

Abstract
The article represents literature review dedicated to molecular and cellular mechanisms underlying clinical manifestations and outcomes of acute myocardial infarction. Extracellular matrix adaptive changes are described in detail as one of the most important factors contributing to healing of damaged myocardium and post-infarction cardiac remodeling. Extracellular matrix is reviewed as dynamic constantly remodeling structure that plays a pivotal role in myocardial repair. The role of matrix metalloproteinases and their tissue inhibitors in fragmentation and degradation of extracellular matrix as well as in myocardium healing is discussed. This review provides current information about fibroblasts activity, the role of growth factors, particularly transforming growth factor β and cardiotrophin-1, colony-stimulating factors, adipokines and gastrointestinal hormones, various matricellular proteins. In conclusion considering the fact that dynamic transformation of extracellular matrix after myocardial ischemic damage plays a pivotal role in myocardial infarction outcomes and prognosis, we suggest a high importance of further investigation of mechanisms underlying extracellular matrix remodeling and cell-matrix interactions in cardiovascular diseases.
Keywords: Myocardial infarction, cardiac remodeling, ECM, cytokines, matricellular proteins, cellular mechanisms
1. INTRODUCTION
Over the past few decades, cardiovascular diseases (CVD) have firmly occupied the first place among all causes of mortality and disability worldwide. Coronary heart disease (CHD) and specifically myocardial infarction (MI) play the leading role in the structure of cardiovascular morbidity, significantly reducing duration and quality of life [1, 2].
Molecular and cellular mechanisms underlying clinical manifestations and outcomes of acute MI determine adaptive and reparative processes and lead to the remodeling of myocardial tissue in the later stages. At the same time with a decrease in a number of functioning cardiomyocytes (due to myocardial damage), extracellular matrix (ECM) undergoes adaptive changes aimed at the maintenance of cardiac output [3]. Therewith, it is well known that maladaptive post-infarction cardiac remodeling unavoidably leads to the development of heart failure (HF) - one of the critical factors determining quality and expectancy of life after MI [4, 5]. Condition of myocardial non-contractile elements is one of the most important factors controlling the healing of damaged myocardium and post-infarction cardiac remodeling [6, 7].
There are two opposite processes in ECM underlying myocardial remodeling after MI. In the early period of MI, protein lysis and degradation prevail, while the later stages are characterized by activation of synthesis, accumulation and deposition of fibrillar collagen. Healing of the infarcted myocardium undergoes 3 phases: (i) inflammatory phase; (ii) proliferative phase or formation of granulation tissue; (iii) maturation phase or formation of collagen scar. Thereby, ECM is considered as a dynamic constantly remodeling structure that plays a pivotal role in cardiac repair [8, 9].
2. MATRIX METALLOPROTEINASES
During the first days after onset of MI in the infarcted area, fragmentation and degradation of ECM take place [10]. Cardiomyocyte necrosis causes prompt activation of matrix metalloproteinases (MMPs) that in normal condition appear to be in the form of inactive proenzymes. This results in the imbalance between MMPs and tissue inhibitors of metalloproteinases (TIMPs) expression [11]. It should be noted that regulation and maintenance of appropriate MMP/TIMP balance in myocardium is critical to ensure effective MI healing and adaptive cardiac remodeling, whereas its imbalance may adversely manifest in both the acute stage of the disease (e.g. development of acute heart failure, formation of acute aneurysm) and in post-MI period (maladaptive remodeling, progressive HF) [11-13]. MMPs can be divided into five groups based on the substrate specificity and main localization: collagenases, gelatinases, stromelysins, matrilysins and membrane-type MMPs (Fig. 1). Earliest destruction of collagen matrix in infarcted myocardium is mostly associated with the activation of collagenases (MMP-1) and gelatinases (MMP-2 and MMP-9), as well as other proteases, such as plasmin, cathepsin G and B [3, 14, 15]. Various experimental studies aimed to investigate post-MI left ventricular function through MMP inhibition presented conflicting results. It has been shown that the suppression of early ECM degradation, on one hand, reduced the risk of myocardium ruptures, but on the other hand, slowed down myocardium healing, angiogenesis, and the formation of collagen scar, and subsequently led to the development of more severe HF [16]. It has also been established that MMP-9 deletion attenuated left ventricular dilation by regulating myocardial collagen and fibronectin cleavage [17]. Another study demonstrated that MMP-9 overexpression attenuated inflammatory and fibrotic response in ECM improving post-MI left ventricular function [18].
Fig. (1).
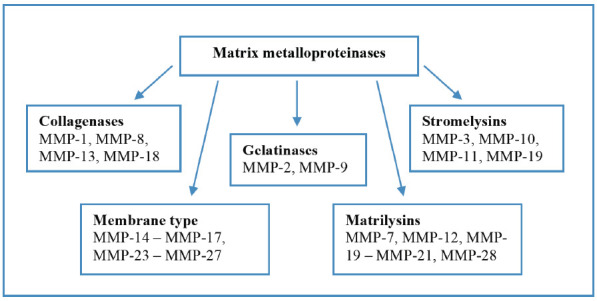
Matrix metalloproteinases’ groups. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
3. FIBROBLASTS
Destruction and fragmentation of native ECM during MI occur due to activation of inflammatory cells, fibroblasts, cardiomyocytes, various cytokines, chemokines, growth factors and hormones [6, 19]. T-lymphocytes also play a certain role in ECM remodeling. T-helpers type 1 cells enhance myocardial stiffness, reduce MMP-9 and MMP-13 expression, increase the total amount of collagen and strengthen cardiac collagen cross-linking. T-helpers type 2 cells have opposite effects resulting in dilatation of heart chambers [20, 21].
Subsequently, a decrease in protease activity and the release of anti-inflammatory cytokines denote the transition to the next phase - fibrogenesis. It is regulated mainly by fibroblasts, that proliferate and acquire myofibroblast phenotype under the cooperative influence of various cytokines, growth and trophic factors [22-24]. There is a reciprocal functional and regulatory interrelation between fibroblasts and MMP/TIMP. On one hand, fibroblasts exert modulating effects on proteases and their inhibitors, on the other hand, MMP and TIMP can regulate fibroblasts state and activity [25, 26]. Moreover, it has been demonstrated that membrane type 1 MMP in addition to collagenase activity, was also able to modulate fibroblasts activity, functional condition of the myocardium, cardiomyocyte survival, and matricellular interactions [27]. MMP activity can also be determined by conditions associated with myocardial ischemia/reperfusion, particularly post conditioning that inhibits both MMP’s activity and myocardial fibrogenesis [28].
Early period of MI is characterized by migration of interstitial fibroblasts from the adjacent intact myocardium to damaged zone and active fibroblast proliferation in this area [29]. Fibrin/fibronectin deposition in the ECM serves as a kind of guide for fibroblasts’ migration into the damaged myocardium [30, 31]. In addition, the infarcted area attracts circulating myofibroblast precursors. As a result, in MI, both types of cells undergo differentiation into mature myofibroblasts, functionally characterized by high synthetic and contractile activity, but poor migration ability [32, 33]. Myofibroblast phenotype is characterized by the expression of various contractile, structural and matricellular proteins (e.g. α-smooth muscle actin, vimentin, embryonic smooth muscle myosin heavy chain (Smemb), fibrillar collagens, periostin, osteopontin, tenascin C), and also cell surface receptors (e.g. transforming growth factor beta (TGF-β) type II receptor, type 1 angiotensin II receptor (AT1), leukemia inhibitory factor receptor (gp130) (Fig. 2) [24, 34]. Furthermore, mature myofibroblasts have rough endoplasmic reticulum, myofilaments with focal densities, collagen-secreting granules and fibronexus junctions [35, 36]. One of the most specific markers of myofibroblasts is Smemb, expressed in the infarcted area and in the hibernating myocardium [37-39]. Unlike dermal fibroblasts that disappear from the infarcted area after transformation of the granulation tissue into the mature scar, myofibroblasts can be identified in the myocardial scar for many years after MI [40]. An important role in inflammatory response suppression and in myofibroblasts’ activation is played by transmembrane CD 44 receptors expressed by leukocytes and endotheliocytes infiltrating MI area [41]. Fibroblasts’ differentiation into myofibroblasts requires additional biochemical stimuli such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, platelet growth factor, stem cell factor, and particularly TGF-β and fibronectin domain ED-A [24, 34, 42-44]. Leukemia inhibitory factor, on the contrary, blocks myofibroblasts transformation and also inhibits angiogenesis [45, 46]. Due to its contractile ability, myofibroblasts embedded in the extracellular matrix produce isometric tension of the granulation tissue, which is mediated by focal adhesions with a connective tissue matrix and is potentiated by TGF-β. This leads to consolidation (reduction in size) of post-infarct scar in the later period of MI [47]. In the early period of MI, normal healing of damaged myocardium requires active migration and proliferation of myofibroblasts. Therefore, formation and dissociation of focal adhesions are accelerated and contacts appear to be not strong enough. This process is mediated by focal adhesion kinase that, in addition to the destruction of cell-matrix connections, is also capable of regulating a cell cycle [48].
Fig. (2).
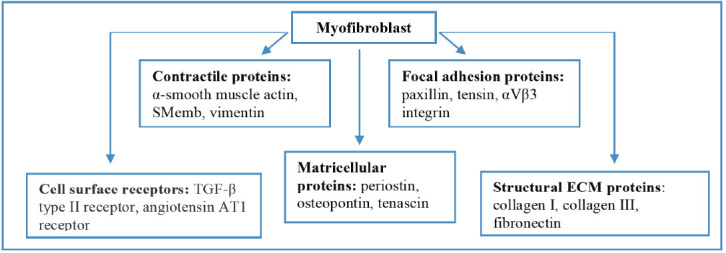
Proteins expressed by myofibroblasts. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
Besides the factors mentioned above, the functions of fibroblasts and myofibroblasts are affected by numerous cytokines and growth factors, including epidermal growth factor, fibroblast growth factor (FGF), heparin-binding epidermal growth factor-like growth factor, connective tissue growth factor, reactive oxygen species and intracellular molecules involved in regulation of free radical oxidation [49-52].
4. GROWTH FACTORS
A large body of evidence suggests that growth factors may play a crucial role in the regulation of cellular mechanisms associated with myocardial repair and remodeling. Growth factors are able to induce matrix protein synthesis and regulate myofibroblast transformation and proliferation during the proliferative phase of myocardial healing [24, 53].
In particular, TGF-β appears to be an essential promoter of myofibroblast transformation. It is important to note that TGF-β has the ability to regulate the function of all cell types engaged in myocardial injury and healing, and on the other hand, most cell types involved in myocardial remodeling are able to secrete TGF-β [54]. TGF-β is secreted as an inactive form that is activated following MI. Reactive oxygen species, integrins, serine and cysteine proteases, cathepsins, metalloproteinases and matricellular proteins contribute to TGF-β activation [55-57]. Besides stimulation of myofibroblast phenotype, TGF-β induces synthesis of ECM proteins, such as fibronectin, collagen I and III. It has been revealed that TGF-β inhibition in the early period of MI reduces stable scar formation resulting in the left ventricular dilation and increased mortality rates, while its inhibition in later period attenuates the severity of myocardial fibrosis [58]. In addition, TGF-β exerts an anti-inflammatory effect, promotes angiogenesis, reduces oxidative stress, and protects cardiomyocytes from apoptosis via p42/44 mitogen-activated protein kinase (p42/44 MAPK) and phosphoinositide 3-kinase/Akt (PI3K/Akt) pathways [54].
FGF-2 is capable of promoting the proliferation of fibroblasts, endothelial cells, and smooth muscle cells. It also appears to be an important regulator of cell survival, migration, differentiation and apoptosis [59]. The beneficial effect of FGF-2 in myocardial healing manifests in attenuation of left ventricle (LV) dilation and scar thinning, development of compensatory cardiomyocyte hypertrophy, an increase of fibroblast proliferation and collagen deposition, cardioprotection and promotion of angiogenesis [25, 26, 53, 60]. Experimental studies demonstrated that deletion of FGF-2 in acute MI resulted in enhanced ventricular dilation, infarct expansion and reduced cardiac function [60].
Platelet-derived growth factors (PDGF) - a family of proteins are composed of four isomers: PDGF-A, -B, -C, and -D that exert their action by binding with two PDGF receptors - α and β [61]. PDGF family has a significant influence on post-infarct remodeling and repair. PDGF regulates fibroblast migration, proliferation and transformation, promoting fibrogenesis [26, 53]. PDGF also induces TGF-β synthesis that exerts potent profibrogenic effects. Moreover, PDGF is involved in post-infarct angiogenesis. It facilitates stabilization and maturation of neovessels leading thereby to inhibition of myocardial inflammation [62].
Another factor implicated in angiogenesis after myocardial damage is vascular endothelial growth factor as it regulates proliferation and migration of endothelial cells and lumen formation [63]. In addition, experimental studies demonstrate that vascular endothelial growth factor may inhibit cardiomyocyte apoptosis and enhance myocardial contractility [64].
5. CARDIOTROPHIN-1
An important role in the coordination of MI healing and ECM remodeling is played by cardiotrophin-1 (CT-1) [65, 66]. CT-1 is a member of the IL-6 family of cytokines, described as the most potent stimulant of cardiomyocyte hypertrophy from IL-6 family [67]. Elevated CT-1 expression is associated with MI, unstable angina, myocardial pressure overload and chronic HF [65, 68-71]. CT-1 expression directly correlates with the left ventricular mass index; therefore CT-1 level could be used as a diagnostic marker of hypertensive heart disease [72]. It has also been shown that elevated CT-1 level is associated with increased risk of atrial fibrillation relapse after successful cardioversion in patients with persistent atrial fibrillation, thus CT-1 could be used as a predictor of sinus rhythm constancy in this cohort of patients [73]. CT-1 secretion is potentiated by mechanical stretch of cardiomyocytes and by various biochemical agents such as catecholamines, angiotensin II, aldosterone, and by myocardial hypoxia (Fig. 3) [65, 74-76].
Fig. (3).
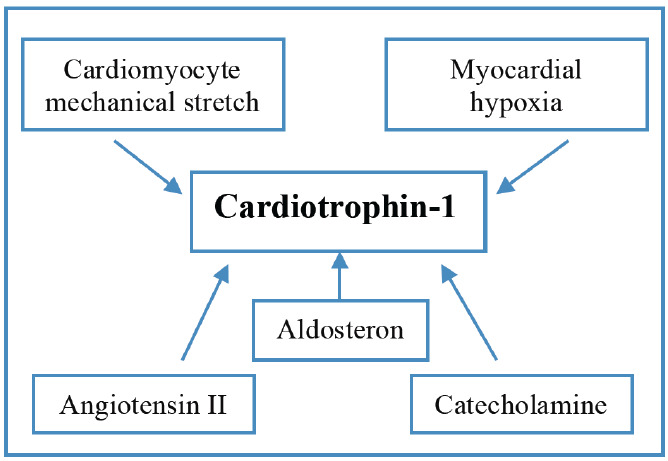
Factors stimulating cardiotrophin-1 expression. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
CT-1 hypertrophic activity is mediated by a complex receptor consisting of leukemia inhibitory factor receptor β and glycoprotein 130 heterodimer receptor, which are phosphorylated by Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3). JAK/STAT 3 pathway activation results in myocardial hypertrophy, while the involvement of other signaling pathways - p42/44 MAPK and PI3K/Akt induces cardioprotection, cardiomyocyte survival and inhibition of apoptosis [76, 77]. Ventricular hypertrophy mediated by CT-1 has certain specific features. Unlike other hypertrophic stimuli that cause predominant enlargement of cardiomyocytes in width due to the assembly of new myofibrils in parallel direction, CT-1 promotes assembly of sarcomere units in series leading to cardiomyocyte enlargement primarily due to an increase in cell length [78]. CT-1 attenuates ischemia/reperfusion myocardium injury, resulting in a decrease in the number of dying cardiomyocytes, it also maintains myocardial contractility by stimulation of cardiomyocyte hypertrophy [66, 79, 80].
CT-1 appears in the infarcted area starting from 24 hours and up to 8 weeks, with being maximum at about 2 weeks after MI. In the non-infarcted myocardium, CT-1 expression is very low and it increases gradually with a significant increase at the second week after the onset of MI [81]. CT-1 stimulates fibroblast migration and proliferation [82]. In addition, CT-1 can induce bone marrow stem cells migration [75]. CT-1 action might be compared to a certain extent with the TGF-β effect on myofibroblasts meaning that while CT-1 promotes proliferation and migration of myofibroblasts, TGF-β induces the formation of stable contractile and secreting phenotype [24, 42]. CT-1 stimulates the synthesis of extracellular matrix proteins - collagen, fibronectin, tenascin; proteins involved in the regulation of cell cycle - cyclins, proliferative nuclear antigen; proteins involved in cell adhesion and migration - mainly integrins. However, the effect of CT-1 on collagen synthesis appears to be modest [81, 83].
6. COLONY-STIMULATING FACTORS
Over the last decades, numerous studies have demonstrated that various colony-stimulating factors play an important role in MI healing. Until recently, it has been considered that regulation of hematopoiesis is the only function of these factors. Later on, it was discovered that granulocyte colony-stimulating factor reduced the size and thickened collagen scar, stimulated hypertrophy of survived cardiomyocytes, diminished severity of fibrosis in non-infarcted myocardium, activated STAT-3 (Signal Transducer and Activator of Transcription) and nuclear factor GATA-4, increased the expression of myosin heavy chain, desmin and troponin T, increased the activity of MMP-2 and MMP-9, reduced TNF-α, TGF-β, angiotensin II type 1 receptor expression [84, 85]. Consequently, its cardioprotective effect results in a reduction of HF severity after MI. The RIGENERA trial showed that the administration of granulocyte colony-stimulating factor in patients with ST-elevation MI was associated with a significant attenuation of adverse left ventricular remodeling [84]. Granulocyte colony-stimulating factor and macrophage colony-stimulating factor accelerate MI healing and suppress the expansion of the peri-infarct border zone. This is associated with the increase of matrix ribonucleic acid (mRNA), TGF-β, procollagen types I and III expressions in the MI zone, but not in non-infarcted myocardium, and results in the improvement of myocardial function [86]. Macrophage colony-stimulating factor also increases the amount of collagen in the MI area and reduces the proportion of collagen fine filaments, thereby, the severity of myocardial dysfunction can be diminished by the acceleration of myocardium healing [87]. At the same time, excessive activation of granulocyte-macrophage-colony-stimulating factor promotes MI expansion, increases the number of mononuclear cells in the damaged area and collagen deposition [88, 89].
7. ADIPOSE TISSUE AND GASTROINTESTINAL HORMONES
An important role in post-infarct ECM remodeling is played by adipo(cyto)kines known as adipose tissue hormones, which regulate lipid and carbohydrate metabolism.
Adiponectin serves as a scaffold for newly formed collagen in MI healing. Its allocation in MI coincides with that of fibronectin and type IV collagen. It is primarily distributed peripherally to survived cardiomyocytes zone adjacent to an infarcted myocardium [90]. Moreover, adiponectin exerts anti-fibrotic activity by increasing MMP-9 expression resulting in enhanced collagen cleavage and thus inhibiting adverse ECM remodeling [91].
Leptin also plays a significant role in pathophysiological changes taking place during the first days of MI. It has been determined that in MI patients, circulating leptin levels are elevated independently of the presence or absence of obesity [92]. Leptin activates STAT3 known to regulate biochemical reactions involved in cardiac remodeling after MI through leptin receptors of cardiomyocytes. Leptin may have a protective effect after myocardial injury, because leptin-null mice in experimentally induced MI setting showed increased mortality rate associated with the development of severe hypertrophy, dilation and reduced contractility of LV [93].
Resistin levels significantly increase 12 hours after the onset of MI and correlate with troponin I and C-reactive protein blood levels. Resistin promotes TNF-α expression and activates NF-κB proinflammatory signaling pathway in cardiac tissue resulting in myocardial inflammation and fibrosis [65].
Another member of the adipokine family - apelin can induce a cardioprotective effect in myocardial injury. Recent studies discovered that apelin reduces the infarct size, attenuates myocardial fibrosis and promotes angiogenesis in an infarcted myocardium [94, 95].
Ghrelin - a growth-hormone releasing peptide that belongs to the gastrointestinal hormones family has also been found to exert a cardioprotective effect. In addition to ghrelin’s ability to regulate appetite, body weight, glucose and lipid metabolism, it also participates in the pathogenesis of MI [96]. Various studies demonstrated that ghrelin reduces adverse cardiac remodeling by inhibiting the inflammatory response and MMP-2 and MMP-9 activity. Moreover, ghrelin administration suppresses cardiomyocyte apoptosis, improves endothelial function, and prevents excessive cardiac sympathetic nerve activity, thus leading to more favorable MI prognosis [97, 98].
8. MATRICELLULAR PROTEINS
An essential part in the regulation of ECM dynamic changes in MI healing and formation of adaptive remodeling in post-infarct period belongs to matricellular proteins. Matricellular proteins are a family of nonstructural matrix components that are able to modulate cell function and activity, regulate cytokine and growth factor responses, promote cardiomyocytes survival and ensure the formation of a structural matrix [55]. The family includes tenascin-C and -X, thrombospondin-1, -2 and -4, osteonectin/SPARC (secreted protein, acidic and rich in cysteine), osteopontin, periostin, and CCN family members [34].
Matricellular proteins are markedly expressed during embryogenesis. Later in healthy adult hearts, their expression is reduced, but it significantly increases in case of tissue damage [3, 99]. Matricellular proteins modulate migration, proliferation and adhesion of cells involved in post-infarct cardiac repair. Their activity in transducing molecular signals is implemented by interacting with cell surface receptors, or modulating the expression of various cytokines, growth factors, proteases, and structural matrix proteins [100, 101].
Tenascin-C is predominantly localized in the infarct border zone - area where the most intensive matrix remodeling is observed. Tenascin-C lessens cardiomyocyte-matrix interactions due to de-adhesive properties that result in cardiomyocytes “slippage”, facilitating the penetration of inflammatory cells and capillaries’ growth between cardiomyocytes [102]. Tenascin-C stimulates the synthesis of ECM structural components, particularly collagens via involvement and activation of myofibroblasts. Tenascin-C also induces fibroblast migration into the remodeling area and myofibroblast transformation promoting myocardial fibrosis and diastolic dysfunction [103]. In addition, tenascin-C possesses elastic properties that help to prevent mechanical overload of the border zone [104]. Some studies show that tenascin-C is cleaved by MMP-2 that unfolds its cryptic adhesive site, and conversely, tenascin-C can induce the expression of MMPs that potentiates structural matrix destruction [105]. Tenascin-X also promotes collagen deposition, facilitates consolidation and elasticity of ECM and contributes to the inhibition of MMP-2 and MMP-9 activity, but numerous studies did not observe any significant role of tenascin-X in the cardiovascular disease [106, 107].
Osteonectin or SPARC is markedly expressed in MI with its peak on 7th-14th day of the disease [108]. SPARC plays a pivotal role in post-infarct cardiac repair and ECM remodeling through several mechanisms: i) modulating activity of growth factors involved in myocardial healing - TGF-β, fibroblasts growth factor, platelets growth factor, vascular endothelial growth factor; ii) inducing myocardial cells de-adhesion; iii) promoting angiogenesis; and iv) facilitating the assembly of newly-formed collagen [55, 109, 110]. SPARC also mediates the expression of large numbers of genes encoding ECM proteins, adhesion molecules, connective tissue growth factor, MMPs and TIMPs. It has been observed that SPARC deficiency significantly affects the severity of left ventricular dilatation and contractile function in the first days of MI [111]. However, defining the role of this protein in post-infarct cardiac repair and remodeling requires further investigation.
Another important mediator in post-infarct myocardial remodeling is osteopontin (Eta-1). Osteopontin is an adhesive glycophosphoprotein that interacts with αvβ1, αvβ3, αvβ5, αvβ6 and α5β1 integrins, transmembrane CD 44 receptors, collagen and fibronectin [112, 113]. Osteopontin induces collagen synthesis and organization regulating ECM assembly in the infarcted myocardium, inhibits cell apoptosis, and acts as a chemoattractant for T-lymphocytes and macrophages. Osteopontin deficiency leads to adverse post-infarct remodeling and left ventricular dilatation due to reduced collagen deposition in the healing scar [114]. Its expression peaks on the 2-3rd day after MI, but then falls on 7-10th days. Osteopontin expression is significantly upregulated in the damaged area compared to non-infarcted myocardium. Osteopontin can be expressed by various cells, in particular by macrophages and T-lymphocytes. Pro-inflammatory cytokines and angiotensin II promote osteopontin synthesis by fibroblasts [55, 115, 116]. Some studies have shown that osteopontin is cleaved by MMP-2, -3, -7, -9, and -12 resulting in the formation of fragments with adhesive properties [113]. Moreover, osteopontin can stimulate MMP-9 expression [117]. In addition, osteopontin modulates growth factor signaling and activates angiogenesis that plays a pivotal role in post-infarction cardiac repair [118, 119].
Thrombospondins 1 and 2 are trimeric proteins that show a very low expression in the normal heart with a gradual increase in thrombospondin-1 mRNA content as early as 1 hour after the onset of MI [55]. The primary localization of thrombospondin-1 in infarcted myocardium is the border zone, which serves as a barrier between the damaged and intact tissue. This localization particularly protects non-infarcted myocardium from adverse remodeling [11]. One of the key functions of thrombospondin-1 is the activation of TGF-β1 resulting in the reduction of an inflammatory response, modulation of fibroblast migration and myofibroblast transdifferentiation, stimulation of fibrogenesis, and thus leading to ECM preservation [120, 56]. Thrombospondin-1 inhibits angiogenesis by suppressing new vessels’ formation and thereby precluding premature granulation tissue growth [121]. Thrombospondin-1 also reduces the expression of monocyte chemoattractant protein-1, macrophage inflammatory protein-1α, interferon-γ-inducible protein 10, IL-1β, IL-6, and decreases the accumulation of macrophages in infarcted myocardium and in border zone which leads to the suppression of inflammatory response [122]. Thrombospondin-2, as well as thrombospondin-1 exert anti-angiogenic and anti-inflammatory effects [121]. Furthermore, it is responsible for fibroblasts’ interactions with matrix proteins, including fibronectin, and their migration into the infarcted area [123]. It has been demonstrated that thrombospondin-2 null mice developed cardiac rupture in 90% of cases within 48 hours following MI, suggesting that thrombospondin is vital in sustaining ECM integrity and structure [124]. Both thrombospondin-1 and -2 can inhibit MMP-2 and -9 thus preventing ECM degradation in MI [121, 123]. Thrombospondin-4 is highly expressed in adults’ healthy heart, but its role in MI is still to be elucidated [55].
Periostin is another matricellular protein related to the fasciclin family involved in cell adhesion and migration [125]. There is a very low expression of periostin in healthy adult heart, but it can be detected in cardiac valves and in case of various CVDs. Periostin is upregulated in myocardium beginning from the 4-5th day and up to 14-28 days after the development of MI [126]. It plays a significant role in collagen scar formation and ECM organization by activating cardiac fibroblasts, promoting their migration and differentiation into myofibroblasts, stimulating fibrillogenesis and promoting fibrosis [127]. Several studies established that periostin deficiency in mice was significantly associated with cardiac rupture in acute MI, although periostin-null mice developed better systolic function in later stages of the post-infarction period [128].
CCN family consists of six prototypic polypeptide molecules ranging from CCN1 to CCN6. The abbreviation of CCN is derived from the names of first three members of the family: cysteine-rich protein 61 (CCN1), connective tissue growth factor (CCN2), and nephroblastoma overexpressed protein (CCN3) [129]. ССNs play an important role in cell differentiation, proliferation, migration and survival via interaction with cell adhesion receptors. CCN2 expression is increased primarily in myofibroblasts and cardiomyocytes of border zone after MI. Its expression in infarcted myocardium is induced by TGF-β1, angiotensin II, and endothelin-1 [130]. CCN2 promotes fibroblast migration and differentiation, stimulates angiogenesis and activates TGF-β1 signaling pathways inducing fibrogenesis [131, 132]. It has also been demonstrated that CCN2 may exert a protective effect on cardiomyocytes [133].
9. PROTEINS WITH MATRICELLULAR FUNCTIONS
There is a considerable amount of various proteins involved in ECM remodeling after MI, in particular, decorin – small leucine-rich proteoglycan (SLRP) plays an important role in collagen fibrillogenesis and in maintenance of ECM structural integrity. Decorin deficiency has no effect on the infarct size, but it causes a wider distribution of collagen fibrils with the formation of less organized and loosely packed large scars, development of aneurysms, ventricular hypertrophy (possibly compensatory) and dilatation with decreased myocardium contractility [100, 134].
Another member of SLRPs family is biglycan that is also implicated in cardiac repair and remodeling after MI. It has been demonstrated that biglycan is essential for adequate collagen cross-linking and formation of a stable scar. Biglycan deficiency leads to enhanced left ventricular dilation and systolic dysfunction. In addition, its deficiency induces fibroblasts differentiation into myofibroblasts [100, 135].
Galectin-3 - a member of the lectin family is mainly expressed by macrophages and is known to be a marker of cardiovascular inflammation, fibrosis and remodeling [136]. Galectin-3 induces redundant collagen synthesis in the myocardium and upregulates fibroblast activity. Elevated circulating galectin-3 levels in post-MI period are positively associated with infarct size, systolic dysfunction and adverse LV remodeling [137].
Various studies discuss the protective role of syndecan family proteins, namely syndecan-1 and -4, in MI outcomes. Absence of syndecan-1 in mice model of MI caused increased inflammation in cardiac tissue, activation of MMP-2 and MMP-9, collagen fragmentation and disorganization, resulting in adverse myocardial remodeling, LV dilation and systolic dysfunction [138]. Syndecan-4 also exerts a cardioprotective effect by promoting angiogenesis, activating endothelial cells, reducing myocardial inflammation and fibrosis, and inhibiting cardiomyocyte apoptosis [139].
Another receptor, which is likely to play a role in MI-associated ECM remodeling, is TNF cytokine superfamily member osteoprotegerin. Osteoprotegerin is involved in atherogenesis and arterial calcification [140]. Its level within 1 hour after the onset of MI is higher than in stable coronary artery disease, in which, in turn, it is slightly higher than in the control. Then, with the course of MI, it gradually, but very slowly decreases. It is significantly upregulated in MI compared to stable coronary artery disease patients and controls. This study also showed the quite interesting balance of the RANKL (Receptor Activator of NF-κB Ligand) in MI, stable coronary artery disease and healthy individuals. It was the lowest in stable coronary artery disease. In MI, it was somewhat higher, but nevertheless 3 times lower than in the control [141]. Moreover, increased serum osteoprotegerin levels in acute MI patients after the percutaneous coronary intervention were strongly associated with the final infarct size. Osteoprotegerin expression has been shown to be associated with increased mortality, but pathophysiological mechanisms underlying this connection require further investigation [142].
Table 1 summarizes the roles of the most important factors regulating molecular-cellular interactions in ECM remodeling during MI.
Table 1. Signaling molecules modulating extracellular matrix dynamic changes in myocardial infarction.
| Molecule | Localization | Expression Time | Effect | Refs. |
|---|---|---|---|---|
| Matricellular Proteins | ||||
| Tenascin-C | Infarcted myocardium, border zone, remodeling myocardium | Early post-infarct period (proliferative phase of myocardial healing) | Stimulates collagen synthesis; Induces fibroblast migration and differentiation; Promotes inflammation and angiogenesis |
[102-105] |
| Tenascin-X | Not known | Not known | Promotes collagen deposition; Maintains ECM preservation; Contributes to inhibition of MMP-2 and MMP-9 activity |
[106, 107] |
| Trombospondin-1 | Infarct border zone | Early post-infarct period (inflammatory phase of myocardial healing) | Activates TGF-β1; Modulates fibroblast migration and differentiation; Promotes fibrogenesis; Suppresses inflammatory response; Inhibits angiogenesis; Inhibits MMPs |
[56, 121, 122] |
| Trombospondin-2 | Not known | Late post-infarct period after trombospondin-1 level decreases | Exerts anti-inflammatory effects; Reduces angiogenesis; Inhibits MMPs; Modulates fibroblast interactions with fibronectin |
[121, 123, 124] |
| Osteonectin | Infarcted myocardium, non-infarcted myocardium | Early and late post-infarct period (3rd - 14th day) | Modulates activity of TGF-β, fibroblasts growth factor, platelets growth factor, vascular endothelial growth factor; Induces myocardial cells de-adhesion; Promotes angiogenesis; Regulates assembly of newly-formed collagen |
[108-111] |
| Osteopontin | Infarcted myocardium, non-infarcted myocardium |
Early and late post-infarct period (1st - 28th day) | Induces collagen synthesis and organization; Inhibits cell apoptosis; Modulates growth factor signaling; Activates angiogenesis |
[114-119] |
| Periostin | Infarcted myocardium, border zone, remodeling myocardium | Early and late post-infarct period (3rd - 28th day) | Promotes fibroblasts migration and differentiation; Stimulates fibrillogenesis |
[125-128] |
| CCN-2 | Infarcted myocardium | Early and late post-infarct period (7th-28th day) | Activates TGF-β1 signaling pathways; Stimulates angiogenesis; Promotes fibroblast migration and differentiation; Exerts cardioprotective effect |
[130-133] |
| Proteins with matricellular function | ||||
| Biglycan | Infarcted myocardium, border zone | Early and late post-infarct period (2nd – 28th day) with peaking at 14th day | Stimulates collagen deposition and appropriate cross-linking; Regulates fibroblast phenotype and function; Decreases ventricular dilation; Attenuates ventricular rupture; Modulates scar organization |
[100, 135] |
| Decorin | Infarcted myocardium, border zone | Early and late post-infarct period (7th-28thday) with peaking at 14th day | Regulates collagen fibrillogenesis and proper matrix assembly; Decreases infarct size and ventricular hypertrophy; Diminishes left ventricular dilation; Promotes scar organization |
[100, 134] |
| Molecule | Localization | Expression Time | Effect | Refs. |
| Syndecans | Infarcted myocardium | Early post-infarct period (24h – 7th day peak) | Prevent excessive matrix degradation; Enhance myofibroblast transdifferentiation; Stimulate growth factor-dependent endothelial cell proliferation; Promote angiogenesis; Reduce myocardial inflammation and fibrosis; Inhibit cardiomyocyte apoptosis |
[138, 139] |
| Galectins | Infarcted myocardium | Early post-infarct period (2nd -14th day) | Induce fibroblast proliferation; Promote matrix deposition; Stimulate macrophage migration |
[136, 137] |
| Growth factors | ||||
| Transforming growth factor beta | Predominantly infarct border zone | Early and late post-infarct period (3rd - 28th day) | Promotes myofibroblast differentiation; Increases collagen and fibronectin synthesis; Activates TIMP; Stimulates cardiomyocyte hypertrophy; Induces mononuclear cells chemotaxis; Promotes angiogenesis; Reduces oxidative stress; Protects cardiomyocytes from apoptosis |
[54, 56, 120] |
| Fibroblast growth factor | Infarcted myocardium, border zone | Early and late post-infarct period (7th-28th day) | Promotes proliferation of fibroblasts, endothelial cells, smooth muscle cells; Regulatesprocesses of cell survival, migration, differentiation and apoptosis; Stimulates angiogenesis; Attenuates fibroblast proliferation and collagen deposition |
[59, 60] |
| Platelet derived growth factor | Infarcted myocardium | Early and late post-infarct period (7th-28th day) | Regulates fibroblast migration, proliferation and transformation; Promotes fibrogenesis; Induces TGF-β synthesis; Facilitates stabilization and maturation of neovessels; Promotes resolution of inflammation |
[53, 62] |
| Vascular endothelial growth factor | Infarcted myocardium, border zone | Early and late post-infarct period (24 hours – 6 weeks) | Promotes angiogenesis and proliferation of endothelial cells; Inhibits cardiomyocyte apoptosis; Enhances myocardial contractility |
[63, 64] |
| Cardiotrophin-1 | Predominantly infarcted myocardium | Early and late pot-infarct period (24 hours – 8 weeks) | Induces cardiomyocyte survival; Inhibits cardiomyocyte apoptosis; Attenuates ischemia/reperfusion myocardium injury; Maintains myocardial contractility by stimulation of cardiomyocyte hypertrophy; Stimulates fibroblast migration and proliferation; Induces bone marrow stem cells migration; Stimulates synthesis of multiple extracellular matrix proteins |
[65, 80, 81] |
10. THERAPEUTIC TARGETING AT ECM REMODELING IN MI
Considering the fact that ECM plays a crucial role in the regulation of molecular and cellular response to cardiac muscle ischemic injury and ischemia (infarction)-induced cardiac remodeling, the search for new possible therapeutic approaches targeting ECM in patients with MI seems to be of great importance.
As mentioned above, a variety of clinical and experimental studies have indicated the important role of MMPs’ inhibition in postinfarct remodeling and progression of HF [16, 17, 143, 144]. It has been demonstrated that the administration of non-selective MMPs inhibitor in rabbits at 4 weeks after MI attenuated LV dilation and reduced the infarcted wall thinning [145]. Another study showed that the inhibition of MMP-2 activity in post-MI mice prevented cardiac rupture and reduced cardiac remodeling by decreasing macrophage infiltration [146]. On the other hand, according to the PREMIER trial data, administration of broad-spectrum MMPs inhibitor for 90 days in patients with ST-segment elevation MI did not improve LV ejection fraction and failed to reduce cardiac remodeling and the incidence of post-MI complications [147]. Another experimental study indicated that the selective MMP-9 inhibition in the early postinfarct period in mice model caused the development of delayed inflammation resolution, resulted consequently in the impairment of cardiac pump function restoration [148]. Therewith a selective MMP-12 inhibition has been shown to worsen post-MI cardiac dysfunction by delaying inflammation resolution [149]. Thus, numerous studies provided controversial results indicating that further research is needed to reveal the complex role of MMPs in targeting post-MI ECM remodeling.
A variety of ECM-targeting mediators being involved in MI-induced cardiac remodeling has been considered to be a promising target for promoting myocardial healing and repair [10]. Some studies demonstrated that the suppression of TGF-β signaling in acute MI mice significantly attenuated ventricular dilation, reduced fibrous tissue formation and improved LV systolic function [150]. However, it should be accentuated that TGF-β has pleiotropic and complex effects on various cell types and the responses to its inhibition differ considerably between individuals. There are some studies suggesting that TGF-β suppression leads to the formation of aortic aneurysm and cardiac rupture [151]. Two large clinical trials demonstrated that TNF-α antagonist - etanercept did not have a clinical effect in patients with HF and did not reduce mortality [152]. Another study indicated that the suppression of TNF-α in mice with acute MI resulted in significant anti-inflammatory effect and improved cardiac function [153]. Some recent studies suggest the suppression of IL-1β activity to be a potential therapeutic target in MI. Therewith, another study has shown that IL-1β inhibition in patients with MI resulted in a lower incidence of HF, but had no significant effect on LV volume and pump function [154]. Contradictory results of the studies on proinflammatory mediator’s inhibition in MI highlighted the complexity of their function, pleiotropy of the effects and involvement of many other co-factors contributing to pathophysiological alterations developing during all phases of MI and post-MI period.
Matricellular proteins also appear to be a promising therapeutic target in MI because of their ability to modulate cell-matrix interactions, growth factors and cytokines signaling. It has been demonstrated that in the rat model of MI, periostin improved ventricular remodeling, reduced fibrosis and infarct size, and increased angiogenesis [155]. Another study showed that periostin treatment in swine model of MI in addition to the improvement of LV systolic function increased myocardial fibrosis [156]. However, matricellular proteins have diverse functions and their effects depend on a variety of factors including cytokines and growth factors profiles and ECM structural condition [34, 157]. Thus, the multifunctional role of matricellular proteins also brings certain difficulties to their therapeutic implementation.
Stem cell therapy seems to be a highly promising therapeutic approach in post-MI patients. Recent meta-analyses evaluating data from 33 randomized control trials established that stem cell treatment in a postinfarct period significantly improved LV ejection fraction and reduced infarct size [158]. Some studies demonstrated that the application of 3D printed pre-vascularized stem cell patch augmented stem cell delivery by promoting vascularization and tissue matrix formation in vivo leading to the reduction of ventricular hypertrophy and fibrosis and to the improvement in cardiac function [159, 160]. A growing body of data suggests a perfusion-decellularization strategy that represents decellularization of the human heart with preserved 3D architecture and vasculature and then its recellularization with cardiomyocytes derived from induced pluripotent stem cells as the novel treatment option [161]. This strategy may be considered as an important step toward the production of extracellular matrix components in cardiovascular diseases and in MI, in particular. But it requires further experimental and clinical investigations.
CONCLUSION
Over the last few decades, knowledge about the role of ECM in MI spread far beyond the earlier concept of ECM providing mainly mechanical scaffolding support for contractile myocardial cells. Nowadays, ECM is recognized also as a dynamic network that transduces key signals essential for survival and function of both cardiomyocytes and non-contractile cardiac cells. Overall, pathological processes and dynamic transformation of ECM after myocardial damage and its post-infarction remodeling determine the course of MI and its short- and long-term prognosis in to a considerable extent than changes occurring in cardiomyocytes. This fact suggests high importance of further investigation of the mechanisms underlying ECM remodeling and cell-matrix interactions in CVDs and in MI particularly that may provide a good fundamental basis for elaboration of new therapeutic approaches to the treatment of such patients.
ACKNOWLEDGEMENTS
Declared none.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Mozaffarian D., Benjamin E.J., Go A.S., et al. Heart disease and stroke statistics--2015 update: A report from the American Heart Association. Circulation. 2015;131(4):e29–e322. doi: 10.1161/CIR.0000000000000152. [http://dx.doi.org/10.1161/CIR.0000000000000152]. [PMID: 25520374]. [DOI] [PubMed] [Google Scholar]
- 2.Nascimento B.R., Brant L.C., Moraes D.N., Ribeiro A.L. Global health and cardiovascular disease. Heart. 2014;100(22):1743–1749. doi: 10.1136/heartjnl-2014-306026. [http://dx.doi.org/10.1136/heartjnl-2014-306026]. [PMID: 25327515]. [DOI] [PubMed] [Google Scholar]
- 3.Dobaczewski M., Gonzalez-Quesada C., Frangogiannis N.G. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J. Mol. Cell. Cardiol. 2010;48(3):504–511. doi: 10.1016/j.yjmcc.2009.07.015. [http://dx.doi.org/10.1016/j.yjmcc.2009.07.015]. [PMID: 19631653]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bahit M.C., Kochar A., Granger C.B. Post-Myocardial Infarction Heart Failure. JACC Heart Fail. 2018;6(3):179–186. doi: 10.1016/j.jchf.2017.09.015. [http://dx.doi.org/10.1016/j.jchf.2017.09.015]. [PMID: 29496021]. [DOI] [PubMed] [Google Scholar]
- 5.Miyoshi H., Oishi Y., Mizuguchi Y., et al. Association of left atrial reservoir function with left atrial structural remodeling related to left ventricular dysfunction in asymptomatic patients with hypertension: evaluation by two-dimensional speckle-tracking echocardiography. Clin. Exp. Hypertens. 2015;37(2):155–165. doi: 10.3109/10641963.2014.933962. [http://dx.doi.org/10.3109/10641963.2014.933962]. [PMID: 25050647]. [DOI] [PubMed] [Google Scholar]
- 6.Deschamps A.M., Spinale F.G. Matrix modulation and heart failure: new concepts question old beliefs. Curr. Opin. Cardiol. 2005;20(3):211–216. doi: 10.1097/01.hco.0000162397.44843.83. [http://dx.doi.org/10.1097/01.hco.0000162397.44843.83]. [PMID: 15861009]. [DOI] [PubMed] [Google Scholar]
- 7.Jugdutt B.I. Ventricular remodeling after infarction and the extracellular collagen matrix: When is enough enough? Circulation. 2003;108(11):1395–1403. doi: 10.1161/01.CIR.0000085658.98621.49. [http://dx.doi.org/10.1161/01.CIR.0000085658.98621.49]. [PMID: 12975244]. [DOI] [PubMed] [Google Scholar]
- 8.Frangogiannis N.G. Pathophysiology of myocardial infarction. Compr. Physiol. 2015;5(4):1841–1875. doi: 10.1002/cphy.c150006. [http://dx.doi.org/10.1002/cphy.c150006]. [PMID: 26426469]. [DOI] [PubMed] [Google Scholar]
- 9.Matsui Y., Morimoto J., Uede T. Role of matricellular proteins in cardiac tissue remodeling after myocardial infarction. World J. Biol. Chem. 2010;1(5):69–80. doi: 10.4331/wjbc.v1.i5.69. [http://dx.doi.org/10.4331/wjbc.v1.i5.69]. [PMID: 21540992]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prabhu S.D., Frangogiannis N.G. The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ. Res. 2016;119(1):91–112. doi: 10.1161/CIRCRESAHA.116.303577. [http://dx.doi.org/10.1161/CIRCRESAHA.116.303577]. [PMID: 27340270]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindsey M.L., Zamilpa R. Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc. Ther. 2012;30(1):31–41. doi: 10.1111/j.1755-5922.2010.00207.x. [http://dx.doi.org/10.1111/j.1755-5922.2010.00207.x]. [PMID: 20645986]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kandalam V., Basu R., Abraham T., et al. Early activation of matrix metalloproteinases underlies the exacerbated systolic and diastolic dysfunction in mice lacking TIMP3 following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2010;299(4):H1012–H1023. doi: 10.1152/ajpheart.00246.2010. [http://dx.doi.org/10.1152/ajpheart.00246.2010]. [PMID: 20675565]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kandalam V., Basu R., Abraham T., et al. TIMP2 deficiency accelerates adverse post-myocardial infarction remodeling because of enhanced MT1-MMP activity despite lack of MMP2 activation. Circ. Res. 2010;106(4):796–808. doi: 10.1161/CIRCRESAHA.109.209189. [http://dx.doi.org/10.1161/CIRCRESAHA.109.209189]. [PMID: 20056917]. [DOI] [PubMed] [Google Scholar]
- 14.Koenig G.C., Rowe R.G., Day S.M., et al. MT1-MMP-dependent remodeling of cardiac extracellular matrix structure and function following myocardial infarction. Am. J. Pathol. 2012;180(5):1863–1878. doi: 10.1016/j.ajpath.2012.01.022. [http://dx.doi.org/10.1016/j.ajpath.2012.01.022]. [PMID: 22464947]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cleutjens J.P.M., Kandala J.C., Guarda E., Guntaka R.V., Weber K.T. Regulation of collagen degradation in the rat myocardium after infarction. J. Mol. Cell. Cardiol. 1995;27(6):1281–1292. doi: 10.1016/s0022-2828(05)82390-9. [http://dx.doi.org/10.1016/S0022-2828(05)82390-9]. [PMID: 8531210]. [DOI] [PubMed] [Google Scholar]
- 16.Heymans S., Luttun A., Nuyens D., et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat. Med. 1999;5(10):1135–1142. doi: 10.1038/13459. [http://dx.doi.org/10.1038/13459]. [PMID: 10502816]. [DOI] [PubMed] [Google Scholar]
- 17.Chiao Y.A., Ramirez T.A., Zamilpa R., et al. Matrix metalloproteinase-9 deletion attenuates myocardial fibrosis and diastolic dysfunction in ageing mice. Cardiovasc. Res. 2012;96(3):444–455. doi: 10.1093/cvr/cvs275. [http://dx.doi.org/10.1093/cvr/cvs275]. [PMID: 22918978]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zamilpa R., Ibarra J., de Castro Brás L.E., et al. Transgenic overexpression of matrix metalloproteinase-9 in macrophages attenuates the inflammatory response and improves left ventricular function post-myocardial infarction. J. Mol. Cell. Cardiol. 2012;53(5):599–608. doi: 10.1016/j.yjmcc.2012.07.017. [http://dx.doi.org/10.1016/j.yjmcc.2012.07.017]. [PMID: 22884843]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma Y., Chiao Y.A., Clark R., et al. Deriving a cardiac ageing signature to reveal MMP-9-dependent inflammatory signalling in senescence. Cardiovasc. Res. 2015;106(3):421–431. doi: 10.1093/cvr/cvv128. [http://dx.doi.org/10.1093/cvr/cvv128]. [PMID: 25883218]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frieler R.A., Mortensen R.M. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation. 2015;131(11):1019–1030. doi: 10.1161/CIRCULATIONAHA.114.008788. [http://dx.doi.org/10.1161/CIRCULATIONAHA.114.008788]. [PMID: 25779542]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu Q., Watson R.R., Marchalonis J.J., Larson D.F. A role for T lymphocytes in mediating cardiac diastolic function. Am. J. Physiol. Heart Circ. Physiol. 2005;289(2):H643–H651. doi: 10.1152/ajpheart.00073.2005. [http://dx.doi.org/10.1152/ajpheart.00073.2005]. [PMID: 16014617]. [DOI] [PubMed] [Google Scholar]
- 22.Hinz B., Phan S.H., Thannickal V.J., et al. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012;180(4):1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [http://dx.doi.org/10.1016/j.ajpath.2012.02.004]. [PMID: 22387320]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krenning G., Zeisberg E.M., Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J. Cell. Physiol. 2010;225(3):631–637. doi: 10.1002/jcp.22322. [http://dx.doi.org/10.1002/jcp.22322]. [PMID: 20635395]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turner N.A., Porter K.E. Function and fate of myofibroblasts after myocardial infarction. Fibrogen Tissue Rep. 2013;6(1):5. doi: 10.1186/1755-1536-6-5. [http://dx.doi.org/10.1186/1755-1536-6-5]. [PMID: 23448358]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan D., Takawale A., Lee J., Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012;5(1):15–28. doi: 10.1186/1755-1536-5-15. [http://dx.doi.org/10.1186/1755-1536-5-15]. [PMID: 22943504]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma Y., Halade G.V., Lindsey M.L. Extracellular matrix and fibroblast communication following myocardial infarction. J. Cardiovasc. Transl. Res. 2012;5(6):848–857. doi: 10.1007/s12265-012-9398-z. [http://dx.doi.org/10.1007/s12265-012-9398-z]. [PMID: 22926488]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zavadzkas J.A., Mukherjee R., Rivers W.T., et al. Direct regulation of membrane type 1 matrix metalloproteinase following myocardial infarction causes changes in survival, cardiac function, and remodeling. Am. J. Physiol. Heart Circ. Physiol. 2011;301(4):H1656–H1666. doi: 10.1152/ajpheart.00141.2011. [http://dx.doi.org/10.1152/ajpheart.00141.2011]. [PMID: 21666120]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z.F., Wang N.P., Harmouche S., et al. Postconditioning promotes the cardiac repair through balancing collagen degradation and synthesis after myocardial infarction in rats. Basic Res. Cardiol. 2013;108(1):318–327. doi: 10.1007/s00395-012-0318-9. [http://dx.doi.org/10.1007/s00395-012-0318-9]. [PMID: 23203208]. [DOI] [PubMed] [Google Scholar]
- 29.Okada M., Oba Y., Yamawaki H. Endostatin stimulates proliferation and migration of adult rat cardiac fibroblasts through PI3K/Akt pathway. Eur. J. Pharmacol. 2015;750:20–26. doi: 10.1016/j.ejphar.2015.01.019. [http://dx.doi.org/10.1016/j.ejphar.2015.01.019]. [PMID: 25620135]. [DOI] [PubMed] [Google Scholar]
- 30.Dobaczewski M., de Haan J.J., Frangogiannis N.G. The extracellular matrix modulates fibroblast phenotype and function in the infarcted myocardium. J. Cardiovasc. Transl. Res. 2012;5(6):837–847. doi: 10.1007/s12265-012-9406-3. [http://dx.doi.org/10.1007/s12265-012-9406-3]. [PMID: 22956156]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Konstandin M.H., Völkers M., Collins B., et al. Fibronectin contributes to pathological cardiac hypertrophy but not physiological growth. Basic Res. Cardiol. 2013;108(5) doi: 10.1007/s00395-013-0375-8. [http://dx.doi.org/10.1007/s00395-013-0375-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van den Borne S.W., Diez J., Blankesteijn W.M., Verjans J., Hofstra L., Narula J. Myocardial remodeling after infarction: The role of myofibroblasts. Nat. Rev. Cardiol. 2010;7(1):30–37. doi: 10.1038/nrcardio.2009.199. [http://dx.doi.org/10.1038/nrcardio.2009.199]. [PMID: 19949426]. [DOI] [PubMed] [Google Scholar]
- 33.Squires C.E., Escobar G.P., Payne J.F., et al. Altered fibroblast function following myocardial infarction. J. Mol. Cell. Cardiol. 2005;39(4):699–707. doi: 10.1016/j.yjmcc.2005.07.008. [http://dx.doi.org/10.1016/j.yjmcc.2005.07.008]. [PMID: 16111700]. [DOI] [PubMed] [Google Scholar]
- 34.Frangogiannis N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Invest. 2017;127(5):1600–1612. doi: 10.1172/JCI87491. [http://dx.doi.org/10.1172/JCI87491]. [PMID: 28459429]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jousset F., Maguy A., Rohr S., Kucera J.P. Myofibroblasts electrotonically coupled to cardiomyocytes alter conduction: Insights at the cellular level from a detailed in silico tissue structure model. Front. Physiol. 2016;7:496. doi: 10.3389/fphys.2016.00496. [http://dx.doi.org/10.3389/fphys.2016.00496]. [PMID: 27833567]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lighthouse J.K., Small E.M. Transcriptional control of cardiac fibroblast plasticity. J. Mol. Cell. Cardiol. 2016;91:52–60. doi: 10.1016/j.yjmcc.2015.12.016. [http://dx.doi.org/10.1016/j.yjmcc.2015.12.016]. [PMID: 26721596]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tian J., An X., Niu L. Myocardial fibrosis in congenital and pediatric heart disease. Exp. Ther. Med. 2017;13(5):1660–1664. doi: 10.3892/etm.2017.4224. [http://dx.doi.org/10.3892/etm.2017.4224]. [PMID: 28565750]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frangogiannis N.G., Michael L.H., Entman M.L. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb). Cardiovasc. Res. 2000;48(1):89–100. doi: 10.1016/s0008-6363(00)00158-9. [http://dx.doi.org/10.1016/S0008-6363(00)00158-9]. [PMID: 11033111]. [DOI] [PubMed] [Google Scholar]
- 39.Frangogiannis N.G., Shimoni S., Chang S.M., et al. Active interstitial remodeling: an important process in the hibernating human myocardium. J. Am. Coll. Cardiol. 2002;39(9):1468–1474. doi: 10.1016/s0735-1097(02)01792-8. [http://dx.doi.org/10.1016/S0735-1097(02)01792-8]. [PMID: 11985909]. [DOI] [PubMed] [Google Scholar]
- 40.Willems I.E., Havenith M.G., De Mey J.G., Daemen M.J. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am. J. Pathol. 1994;145(4):868–875. [PMID: 7943177]. [PMC free article] [PubMed] [Google Scholar]
- 41.Huebener P., Abou-Khamis T., Zymek P., et al. CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. J. Immunol. 2008;180(4):2625–2633. doi: 10.4049/jimmunol.180.4.2625. [http://dx.doi.org/10.4049/jimmunol.180.4.2625]. [PMID: 18250474]. [DOI] [PubMed] [Google Scholar]
- 42.Serini G., Gabbiani G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp. Cell Res. 1999;250(2):273–283. doi: 10.1006/excr.1999.4543. [http://dx.doi.org/10.1006/excr.1999.4543]. [PMID: 10413583]. [DOI] [PubMed] [Google Scholar]
- 43.Nah D.Y., Rhee M.Y. The inflammatory response and cardiac repair after myocardial infarction. Korean Circ. J. 2009;39(10):393–398. doi: 10.4070/kcj.2009.39.10.393. [http://dx.doi.org/10.4070/kcj.2009.39.10.393]. [PMID: 19949583]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bujak M., Dobaczewski M., Chatila K., et al. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am. J. Pathol. 2008;173(1):57–67. doi: 10.2353/ajpath.2008.070974. [http://dx.doi.org/10.2353/ajpath.2008.070974]. [PMID: 18535174]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang F., Trial J., Diwan A., et al. Regulation of cardiac fibroblast cellular function by leukemia inhibitory factor. J. Mol. Cell. Cardiol. 2002;34(10):1309–1316. doi: 10.1006/jmcc.2002.2059. [http://dx.doi.org/10.1006/jmcc.2002.2059]. [PMID: 12392991]. [DOI] [PubMed] [Google Scholar]
- 46.Pepper M.S., Ferrara N., Orci L., Montesano R. Leukemia inhibitory factor (LIF) inhibits angiogenesis in vitro. J. Cell Sci. 1995;108(Pt 1):73–83. doi: 10.1242/jcs.108.1.73. [PMID: 7537748]. [DOI] [PubMed] [Google Scholar]
- 47.Sun Y., Weber K.T. Infarct scar: a dynamic tissue. Cardiovasc. Res. 2000;46(2):250–256. doi: 10.1016/s0008-6363(00)00032-8. [http://dx.doi.org/10.1016/S0008-6363(00)00032-8]. [PMID: 10773228]. [DOI] [PubMed] [Google Scholar]
- 48.Zhang J., Fan G., Zhao H., et al. Targeted inhibition of focal adhesion kinase attenuates cardiac fibrosis and preserves heart function in adverse cardiac remodeling. Sci. Rep. 2017;7:43146. doi: 10.1038/srep43146. [http://dx.doi.org/10.1038/srep43146]. [PMID: 28225063]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moore J.B., IV, Zhao J., Fischer A.G., et al. Histone deacetylase 1 depletion activates human cardiac mesenchymal stromal cell proangiogenic paracrine signaling through a mechanism requiring enhanced basic fibroblast growth factor synthesis and secretion. J. Am. Heart Assoc. 2017;6(7):e006183. doi: 10.1161/JAHA.117.006183. [http://dx.doi.org/10.1161/JAHA.117.006183]. [PMID: 28679560]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ushikoshi H., Takahashi T., Chen X., et al. Local overexpression of HB-EGF exacerbates remodeling following myocardial infarction by activating noncardiomyocytes. Lab. Invest. 2005;85(7):862–873. doi: 10.1038/labinvest.3700282. [http://dx.doi.org/10.1038/labinvest.3700282]. [PMID: 15856048]. [DOI] [PubMed] [Google Scholar]
- 51.Koshman YE, Sternlicht MD, Kim T, et al. Connective tissue growth factor regulates cardiac function and tissue remodeling in a mouse model of dilated cardiomyopathy. J Mol Cell Cardiol. 2015;89(Pt B):214–222.. doi: 10.1016/j.yjmcc.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu X.H., Pan L.L., Deng H.Y., et al. Leonurine (SCM-198) attenuates myocardial fibrotic response via inhibition of NADPH oxidase 4. Free Radic. Biol. Med. 2013;54:93–104. doi: 10.1016/j.freeradbiomed.2012.10.555. [http://dx.doi.org/10.1016/j.freeradbiomed.2012.10.555]. [PMID: 23127783]. [DOI] [PubMed] [Google Scholar]
- 53.Shinde A.V., Frangogiannis N.G. Fibroblasts in myocardial infarction: a role in inflammation and repair. J. Mol. Cell. Cardiol. 2014;70:74–82. doi: 10.1016/j.yjmcc.2013.11.015. [http://dx.doi.org/10.1016/j.yjmcc.2013.11.015]. [PMID: 24321195]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frangogiannis N.G. The role of transforming growth factor (TGF)-β in the infarcted myocardium. J. Thorac. Dis. 2017;9(1):S52–S63. doi: 10.21037/jtd.2016.11.19. [http://dx.doi.org/10.21037/jtd.2016.11.19]. [PMID: 28446968]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frangogiannis N.G. Matricellular proteins in cardiac adaptation and disease. Physiol. Rev. 2012;92(2):635–688. doi: 10.1152/physrev.00008.2011. [http://dx.doi.org/10.1152/physrev.00008.2011]. [PMID: 22535894]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dobaczewski M., Chen W., Frangogiannis N.G. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011;51(4):600–606. doi: 10.1016/j.yjmcc.2010.10.033. [http://dx.doi.org/10.1016/j.yjmcc.2010.10.033]. [PMID: 21059352]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sarrazy V., Koehler A., Chow M.L., et al. Integrins αvβ5 and αvβ3 promote latent TGF-β1 activation by human cardiac fibroblast contraction. Cardiovasc. Res. 2014;102(3):407–417. doi: 10.1093/cvr/cvu053. [http://dx.doi.org/10.1093/cvr/cvu053]. [PMID: 24639195]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frantz S., Hu K., Adamek A., et al. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res. Cardiol. 2008;103(5):485–492. doi: 10.1007/s00395-008-0739-7. [http://dx.doi.org/10.1007/s00395-008-0739-7]. [PMID: 18651091]. [DOI] [PubMed] [Google Scholar]
- 59.Svystonyuk D.A., Ngu J.M., Mewhort H.E., et al. Fibroblast growth factor-2 regulates human cardiac myofibroblast-mediated extracellular matrix remodeling. J. Transl. Med. 2015;13:147. doi: 10.1186/s12967-015-0510-4. [http://dx.doi.org/10.1186/s12967-015-0510-4]. [PMID: 25948488]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Virag J.A., Rolle M.L., Reece J., Hardouin S., Feigl E.O., Murry C.E. Fibroblast growth factor-2 regulates myocardial infarct repair: Effects on cell proliferation, scar contraction, and ventricular function. Am. J. Pathol. 2007;171(5):1431–1440. doi: 10.2353/ajpath.2007.070003. [http://dx.doi.org/10.2353/ajpath.2007.070003]. [PMID: 17872976]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao T., Zhao W., Chen Y., Li V.S., Meng W., Sun Y. Platelet-derived growth factor-D promotes fibrogenesis of cardiac fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2013;304(12):H1719–H1726. doi: 10.1152/ajpheart.00130.2013. [http://dx.doi.org/10.1152/ajpheart.00130.2013]. [PMID: 23585135]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zymek P., Bujak M., Chatila K., et al. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J. Am. Coll. Cardiol. 2006;48(11):2315–2323. doi: 10.1016/j.jacc.2006.07.060. [http://dx.doi.org/10.1016/j.jacc.2006.07.060]. [PMID: 17161265]. [DOI] [PubMed] [Google Scholar]
- 63.Awada H.K., Johnson N.R., Wang Y. Sequential delivery of angiogenic growth factors improves revascularization and heart function after myocardial infarction. J. Control. Release. 2015;207:7–17. doi: 10.1016/j.jconrel.2015.03.034. [http://dx.doi.org/10.1016/j.jconrel.2015.03.034]. [PMID: 25836592]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bry M., Kivelä R., Leppänen V.M., Alitalo K. Vascular endothelial growth factor-B in physiology and disease. Physiol. Rev. 2014;94(3):779–794. doi: 10.1152/physrev.00028.2013. [http://dx.doi.org/10.1152/physrev.00028.2013]. [PMID: 24987005]. [DOI] [PubMed] [Google Scholar]
- 65.Hogas S., Bilha S.C., Branisteanu D., et al. Potential novel biomarkers of cardiovascular dysfunction and disease: Cardiotrophin-1, adipokines and galectin-3. Arch. Med. Sci. 2017;13(4):897–913. doi: 10.5114/aoms.2016.58664. [http://dx.doi.org/10.5114/aoms.2016.58664]. [PMID: 28721158]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abdul-Ghani M., Suen C., Jiang B., et al. Cardiotrophin 1 stimulates beneficial myogenic and vascular remodeling of the heart. Cell Res. 2017;27(10):1195–1215. doi: 10.1038/cr.2017.87. [http://dx.doi.org/10.1038/cr.2017.87]. [PMID: 28785017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pennica D., King K.L., Shaw K.J., et al. Expression cloning of cardiotrophin 1, a cytokine that induces cardiac myocyte hypertrophy. Proc. Natl. Acad. Sci. USA. 1995;92(4):1142–1146. doi: 10.1073/pnas.92.4.1142. [http://dx.doi.org/10.1073/pnas.92.4.1142]. [PMID: 7862649]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.López B., González A., Querejeta R., Larman M., Rábago G., Díez J. Association of cardiotrophin-1 with myocardial fibrosis in hypertensive patients with heart failure. Hypertension. 2014;63(3):483–489. doi: 10.1161/HYPERTENSIONAHA.113.02654. [http://dx.doi.org/10.1161/HYPERTENSIONAHA.113.02654]. [PMID: 24366078]. [DOI] [PubMed] [Google Scholar]
- 69.Schillaci G., Pucci G., Perlini S. From hypertension to hypertrophy to heart failure: the role of cardiotrophin-1. J. Hypertens. 2013;31(3):474–476. doi: 10.1097/HJH.0b013e32835ed4bb. [http://dx.doi.org/10.1097/HJH.0b013e32835ed4bb]. [PMID: 23615209]. [DOI] [PubMed] [Google Scholar]
- 70.Tecimer M.E., Yuksel A., Bicer M., et al. The role of cardiotrophin-1 in the evaluation of myocardial ischemia in patients undergoing off-pump and on-pump coronary artery bypass surgery. Scientific Pages Heart. 2016;1(1):14–20. [Google Scholar]
- 71.Aoyama T., Takimoto Y., Pennica D., et al. Augmented expression of cardiotrophin-1 and its receptor component, gp130, in both left and right ventricles after myocardial infarction in the rat. J. Mol. Cell. Cardiol. 2000;32(10):1821–1830. doi: 10.1006/jmcc.2000.1218. [http://dx.doi.org/10.1006/jmcc.2000.1218]. [PMID: 11013126]. [DOI] [PubMed] [Google Scholar]
- 72.López B., González A., Lasarte J.J., et al. Is plasma cardiotrophin-1 a marker of hypertensive heart disease? J. Hypertens. 2005;23(3):625–632. doi: 10.1097/01.hjh.0000160221.09468.d3. [http://dx.doi.org/10.1097/01.hjh.0000160221.09468.d3]. [PMID: 15716706]. [DOI] [PubMed] [Google Scholar]
- 73.Altun I., Pamukcu B., Yildiz C.E., et al. Cardiotrophin-1: A new predictor of atrial fibrillation relapses after successful cardioversion. Bosn. J. Basic Med. Sci. 2015;15(3):68–73. doi: 10.17305/bjbms.2015.503. [http://dx.doi.org/10.17305/bjbms.2015.503]. [PMID: 26295297]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Al-Mazroua H.A., Al-Rasheed N.M., Korashy H.M. Downregulation of the cardiotrophin-1 gene expression by valsartan and spironolactone in hypertrophied heart rats in vivo and rat cardiomyocyte H9c2 cell line in vitro: A novel mechanism of cardioprotection. J. Cardiovasc. Pharmacol. 2013;61(4):337–344. doi: 10.1097/FJC.0b013e318283a565. [http://dx.doi.org/10.1097/FJC.0b013e318283a565]. [PMID: 23288202]. [DOI] [PubMed] [Google Scholar]
- 75.González A., López B., Ravassa S., et al. Cardiotrophin-1 in hypertensive heart disease. Endocrine. 2012;42(1):9–17. doi: 10.1007/s12020-012-9649-4. [http://dx.doi.org/10.1007/s12020-012-9649-4]. [PMID: 22418690]. [DOI] [PubMed] [Google Scholar]
- 76.Robador P.A., San José G., Rodríguez C., et al. HIF-1-mediated up-regulation of cardiotrophin-1 is involved in the survival response of cardiomyocytes to hypoxia. Cardiovasc. Res. 2011;92(2):247–255. doi: 10.1093/cvr/cvr202. [http://dx.doi.org/10.1093/cvr/cvr202]. [PMID: 21771897]. [DOI] [PubMed] [Google Scholar]
- 77.Song K., Wang S., Huang B., Luciano A., Srivastava R., Mani A. Plasma cardiotrophin-1 levels are associated with hypertensive heart disease: A meta-analysis. J. Clin. Hypertens. (Greenwich) 2014;16(9):686–692. doi: 10.1111/jch.12376. [http://dx.doi.org/10.1111/jch.12376]. [PMID: 25052897]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wollert K.C., Taga T., Saito M., et al. Cardiotrophin-1 activates a distinct form of cardiac muscle cell hypertrophy. Assembly of sarcomeric units in series VIA gp130/leukemia inhibitory factor receptor-dependent pathways. J. Biol. Chem. 1996;271(16):9535–9545. doi: 10.1074/jbc.271.16.9535. [http://dx.doi.org/10.1074/jbc.271.16.9535]. [PMID: 8621626]. [DOI] [PubMed] [Google Scholar]
- 79.Aguilar-Melero P., Luque A., Machuca M.M., et al. Cardiotrophin-1 reduces ischemia/reperfusion injury during liver transplant. J. Surg. Res. 2013;181(2):e83–e91. doi: 10.1016/j.jss.2012.07.046. [http://dx.doi.org/10.1016/j.jss.2012.07.046]. [PMID: 22906559]. [DOI] [PubMed] [Google Scholar]
- 80.Calabrò P., Limongelli G., Riegler L., et al. Novel insights into the role of cardiotrophin-1 in cardiovascular diseases. J. Mol. Cell. Cardiol. 2009;46(2):142–148. doi: 10.1016/j.yjmcc.2008.11.002. [http://dx.doi.org/10.1016/j.yjmcc.2008.11.002]. [PMID: 19059413]. [DOI] [PubMed] [Google Scholar]
- 81.Freed D.H., Cunnington R.H., Dangerfield A.L., Sutton J.S., Dixon I.M. Emerging evidence for the role of cardiotrophin-1 in cardiac repair in the infarcted heart. Cardiovasc. Res. 2005;65(4):782–792. doi: 10.1016/j.cardiores.2004.11.026. [http://dx.doi.org/10.1016/j.cardiores.2004.11.026]. [PMID: 15721858]. [DOI] [PubMed] [Google Scholar]
- 82.Freed D.H., Chilton L., Li Y., et al. Role of myosin light chain kinase in cardiotrophin-1-induced cardiac myofibroblast cell migration. Am. J. Physiol. Heart Circ. Physiol. 2011;301(2):H514–H522. doi: 10.1152/ajpheart.01041.2010. [http://dx.doi.org/10.1152/ajpheart.01041.2010]. [PMID: 21572008]. [DOI] [PubMed] [Google Scholar]
- 83.Freed D.H., Moon M.C., Borowiec A.M., Jones S.C., Zahradka P., Dixon I.M. Cardiotrophin-1: expression in experimental myocardial infarction and potential role in post-MI wound healing. Mol. Cell. Biochem. 2003;254(1-2):247–256. doi: 10.1023/a:1027332504861. [http://dx.doi.org/10.1023/A:1027332504861]. [PMID: 14674704]. [DOI] [PubMed] [Google Scholar]
- 84.D’Amario D., Leone A.M., Cannata F., et al. Granulocyte colony-stimulating factor in patients with a large anterior wall acute myocardial infarction to prevent left ventricular remodeling (the RIGENERA trial): 10 years follow-up - Final results. Eur. Heart J. 2017;38(1) [Google Scholar]
- 85.D’Amario D., Leone A.M., Borovac J.A., et al. Granulocyte colony-stimulating factor for the treatment of cardiovascular diseases: An update with a critical appraisal. Pharmacol. Res. 2018;127:67–76. doi: 10.1016/j.phrs.2017.06.001. [http://dx.doi.org/10.1016/j.phrs.2017.06.001]. [PMID: 28602846]. [DOI] [PubMed] [Google Scholar]
- 86.Sugano Y., Anzai T., Yoshikawa T., et al. Granulocyte colony-stimulating factor attenuates early ventricular expansion after experimental myocardial infarction. Cardiovasc. Res. 2005;65(2):446–456. doi: 10.1016/j.cardiores.2004.10.008. [http://dx.doi.org/10.1016/j.cardiores.2004.10.008]. [PMID: 15639484]. [DOI] [PubMed] [Google Scholar]
- 87.Yano T., Miura T., Whittaker P., et al. Macrophage colony-stimulating factor treatment after myocardial infarction attenuates left ventricular dysfunction by accelerating infarct repair. J. Am. Coll. Cardiol. 2006;47(3):626–634. doi: 10.1016/j.jacc.2005.09.037. [http://dx.doi.org/10.1016/j.jacc.2005.09.037]. [PMID: 16458148]. [DOI] [PubMed] [Google Scholar]
- 88.Maekawa Y., Anzai T., Yoshikawa T., et al. Effect of granulocyte-macrophage colony-stimulating factor inducer on left ventricular remodeling after acute myocardial infarction. J. Am. Coll. Cardiol. 2004;44(7):1510–1520. doi: 10.1016/j.jacc.2004.05.083. [http://dx.doi.org/10.1016/j.jacc.2004.05.083]. [PMID: 15464336]. [DOI] [PubMed] [Google Scholar]
- 89.Morishita K., Takemura G., Tsujimoto A., et al. Postinfarction cardiac remodeling proceeds normally in granulocyte colony-stimulating factor knockout mice. Am. J. Pathol. 2015;185(7):1899–1911. doi: 10.1016/j.ajpath.2015.03.018. [http://dx.doi.org/10.1016/j.ajpath.2015.03.018]. [PMID: 25976246]. [DOI] [PubMed] [Google Scholar]
- 90.Ishikawa Y., Akasaka Y., Ishii T., et al. Changes in the distribution pattern of gelatin-binding protein of 28 kDa (adiponectin) in myocardial remodelling after ischaemic injury. Histopathology. 2003;42(1):43–52. doi: 10.1046/j.1365-2559.2003.01518.x. [http://dx.doi.org/10.1046/j.1365-2559.2003.01518.x]. [PMID: 12493024]. [DOI] [PubMed] [Google Scholar]
- 91.Jenke A., Schur R., Röger C., et al. Adiponectin attenuates profibrotic extracellular matrix remodeling following cardiac injury by up-regulating matrix metalloproteinase 9 expression in mice. Physiol. Rep. 2017;5(24):e13523. doi: 10.14814/phy2.13523. [http://dx.doi.org/10.14814/phy2.13523]. [PMID: 29263115]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ghantous C.M., Azrak Z., Hanache S., Abou-Kheir W., Zeidan A. Differential role of leptin and adiponectin in cardiovascular system. Int. J. Endocrinol. 2015;•••:2015534320. doi: 10.1155/2015/534320. [http://dx.doi.org/10.1155/2015/534320]. [PMID: 26064110]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McGaffin K.R., Sun C-K., Rager J.J., et al. Leptin signalling reduces the severity of cardiac dysfunction and remodelling after chronic ischaemic injury. Cardiovasc. Res. 2008;77(1):54–63. doi: 10.1093/cvr/cvm023. [http://dx.doi.org/10.1093/cvr/cvm023]. [PMID: 18006469]. [DOI] [PubMed] [Google Scholar]
- 94.Zhang B.H., Guo C.X., Wang H.X., et al. Cardioprotective effects of adipokine apelin on myocardial infarction. Heart Vessels. 2014;29(5):679–689. doi: 10.1007/s00380-013-0425-z. [http://dx.doi.org/10.1007/s00380-013-0425-z]. [PMID: 24141989]. [DOI] [PubMed] [Google Scholar]
- 95.Zhang X., Hu W., Feng F., Xu J., Wu F. Apelin-13 protects against myocardial infarction-induced myocardial fibrosis. Mol. Med. Rep. 2016;13(6):5262–5268. doi: 10.3892/mmr.2016.5163. [http://dx.doi.org/10.3892/mmr.2016.5163]. [PMID: 27109054]. [DOI] [PubMed] [Google Scholar]
- 96.Pradhan G., Samson S.L., Sun Y. Ghrelin: Much more than a hunger hormone. Curr. Opin. Clin. Nutr. Metab. Care. 2013;16(6):619–624. doi: 10.1097/MCO.0b013e328365b9be. [http://dx.doi.org/10.1097/MCO.0b013e328365b9be]. [PMID: 24100676]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yuan M.J. He-Huang, Hu HY, Li-Quan, Hong-Jiang, Huang CX. Myocardial angiogenesis after chronic ghrelin treatment in a rat myocardial infarction model. Regul. Pept. 2012;179(1-3):39–42. doi: 10.1016/j.regpep.2012.08.013. [http://dx.doi.org/10.1016/j.regpep.2012.08.013]. [PMID: 22960289]. [DOI] [PubMed] [Google Scholar]
- 98.Mao Y., Tokudome T., Otani K., et al. Ghrelin prevents incidence of malignant arrhythmia after acute myocardial infarction through vagal afferent nerves. Endocrinology. 2012;153(7):3426–3434. doi: 10.1210/en.2012-1065. [http://dx.doi.org/10.1210/en.2012-1065]. [PMID: 22535766]. [DOI] [PubMed] [Google Scholar]
- 99.Schellings M.W., Pinto Y.M., Heymans S. Matricellular proteins in the heart: possible role during stress and remodeling. Cardiovasc. Res. 2004;64(1):24–31. doi: 10.1016/j.cardiores.2004.06.006. [http://dx.doi.org/10.1016/j.cardiores.2004.06.006]. [PMID: 15364610]. [DOI] [PubMed] [Google Scholar]
- 100.Ma Y., de Castro Brás L.E., Toba H., et al. Myofibroblasts and the extracellular matrix network in post-myocardial infarction cardiac remodeling. Pflugers Arch. 2014;466(6):1113–1127. doi: 10.1007/s00424-014-1463-9. [http://dx.doi.org/10.1007/s00424-014-1463-9]. [PMID: 24519465]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bornstein P. Matricellular proteins: An overview. J. Cell Commun. Signal. 2009;3(3-4):163–165. doi: 10.1007/s12079-009-0069-z. [http://dx.doi.org/10.1007/s12079-009-0069-z]. [PMID: 19779848]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Imanaka-Yoshida K., Hiroe M., Nishikawa T., et al. Tenascin-C modulates adhesion of cardiomyocytes to extracellular matrix during tissue remodeling after myocardial infarction. Lab. Invest. 2001;81(7):1015–1024. doi: 10.1038/labinvest.3780313. [http://dx.doi.org/10.1038/labinvest.3780313]. [PMID: 11454990]. [DOI] [PubMed] [Google Scholar]
- 103.Nishioka T., Onishi K., Shimojo N., et al. Tenascin-C may aggravate left ventricular remodeling and function after myocardial infarction in mice. Am. J. Physiol. Heart Circ. Physiol. 2010;298(3):H1072–H1078. doi: 10.1152/ajpheart.00255.2009. [http://dx.doi.org/10.1152/ajpheart.00255.2009]. [PMID: 20081106]. [DOI] [PubMed] [Google Scholar]
- 104.Imanaka-Yoshida K. Extracellular matrix remodeling in vascular development and disease. 2016 https://europepmc.org/books/NBK500279;jsessionid=A0E7D81C6A80CC5CF3186C92A2F3B60A [PubMed]
- 105.Midwood K.S., Orend G. The role of tenascin-C in tissue injury and tumorigenesis. J. Cell Commun. Signal. 2009;3(3-4):287–310. doi: 10.1007/s12079-009-0075-1. [http://dx.doi.org/10.1007/s12079-009-0075-1]. [PMID: 19838819]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mao J.R., Taylor G., Dean W.B., et al. Tenascin-X deficiency mimics Ehlers-Danlos syndrome in mice through alteration of collagen deposition. Nat. Genet. 2002;30(4):421–425. doi: 10.1038/ng850. [http://dx.doi.org/10.1038/ng850]. [PMID: 11925569]. [DOI] [PubMed] [Google Scholar]
- 107.Matsumoto K., Takayama N., Ohnishi J., et al. Tumour invasion and metastasis are promoted in mice deficient in tenascin-X. Genes Cells. 2001;6(12):1101–1111. doi: 10.1046/j.1365-2443.2001.00482.x. [http://dx.doi.org/10.1046/j.1365-2443.2001.00482.x]. [PMID: 11737270]. [DOI] [PubMed] [Google Scholar]
- 108.Bradshaw A.D. The role of secreted protein acidic and rich in cysteine (SPARC) in cardiac repair and fibrosis: Does expression of SPARC by macrophages influence outcomes? J. Mol. Cell. Cardiol. 2016;93:156–161. doi: 10.1016/j.yjmcc.2015.11.014. [http://dx.doi.org/10.1016/j.yjmcc.2015.11.014]. [PMID: 26582465]. [DOI] [PubMed] [Google Scholar]
- 109.Schellings M.W., Vanhoutte D., Swinnen M., et al. Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J. Exp. Med. 2009;206(1):113–123. doi: 10.1084/jem.20081244. [http://dx.doi.org/10.1084/jem.20081244]. [PMID: 19103879]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rienks M., Papageorgiou A.P., Frangogiannis N.G., Heymans S. Myocardial extracellular matrix: An ever-changing and diverse entity. Circ. Res. 2014;114(5):872–888. doi: 10.1161/CIRCRESAHA.114.302533. [http://dx.doi.org/10.1161/CIRCRESAHA.114.302533]. [PMID: 24577967]. [DOI] [PubMed] [Google Scholar]
- 111.McCurdy S.M., Dai Q., Zhang J., et al. SPARC mediates early extracellular matrix remodeling following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2011;301(2):H497–H505. doi: 10.1152/ajpheart.01070.2010. [http://dx.doi.org/10.1152/ajpheart.01070.2010]. [PMID: 21602472]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yokosaki Y., Tanaka K., Higashikawa F., Yamashita K., Eboshida A. Distinct structural requirements for binding of the integrins alphavbeta6, alphavbeta3, alphavbeta5, alpha5beta1 and alpha9beta1 to osteopontin. Matrix Biol. 2005;24(6):418–427. doi: 10.1016/j.matbio.2005.05.005. [http://dx.doi.org/10.1016/j.matbio.2005.05.005]. [PMID: 16005200]. [DOI] [PubMed] [Google Scholar]
- 113.Scatena M., Liaw L., Giachelli C.M. Osteopontin: A multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler. Thromb. Vasc. Biol. 2007;27(11):2302–2309. doi: 10.1161/ATVBAHA.107.144824. [http://dx.doi.org/10.1161/ATVBAHA.107.144824]. [PMID: 17717292]. [DOI] [PubMed] [Google Scholar]
- 114.Trueblood N.A., Xie Z., Communal C., et al. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ. Res. 2001;88(10):1080–1087. doi: 10.1161/hh1001.090842. [http://dx.doi.org/10.1161/hh1001.090842]. [PMID: 11375279]. [DOI] [PubMed] [Google Scholar]
- 115.Xie Z., Singh M., Singh K. ERK1/2 and JNKs, but not p38 kinase, are involved in reactive oxygen species-mediated induction of osteopontin gene expression by angiotensin II and interleukin-1beta in adult rat cardiac fibroblasts. J. Cell. Physiol. 2004;198(3):399–407. doi: 10.1002/jcp.10419. [http://dx.doi.org/10.1002/jcp.10419]. [PMID: 14755545]. [DOI] [PubMed] [Google Scholar]
- 116.Lindsey M.L., Saucerman J.J., DeLeon-Pennell K.Y. Knowledge gaps to understanding cardiac macrophage polarization following myocardial infarction. Biochim. Biophys. Acta. 2016;1862(12):2288–2292. doi: 10.1016/j.bbadis.2016.05.013. [http://dx.doi.org/10.1016/j.bbadis.2016.05.013]. [PMID: 27240543]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dahiya S., Givvimani S., Bhatnagar S., Qipshidze N., Tyagi S.C., Kumar A. Osteopontin-stimulated expression of matrix metalloproteinase-9 causes cardiomyopathy in the mdx model of Duchenne muscular dystrophy. J. Immunol. 2011;187(5):2723–2731. doi: 10.4049/jimmunol.1101342. [http://dx.doi.org/10.4049/jimmunol.1101342]. [PMID: 21810612]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sponder M., Fritzer-Szekeres M., Marculescu R., Litschauer B., Strametz-Juranek J. Physical inactivity increases endostatin and osteopontin in patients with coronary artery disease. Heart Vessels. 2016;31(10):1603–1608. doi: 10.1007/s00380-015-0778-6. [http://dx.doi.org/10.1007/s00380-015-0778-6]. [PMID: 26661073]. [DOI] [PubMed] [Google Scholar]
- 119.Fujita N., Fujita S., Okada Y., et al. Impaired angiogenic response in the corneas of mice lacking osteopontin. Invest. Ophthalmol. Vis. Sci. 2010;51(2):790–794. doi: 10.1167/iovs.09-3420. [http://dx.doi.org/10.1167/iovs.09-3420]. [PMID: 19741245]. [DOI] [PubMed] [Google Scholar]
- 120.Biernacka A., Dobaczewski M., Frangogiannis N.G. TGF-β signaling in fibrosis. Growth Factors. 2011;29(5):196–202. doi: 10.3109/08977194.2011.595714. [http://dx.doi.org/10.3109/08977194.2011.595714]. [PMID: 21740331]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kirk J.A., Cingolani O.H. Thrombospondins in the transition from myocardial infarction to heart failure. J. Mol. Cell. Cardiol. 2016;90:102–110. doi: 10.1016/j.yjmcc.2015.12.009. [http://dx.doi.org/10.1016/j.yjmcc.2015.12.009]. [PMID: 26686988]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mustonen E., Ruskoaho H., Rysä J. Thrombospondins, potential drug targets for cardiovascular diseases. Basic Clin. Pharmacol. Toxicol. 2013;112(1):4–12. doi: 10.1111/bcpt.12026. [http://dx.doi.org/10.1111/bcpt.12026]. [PMID: 23074998]. [DOI] [PubMed] [Google Scholar]
- 123.Calabro N.E., Kristofik N.J., Kyriakides T.R. Thrombospondin-2 and extracellular matrix assembly. Biochim. Biophys. Acta. 2014;1840(8):2396–2402. doi: 10.1016/j.bbagen.2014.01.013. [http://dx.doi.org/10.1016/j.bbagen.2014.01.013]. [PMID: 24440155]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cleutjens J., Huynen F., Smits J., et al. Thrombospondin-2 deficiency in mice results in cardiac rupture early after myocardial infarction. Circ. Res. 1999;100:156. [Google Scholar]
- 125.Shimazaki M., Nakamura K., Kii I., et al. Periostin is essential for cardiac healing after acute myocardial infarction. J. Exp. Med. 2008;205(2):295–303. doi: 10.1084/jem.20071297. [http://dx.doi.org/10.1084/jem.20071297]. [PMID: 18208976]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Taniyama Y., Katsuragi N., Sanada F., et al. Selective blockade of periostin exon 17 preserves cardiac performance in acute myocardial infarction. Hypertension. 2016;67(2):356–361. doi: 10.1161/HYPERTENSIONAHA.115.06265. [http://dx.doi.org/10.1161/HYPERTENSIONAHA.115.06265]. [PMID: 26644236]. [DOI] [PubMed] [Google Scholar]
- 127.Kaur H., Takefuji M., Ngai C.Y., et al. Targeted ablation of periostin-expressing activated fibroblasts prevents adverse cardiac remodeling in mice. Circ. Res. 2016;118(12):1906–1917. doi: 10.1161/CIRCRESAHA.116.308643. [http://dx.doi.org/10.1161/CIRCRESAHA.116.308643]. [PMID: 27140435]. [DOI] [PubMed] [Google Scholar]
- 128.Oka T., Xu J., Kaiser R.A., et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ. Res. 2007;101(3):313–321. doi: 10.1161/CIRCRESAHA.107.149047. [http://dx.doi.org/10.1161/CIRCRESAHA.107.149047]. [PMID: 17569887]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chen C.C., Lau L.F. Functions and mechanisms of action of CCN matricellular proteins. Int. J. Biochem. Cell Biol. 2009;41(4):771–783. doi: 10.1016/j.biocel.2008.07.025. [http://dx.doi.org/10.1016/j.biocel.2008.07.025]. [PMID: 18775791]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dobaczewski M., Bujak M., Li N., et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ. Res. 2010;107(3):418–428. doi: 10.1161/CIRCRESAHA.109.216101. [http://dx.doi.org/10.1161/CIRCRESAHA.109.216101]. [PMID: 20522804]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lee C.H., Shah B., Moioli E.K., Mao J.J. CTGF directs fibroblast differentiation from human mesenchymal stem/stromal cells and defines connective tissue healing in a rodent injury model. J. Clin. Invest. 2010;120(9):3340–3349. doi: 10.1172/JCI43230. [http://dx.doi.org/10.1172/JCI43230]. [PMID: 20679726]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Twigg S.M. Regulation and bioactivity of the CCN family of genes and proteins in obesity and diabetes. J. Cell Commun. Signal. 2018;12(1):359–368. doi: 10.1007/s12079-018-0458-2. [http://dx.doi.org/10.1007/s12079-018-0458-2]. [PMID: 29411334]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ahmed M.S., Gravning J., Martinov V.N., et al. Mechanisms of novel cardioprotective functions of CCN2/CTGF in myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2011;300(4):H1291–H1302. doi: 10.1152/ajpheart.00604.2010. [http://dx.doi.org/10.1152/ajpheart.00604.2010]. [PMID: 21186275]. [DOI] [PubMed] [Google Scholar]
- 134.Weis S.M., Zimmerman S.D., Shah M., et al. A role for decorin in the remodeling of myocardial infarction. Matrix Biol. 2005;24(4):313–324. doi: 10.1016/j.matbio.2005.05.003. [http://dx.doi.org/10.1016/j.matbio.2005.05.003]. [PMID: 15949932]. [DOI] [PubMed] [Google Scholar]
- 135.Melchior-Becker A., Dai G., Ding Z., et al. Deficiency of biglycan causes cardiac fibroblasts to differentiate into a myofibroblast phenotype. J. Biol. Chem. 2011;286(19):17365–17375. doi: 10.1074/jbc.M110.192682. [http://dx.doi.org/10.1074/jbc.M110.192682]. [PMID: 21454527]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Weir R.A., Petrie C.J., Murphy C.A., et al. Galectin-3 and cardiac function in survivors of acute myocardial infarction. Circ Heart Fail. 2013;6(3):492–498. doi: 10.1161/CIRCHEARTFAILURE.112.000146. [http://dx.doi.org/10.1161/CIRCHEARTFAILURE.112.000146]. [PMID: 23505301]. [DOI] [PubMed] [Google Scholar]
- 137.Mayr A., Klug G., Mair J., et al. Galectin-3: relation to infarct scar and left ventricular function after myocardial infarction. Int. J. Cardiol. 2013;163(3):335–337. doi: 10.1016/j.ijcard.2012.06.087. [http://dx.doi.org/10.1016/j.ijcard.2012.06.087]. [PMID: 22795719]. [DOI] [PubMed] [Google Scholar]
- 138.Vanhoutte D., Schellings M.W., Götte M., et al. Increased expression of syndecan-1 protects against cardiac dilatation and dysfunction after myocardial infarction. Circulation. 2007;115(4):475–482. doi: 10.1161/CIRCULATIONAHA.106.644609. [http://dx.doi.org/10.1161/CIRCULATIONAHA.106.644609]. [PMID: 17242279]. [DOI] [PubMed] [Google Scholar]
- 139.Xie J., Wang J., Li R., et al. Syndecan-4 over-expression preserves cardiac function in a rat model of myocardial infarction. J. Mol. Cell. Cardiol. 2012;53(2):250–258. doi: 10.1016/j.yjmcc.2012.04.014. [http://dx.doi.org/10.1016/j.yjmcc.2012.04.014]. [PMID: 22561100]. [DOI] [PubMed] [Google Scholar]
- 140.Shetelig C., Limalanathan S., Eritsland J., et al. Osteoprotegerin levels in ST-elevation myocardial infarction: Temporal profile and association with myocardial injury and left ventricular function. PLoS One. 2017;12(3):e0173034. doi: 10.1371/journal.pone.0173034. [http://dx.doi.org/10.1371/journal.pone.0173034]. [PMID: 28253327]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Crisafulli A., Micari A., Altavilla D., et al. Serum levels of osteoprotegerin and RANKL in patients with ST elevation acute myocardial infarction. Clin. Sci. (Lond.) 2005;109(4):389–395. doi: 10.1042/CS20050058. [http://dx.doi.org/10.1042/CS20050058]. [PMID: 15926884]. [DOI] [PubMed] [Google Scholar]
- 142.Andersen G.O., Knudsen E.C., Aukrust P., et al. Elevated serum osteoprotegerin levels measured early after acute ST-elevation myocardial infarction predict final infarct size. Heart. 2011;97(6):460–465. doi: 10.1136/hrt.2010.206714. [http://dx.doi.org/10.1136/hrt.2010.206714]. [PMID: 21270073]. [DOI] [PubMed] [Google Scholar]
- 143.DeLeon-Pennell K.Y., Tian Y., Zhang B., et al. Cd36 is a matrix metalloproteinase-9 substrate that stimulates neutrophil apoptosis and removal during cardiac remodeling. Circ Cardiovasc Genet. 2016;9(1):14–25. doi: 10.1161/CIRCGENETICS.115.001249. [http://dx.doi.org/10.1161/CIRCGENETICS.115.001249]. [PMID: 26578544]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.DeLeon-Pennell K.Y., Meschiari C.A., Jung M., Lindsey M.L. Matrix metalloproteinases in myocardial infarction and heart failure. Prog. Mol. Biol. Transl. Sci. 2017;147:75–100. doi: 10.1016/bs.pmbts.2017.02.001. [http://dx.doi.org/10.1016/bs.pmbts.2017.02.001]. [PMID: 28413032]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lindsey M.L., Gannon J., Aikawa M., et al. Selective matrix metalloproteinase inhibition reduces left ventricular remodeling but does not inhibit angiogenesis after myocardial infarction. Circulation. 2002;105(6):753–758. doi: 10.1161/hc0602.103674. [http://dx.doi.org/10.1161/hc0602.103674]. [PMID: 11839633]. [DOI] [PubMed] [Google Scholar]
- 146.Matsumura S., Iwanaga S., Mochizuki S., Okamoto H., Ogawa S., Okada Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J. Clin. Invest. 2005;115(3):599–609. doi: 10.1172/JCI22304. [http://dx.doi.org/10.1172/JCI22304]. [PMID: 15711638]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hudson M.P., Armstrong P.W., Ruzyllo W., et al. Effects of selective matrix metalloproteinase inhibitor (PG-116800) to prevent ventricular remodeling after myocardial infarction: Results of the PREMIER (Prevention of Myocardial Infarction Early Remodeling) trial. J. Am. Coll. Cardiol. 2006;48(1):15–20. doi: 10.1016/j.jacc.2006.02.055. [http://dx.doi.org/10.1016/j.jacc.2006.02.055]. [PMID: 16814643]. [DOI] [PubMed] [Google Scholar]
- 148.Iyer R.P., de Castro Brás L.E., Patterson N.L., et al. Early matrix metalloproteinase-9 inhibition post-myocardial infarction worsens cardiac dysfunction by delaying inflammation resolution. J. Mol. Cell. Cardiol. 2016;100:109–117. doi: 10.1016/j.yjmcc.2016.10.005. [http://dx.doi.org/10.1016/j.yjmcc.2016.10.005]. [PMID: 27746126]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Iyer R.P., Patterson N.L., Zouein F.A., et al. Early matrix metalloproteinase-12 inhibition worsens post-myocardial infarction cardiac dysfunction by delaying inflammation resolution. Int. J. Cardiol. 2015;185:198–208. doi: 10.1016/j.ijcard.2015.03.054. [http://dx.doi.org/10.1016/j.ijcard.2015.03.054]. [PMID: 25797678]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Okada H., Takemura G., Kosai K., et al. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. 2005;111(19):2430–2437. doi: 10.1161/01.CIR.0000165066.71481.8E. [http://dx.doi.org/10.1161/01.CIR.0000165066.71481.8E]. [PMID: 15867170]. [DOI] [PubMed] [Google Scholar]
- 151.Biernacka A., Cavalera M., Wang J., et al. Smad3 signaling promotes fibrosis while preserving cardiac and aortic geometry in obese diabetic mice. Circ Heart Fail. 2015;8(4):788–798. doi: 10.1161/CIRCHEARTFAILURE.114.001963. [http://dx.doi.org/10.1161/CIRCHEARTFAILURE.114.001963]. [PMID: 25985794]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mann D.L., McMurray J.J., Packer M., et al. Targeted anticytokine therapy in patients with chronic heart failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation. 2004;109(13):1594–1602. doi: 10.1161/01.CIR.0000124490.27666.B2. [http://dx.doi.org/10.1161/01.CIR.0000124490.27666.B2]. [PMID: 15023878]. [DOI] [PubMed] [Google Scholar]
- 153.Somasuntharam I., Yehl K., Carroll S.L., et al. Knockdown of TNF-α by DNAzyme gold nanoparticles as an anti-inflammatory therapy for myocardial infarction. Biomaterials. 2016;83:12–22. doi: 10.1016/j.biomaterials.2015.12.022. [http://dx.doi.org/10.1016/j.biomaterials.2015.12.022]. [PMID: 26773660]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Abbate A., Van Tassell B.W., Biondi-Zoccai G., et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction. Am. J. Cardiol. 2013;111(10):1394–1400. doi: 10.1016/j.amjcard.2013.01.287. [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study]. [http://dx.doi.org/10.1016/j.amjcard.2013.01.287]. [PMID: 23453459]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kühn B., del Monte F., Hajjar R.J., et al. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat. Med. 2007;13(8):962–969. doi: 10.1038/nm1619. [http://dx.doi.org/10.1038/nm1619]. [PMID: 17632525]. [DOI] [PubMed] [Google Scholar]
- 156.Ladage D., Yaniz-Galende E., Rapti K., et al. Stimulating myocardial regeneration with periostin Peptide in large mammals improves function post-myocardial infarction but increases myocardial fibrosis. PLoS One. 2013;8(5):e59656. doi: 10.1371/journal.pone.0059656. [http://dx.doi.org/10.1371/journal.pone.0059656]. [PMID: 23700403]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sawyer A.J., Kyriakides T.R. Matricellular proteins in drug delivery: Therapeutic targets, active agents, and therapeutic localization. Adv. Drug Deliv. Rev. 2016;97:56–68. doi: 10.1016/j.addr.2015.12.016. [http://dx.doi.org/10.1016/j.addr.2015.12.016]. [PMID: 26763408]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Clifford D.M., Fisher S.A., Brunskill S.J., et al. Stem cell treatment for acute myocardial infarction. Cochrane Database Syst. Rev. 2012;2(2):CD006536. doi: 10.1002/14651858.CD006536.pub3. [http://dx.doi.org/10.1002/14651858.CD006536.pub3]. [PMID: 22336818]. [DOI] [PubMed] [Google Scholar]
- 159.Jang J., Park H.J., Kim S.W., et al. 3D printed complex tissue construct using stem cell-laden decellularized extracellular matrix bioinks for cardiac repair. Biomaterials. 2017;112:264–274. doi: 10.1016/j.biomaterials.2016.10.026. [http://dx.doi.org/10.1016/j.biomaterials.2016.10.026]. [PMID: 27770630]. [DOI] [PubMed] [Google Scholar]
- 160.Gaetani R., Feyen D.A., Verhage V., et al. Epicardial application of cardiac progenitor cells in a 3D-printed gelatin/hyaluronic acid patch preserves cardiac function after myocardial infarction. Biomaterials. 2015;61:339–348. doi: 10.1016/j.biomaterials.2015.05.005. [http://dx.doi.org/10.1016/j.biomaterials.2015.05.005]. [PMID: 26043062]. [DOI] [PubMed] [Google Scholar]
- 161.Guyette J.P., Charest J.M., Mills R.W., et al. Bioengineering human myocardium on native extracellular matrix. Circ. Res. 2016;118(1):56–72. doi: 10.1161/CIRCRESAHA.115.306874. [http://dx.doi.org/10.1161/CIRCRESAHA.115.306874]. [PMID: 26503464]. [DOI] [PMC free article] [PubMed] [Google Scholar]