

Abstract
Neonatal hypoxia-ischemia (HI) is a common cause of brain injury in infants. Acute kidney injury frequently occurs after birth asphyxia and is associated with adverse outcome. Treatment with acetyl-L-carnitine (ALCAR) after HI protects brain and improves outcome. Rat pups underwent carotid ligation and 75 min hypoxia on postnatal day 7 to determine effects of HI on kidney which is understudied in this model. HI + ALCAR pups were treated at 0, 4 and 24 hr after HI. The organic cation/carnitine transporter 2 (OCTN2), transports ALCAR and functions to reabsorb carnitine and acylcarnitines from urine. At 24 hours after injury OCTN2 levels were significantly decreased in kidney from HI pups, 0.80 ± 0.04 (p <0.01), compared to sham controls 1.03 ± 0.04, and HI + ALCAR pups 1.11 ± 0.06. The effect of HI on the level of pyruvate dehydrogenase (PDH) was determined since kidney has high energy requirements. At 24 hr after HI, kidney PDH/β-actin ratios were significantly lower in HI pups, 0.98 ± 0.05 (p <0.05); compared to sham controls 01.16 ± 0.06, and HI + ALCAR pups 1.24 ± 0.03. Treatment of pups with ALCAR after HI injury prevented the decrease in renal OCTN2 and PDH levels at 24 hr after injury. Protection of PDH and OCTN2 after HI would improve energy metabolism in kidney, maintain tissue carnitine levels and overall carnitine homeostasis which is essential for neonatal health.
Keywords: hypoxia-ischemia, acetyl-L-carnitine, kidney, neonatal, OCTN2 transporter, pyruvate dehydrogenase
Introduction
Neonatal hypoxic ischemic encephalopathy (HIE) is a leading cause of neonatal brain damage resulting from birth asphyxia which occurs at a rate of approximately 1.5/1000 live births in developed nations with substantially higher incidence in developing nations (Kurinczuk et al., 2010). Mild therapeutic hypothermia remains the primary treatment option for infants with HIE, but death or significant neurological disability still occurs in 40–50% of treated infants (Higgins et al., 2011). Male infants are more likely to suffer from birth complication which result in HIE and suffer from worse outcomes after insult (Hill and Fitch, 2012).
A number of recent studies have documented the high prevalence of acute kidney injury in infants with HIE, and association of kidney injury with poor neurologic outcome and increased risk of mortality (Alaro et al., 2014; Hadzimuratovic et al., 2014; Sarkar et al., 2014; Shellhaas et al., 2015). Up to 56% of infants with birth asphyxia also have acute kidney injury (Durkan and Alexander, 2011). Recent studies report that 38% of the neonates treated with hypothermia still exhibit acute kidney injury, and asphyxiated neonates with acute kidney injury have poorer outcomes and increased mortality (Aggarwal et al., 2005; Selewski et al., 2013; Shellhaas et al., 2015). Infants with HI and acute kidney injury required longer mechanical ventilation and had longer stays in the NICU (Selewski et al., 2013). An acute kidney injury in the neonatal period can result in residual renal dysfunction and a greater risk of developing chronic kidney disease later in life (Durkan and Alexander, 2011). There is very little research in models of neonatal hypoxia ischemia assessing effects on kidney. Xu et al. (Xu et al., 2017) recently reported swelling in renal cortex, membrane alterations in glomerular barriers, and changes in protein levels in kidney from rat pups at 24 hours after HI. Despite the prevalence of kidney injury as a secondary injury in infants with HIE and the significant long-term implications of such damage, few studies have assessed changes in kidney or evaluated potential treatments for kidney injury in infants with HIE.
Acute ATP shortages and damage to energy metabolism pathways due to oxidative stress and other mechanisms contribute to both HIE (Robertson et al., 2009) and acute kidney injury (Sharfuddin and Molitoris, 2011). Pyruvate dehydrogenase (PDH), the key enzyme linking glycolysis to the tricarboxylic acid (TCA) cycle, is particularly vulnerable to damage from oxidative stress (Fiskum et al., 2004). Loss of PDH would prevent glucose from being incorporated into oxidative metabolism and exacerbate the initial ATP shortage that occurs after HI injury.
Acetyl-L-carnitine (ALCAR) is a naturally occurring ester of carnitine which consists of an acetyl moiety bound to an L-carnitine moiety and is naturally synthesized in low levels by the liver (Aureli et al., 1999). It enters cells primarily through the sodium-dependent transport protein organic carnitine/cation transporter 2 (OCTN2), one of a family of organic cation transporters (Wu et al., 1999), where it is metabolized to acetyl CoA and L-carnitine as shown in Figure 1. Treatment with ALCAR is neuroprotective in animal models of brain injury including neonatal hypoxia ischemia (Demarest et al., 2016a; Demarest et al., 2016b; Demarest et al., 2016c; Ferreira and McKenna, 2017; Tang et al., 2017; Xu et al., 2015), global cerebral ischemia (Zanelli et al., 2005), and pediatric traumatic brain injury (Scafidi et al., 2010b). Clinical studies show protection in a variety of conditions including Alzheimer’s disease (Parnetti et al., 1993), depression (Wang et al., 2014), peripheral neuropathy (Sima et al., 2005), and improved neurological function in hepatic encephalopathy (Malaguarnera et al., 2011). Our group has demonstrated that administration of ALCAR after HI in the postnatal day 7 rat pup decreased lesion size, improved mitochondrial function and improved motor function and learning (Demarest et al., 2016a; Demarest et al., 2016b; Demarest et al., 2016c; Tang et al., 2017; Xu et al., 2015).
Figure 1.
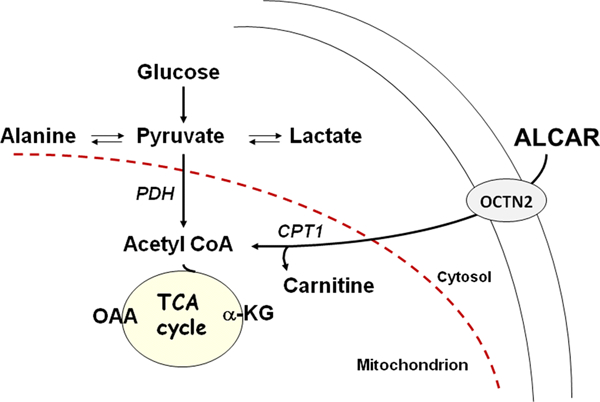
ALCAR enters kidney via the OCTN2 transporter. In the mitochondria ALCAR can be cleaved by the enzyme carnitine acetyltransferase (CAT) to carnitine and acetyl CoA which can be metabolized to provide energy. The OCTN2 transporter in kidney releases carnitine into the circulation thus helping to maintain carnitine status.
It is critically important that potential treatments for HIE do not harm the body or exacerbate other complications of hypoxia ischemia, such as renal injury. The few studies that have been conducted show that ALCAR reduced oxidative damage in kidney in a carbon tetrachloride model of oxidative stress (Annadurai et al., 2011) and protected renal function against cisplatin-induced caspase-3 activity, apoptosis, and inflammation (Tufekci et al., 2009). ALCAR may protect against injury through the conversion of its acetyl moiety to acetyl-CoA and serve as an alternative metabolic substrate when glucose metabolism is impaired (Aureli et al., 1998; Aureli et al., 1999; Scafidi et al., 2010a). L-carnitine facilitates uptake of free fatty acids into the mitochondria for β-oxidation of free fatty acids, potentially protecting against oxidative stress (Calabrese and Rizza, 1999; Jones et al., 2010). These characteristics of ALCAR suggest that it could be protective against both HIE and related kidney injury.
OCTN2, the transporter for ALCAR, is found throughout the kidneys, but is most abundant in renal tubular epithelial cells, where the transporter functions to reabsorb L-carnitine and acylcarnitines from urine after glomerular filtration to maintain the plasma concentration of L-carnitine (Tamai et al., 2001; Wu et al., 1999). It has been reported that the increases in serum L-carnitine during early development were positively correlated with the postnatal increases in OCTN2 expression in kidney (Ling et al., 2012). A secondary carnitine deficiency can occur in infants with severe neonatal asphyxia (Bayes et al., 2001; Cam et al., 2005; Ezgü et al., 2004).
Despite the prevalence of research regarding the protective effects of ALCAR on a variety of neurological and renal disorders, little research has involved the interaction between injury, ALCAR treatment, and its transporter, OCTN2. Few studies have discussed the effect of renal injury on metabolic changes and PDH in particular. Our study sought to determine the effect of neonatal hypoxic ischemic injury and subsequent ALCAR treatment on levels of the OCTN2 transporter and PDH enzymes in kidney.
2. Materials and Methods
2.1. Animals
This study was approved by the University of Maryland, Baltimore, Animal Care and Use Committee. All care and handling of rats were in compliance with the National Institutes of Health guidelines. Timed-pregnant female Sprague–Dawley rats were obtained from Charles River Laboratories (Frederick, MD, DE, USA) and were housed in the central animal facility.
2.2. Surgery and Hypoxia-Ischemia
Seven day old pups were anesthetized with 3% isoflurane for induction (and 1.5% for maintenance) and underwent a modification of the Rice-Vanucci model of HI (right carotid artery ligation, 75 minutes 8% oxygen at 37°C), or sham surgery (Rice et al., 1981; Tang et al., 2017; Xu et al., 2015). After surgery pups were placed in an open jar in a 37°C water bath for 25 min to recover. Pups were returned to the dam for 60 minutes before the hypoxia. Hypoxia was performed in pre-warmed glass jars (2 pups per jar) in a 37°C water bath. Jars were sealed, flushed with warmed gas mixture (8% oxygen and 92% nitrogen) at 1 liter/minute and placed in a 37°C water bath for 75 minutes. After hypoxia the pups recovered at 37°C in room air for 2 hours prior to being returned to the dam. Sham treated controls were anesthetized for 4.5 minutes and received an incision without any artery manipulation or hypoxia. Sham control pups were taken away from the dam for the same time as the HI pups, but did not undergo the operation or hypoxia. ALCAR treated animals received 3 doses of ALCAR (100 mg/kg in 8% sodium bicarbonate to control pH) subcutaneously immediately after HI, 4 hr after, and 24 hr after HI (Scafidi et al., 2010b). Control animals were injected with saline. ALCAR and saline treated animals were euthanized two hours after the final injection. After rat pups were euthanized the kidneys were removed, flash frozen in liquid nitrogen, and stored at −80°C until usage.
2.3. Tissue Preparation
Renal cortex tissue samples were taken on dry ice, minced and placed in 400ul immunoprecipitation assay lysis buffer (Cell Signaling Technology, Danvers, MA) supplemented with a cocktail of protease and phosphatase inhibitors (Roche Diagnostic, Indianapolis, IN) on ice for 25 minutes to begin homogenization. Tissue samples were fully homogenized by sonication and centrifuged for 10 minutes at 10,000 × g to remove nuclear and cellular debris. The supernatant was removed from the pellet and aliquots frozen at −80°C until further use for western blots and protein determination. Protein concentrations were determined by the Pierce BCA microreagent assay (Smith et al., 1985).
2.4. Gel Electrophoresis and Transfer
Samples were supplemented with 10 μl 4x lithium dodecyl sulfate sample buffer (Invitrogen, Carlsbad, CA) and deionized water to reach a desired protein concentration of 30μg/40μl solution. One lane in each gel was used for a combined protein standard (Novex® Sharp Pre-Stained Protein Standard, Invitrogen Carlsbad, CA) and biotinylated protein molecular weight ladder (Cell Signaling Technology, Danvers, MA). Tissue samples were incubated at 37° C for 15 minutes to prepare for electrophoresis of membrane proteins (Hanu et al., 2000) before being loaded onto a 4–12% polyacrylamide gel buffered with Bis-Tris-HCl, pH 6.4 and subjected to gel electrophoresis with MOPS SDS running buffer (Invitrogen, Carlsbad, CA) for 120 minutes at 120V on ice. The gel was then transferred to a PVDF membrane in NuPAGE® Transfer Buffer (Invitrogen, Carlsbad, CA) for 60 minutes at 30V.
2.5. Western Blots
The membranes were blocked with 5% skim milk for 1 hr at room temperature prior to incubation with primary antibody. The rabbit polyclonal anti-OCTN2 primary antibody (Alpha Diagnostic, San Antonio, TX) was used at concentrations of 2μg/ml in 2.5% skim milk overnight at 4°C. Following primary antibody exposure, the membranes were washed in TBST (2.42g Trizma-HCl, 8.01g NaCl, 0.1% Tween20, pH 7.6) before being incubated in a donkey anti-rabbit IgG Horseradish Peroxidase linked F(ab’)2 secondary antibody (GE Healthcare UK, Little Chalfont, Buckinghamshire, UK) for 1.5 hours at a 1/3000 dilution in 2.5% skim milk at room temperature. For pyruvate dehydrogenase (PDH) the membranes were incubated with a mouse monoclonal anti-PDH E1-α subunit primary antibody (Abcam, Cambridge, MA) at concentrations of 2μg/ml in 2.5% skim milk overnight at 4°C. Following primary antibody exposure, the membrane was washed in TBST as described above and incubated in a d goat anti-mouse IgG (H+L) HRP conjugate (Biorad, US) for 1.5 hours at a 1/3000 dilution in 2.5% skim milk at room temperature. After washing, the membranes were treated with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, IL) and exposed with an aperture of .95 on a Multilmage II machine (Alpha-Innotech). After exposure, the membrane was stored at 4°C until further analysis for β-actin.
For the β-actin Western blots the membranes were stripped with Restore Western Blot Stripping Buffer (Thermo Scientific, Rockford, IL) for 15 minutes, washed, re-blocked in 5% skim milk for 1hr at room temperature, incubated with a mouse monoclonal anti-β-Actin primary antibody (SIGMA Aldrich, St. Louis, MO) at concentrations of 0.05μg/ml in 2.5% skim milk for 1hr at room temperature, washed again, and incubated with goat anti-mouse IgG (H+L) HRP conjugate (Biorad, US) at a 1/3000 concentration in 2.5% skim milk for 1hr at room temperature. The membranes were then treated with ECL and exposed at a 2.5 aperture on the MultiImage II machine.
2.6. Statistical Analysis
Data are presented as mean ± S.E.M. Statistical significance was determined by one way ANOVA followed by Bonferroni’s Multiple Comparison Test. A value of p < 0.05 was considered significant.
3. Results
3.1. Changes in OCTN2 from HI and ALCAR treatment
As shown in Figure 2, OCTN2 bands were observed at approximately 70–75 kDa, with an additional band at ~130 kDa that is likely a dimer or most postranslationally modified form of the protein. These two bands are consistent with the major bands of 80 and ~160 kD observed by Nagai et al. (Nagai et al., 2006) although the lighter band at 60 kDa which they observed was not visible in our blots. As shown in Figure 2, OCTN2 levels were significantly decreased at 24 hours after injury in kidney from animals subjected to HI compared to shams and HI + ALCAR pups, p <0.01. The HI pups treated with ALCAR had OCTN2 levels comparable to kidney from sham controls. OCTN2/β-actin ratios were control, 1.03 ± 0.04; HI + Saline, 0.80 ± 0.04 and HI + ALCAR 1.11 ± 0.06. Renal OCTN2 decreased after HI but pups treated with ALCAR did not exhibit this decrease.
Figure 2.
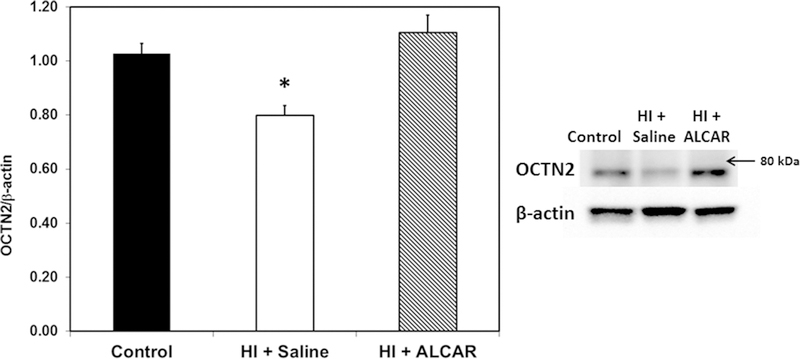
Ratio of OCTN2 transporter to β-actin in kidney from sham control (black bar), HI + saline (white bar) and HI + ALCAR (hatched bar) rat pups 24 h after HI. OCTN2 was significantly decreased in HI + saline pups. Treatment with ALCAR after HI led to significantly increased OCTN2 levels compared to HI + saline pups. Western blot of proteins is shown on the right. Values are mean ± SEM for 13 controls, 14 HI + saline and 6 HI + ALCAR pups. Data were analyzed by ANOVA and Bonferroni post hoc test. *p < 0.01
3.2. Changes in PDH protein from HI and ALCAR treatment
The PDH complex is comprised of several subunits. The target of the primary antibody used was the E1-α subunit of the complex, which has been shown to be vulnerable to oxidative damage (Richards et al., 2006). As shown in Figure 3, PDH bands were observed at 40 kDa. PDH E1-α levels in kidney from HI + saline pups were significantly lower than PDH levels in sham control pups at 24 hours after injury, p < 0.05. PDH levels in kidney from HI+ ALCAR pups were comparable to levels in sham controls, and higher than levels in HI + saline pups, p <0.01. PDH/β-actin ratios were 1.16 ± .06 in kidneys from sham animals, 0.98 ± .05 for HI + saline animals, and 1.24 ± 0.03 for HI + ALCAR animals. This suggests that HI may damage the E1-α subunit of PDH and that ALCAR may prevent this damage.
Figure 3.
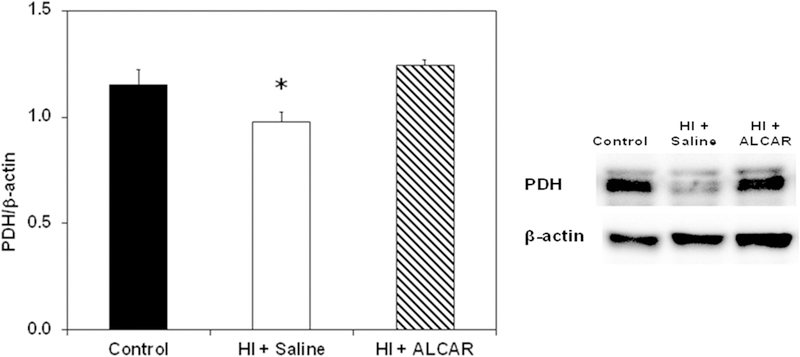
Ratio of pyruvate dehydrogenase (PDH) to β-actin in kidney from sham control (black bar), HI + saline (white bar) and HI + ALCAR (hatched bar) rat pups 24 h after HI. Western blot of proteins is shown on the right. The primary antibody was against the PDH E1-α subunit. PDH levels in HI + saline pups were different than control (p < 0.05) and HI + ALCAR (p < 0.01) pups. Values are mean ± SEM for 14 controls, 14 HI + saline and 6 HI + ALCAR pups. Data were analyzed by ANOVA and Bonferroni post hoc test
3.3. OCTN2 and PDH levels in male and female kidney
The ratios of PDH and OCTN2/β-actin were determined in kidney from both male and female rat pups. No significant sex differences were observed in either OCTN2 and PDH levels as shown in Figures 4 and 5, respectively. However, OCTN2 levels tended to be higher in kidney from rat pups treated with ALCAR after HI, compared to HI + saline animals (Fig. 4). PDH levels tended to be higher in kidney from male rat pups treated with ALCAR after HI, compared to male pups treated with saline after HI (Fig.5 ).
Figure 4.
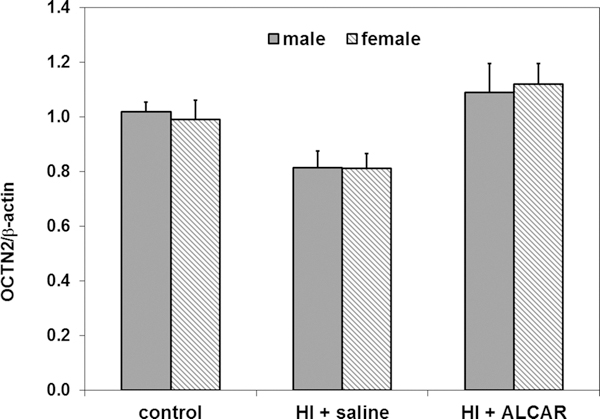
Ratio of OCTN2 transporter to β-actin in kidney from male (solid grey bars) and female (hatched grey bars) control, HI + saline and HI + ALCAR rat pups 24 hours after HI. There were no significant differences in OCTN2 levels in kidneys from male and female rats under any of the conditions studied. Values are means ± SEM for 5 male and 8 female controls, 5 male and 9 female HI + saline pups, and 3 male and 3 female HI + ALCAR pups. Data were analyzed by ANOVA.
Figure 5.
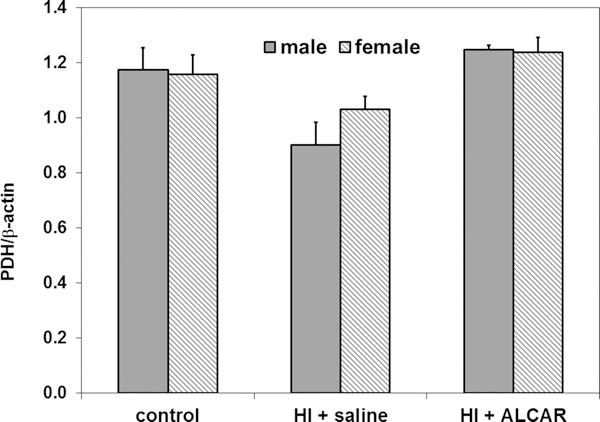
Ratio of PDH E1-α subunit to β-actin in kidney from male (solid grey bars) and female (hatched grey bars) control, HI + saline and HI + ALCAR rat pups 24 hours after HI. There were no significant differences in PDH levels in kidney from male and female rat pups under any of the conditions studied. Values are means ± SEM for 5 male and 9 female controls, 6 male and 8 female HI + saline pups, and 3 male and 3 female HI + ALCAR pups. Data were analyzed by ANOVA.
4. Discussion
The widely used Rice-Vannucci method of neonatal hypoxic ischemic brain injury was used in the current study (Rice et al., 1981; Tang et al., 2017; Xu et al., 2015). Although alterations in brain after injury have been widely studied in this model, to our knowledge there is only one report of changes in kidney after HI in this model (Xu et al., 2017). Xu et al. (Xu et al., 2017) recently reported swelling in renal cortex, membrane alterations in glomerular barriers, and changes in the expression of zonulin, occludin and aquaporin-4 in kidney from rat pups at 24 hours after severe HI injury, and some attenuation of damage by an intraperitoneal injection of melatonin after injury. The present study shows that a neonatal HI injury procedure that results in a moderate brain injury led to a decrease in renal proteins involved in carnitine homeostasis and energy metabolism. Treatment with ALCAR after HI injury, which attenuates brain injury and improves behavioral outcomes (Demarest et al., 2016a; Demarest et al., 2016b; Demarest et al., 2016c; Tang et al., 2017; Xu et al., 2015), prevented the loss of these key proteins in kidney. These results suggest that ALCAR, a proposed treatment for HIE, does not have an adverse effect on the kidney and thus could also be a potential treatment for kidney injury associated with neonatal HI.
A decrease in the renal OCTN2/β-actin ratio observed in HI + Saline animals 24 hours after injury is consistent with reports of decreased OCTN2 levels in placental cells following 48 hours of hypoxia (Chang et al., 2011; Rytting and Audus, 2007), however the mechanism leading to decreased OCTN2 levels is unclear. One possibility is through nitric oxide-mediated inhibition of OCTN2 expression. Schneider et al. (2011) found that renal ischemia-reperfusion in adult rats and the subsequent increase in inducible nitric oxide synthase generated nitric oxide decreased the expression and levels of OCT1 and OCT2, two other proteins in the organic cation transporter family which exhibit significant similarity to OCTN2 (Wu et al., 1998).
Decreased OCTN2 levels are clinically relevant because of the role of OCTN2 in maintaining carnitine homeostasis (Jones et al., 2010). Placental uptake of carnitine is needed to provide adequate levels during gestation. Thus, it is significant to note that OCTN2 levels were found to be profoundly decreased in human placental tissue from mothers with preeclampsia Chang et al., 2011). Normal placental explants had decreased OCTN2 levels after 48 hours of severe experimental hypoxia (Chang et al., 2011). In young rat pups OCTN2 levels are closely linked with serum L-carnitine levels (Ling et al., 2012). Several groups have observed decreased free carnitine levels and increased long chain acylcarnitine levels in infants with HIE (Bayes et al., 2001; Cam et al., 2005; Solberg et al., 2010). A decrease in renal OCTN2 levels leading to increased excretion of serum carnitine and/or decreased metabolism of acyl-carnitines could explain these differences
In the present study, animals treated with ALCAR after HI, the ratio of renal OCTN2/ β-actin was comparable to that of the control animals at 24 hours after injury, and significantly higher than the HI + saline animals. This suggests that OCTN2 levels respond to changes in levels of the substrate acetyl-L-carnitine. Furuichi et al. (2012) observed a similar effect in skeletal muscle, where OCTN2 levels increased in response to increased intracellular carnitine levels. Prolonged (two weeks) chemically induced depletion of carnitine led to severely decreased plasma and tissue carnitine levels and increased expression of OCTN2 in kidney (Schurch et al., 2010). In contrast, Gomez-Amores et al. (2003) reported a decrease in kidney OCTN2 levels after long term (six weeks) carnitine treatment. To our knowledge, the present study is the first to report an effect on OCTN2 transporter levels after ALCAR administration. Due to the antioxidant and possibly antiapoptotic effects of carnitine (Calandrella et al., 2010; Lango et al., 2001; Liu et al., 2012), it could be beneficial to maintain a high carnitine level in infants subjected to HI. The would be particularly important since infants are more vulnerable to carnitine deficiency due immature carnitine biosynthesis pathways (Bayes et al., 2001; Ferreira and McKenna, 2017). ALCAR treatment could increase the level of carnitine in two ways, as a direct external source of supplemental carnitine and through raising the renal OCTN2 level, thus increasing the efficiency of carnitine resorption.
Decreased levels of PDH in the present study suggest potential damage to renal energy metabolism in rat kidney at 24 hours after HI injury. Studies have identified damage to the mitochondrial complex I of the electron transport chain in adult kidney during hypoxia (Weinberg et al., 2000), but to our knowledge, this is the first to determine renal PDH levels in a model of neonatal HI. This enzyme, which catalyzes the oxidative decarboxylation of pyruvate to acetyl-CoA, NADH and CO2, is the essential link between glycolysis and oxidative glucose metabolism. The E1-α subunit, which was determined in this study, is the regulatory subunit of the PDH enzyme complex (Linn et al., 1969), and is vulnerable to damage by oxidative stress (Fiskum et al., 2004). Decreased PDH E1-α suggests the presence of oxidative damage in kidneys after HI. Damage to this metabolic enzyme has been shown in the brain as the result of global cerebral ischemia with hyperoxic reperfusion (Richards et al., 2006) and traumatic brain injury (Sharma et al., 2009).
ALCAR treatment holds potential for reducing the effects of HI and oxidative stress injury on renal metabolism. No studies have determined the effect of ALCAR treatment on metabolism in the kidneys. However, Annandurai et al. (2011) showed that ALCAR treatment after a model of oxidative stress reduced the level of lipid peroxidation and increased the amounts of antioxidants in kidney tissue. Another possible explanation for the protection of PDH levels after treatment with ALCAR is through the action of carnitine, which has been found to reverse depressed PDH activity and mitochondrial function in hypoxic myocardial cells, though the mechanism is not known (Yamada et al., 1990). These possibilities suggest that ALCAR does not just bypass PDH as an alternative source of energy; but may also play a role in protecting or restoring PDH-mediated glucose metabolism.
In summary, the results of the current study show that treatment with 100 mg/kg ALCAR after HI in the 7 day old rat pup prevented the loss of two key proteins in kidney at 24 hr after injury. These results, in conjunction with studies demonstrating that ALCAR decreases lesion size in brain (Tang et al., 2017; Xu et al., 2015), protects mitochondrial function (Demarest et al., 2016a; Xu et al., 2015) and improves behavioral outcome (Tang et al., 2017) provides additional evidence supporting the potential of ALCAR as a treatment for neonatal hypoxia-ischemia.
Acknowledgements
These studies were supported by NIH grant 5P01 HD016596 to M.C.M. This research was performed through the Gifted and Talented Intern/Mentor Program at Mount Hebron High School, Ellicott City, MD.
References
- Aggarwal A, Kumar P, Chowdhary G, Majumdar S, Narang A. (2005) Evaluation of renal functions in asphyxiated newborns. J Trop Pediatr 51:295–299 [DOI] [PubMed] [Google Scholar]
- Alaro D, Bashir A, Musoke R, Wanaiana L. (2014) Prevalence and outcomes of acute kidney injury in term neonates with perinatal asphyxia. Afr Health Sci 14:682–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annadurai T, Vigneshwari S, Thirukumaran R, Thomas PA, Geraldine P. (2011) Acetyl-l-carnitine prevents carbon tetrachloride-induced oxidative stress in various tissues of wistar rats. J Physiol Biochem 67:519–530 [DOI] [PubMed] [Google Scholar]
- Aureli T, Di Cocco ME, Puccetti C, Ricciolini R, Scalibastri M, Miccheli A, Manetti C, Conti F. (1998) Acetyl-l-carnitine modulates glucose metabolism and stimulates glycogen synthesis in rat brain. Brain Res 796:75–81 [DOI] [PubMed] [Google Scholar]
- Aureli T, Puccetti C, Di Cocco ME, Arduini A, Ricciolini R, Scalibastri M, Manetti C, Conti F. (1999) Entry of [(1,2–13c2)acetyl]-l-carnitine in liver tricarboxylic acid cycle and lipogenesis: A study by 13c nmr spectroscopy in conscious, freely moving rats. Eur J Biochem 263:287–293 [DOI] [PubMed] [Google Scholar]
- Bayes R, Campoy C, Goicoechea A, Peinado JM, Pedrosa T, Baena RM, Lopez C, Rivero M, Molina-Font JA. (2001) Role of intrapartum hypoxia in carnitine nutritional status during the early neonatal period. Early Hum Dev 65 Suppl:S103–110 [DOI] [PubMed] [Google Scholar]
- Calabrese V and Rizza V. (1999) Formation of propionate after short-term ethanol treatment and its interaction with the carnitine pool in rat. Alcohol 19:169–176 [DOI] [PubMed] [Google Scholar]
- Calandrella N, De Seta C, Scarsella G, Risuleo G. (2010) Carnitine reduces the lipoperoxidative damage of the membrane and apoptosis after induction of cell stress in experimental glaucoma. Cell Death Dis 1:e62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cam H, Yildirim B, Aydin A, Say A. (2005) Carnitine levels in neonatal hypoxia. J Trop Pediatr 51:106–108 [DOI] [PubMed] [Google Scholar]
- Chang TT, Shyu MK, Huang MC, Hsu CC, Yeh SY, Chen MR, Lin CJ. (2011) Hypoxia-mediated down-regulation of octn2 and pparalpha expression in human placentas and in bewo cells. Mol Pharm 8:117–125 [DOI] [PubMed] [Google Scholar]
- Demarest TG, Schuh RA, Waddell J, McKenna MC, Fiskum G. (2016a) Sex-dependent mitochondrial respiratory impairment and oxidative stress in a rat model of neonatal hypoxic-ischemic encephalopathy. J Neurochem 137:714–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarest TG, Schuh RA, Waite EL, Waddell J, McKenna MC, Fiskum G. (2016b) Sex dependent alterations in mitochondrial electron transport chain proteins following neonatal rat cerebral hypoxic-ischemia. J Bioenerg Biomembr [DOI] [PubMed] [Google Scholar]
- Demarest TG, Waite EL, Kristian T, Puche AC, Waddell J, McKenna MC, Fiskum G. (2016c) Sex-dependent mitophagy and neuronal death following rat neonatal hypoxia-ischemia. Neuroscience 335:103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkan AM and Alexander RT. (2011) Acute kidney injury post neonatal asphyxia. J Pediatr 158:e29–33 [DOI] [PubMed] [Google Scholar]
- Ferreira GC and McKenna MC. (2017) L-carnitine and acetyl-l-carnitine roles and neuroprotection in developing brain. Neurochem Res 42:1661–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskum G, Rosenthal RE, Vereczki V, Martin E, Hoffman GE, Chinopoulos C, Kowaltowski A. (2004) Protection against ischemic brain injury by inhibition of mitochondrial oxidative stress. J Bioenerg Biomembr 36:347–352 [DOI] [PubMed] [Google Scholar]
- Furuichi Y, Sugiura T, Kato Y, Takakura H, Hanai Y, Hashimoto T, Masuda K. (2012) Muscle contraction increases carnitine uptake via translocation of octn2. Biochem Biophys Res Commun 418:774–779 [DOI] [PubMed] [Google Scholar]
- Gomez-Amores L, Mate A, Vazquez CM. (2003) L-carnitine transport in kidney of normotensive, wistar-kyoto rats: Effect of chronic l-carnitine administration. Pharm Res 20:1133–1140 [DOI] [PubMed] [Google Scholar]
- Hadzimuratovic E, Skrablin S, Hadzimuratovic A, Dinarevic SM. (2014) Postasphyxial renal injury in newborns as a prognostic factor of neurological outcome. J Matern Fetal Neonatal Med 27:407–410 [DOI] [PubMed] [Google Scholar]
- Hanu R, McKenna M, O’Neill A, Resneck WG, Bloch RJ. (2000) Monocarboxylic acid transporters, mct1 and mct2, in cortical astrocytes in vitro and in vivo. Am J Physiol Cell Physiol 278:C921–930 [DOI] [PubMed] [Google Scholar]
- Higgins RD, Raju T, Edwards AD, Azzopardi DV, Bose CL, Clark RH, Ferriero DM, Guillet R, Gunn AJ, Hagberg H, Hirtz D, Inder TE, Jacobs SE, Jenkins D, Juul S, Laptook AR, Lucey JF, Maze M, Palmer C, Papile L, Pfister RH, Robertson NJ, Rutherford M, Shankaran S, Silverstein FS, Soll RF, Thoresen M, Walsh WF. (2011) Hypothermia and other treatment options for neonatal encephalopathy: An executive summary of the eunice kennedy shriver nichd workshop. J Pediatr 159:851–858 e851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CA and Fitch RH. (2012) Sex differences in mechanisms and outcome of neonatal hypoxia-ischemia in rodent models: Implications for sex-specific neuroprotection in clinical neonatal practice. Neurol Res Int 2012:867531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LL, McDonald DA, Borum PR. (2010) Acylcarnitines: Role in brain. Prog Lipid Res 49:61–75 [DOI] [PubMed] [Google Scholar]
- Kurinczuk JJ, White-Koning M, Badawi N. (2010) Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum Dev 86:329–338 [DOI] [PubMed] [Google Scholar]
- Lango R, Smolenski RT, Narkiewicz M, Suchorzewska J, Lysiak-Szydlowska W. (2001) Influence of l-carnitine and its derivatives on myocardial metabolism and function in ischemic heart disease and during cardiopulmonary bypass. Cardiovasc Res 51:21–29 [DOI] [PubMed] [Google Scholar]
- Ling B, Aziz C, Alcorn J. (2012) Systematic evaluation of key l-carnitine homeostasis mechanisms during postnatal development in rat. Nutr Metab (Lond) 9:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linn TC, Pettit FH, Reed LJ. (1969) Alpha-keto acid dehydrogenase complexes. X. Regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc Natl Acad Sci U S A 62:234–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Yan S, Ji C, Dai W, Hu W, Zhang W, Mei C. (2012) Metabolomic changes and protective effect of (l)-carnitine in rat kidney ischemia/reperfusion injury. Kidney Blood Press Res 35:373–381 [DOI] [PubMed] [Google Scholar]
- Malaguarnera M, Vacante M, Motta M, Giordano M, Malaguarnera G, Bella R, Nunnari G, Rampello L, Pennisi G. (2011) Acetyl-l-carnitine improves cognitive functions in severe hepatic encephalopathy: A randomized and controlled clinical trial. Metab Brain Dis 26:281–289 [DOI] [PubMed] [Google Scholar]
- Nagai K, Takikawa O, Kawakami N, Fukao M, Soma T, Oda A, Nishiya T, Hayashi M, Lu L, Nakano M, Kajita E, Fujita H, Miwa S. (2006) Cloning and functional characterization of a novel up-regulator, cartregulin, of carnitine transporter, octn2. Arch Biochem Biophys 452:29–37 [DOI] [PubMed] [Google Scholar]
- Parnetti L, Abate G, Bartorelli L, Cucinotta D, Cuzzupoli M, Maggioni M, Villardita C, Senin U. (1993) Multicentre study of l-alpha-glyceryl-phosphorylcholine vs st200 among patients with probable senile dementia of alzheimer’s type. Drugs Aging 3:159–164 [DOI] [PubMed] [Google Scholar]
- Rice JE 3rd, Vannucci RC, Brierley JB. (1981) The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9:131–141 [DOI] [PubMed] [Google Scholar]
- Richards EM, Rosenthal RE, Kristian T, Fiskum G. (2006) Postischemic hyperoxia reduces hippocampal pyruvate dehydrogenase activity. Free Radic Biol Med 40:1960–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CL, Scafidi S, McKenna MC, Fiskum G. (2009) Mitochondrial mechanisms of cell death and neuroprotection in pediatric ischemic and traumatic brain injury. Exp Neurol 218:371–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rytting E and Audus KL. (2007) Effects of low oxygen levels on the expression and function of transporter octn2 in bewo cells. J Pharm Pharmacol 59:1095–1102 [DOI] [PubMed] [Google Scholar]
- Sarkar S, Askenazi DJ, Jordan BK, Bhagat I, Bapuraj JR, Dechert RE, Selewski DT. (2014) Relationship between acute kidney injury and brain mri findings in asphyxiated newborns after therapeutic hypothermia. Pediatr Res 75:431–435 [DOI] [PubMed] [Google Scholar]
- Scafidi S, Fiskum G, Lindauer SL, Bamford P, Shi D, Hopkins I, McKenna MC. (2010a) Metabolism of acetyl-l-carnitine for energy and neurotransmitter synthesis in the immature rat brain. J Neurochem 114:820–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scafidi S, Racz J, Hazelton J, McKenna MC, Fiskum G. (2010b) Neuroprotection by acetyl-l-carnitine after traumatic injury to the immature rat brain. Dev Neurosci 32:480–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R, Meusel M, Betz B, Kersten M, Moller-Ehrlich K, Wanner C, Koepsell H, Sauvant C. (2011) Nitric oxide-induced regulation of renal organic cation transport after renal ischemia-reperfusion injury. Am J Physiol Renal Physiol 301:F997–F1004 [DOI] [PubMed] [Google Scholar]
- Selewski DT, Jordan BK, Askenazi DJ, Dechert RE, Sarkar S. (2013) Acute kidney injury in asphyxiated newborns treated with therapeutic hypothermia. J Pediatr 162:725–729 e721 [DOI] [PubMed] [Google Scholar]
- Sharfuddin AA and Molitoris BA. (2011) Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol 7:189–200 [DOI] [PubMed] [Google Scholar]
- Sharma P, Benford B, Li ZZ, Ling GS. (2009) Role of pyruvate dehydrogenase complex in traumatic brain injury and measurement of pyruvate dehydrogenase enzyme by dipstick test. J Emerg Trauma Shock 2:67–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shellhaas RA, Kushwaha JS, Plegue MA, Selewski DT, Barks JD. (2015) An evaluation of cerebral and systemic predictors of 18-month outcomes for neonates with hypoxic ischemic encephalopathy. J Child Neurol 30:1526–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sima AA, Calvani M, Mehra M, Amato A. (2005) Acetyl-l-carnitine improves pain, nerve regeneration, and vibratory perception in patients with chronic diabetic neuropathy: An analysis of two randomized placebo-controlled trials. Diabetes Care 28:89–94 [DOI] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150:76–85 [DOI] [PubMed] [Google Scholar]
- Solberg R, Enot D, Deigner HP, Koal T, Scholl-Burgi S, Saugstad OD, Keller M. (2010) Metabolomic analyses of plasma reveals new insights into asphyxia and resuscitation in pigs. PLoS One 5:e9606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai I, China K, Sai Y, Kobayashi D, Nezu J, Kawahara E, Tsuji A. (2001) Na(+)-coupled transport of l-carnitine via high-affinity carnitine transporter octn2 and its subcellular localization in kidney. Biochim Biophys Acta 1512:273–284 [DOI] [PubMed] [Google Scholar]
- Tang S, Xu S, Lu X, Gullapalli RP, McKenna MC, Waddell J. (2017) Neuroprotective effects of acetyl-l-carnitine on neonatal hypoxia-ischemia induced brain injury in rats. Dev. Neurosci. Under review: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufekci O, Gunes D, Ozogul C, Kolatan E, Altun Z, Yilmaz O, Aktas S, Erbayraktar Z, Kirkim G, Mutafoglu K, Soylu A, Serbetcioglu B, Guneri EA, Olgun N. (2009) Evaluation of the effect of acetyl l-carnitine on experimental cisplatin nephrotoxicity. Chemotherapy 55:451–459 [DOI] [PubMed] [Google Scholar]
- Wang SM, Han C, Lee SJ, Patkar AA, Masand PS, Pae CU. (2014) A review of current evidence for acetyl-l-carnitine in the treatment of depression. J Psychiatr Res 53:30–37 [DOI] [PubMed] [Google Scholar]
- Weinberg JM, Venkatachalam MA, Roeser NF, Nissim I. (2000) Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc Natl Acad Sci U S A 97:2826–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Prasad PD, Leibach FH, Ganapathy V. (1998) Cdna sequence, transport function, and genomic organization of human octn2, a new member of the organic cation transporter family. Biochem Biophys Res Commun 246:589–595 [DOI] [PubMed] [Google Scholar]
- Wu X, Huang W, Prasad PD, Seth P, Rajan DP, Leibach FH, Chen J, Conway SJ, Ganapathy V. (1999) Functional characteristics and tissue distribution pattern of organic cation transporter 2 (octn2), an organic cation/carnitine transporter. J Pharmacol Exp Ther 290:1482–1492 [PubMed] [Google Scholar]
- Xu LX, Lv Y, Li YH, Ding X, Wang Y, Han X, Liu MH, Sun B, Feng X. (2017) Melatonin alleviates brain and peripheral tissue edema in a neonatal rat model of hypoxic-ischemic brain damage: The involvement of edema related proteins. BMC Pediatr 17:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Waddell J, Zhu W, Shi D, Marshall AD, McKenna MC, Gullapalli RP. (2015) In vivo longitudinal proton magnetic resonance spectroscopy on neonatal hypoxic-ischemic rat brain injury: Neuroprotective effects of acetyl-l-carnitine. Magn Reson Med 74:1530–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Hironaka Y, Hama T. (1990) [effects of levocarnitine chloride, a new mitochondrial function reactivating agent, on fatty acid and glucose oxidation under hypoxic condition in homogenates from rat heart]. Yakugaku Zasshi 110:225–234 [DOI] [PubMed] [Google Scholar]
- Zanelli SA, Solenski NJ, Rosenthal RE, Fiskum G. (2005) Mechanisms of ischemic neuroprotection by acetyl-l-carnitine. Ann N Y Acad Sci 1053:153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezgü FS, Atalay Y, Hasanoğlu A, Gücüyener K, Biberoğlu G, Koç E, Ergenekon E, Tümer L. (2004) Serum carnitine levels in newborns with perinatal asphyxia and relation to neurologic prognosis. Nutr Neurosci. 7:351–356. [DOI] [PubMed] [Google Scholar]
- Schürch R, Todesco L, Novakova K, Mevissen M, Stieger B, Krähenbühl S. (2010) The plasma carnitine concentration regulates renal OCTN2 expression and carnitine transport in rats. 635:171–176. [DOI] [PubMed] [Google Scholar]