

Abstract
Oncogenic RAS proteins, which are mutated in approximately 24% of all human cancers, have earned a well-deserved reputation as being “undruggable.” However, several studies have challenged that reputation. With the first small molecules that directly target one oncogenic RAS mutant (G12C) undergoing clinical evaluation, there have been substantial advances in finding new anti-RAS therapeutic strategies. Furthermore, new insights have come from the growing appreciation that neither all RAS proteins (HRAS, NRAS, and KRAS4A/KRAS4B) nor all oncogenic RAS mutations (such as at residues Gly12, Gly13, and Gln61) have the same impact on RAS signaling and function. The role of the nonmutated, wild-type RAS proteins in the context of mutant RAS is increasingly considered to be targetable, with reports of strategies that directly disrupt either the RAS interaction with activating guanine nucleotide exchange factors (GEFs) or receptor tyrosine kinase–mediated and GEF-dependent RAS activation (such as by targeting the scaffolding phosphatase SHP2). Lastly, the development of new agents that target downstream effectors of RAS signaling has advanced substantially. In this review, we highlight some important trends in the targeting of RAS proteins in cancer.
Introduction to RAS
Activating mutations in RAS proteins are found in ~24% of all cancers [as reported in the Catalog of Somatic Mutated in Cancer (COSMIC) database, v89] and are commonly associated with resistance to frontline therapies1. The three human RAS genes encode four highly identical proteins (83–85% identity): HRAS, NRAS, and KRAS4A and KRAS4B, with KRAS encoding splice variants due to alternative exon 4 utilization. Wild-type (WT) RAS proteins are GTP-hydrolyzing proteins (GTPases) that cycle between GTP- and GDP-bound states. In the GDP-bound state, they are unable to engage effector proteins and are considered inactive. RAS is activated by guanine nucleotide exchange factors (GEFs, including SOS1 and SOS2) that promote exchange of GDP for GTP. In the GTP-bound state, RAS engages downstream effectors through its dynamic conformation switch regions (SWI and SWII) to stimulate intracellular signaling. RAS is inactivated by the hydrolysis of bound GTP by GTPase-activating proteins (GAPs; such as neurofibromin) that stimulate the otherwise inefficient intrinsic GTPase activity. The activating single missense mutations that have been found in human cancers occur primarily at codons encoding glycines at residues 12 (Gly12, commonly referred to clinically and per nomenclature as G12) and 13 (G13) or glutamine at residue 61 (Q61). These single amino acid mutations impair intrinsic and GAP-stimulated GTP hydrolysis, favoring formation of RAS-GTP. Despite the similar functional consequences of these mutations, they are found at disparate frequencies among cancer types and among RAS genes. For example, in solid tumors KRAS mutations occur most commonly at G12, whereas NRAS mutations occur most commonly at Q611. Similarly, RAS mutations in pancreatic ductal adenocarcinoma (PDAC) are almost exclusively found in KRAS (~98%), whereas in melanoma mutations in NRAS predominate (94%)1. These findings suggest that the different RAS isoforms, as well as individual mutations within one RAS protein, have distinct properties despite their high sequence similarity and conserved function.
After almost four decades of intensive research, no clinically effective therapies for RAS-mutant cancers have been developed, placing RAS at the top of the therapeutic “most-wanted” list. Past challenges in therapeutic targeting of RAS-mutant cancers can be ascribed to multiple causes. First, the RAS structure does not display an attractive topology for the design of high affinity and selective small molecule recognition, prompting the widely held perception that RAS itself is “undruggable”. Second, whereas potent and selective inhibitors of the RAF-MEK-ERK protein kinase cascade have been developed to block this key RAS effector pathway, the development and efficacy of MEK inhibitors have been limited by resistance caused by the relief of ERK-mediated feedback inhibition, leading to compensatory reactivation of ERK. Third, inhibitors of farnesyltransferase and thereby of RAS membrane association were rendered ineffective by cells’ unanticipated compensatory use of the related enzyme geranylgeranyltransferase-I, restoring KRAS and NRAS (but not HRAS) membrane association. Fourth, searches for synthetic lethal genetic interactors with mutant RAS proteins were compromised by off-target activities of RNA interference (RNAi) constructs as well as the use of flawed cell models. An additional complicating issue has been an assumption that all RAS-mutant cancers share the same therapeutic vulnerabilities. With lessons learned from past failures, recent studies have reinvigorated interest in pursuing directions once left for dead.
Direct RAS targeting
Recent findings have begun to challenge the perception that RAS is undruggable. A breakthrough that has seen tremendous progress is the development of direct RAS inhibitors that specifically target the glycine to cysteine mutation at residue 12 (G12C) in KRAS. This often smoking-associated G12C mutation represents ~12% of all KRAS mutations (per COSMIC v89) and is the most prevalent KRAS mutation (46%) in non-small cell lung cancer (NSCLC), the most deadly cancer type across both sexes in the US2. The first cysteine-directed tethering of a small molecule to KRASG12C identified a previously unknown pocket beneath the dynamic SWII region of RAS-GDP, but not RAS-GTP. Therefore, such molecules can lock KRASG12C in the inactive GDP state, thus preventing effector engagement3. It has been established that substitutions at residues 12, 13, or 61 render RAS insensitive to GAP-catalyzed hydrolysis of GTP to GDP4. However, the G12C mutation does not significantly impair intrinsic GTP hydrolysis compared to other G12/G13 mutations5. Consequently, KRASG12C occupies the GDP-bound state more frequently than other mutants and is still dependent on GEF stimulation to achieve full activation. This existence of KRASG12C in the GDP-bound state, together with cysteine-reactive warheads, presented a unique therapeutic vulnerability for G12C-directed inhibitors that recognize only the GDP-bound protein. As a result, first-in-class RASG12C-specific inhibitors have entered phase I/II clinical trials in solid tumors that have a KRASG12C mutation: AMG 510 (NCT03600883, NCT04185883), MRTX-849 (NCT03785249), JNJ-74699157 (NCT03114319), and LY3499446 (NCT04165031) with more in the pipeline. Early clinical trial observations have been promising, in particular for NSCLC6.
Nevertheless, treatment-induced acquired resistance will almost certainly reduce the long-term effectiveness of these inhibitors. Lou and colleagues (2019) set out to address this concern by performing a genome-wide CRISPR interference (CRISPRi) screen in search of genes that sensitize cells to KRASG12C inhibition7 (Figure 1). Using the recently reported G12C inhibitor ARS-16208 (G12Ci), they describe genetic dependencies revealed upon KRASG12C inhibition. As expected, they found that G12Ci lethality was enhanced by silencing upstream components of receptor tyrosine kinase (RTK)-mediated signaling that lead to RAS activation, thereby increasing RAS-GDP and making available more SII pockets for G12Ci binding. These components included RTKs themselves (such as AXL and FGFR1), scaffolding proteins between RTKs and the RASGEF SOS1 (such as GRB2 and SHP2), as well as SOS1 itself. These results fit with recent studies in preclinical models of mutant KRAS-driven NSCLC and PDAC that found a significant therapeutic benefit from combining the MEK inhibitors selumetinib or trametinib with the SHP2 inhibitor SHP-0999,10. Lou et al. also observed sensitization to KRASG12C inhibition by loss of genes encoding components of the PI3K-AKT-mTOR pathway (e.g., PI3KCA, AKT1, RHEB, RPTOR) and cell cycle regulators (CDK4 and CCND1), consistent with previous findings with RAF and MEK inhibitors11,12. Together, these studies suggest that, as KRASG12C inhibitors make their way through the clinic, rational combinations with both upstream and downstream signaling components (such as MEK; NCT04185883) would be beneficial to delay onset of resistance and obtain long-term responses.
Figure 1. Defining novel RAS vulnerabilities for the development of anti-RAS strategies.
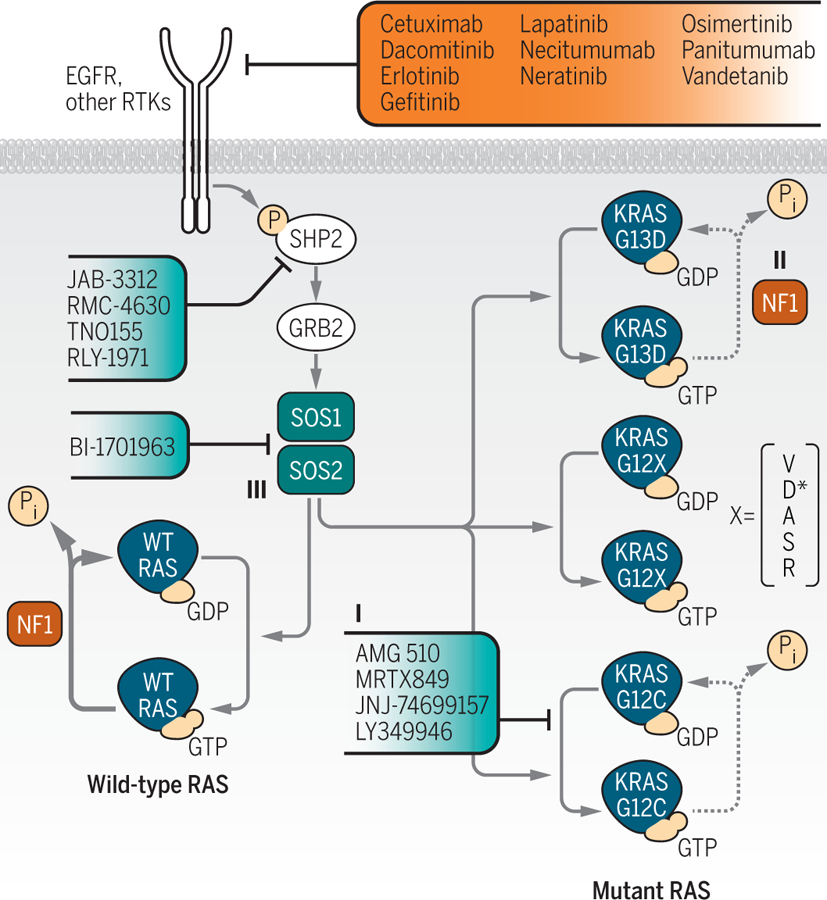
A series of recent studies, addressing different aspects of RAS function, have provided potential new clues to targeting RAS for cancer treatment. Where appropriate, clinically approved (red) and investigational (blue) therapeutics indicated for targeting RAS activation and signaling in cancer are noted. Lou et al. applied a CRISPR genetic loss-of-function screen to identify signaling modulators of a small molecule inhibitor selective for the KRASG12C mutant (I) found predominantly in lung cancer7. In contrast to other G12 mutants, KRASG12C retains intrinsic GTP-hydrolysis (*with the exception of G12D which retains minor GTP-hydrolysis), thereby enabling its’ targeting by GDP-KRASG12C-specific small molecules. The findings revealed various pathways to target to enhance the potency and durable efficacy of the inhibitor. McFall et al. proposed a mechanism to explain the EGFR-dependence of KRASG13D-mutant colorectal cancer22. Decreased affinity for the RAS-GAP NF1 (II) by the G13D mutant protein enables GAP- and EGFR-dependent regulation of wild-type RAS, thereby retaining sensitivity to EGFR inhibitors in KRASG13D-mutant cells. And Sheffels et al. identified a role for the RAS-GEF SOS2 (III) in promoting WT HRAS activation of AKT to support mutant KRAS-induced transformation of mouse fibroblasts in 3D growth culture conditions.
This approach to directly targeting KRASG12C is an example of an anti-RAS strategy that cannot be readily applied to other RAS mutants. Although the SWII pocket is present on all GDP-bound RAS proteins, compounds that target KRASG12C rely on the reactive cysteine substitution and the continued dependency of this mutant on GEF activity to achieve its full activated state. Further, the G12C mutation is found at significantly lower frequencies in colorectal (CRC; 8%) and pancreatic ductal (PDAC; 2%) adenocarcinomas1, the second and third most common causes of US cancer deaths. Therefore, new strategies need to be considered for the more prevalent KRAS mutations, G12D and G12V. With momentum coming from the first KRASG12C-specific drug entering the clinic, there is hope that the perception of targeting the other RAS mutations becomes widely regarded as “not-yet-druggable”.
Aside from cysteine-tethering screens used to find KRASG12C inhibitors, to date two alternative approaches have been described to identify druggable surfaces on RAS: (1) fragment library screens13,14, and (2) small antibody-like protein screens15,16. These methods have yielded promising results, including a new generation of RAS:SOS inhibitors, as well as small molecules and protein binding domains that disrupt effector binding.
In addition to these strategies, a recent discovery has identified a novel approach for directly inhibiting RAS proteins. Biancucci et al. (2019) reported the mechanism by which a bacterial endopeptidase, termed the Ras/Rap1-specific endopeptidase (RRSP), disrupts RAS function17 (Figure 2). RRSP was reported previously to cause proteolytic processing of RAS proteins between residues Tyr-32- and Asp-33 in SWI. Importantly, this cleavage site is present in all RAS proteins and oncogenic mutants, although also found in the related RAP1 small GTPase. Proteolytic processing by RRSP did not cause the degradation of RAS or release of nucleotide, but instead locally altered RAS SWI structure that impaired both SOS1 as well as RAF effector binding. While the 494-residue RRSP peptide itself is not a feasible therapeutic, it may be a useful tool reagent for defining a novel RAS vulnerability. That RRSP can disable both WT and mutant RAS proteins can help discern the therapeutic advantages and disadvantages of pan-RAS versus RAS mutant- or isoform-selective inhibitors. Together these strategies have presented clues towards building a growing arsenal of anti-RAS approaches that will inform the development of translatable therapies.
Figure 2. More RAS vulnerabilities, downstream.
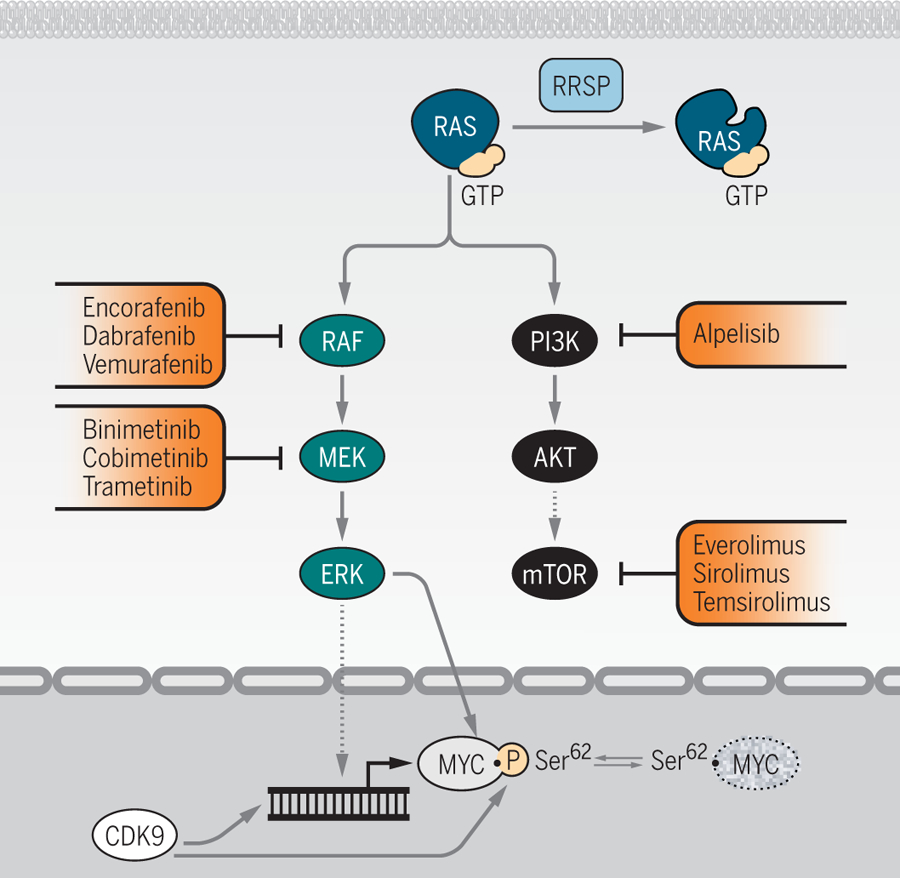
Insight into additional aspects of RAS protein and pathway regulation reveal more ways to potentially target RAS. Biancucci et al. showed that endopeptidase RRSP modification of RAS (I) impairs its interaction with downstream kinase RAF17. Looking further downstream still, Blake et al. applied a MYC degradation screen to identify the kinase CDK9 as a positive regulator of MYC protein stability (II) and, consequently, cell growth and survival39. Some clinically approved therapeutics indicated for targeting RAS activation and signaling in cancer are noted (red).
Targeting upstream regulators
At present, the clinical value of identifying a RAS mutation is largely to guide against the use of certain therapies. Thus, while EGFR inhibitors are approved for the treatment of CRC, the FDA recommendation is that patients with KRAS mutations should not be candidates for this therapy. In particular, some studies of CRC patients treated with anti-EGFR monoclonal antibodies have shown that patients harboring KRASG13D mutations fare better in overall and/or progression free survival than those harboring other RAS mutations18. While subsequent studies did not support this premise20,21, nevertheless, there is growing appreciation that all KRAS-mutant cancers should not be considered one homogeneous patient group21. The idea that any KRAS mutant cancer would still be responsive to EGFR inhibitor therapy is counterintuitive, given that a key cancer driving consequence of aberrant EGFR signaling is hyperactivation of RAS and ERK MAPK signaling. To address this concept, McFall and colleagues (2019) used computational modeling to identify a mechanism whereby KRASG13D mutant CRC may still be uniquely dependent on EGFR signaling22. This invokes the active role that the remaining WT RAS isoforms, NRAS and HRAS, play in supporting the cancer driver function of KRASG13D. Previous work established that WT RAS can have tumor promoting activity in cells containing mutant (MT) RAS23–25. Further, MT RAS can activate WT RAS by binding to the allosteric activation site of the RASGEF SOS126 or by sequestration of RASGAPs, both of which lead to increased GTP-bound WT RAS but by different mechanisms. McFall et al. report that other KRAS mutants are highly effective at sequestering the RASGAP neurofibromin (NF1) and thereby promote formation of WT RAS-GTP independent of upstream RTK activity. In contrast, they report that the KRASG13D mutant is much less effective at NF1 sequestration, so only the KRASG13D mutant CRC cells remain dependent upon EGFR-stimulated, SOS1-dependent formation of GTP-bound WT RAS (Figure 1). The authors therefore speculate that this provides a mechanistic basis for a therapeutic benefit of EGFR inhibition in KRASG13D mutant CRC. Another recent study also determined that KRASG13D has distinct structural and biochemical properties, with reduced engagement of RAF and oncogenic potency27. These findings with KRASG13D further exemplifies the importance of considering each RAS mutation as distinct.
An early misconception due to the high sequence identity of the four RAS proteins was that they have essentially identical roles as cancer drivers. The focus of early RAS studies on HRAS, based largely on convenience of available reagents, in turn led to additional misconceptions. Beginning with the unexpected finding that farnesyltransferase inhibitors were effective in blocking HRAS, but not KRAS or NRAS, membrane association, the field has refocused on studying the most commonly mutated RAS isoform and its most widely expressed splice variant, KRAS4B. RAS isoform differences in effector utilization have also been described. For example, HRAS, NRAS, and KRAS are reported to have varying specificities for the activation of the downstream effectors PI3K and RAF, with HRAS activating PI3K more strongly than K/NRAS and, conversely, KRAS activating RAF more strongly than H/NRAS28. This effector preference could explain tumor promoting roles of WT RAS in the context of MT RAS. Sheffels and colleagues (2018) have shed new light on this topic by reporting that, in a model mouse fibroblast cell system, MT KRAS, but not MT NRAS or HRAS, was more dependent on activation of WT HRAS for growth and oncogenic transformation. They report that MT KRAS dependence on WT RAS activity was due to a requirement for activation of PI3K-AKT, which is accomplished principally by WT HRAS and NRAS. Interestingly, the activation of WT RAS was SOS2-dependent, and did not depend on the feed-forward activity of MT RAS binding to the allosteric region, as has been reported for SOS126 (Figure 1). In this model, upstream RTK activation led to the SOS2-dependent activation of PI3K by WT RAS to promote MT KRAS-driven growth. Furthermore, it presents targeting SOS2 as a general strategy for inhibiting WT RAS isozymes in the context of MT KRAS. Targeting SOS2 alone as opposed to both SOS1 and SOS2 might be more therapeutically tractable, as previous reports found double knockout of SOS1 and SOS2 in adult mice to be lethal, whereas mice with single knockout of either SOS1 or SOS2 were viable29. This is a timely observation as a new SOS1 therapeutic recently entered phase I clinical trials in KRAS mutant solid tumors (BI-1701963, NCT04111458).
Targeting downstream effector signaling
Extensive efforts have been devoted to the targeting of RAS downstream effector signaling, with inhibitors already approved or in advanced clinical candidacy. The RAF-MEK-ERK signaling network, followed by PI3K-AKT-mTOR, are considered the most important mediators of MT RAS-dependent cancer growth. This prominence is supported by findings in mouse models of KRAS-driven cancer as well as by the frequent mutational activation of these RAS effectors (such as PIK3CAH1047R/E545K and BRAFV600E) in cancer. Fortunately, these signaling components are readily druggable kinases, with inhibitors for both BRAF and PI3Kα approved for cancer treatment. However, because of the central roles of these kinase networks in normal cell physiology, and of inhibitor-driven induction of compensatory signaling resistance mechanisms, the clinical advancement of effector inhibitors has been met with both expected and unexpected complications.
Perhaps the most progress has been made in the development of RAF and MEK inhibitors for BRAF-mutant melanoma, with three approved inhibitors each for BRAF and for MEK. Mutations in BRAF were identified in 2002 in several tumor types, leading to initial efforts focused on targeting the then lesser-studied RAF isoform, BRAF. The three RAF family proteins (ARAF, BRAF and CRAF/RAF1) are serine/threonine kinases that are activated upon binding to RAS-GTP through translocation to the plasma membrane as well as through relief of N-terminal autoinhibition of the C-terminal kinase domain. Additionally, RAF hetero-/homo-dimerization mediated by association with RAS is required for full activation of RAF catalytic activity towards their only widely accepted substrates, MEK1 and MEK2. MEK1/2 are dual specificity kinases that, like RAF, have only two well-validated substrates, the highly related ERK1 and ERK2 MAPKs. This substrate stringency led to the earlier misconception that the RAF-MEK-ERK kinase cascade acts as a simple linear and unidirectional pathway that culminates in ERK activation, in turn leading the field to focus initially on therapeutics targeting BRAF and MEK, rather than ERK. The first clinically approved BRAF inhibitors, vemurafenib and dabrafenib, were effective in treating BRAF-mutant melanoma. However, these inhibitors led to an unexpected paradoxical activation of CRAF through their ability to activate RAS-dependent formation of RAF homo-/hetero-dimers, reviewed elsewhere30.
The paradoxical activation of RAF by early BRAF inhibitors prompted the development of RAF inhibitors that do not lead to this unwanted activation, and hence are termed ‘paradox-breakers’. These paradox-breakers are now in clinical development. There is still a concern, however, that even pan-RAF inhibitors will meet challenges like those of BRAF inhibitors, where pathway reactivation occurs through the upregulation of RTKs and relief of negative feedback. To address this, combinations with MEK1/2 inhibitors, such as the clinically approved MEK inhibitors trametinib, cobimetinib and binimetinib, can aid in preventing pathway reactivation that leads to the activation of ERK1/2. The vertical inhibition strategy of targeting BRAF and MEK together has been evaluated in BRAF-mutant melanoma, where combination BRAF and MEK inhibitor treatment is now the standard-of-care31. Since ERK1/2 has a broad signaling network of over 1,000 intracellular targets, and since the RAS-RAF-MEK cascade results in the activation of ERK, perhaps inhibiting ERK1/2 would be even more effective. Preclinical models have suggested that this could be the case32. Although the development of ERK1/2 inhibitors has lagged behind RAF and MEK inhibitors, appreciation that ERK reactivation is a major basis for acquired resistance to RAF and MEK inhibitors prompted renewed interest. Several selective ERK1/2 inhibitors are now in clinical evaluation (NCT02857270, NCT03520075, NCT03415126 and NCT03698994).
The PI3K-AKT-mTOR pathway is complementary to the RAF-MEK-ERK cascade and its targeting has been an attractive strategy in RAS mutant cancers. However, in some tissues this pathway seems to play a lesser role in RAS-dependent cancer growth compared to the MAPK pathway. Inhibition of the PI3K pathway, either directly or indirectly, often leads to the upregulation of the MAPK pathway. Disappointingly, the combination of PI3K-AKT-mTOR inhibitors and RAF-MEK inhibitors, despite impressive activities in preclinical mouse models, has been limited by toxicity in cancer patients. Nonetheless, there is hope that some of these challenges will be overcome with more selective, isoform-specific inhibitors. For example, combinations of KRASG12C inhibitors with PI3K-AKT-mTOR inhibitors should be better tolerated because they would not block activation of ERK MAPK signaling by WT RAS33. In a complementary manner, recent reports suggest that RASGEF inhibitors would effectively act as RAS-dependent PI3K and MAPK inhibitors through the impairment of their activation by WT RAS34. As progress is made in our understanding of the complex compensatory signaling mechanisms that limit the effectiveness of single agent inhibition of RAS effector signaling, it is hoped that combination inhibitor strategies will be the key to success for therapeutic targeting of the RAF and PI3K effector signaling networks.
In recent years, efforts to find synthetic lethal interactors with mutant KRAS have not uncovered any ‘silver bullets’, as was hoped for at the onset of these studies35. In search of genes that selectively kill MT RAS-driven cells, these efforts largely depend on genetic loss-of-function screens, such as RNAi or more recently CRISPRi/CRISPR-KO, in both MT RAS and WT RAS cells. Project DRIVE, collecting perhaps the largest set of cell line dependencies following genome-wide shRNA knock-down, indicated that some of the commonly used MT KRAS cell line models were not dependent on continued expression of KRAS36. This observation reminds us that not all MT-RAS driven cells exhibit the same oncogene dependencies, and that synthetic lethal interactions with MT-RAS are highly dependent on specific contexts of tissue, cell type, RAS allele, and RAS mutation.
Unlike the restricted number of substrates of RAF and MEK, hundreds of validated/putative ERK substrates have been identified. Of these substrates, the MYC transcription factor, another ‘undruggable’ cancer target, is arguably the critical component of ERK-dependent cancer growth37. The critical interdependency of RAS and MYC in oncogenesis is well-established. While many of the efforts to target MYC have centered on blocking MYC gene transcription, a recent study demonstrated the critical role of KRAS in maintaining MYC protein stability, through protein kinases that have yet to be identified38. Motivated by this, Blake et al. (2019) characterized and applied a MYC degradation screen in PDAC cells to identify protein kinase modulators of MYC protein stability39. Using this screen, they identified cyclin-dependent kinase 9 (CDK9) and determined that CDK9 regulated MYC protein expression at both the transcriptional and post-translational levels (Figure 2). With KRAS dependent on MYC to drive cancer growth, disruption of the mechanisms by which KRAS maintains MYC protein stability represents a tractable therapeutic strategy.
As with conventional cytotoxic oncology drugs, where combinations are the rule (e.g., FOLFIRINOX for PDAC), cocktails of signaling inhibitors will likely be required to effectively target RAS effector signaling. Sun et al. (2017) identified an initially surprising synergistic combination in NSCLC of MEK inhibition and PARP inhibition40. Their finding that inhibition of MEK increased expression and reliance on PARP then enabled rational targeting of this combination. Similarly, Chen et al. (2018) reported the synergistic combination of a pan-RAF inhibitor with a CDK4/6 inhibitor, where RAF inhibition could ablate the upregulation of cyclin D1 that occurs upon CDK4/6 inhibition41. More recently, two studies independently determined that ERK MAPK inhibition increased the dependency of KRAS-mutant PDAC on autophagy, leading to clinical evaluation of combination MEK and autophagy inhibition for PDAC42,43. These and other findings suggest that effective signaling inhibitor combinations may not always be logically deduced a priori, but once identified, can be mechanistically understood and exploited.
Conclusions and future perspectives
The RAS research field and the pharmaceutical industry have struggled mightily for nearly four decades in search of an elusive anti-RAS therapy. At times, the prospects seemed dim, with past failures creating a perception that RAS is undruggable. However, recent findings reveal promising new directions for these efforts. The advancement of KRASG12C-selective inhibitors to early clinical promise has stimulated considerable excitement that at long last RAS can be targeted. For other RAS mutants, combination strategies that target components upstream or downstream of RAS might open new therapeutic windows. The long history of developing RAS-targeted therapeutics has seen ups and downs, twists and turns, but is ultimately heading towards effective therapeutic strategies.
Acknowledgments:
We thank Adrienne D. Cox for critical comments on the manuscript.
Funding: Related work by C.J.D. is funded by the National Cancer Institute (NCI) (CA42978, CA199235, CA196510 and CA203657), the Department of Defense, the Lustgarten Foundation and the Pancreatic Cancer Action Network/AACR; work by C.A.S. is supported by the NCI (T32CA009156 and F32CA232529).
Footnotes
Competing interests: C.J.D. was a consultant for Eli Lilly and has also consulted for and received funding from Mirati Therapeutics and Deciphera Pharmaceuticals.
REFERENCES AND NOTES
- 1.Cox AD, Fesik SW, Kimmelman AC, Luo J & Der CJ Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov 13, 828–851 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD & Jemal A Cancer statistics, 2019. CA. Cancer J. Clin 69, 7–34 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Ostrem JM, Peters U, Sos ML, Wells JA & Shokat KM K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scheffzek K, Ahmadian MR, Kabsch W, Wiesmüller L, Lautwein A, Schmitz F & Wittinghofer A The Ras-RasGAP Complex: Structural Basis for GTPase Activation and Its Loss in Oncogenic Ras Mutants. Science 277, 333–339 (1997). [DOI] [PubMed] [Google Scholar]
- 5.Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S & Westover KD Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res 13, 1325–1335 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Fakih M, O’Neil B, Price TJ, Falchook GS, Desai J, Kuo J, Govindan R, Rasmussen E, Morrow PKH, Ngang J, Henary HA & Hong DS Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRASG12C inhibitor, in advanced solid tumors. J. Clin. Oncol 37, 3003–3003 (2019). [Google Scholar]
- 7.Lou K, Steri V, Ge AY, Hwang YC, Yogodzinski CH, Shkedi AR, Choi ALM, Mitchell DC, Swaney DL, Hann B, Gordan JD, Shokat KM & Gilbert LA KRASG12C inhibition produces a driver-limited state revealing collateral dependencies. Sci. Signal 12, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Janes MR, Zhang J, Li L-S, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, Feng J, Chen JH, Li S, Li S, Long YO, Thach C, Liu Y, Zarieh A, Ely T, Kucharski JM, Kessler LV, Wu T, Yu K, Wang Y, Yao Y, Deng X, Zarrinkar PP, Brehmer D, Dhanak D, Lorenzi MV, Hu-Lowe D, Patricelli MP, Ren P & Liu Y Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 172, 578–589 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Mainardi S, Mulero-Sánchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, Lieftink C, Steinberg JD, Wit N. de, Gonçalves-Ribeiro S, Nadal E, Bardelli A, Villanueva A & Bernards R SHP2 is required for growth of KRAS -mutant non-small-cell lung cancer in vivo. Nat. Med 24, 961–967 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, Görgülü K, Dantes Z, Wörmann SM, Diakopoulos KN, Karpathaki AF, Kowalska M, Kaya-Aksoy E, Song L, Laan E. A. Z. van der, López-Alberca MP, Nazaré M, Reichert M, Saur D, Erkan MM, Hopt UT, Sainz B, Birchmeier W, Schmid RM, Lesina M & Algül H Mutant KRAS -driven cancers depend on PTPN11 /SHP2 phosphatase. Nat. Med 24, 954–960 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Knudsen ES & Witkiewicz AK The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 3, 39–55 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, Gunn S, Smetzer L, Mays TA, Kaiser B, Wick MJ, Alvarez C, Cavazos A, Mangold GL & Patnaik A The Clinical Effect of the Dual-Targeting Strategy Involving PI3K/AKT/mTOR and RAS/MEK/ERK Pathways in Patients with Advanced Cancer. Clin. Cancer Res 18, 2316–2325 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M, Gollner A, Covini D, Fischer S, Gerstberger T, Gmaschitz T, Goodwin C, Greb P, Häring D, Hela W, Hoffmann J, Karolyi-Oezguer J, Knesl P, Kornigg S, Koegl M, Kousek R, Lamarre L, Moser F, Munico-Martinez S, Peinsipp C, Phan J, Rinnenthal J, Sai J, Salamon C, Scherbantin Y, Schipany K, Schnitzer R, Schrenk A, Sharps B, Siszler G, Sun Q, Waterson A, Wolkerstorfer B, Zeeb M, Pearson M, Fesik SW & McConnell DB Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci 116, 15823–15829 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hillig RC, Sautier B, Schroeder J, Moosmayer D, Hilpmann A, Stegmann CM, Werbeck ND, Briem H, Boemer U, Weiske J, Badock V, Mastouri J, Petersen K, Siemeister G, Kahmann JD, Wegener D, Böhnke N, Eis K, Graham K, Wortmann L, Nussbaum F. von & Bader B Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 interaction. Proc. Natl. Acad. Sci 116, 2551–2560 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spencer-Smith R, Koide A, Zhou Y, Eguchi RR, Sha F, Gajwani P, Santana D, Gupta A, Jacobs M, Herrero-Garcia E, Cobbert J, Lavoie H, Smith M, Rajakulendran T, Dowdell E, Okur MN, Dementieva I, Sicheri F, Therrien M, Hancock JF, Ikura M, Koide S & O’Bryan JP Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol 13, 62–68 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guillard S, Kolasinska-Zwierz P, Debreczeni J, Breed J, Zhang J, Bery N, Marwood R, Tart J, Overman R, Stocki P, Mistry B, Phillips C, Rabbitts T, Jackson R & Minter R Structural and functional characterization of a DARPin which inhibits Ras nucleotide exchange. Nat. Commun 8, 16111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biancucci M, Minasov G, Banerjee A, Herrera A, Woida PJ, Kieffer MB, Bindu L, Abreu-Blanco M, Anderson WF, Gaponenko V, Stephen AG, Holderfield M & Satchell KJF The bacterial Ras/Rap1 site-specific endopeptidase RRSP cleaves Ras through an atypical mechanism to disrupt Ras-ERK signaling. Sci. Signal 11, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roock WD, Jonker DJ, Nicolantonio FD, Sartore-Bianchi A, Tu D, Siena S, Lamba S, Arena S, Frattini M, Piessevaux H, Cutsem EV, O’Callaghan CJ, Khambata-Ford S, Zalcberg JR, Simes J, Karapetis CS, Bardelli A & Tejpar S Association of KRAS p.G13D Mutation With Outcome in Patients With Chemotherapy-Refractory Metastatic Colorectal Cancer Treated With Cetuximab. JAMA 304, 1812–1820 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Rowland A, Dias MM, Wiese MD, Kichenadasse G, McKinnon RA, Karapetis CS & Sorich MJ Meta-analysis comparing the efficacy of anti-EGFR monoclonal antibody therapy between KRAS G13D and other KRAS mutant metastatic colorectal cancer tumours. Eur. J. Cancer Oxf. Engl 1990 55, 122–130 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Segelov E, Thavaneswaran S, Waring PM, Desai J, Robledo KP, Gebski VJ, Elez E, Nott LM, Karapetis CS, Lunke S, Chantrill LA, Pavlakis N, Khasraw M, Underhill C, Ciardiello F, Jefford M, Wasan H, Haydon A, Price TJ, van Hazel G, Wilson K, Simes J & Shapiro JD Response to Cetuximab With or Without Irinotecan in Patients With Refractory Metastatic Colorectal Cancer Harboring the KRAS G13D Mutation: Australasian Gastro-Intestinal Trials Group ICECREAM Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 34, 2258–2264 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Haigis KM KRAS Alleles: The Devil Is in the Detail. Trends Cancer 3, 686–697 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McFall T, Diedrich JK, Mengistu M, Littlechild SL, Paskvan KV, Sisk-Hackworth L, Moresco JJ, Shaw AS & Stites EC A Systems Mechanism for KRAS Mutant Allele Specific Responses to Targeted Therapy. Sci. Signal 12, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou B, Der CJ & Cox AD The role of wild type RAS isoforms in cancer. Semin. Cell Dev. Biol 58, 60–69 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim K-H, Ancrile BB, Kashatus DF & Counter CM Tumour maintenance is mediated by eNOS. Nature 452, 646 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grabocka E, Pylayeva-Gupta Y, Jones MJ, Lubkov V, Yemanaberhan E, Taylor L, Jeng HH & Bar-Sagi D Wild-type H- and N-Ras promote mutant K-Ras driven tumorigenesis by modulating the DNA damage response. Cancer Cell 25, 243–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boykevisch S, Zhao C, Sondermann H, Philippidou P, Halegoua S, Kuriyan J & Bar-Sagi D Regulation of Ras Signaling Dynamics by Sos-Mediated Positive Feedback. Curr. Biol 16, 2173–2179 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Johnson CW, Lin Y-J, Reid D, Parker J, Pavlopoulos S, Dischinger P, Graveel C, Aguirre AJ, Steensma M, Haigis KM & Mattos C Isoform-Specific Destabilization of the Active Site Reveals a Molecular Mechanism of Intrinsic Activation of KRas G13D. Cell Rep 28, 1538–1550 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan J, Roy S, Apolloni A, Lane A & Hancock JF Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J. Biol. Chem 273, 24052–24056 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Baltanás FC, Pérez-Andrés M, Ginel-Picardo A, Diaz D, Jimeno D, Liceras-Boillos P, Kortum RL, Samelson LE, Orfao A & Santos E Functional redundancy of Sos1 and Sos2 for lymphopoiesis and organismal homeostasis and survival. Mol. Cell. Biol 33, 4562–4578 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lito P, Rosen N & Solit DB Tumor adaptation and resistance to RAF inhibitors. Nat. Med 19, 1401–1409 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Chapman PB, Solit DB & Rosen N Combination of RAF and MEK inhibition for the treatment of BRAF-mutated melanoma: feedback is not encouraged. Cancer Cell 26, 603–604 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Herrero A, Pinto A, Colón-Bolea P, Casar B, Jones M, Agudo-Ibáñez L, Vidal R, Tenbaum SP, Nuciforo P, Valdizán EM, Horvath Z, Orfi L, Pineda-Lucena A, Bony E, Keri G, Rivas G, Pazos A, Gozalbes R, Palmer HG, Hurlstone A & Crespo P Small Molecule Inhibition of ERK Dimerization Prevents Tumorigenesis by RAS-ERK Pathway Oncogenes. Cancer Cell 28, 170–182 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Misale S, Fatherree JP, Cortez E, Li C, Bilton S, Timonina D, Myers DT, Lee D, Gomez-Caraballo M, Greenberg M, Nangia V, Greninger P, Egan RK, McClanaghan J, Stein GT, Murchie E, Zarrinkar PP, Janes MR, Li L-S, Liu Y, Hata AN & Benes CH KRAS G12C NSCLC Models Are Sensitive to Direct Targeting of KRAS in Combination with PI3K Inhibition. Clin. Cancer Res 25, 796–807 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Murillo MM, Rana S, Spencer-Dene B, Nye E, Stamp G & Downward J Disruption of the Interaction of RAS with PI 3-Kinase Induces Regression of EGFR-Mutant-Driven Lung Cancer. Cell Rep 25, 3545–3553 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Downward J RAS Synthetic Lethal Screens Revisited: Still Seeking the Elusive Prize? Clin. Cancer Res 21, 1802–1809 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McDonald ER, Weck A. de, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E, Gampa K, Golji J, Kao I, Li L, Megel P, Perkins TA, Ramadan N, Ruddy DA, Silver SJ, Sovath S, Stump M, Weber O, Widmer R, Yu J, Yu K, Yue Y, Abramowski D, Ackley E, Barrett R, Berger J, Bernard JL, Billig R, Brachmann SM, Buxton F, Caothien R, Caushi JX, Chung FS, Cortés-Cros M, deBeaumont RS, Delaunay C, Desplat A, Duong W, Dwoske DA, Eldridge RS, Farsidjani A, Feng F, Feng J, Flemming D, Forrester W, Galli GG, Gao Z, Gauter F, Gibaja V, Haas K, Hattenberger M, Hood T, Hurov KE, Jagani Z, Jenal M, Johnson JA, Jones MD, Kapoor A, Korn J, Liu J, Liu Q, Liu S, Liu Y, Loo AT, Macchi KJ, Martin T, McAllister G, Meyer A, Mollé S, Pagliarini RA, Phadke T, Repko B, Schouwey T, Shanahan F, Shen Q, Stamm C, Stephan C, Stucke VM, Tiedt R, Varadarajan M, Venkatesan K, Vitari AC, Wallroth M, Weiler J, Zhang J, Mickanin C, Myer VE, Porter JA, Lai A, Bitter H, Lees E, Keen N, Kauffmann A, Stegmeier F, Hofmann F, Schmelzle T & Sellers WR Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 170, 577–592 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Farrell AS, Joly MM, Allen-Petersen BL, Worth PJ, Lanciault C, Sauer D, Link J, Pelz C, Heiser LM, Morton JP, Muthalagu N, Hoffman MT, Manning SL, Pratt ED, Kendsersky ND, Egbukichi N, Amery TS, Thoma MC, Jenny ZP, Rhim AD, Murphy DJ, Sansom OJ, Crawford HC, Sheppard BC & Sears RC MYC regulates ductal-neuroendocrine lineage plasticity in pancreatic ductal adenocarcinoma associated with poor outcome and chemoresistance. Nat. Commun 8, 1–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vaseva AV, Blake DR, Gilbert TSK, Ng S, Hostetter G, Azam SH, Ozkan-Dagliyan I, Gautam P, Bryant KL, Pearce KH, Herring LE, Han H, Graves LM, Witkiewicz AK, Knudsen ES, Pecot CV, Rashid N, Houghton PJ, Wennerberg K, Cox AD & Der CJ KRAS Suppression-Induced Degradation of MYC Is Antagonized by a MEK5-ERK5 Compensatory Mechanism. Cancer Cell 34, 807–822 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blake DR, Vaseva AV, Hodge RG, Kline MP, Gilbert TSK, Tyagi V, Huang D, Whiten GC, Larson JE, Wang X, Pearce KH, Herring LE, Graves LM, Frye SV, Emanuele MJ, Cox AD & Der CJ Application of a MYC degradation screen identifies sensitivity to CDK9 inhibitors in KRAS-mutant pancreatic cancer. Sci. Signal 12, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, Graham SH, Carcillo JA, Szabó C & Clark RSB Intra-mitochondrial Poly(ADP-ribosylation) Contributes to NAD+ Depletion and Cell Death Induced by Oxidative Stress. J. Biol. Chem 278, 18426–18433 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Chen S-H, Gong X, Zhang Y, Van Horn RD, Yin T, Huber L, Burke TF, Manro J, Iversen PW, Wu W, Bhagwat SV, Beckmann RP, Tiu RV, Buchanan SG & Peng S-B RAF inhibitor LY3009120 sensitizes RAS or BRAF mutant cancer to CDK4/6 inhibition by abemaciclib via superior inhibition of phospho-RB and suppression of cyclin D1. Oncogene 37, 821–832 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM, George SD, Tomar G, Papke B, Hobbs GA, Yan L, Hayes TK, Diehl JN, Goode GD, Chaika NV, Wang Y, Zhang G-F, Witkiewicz AK, Knudsen ES, Petricoin EF, Singh PK, Macdonald JM, Tran NL, Lyssiotis CA, Ying H, Kimmelman AC, Cox AD & Der CJ Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med 25, 628–640 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT, Yap JT, Burrell LD, Lum DH, Whisenant JR, Gilcrease GW, Cavalieri CC, Rehbein KM, Cutler SL, Affolter KE, Welm AL, Welm BE, Scaife CL, Snyder EL & McMahon M Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med 25, 620–627 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]