

SUMMARY
Background:
Accumulating data support a protective role of Helicobacter pylori against inflammatory bowel diseases (IBD), which might be mediated by strain-specific constituents, specifically cagA expression.
Aim:
To perform a systematic review and meta-analysis to more clearly define the association between CagA seropositivity and IBD.
Methods:
We identified comparative studies that included sufficient detail to determine the odds or risk of IBD, Crohn’s disease (CD) or ulcerative colitis (UC) among individuals with versus without evidence of cagA expression (e.g. CagA seropositivity). Estimates were pooled using a random-effects model.
Results:
Three clinical studies met inclusion criteria. cagA expression was represented by CagA seropositivity in all studies. Compared to CagA seronegativity overall, CagA seropositivity was associated with lower odds of IBD (OR 0.31, 95% CI:0.21-0.44) and CD (OR 0.25, 95% CI:0.17-0.38), and statistically nonsignificant lower odds for UC (OR 0.68, 95% CI:0.35-1.32). Similarly, compared to H pylori non-exposed individuals, H pylori exposed, CagA seropositive individuals had lower odds of IBD (OR 0.26, 95% CI: 0.16-0.41) and CD (OR 0.23, 95% CI: 0.15-0.35), but not UC (OR 0.66, 0.34-1.27). However, there was no significant difference in the odds of IBD, CD, or UC between H pylori exposed, CagA seronegative and H pylori non-exposed individuals.
Conclusion:
We found evidence for a significant association between CagA seropositive H pylori exposure and reduced odds of IBD, particularly CD, but not for CagA seronegative H pylori exposure. Additional studies are needed to confirm these findings and define underlying mechanisms.
Keywords: bacteria, digestive system, immune system diseases, epidemiology, allergy
INTRODUCTION
The inflammatory bowel diseases (IBD), ulcerative colitis (UC) and Crohn’s disease (CD), are chronic inflammatory diseases of the intestinal tract.1,2 The pathophysiology of IBD is complex and implicates interactions between genetic predisposition, immune dysregulation, environmental factors, and likely gut dysbiosis with altered host response to commensal microbiota.1,2 While much attention is focused on risk factors for IBD, factors that may attenuate or prevent IBD are largely undefined.
Helicobacter pylori (H pylori) is the most common chronic bacterial infection in the world and is estimated to infect approximately 4.4 billion people.3 Although considered the primary risk factor for gastric cancer, H pylori has co-evolved with humans for over 100,000 years4,5, and accumulating data support a potential protective role of H pylori against certain immune-mediated diseases, including IBD.6,7 Certain H pylori strain specific virulence constituents, including cytotoxin-associated gene A (cagA) and certain alleles of vacuolating cytotoxin (vacA), while associated with an increased risk of gastric cancer, might also mediate beneficial effects of H pylori through modulation of host immune defenses and other mechanisms. As support, in experimental models, H pylori-induced immunomodulation appears to be a critical mechanism for protection and this protection is at least partly mediated by H pylori proteins.8-14 Preliminary data implicate specific H pylori constituents14-18, although experimental data in colitis are inconsistent and generally limited to cagA gene or CagA protein expression.8 Epidemiologic literature overall supports an inverse association between H pylori and IBD7, but the impact of strain-specific H pylori constituents on this association is incompletely investigated. We hypothesize that the host immune response to CagA critically mediates the observed protective association between H pylori exposure and IBD. CagA seropositivity is a surrogate serological marker of this host immune response.
Thus, the primary objective of the present study was to perform a comprehensive systematic review to identify both clinical and experimental studies analyzing the association between strain-specific H pylori constituents and IBD (CD, UC) or experimental colitis, respectively. A meta-analysis of clinical studies was planned a priori to test the hypothesis that seropositivity to certain strain-specific factors, namely CagA, is associated with a decreased likelihood of IBD.
METHODS
The current study follows the methodology stipulated in the Cochrane Handbook19 and the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement guidelines.20
Data Sources and Searches
We searched three databases—PubMed, Embase, and Web of Science—for relevant literature in conjunction with a certified biomedical librarian at Vanderbilt University (initial search: November 30, 2018; updated search: March 3, 2019). The complete search string is included in the supplemental material. No restrictions were applied based on language, publication date, or peer-reviewed publication type, including published abstracts. Human and animal studies were both included. We also hand-searched references from included studies and relevant review articles.
Inclusion/Exclusion Criteria – Clinical Studies (Qualitative Synthesis and Quantitative Analysis)
All clinical trials (randomized, nonrandomized), cohort (prospective, retrospective), case-control, and cross-sectional studies were considered eligible if they met the following criteria: (1) diagnosis of IBD, inclusive of CD, UC, or IBD-U, according to standard diagnostic criteria; (2) comparative study design with distinct comparison between individuals with versus without IBD; (3) H pylori testing in both case and comparator groups, along with testing modality and test results; (4) determination of the expression of H pylori strain-specific constituents and testing modality (e.g. serologic response to CagA) among individuals with versus without IBD; and (5) sufficient detail to calculate effect estimates.
Studies not meeting these criteria were excluded for quantitative analysis (i.e. meta-analysis). Of note, while we were interested in serological responses to H pylori strain-specific constituents since this at least reflects host immune recognition, we did not exclude other testing modalities a priori, although sensitivity analyses were planned for testing modality. For example, some people who are infected with cagA gene positive H pylori strains do not mount a serologic immune response and these strains are only identified via biopsy of the gastric mucosa and molecular analyses.21
Inclusion/Exclusion Criteria – Experimental Studies (Qualitative Synthesis Only)
Because there were only limited experimental data on H pylori exposure and experimental colitis/IBD, we elected to be more inclusive of experimental studies, even if data on strain-specific factors per se were not reported.
Experimental studies were included for qualitative synthesis if they met the following criteria: (1) use of an accepted (contemporaneous) experimental model of colitis (e.g. dextran sulfate sodium (DSS) colitis) or IBD (e.g. SAMP1/YitFc ileitis)22; (2) comparative study design with distinct comparison between animals with versus without experimental colitis/IBD stratified by H pylori or H pylori extract exposure versus no exposure.
Data Extraction
Eligibility assessment and data extraction were carried out independently by SCS and AT with discrepancies resolved by a third investigator (NN). Two separate data collection forms (clinical, experimental) were designed by SCS. For the clinical studies, the following information was extracted as available: first author’s last name; publication date; study design; country of origin; study time period; number of people in the study; study demographics; study setting and study-specific inclusion/exclusion criteria; H pylori related details (number infected; current vs. former vs. never infected; diagnostic method; duration of infection; treatment details including regimen and eradication success or failure); IBD-type (CD, UC, IBD-U) and IBD-related details (age of diagnosis, disease duration, disease location, disease therapy). For experimental studies, study information was extracted along with the following: animal type and number, experimental model of colitis/IBD, method of H pylori infection, timing of H pylori infection (or exposure to H pylori extract if relevant), strain of H pylori, duration of infection prior to animal sacking, and overall findings.
Data extraction was performed in duplicate by SCS and AT.
Primary exposure and outcomes (quantitative analysis)
The primary exposure was serological response to strain-specific H pylori antigens (e.g. CagA seropositivity). None of the included clinical studies used alternative (non-serological) diagnostic modalities for exposure assessment. The primary outcome was IBD (CD, UC, IBD-U).
Quality and Risk of Bias Assessment (Clinical Studies only)
Quality assessment was performed independently by SCS and AT using the Newcastle-Ottawa Scale (NOS)23 for nonrandomized studies and the Cochrane Risk of Bias tool for randomized trials, although there were no studies fitting the latter category. Discrepancies were resolved by consensus with a third investigator (NN). For the NOS, studies scored as >/= 7 (of maximum score 9, or 10 for cross-sectional studies) were considered high-quality.23 Separate NOS rubrics were used for case-control, cohort, and cross-sectional studies (modified NOS cohort rubric) as appropriate.24,25
Qualitative Synthesis and Quantitative Statistical Analysis
Details of each included study were summarized and synthesized. Studies meeting inclusion criteria only included information on serological response to CagA and no other H pylori strain-specific constituents; thus, we focused our analysis on CagA seropositivity as the primary exposure of interest. The odds of IBD, CD, or UC (primary outcomes; no studies included IBD-U) among individuals who were CagA seropositive versus CagA seronegative was calculated and reported as odds ratios (ORs) with 95% confidence intervals (95% CI) for each individual study. Of note, for the primary analysis, the CagA seronegative reference group included both H pylori exposed individuals who were seronegative for CagA and H pylori non-exposed individuals. As secondary analyses, the odds of IBD, CD, or UC among 1) H pylori exposed individuals who were CagA seropositive, and 2) H pylori exposed individuals who were CagA seronegative versus the reference category of H pylori non-exposed individuals overall was calculated for each individual study. In keeping with the primary objective of our study, our inclusion criteria were strictly limited to studies that included additional data on H pylori strain-specific antigens and the odds or risk of IBD, as opposed to just H pylori exposure alone, the latter of which has already been evaluated in multiple studies and meta-analyses; accordingly, we did not calculate the odds of IBD among H pylori exposed versus H pylori non-exposed as the data profile from our search might not be representative. Corresponding authors were contacted for additional details as needed.
Individual study ORs were combined into a pooled OR by using a random effects model. Egger test26 and a funnel plot were planned for assessment of publication bias for the primary outcome, although these tests are not robust when <10 studies are included for meta-analysis.27 Heterogeneity was estimated with chi-squared and I2 test statistics. The chi-squared test suggests heterogeneity between studies when the P-value is less than 0.15. We further used I2 cut-offs of <30%, 30–59%, 60–75%, and >75% to for low, moderate, substantial, and considerable heterogeneity, respectively.28 The follow meta-regression analyses were planned a priori to adjust for: study type (cohort versus case-control studies, and prospective versus retrospective studies), study geography (Asia-Pacific versus Western geography), pediatric only versus adult only populations, diagnostic modalities, and duration of H pylori infection prior to IBD diagnosis (or matched time point in comparator group). However, due to the limited number of studies meeting inclusion criteria and details provided in the studies, no meta-regressions were feasible.
All analyses were performed with RevMan 5.1 (Review Manager Version 5.1, Copenhagen, Denmark) and Comprehensive MetaAnalysis (version 2.0; Biostat, Englewood, NJ).
RESULTS
The initial search yielded 501 studies. After removal of duplicates, 487 studies remained. Of these, 469 were removed via screening of title or abstract, which provided enough detail to determine that the study did not include information relevant to the present analysis. No studies were removed based on language. From 18 full-text articles reviewed, 9 studies were excluded for the reasons detailed in the flow diagram (Figure 1). One study7 was identified based on search of reference lists, but was excluded after full text review, as this study did not include information on H pylori strain-specific constituents. We performed an updated search on March 3, 2019, which yielded no additional studies for inclusion (Supplemental material).
FIGURE 1.
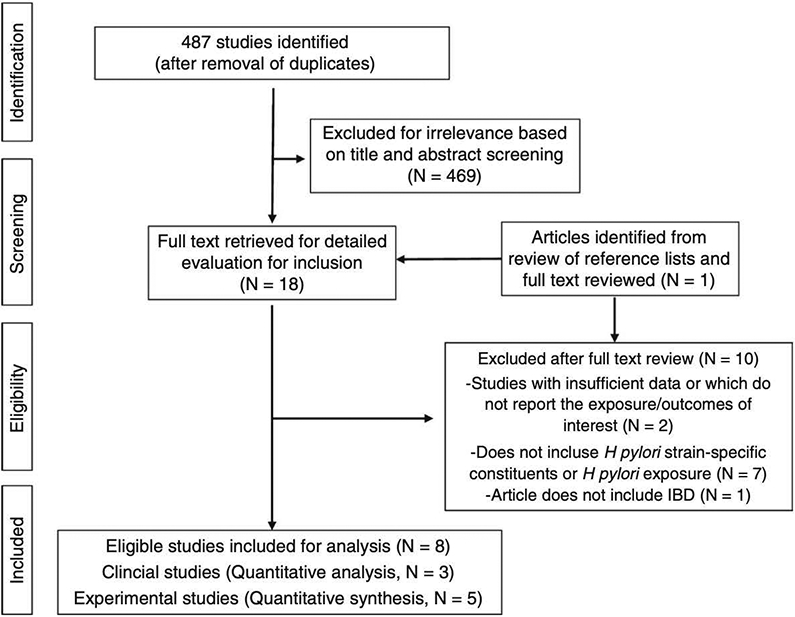
PRISMA diagram of study selection
Of the 9 relevant articles, 3 were human studies29-31, all of which included information on CagA serostatus only and no other H pylori strain-specific constituents. The remaining 6 studies were all experimental studies conducted in animals. One experimental study was excluded because it used a transgenic murine model constitutively expressing CagA and did not involve natural H pylori exposure.8 Of the 5 included experimental studies, only one specifically investigated CagA-dependent effects32 while the remaining 4 studies did not distinctly investigate H pylori strain-specific constituents.12,13,33,34
Clinical Data (Table 1 and Table 2a-c)
Table 1:
Characteristics of clinical studies meeting inclusion criteria
| Author, Year |
Study Design |
Country/Study Period |
Prevalence HP (%) |
Total, N |
IBD, N |
CD, N |
UC, N |
Controls, N |
Mean Age, IBD |
Mean Age, Control |
Sex (%, Female) |
HP Prevalence, Control (%) |
HP Prevalence, IBD (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lord, 2018a | Cross Sectional | Australia 1996-2009 | 10.8 | 704 | 447 | 212 | 235 | 257 | 29.9 (SD ±13.2) | 57.2 (SD +13.5) | 53.8 | 13.2 | 9.4 |
| Rosania, 2018 | Case-Control, age and sex-matched | Germany 2016-2017 | 24.7 | 381 | 127 | 90 | 37 | 254b | 42 (SD ±12) | 41 (SD ±12.4) | 53.3c | 28.7 | 16.5 |
| Wagtmans, 1997e | Case-control, age-matched | Netherlands 1974-1975 | 21.9 | 663 | 386 | 386 | 0 | 277 | 37 | 36.9 | 44.9 | 35.4 | 12.2 |
This study included H. pylori (HP) positive, trace, and negative samples. No information was provided delineating positive from trace positive, but the study considered trace to be a “non-positive” result. Accordingly, we considered “trace” to be HP negative for this analysis.
Rosania et al included control groups for each set: CD, UC, and IBD. 254 was the number of controls for the IBD group. Separate UC and CD control groups contained 180 and 74, respectively.
Overall breakdown of sex was calculated using IBD group added to control group for IBD. Each subset, UC and CD, was compared to separate controls not included in this calculation. In addition, this study contained an error in its tables where totals for sex for IBD was reversed. This was corrected in our calculation and the authors were contacted.
Serologic evidence included ELISA and/or Western Blot immunoassays
This study only included patients with CD and not UC. Therefore, IBD total is equal to CD total.
Both IgG and IgA were tested. However, CagA was only tested in the samples where IgG was positive. In addition, the study determined seroprevalence by IgG alone and not IgA positivity. Therefore, IgA positivity was not included for this analysis.
Table 2a.
H pylori exposure and CagA serostatus among individuals with IBD compared to individuals without IBD
| Author, Year | IBD | No IBD | IBD | No IBD | ||||
|---|---|---|---|---|---|---|---|---|
| HP- | HP+ | HP− | HP+ | CagA− | CagA+ | CagA− | CagA+ | |
| Lord, 2018a | 405 | 42 | 223 | 34 | 430 | 14 | 235 | 19 |
| Rosania, 2018b | 106 | 21 | 181 | 73 | 121 | 6 | 223 | 31 |
| Wagtmans, 1997c | 339 | 47 | 179 | 98 | 355 | 31 | 209 | 68 |
This study included H. pylori (HP) positive, trace, and negative samples. No information was provided delineating positive from trace positive, but the study considered trace to be a “non-positive” result. Accordingly, we considered “trace” to be HP negative for this analysis. In addition, CagA samples were “missing” for 6 samples total (1 in CD, 2 in UC, and 3 in controls).
Based on data presented in this study, the HP positive subjects in the UC and CD subgroups do not add up to the total HP positive subjects in the IBD group and the reason for the discrepancy is not provided by the authors.
This study only included patients with CD and not UC
Table 2c.
H pylori exposure and CagA serostatus among individuals with UC compared to individuals without IBD
| Author, Year | UC | No IBD | UC | No IBD | |||||
|---|---|---|---|---|---|---|---|---|---|
| HP− | HP+ | HP− | HP+ | CagA− | CagA+ | CagA− | CagA+ | ||
| Lord, 2018a | 211 | 24 | 223 | 34 | 221 | 12 | 235 | 19 | |
| Rosania, 2018b | 32 | 5 | 54 | 20 | 34 | 3 | 66 | 8 | |
| Wagtmans, 1997c | |||||||||
This study included H. pylori (HP) positive, trace, and negative samples. No information was provided delineating positive from trace positive, but the study considered trace to be a “non-positive” result. Accordingly, we considered “trace” to be HP negative for this analysis. In addition, CagA samples were “missing” for 6 samples total (1 in CD, 2 in UC, and 3 in controls).
Based on data presented in this study, the HP positive subjects in the UC and CD subgroups do not add up to the total HP positive subjects in the IBD group and the reason for the discrepancy is not provided by the authors.
This study only included patients with CD and not UC
A total of 3 studies met inclusion criteria and included 1748 people, 960 of whom were diagnosed with IBD (688 CD, 272 UC; one study29 included only individuals with CD and not UC). These included 2 case-control studies and 1 cross-sectional study. All 3 studies determined H pylori exposure status using ELISA or Western blot serological testing; the study by Rosania et al.30 also included evidence of prior H pylori eradication therapy as a diagnostic criterion for H pylori exposure (see sensitivity analysis below). Each study determined CagA status using ELISA or Western blot serological analysis; no alternative (i.e. non-serological) modalities were utilized.
Baseline characteristics of the patient population, including age and gender breakdown, for each study as well other study details are provided in Table 1. All studies reported on the type of IBD and, as noted, Wagtmans et al. analyzed only individuals with CD and not UC.29 Details regarding age of IBD diagnosis and duration of IBD at the time of serologic testing were not provided in any study. Only the Rosania et al. study provided details of H pylori eradication therapy. Each study provided sufficient information for determining the odds of IBD, CD, or UC according to CagA serostatus. The two case-control studies, Wagtmans et al. and Rosania et al. included age-matched only and age- and sex-matched controls, respectively, but did not adjust the odds ratio estimates for additional factors. Lord et al., a cross-sectional study, reported odds ratios but did not comment on factors adjusted for, if any, in these estimates; attempts at contacting the authors for details were unsuccessful.
For the primary analysis, the CagA seronegative reference group was comprised of H pylori exposed, CagA seronegative and H pylori non-exposed (CagA seronegative) individuals. Compared to CagA seronegativity, CagA seropositivity was associated with 69% lower odds of IBD (OR 0.31, 95% CI: 0.21–0.44; I2=0%) and 75% lower odds of CD (OR 0.25, 95% CI: 0.17–0.38; I2=0%) (Figure 2a-2b). While there was a similar suggestive inverse association between CagA seropositivity and odds of UC (OR 0.68, 95% CI: 0.35–1.32; I2=0%), this was not statistically significant based on the two studies that included UC data (Figure 2c). The directionality and magnitude of odds of IBD (OR 0.26, 95% CI: 0.16–0.41; I2=34.4%), CD (OR 0.23, 95% CI: 0.15–0.35; I2=0%), and UC (OR 0.66, 95% CI: 0.34–1.27; I2=0%) paralleled the primary analysis when comparing H pylori exposed, CagA seropositive individuals to H pylori non-exposed individuals as the reference group (Figures 3a-3c). By contrast, H pylori exposed, CagA seronegative individuals had similar odds of IBD (OR 0.74, 95% CI: 0.46–1.17; I2=88.3%), CD (OR 0.97, 95% CI: 0.82–1.14; I2=0%), and UC (OR 0.97, 95% CI: 0.76–1.24; I2=0%) compared to H pylori non-exposed individuals (Figures 4a-4c).
FIGURE 2.

A, Odds of IBD associated with CagA seropositive vs CagA seronegative status (reference group: Helicobacter pylori exposed, CagA seronegative and H pylori non-exposed individuals). B, Odds of CD associated with CagA seropositive vs CagA seronegative status (reference group: H pylori exposed, CagA seronegative and H pylori non-exposed individuals). C, Odds of UC associated with CagA seropositive vs CagA seronegative status (reference group: H pylori exposed, CagA seronegative and H pylori non-exposed individuals)
FIGURE 3.

A, Odds of IBD associated with Helicobacter pylori exposed, CagA seropositive vs H pylori non-exposed status. B, Odds of CD associated with H pylori exposed, CagA seropositive vs H pylori non-exposed status. C, Odds of UC associated with H pylori exposed, CagA seropositive vs H pylori non-exposed status
FIGURE 4.

A, Odds of IBD associated with Helicobacter pylori exposed, CagA seronegative vs H pylori non-exposed status. B, Odds of CD associated with H pylori exposed, CagA seronegative vs H pylori non-exposed status. C, Odds of UC associated with H pylori exposed, CagA seronegative vs H pylori non-exposed status
A sensitivity analysis was performed removing the study by Rosania et al. given that this study also included prescription of H pylori eradication therapy as diagnostic for H pylori exposure. Removing this study did not significantly affect the directionality, magnitude, or statistical significance of the primary outcome (Supplemental figure 1).
Qualitative Synthesis of Experimental Data (Supplemental Table 1)
The five experimental studies included for qualitative synthesis are summarized in Supplemental Table 1. All studies included mouse models and all were limited to colonic pathology as opposed to other parts of the luminal GI tract. Four of the experimental colitis models investigated chemical colitis (dextran sulfate sodium (DSS), N=3; 2,4,6-trinitrobenzene sulfonic acid (TNBS), N=1), one study additionally induced colitis via T-cell transfer12, and one study induced colitis via Salmonella typhimurium inoculation.33 In all studies, mice were infected with H pylori according to various protocols or exposed to H pylori DNA extract.33 Three of these 4 studies inoculated mice with the SS1 strain of H pylori, which has a nonfunctional cag pathogenicity island, or its DNA extract, while one study12 inoculated mice with the PMSS1 strain of H pylori, which has a functional cag pathogenicity island. Two studies32,33 also included strain 26695 and one study included strain J9933, both of which have functional cag pathogenicity islands.
In all studies, H pylori exposure attenuated experimentally-induced colitis, which, based on four studies12,13,32,34, correlated with a blunted Th17 response. Two studies13,32 additionally demonstrated enhanced Treg and Th2 responses in mice with H pylori exposure versus uninfected controls. The study by Luther et al. further defined the immunoregulatory to immunostimulatory ratio (IRS:ISS) of H pylori DNA compared to other common intestinal flora that are able to cause acute infection (e.g. E coli K27 and E faecalis), as well as Mycobacterium paratuberculosis (implicated in IBD pathogenesis35). While H pylori had an IRS:ISS >1 (mean: 29.4), all others had an IRS:ISS<1 (E coli K27 (mean: 0.36), E faecalis (mean: 0.36) and Mycobacterium paratuberculosis (mean: 0.70)). Notably, Helicobacter hepaticus had an IRS:ISS>1 (mean: 3.08) but it was significantly lower than the IRS:ISS for H pylori.
Only one study32 specifically investigated CagA-dependent mechanisms. Zhang and colleagues demonstrated that H pylori SS1 infection was associated with less severe DSS-colitis. They showed that this was at least in part due to CagA-dependent polarization of macrophages from the M1 (pro-inflammatory) to M2 (anti-inflammatory) subtypes, along with increased concentration of M2 macrophages in the colonic lamina propria, spleen, and mesenteric lymph nodes. M2 macrophages also produce TGF-beta and thus promote Treg differentiation.
Publication bias and heterogeneity
Egger’s test confirmed no publication bias (P-value=0.36), but as noted above this test is not as robust for meta-analyses of less than 10 studies.26 Statistical tests of heterogeneity demonstrated no heterogeneity for our primary analyses assessing CagA seropositivity versus seronegativity and odds of IBD, CD, or UC (I2 = 0%, chi-squared test P=0.57–0.92). There was also no heterogeneity for the secondary analyses assessing CagA seropositive H pylori exposure versus H pylori non-exposure and odds of CD or UC (I2 = 0%, chi-squared test P=0.63–0.95), nor for CagA seronegative H pylori exposure versus H pylori non-exposure and odds of CD or UC (I2 = 0%, chi-squared test P=0.69–0.74). However, there was moderate to considerable heterogeneity for the secondary analyses assessing CagA seropositive H pylori exposure (I2 = 34.4%, chi-squared test P=0.22) and CagA seronegative H pylori exposure (I2 = 88.3%, chi-squared test P<0.001), respectively, versus H pylori non-exposure and odds of IBD overall.
Risk of Bias Assessment according to the Newcastle Ottawa Scale (NOS)
All studies were rated as high-quality according to the respective NOS scale based on study design. Factors determining quality included information provided by each study regarding the selection of cases and controls, comparability of cases and controls, and the methods of outcome measurements. Greater levels of information provided, higher comparability of groups, and more objective methods led to higher scores on quality assessment. (Supplemental table 2)
DISCUSSION
The findings of this comprehensive systematic review and meta-analysis are consistent with the hypothesis that H pylori strain-specific factors, namely CagA, might be key determinants in the protective association with IBD, and especially CD. Based on meta-analysis, we found that serologic response to CagA was associated with a markedly reduced odds of IBD overall, which was driven by a strong inverse association with CD as opposed to UC; the results were similar in magnitude irrespective of whether the comparison group was CagA seronegative individuals—defined for the primary analysis by the composite of H pylori exposed, CagA seronegative and all H pylori non-exposed individuals—or whether the reference group was H pylori non-exposed individuals alone. As additional support, we demonstrated that individuals exposed to H pylori but who were CagA seronegative had similar odds of IBD, CD, and UC as individuals without H pylori exposure. We further compiled a complementary synthesis of relevant experimental data, which provide a platform of support for biological plausibility of CagA as one critical mediator of the protective association observed clinically between H pylori exposure and odds of IBD, as well as very likely other immune-mediated diseases.6
The acute increase in IBD globally over the last few decades suggests that a relatively abrupt change in environmental exposures is most likely responsible, as opposed to a shift in genetic predisposition.36-38 In areas where the rise in IBD has been most striking, namely the Asia-Pacific region, the opposite trend is observed for H pylori.3 The decreasing H pylori prevalence is due in large part to industrialization, improved sanitation, and H pylori eradication efforts as part of gastric cancer reduction campaigns primarily focused in Japan and Korea.39 A substantial body of literature has clearly established the heightened pathogenic and carcinogenic potential of H pylori strains harboring the cagA gene, with older studies demonstrating that H pylori strains isolated from biopsies of individuals with severe gastric diseases, including peptic ulcer disease, mucosa-associated lymphoid tissue lymphoma and gastric cancer, harbored the cagA gene in over 90%.40-42 Our data, though, also suggest a protective role of CagA seropositivity against IBD in susceptible people. Interestingly, the rate of loss of cagA gene positive H pylori strains may vary between populations, as H pylori strains isolated from people in the Asia-Pacific region (and also developing countries) nearly universally contain the cagA gene, as compared to H pylori strains isolated from most developed countries where the proportion of H pylori strains harboring versus not harboring the cagA gene is more balanced.43-46 The impressive rate of increase of IBD in the Asia-Pacific region, which is disproportionate when compared to other developed regions47, may be one consequence of the rapid reduction in exposure to cagA gene positive H pylori strains, especially childhood exposure; as a consequence, there is less opportunity for developing the host immune response to CagA with each successive generation. When present, host responses to H pylori strains expressing CagA are more robust in children, which is when H pylori exposure most often occurs.9,48,49 Indeed, these early H pylori associated immunomodulatory effects might also protect against immune-mediated diseases other than IBD, such as asthma and allergy.6,9,10 One cross-sectional analysis of 7,663 adults from the Third National Health and Nutrition Examination Survey found that the inverse association between H pylori and asthma, allergic rhinitis, and allergies was more pronounced among younger individuals and among those who were CagA seropositive.50
The protective mechanisms specifically associated with host serologic response to CagA, however, are not defined in IBD, although several hypotheses can be generated based on knowledge of differences in host response in the presence of infection with cagA gene positive H pylori strains. For example, colonization with H pylori strains harboring a functional cag pathogenicity island, which contains the cagA gene, resulted in higher production of beta-defensins and other antimicrobial effectors more so than strains without a functioning cag pathogenicity island.45,51 This is relevant since it is well-established that altered human defensin production is associated with IBD pathogenesis, specifically CD.52-55 Only one experimental study in our analysis investigated the effect of infection with wild type H pylori strains producing CagA versus mutant H pylori strains not producing CagA on colitis in murine models; this study demonstrated that the Th2 cytokine response, which protects against experimental colitis, was CagA-dependent.32 Generally speaking, H pylori strains that express CagA also express other factors like BabA and certain alleles of VacA; these strains tend to interact more with the host and are thus more likely to influence host immune responses.56,57 Along these lines, even though individuals might be colonized with H pylori strains containing the cagA gene, as determined via molecular analysis of gastric biopsies for example, a proportion will not mount a serologic immune response and thus would not be identified as CagA seropositive. One study demonstrated that certain vacA polymorphisms were strong determinants for the serologic immune response to cagA gene positive H pylori strains. Thus, it is possible that there are other unmeasured factors mediating the observed association between CagA seropositivity and decreased odds of IBD, of which we are not able to comment on in this study, but should be a major focus of future investigations. That said, CagA seropositivity is potentially a very useful surrogate marker, which is why we selected this as the primary exposure of interest a priori; although, ultimately this criterion was moot as no studies that otherwise met inclusion criteria analyzed non-serological diagnostic modalities.
This study is the most comprehensive analysis of CagA seropositivity and odds of IBD. All included clinical studies were high-quality, without significant heterogeneity for our primary analyses and the majority of secondary analyses. Our study does have limitations, which mostly reflect the limited data. Because of the designs of the included studies, direction of causality cannot be determined with certainty. Generally speaking, though, H pylori exposure occurs early in childhood9,48,49 and IBD most often presents in early adulthood. Moreover, biological plausibility for IBD altering the prevalence or likelihood of H pylori infection has not been established, which is in contrast to the growing body of literature supporting the converse. Other than the Rosania et al. study, details regarding H pylori eradication therapy were not provided. Eradication therapy typically involves at least 2–3 antibiotics and high-dose acid suppression for a set duration, with or without repeated courses, and it is plausible that this might modify the risk of IBD in genetically susceptible individuals, such as via inducing gut dysbiosis or other mechanisms. Although the sensitivity analysis excluding the Rosania et al. study yielded unchanged findings for our primary analysis, we are limited in our ability to make any strong conclusions regarding the effect of eradication therapy on the observed association between CagA seropositivity and odds of IBD. Another limitation is that CagA was the only strain-specific factor analyzed in clinical studies. cagA gene positive H pylori strains do not exist in isolation and these strains more commonly express other strain-specific factors, which might also modulate the host immune response;21,58 how these other constituents modify the risk of IBD has not been investigated. Our study might be underpowered for smaller effect sizes, for example with respect to UC, since unmeasured confounders would bias towards a null association. Another consideration is that all three clinical studies were from Western industrialized countries (H pylori prevalence 11–25%) and these findings may therefore not be generalizable to other populations. Along these lines, emerging literature demonstrates nuances in IBD pathogenesis and disease course in Asian-Pacific versus Western populations59-62 not to mention differences in H pylori disease manifestations based on ethnic concordance of host and H pylori strain.5 The cagA gene positive H pylori strain prevalence in the included clinical studies might also be lower than some endemic geographic areas; that said, the lower prevalence would expectedly bias towards a null association.
In conclusion, based on meta-analysis of clinical studies that included 1748 people, we found that CagA seropositivity was associated with decreased odds of IBD, particularly CD. These findings mirror those of some other immune- and allergen-mediated diseases and are supported by biologically plausible mechanisms based on experimental data. Larger, well-designed prospective clinical studies, complemented by studies in models that more faithfully recapitulate human IBD (i.e. non-chemical colitis models) are needed, including studies defining the role of other H pylori strain-specific factors apart from CagA.
Supplementary Material
Table 2b.
H pylori exposure and CagA serostatus among individuals with CD compared to individuals without IBD
| Author, Year | CD | No IBD | CD | No IBD | ||||
|---|---|---|---|---|---|---|---|---|
| HP− | HP+ | HP− | HP+ | CagA− | CagA+ | CagA− | CagA+ | |
| Lord, 2018a | 194 | 18 | 223 | 34 | 209 | 2 | 235 | 19 |
| Rosania, 2018b | 73 | 17 | 138 | 42 | 87 | 3 | 161 | 19 |
| Wagtmans, 1997c | 339 | 47 | 179 | 98 | 355 | 31 | 209 | 68 |
This study included H. pylori (HP) positive, trace, and negative samples. No information was provided delineating positive from trace positive, but the study considered trace to be a “non-positive” result. Accordingly, we considered “trace” to be HP negative for this analysis. In addition, CagA samples were “missing” for 6 samples total (1 in CD, 2 in UC, and 3 in controls).
Based on data presented in this study, the HP positive subjects in the UC and CD subgroups do not add up to the total HP positive subjects in the IBD group and the reason for the discrepancy is not provided by the authors.
This study only included patients with CD and not UC
Acknowledgements Section
(i) Guarantor of the article: Shailja C. Shah
The authors would like to thank the Vanderbilt University librarians, Rachel Walden, MLIS and Heather Laferriere, MLIS for their assistance in the literature search.
(iii): All authors approved the final version of the manuscript
Grant Support: none
Footnotes
Disclosures/conflict of interest statement: The authors have no potential conflicts (financial, professional, nor personal) that are relevant to this manuscript.
Writing assistance: No additional writing assistance was used for this manuscript
Publisher's Disclaimer: This is the peer reviewed version of the following article: Tepler A, Narula N, Peek RM Jr, Patel A, Edelson C, Colombel JF, Shah SC. Systematic review with meta-analysis: association between Helicobacter pylori CagA seropositivity and odds of inflammatory bowel disease. Aliment Pharmacol Ther 2019 Jul;50(2):121–131. doi: 10.1111/apt.15306. Epub 2019 Jun 5. PubMed PMID: 31165513, which has been published in final form at https://doi.org/10.1111/apt.15306. This article may be used for non-commercial purposes in accordance with Wiley Terms and Conditions for Use of Self-Archived Versions
References
- 1.Ungaro R, Mehandru S, Allen PB, Peyrin-Biroulet L, Colombel J-F. Ulcerative colitis. The Lancet 2017; 389: 1756–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torres J, Mehandru S, Colombel J-F, Peyrin-Biroulet L. Crohn’s disease. The Lancet 2017; 389: 1741–1755. [DOI] [PubMed] [Google Scholar]
- 3.Hooi JKY, Lai WY, Ng WK, et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017; 153: 420–429. [DOI] [PubMed] [Google Scholar]
- 4.Breurec S, Raymond J, Thiberge J-M, et al. Impact of human migrations on diversity of Helicobacter pylori in Cambodia and New Caledonia. Helicobacter 2013; 18: 249–261. [DOI] [PubMed] [Google Scholar]
- 5.Kodaman N, Pazos A, Schneider BG, et al. Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc Natl Acad Sci U S A 2014; 111: 1455–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah SC, Tepler A, Peek RM, Colombel J-F, Hirano I, Narula N. Association Between Helicobacter pylori Exposure and Decreased Odds of Eosinophilic Esophagitis-a Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol 2019; published online January 16 DOI: 10.1016/j.cgh.2019.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castaño-Rodríguez N, Kaakoush NO, Lee WS, Mitchell HM. Dual role of Helicobacter and Campylobacter species in IBD: a systematic review and meta-analysis. Gut 2017; 66: 235–249. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki N, Murata-Kamiya N, Yanagiya K, et al. Mutual reinforcement of inflammation and carcinogenesis by the Helicobacter pylori CagA oncoprotein. Sci Rep 2015; 5: 10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnold IC, Hitzler I, Müller A. The immunomodulatory properties of Helicobacter pylori confer protection against allergic and chronic inflammatory disorders. Front Cell Infect Microbiol 2012; 2: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arnold IC, Dehzad N, Reuter S, et al. Helicobacter pylori infection prevents allergic’ ’ asthma in mouse models through the induction of regulatory T cells. J Clin Invest 2011; 121: 3088–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Owyang SY, Luther J, Owyang CC, Zhang M, Kao JY. Helicobacter pylori DNA’s anti-inflammatory effect on experimental colitis. Gut Microbes 2012; 3: 168–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engler DB, Leonardi I, Hartung ML, et al. Helicobacter pylori-specific protection against inflammatory bowel disease requires the NLRP3 inflammasome and IL-18. Inflamm Bowel Dis 2015; 21: 854–861. [DOI] [PubMed] [Google Scholar]
- 13.Wu Y-Z, Tan G, Wu F, Zhi F-C. H. pylori attenuates TNBS-induced colitis via increasing mucosal Th2 cells in mice. Oncotarget 2017; 8: 73810–73816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kyburz A, Urban S, Altobelli A, et al. Helicobacter pylori and its secreted immunomodulator VacA protect against anaphylaxis in experimental models of food allergy. Clin Exp Allergy 2017; 47: 1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oertli M, Noben M, Engler DB, et al. Helicobacter pylori γ-glutamyl transpeptidase and vacuolating cytotoxin promote gastric persistence and immune tolerance. Proc Natl Acad Sci U S A 2013; 110: 3047–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricci V, Giannouli M, Romano M, Zarrilli R. Helicobacter pylori gamma-glutamyl transpeptidase and its pathogenic role. World J Gastroenterol 2014; 20: 630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wüstner S, Anderl F, Wanisch A, et al. Helicobacter pylori γ-glutamyl transferase contributes to colonization and differential recruitment of T cells during persistence. Sci Rep 2017; 7: 13636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lina TT, Pinchuk IV, House J, et al. CagA-dependent downregulation of B7-H2 expression on gastric mucosa and inhibition of Th17 responses during Helicobacter pylori infection. J Immunol 2013; 191: 3838–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Higgins JPT, Green S. Cochrane Handbook for Systematic Reviews of Interventions (version 5.0.1). London: The Cochrane Collaboration, 2011. [Google Scholar]
- 20.Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med 2009; 6: e1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Link A, Langner C, Schirrmeister W, et al. Helicobacter pylori vacA genotype is a predominant determinant of immune response to Helicobacter pylori CagA. World J Gastroenterol 2017; 23: 4712–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cominelli F, Arseneau KO, Rodriguez-Palacios A, Pizarro TT. Uncovering Pathogenic Mechanisms of Inflammatory Bowel Disease Using Mouse Models of Crohn’s Disease-Like Ileitis: What is the Right Model? Cell Mol Gastroenterol Hepatol 2017; 4: 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ottawa Hospital Research Institute. http://www.ohri.ca/programs/clinical_epidemiology/oxford.asp (accessed Nov 7, 2018).
- 24.Herzog R, Álvarez-Pasquin MJ, Díaz C, Del Barrio JL, Estrada JM, Gil Á. Are healthcare workers’ intentions to vaccinate related to their knowledge, beliefs and attitudes? A systematic review. BMC Public Health 2013; 13: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Modesti PA, Reboldi G, Cappuccio FP, et al. Panethnic Differences in Blood Pressure in Europe: A Systematic Review and Meta-Analysis. PLoS ONE 2016; 11: e0147601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ 1997; 315: 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sterne JAC, Sutton AJ, Ioannidis JPA, et al. Recommendations for examining and interpreting funnel plot asymmetry in meta-analyses of randomised controlled trials. BMJ 2011; 343: d4002. [DOI] [PubMed] [Google Scholar]
- 28.Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003; 327: 557–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagtmans MJ, Witte AM, Taylor DR, et al. Low seroprevalence of Helicobacter pylori antibodies in historical sera of patients with Crohn’s disease. Scand J Gastroenterol 1997; 32: 712–718. [DOI] [PubMed] [Google Scholar]
- 30.Rosania R, Von Arnim U, Link A, et al. Helicobacter pylori eradication therapy is not associated with the onset of inflammatory bowel diseases. A case-control study. J Gastrointestin Liver Dis 2018; 27: 119–125. [DOI] [PubMed] [Google Scholar]
- 31.Lord AR, Simms LA, Hanigan K, Sullivan R, Hobson P, Radford-Smith GL. Protective effects of Helicobacter pylori for IBD are related to the cagA-positive strain. Gut 2018; 67: 393–394. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H, Dai Y, Liu Y, et al. Helicobacter pylori Colonization Protects Against Chronic Experimental Colitis by Regulating Th17/Treg Balance. Inflamm Bowel Dis 2018; 24: 1481–1492. [DOI] [PubMed] [Google Scholar]
- 33.Luther J, Owyang SY, Takeuchi T, et al. Helicobacter pylori DNA decreases pro-inflammatory cytokine production by dendritic cells and attenuates dextran sodium sulphate-induced colitis. Gut 2011; 60: 1479–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Higgins PDR, Johnson LA, Luther J, et al. Prior Helicobacter pylori infection ameliorates Salmonella typhimurium-induced colitis: mucosal crosstalk between stomach and distal intestine. Inflamm Bowel Dis 2011; 17: 1398–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiodini RJ, Van Kruiningen HJ, Thayer WR, Merkal RS, Coutu JA. Possible role of mycobacteria in inflammatory bowel disease. I. An unclassified Mycobacterium species isolated from patients with Crohn’s disease. Dig Dis Sci 1984; 29: 1073–1079. [DOI] [PubMed] [Google Scholar]
- 36.Ng WK, Wong SH, Ng SC. Changing epidemiological trends of inflammatory bowel disease in Asia. Intest Res 2016; 14: 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ng SC, Bernstein CN, Vatn MH, et al. Geographical variability and environmental risk factors in inflammatory bowel disease. Gut 2013; 62: 630–649. [DOI] [PubMed] [Google Scholar]
- 38.Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012; 142: 46–54.e42; quiz e30. [DOI] [PubMed] [Google Scholar]
- 39.Peleteiro B, Bastos A, Ferro A, Lunet N. Prevalence of Helicobacter pylori infection worldwide: a systematic review of studies with national coverage. Dig Dis Sci 2014; 59: 1698–1709. [DOI] [PubMed] [Google Scholar]
- 40.Kuipers EJ, Pérez-Pérez GI, Meuwissen SG, Blaser MJ. Helicobacter pylori and atrophic gastritis: importance of the cagA status. J Natl Cancer Inst 1995; 87: 1777–1780. [DOI] [PubMed] [Google Scholar]
- 41.Blaser MJ, Perez-Perez GI, Kleanthous H, et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res 1995; 55: 2111–2115. [PubMed] [Google Scholar]
- 42.Stein M, Rappuoli R, Covacci A. The cag pathogenicity island In: Mobley HL, Mendz GL, Hazell SL, eds. Helicobacter pylori: Physiology and Genetics. Washington (DC): ASM Press, 2001. [PubMed] [Google Scholar]
- 43.Parsonnet J, Replogle M, Yang S, Hiatt R. Seroprevalence of CagA-positive strains among Helicobacter pylori-infected, healthy young adults. J Infect Dis 1997; 175: 1240–1242. [DOI] [PubMed] [Google Scholar]
- 44.Van Doorn LJ, Figueiredo C, Mégraud F, et al. Geographic distribution of vacA allelic types of Helicobacter pylori. Gastroenterology 1999; 116: 823–830. [DOI] [PubMed] [Google Scholar]
- 45.Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology 2009; 136: 1863–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hatakeyama M Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe 2014; 15: 306–316. [DOI] [PubMed] [Google Scholar]
- 47.Ng SC. Inflammatory bowel disease in Asia. Gastroenterol Hepatol (N Y) 2013; 9: 28–30. [PMC free article] [PubMed] [Google Scholar]
- 48.Dzierzanowska-Fangrat K, Raeiszadeh M, Dzierzanowska D, Gladkowska-Dura M, Celinska-Cedro D, Crabtree JE. IgG subclass response to Helicobacter pylori and CagA antigens in children. Clin Exp Immunol 2003; 134: 442–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Torres J, Camorlinga-Ponce M, Perez-Perez G, Muñoz L, Muñoz O. Specific serum immunoglobulin G response to urease and CagA antigens of Helicobacter pylori in infected children and adults in a country with high prevalence of infection. Clin Diagn Lab Immunol 2002; 9: 97–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Y, Blaser MJ. Inverse associations of Helicobacter pylori with asthma and allergy. Arch Intern Med 2007; 167: 821–827. [DOI] [PubMed] [Google Scholar]
- 51.Hornsby MJ, Huff JL, Kays RJ, Canfield DR, Bevins CL, Solnick JV. Helicobacter pylori induces an antimicrobial response in rhesus macaques in a cag pathogenicity island-dependent manner. Gastroenterology 2008; 134: 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wehkamp J, Fellermann K, Stange EF. Human defensins in Crohn’s disease. Chem Immunol Allergy 2005; 86: 42–54. [DOI] [PubMed] [Google Scholar]
- 53.Ho S, Pothoulakis C, Koon HW. Antimicrobial peptides and colitis. Curr Pharm Des 2013; 19: 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramasundara M, Leach ST, Lemberg DA, Day AS. Defensins and inflammation: the role of defensins in inflammatory bowel disease. J Gastroenterol Hepatol 2009; 24: 202–208. [DOI] [PubMed] [Google Scholar]
- 55.Wehkamp J, Harder J, Weichenthal M, et al. Inducible and constitutive beta-defensins are differentially expressed in Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis 2003; 9: 215–223. [DOI] [PubMed] [Google Scholar]
- 56.Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest 2004; 113: 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peek RM, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer 2002; 2: 28–37. [DOI] [PubMed] [Google Scholar]
- 58.Atherton JC, Tham KT, Peek RM, Cover TL, Blaser MJ. Density of Helicobacter pylori infection in vivo as assessed by quantitative culture and histology. J Infect Dis 1996; 174: 552–556. [DOI] [PubMed] [Google Scholar]
- 59.Ng SC, Tang W, Ching JY, et al. Incidence and phenotype of inflammatory bowel disease based on results from the Asia-pacific Crohn’s and colitis epidemiology study. Gastroenterology 2013; 145: 158–165.e2. [DOI] [PubMed] [Google Scholar]
- 60.Burisch J, Pedersen N, Čuković-Čavka S, et al. East-West gradient in the incidence of inflammatory bowel disease in Europe: the ECCO-EpiCom inception cohort. Gut 2014; 63: 588–597. [DOI] [PubMed] [Google Scholar]
- 61.Shi HY, Levy AN, Trivedi HD, Chan FKL, Ng SC, Ananthakrishnan AN. Ethnicity Influences Phenotype and Outcomes in Inflammatory Bowel Disease: A Systematic Review and Meta-analysis of Population-based Studies. Clin Gastroenterol Hepatol 2018; 16: 190–197.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ng SC, Kaplan GG, Tang W, et al. Population Density and Risk of Inflammatory Bowel Disease: A Prospective Population-Based Study in 13 Countries or Regions in Asia-Pacific. Am J Gastroenterol 2018; published online September 3 DOI: 10.1038/s41395-018-0233-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.