

Abstract
Bromodomain-containing proteins regulate transcription through protein-protein interactions with chromatin and serve as scaffolding proteins for recruiting essential members of the transcriptional machinery. One such protein is the bromodomain and PHD-containing transcription factor (BPTF), the largest member of the nucleosome remodeling complex, NURF. Despite an emerging role for BPTF in regulating a diverse set of cancers, small molecule development for inhibiting the BPTF bromodomain has been lacking. Here we cross-validate three complementary biophysical assays to further the discovery of BPTF bromodomain inhibitors for chemical probe development: two direct binding assays (protein-observed 19F (PrOF) NMR and surface plasmon resonance (SPR)) and a competitive inhibition assay (AlphaScreen). We first compare the assays using three small molecules and acetylated histone peptides with reported affinity for the BPTF bromodomain. Using SPR with both unlabeled and fluorinated BPTF, we further determine that there is a minimal effect of 19F incorporation on ligand binding for future PrOF NMR experiments. To guide medicinal chemistry efforts towards chemical probe development, we subsequently evaluate two new BPTF inhibitor scaffolds with our suite of biophysical assays and rank-order compound affinities which could not otherwise be determined by PrOF NMR. Finally, we cocrystallize a subset of small molecule inhibitors and present the first small molecule-protein structures with the BPTF bromodomain. We envision the biophysical assays described here and the structural insights from the crystallography will guide researchers towards developing selective and potent BPTF bromodomain inhibitors.
Graphical Abstract
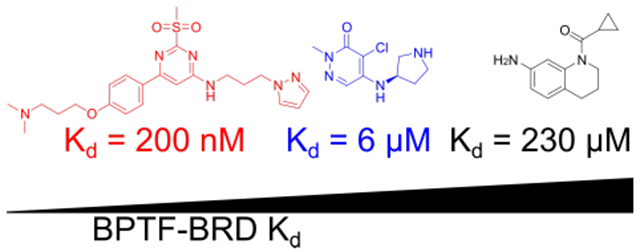
We report the first set of small molecule co-crystal structures with the bromodomain of BPTF and describe several new leads for chemical probe development.
Introduction
Lysine acetylation of histone tails is a dynamic post-translational modification associated with an increase in gene expression.1 Bromodomains are protein motifs that bind to acetylated lysine residues on chromatin and modulate transcription by recruiting transcription factors, enzymes, and nucleosome remodeling complexes to distinct genomic loci.2 Inhibition of several bromodomain-containing proteins is currently being investigated as a viable therapeutic approach for treating cancer, inflammation, and heart disease.3 These inhibitors function through multiple mechanisms including suppressing oncogenes, cytokine secretion, and inducing terminal cell differentiation of self-renewing cancer cells.4–6 The medicinal chemistry efforts for inhibiting a select set of bromodomains has benefitted from disclosure of potent chemical probes.7 The structure-activity relationship studies resulting in these chemical probes has been enabled by robust biophysical assays, particularly for the bromodomain and extraterminal (BET) family proteins.7 However, without cross-validation of assays, conflicting data has been reported in the literature for bromodomain inhibitor selectivity and affinity.8
The bromodomain and PHD-finger containing transcription factor, (BPTF) is a non-BET bromodomain-containing protein, and the largest protein component of the nucleosome remodeling factor (NURF).9 Unlike additional chromatin remodeling complexes, such as SWI/SNF,10–12 BPTF chemical probes have been lacking in the literature. Dysregulation of BPTF is implicated in a number of diseases including melanoma,13 hepatocellular carcinoma,14 colorectal,15 bladder,16 lung,17 and breast cancers.18 Due to the importance of BPTF in these diseases, we and others have begun to develop small molecule inhibitors of the BPTF bromodomain.19–22 One limitation in the development of these inhibitors is the lack of biophysical tools to study interactions with the bromodomain, and x-ray cocrystal structures to guide structure-based design. Several biophysical tools for BPTF interaction analysis have been described in the literature including NMR, AlphaScreen, surface plasmon resonance (SPR), and homogenous time-resolved fluorescence (HTRF), however, few have been systematically investigated.21, 23–24 Here we report several of the first BPTF bromodomain-small molecule cocrystal structures, and cross-validate several biophysical assays to guide structure-based design of new BPTF bromodomain inhibitors.
We previously reported on a protein-observed 19F (PrOF) NMR assay using the BPTF bromodomain for quantifying BPTF-ligand interactions.25 In this case, a single tryptophan is replaced with 5-fluorotryptophan (5FW) in a region near the histone binding site, termed the WPF shelf. Due to the responsiveness of fluorine nuclei to subtle changes in chemical environment, PrOF NMR can quantify the affinity for moderate to weak-binding ligands, and characterize the binding site.26 When analyzed by 19F NMR, the chemical shift of the fluorinated amino acid in the protein is measured in the presence and absence of ligand (Figure 1A). A change in a chemical shift indicates a binding event, or a protein conformational change near the observed fluorinated amino acid.19, 27–29 An advantage of PrOF NMR is the ability to quantify the affinity of ligands that are in rapid chemical exchange with the free and bound state of the protein. This is typically observed for compounds with dissociation constants in the mM to mid μM range.26, 30 However, higher affinity ligands exhibiting slower exchange rates are more challenging to quantify with NMR, due to significant resonance broadening in some cases (intermediate exchange), or resolved bound and unbound states due to slow chemical exchange rates (e.g. Figure 1A). In many cases of slow exchange, affinity values are significantly below the concentration of the fluorine-labeled protein leading to stoichiometric binding, preventing Kd determinations.26
Figure 1:
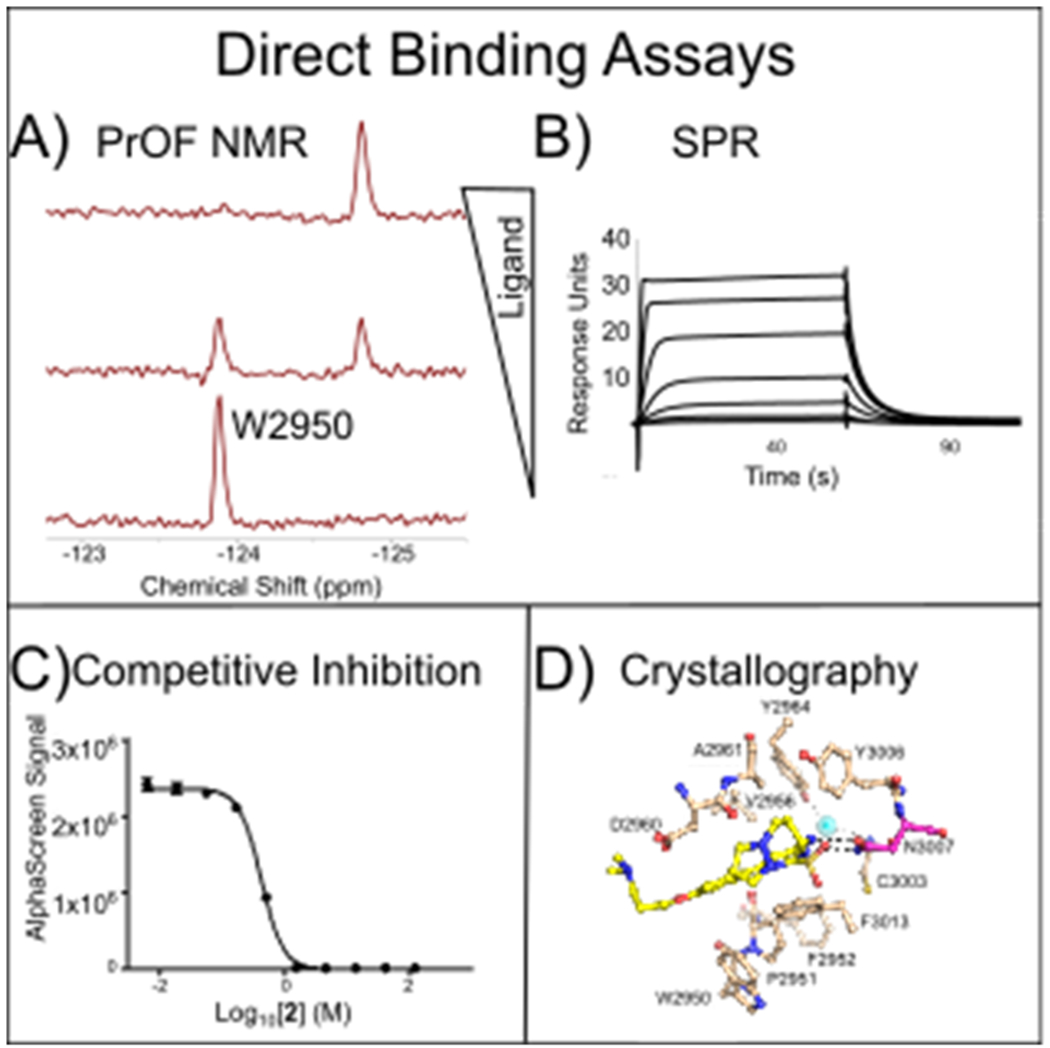
Methods developed for BPTF bromodomain-ligand interaction analyses. A) PrOF NMR is used to assess binding site interactions and affinity B) Kd values are determined by SPR C) Competition with native histone peptides is verified with AlphaScreen. D) Inhibitor binding modes are evaluated using BPTF cocrystal structures.
To quantify the affinity of more potent ligands, complementary binding assays are necessary. Surface plasmon resonance (SPR) and AlphaScreen assays are compatible with both fluorine-labeled and unlabeled proteins to quantify binding affinities (Figures 1B, C, respectively). For AlphaScreen, IC50 values can approximate the dissociation constant of the inhibitor, Ki, due to the low levels of protein and peptide used.31 AlphaScreen is also used to demonstrate functional effects from inhibition of native histone interactions.
Prior to our structural analysis of small molecule-bromodomain interactions, we first cross-validate these three biophysical assays with a panel of recently reported BPTF ligands and discuss the advantages of each approach. We next characterize a series of new molecules based on two chemical scaffolds as starting points for developing selective BPTF inhibitors. Finally, we provide several of the first x-ray cocrystal structures of small molecules bound to the BPTF bromodomain with affinities ranging from Kd= 0.20-290 μM. We envision these validated approaches and structural biology tools will enable rapid chemical probe development for the BPTF bromodomain using structure-based design.
Experimental
Protein expression-
The His6-BPTF gene (addgene plasmid #39111) was first modified by addition of three histidines to the hexahistidine tag via site-directed mutagenesis (described further in the supporting information). The resulting gene, His9-BPTF, in a pNIC-BSA4 vector was cotransformed with pRARE into BL21(DE3) E. Coli. Cells were grown on Luria-Bertani (LB) agar plates containing kanamycin (100 μg/mL) at 37 °C for 12 h. Individual colonies were picked and grown for 12 h in 5 mL of LB containing kanamycin. Four 5 mL cultures were diluted into 1 L of LB media containing kanamycin and the culture was grown by shaking at 220 RPM at 37 °C until an OD600 of 0.6-0.8 was reached. 5-Fluorotryptophan (5FW) was incorporated into BPTF following centrifugation of the culture at 7,000 g for 0.5 h, resuspension in previously described media containing 5-fluoroindole,26 and subsequent induction of protein expression with 1 mM IPTG at 20 °C for 12-18 h. The media exchange step was omitted for expression of unlabeled BPTF. Cultures were centrifuged at 7,000 g and pellets were stored at −20 °C.
His9-BPTF Protein purification-
Cells were thawed at RT in 45 mL of lysis buffer (50 mM sodium phosphate, 300 mM sodium chloride, pH 7.4) followed by addition of 12 mg of PMSF. Cells were then set on ice and lysed using sonication for 15 min in cycles of 30 s of sonication followed by 60 s of cooling. The lysed cells were centrifuged at 10,000 g for 10 minutes and cellular debris was removed by decanting the cell lysate and filtered using Whatman filters. His9-BPTF was purified on a HisTrap FF 5 mL column (GE Healthcare) using a gradient of 40 mM to 400 mM imidazole. Purified protein was then buffer exchanged into 50 mM Tris, 100 mM NaCl, pH 7.4 using a HiPrep desalting column (GE Healthcare). Protein was concentrated to 40-50 μM for NMR studies or 20 μM for SPR/AlphaScreen studies, flash-frozen, and stored at −20 °C.
His6-BPTF Protein purification for crystallography-
Protein purification was performed at 4 °C by FPLC using columns and chromatography resins from GE Healthcare. Cell pellets were re-suspended in 50 mM Na/K Phosphate buffer (pH 7.4) containing 100 mM NaCl, 20 mM imidazole, 0.01% w/v lysozyme, 0.01% v/v Triton X-100, and 1 mM DTT. Cells were lysed using a homogenizer, the lysate was clarified by centrifugation and subjected to purification on immobilized Ni2+-affinity chromatography (Qiagen) using a linear gradient of 20 - 500 mM imidazole. Fractions containing BPTF were pooled and incubated overnight with TEV protease at 4 °C. Cleaved BPTF was subjected to a second Ni2+-affinity chromatography run to remove His-TEV and the cleaved His-tag. The flow-through containing BPTF was concentrated and purified to homogeneity by size exclusion chromatography using a Superdex 26/60 column. Protein was eluted using 50 mM Tris/HCl (pH 8.0) containing 100 mM NaCl and 1 mM DTT. Peak fractions were combined, concentrated to 5 mg/mL, flash-frozen in liquid N2 and stored at −80 °C.
PrOF NMR-
5FW-BPTF (40-50 μM) was diluted by addition of 25 μL of D2O and 2 μL of 0.1% TFA for NMR locking and referencing purposes, respectively. Two spectra were taken of the control protein sample in the presence of 5 μL of DMSO (1% final concentration) at an O1P of −75 ppm, ns = 16, d1 = 1 s, AQ = 0.5 s (samples were referenced to trifluoroacetate at −75.25 ppm) and an O1P of −125 ppm, ns = 256-512, d1 = 0.7 s, AQ = 0.05 s (protein resonances). Ligands were titrated and the change in chemical shift relative to the control sample was plotted as a function of ligand concentration to generate binding isotherms. Data was processed in Mestrenova and isotherms were fit using Prism with Equation 1, whose derivation we described previously.26 Δδobs is the change in chemical shift, [L] is the ligand concentration, and [P] is the protein concentration.
| Equation 1: |
SPR-
His9 unlabeled BPTF or 5FW-BPTF was diluted to ~1-2 μM in SPR running buffer (50 mM HEPES-Na+) (ChemImpex), 150 mM NaCl (SigmaAldrich), 0.05% Tween-20 (SigmaAldrich)), pH 7.4, 1% DMSO. All analyses were performed on a Biacore S200 (GE Healthcare) at 15 °C. Protein was immobilized on a series S NTA chip (GE Healthcare) by first cleaning the chip surface with 500 mM EDTA (pH 8.2) for 120 s at 60 μL/min, followed by activation of the surface with 500 μM NiCl2 (SigmaAldrich) for 60 s at 10 μL/min. Protein was injected over the activated surface for 10 s at 10 μL/min, resulting in immobilization values of 700-1500 response units (RU). Isotherms were created by immobilizing protein, titrating the molecule diluted in running buffer for a 60 s association time, followed by a 60 s dissociation time. Protein was reimmobilized after each point in the titration to ensure equal levels of protein. A DMSO solvent correction was run for each titration to correct for RU changes due to alterations in DMSO concentration. For analysis of histone peptides, anti-GST was immobilized on a CM5 chip using the GST capture kit (GE Healthcare) according to manufacturer’s specifications. GST-BPTF was immobilized by injecting 10 μg/mL of protein for 180 s; typical protein immobilization levels were 900 RU. Peptide-protein interactions were analyzed with a 60 s association time followed by a 90 s dissociation time. The surface was regenerated with 10 mM glycine-HCl (pH 2.1) solution for 120 s, followed by reimmobilization before the beginning of each eight-point titration. Data was analyzed using the Biaevaluation software. All dissociation constants were determined using a steady-state model. An example of the immobilization workflow is demonstrated in Figure S1.
AlphaScreen-
The AlphaScreen assay procedure for BPTF was adapted from the manufacturers protocol (PerkinElmer, USA). Nickel chelate (Ni-NTA) acceptor beads and streptavidin donor beads were purchased from PerkinElmer (Cat. #: 6760619M). The biotinylated Histone H4 KAc5,8,12,16 peptide was purchased from EpiCypher, with the sequence:
Ac-SGRGK(Ac)GGK(Ac)GLGK(Ac)GGAK(Ac)RHRKVLR-Peg(Biot) All reagents were diluted in the assay buffer (50 mM HEPES-Na+ (ChemImpex), 100 mM NaCl (SigmaAldrich), 0.05% CHAPS (RPI), 0.1% BSA (SigmaAldrich), pH 7.4). Final assay concentrations (after the addition of all assay components) of 30 nM for His9-tagged BPTF bromodomain and 50 nM for the biotinylated peptide were used. 3-fold serial dilutions were prepared with varying concentrations of the compounds and a fixed protein concentration, keeping the final DMSO concentration at either 0.25% or 0.5% v/v, depending upon the solubility of the compounds. 5 μL of these solutions were added to a 384-well plate (ProxiPlate-384, PerkinElmer). The plate was sealed and kept at room temperature for 30 min, followed by the addition of 5 μL of the biotinylated peptide. 5 μL of nickel chelate acceptor beads was added to each well under low light conditions (<100 lux), to a final concentration of 20 μg/mL, and the plate was incubated at room temperature in the dark for 30 minutes. This was followed by the addition of 5 μL (20 μg/mL final concentration) of streptavidin donor beads in low light conditions. After incubation for 30 min in the dark, the plate was read in AlphaScreen mode using a PerkinElmer EnSpire plate reader. Each compound was run in duplicate and IC50 values were calculated in GraphPad Prism 5.
Crystallization and structure determination-
Crystallization screening campaigns were performed at 18 °C with precipitant solutions from Hampton Research using a Mosquito liquid handler (TTP Labtech). Robust crystallization conditions were established using 25% PEG 3,350 (w/v), 0.2 M lithium sulfate monohydrate, 0.1 M Bis-Tris (pH 6.5) mixed with an equal volume of protein in hanging droplets (2.5 mg/ml final concentration of BPTF). Inhibitors were cocrystallized with BPTF at 1 mM final concentration. Crystals were cryoprotected by addition of 20% ethylene glycol in the precipitant and flash-frozen in liquid N2. During data collection, crystals were maintained under a constant stream of N2 gas (−180 ° x-ray diffraction data were recorded at beamlines 22-ID, 22-BM, 21-ID-D and GM/CA of Argonne National Laboratories. Data were indexed and scaled with XDS32. Phasing and refinement was performed using PHENIX33 and model building with Coot34. PDB entry 3UV2 served as the search model for molecular replacement. Initial models for small molecule ligands were generated through MarvinSketch (ChemAxon, Cambridge, MA) and ligands restraints through eLBOW of the PHENIX suite. All structures have been validated by MolProbity. Figures were prepared using PyMOL (Schrödinger, LLC).
Results and Discussion
Our prior efforts to design BPTF chemical probes using PrOF NMR were hampered by the lack of complementary biophysical tools to rapidly assess high affinity molecules.19–20To address this need, we first sought to develop an SPR assay, which measures interactions of immobilized protein with ligands in solution, based on an NTA protein immobilization strategy. This binding assay is compatible with the commonly used N-terminal His-tag used for purification of the BPTF bromodomain which would be compatible in both the SPR and PrOF NMR experiments. Initial attempts at immobilizing a His6-bromodomain resulted in fast dissociation of the protein from the chip surface, which could not be used to determine ligand affinities. We inferred from this experiment that our His6 affinity tag was not suitable for ligand-protein binding analysis by SPR. This result was consistent with our failed attempts at immobilizing our His6-BPTF bromodomain for bead-based assays, HTRF and AlphaScreen (data not shown). To increase the affinity of the His tag-NTA interaction, we designed a new protein construct with an elongated His9 tag. Protein immobilization with this affinity tag decreased the time-dependent protein loss substantially. A comparison of the binding data for His6 and the His9-tagged bromodomains is displayed in Figure S2. Using these immobilization conditions, we could subsequently test ligand binding to both unlabeled and 5FW-labeled BPTF bromodomains to compare binding affinity with literature reported values, as well as perturbation in binding affinity from fluorine incorporation.
As an initial comparison of our SPR-derived Kd values to literature-reported values, we chose three compounds with reported Kd values for the BPTF bromodomain to benchmark our SPR assay (Table 1). Bromosporine, 1, is a pan-bromodomain inhibitor, with a reported 1.8 μM Kd for BPTF as determined by ITC.35 TP-238, 2, is a dual CECR2/BPTF bromodomain ligand, which binds to BPTF with a Kd of 120 nM, a 10-fold weaker affinity over CECR2 as determined by ITC.37 GSK4027, 3, is a PCAF/GCN5 inhibitor with off-target binding to BPTF. An estimated affinity for 3 of 130 nM was determined by a single point measurement in a phage display competition experiment.36 We chose these molecules due to their relatively high affinity for BPTF, which were otherwise difficult to determine using PrOF NMR. An example of the SPR sensorgrams for 2, with unlabeled and 5FW-BPTF is shown in Figure 2.
Table 1:
Comparison of PrOF NMR and SPR Kd with AlphaScreen IC50 values for the BPTF bromodomain
| Ligand |
 1, Bromosporine |
 2, TP-238 |
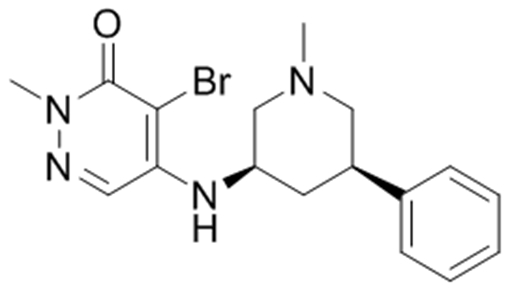 3, GSK4027 |
|---|---|---|---|
| aReported BPTF Kd | 1.8 μM35 | 120 nM† | 130 nM36 |
| 5FW-BPTF PrOF NMR Kd | 37 μM | * | * |
| 5FW-BPTF SPR Kd | 19 ± 3 μM | 250 ± 20 nM | 2.3 ± 0.2 μM |
| aBPTF SPR Kd | 9 ± 1 μM | 200 ± 10 nM | 1.7 ± 0.1 μM |
| aBPTF AlphaScreen IC50 | 10 μM | 430 nM | 1.5 ± 0.2 μM |
AlphaScreen and SPR values were an average of duplicate measurements, except for TP-238 (2) where the SPR value was an average of triplicate measurements and the AlphaScreen value for 3 which is an average of six experimental replicates.
Indicates the unlabeled BPTF protein was used, otherwise the 5FW-labeled protein was used.
Indicates molecules entered intermediate/slow exchange and Kd values could not be determined.
Value reported on thesgc.org.
Figure 2:
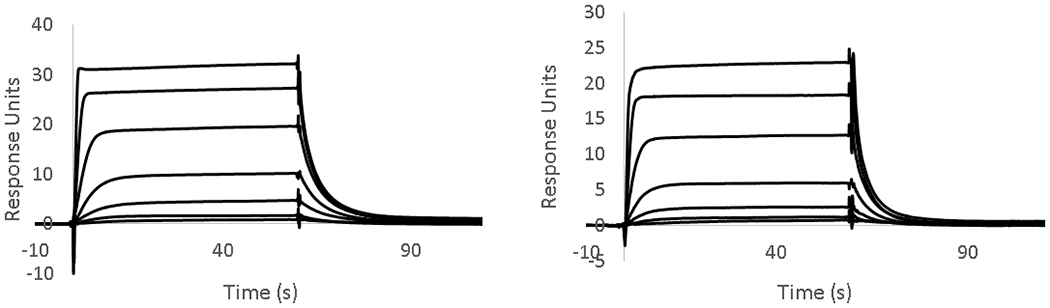
Comparison of SPR sensorgrams for TP-238 (2) using His9-tagged proteins: with (A) unlabeled-BPTF and (B) 5FW-BPTF immobilized on an NTA surface. (2) was titrated in a 7 point 3-fold serial dilution beginning at 2 μM. Kd values are listed in Table 1.
In these cases, using the non-fluorine labeled His9-BPTF construct, we found that the values measured for 1 and 3 of 9 μM and 1.7 μM, respectively, were five- and thirteen-fold higher than their respective literature reports. For 2 the Kd of 200 nM was similar to the reported value of 120 nM. A kinetic-fit of the data also yielded a similar but higher Kd of 262 nM (Fig S3). In the case of 1 and 3, The Kd values determined by SPR were repeatable to within 1.1-2.1 fold of the affinity (Table S2). The weaker affinity values obtained by SPR for 1 and 3 versus literature values may reflect the differences in assay format and protein constructs used between the solution phase ITC experiment, phage display assays, and the surface-bound protein by SPR. One potential assay difference was the organic co-solvent used. The SPR assay uses 1% DMSO, which is known to also compete for bromodomain binding sites.24 Therefore, the effect of DMSO on ligand Kd determination was investigated with 1 using 0.5 and 2% DMSO (Supplementary Table S1). However, in this case there was negligible difference in Kd values (7 - 8 μM).
We next sought to probe differences in ligand-protein interactions due to fluorine incorporation in an assay-consistent manner using SPR. Following labeling His9-BPTF with 5FW, we immobilized the fluorinated protein and compared ligand interactions with the unlabeled His9-BPTF construct. These interactions are also compared in Table 1. For molecules 1-3, we determined affinity values leading to perturbations from 1.3-2.1 fold, which are similar to our reproducibility error (Table S2). From these results, we conclude that 5FW-labeling of BPTF leads to a minimal reduction in ligand binding.
In addition to small molecule ligands, we also evaluated mimics of the native protein-protein interaction by synthesizing and testing histone tail peptides and comparing affinity values we obtained via PrOF NMR and our SPR assay to literature values obtained using ITC, FP and PrOF NMR (Table 2). Here we altered the immobilization of the BPTF bromodomain by using a glutathione S-transferase (GST)-BPTF fusion construct due to non-specific binding of the peptides to the NTA chip surface. This construct yielded similar affinity values for 1-3 (Table S4) reducing concerns from any artifacts introduced by the GST tag. We first tested an H4 histone peptide acetylated at lysine 16, (H4 K16ac). This acetylated histone is reported to associate with BPTF on chromatin.38 We also tested a promiscuous tetraacylated peptide H4 K5ac,K8ac,K12ac,K16ac and a weak binding histone variant, H2AZ.1 K4ac,K11ac reported to bind to the BPTF bromodomain (Table 2).39 Our SPR and PrOF NMR results agree well with each other and literature reported values over a range of affinities from 65-720 μM and provide an additional method for verifying histone-bromodomain interactions.
Table 2:
Comparison of Kd values with PrOF NMR and SPR using acetylated histones.
To further verify the Kd values we obtained via SPR due to discrepancies in our determined values versus reported affinities described above for 1 and 3, we optimized an AlphaScreen assay. The conditions of this assay utilize the interaction of the same His9-BPTF immobilized on an Ni-NTA acceptor bead and a biotinylated tetraacetylated H4 peptide immobilized using streptavidin conjugated to a donor bead. In these cases, IC50 values obtained with our AlphaScreen conditions agreed closer (~2-fold or less) with the Kd values observed by SPR using the unlabeled BPTF bromodomain (Table 1) than with the techniques previously published. We conclude from these studies that both SPR and AlphaScreen can be used to quantify ligand affinities over a wide affinity range. These assays also complement PrOF NMR binding experiments, which are quantitative for weak binding ligands such as acetylated histones, but can only qualitatively providing binding information for higher affinity ligands, such as those in slow exchange (e.g. 2, Figures 3, S10, and S11).
Figure 3:
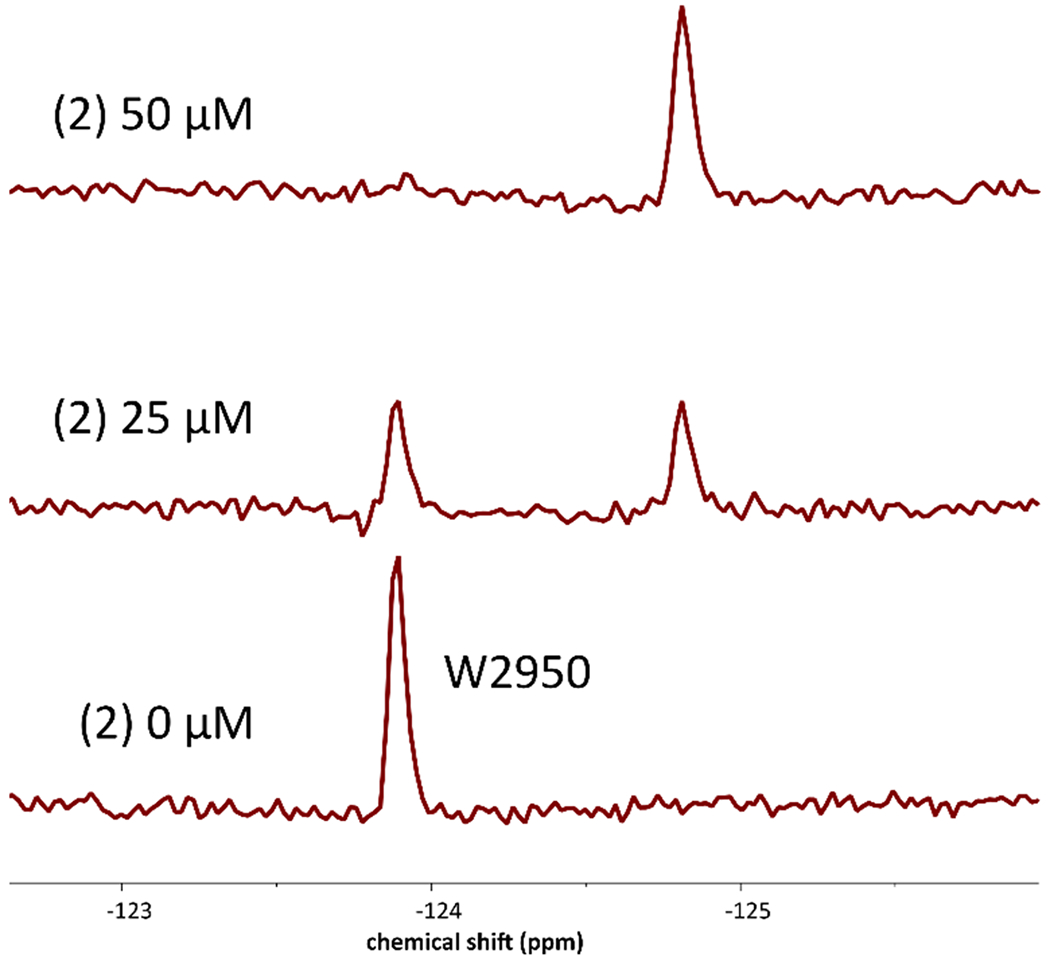
PrOF NMR spectra of 50 μM 5FW-BPTF at various concentrations of 2, a stoichiometric binder.
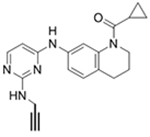
Since the publication of our BPTF bromodomain inhibitor (AU1),19 and the difficulty in enhancing the affinity and selectivity of this molecule, we initiated efforts towards the discovery of new chemical scaffolds for BPTF. We briefly investigate two scaffolds for BPTF inhibitor development here, the tetrahydroquinoline scaffold (4-5) discovered as an off-target in a fragment screen against BRD4 and a pyridazinone scaffold based on 3, (6-9).36, 41 We used these scaffolds to further test the influence of 19F incorporation on ligand binding and to cross-validate our biophysical assays (Table 3). We previously demonstrated that the 19F- and unlabeled-protein have similar stability and only moderately perturbed ligand binding for both histone peptides and 1-3.25 Here, we determine the effect of fluorine incorporation on the affinity of a broader set of ligands (4-9). Using SPR we tested two tetrahydroquinoline ligands and four pyridazinones with both unlabeled and 19F-labeled proteins. Encouragingly, affinity values for all compounds tested using SPR are within error of each other, further supporting a minimal affinity difference between the labeled and unlabeled protein. Additionally, molecules 4, 6, and 8 for which PrOF NMR and SPR Kd values could both be quantified generally showed good agreement, with the largest discrepancy being a 6.7-fold difference for 8. The affinity of 6 and 8 could only be estimated by PrOF NMR due to binding approaching an intermediate exchange rate which leads to significant resonance broadening and may give rise to the error in the measurement for 8. This highlights a limitation in PrOF NMR for moderate affinity ligands.42 Nevertheless, with an SPR-derived Kd of 3 μM for BPTF, compound 8 matches the affinity of our prior lead inhibitor AU1 (Kd = 2.8),19 but with a significantly improved ligand efficiency (0.22 (AU1) vs 0.50 (8)). For molecules whose binding could not be quantified by either PrOF NMR or SPR (due to either broadening in NMR or non-specific binding in SPR), such as 5, we used AlphaScreen assay as an alternative method to determine affinity values based on competitive inhibition. Using 3 and 8 as internal controls in each assay to account for assay variability, we found AlphaScreen IC50 values to result in low variability between assays (Tables 1, 3). Binding of 5 was further confirmed by x-ray crystallography and is described below. We conclude from these analyses that our three assays are compatible with characterizing BPTF bromodomain ligand affinity over a broad affinity range, and that the pyridazinone scaffold represents a ligand-efficient starting point for BPTF inhibitor development.
Table 3:
Panel of small molecules tested in PrOF NMR, SPR, and AlphaScreen assays.
| Ligand | PrOF NMR Kd (μM) | BPTF SPR Kd (μM) | 19F BPTF SPR Kd (μM) | BPTF AlphaScreen IC50 (μM) |
|---|---|---|---|---|
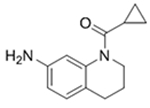
4
|
175 | 230 ± 50 | 280 ± 70 | 200a |
5
|
* | NSB | NSB | 36a |
6
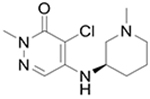
|
7† | 41 ± 9 | 26 ± 10 | 19b |
7
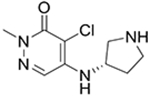
|
* | 25 ± 10 | 19 ± 13 | 31b |
8
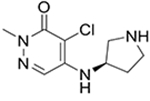
|
20† | 6 ± 2 | 3 ± 1 | 10 ± 2b |
9
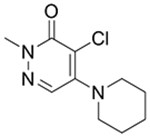
|
330† | Bound to reference | Bound to reference | NB |
indicates molecules entered intermediate exchange and Kd values could not be determined
represents molecules with broadened resonances or a small dynamic range making quantification less accurate.
indicates compound was insoluble at high concentration. PrOF NMR Kd values were determined from a single titration. SPR Kd values were based on data collected in triplicate using His9-tagged BPTF bromodomain. NSB indicates that molecules bound non-specifically and 1:1 binding isotherms could not be generated. Bound to reference indicates molecule bound to chip surface at high ligand concentrations and Kd could not be determined. AlphaScreen IC50 values were reported as the average of technical duplicates except for 8 which is an average of three experimental replicates.
Molecules were tested in the presence of 0.5% DMSO.
Molecules were tested at 0.25% DMSO. NB indicates non-binding up to 250 μM of compound.
Biophysical assays are essential for supporting structure-based design, However, an additional challenge for developing BPTF bromodomain inhibitors has been a lack of small molecule cocrystal structures. Here we report five of the first such examples. Previously, information on the structure of BPTF was limited to the unliganded and liganded states with acetylated histone peptides. To determine the structural basis of BPTF inhibition by known and new small molecule inhibitors, we established cocrystal structure conditions. Following optimization of crystallization conditions, cocrystal structures were determined with 1-3 and tetrahydroquinolines 4 and 5 between 1.23 and 1.77 Å resolution (Supplementary Table S5). All inhibitors bind to the acetylated lysine binding site of BPTF through canonical H-bonding interactions with N3007 (Figure 4 and S41 for 1). Additionally, several hydrophobic van der Waals (VDW) contacts within this site hold the inhibitors in place. The core ring systems of all inhibitors establish pi-stacking interactions with the side chain of F3013, albeit 4 and 5 assume positions that appear less suited for enhanced interactions with F3013, consistent with their weaker binding affinity relative to the pyridazinone ligands 2 and 8 which bind with higher ligand efficiency. Both BPTF and BET bromodomains contain a WPF shelf, which is critical for the high affinity binding of other bromodomain inhibitors.43 For 3, additional pi-stacking T-shaped interactions exist with the side chain of W2950 of the WPF shelf in BPTF (Fig. 4B). 2 is in VDW contact with W2950 (Fig. 4A) while 4 and 5 are in VDW contact only with P2951 and F2952 of the WPF shelf through the cyclopropyl group Fig. 4C, D). Neither 1 or 2 make hydrogen bonds with the backbone carbonyl of P2951, which may lead to the low affinity of 1. Given that these structures represent the first reported cocrystal structure analyses for BPTF-small molecule interactions, we anticipate they will provide a framework for future rational structure-based drug design and complement our biophysical methods.
Figure 4.
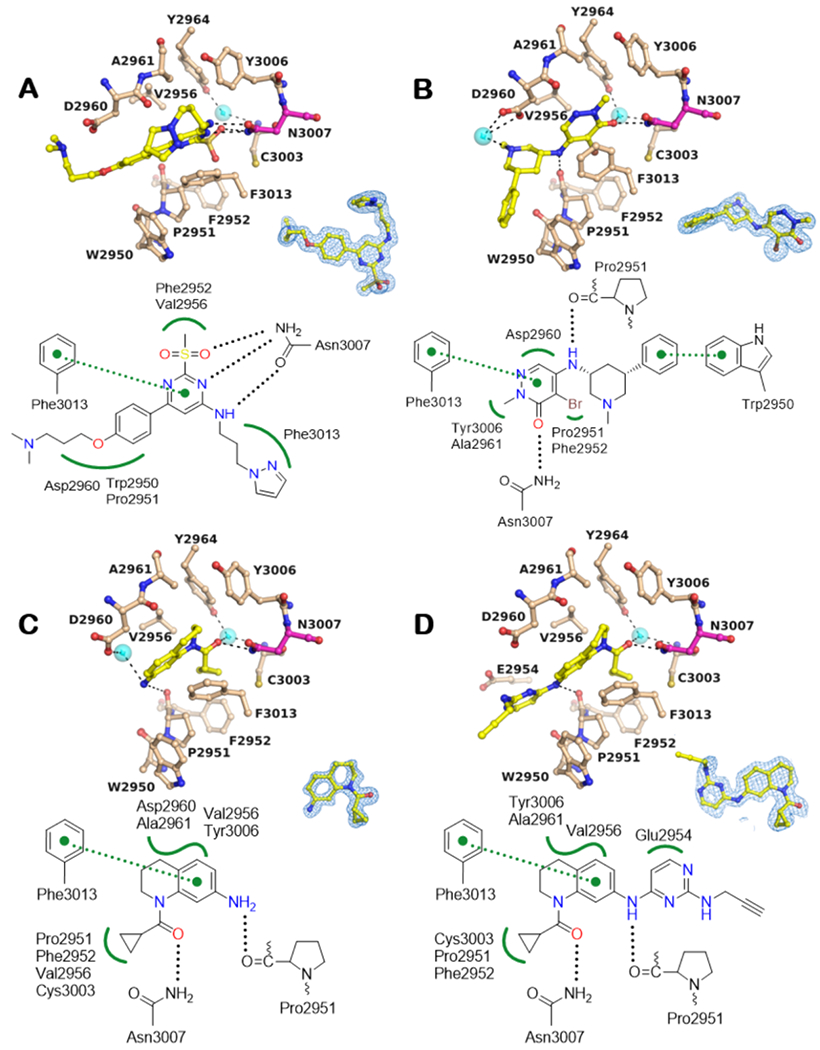
BPTF Bromodomain cocrystal structures. A -D: Cocrystal structures with compounds 2, 3, 4 and 5, respectively.
BPTF residues and small molecules are depicted in beige and yellow, respectively. Potential H-bonding interactions include conserved N3007 (magenta) in the acetylated lysine binding pocket and the main chain carbonyl oxygen of Pro2951 (black dotted lines, 2.2 < d < 3.5 Å). Potential hydrophobic VDW interactions include residues of the WPF shelf (2950-2952) among other residues (green wiggled lines) and Pi-Pi interactions with F3013 green dotted lines with distance cut-off 3.3 < d < 4.0 Å. Cyan spheres show bound water molecules. The blue mesh around the inhibitor shows the corresponding 2Fo-Fc electron density map contoured at 1α. Water mediated H-bonds were excluded from the interaction schematics for clarity.
Conclusions
The biophysical assays explored here have been cross-validated as complementary quantitative techniques for enabling ligand discovery for the BPTF bromodomain. Through comparing the same molecules with PrOF NMR and SPR, we conclude that dissociation constants determined between these different assays lead to affinities that are close agreement with minimal perturbation of ligand binding due to fluorine incorporation. Ligand affinities measured by SPR and AlphaScreen were within ~2 fold, indicating that affinities determined between these two assays are also comparable under these conditions, establishing three robust methods for readily quantifying ligand affinities over millimolar to nanomolar affinity ranges. Finally, for future medicinal chemistry optimization, the new BPTF ligands presented in Table 3, tetrahydroquinolines 4-5 and pyridazinones 6-9, provide starting points for the development of BPTF inhibitors with pyridazinone, 8, possessing high ligand efficiency and potency comparable to AU1. We anticipate the first high-resolution small molecule-BPTF cocrystal complexes will enable rational probe development towards higher affinity and higher selectivity chemical probes for the BPTF bromodomain.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences, R01GM121414. JAJ and NMO were supported by a National Institutes of Health Biotechnology training grant 5T32GM008347-27 5T32GM008347-29 respectively. SPR usage was supported by Office of Research Infrastructure Programs (ORIP) / NIH grant 1S10OD021539-01. GSK4027 and TP238 were acquired from the structural genomics consortium (SGC) through chemicalprobes.org. We would like to acknowledge the Leibniz-Forschungsinstitut for Molekulare Pharmakologie for analytical chemistry support.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
References
- 1.Allfrey VG; Faulkner R; Mirsky AE, Proc. Natl. Acad. Sci. U. S. A. 1964, 51,786–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kouzarides T, Cell 2007, 128,693–705. [DOI] [PubMed] [Google Scholar]
- 3.Liu Z; Wang P; Chen H; Wold EA; Tian B; Brasier AR; Zhou J, J. Med. Chem. 2017, 60,4533–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delmore JE; Issa GC; Lemieux ME; Rahl PB; Shi J; Jacobs HM; Kastritis E; Gilpatrick T; Paranal RM; Qi J; Chesi M; Schinzel AC; McKeown MR; Heffernan TP; Vakoc CR; Bergsagel PL; Ghobrial IM; Richardson PG; Young RA; Hahn WC; Anderson KC; Kung AL; Bradner JE; Mitsiades CS, Cell 2011, 146,904–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fu L.-l.; Tian M; Li X; Li J.-j.; Huang J; Ouyang L; Zhang Y; Liu B, Oncotarget 2015, 6,5501–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Filippakopoulos P; Qi J; Picaud S; Shen Y; Smith WB; Fedorov O; Morse EM; Keates T; Hickman TT; Felletar I; Philpott M; Munro S; McKeown MR; Wang Y; Christie AL; West N; Cameron MJ; Schwartz B; Heightman TD; La Thangue N; French CA; Wiest O; Kung AL; Knapp S; Bradner JE, Nature 2010, 468,1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Filippakopoulos P; Knapp S, Nature Reviews Drug Discovery 2014, 13,337. [DOI] [PubMed] [Google Scholar]
- 8.Gilan O; Rioja I; Knezevic K; Bell MJ; Yeung MM; Harker NR; Lam EYN; Chung C.-w.; Bamborough P; Petretich M; Urh M; Atkinson SJ; Bassil AK; Roberts EJ; Vassiliadis D; Burr ML; Preston AGS; Wellaway C; Werner T; Gray JR; Michon A-M; Gobbetti T; Kumar V; Soden PE; Haynes A; Vappiani J; Tough DF; Taylor S; Dawson S-J; Bantscheff M; Lindon M; Drewes G; Demont EH; Daniels DL; Grandi P; Prinjha RK; Dawson MA, Science 2020, 368,387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mayes K; Qiu Z; Alhazmi A; Landry JW, Chapter Five - ATP-Dependent Chromatin Remodeling Complexes as Novel Targets for Cancer Therapy In Advances in Cancer Research, Tew KD; Fisher PB, Eds. Academic Press: 2014; Vol. 121, pp 183–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fedorov O; Castex J; Tallant C; Owen DR; Martin S; Aldeghi M; Monteiro O; Filippakopoulos P; Picaud S; Trzupek JD; Gerstenberger BS; Bountra C; Willmann D; Wells C; Philpott M; Rogers C; Biggin PC; Brennan PE; Bunnage ME; Schüle R; Günther T; Knapp S; Müller S, Science Advances 2015, 1,e1500723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hohmann AF; Martin LJ; Minder JL; Roe J-S; Shi J; Steurer S; Bader G; McConnell D; Pearson M; Gerstberger T; Gottschamel T; Thompson D; Suzuki Y; Koegl M; Vakoc CR, Nature Chemical Biology 2016, 12,672–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marian CA; Stoszko M; Wang L; Leighty MW; de Crignis E; Maschinot CA; Gatchalian J; Carter BC; Chowdhury B; Hargreaves DC; Duvall JR; Crabtree GR; Mahmoudi T; Dykhuizen EC, Cell Chemical Biology 2018, 25,1443–1455.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dar AA; Nosrati M; Bezrookove V; de Semir D; Majid S; Thummala S; Sun V; Tong S; Leong SPL; Minor D; Billings PR; Soroceanu L; Debs R; Miller JR; Sagebiel RW; Kashani-Sabet M, Journal of the National Cancer Institute 2015, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao S; Liu L; Fang M; Zhou X; Peng X; Long J; Lu X, Dig Dis Sci 2015, 60,910–8. [DOI] [PubMed] [Google Scholar]
- 15.Xiao S; Liu L; Lu X; Long J; Zhou X; Fang M, J Cancer Res Clin Oncol 2015, 141,1465–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim K; Punj V; Choi J; Heo K; Kim J-M; Laird PW; An W, Epigenetics Chromatin 2013, 6,34–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai M; Lu JJ; Guo W; Yu W; Wang Q; Tang R; Tang Z; Xiao Y; Li Z; Sun W; Sun X; Qin Y; Huang W; Deng WG; Wu T, Oncotarget 2015, 6,33878–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frey WD; Chaudhry A; Slepicka PF; Ouellette AM; Kirberger SE; Pomerantz WCK; Hannon GJ; dos Santos CO, Stem Cell Reports 2017, 9,23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Urick AK; Hawk LM; Cassel MK; Mishra NK; Liu S; Adhikari N; Zhang W; Dos Santos CO; Hall JL; Pomerantz WC, ACS Chem Biol 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirberger SE; Ycas PD; Johnson JA; Chen C; Ciccone MF; Woo RWL; Urick AK; Zahid H; Shi K; Aihara H; McAllister SD; Kashani-Sabet M; Shi J; Dickson A; dos Santos CO; Pomerantz WCK, Organic & Biomolecular Chemistry 2019, 17,2020–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang D; Han J; Lu W; Lian F; Wang J; Lu T; Tao H; Xiao S; Zhang F; Liu YC; Liu R; Zhang N; Jiang H; Chen K; Zhao C; Luo C, Bioorganic chemistry 2019, 86,494–500. [DOI] [PubMed] [Google Scholar]
- 22.Xu J; Wang Q; Leung ELH; Li Y; Fan X; Wu Q; Yao X; Liu L; Leung ELH; Leung ELH, Front Med 2019. [Google Scholar]
- 23.Urick AK; Calle LP; Espinosa JF; Hu H; Pomerantz WC, ACS Chem Biol 2016, 11,3154–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Philpott M; Yang J; Tumber T; Fedorov O; Uttarkar S; Filippakopoulos P; Picaud S; Keates T; Felletar I; Ciulli A; Knapp S; Heightman TD, Mol. BioSyst. 2011, 7,2899–2908. [DOI] [PubMed] [Google Scholar]
- 25.Urick AK; Hawk LM; Cassel MK; Mishra NK; Liu S; Adhikari N; Zhang W; dos Santos CO; Hall JL; Pomerantz WC, ACS Chem Biol 2015, 10,2246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gee CT; Arntson KE; Urick AK; Mishra NK; Hawk LML; Wisniewski AJ; Pomerantz WCK, Nat. Protocols 2016, 11,1414–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mishra NK; Urick AK; Ember SW; Schonbrunn E; Pomerantz WC, ACS Chem Biol 2014, 9,2755–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gee CT; Koleski EJ; Pomerantz WC, Angew Chem Int Ed Engl 2015, 54,3735–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pomerantz WC; Wang N; Lipinski AK; Wang R; Cierpicki T; Mapp AK, ACS Chem Biol 2012, 7,1345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Divakaran A; Kirberger SE; Pomerantz WCK, Accounts of Chemical Research 2019, 52,3407–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pomerantz WCK; Johnson JA; Ycas PD, Applied Biophysics for Bromodomain Drug Discovery. Springer: Berlin, 2019. [Google Scholar]
- 32.Kabsch W, Acta Crystallogr D Biol Crystallogr 2010, 66,125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH, Acta Crystallogr D Biol Crystallogr 2010, 66,213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P; Lohkamp B; Scott WG; Cowtan K, Acta Crystallogr D Biol Crystallogr 2010, 66,486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Picaud S, Leonards K, Lambert J, Dovey O, Wells C, Fedorov O, Monteiro O, et al. , Science Advances 2016, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Humphreys PG; Bamborough P; Chung C.-w.; Craggs PD; Gordon L; Grandi P; Hayhow TG; Hussain J; Jones KL; Lindon M; Michon A-M; Renaux JF; Suckling CJ.; Tough DF; Prinjha RK, Journal of Medicinal Chemistry 2017, 60,695–709. [DOI] [PubMed] [Google Scholar]
- 37.Dickson A; Brooks CL, The Journal of Physical Chemistry B 2014, 118,3532–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruthenburg AJ; Li H; Milne TA; Dewell S; McGinty RK; Yuen M; Ueberheide B; Dou Y; Muir TW; Patel DJ; Allis CD, Cell 2011, 145,692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perell GT; Mishra NK; Sudhamalla B; Ycas PD; Islam K; Pomerantz WCK, Biochemistry 2017, 56,4607–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sudhamalla B; Dey D; Breski M; Nguyen T; Islam K, Chemical Science 2017, 8,4250–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson JA; Nicolaou CA; Kirberger SE; Pandey AK; Hu H; Pomerantz WCK, ACS Medicinal Chemistry Letters 2019, 10,1648–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stadmiller SS; Aguilar JS; Waudby CA; Pielak GJ, Biophysical Journal 2020, 118,2537–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ember SWJ; Zhu J-Y; Olesen SH; Martin MP; Becker A; Berndt N; Georg GI; Schönbrunn E, ACS Chemical Biology 2014, 9,1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.