

Abstract
Purpose of Review
Disaccharidase testing, as applied to the evaluation of gastrointestinal disturbances is available, but it is not routinely considered in the diagnostic work-up. The purpose of this review was to determine if disaccharidase testing is clinically useful and to consider how the results could alter patient management.
Recent Findings:
indicate that carbohydrate maldigestion could contribute functional bowel disorders and negatively impact the fecal microbiome. Diagnostic techniques include enzyme activity assays performed on random endoscopically-obtained small intestinal biopsies, immunohistochemistry, stable isotope tracer and non-enriched substrate load breath testing, and genetic testing for mutations. More than 40 sucrase-isomaltase-gene variants coding for defective or reduced enzymatic activity have been reported and deficiency conditions are more common than previously thought.
Summary
The rationale for disaccharidase activity testing relates to a need to fully assess unexplained recurrent abdominal discomfort and associated symptoms. All disaccharidases share the same basic mechanism of mucosal expression and deficiency has far reaching consequences. Testing for disaccharidase expression appears to have an important role in symptom evaluation, but there are accuracy and logistical issues that should be considered. It is likely that specific recommendations for patient management, dietary modification and enzyme supplementation would come from better testing methods.
Keywords: lactase, sucrase, isomaltase, palatinase, breath testing, immunohistochemistry, enzyme supplementation, sacrosidase, amyloglucosidase
Introduction
Carbohydrates constitute a large portion of the human diet and consist of starches, dextrins, oligosaccharides, and disaccharides [1, 2]. These substrates must be completely hydrolyzed by small intestinal, apical-surface-mucosal disaccharidases (on the ‘brush border’), and by pancreatic alpha-amylase (in the cases of starches) to ultimately be absorbed as monomeric sugars (e.g., glucose, galactose, and fructose) by transporter-dependent means. Larger starch molecules must first be partially hydrolyzed by alpha-amylase to be made sufficiently soluble to engage the mucosal-anchored disaccharidases. The disaccharidases lactase, sucrase, maltase, and trehalase are embedded on the microvilli ultrastructure of small intestinal enterocytes and function to complete the luminal glycolysis to monomers from their respective substrates [3]. This process ideally permits rapid, transporter-mediated absorption before any microorganisms can theoretically and excessively proliferate (overgrowth) in the small intestine and competitively consume the freed sugars or be passed on to the colon [4].
When small intestinal, carbohydrate-dense chyme exceeds the disaccharidase glycolytic ability and enterocyte absorptive capacity, the maldigested oligosaccharides that pass to the colon become subject to excessive fermentation. In excess, this epiphenomenon could affect colonic homeostasis and adversely alter the microbiota (dysbiosis) [5]. Symptoms thought to be caused by dysbiosis have been linked to increased by-products of colonic fermentation (including osmotically active particles) and a change in luminal pH known to have neuromotor consequences [6]. Resultant clinical features include abdominal distention to luminal retention of water and gases, discomfort, variable somatic complaints, and bothersome changes in bowel habits. There is extensive symptom-overlap with maldigestion and other potentially serious conditions that are frequently considered before the evaluation of mucosal disaccharidase activity is undertaken.
There appears to be a balance between tolerable (and potentially beneficial) and incomplete carbohydrate digestion and intolerable or noxious maldigestion due in part to disaccharidase deficiency or other conditions such as pancreatic insufficiency or motility issues. In the former and favorable case, some short-chain fatty acids (SCFA), such as acetate, butyrate, and propionate esters, are produced in moderation from carbohydrates by fecal species in the Firmicutes, Bacteroides phyla and others. The mix of SCFA appears to be dependent upon the diversity of the colonic microbiota, which in turn is dependent upon released nutrients that pass to the colon [7, 8]. Butyrate and propionate are typically absorbed and constitute a caloric salvage mechanism, provide nourishment to the colonic mucosa, and regulate inflammatory responses [9, 10]. It also appears that the SCFA mix also plays an important key role in the prevention and treatment of the metabolic syndrome, functional bowel disorders, and cancer through inhibition of nuclear factor kappa B activation and histone deacetylation [11]. For example, the inhibition of histone deacetylases (HDACs) and activation of G-protein-coupled receptors (GPCRs) have been linked to two SCFA signaling mechanisms [12]. Since the optimal production of SCFA is, in part, dependent upon upstream disaccharidase digestion, repercussions from any deficiency conditions could have harmful consequences on how HDACs regulate carcinogenic gene expression in the colon [13].
Maldigestion can indirectly lead to decreased reabsorption of bile acids, which also can deleteriously alter the colonic flora [14, 15]. Furthermore, the preferment of certain strains of Bacteroides fragilis may result in inflammatory diarrhea and may interfere with DNA-repair mechanisms that are important to the prevention of colorectal cancer development [16]. Nevertheless, more understandings are needed about the modulation of colonic floral milieu by dietary substrate diversity with attention given to disease prevention or pathogenesis. Specifically, how the various proximally maldigestion conditions, including disaccharidase deficiency, could influence the disease cascade is the subject of ongoing investigations.
It appears that diets that are low in fermentable substrates [17] are associated with decreased frequency of abdominal pain in adults and children with irritable bowel syndrome [18, 19]. The putative dietary substrates that are thought to contribute to increased ‘functional’ symptoms include maldigested fructans [20], lactose, sucrose, [21], fructose [22], oligosaccharides and starch using processes that can be dependent on effective disaccharidase activity. Excessive colonic carbohydrate fermentation can also result in the production of lactate that results in a decrease in colonic luminal pH and potentially alter 5-HT-related biological processes [23, 24]. This is accompanied by the production of gases such as hydrogen and methane [25, 26], which have been linked with bloating and altered colonic motility [27–29]. Also, an excessive release of various small-molecules in the colon may evoke inflammatory responses and elicit or suppress reflexive motor actions [30, 31]. As such, disaccharidase activity appears to be a critical factor in health and digestive pathophysiology.
Disaccharidase Insufficiency
Disaccharidase insufficiency, as a cause for maldigestion-related complaints, has been recognized for more than a century [32] and can co-exist with organic diseases or manifest with the natural aging processes. The clinical manifestations are highly variable, although none of the features are substrate-specific. Lactase expression, in part, defines the mammalian class of vertebrates and lactase insufficiency is the quintessential clinical model for this mucosal disaccharidase shortfall. It calls attention to the importance of the other types of disaccharidase insufficiency. Typically, symptoms of abdominal discomfort first suggest other organic causes for symptoms including small intestinal bacterial overgrowth (SIBO) [33], celiac disease [34], infectious enteritis [35], radiation enteritis [36], somatostatinoma [37], and ‘functional’ bowel syndrome [38, 39]; and any of these conditions could co-exist with disaccharidase insufficiency.
Lactose and Lactase Insufficiency
Lactose-containing dairy product consumption commonly occurs in modern western society, and lactose is often hidden within many modern foodstuffs. Modern dairy farming, particularly among the temperate latitudes, became commonplace once routine pasteurization came in to being and, as such, lactose consumption increased in many societies. However, the evolutionary nutrient ‘blueprint’ for the human species does not always appear to be well-suited for ongoing milk ingestion beyond infancy [40]. For more than half of the world’s population, adult-type hypolactasia begins after weaning and is somewhat dependent upon the regulatory factors expressed by the autosomal dominant MCM6 gene [41]. For reasons not understood, some people with adult-type hypolactasia may become symptomatic after ingestion of small amounts of dairy products, and others may tolerate 250 mL or more before admitting to significant symptoms [42–43].
Symptomatic lactose maldigestion can occur in unsuspecting people when tolerance thresholds are unknowingly exceeded or when acute comorbidities present. The symptom disparity may be due, in part, to maladaptive colonic microbiota [44, 45]. Sustained viral infections may alter the enterocyte membrane ultrastructure and downregulate lactase expression [46]. After a giardia infection and viral gastroenteritis, an affected patient may acquire lactose intolerance [47] and post-infectious chronic diarrhea may be especially problematic among the elderly and among those who are immunologically compromised [48, 49].
Infantile lactase deficiency is rare, and due to autosomal recessive mutations in the LCT gene located on chromosome 2 at position q21.3 [41]. However, malnourished infants have reduced lactase activity because of epigenetic suppression or from acquired conditions [50], such as iatrogenic mucositis, malnutrition, and short-gut syndrome. Since the mechanism for protein expression on the apical membranes is similar among all disaccharidases [51], symptoms may indicate the need for broad disaccharidase testing, and/or the reflexive institution of restriction diets or enzyme supplementation or parenteral nutritional support in severe cases. To provide symptom relief from lactase insufficiency conditions, a multi-billion dollar industry has emerged. Over-the-counter approaches include consumption of hard cheese and yogurt, use of probiotics, and per-oral lactase supplementation to serve the needs of those unable to completely avoid lactose ingestion and to help meet micronutrient needs [52, 53].
Lactase persistence is not uncommon among approximately one-third of the human population, and the reason selective lactase persistence remains poorly understood [54]. Several dominant allele genotypes have been associated with the lactase-persistence phenotype (including LCT-13910CT and LCT-13910TT) [55, 56] and epigenetic factors may play an important role in the influence of phenotypic outcomes [57]. Lactase persistence polymorphisms have been associated with increased BMI and obesity [58, 59], and, in turn, lactose tolerance may facilitate an increased risk for obesity-associated malignancies [60], but this concept remains controversial [61]. Conversely, lactase persistence may be an evolutionary flaw. The Kuopio Ischaemic Heart Disease Risk Factor Study [62] found that low-fat (<3·5 %), fermented (aka: lactose free) dairy product intake was associated with a lower risk of heart disease (hazard ratio in the highest quartile=0·74; 95 % CI 0·57, 0·97; P-trend=0·03). As such, dairy ingestion and lactase ‘insufficiency’ remain clinically important considerations beyond the realm of abdominal discomfort and as a nutritional calcium source. However, when persistent lactase activity is confirmed, alternative explanations of adverse abdominal symptoms might be sought.
Lactase Testing
When results from adequate adherence to the exclusionary-lactose diet are ambiguous, diagnostic testing might include breath hydrogen testing and perhaps in vitro enzyme assay of endoscopic mucosal biopsies. This approach may be warranted along when consideration of secondary lactase insufficiency could be due to pathogenic defects as caused by parasitic states, bacterial infections, viral infections, and iatrogenic chemo-radiotherapy. It seems unlikely that genetic testing for lactase polymorphisms could be helpful in this setting.
Breath hydrogen testing [63] may be used to establish a diagnosis of lactase insufficiency, but a diagnosis of small intestinal bacterial overgrowth (SIBO) should first be excluded. Some centers advocate the use of a preliminary glucose breath test (up to 100 gram per-oral load, which equates to the consumption of a quart of milk) to exclude SIBO prior to lactose breath testing [64]. However, by consensus, a loading meal of 75 grams of glucose may be sufficient. The theory behind the glucose breath test is that an early rise in blood glucose concentration and the absence of significant breath hydrogen changes would point against SIBO as a cause for symptoms. There are several nuances practiced in different regions, and the techniques used often vary. We follow recently published guidelines which consider that accept a 20 PPM rise in breath hydrogen expression by 90 minutes after glucose substrate ingestion points toward a diagnosis of SIBO [65]. If the glucose breath test is negative before 90 for SIBO, disaccharidase insufficiency might then be considered [66], and a per-oral lactose challenge might be performed in an attempt to reproduce symptoms.
With the combined breath testing and blood lactose tolerance testing, a 25g meal of lactose is administered and timed breath samples are obtained for hydrogen analyses. A blood glucose concentration may be measured at 1 and 2 hours after substrate ingestion [65]. An early-rise in blood glucose concentration of more than 20 mg/dL would suggest that lactose digestion occurred, but the appearance of a late-rise in breath hydrogen would indicate that some lactose was not digested proximally. The appearance of such a late breath hydrogen rise (delta >20 PPM) is also dependent upon transit time to the cecum. It is of interest that a minority of patients are colonized with a methane-producing species that rapidly convert hydrogen to methane at a 4:1 ratio [67]. As such, concurrent analysis for methane expression may identify those who are lactase deficient but do not demonstrate a positive breath hydrogen test (hydrogen non-excretors). It is unlikely that immunohistochemistry or genetic testing could offer much further information or perspective when considering lactase deficiency and the response to per-oral lactase supplementation.
Sucrose and Sucrase Testing
The majority of the habitual western diet is composed of starch and augmented with high sucrose consumption. Sucrose role in the human diet increased with human agriculture production and trade. Sucrose consumption in the United States currently approximates 11-million metric tons, which is a 1-million metric ton increase since 2009 [68]. Sucrose is hydrolyzed by sucrase to free glucose and fructose by the distal aspect of the sucrase-isomaltase gene product [69]. The release of free fructose can be problematic for some people with decreased transport capacity, a mechanistic process not well understood [70]. Sucrose ingestion is a prime source of fructose and is dependent upon small intestinal glycolysis for absorption. The major fructose co-transporter in the small intestine is thought to be GLUT5 and is subject to up- and down-regulation depending upon load conditions. Most adults can tolerate up to 25 grams of fructose, [71] but incomplete absorption and symptoms of intolerance may suggest that other factors, such as female gender, may be involved [72]. Very few cases of genetically determined fructose malabsorption have been reported to date [73]. The extreme rarity of GLUT5 mutation suggests that fructose intolerance, when present, is probably multifactorial. Over-the-counter glucose-isomerase supplementation is available to attempt symptom relief, but clinical efficacy has not yet been established.
Sucrase-isomaltase (SI) is expressed as a dimeric gene product [cytogenetic location: 3q26.1] that is anchored to the mucosal brush border with the n-terminus of proximal isomaltase component and the sucrase component vulnerable to luminal proteolytic cleavage [74, 75]. Congenital sucrase-isomaltase deficiency (CSID) syndrome, like lactase deficiency, has been linked to dietary sucrose and starch intolerance [69]. Since dietary sucrose and starches are pervasive, the clinical consequences of this type of disaccharidase insufficiency are, for some, difficult to avoid. Failure to proximally hydrolyze sucrose and starches effectively relegates them to prebiotic status and may have deleterious effects that could alter the colonic microbiota and/or cause symptoms. CSID is also a model syndrome to study the maldigestion of carbohydrates at the mucosal level [76]. The condition has significant symptom similarity with enteritis and in severe cases, can result in malnutrition [77].
With considerable diligence, sucrose can be significantly reduced from the habitual diet as a means of alleviating CSID symptoms [78, 79], but supplemental sacrosidase is available and may have some utility beyond restriction diets [80–82]. The restriction of fructose might also be considered, but there is a general lack of strong evidence supporting the restriction of individual carbohydrates [20, 83]. Fructose load breath tests may be helpful, but this is a challenging consideration to make since the test reproducibility may be poor due to rapid metabolic considerations [84]. Tracer labeled (13C) fructose substrates are available, but are too costly to use early in the screening algorithm.
Starch, Dextrins, Isomaltase and Glucoamylase Testing
Starch maldigestion is another aspect of the sucrase-isomaltase insufficiency condition for consideration. The incomplete digestion of starch-derived dextrins was recognized as an important factor in the CSID condition more than forty years ago [85]. Since then, more than 40 different CSID mutations have been identified, and there is recent evidence to indicate that variable phenotypes may be more common than are currently recognized [86]. Commercial genetic screening is available to confirm genetic forms, but routine use of genetic screening remains cost-prohibitive. If a restriction diet were to be adhered to and followed by a dietary challenge that yielded a clinical response, disaccharidase testing could be performed to provide patients with a concrete diagnosis. Specifically, a sucrose-load breath hydrogen test or a stable isotope tracer (13C-carbon) breath test might be useful to support the diagnosis since isomaltase is co-expressed with sucrase [87]. Assay of sucrase activity should proportionally parallel palatinase activity (the in vitro surrogate α1–6 substrate for isomaltose) since sucrase and isomaltase are initially expressed as a single gene product [88]. Immunohistochemistry might also be considered as a semi-quantitative means to assess SI expression, but that would not completely address glycolytic capacity in all cases. A visual analog scale that is similar to the Visual Analog Pain Scale and the Bristol Stool Form Scale [89] is suggested for immunohistochemistry assessment SI expression. Figure 1 proposes a scale similar to where zero represents an absence of detectable SI expression by immunohistochemistry, and +4 equates to robust SI expression from the intestinal crypts to the tips of the small intestinal villae. Further validation work is needed before this grading approach might be considered.
Figure 1.
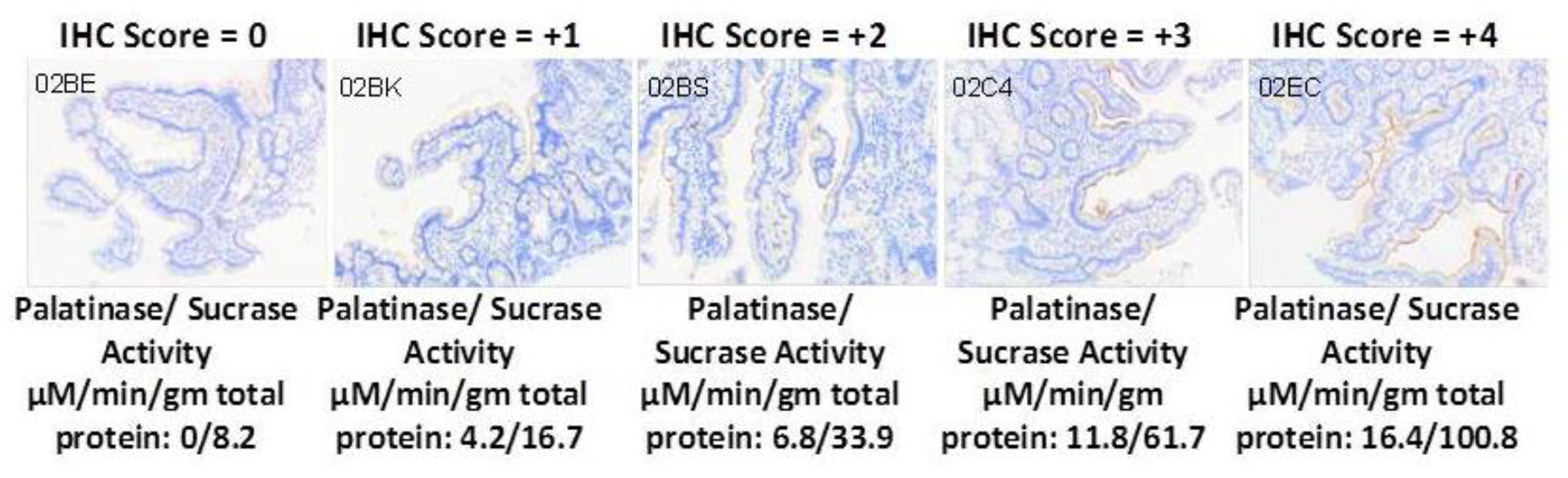
A Semi-quantitative analog grading scale of immunohistochemistry staining is proposed for sucrase-isomaltase expression along the apical mucosal surface (brown). The relatively unavailability of activity assays makes immunohistochemistry staining and attractive alternative and worthy of further development. The images selected about are representative of correctly handled samples and were compared with the reported Dahlqvist assay outcomes (μM/min/gram-protein). Enzyme activity results were normalized using total protein content determinations which are problematic since biopsies may contain various amounts of protein depending upon the forceps depth and concurrent retrieval of adventitia. Such adjustments that could easily skew the reported disaccharidase activity outcomes; immunohistochemistry staining may offer a more practical and cost-effective alternative.
Isomaltase has considerable starchy oligosaccharide hydrolytic capacity, and the spectrum of clinical impairment is varied, much like lactose intolerance [90]. Several recent findings suggest that certain CSID variants, with reduced SI enzymatic activity, may contribute to the development of IBS symptoms [91, 92]. However, in a recent small study of patients with IBS with diarrhea (IBS-D) [93], SI mutation carriers only experienced a modest benefit from the use of the low FODMAP (Fermentable Oligosaccharide, Disaccharide, Monosaccharide, and Polyols) diet when compared with non-SI carriers. This finding should have been expected since most starch substrates are not part of the FODMAP constellation [94]. The SI C-terminus plays the role of the sustaining but limiting mucosal glucosidase activity [69. 75]. The two gene products carefully orchestrate starch digestion and determining how much fermentable substrate reaches the colon; therefore, gene product aberrancies are expected to have far reaching clinical manifestations. The importance of homozygous or heterozygous SI gene variants in the pathogenesis of IBS or other functional gastrointestinal disorders remains to be validated.
Maldigestion of starch is seldom assessed in adults, perhaps because it is widely assumed that pancreatic α-amylase secretion and its associated activity is ample, especially when fat absorption is concurrently normal. Limited testing for starch hydrolysis is available, such as secretin or breath testing [95, 96], and empiric amylase supplementation has been shown to successfully treat cases of post-prandial diarrhea [97]. After excluding pancreatic insufficiency, inflammatory conditions and celiac disease, indirect disaccharidase substrate testing for maltose, palatinose (used in lieu of isomaltose), sucrose, and dextrins (very-short-chain, soluble fragments derived from amylase action on starch) could be considered for still unexplained symptoms in adults.
Maltase-glucoamylase is a chromosome-7 gene product (band location 7q34) with significant sequence homology with sucrase-isomaltase [98]. It is capable of hydrolyzing longer maltooligosaccharides, such as maltotetrose and maltopentose [69, 99]. The C-terminal of the enzyme is the most highly glucosidase but is inhibited by starch dextrins produced by alpha amylase [69]. It appears to be somewhat of a fail-safe enzyme that provides scavenger functionality to complete starch maldigestion. This mechanism appears to explain the antidotal reports of improved symptoms with dietary titration of dietary starch to match personal capacity. Unlike sucrase [92], with more than 700 coding single-nucleotide polymorphisms and mutations reported, isolated maltase-glucoamylase (MGAM) deficiency has not been confirmed to date. However, a second pseudogene (MGAM2) has been reported with significant base-pair homology to MGAM, [100], but it is unclear if a complimentary functional gene product can be ascribed. Aberrant maltase activities are difficult to interpret because the maltase-glucoamylase gene-product and the isomaltase component of the sucrase-isomaltase have overlapping maltase (glucosidase) activities. However, decreased maltase activity, with preserved sucrase and palatinase activities, might suggest MGAM mutations if the quality of the sample is good.
The pan-disaccharidase-deficiency condition has been rarely reported without clear etiologies [101, 102]. Mucosal injury from infection, antineoplastic agents, and acarbose (an antidiabetic alpha glucosidase inhibitor) would be expected to result in a transient condition that affects all ingested food substrates [103]. Dietary management approaches, including elemental diets, have been used until the condition improved. N-butyl-deoxynojirimycin (miglustat), an imino sugar and synthetic analog of D-glucose, is a medication used for treatment of lysosomal storage diseases. Miglustat has been shown to interfere with N-glycosylation of the proteins in the ER that delays intracellular trafficking of disaccharidase molecules to the apical surface [104]; and this epiphenomenon suggests a host of investigative models for maldigestion worthy of future investigation of disaccharidase deficiency should specific replacement enzyme therapy become available for clinical testing.
One might argue that starch and oligosaccharide maldigestion is the greater issue than sucrose in CSID, but this assessment is too often overlooked since starch has such an important role in the habitual diet and there is no recognized approach to addressing glucose polymer oligosaccharide maldigestion in humans. Stable 13C-isotope labeled substrates composed of sucrose or alpha-limit dextrins (soluble oligosaccharide substrates no longer vulnerable to alpha-amylase), may be prepared and used as a more specific breath test substrate [76]. Assay of small intestinal biopsies are difficult to interpret due to substrate-activity overlap. A pediatric study demonstrated that assay outcomes did not correlated with disaccharidase tolerance [105]. Furthermore, the samples taken by forceps are likely to vary in depth and breadth of mucosa such that reported activity ratio outcomes may not be truly representative due to variable total protein content (divisor). Breath testing with stable isotope-tracer-tagged alpha limit dextrin provides oxidative values that represent combined digestive capacity of isomaltase (palatinase) and maltase-glucoamylase. Crude starch substrate would be dependent upon alpha-amylase action as well as disaccharidases. Sucrose substrate breath tests results also reflect in vivo activity that test could be most useful to assess the complimentary portion of the SI dimer. Sacrosidase supplementation therapy is available to be used in concert with strict dietary fructose restriction for best results [106]. Should other candidate therapies advance to replace mucosal isomaltase and glucoamylase activities in vivo [107, 108], per-oral therapy activity could be easily assessed and efficacy might be determined.
Summary/conclusion
This communiqué reviewed the rationale for disaccharidase testing in clinical practice when there is a need to assess unexplained chronic and recurrent abdominal discomfort and associated symptoms. Disaccharidase insufficiency hides in plain-sight and lies just beyond the reach of most routine endoscopist’s forceps, but sample integrity and representation remain obstacles to routine use enzyme activity assays. Disaccharidase insufficiency is a real and underappreciated problem for which technology has advanced the evaluative tools to investigate with greater specificity. Immunohistochemistry staining is an attractive and practical alternative and worthy of further development. Commercial microscopes and intensity algorithms are available to quantify enzyme expression and implementation would not significantly extend procedure time or increase clinical risks. However, it is yet unclear how these tools should be accommodated into mainstream practice since therapies beyond dietary restrictions are few. Furthermore, the microbiome response to disaccharidase deficiencies is an involving field and should provide insights into maladies beyond functional bowel syndrome and mucositis.
If one believes that lactase insufficiency frequently exists, and the associated gastrointestinal symptoms of lactose intolerance impact the activities of daily living for millions of patients, the same credence should be paid to the other disaccharidases such as sucrase-isomaltase that relate to more important foodstuffs. One should consider that all disaccharidases share the same basic mechanism of protein expression when unexplained signs and symptoms appear that resemble lactase insufficiency. Therefore, testing for SI and maltase-glucoamylase expression appears to have an important role in the evaluation of unexplained abdominal discomfort and related symptoms and enzyme testing should have a great role in the evaluation of patients. Unfortunately, the biopsy enzyme activity assays cannot universally be relied upon for definitive assessments and substrate-specific breath testing is not widely available, but would be welcome. The dilemma is ‘what would one do with the disaccharidase activity data?’ since, at present, there is relatively little that can be done beyond dietary modifications to address any specific maldigestive issues. Recommendations for dietary modification have a role in patient management and disaccharidase testing outcomes would justify such testing.
Summary Bullet Points.
Lactase persistence beyond early childhood with several dominant alleles is frequent, but recent studies point to such persistence being a marker for increased risk for obesity and obesity-associated malignancies.
More than 40 sucrase-isomaltase-gene variants coding for reduced enzymatic activity have been reported and indicates deficiency conditions are more common than previously thought may contribute to the development of IBS-like symptoms.
Disaccharidase testing is challenging to perform routinely, and outcomes currently rely on non-standardized mucosal samples snap-frozen on liquid nitrogen or indirect estimates of activity that are based upon by-products of maldigestion.
Disaccharidase testing, especially for sucrase-isomaltase, that might be better served by the use of immunohistochemistry with results applied to a semi-quantitative comparative scale, but further validation work is needed.
Acknowledgements:
This work was supported, in part, by a gift from DR and GL Laws and Public Health Service Grant P30 DK56388, which funds the Texas Medical Center Digestive Diseases Center. Financial and/or intellectual support was also provided by National Institutes of Health K23DK101688 and R03DK117219.
Footnotes
Author’s Disclosures or Potential Conflicts of Interest: none of the authors report has any conflicting disclosures to make with regard to consultancies, advisory roles, stock ownership, honoraia, expert testimony, royalties or awarded patents.
References
- 1.Willett WC. Overview and perspective in human nutrition. Asia Pac J Clin Nutr. 2008; 17 Suppl 1:1–4. [PubMed] [Google Scholar]
- 2.Oza-Frank R, Cheng YJ, Narayan KM, Gregg EW. Trends in nutrient intake among adults with diabetes in the united states: 1988–2004. J Am Diet Assoc. 2009; 109:1173–1178. [DOI] [PubMed] [Google Scholar]
- 3.Hauser H, Semenza G. Sucrase-isomaltase: A stalked intrinsic protein of the brush border membrane. CRC Crit Rev Biochem. 1983; 14:319–345. [DOI] [PubMed] [Google Scholar]
- 4.Gregg CR. Enteric bacterial flora and bacterial overgrowth syndrome. Semin Gastrointest Dis. 2002; 13:200–9 [PubMed] [Google Scholar]
- 5.Talbotec C, Schmitz J. Intestinal malabsorption in the child. Rev Prat. 2001; 51:983–7. [PubMed] [Google Scholar]
- 6.Olesen M, Gudmand-Høyer E. Maldigestion and colonic fermentation of wheat bread in humans and the influence of dietary fat. Am J Clin Nutr. 1997; 66: 62–6. [DOI] [PubMed] [Google Scholar]
- 7.Tuncil YE, Thakkar RD, Marcia ADR, Hamaker BR, Lindemann SR. Divergent short-chain fatty acid production and succession of colonic microbiota arise in fermentation of variously-sized wheat bran fractions. Sci Rep. 2018; 8:16655.-018-34912-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krishnareddy S. The microbiome in celiac disease. Gastroenterol Clin North Am. 2019; 48(1):115–126. [DOI] [PubMed] [Google Scholar]
- 9.Simpson HL, Campbell BJ. Review article: Dietary fibre-microbiota interactions. Aliment Pharmacol Ther. 2015; 42:158–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furusawa Y, Obata Y, Fukuda S, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013; 504(7480):446–450. [DOI] [PubMed] [Google Scholar]
- 11.Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2008; 27:104–19. [DOI] [PubMed] [Google Scholar]
- 12.Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol. 2014;121:91–119. [DOI] [PubMed] [Google Scholar]
- 13.Tampakis A, Tampaki EC, Nebiker CA, Kouraklis G. Histone deacetylase inhibitors and colorectal cancer: What is new? Anticancer Agents Med Chem. 2014; 14:1220–1227. [DOI] [PubMed] [Google Scholar]
- 14.Camilleri M, Busciglio I, Acosta A, Shin A, Carlson P+G21, Burton D et al., Effect of increased bile acid synthesis or fecal excretion in irritable bowel syndrome-diarrhea. Am J Gastroenterol. 2014; 109:1621–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harewood GC, Murray JA. Approaching the patient with chronic malabsorption syndrome. Semin Gastrointest Dis. 1999; 10:138–44. [PubMed] [Google Scholar]
- 16.Candela M, Turroni S, Biagi E, et al. Inflammation and colorectal cancer, when microbiota-host mutualism breaks. World J Gastroenterol. 2014; 20:908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halmos EP, Power VA, Shepherd SJ, Gibson PR, Muir JG. A diet low in FODMAPs reduces symptoms of irritable bowel syndrome. Gastroenterology 2014; 146:67–75.e5. [DOI] [PubMed] [Google Scholar]
- 18.Nanayakkara WS, Skidmore PM, O’Brien L, Wilkinson TJ, Gearry RB. Efficacy of the low FODMAP diet for treating irritable bowel syndrome: The evidence to date. Clin Exp Gastroenterol. 2016;9:131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chumpitazi BP, Hollister EB, Oezguen N, et al. Gut microbiota influences low fermentable substrate diet efficacy in children with irritable bowel syndrome. Gut Microbes. 2014;5(2):165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandez-Banares F, Esteve M, Viver JM. Fructose-sorbitol malabsorption. Curr Gastroenterol Rep. 2009; 11:368–374. [DOI] [PubMed] [Google Scholar]
- 21.Kim SB, Calmet FH, Garrido J, Garcia-Buitrago MT, Moshiree B. Sucrase-Isomaltase Deficiency as a Potential Masquerader in Irritable Bowel Syndrome. Dig Dis Sci. 2019. [DOI] [PubMed] [Google Scholar]; Describes the potential importance of disaccharidase deficiency in the pathogenesis of functional bowel disorders.
- 22.Born P. Carbohydrate malabsorption in patients with non-specific abdominal complaints. World J Gastroenterol. 2007; 13:5687–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao H, Liu X, An Y, et al. Dysbiosis contributes to chronic constipation development via regulation of serotonin transporter in the intestine. Sci Rep. 2017; 7+G71:10322.-017-10835-8.G87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yano JM, Yu K, Donaldson GP, et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell. 2015; 161:264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mortensen PB, Clausen MR. Short-chain fatty acids in the human colon: Relation to gastrointestinal health and disease. Scand J Gastroenterol Suppl. 1996; 216:132–148. [DOI] [PubMed] [Google Scholar]
- 26.Olesen M, Gudmand-Hoyer E. Maldigestion and colonic fermentation of wheat bread in humans and the influence of dietary fat. Am J Clin Nutr. 1997; 66:62–66. [DOI] [PubMed] [Google Scholar]
- 27.Chatterjee S, Park S, Low K, Kong Y, Pimentel M. The degree of breath methane production in IBS correlates with the severity of constipation. Am J Gastroenterol. 2007; 102:837–841. [DOI] [PubMed] [Google Scholar]
- 28.Pimentel M, Lin HC, Enayati P, et al. Methane, a gas produced by enteric bacteria, slows intestinal transit and augments small intestinal contractile activity. Am J Physiol Gastrointest Liver Physiol. 2006; 290:G1089–95. [DOI] [PubMed] [Google Scholar]
- 29.Anitha M, Reichardt F, Tabatabavakili S, et al. Intestinal dysbiosis contributes to the delayed gastrointestinal transit in high-fat diet fed mice. Cell Mol Gastroenterol Hepatol. 2016; 2:328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Camilleri M. Peripheral mechanisms in irritable bowel syndrome. N Engl J Med. 2012; 367:1626–1635. [DOI] [PubMed] [Google Scholar]
- 31.Yang J, Rose DJ. The impact of long-term dietary pattern of fecal donor on in vitro fecal fermentation properties of inulin. Food Funct. 2016; 7:1805–13. [DOI] [PubMed] [Google Scholar]
- 32.Semenza G. Structure-function relationships in the small-intestinal brush border membrane: A site of the merging of biochemistry, physiology and nutrition which bottazzi indicated two thirds of a century ago. Boll Soc Ital Biol Sper. 1979; 55:597–639. [PubMed] [Google Scholar]
- 33.Quigley EMM. The spectrum of small intestinal bacterial overgrowth (SIBO). Curr Gastroenterol Rep. 2019; 21:3.-019–0671-z. [DOI] [PubMed] [Google Scholar]
- 34.König J, Wells J, Cani PD, et al. Human intestinal barrier function in health and disease. Clin Transl Gastroenterol. 2016; 7:e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fromhold-Treu S, Lamprecht G. Gastrointestinal causes of weight loss: Clinical presentation, diagnostic workup and therapy. Dtsch Med Wochenschr. 2016; 141:253–260. [DOI] [PubMed] [Google Scholar]
- 36.Grabenbauer GG, Holger G. Management of radiation and chemotherapy related acute toxicity in gastrointestinal cancer. Best Pract Res Clin Gastroenterol. 2016; 30:655–664. [DOI] [PubMed] [Google Scholar]
- 37.Harris GJ, Tio F, Cruz AB Jr., Somatostatinoma: A case report and review of the literature. J Surg Oncol. 1987; 36:8–16. [DOI] [PubMed] [Google Scholar]
- 38.Mearin F, Lacy BE, Chang L, Chey WD, Lembo AJ, Simren M, Spiller R. Bowel disorders. Gastroenterology 2016; 150:1393–1407; e5. [DOI] [PubMed] [Google Scholar]
- 39.Puertolas MV, Fifi AC. The Role of Disaccharidase Deficiencies in Functional Abdominal Pain Disorders-A Narrative Review. Nutrients 2018; 10, e1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mattar R, de Campos Mazo DF, Carrilho FJ. Lactose intolerance: diagnosis, genetic, and clinical factors. Clin Exp Gastroenterol. 2012; 5:113–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Järvelä I, Torniainen S, Kolho KL. Molecular genetics of human lactase deficiencies. Ann Med. 2009; 41:568–575. [DOI] [PubMed] [Google Scholar]
- 42.Mitchell KJ, Bayless TM, Paige DM, Goodgame RW, Huang SS. Intolerance of eight ounces of milk in healthy lactose-intolerant teen-agers. Pediatrics 1975; 56:718–721. [PubMed] [Google Scholar]
- 43.Levitt M, Wilt T, Shaukat A. Clinical implications of lactose malabsorption versus lactose intolerance. J Clin Gastroenterol. 2013; 47:471–480. [DOI] [PubMed] [Google Scholar]
- 44.Mobassaleh M, Montgomery RK, Biller JA, Grand RJ. Development of carbohydrate absorption in the fetus and neonate. Pediatrics 1985; 75(1 Pt 2):160–166. [PubMed] [Google Scholar]
- 45.Forsgard RA. Lactose digestion in humans: Intestinal lactase appears to be constitutive whereas the colonic microbiome is adaptable. Am J Clin Nutr. 2019; 110:273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonani M, Pereira RM, Misselwitz B, Fehr T, Wuthrich RP, Franzen D. Chronic norovirus infection as a risk factor for secondary lactose maldigestion in renal transplant recipients: A prospective parallel cohort pilot study. Transplantation 2017; 101:1455–1460. [DOI] [PubMed] [Google Scholar]
- 47.Lifshitz F, Fagundes-Neto U, Ferreira VC, Cordano A, Ribeiro Hda C. The response to dietary treatment of patients with chronic post-infectious diarrhea and lactose intolerance. J Am Coll Nutr. 1990; 9:231–40. [DOI] [PubMed] [Google Scholar]
- 48.Pironti A, Tadeu V, Pedroni A, Porcu A, Manca A, Massarelli G, et al. Role of routine small intestinal biopsy in adult patient with irritable bowel syndrome-like symptoms. Minerva Med. 2010; 101:129–34. [PubMed] [Google Scholar]
- 49.Misselwitz B, Butter M, Verbeke K, Fox MR. Update on lactose malabsorption and intolerance: pathogenesis, diagnosis and clinical management. Gut 2019; 68:2080–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]; Excellent current review on lactose malabsorption and intolerance.
- 50.Nichols BL, Dudley MA, Nichols VN, et al. Effects of malnutrition on expression and activity of lactase in children. Gastroenterology 1997; 112:742–751. [DOI] [PubMed] [Google Scholar]
- 51.Amiri M, Diekmann L, von Kockritz-Blickwede M, Naim HY. The diverse forms of lactose intolerance and the putative linkage to several cancers. Nutrients 2015; 7:7209–7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McDonough FE, Hitchins AD, Wong NP, Wells P, Bodwell CE. Modification of sweet acidophilus milk to improve utilization by lactose-intolerant persons. Am J Clin Nutr. 1987; 45:570–574. [DOI] [PubMed] [Google Scholar]
- 53.Savaiano DA. Lactose digestion from yogurt: Mechanism and relevance. Am J Clin Nutr. 2014; 99(5 Suppl):1251S–5S. [DOI] [PubMed] [Google Scholar]
- 54.Baffour-Awuah NY, Fleet S, Montgomery RK, Baker SS, Butler JL, Campbell C, et al. Functional significance of single nucleotide polymorphisms in the lactase gene in diverse US patients and evidence for a novel lactase persistence allele at -13909 in those of European ancestry. J Pediatr Gastroenterol Nutr. 2015; 60:182–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raz M, Sharon Y, Yerushalmi B, Birk R. Frequency of LCT-13910C/T and LCT-22018G/A single nucleotide polymorphisms associated with adult-type hypolactasia/lactase persistence among Israelis of different ethnic groups. Gene 2013; 519:67–70. [DOI] [PubMed] [Google Scholar]
- 56.Mattar R, de Campos Mazo DF, Carrilho FJ. Lactose intolerance: Diagnosis, genetic, and clinical factors. Clin Exp Gastroenterol. 2012; 5:113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Labrie V, Buske OJ, Oh E, et al. Lactase nonpersistence is directed by DNA-variation-dependent epigenetic aging. Nat Struct Mol Biol. 2016; 23:566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corella D, Arregui M, Coltell O, et al. Association of the LCT-13910C>T polymorphism with obesity and its modulation by dairy products in a mediterranean population. Obesity (Silver Spring) 2011; 19:1707–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rautiainen S, Wang L, Lee IM, Manson JE, Buring JE, Sesso HD. Dairy consumption in association with weight change and risk of becoming overweight or obese in middle-aged and older women: A prospective cohort study. Am J Clin Nutr. 2016; 103:979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jeyaraman MM, Abou-Setta AM, Grant L, et al. Dairy product consumption and development of cancer: An overview of reviews. BMJ Open 2019; 9:e023625.-2018–023625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gencdal G, Salman E, Ozutemiz O, Akarca US. Association of LCT-13910 C/T polymorphism and colorectal cancer. Ann Coloproctol. 2017; 33:169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koskinen TT, Virtanen HEK, Voutilainen S, Tuomainen TP, Mursu J, Virtanen JK. Intake of fermented and non-fermented dairy products and risk of incident CHD: the Kuopio Ischaemic Heart Disease Risk Factor Study. Br J Nutr. 2018; 120:1288–1297. [DOI] [PubMed] [Google Scholar]
- 63.Lomer MC, Parkes GC, Sanderson JD. Review article: Lactose intolerance in clinical practice--myths and realities. Aliment Pharmacol Ther. 2008; 27:93–103. [DOI] [PubMed] [Google Scholar]
- 64.Ghoshal UC, Shukla R, Ghoshal U. Small intestinal bacterial overgrowth and irritable bowel syndrome: A bridge between functional organic dichotomy. Gut Liver. 2017; 11:196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rezaie A, Buresi M, Lembo A, et al. Hydrogen and methane-based breath testing in gastrointestinal disorders: The north american consensus. Am J Gastroenterol. 2017; 112:775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ghoshal UC, Srivastava D, Ghoshal U, Misra A. Breath tests in the diagnosis of small intestinal bacterial overgrowth in patients with irritable bowel syndrome in comparison with quantitative upper gut aspirate culture. Eur J Gastroenterol Hepatol. 2014; 26:753–760. [DOI] [PubMed] [Google Scholar]
- 67.Ghoshal UC. How to interpret hydrogen breath tests. J Neurogastroenterol Motil. 2011; 17:312–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. https://www.statista.com/statistics/249692/us-sugar-consumption/
- 69.Diaz-Sotomayor M, Quezada-Calvillo R, Avery SE, et al. Maltase-glucoamylase modulates gluconeogenesis and sucrase-isomaltase dominates starch digestion glucogenesis. J Pediatr Gastroenterol Nutr. 2013; 57:704–712. [DOI] [PubMed] [Google Scholar]
- 70.Jones HF, Butler RN, Brooks DA. Intestinal fructose transport and malabsorption in humans. Am J Physiol Gastrointest Liver Physiol. 2011; 300:G202–6. [DOI] [PubMed] [Google Scholar]
- 71.Rao SS, Attaluri A, Anderson L, Stumbo P. Ability of the normal human small intestine to absorb fructose: Evaluation by breath testing. Clin Gastroenterol Hepatol. 2007; 5(8):959–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Szilagyi A, Malolepszy P, Yesovitch S, et al. Fructose malabsorption may be gender dependent and fails to show compensation by colonic adaptation. Dig Dis Sci. 2007; 52(11):2999–3004. [DOI] [PubMed] [Google Scholar]
- 73.Păcurar D, Leşanu G, Dijmărescu I, Ţincu IF, Gherghiceanu M, Orăşeanu D. Genetic disorder in carbohydrates metabolism: hereditary fructose intolerance associated with celiac disease. Rom J Morphol Embryol. 2017; 58:1109–1113 [PubMed] [Google Scholar]
- 74.Ritz V, Alfalah M, Zimmer KP, Schmitz J, Jacob R, Naim HY. Congenital sucrase-isomaltase deficiency because of an accumulation of the mutant enzyme in the endoplasmic reticulum. Gastroenterology 2003; 125:1678–1685. [DOI] [PubMed] [Google Scholar]
- 75.Shapiro GL, Bulow SD, Conklin KA, Scheving LA, Gray GM. Postinsertional processing of sucrase-alpha-dextrinase precursor to authentic subunits: Multiple step cleavage by trypsin. Am J Physiol. 1991; G80261(5 Pt 1):G847–57. [DOI] [PubMed] [Google Scholar]
- 76.Robayo-Torres CC, Baker SS, Chumpitazi BP, Lecea CE, Nichols BL,Jr, Opekun AR. Poor starch digestion in children with CSID and recurrent abdominal pain. J Pediatr Gastroenterol Nutr. 2012;55 Suppl 2:S32–4. doi: 10.1097/01.mpg.0000421407.88128.5c [doi]. [DOI] [PubMed] [Google Scholar]
- 77.Van Beers EH, Rings EH, Taminiau JA, et al. Regulation of lactase and sucrase-isomaltase gene expression in the duodenum during childhood. J Pediatr Gastroenterol Nutr. 1998; 27:37–46. [DOI] [PubMed] [Google Scholar]
- 78.Nichols BL, Avery SE, Karnsakul W, et al. Congenital maltase-glucoamylase deficiency associated with lactase and sucrase deficiencies. J Pediatr Gastroenterol Nutr. 2002; 35+G87:573–579. [DOI] [PubMed] [Google Scholar]
- 79.Kilby A, Burgess EA, Wigglesworth S, Walker-Smith JA. Sucrase-isomaltase deficiency. A follow-up report. Arch Dis Child. 1978; 53:677–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Treem WR, McAdams L, Stanford L, Kastoff G, Justinich C, Hyams J. Sacrosidase therapy for congenital sucrase-isomaltase deficiency. J Pediatr Gastroenterol Nutr. 1999; 28:137–142. [DOI] [PubMed] [Google Scholar]
- 81.Boney A, Elser HE, Silver HJ. Relationships among dietary intakes and persistent gastrointestinal symptoms in patients receiving enzyme treatment for genetic sucrase-isomaltase deficiency. J Acad Nutr Diet. 2018; 118:440–447. [DOI] [PubMed] [Google Scholar]
- 82.Puntis JW, Zamvar V. Congenital sucrase-isomaltase deficiency: Diagnostic challenges and response to enzyme replacement therapy. Arch Dis Child. 2015; 100:869–871. [DOI] [PubMed] [Google Scholar]
- 83.Chumpitazi BP, Shulman RJ. Dietary carbohydrates and childhood functional abdominal pain. Ann Nutr Metab. 2016; G8968 Suppl 1:8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yao CK, Tuck CJ, Barrett JS, Canale KE, Philpott HL, Gibson PR. Poor reproducibility of breath hydrogen testing: Implications for its application in functional bowel disorders. United European Gastroenterol J. 2017; 5:284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Auricchio S, Ciccimarra F, Moauro L, Rey F, Jos J, Rey J. Intraluminal and mucosal starch digestion in congenital deficiency of intestinal sucrase and isomaltase activities. Pediatr Res. 1972; 6:832–839. [DOI] [PubMed] [Google Scholar]
- 86.Gericke B, Amiri M, Scott CR, Naim HY. Molecular pathogenicity of novel sucrase-isomaltase mutations found in congenital sucrase-isomaltase deficiency patients. Biochim Biophys Acta Mol Basis Dis. 2017; 1863:817–826. [DOI] [PubMed] [Google Scholar]
- 87.Opekun AR, Balesh AM, Shelby HT. Use of the biphasic (13)C-Sucrose/Glucose breath test to assess sucrose maldigestion in adults with functional bowel disorders. Biomed Res Int. 2016; 2016:7952891. [DOI] [PMC free article] [PubMed] [Google Scholar]; Details the use and models stable isotope-labeled substrates in the evaluation of disaccharidase deficiency syndromes.
- 88.National Center for Biotechnology Information. SI sucrase-isomaltase [Homo sapiens (human)]. National Library of Medicine; 2019. [cited 2019 Oct 12]; Available from: https://www.ncbi.nlm.nih.gov/gene/6476 [Google Scholar]
- 89.Chumpitazi BP, Self MM, Czyzewski DI, Cejka S, Swank PR, Shulman RJ. Bristol stool form scale reliability and agreement decreases when determining rome III stool form designations. Neurogastroenterol Motil. 2016; 28:443–448. G94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Treem WR. Clinical aspects and treatment of congenital sucrase-isomaltase deficiency. J Pediatr Gastroenterol Nutr. 2012; G10455 Suppl 2:S7–13. [DOI] [PubMed] [Google Scholar]
- 91.Garcia-Etxebarria K, Zheng T, Bonfiglio F, et al. Increased prevalence of rare sucrase-isomaltase pathogenic variants in irritable bowel syndrome patients. Clin Gastroenterol Hepatol. 2018; 16:1673–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Henstrom M, Diekmann L, Bonfiglio F, et al. Functional variants in the sucrase-isomaltase gene associate with increased risk of irritable bowel syndrome. Gut. 2018; 67:263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]; Details the use and models genomics in the evaluation of disaccharidase deficiency syndromes.
- 93.Zheng T, Eswaran S, Photenhauer AL, Merchant JL, Chey WD, D’Amato M. Reduced efficacy of low FODMAPs diet in patients with IBS-D carrying sucrase-isomaltase (SI) hypomorphic variants. Gut. 2019; pii: gutjnl-2018–318036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dionne J, Ford AC, Yuan Y, et al. A systematic review and meta-analysis evaluating the efficacy of a gluten-free diet and a low FODMAPs diet in treating symptoms of irritable bowel syndrome. Am J Gastroenterol. 2018; 113:1290–1300. [DOI] [PubMed] [Google Scholar]
- 95.Lankisch PG. Exocrine pancreatic function tests. Gut. 1982; 23:777–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Löser C, Möllgaard A, Aygen S, Hennemann O, Fölsch UR. 13C-starch breath test--comparative clinical evaluation of an indirect pancreatic function test. Z Gastroenterol. 1997; 35:187–94. [PubMed] [Google Scholar]
- 97.Money ME, Camilleri M. Review: Management of postprandial diarrhea syndrome. Am J Med. 2012;125(6):538–544. [DOI] [PubMed] [Google Scholar]
- 98.National Center for Biotechnology Information. MGAM maltase-glucoamylase [Homo sapiens (human)]. National Library of Medicine; 2019. [cited 2019 Oct 12]; Available from: https://www.ncbi.nlm.nih.gov/gene/8972 [Google Scholar]
- 99.Nichols BL, Avery S, Sen P, Swallow DM, Hahn D, Sterchi E. The maltase-glucoamylase gene: Common ancestry to sucrase-isomaltase with complementary starch digestion activities. Proc Natl Acad Sci U S A. 2003;100(3):1432–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li M, Maruthur NM, Loomis SJ, et al. Genome-wide association study of 1,5-anhydroglucitol identifies novel genetic loci linked to glucose metabolism. Sci Rep. 2017;7(1):2812.-017–02287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chumpitazi BP, Robayo-Torres CC, Tsai CM, et al. Demographic and clinical correlates of mucosal disaccharidase deficiencies in children with functional dyspepsia. J Pediatr Gastroenterol Nutr. 2018;66 Suppl 3:S52–S55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rao SS, Viswanathan L, Sharma A. Pan-Disaccharidase Deficiency: Symptom Patterns and Association With Small Intestinal Bacterial Overgrowth: Presidential Poster Award. Am J Gastroenterol. 2018; 113: (Suppl.): S666. [Google Scholar]
- 103.Zhang W, Kim D, Philip E, et al. A multinational, observational study to investigate the efficacy, safety and tolerability of acarbose as add-on or monotherapy in a range of patients: The gluco VIP study. Clin Drug Investig. 2013; 33:263–274. [DOI] [PubMed] [Google Scholar]
- 104.Amiri M, Naim HY. Long term differential consequences of miglustat therapy on intestinal disaccharidases. J Inherit Metab Dis. 2014; 37(6):929–937. [DOI] [PubMed] [Google Scholar]
- 105.Calvin RT, Klish WJ, Nichols BL. Disaccharidase activities, jejunal morphology, and carbohydrate tolerance in children with chronic diarrhea. J Pediatr Gastroenterol Nutr. 1985; 4:949–953. [DOI] [PubMed] [Google Scholar]
- 106.Lücke T, Keiser M, Illsinger S, Lentze MJ, Naim HY, Das AM. Congenital and putatively acquired forms of sucrase-isomaltase deficiency in infancy: Effects of sacrosidase therapy. J Pediatr Gastroenterol Nutr. 2009; 49:485–487.G110 [DOI] [PubMed] [Google Scholar]
- 107.Opekun AR, Abdulsada MM, DeJonge Am, Chumpitazi BP, Graham DY. 1105 - Amyloglucosidase Supplementation Corrects Small Intestinal Isomaltase Insufficiency in Symptomatic Patients. Gastroenterology 2018; 104 (Suppl. 1):S218 [Google Scholar]
- 108.Graham DY, Ketwaroo GA, Money ME, Opekun AR. Enzyme therapy for functional bowel disease-like post-prandial distress. J Dig Dis. 2018; 19:650–656. [DOI] [PMC free article] [PubMed] [Google Scholar]