cell migration captured by phase contrast using the IncuCyte HD system. Frames were captured at 4-h intervals from time 0 to 72 h from two separate regions/well using a 10× objective. Image processing involved fluorescent color channel separation, data resizing, illumination adjustment, threshold setting, and data extraction for either GBM#46 PDOs or GBM50 PDOs. Representative results of three separate experiments are shown for GBM#46 and GBM#50 as percentages of cell migration relative to 72-h values of untreated PDO cells.
(D) GBM-PDO cell invasion images were captured similarly with the IncuCyte HD system and shown as percentage of migrated cells relative to 72-h values of untreated GBM-PDO cells. Values determined by two-way ANOVA with Bonferroni post-hoc test (∗∗∗p < 0.001).
(E and F) (E) Three key on-target activity markers were detected in PDOs in response to drug treatments: GFAP (hallmark of GBM), BMI1 (GSCs), and phospho-S6 (PI3K/mTOR activity). Representative confocal images were taken after the indicated treatments in fixed PDOs and represent overlay of green (GFAP on the right, BMI1 in center, and pS6 on the left), red (phalloidin), and blue (DAPI for nuclei). (F) Data represent quantitation of marker expression upon treatment from six independent experiments utilizing GBM-PDOs, GBM#46, and GBM#50. Results are mean ± SD of eight fields of at least 1,000 cells. Comparison of marker expression between combined versus single therapy groups was determined by two-way ANOVA with Bonferroni post-hoc test (∗∗∗p < 0.001). Scale bars, 100 μm in (A and C).
Discussion
We developed the first example of 4D printed programmable cell-culture arrays using a thermo-responsive SMP for cancer drug testing. We used these arrays for histological assessment and drug testing of targeted and combination therapies in single cell-derived GBM-PDOs. 4D printing, which is defined as 3D printing with smart materials that are responsive to stimuli, offers the facile advantages of free-form fabrication and microfluidic approaches for drug discovery (Lee and Cho, 2016). We demonstrated the printability and biocompatibility of the SMP material in 4D printing. We utilized a high-precision additive manufacturing technique, PμSL (Zheng et al., 2012), for 3D printing to fabricate the SMP constructs that self-transform between 3D cell-culture insert arrays and 3D histology cassettes. Our 4D printing approach addresses the current limitations in processing 3D models by allowing for the scaling of screening capacity and simultaneous processing of organoid models for higher-throughput analysis, as recently done with cerebral organoids (Parker et al., 2020). Added advantages of our 4D printing approach is the programmable self-transformation between 3D arrays and 3D histological cassettes, therefore eliminating the time-consuming manual transfers, with their associated possibility of distortion or damage of delicate PDS and PDO biological samples. Moreover, the biological samples remain in the same configuration within the cell-culture compartment of our arrays during the entire process, therefore expediting this process and increasing compatibility with high-throughput assays.
Other advanced 3D printing approaches used to generate GBM models included the bioprinting of GBM-on-a-chip (Yi et al., 2019) or hydrogel meshes laden with polymeric particles (Mirani et al., 2019). Overall, these approaches including organ-on-a-chip technologies are being explored to advance organoid studies for the development and application of organoid-on-a-chip technologies. The transformable SMP cell-culture insert arrays allowed the rapid generation of GBM-PDOs for drug testing and assessing synergy between targeted therapies in a single assay.
We utilized GBM tumors from treatment-naive patients and cultured the cells in 3D serum-free conditions to maintain GBM-PDSs (Mehta et al., 2015), and/or generated organoid-like GBM-PDOs (Hubert et al., 2016; Lancaster et al., 2013). We demonstrated that GBM-PDOs contain highly heterogeneous cellular subtype populations, recapitulating the key expression profiles and tumor cell phenotypes in patients with GBM. The potential of PDO models to recapitulate aspects of GBM biology and heterogeneity is valuable. PDXs in mice, although may be useful in predicting clinical responses (Rokita et al., 2019), remain expensive, are variable in take rates, are time consuming to generate, and may become clonally distinct from originating GBMs (Patrizii et al., 2018). In addition, the phenotypes of GSCs remain debated, with single markers being variable in their expression among different tumors and PDS or PDX conditions. The use of combinations of stem cell markers in these models, although preferable, still might present differently than in human GBM. Cerebral organoids derived from human ESCs/iPSCs were genetically engineered to develop tumors with distinct oncogenic and tumor suppressor alterations and originating cellular identities (Bian et al., 2018), or by inducing mesenchymal GBM-like organoids with HRasG12V activation and p53 disruption (Ogawa et al., 2018). GBM heterogeneity was also modeled by co-culture of patient-derived GSCs with cerebral organoids, where GSCs proliferated over time and integrated into the normal cerebral organoids (Linkous et al., 2019). Most recently, propagating GBM tissues into smaller pieces has been shown to maintain the genetic and molecular signatures of GBM in 3D culture models, when tested within the first 2 weeks (Jacob et al., 2020). We utilized GBM-PDOs propagated for 2 weeks for drug testing, as we demonstrated that they contain heterogeneous cellular subtype populations, recapitulating the expression profiles and tumor cell phenotypes in patients with GBM. Although we could identify potential contributions of the TME cellular components to the expression profiles of our organoid-like GBM-PDOs, a comprehensive assessment of these TME cellular components will be performed in future studies to conclude their phenotypic nature while considering GBM plasticity (Neftel et al., 2019). Unlike the largely epithelial-only PDO models developed, a platform for generating PDOs using an air-liquid interface (ALI) method allowed retaining the native myoepithelial and immune cells, thereby preserving the immune contexture and recapitulating key elements of TME diversity (Neal et al., 2018). These ALI PDOs could be used to assess responses to immune checkpoint inhibition (Neal et al., 2018), therefore advancing PDO modeling toward more complete representation of the tumor and TME cellular components. Our cell-culture arrays may be utilized with many of these models and may be used to validate the potential of pharmacological, targeted, and immunotherapy for GBM.
Despite extensive efforts in defining morphological characteristics, complex molecular signatures, biomarkers, and imaging parameters of GBM that could provide biological tools for selecting GBM therapies, most treatment decisions for patients with GBM are still based on age and performance criteria. Moreover, targeted therapy agents have shown minimal efficacy in molecularly informed clinical trials, largely because of the inadequate consideration of the multitude of biological differences existing between individual patients with GBM. Integrated sequencing strategies provided a broader understanding of the genomic landscape and selective pressure occurring both during early GBM development and with current therapies (Barthel et al., 2019). On the other hand, single-cell sequencing analyses revealed multiple patterns of intratumor heterogeneity and inherent molecular complexity of GBM (Neftel et al., 2019; Patel et al., 2014). Clearly, it will be necessary to cotarget multiple alterations in GBM using combination therapy (Sabaawy, 2013). Indeed, PMO studies provided recent evidence that personalized treatment with combination of customized agents could improve outcomes in patients with refractory malignancies (Rodon et al., 2019; Sicklick et al., 2019). To rationally select agents in combination with standard-of-care TMZ-based chemoradiotherapy for GBM, other US Food and drug Administration-approved therapies targeting pathways that contribute to GBM heterogeneity should be considered. These include drugs (1) with direct targets (e.g., EGFR, PDGFRA, BRAF) that are subsets of significantly mutated genes identified in GBM TCGA and/or with integrated analyses (Barthel et al., 2019), (2) that map to a pathway that is targeted by an approved drug or investigational agents (e.g., PTEN, PIK3CA, NF1, ATRX, and other DDR targets), or (3) that influence cellular plasticity or subclonal dynamics (Vinci et al., 2018). We utilized 4D printing of SMP arrays that were used to derive single-cell clonal GBM-PDOs to assess the effects of treatment with TMZ and/or combinations of molecularly targeted agents, targeting mTOR, PI3K, PTEN, ATRX, NF1 or DDR. Treatment of the GBM-PDOs in our 4D arrays allowed the identification of possibly effective combination therapies. Moreover, our results suggest that targeted therapy against multiple pathways simultaneously, at least in vitro in the 3D GBM-PDOs, may be more advantageous than monotherapy, although this would need to be tested more rigorously. Recent studies utilizing PDC cultures could predict resistance to EGFR inhibitors and test the repurposing of ibrutinib for EGFR-specific therapy in GBM (Lee et al., 2018). Also, PDOs from gastrointestinal cancers could recapitulate patient responses in clinical trials of personalized medicine (Vlachogiannis et al., 2018). Our studies utilizing GBM-PDOs revealed that niraparib (targeting DDR) and BEZ235 (targeting PI3K/mTOR) could act synergistically, whereas BEZ235 may protect from TMZ toxicity, therefore substantiating another recent report on utilizing PDOs for initiating PI3K/mTOR-targeting therapy in a patient with GBM (Loong et al., 2020) and providing new inroads for combination therapy. Further validations of the synergistic effects in vivo, tailoring combinations against clonal cell subpopulations for each GBM, and including other combinations, e.g., with immune checkpoint inhibitors could support future clinical translation. Intriguingly, we identified synergistic activities at lower-than-expected growth inhibitory drug concentrations, therefore suggesting that our 4D printing approach provides a platform to identify potential thresholds for combination therapy to spare the dose-limiting toxicities in GBM (Mehta et al., 2015). In addition, our platform incorporated the assessment of on-target activity offering a validation strategy for personalized therapy (Bartucci et al., 2016; Loong et al., 2020; Vlachogiannis et al., 2018). We generated a 4D platform using SMP that self-transforms from 3D arrays, for PDO culture, into histological cassettes for rapid assessment of drug sensitivity, on-target activity, and synergy. When including exome and/or single-cell sequencing, and histological and targeted therapy assays, we propose that this system could potentially be used for evaluation of therapeutic responses in future PMO clinical trials.
Limitations of the Study
This platform is not without limitations. First, we utilized Matrigel droplets as an ECM source for PDOs. Variability among animal-derived or chemically defined synthetic ECMs could confound the cellular responses in drug testing. Second, culture conditions and growth factor components might limit the prolonged preservation of TME components. Although these stromal components, such as immune, endothelial, and nerve cells, may be reintroduced in coculture to reproduce the natural tissue architecture, modeling of their native architecture and precise bidirectional communication with tumor cells are key for modeling tumor heterogeneity and predicting drug sensitivity. Third, biomechanical variables and bioactive molecules may be reproduced by using biomaterials or bionics. Our future improvements will integrate the SMP 4D printing with different PDOs and TME, ECM bioinks, microfluidics, and/or ALI conditions to provide more precise monitoring of tumor multicellular and clonal interactions and assess metabolic responses to therapy at the native tissue level. Fourth, we utilized GBM PDOs for drug responses after 2 weeks of culture eying the rapid assessment in a time frame that could be feasible for clinical therapeutic decision making. Additional studies are needed to determine the extent of changes in drug responses when allowing PDOs to produce more complex connections and to study the contribution of cellular TME components toward generating full (organ)-mimicking organoids while exploiting the key differences between GBM- and ESC/iPSC-derived organoids. Finally, further research and more patient samples will be required to assure the clinical application and scaling of the developed assay. Improving on these limitations in future studies will allow the discovery of novel therapeutics that may be assessed in PMO clinical trials and used to select more effective personalized therapies.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hatem E. Sabaawy (sabaawhe@cinj.rutgers.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and Code Availability
The published article includes all datasets generated or analyzed during this study.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank our patients for consenting to donating their tissues for use in our studies. We thank Rutgers Cancer Institute of New Jersey Biorepository Services and Tissue Analytical Services (Shafiq Bhat, Lucyann Franciosa, Kelly Walton, and Lei Cong) for assistance with tissue acquisition, histology, immunohistochemistry, and sample processing. We also thank Diane Hanahan and Dr. Parisa Javidian for access to the tissue banking protocols and clinical assessments. We thank Sohan Ganguli and Christopher Ragusa for their help on prototyping of the biaxial stretching device.
This project was supported in part with Federal funds from the National Cancer Institute (NCI), National Institutes of Health (NIH), under Contract No. HHSN2612008000001E (to H.E.S.); NCI R01 award (CA226746 to H.E.S.); Rutgers Cancer Institute of New Jersey Shared Resources, supported, in part, with funding from NCI-CCSG (P30CA072720); and New Jersey Health Foundation Innovation Award (ISFP 7-16 to H.E.S.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. We thank Dr. Ralph Parchment of Division of Cancer Treatment and Diagnosis Laboratory, Frederick National Laboratory for Cancer Research sponsored by NCI, for discussion throughout this project.
Author Contributions
H.L. and H.E.S. conceived the study. C.Y. and H.L. designed, fabricated and tested the self-transformation of the cell-culture insert array. M.C., L.L., C.M.G., and K.J. performed the PDS and PDO experiments. M.C., L.Q., C.M.G., K.J., and H.E.S. analyzed the data, H.E.S. wrote the manuscript, and all authors contributed to editing the manuscript.
Declaration of Interests
Rutgers University has filed two patent applications (PCT/US2018/039994 and PCT/US2019/029931) on the methods described in the manuscript. H.E.S. is the scientific founder of Celvive, Inc.
Published: August 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101365.
Contributor Information
Howon Lee, Email: howon.lee@rutgers.edu.
Hatem E. Sabaawy, Email: sabaawhe@cinj.rutgers.edu.
Supplemental Information
References
- Bansal N., Bartucci M., Yusuff S., Davis S., Flaherty K., Huselid E., Patrizii M., Jones D., Cao L., Sydorenko N. BMI-1 targeting interferes with patient-derived tumor-initiating cell survival and tumor growth in prostate cancer. Clin. Cancer Res. 2016;22:6176–6191. doi: 10.1158/1078-0432.CCR-15-3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel F.P., Johnson K.C., Varn F.S., Moskalik A.D., Tanner G., Kocakavuk E., Anderson K.J., Abiola O., Aldape K., Alfaro K.D. Longitudinal molecular trajectories of diffuse glioma in adults. Nature. 2019;576:112–120. doi: 10.1038/s41586-019-1775-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartucci M., Ferrari A.C., Kim I.Y., Ploss A., Yarmush M., Sabaawy H.E. Personalized medicine approaches in prostate cancer employing patient derived 3D organoids and humanized mice. Front. Cell Dev. Biol. 2016;4:64. doi: 10.3389/fcell.2016.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartucci M., Hussein M.S., Huselid E., Flaherty K., Patrizii M., Laddha S.V., Kui C., Bigos R.A., Gilleran J.A., El Ansary M.M.S. Synthesis and characterization of novel BMI1 inhibitors targeting cellular self-renewal in hepatocellular carcinoma. Target Oncol. 2017;12:449–462. doi: 10.1007/s11523-017-0501-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian S., Repic M., Guo Z., Kavirayani A., Burkard T., Bagley J.A., Krauditsch C., Knoblich J.A. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods. 2018;15:631–639. doi: 10.1038/s41592-018-0070-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiraku M., Watanabe K., Matsuo-Takasaki M., Kawada M., Yonemura S., Matsumura M., Wataya T., Nishiyama A., Muguruma K., Sasai Y. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell. 2008;3:519–532. doi: 10.1016/j.stem.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Ge Q., Sakhaei A.H., Lee H., Dunn C.K., Fang N.X., Dunn M.L. Multimaterial 4D printing with tailorable shape memory polymers. Sci. Rep. 2016;6:31110. doi: 10.1038/srep31110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimple R.C., Bhargava S., Dixit D., Rich J.N. Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019;33:591–609. doi: 10.1101/gad.324301.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert C.G., Rivera M., Spangler L.C., Wu Q., Mack S.C., Prager B.C., Couce M., McLendon R.E., Sloan A.E., Rich J.N. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016;76:2465–2477. doi: 10.1158/0008-5472.CAN-15-2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianevski A., He L., Aittokallio T., Tang J. SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics. 2017;33:2413–2415. doi: 10.1093/bioinformatics/btx162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F., Salinas R.D., Zhang D.Y., Nguyen P.T.T., Schnoll J.G., Wong S.Z.H., Thokala R., Sheikh S., Saxena D., Prokop S. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell. 2020;180:188–204.e22. doi: 10.1016/j.cell.2019.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X., Kim L.J.Y., Wu Q., Wallace L.C., Prager B.C., Sanvoranart T., Gimple R.C., Wang X., Mack S.C., Miller T.E. Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat. Med. 2017;23:1352–1361. doi: 10.1038/nm.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster M.A., Renner M., Martin C.A., Wenzel D., Bicknell L.S., Hurles M.E., Homfray T., Penninger J.M., Jackson A.P., Knoblich J.A. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H., Cho D.W. One-step fabrication of an organ-on-a-chip with spatial heterogeneity using a 3D bioprinting technology. Lab Chip. 2016;16:2618–2625. doi: 10.1039/c6lc00450d. [DOI] [PubMed] [Google Scholar]
- Lee J.K., Liu Z., Sa J.K., Shin S., Wang J., Bordyuh M., Cho H.J., Elliott O., Chu T., Choi S.W. Pharmacogenomic landscape of patient-derived tumor cells informs precision oncology therapy. Nat. Genet. 2018;50:1399–1411. doi: 10.1038/s41588-018-0209-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkous A., Balamatsias D., Snuderl M., Edwards L., Miyaguchi K., Milner T., Reich B., Cohen-Gould L., Storaska A., Nakayama Y. Modeling patient-derived glioblastoma with cerebral organoids. Cell Rep. 2019;26:3203–3211.e5. doi: 10.1016/j.celrep.2019.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loong H.H., Wong A.M., Chan D.T., Cheung M.S., Chow C., Ding X., Chan A.K., Johnston P.A., Lau J.Y., Poon W.S. Patient-derived tumor organoid predicts drugs response in glioblastoma: a step forward in personalized cancer therapy? J. Clin. Neurosci. 2020;20:S0967–S5868. doi: 10.1016/j.jocn.2020.04.107. [DOI] [PubMed] [Google Scholar]
- Mehta M., Khan A., Danish S., Haffty B.G., Sabaawy H.E. Radiosensitization of primary human glioblastoma stem-like cells with low-dose AKT inhibition. Mol. Cancer Ther. 2015;14:1171–1180. doi: 10.1158/1535-7163.MCT-14-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirani B., Pagan E., Shojaei S., Duchscherer J., Toyota B.D., Ghavami S., Akbari M. A 3D bioprinted hydrogel mesh loaded with all-trans retinoic acid for treatment of glioblastoma. Eur. J. Pharmacol. 2019;854:201–212. doi: 10.1016/j.ejphar.2019.04.007. [DOI] [PubMed] [Google Scholar]
- Neal J.T., Li X., Zhu J., Giangarra V., Grzeskowiak C.L., Ju J., Liu I.H., Chiou S.H., Salahudeen A.A., Smith A.R. Organoid modeling of the tumor immune microenvironment. Cell. 2018;175:1972–1988.e16. doi: 10.1016/j.cell.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neftel C., Laffy J., Filbin M.G., Hara T., Shore M.E., Rahme G.J., Richman A.R., Silverbush D., Shaw M.L., Hebert C.M. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019;178:835–849.e21. doi: 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforova M.N., Wald A.I., Melan M.A., Roy S., Zhong S., Hamilton R.L., Lieberman F.S., Drappatz J., Amankulor N.M., Pollack I.F. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol. 2016;18:379–387. doi: 10.1093/neuonc/nov289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa J., Pao G.M., Shokhirev M.N., Verma I.M. Glioblastoma model using human cerebral organoids. Cell Rep. 2018;23:1220–1229. doi: 10.1016/j.celrep.2018.03.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R.N., Cairns D.M., Wu W.A., Jordan K., Guo C., Huang W., Martin-Moldes Z., Kaplan D.L. Smart material hydrogel transfer devices fabricated with stimuli-responsive silk-elastin-like proteins. Adv. Healthc. Mater. 2020;9:e2000266. doi: 10.1002/adhm.202000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca A.M., Sloan S.A., Clarke L.E., Tian Y., Makinson C.D., Huber N., Kim C.H., Park J.Y., O'Rourke N.A., Nguyen K.D. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods. 2015;12:671–678. doi: 10.1038/nmeth.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrizii M., Bartucci M., Pine S.R., Sabaawy H.E. Utility of glioblastoma patient-derived orthotopic xenografts in drug discovery and personalized therapy. Front. Oncol. 2018;8:23. doi: 10.3389/fonc.2018.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prados M.D., Byron S.A., Tran N.L., Phillips J.J., Molinaro A.M., Ligon K.L., Wen P.Y., Kuhn J.G., Mellinghoff I.K., de Groot J.F. Toward precision medicine in glioblastoma: the promise and the challenges. Neuro Oncol. 2015;17:1051–1063. doi: 10.1093/neuonc/nov031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakulendran N., Rowland K.J., Selvadurai H.J., Ahmadi M., Park N.I., Naumenko S., Dolma S., Ward R.J., So M., Lee L. Wnt and Notch signaling govern self-renewal and differentiation in a subset of human glioblastoma stem cells. Genes Dev. 2019;33:498–510. doi: 10.1101/gad.321968.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds B.A., Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Rodon J., Soria J.C., Berger R., Miller W.H., Rubin E., Kugel A., Tsimberidou A., Saintigny P., Ackerstein A., Brana I. Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat. Med. 2019;25:751–758. doi: 10.1038/s41591-019-0424-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokita J.L., Rathi K.S., Cardenas M.F., Upton K.A., Jayaseelan J., Cross K.L., Pfeil J., Egolf L.E., Way G.P., Farrel A. Genomic profiling of childhood tumor patient-derived xenograft models to enable rational clinical trial design. Cell Rep. 2019;29:1675–1689.e9. doi: 10.1016/j.celrep.2019.09.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabaawy H.E. Genetic heterogeneity and clonal evolution of tumor cells and their impact on precision cancer medicine. J. Leuk. 2013;1:1000124. doi: 10.4172/2329-6917.1000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicklick J.K., Kato S., Okamura R., Schwaederle M., Hahn M.E., Williams C.B., De P., Krie A., Piccioni D.E., Miller V.A. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat. Med. 2019;25:744–750. doi: 10.1038/s41591-019-0407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- Tuveson D., Clevers H. Cancer modeling meets human organoid technology. Science. 2019;364:952–955. doi: 10.1126/science.aaw6985. [DOI] [PubMed] [Google Scholar]
- Vinci M., Burford A., Molinari V., Kessler K., Popov S., Clarke M., Taylor K.R., Pemberton H.N., Lord C.J., Gutteridge A. Functional diversity and cooperativity between subclonal populations of pediatric glioblastoma and diffuse intrinsic pontine glioma cells. Nat. Med. 2018;24:1204–1215. doi: 10.1038/s41591-018-0086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachogiannis G., Hedayat S., Vatsiou A., Jamin Y., Fernandez-Mateos J., Khan K., Lampis A., Eason K., Huntingford I., Burke R. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science. 2018;359:920–926. doi: 10.1126/science.aao2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller M., Wick W., Aldape K., Brada M., Berger M., Pfister S.M., Nishikawa R., Rosenthal M., Wen P.Y., Stupp R. Glioma. Nat. Rev. Dis. Primers. 2015;1:15017. doi: 10.1038/nrdp.2015.17. [DOI] [PubMed] [Google Scholar]
- Yang C., Boorugu M., Dopp A., Ren J., Martin R., Han D., Choi W., Lee H. 4D printing reconfigurable, deployable and mechanically tunable metamaterials. Mater. Horiz. 2019;6:1244–1250. [Google Scholar]
- Yi H.G., Jeong Y.H., Kim Y., Choi Y.J., Moon H.E., Park S.H., Kang K.S., Bae M., Jang J., Youn H. A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nat. Biomed. Eng. 2019;3:509–519. doi: 10.1038/s41551-019-0363-x. [DOI] [PubMed] [Google Scholar]
- Zheng X., Deotte J., Alonso M.P., Farquar G.R., Weisgraber T.H., Gemberling S., Lee H., Fang N., Spadaccini C.M. Design and optimization of a light-emitting diode projection micro-stereolithography three-dimensional manufacturing system. Rev. Sci. Instr. 2012;83:125001. doi: 10.1063/1.4769050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The printed insert that was stretched to the 96-well plate configuration transforms into the histological cassette configuration when heated to 50oC while maintaining the registry of the insert array. In the cassette configuration, the insert is ready for histological processing to obtain the histology of the entire 3D cell-culture insert array contents with a single histological processing and sectioning step. The movie was taken during the 20-min heat-responsive transformation and was set to 200X playback.
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.