

Abstract
Our developmental studies provide an insight into the pathogenesis of heart failure in adults. These studies reveal a mechanistic link between fetal cardiomyocytes and pathologically stressed adult cardiomyocytes at the level of chromatin regulation. In embryos, chromatin-regulating factors within the cardiomyocytes respond to developmental signals to program cardiac gene expression to promote cell proliferation and inhibit premature cell differentiation. In the neonatal period, the activity of these developmental chromatin regulators is quickly turned off in cardiomyocytes, coinciding with the cessation of cell proliferation and advance in cell differentiation toward adult maturity. When the mature hearts are pathologically stressed, those chromatin regulators essential for cardiomyocyte development in embryos are reactivated, triggering gene reprogramming to a fetal-like state and pathological cardiac hypertrophy. Furthermore, in the study of chromatin regulation and cardiac gene expression, we identified a long noncoding RNA that interacts with chromatin remodeling factor to regulate the cardiac response to environmental changes. This article is part of a Special Issue entitled: Cardiomyocyte Biology: Integration of Developmental and Environmental Cues in the Heart edited by Marcus Schaub and Hughes Abriel.
Keywords: Chromatin, Brg1, BAF, Myheart, Mhrt, Development, Gene expression, Hypertrophy, Heart failure
1. Introduction
The genetic information is encoded by DNA sequence, and epigenetic alternations of chromatin can regulate the expression of genetic information. Chromatin is evolved to associate DNA with histone proteins, providing a scaffold to package the long 1.7-meter DNA strands into a small 6-μm nucleus. The basic packaging unit of chromatin is the nucleosome, which contains ~147 bp DNA enveloping an octamer core of histones (two molecules of each of the canonical H2A, H2B, H3 and H4 histones) (Fig. 1). Chemical interactions between histones and DNA, as well as the position of nucleosomes in the genome, can alter the accessibility of DNA sequence to transcription regulation. These changes of chromatin are mediated by three major mechanisms: covalent modifications of histone protein side chains, covalent modifications of DNA bases, and non-covalent changes of the composition and positioning of nucleosomes (Fig. 1) [1]. Such covalent and non-covalent changes of nucleosomes enable the chromatin scaffold to respond to environmental signals to modulate gene expression and alter the physiological state of cells.
Fig. 1.
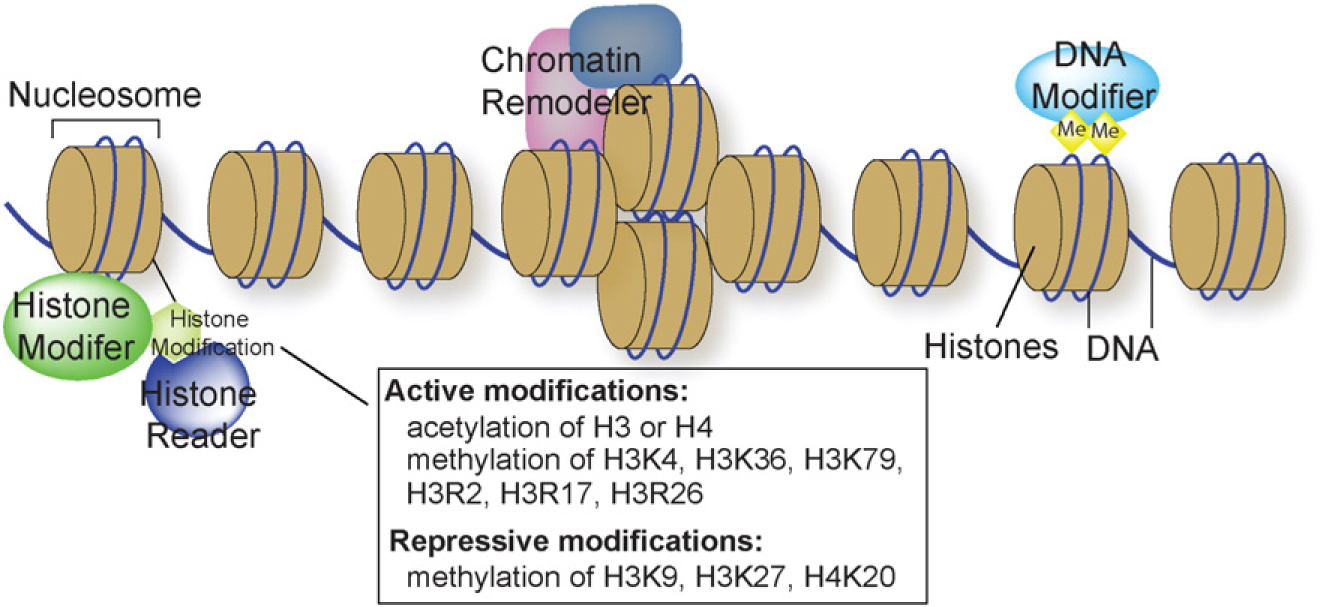
Chromatin structure and major regulatory mechanisms. DNA is packaged by nucleosome units, each composed of ~147 bp of DNA (blue line) that wrap around histones (yellow). Such chromatin structure is controlled by three major regulatory mechanisms. Chromatin remodelers control the composition and position of nucleosomes. Histone modifiers catalyze covalent modifications of histones, which are recognized by histone readers. DNA modifiers, primarily DNA methyltransferases, are responsible for DNA methylation.
Histones can be modified by various histone-modifying enzymes that catalyze methylation, acetylation, or other chemical processes, whereas the cytosine base of DNA can be methylated by DNA methyltransferases. On the other hand, the position and histone composition (canonical vs non-canonical histones) of nucleosomes are regulated by a group of ATP-dependent nucleosome/chromatin-remodeling complexes. A critical component of the chromatin-remodeling complex is the ATPase subunit, which hydrolyzes ATP to mobilize nucleosomes or change their histone composition. In vertebrates, there are four families of chromatin-remodeling complexes defined by the evolutionarily conserved ATPase subunit: SWI/SNF (Switch defective/sucrose nonfermenting), ISWI (imitation switch), and CHD (chromodomain, helicase, DNA binding), and INO80 (inositol requiring 80) (reviewed in [2–5]). Each ATPase protein consists of a conserved catalytic domain (for ATP hydrolysis) and various functional domains that recognize modified histones, regulate ATP catalytic activity, or mediate protein–protein interactions. These specific domains characterize the ATPase from each of the four families of chromatin-remodeling complexes.
Recent studies revealed that long-noncoding RNAs (lncRNAs) can partner with or counteract epigenetic regulators to control chromatin and gene expression. lncRNAs are defined as non-peptide/protein-coding RNA transcripts that are longer than 200 nucleotides, and these RNAs are largely discovered by next generation sequencing-based transcriptome profiling. The expression of lncRNAs is highly tissue-specific and is dynamically regulated by environmental cues from physiological signals or pathological stimuli. lncRNA promoters are enriched with histone 3 lysine 4 trimethylation (H3K4me3), RNA polymerase II (Pol II), and DNase I hypersensitivity sites [6], suggesting active tissue regulation of lncRNA expression. However, it remains largely unknown how lncRNAs function in vivo and how they are regulated.
lncRNAs have biochemical abilities to interact with a wide range of molecules to form RNA–RNA, RNA–DNA, or RNA–protein complexes [7]. With this capacity, lncRNAs show broad associations with the epigenetic machinery to control covalent modifications of chromatin [8–10]. More recently, the epigenetic roles of lncRNAs are extended to the regulation of non-covalent nucleosome remodeling process through a direct physical interaction with the ATP-dependent chromatin-remodeling factor [11]. Here, we use the chromatin remodeler Brg1 and cardiac-specific lncRNA Myheart (Mhrt) as an example to illustrate how cardiomyocytes respond to pathophysiological signals by changing the chromatin-remodeling factor–lncRNA interactions to reprogram gene expression.
1.1. Pathophysiological roles of Brg1 in heart development and disease
Brg1 is an essential ATPase component of the SWI/SNF-like BAF (brahma-associated factor) chromatin-remodeling complex. In embryos, the Brg1/BAF complex responds to developmental signals to regulate the proliferation and differentiation of fetal cardiomyocytes [12]. Tissue-specific disruption of Brg1 in fetal cardiomyocytes results in abnormal heart development with thin myocardium and absence of interventricular septum [12]. These mutant embryos die around embryonic day 11.5 (E11.5) [12], likely due to pump failure of the heart. In fetal cardiomyocytes, Brg1 promotes cell proliferation by maintaining Bmp10 and suppressing p57kip2 (Cdkn1c) expression [12]. Bmp10 is a cardiomyocyte growth factor [13], whereas p57kip2 is a cyclin-dependent kinase inhibitor that suppresses cell cycle progression. In embryonic hearts, Bmp10 is abundantly expressed in cardiomyocytes, and there is no p57kip2 expression. In the absence of Brg1, Bmp10 expression is compromised, with p57kip2 ectopically expressed in cardiomyocytes to inhibit cell proliferation, leading to thin myocardium and absence of interventricular septum. Restoration of Bmp10 in Brg1 mutants rescues such cardiomyocyte proliferation defects. The Brg1– Bmp10 pathway is therefore an essential mechanism that respond to developmental signals to control cardiomyocyte proliferation. Brg1 does not regulate Bmp10 expression at the promoter level [12], suggesting that Brg1 may signal through certain genetic pathways to control Bmp10. The exact mechanism of how Brg1 maintains Bmp10 expression in developing cardiomyocytes will require further investigations.
Although the Brg1-null cardiomyocytes fail to proliferate, these cells undergo premature differentiation, as evidenced by increased thickness and segmentation of myofibrils and by gene expression switch from fetal to adult isoform, such as the switch from predominantly β-myosin heavy chain (β-MHC or Myh7) to predominantly -MHC (Myh6) expression [12]. In mice, Myh6 is the dominant isoform of the MHC molecular motor gene expressed in adult heart ventricles, while Myh7 is the primary isoform in embryonic ventricles. In response to developmental signals that trigger cardiomyocyte proliferation, Brg1 suppresses premature differentiation of fetal cardiomyocytes. Within fetal cardiomyocytes, Brg1 complexes with histone deacetylase (HDAC) and poly (ADP ribose) polymerases (PARP) on the promoters of Myh genes to concurrently repress Myh6 and activate Myh7 expression [12]. The Brg1/HDAC/PARP chromatin complex thus maintains an expression of the fetal isoform of MHC in developing cardiomyocytes to preserve the fetal state of cardiomyocyte differentiation.
In fetal cardiomyocytes, Brg1 controls cell proliferation and differentiation in parallel through two independent pathways [12]. In whole embryo culture studies, recombinant Bmp10 proteins rescue the proliferation defects of Brg1-null cardiomyocytes, without changing the premature switch to Myh6 expression in those cells. Conversely, pharmacological inhibition of HDAC or PARP in cultured wild-type embryos produces the premature Myh switch but without affecting cardiomyocyte proliferation. These observations suggest that Brg1 governs two independent pathways (Bmp10 and HDAC/PARP) to simultaneously control fetal cardiomyocyte proliferation and differentiation. Brg1 therefore serves as a critical switch that is turned on by developmental signals to control cardiomyocyte proliferation and differentiation.
Although highly expressed in the fetal heart, Brg1 is down-regulated in the neonatal heart [12], coinciding with the physiological Myh7-to-Myh6 switch and the maturation of fetal cardiomyocyte into postmitotic, highly-differentiated adult cardiomyocytes. The molecular actions of Brg1 in fetal cardiomyocytes suggest that such Brg1 turnoff in the neonatal period is essential for neonatal cardiomyocytes to cease proliferation and undergo differentiation to become mature cardiomyocytes. The mature adult hearts express low level of Brg1 in cardiomyocytes; however, when the mature cardiomyocytes are pathologically stressed, Brg1 expression is reactivated, triggering pathological Myh6-to-Myh7 switch and adverse cardiac remodeling with hypertrophy and interstitial fibrosis [12]. Genetic disruption of Brg1 in stressed cardiomyocytes prevents the development of cardiac hypertrophy, fibrosis, and failure. Once reactivated by cardiac stress, Brg1 complexes with its embryonic partners HDAC and PARP, and this complex binds to Myh promoters to simultaneously repress Myh6 and activate Myh7 as it does in fetal cardiomyocytes. Besides triggering pathological Myh switch, Brg1 binds to the promoter and activates the expression of Osteopontin (Opn), a pro-fibrotic factor secreted by cardiomyocytes to induce tissue fibrosis [11]. Brg1 also triggers the switch from a predominant fatty acid to glucose metabolism in the stressed hearts by controlling the expression of many metabolic genes (Chang lab, unpublished data). All these Brg1-mediated changes of gene expression in the stressed hearts contribute to the development of pathological hypertrophy and heart failure. BRG1 expression also increases in human hypertrophic hearts, with BRG1 level correlating strongly with the degree of pathological MYH switch and the severity of hypertrophy [12]. The mouse and human tissue studies suggest an evolutionarily conserved Brg1-mediated mechanism in human heart disease.
1.2. Stress-induced chromatin changes in the heart
Brg1 is a chromatin remodeler that is induced environmental stress to reprogram cardiac gene expression and trigger hypertrophy and heart failure [12]. The stress-induced assembly of three classes of chromatin-regulating factors—Brg1, HDAC, and PARP—on the Myh gene promoters suggests that chromatin is a place where stress-response signals converge to reprogram gene expression. However, it remains largely unknown how chromatin-regulating factors act in concert to modulate chromatin scaffold in the heart. The Myh6 and Myh7 gene loci, antithetically regulated under different pathophysiological conditions, contain crucial information about epigenetic regulation of cardiac gene expression. Encoded within the Myh gene loci are the cardiac epigenetic regulator miR208a (within Myh6 loci) [15–17], miR208b (within Myh7 loci) [18], and Myheart (within Myh7 loci) [11] (Fig. 2a). Further analysis of chromatin scaffold changes of Myh6 loci at different pathophysiological states revealed two new classes of chromatin modifiers—G9a/GLP histone methyltransferase and DNA methyltransferase (DNMT)—that are necessary for stress-induced cardiac hypertrophy and failure (Chang lab, unpublished results). In mouse hearts, G9a/Glp and Dnmt3 have expression patterns similar to that of Brg1: they are highly expressed in fetal hearts, their expression down-regulated in mature adult hearts but reactivated in pathologically stressed hearts. Once activated in stressed hearts, Brg1, G9a/Glp, and Dnmt3 proteins form a physical complex in the cardiomyocytes. Brg1 sequentially recruits G9a/Glp and Dnmt3 to Myh6 (but not Myh7) promoter to assemble a repressive chromatin scaffold, composed of methylated histone 3 lysine 9 (H3K9) catalyzed by G9a/Glp as well as methylated CpG dinucleotides catalyzed by Dnmt3. This repressive chromatin silences Myh6 expression in stressed hearts, contributing to the decline of cardiac contractility.
Fig. 2.
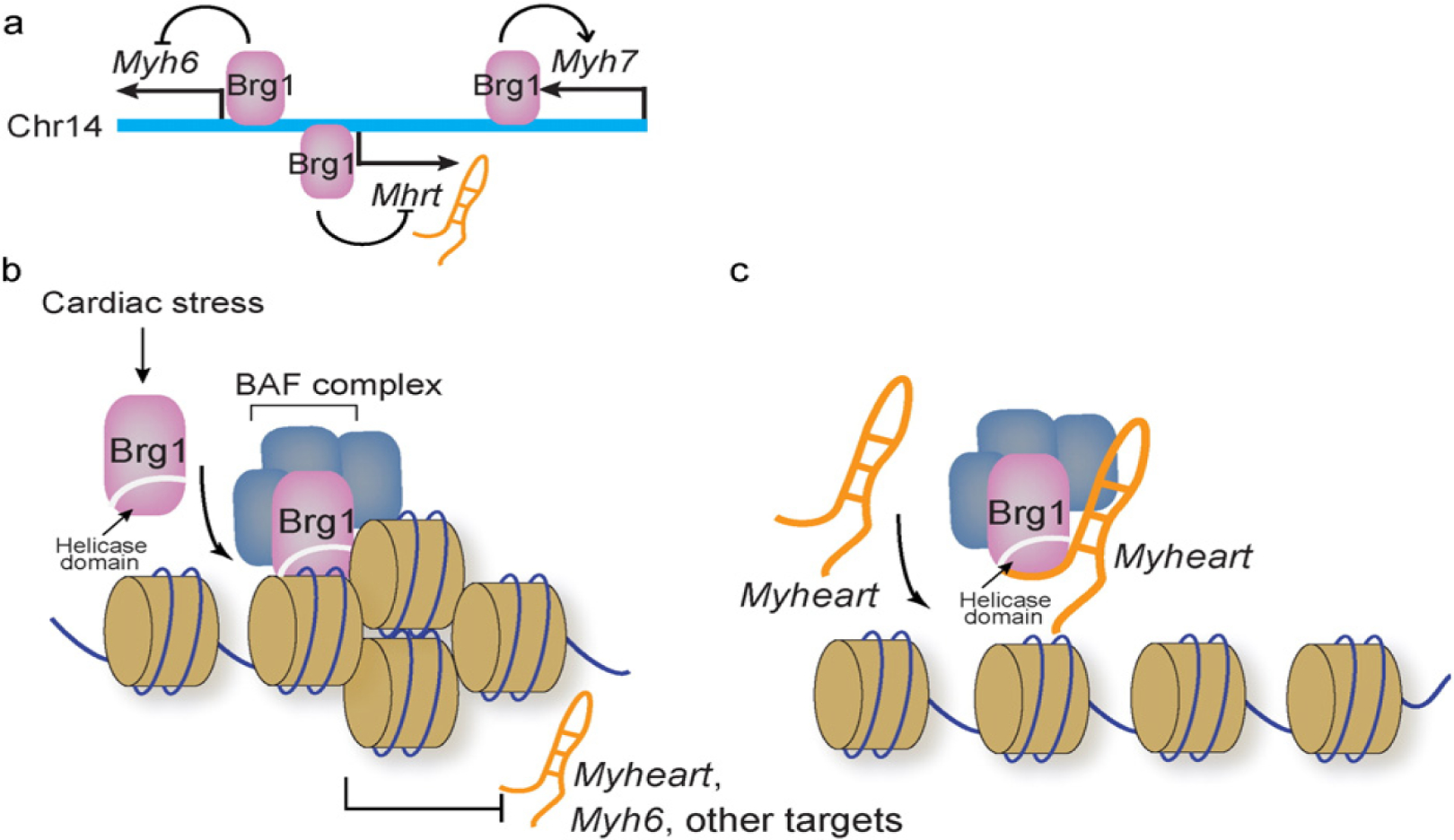
Molecular feedback circuit between Mhrt and Brg1. a. Relative genomic location of Myh6, Myh7, and Mhrt. All three genes are regulated by Brg1.
Genetic disruption of G9a/Glp or Dnmt3a in mouse adult cardiomyocytes or pharmacological inhibition of G9a/Glp or Dnmt reduces the repressive chromatin marks on Myh6, lessens cardiac hypertrophy, and prevents the decline of heart function. The repressive chromatin assembly on Myh6 loci requires sequential molecular interactions between Brg1, G9a/Glp, and Dnmt3. Brg1 first recruits G9a/Glp, which then recruits Dnmt3 to the Myh6 promoter to successively catalyze histone and DNA methylation. G9a/GLP and DNMT, like BRG1, are also increased in human hypertrophic hearts, and their expression levels correlate with disease severity and with the degree of H3K9 and CpG methylation of Myh6 promoter. These findings suggest a conserved chromatin mechanism in human cardiomyopathy.
Brg1-governed sequential recruitment of G9a/Glp and Dnmt3 to Myh6 loci demonstrates a new mechanistic link from chromatin-remodeling factor to H3K9 and CpG methylation, which is critical for the pathogenesis of heart failure. The repressive chromatin environment on Myh6 promoter is created by at least five different classes of chromatin-regulating factors: Brg1, G9a/GLP, Dnmt3, Hdac, and Parp [12] (Chang lab, unpublished results). Among these factors, only the expression of Brg1, G9a/Glp, and Dnmt3—but not Hdac (Hdac1, 2, 3) or Parp1—is activated by pathological stress in mouse or human hearts. To further define the chromatin mechanism that responds to environmental cues, it will be important to identify new components of the stress-induced epigenetic complex and determine how these components biochemically cooperate to target specific genomic sites to orchestrate cardiac epigenomic response to environmental stress.
1.3. Genome-targeting mechanism of Brg1 in the heart
It is physiologically crucial for chromatin-remodeling complexes to localize to specific regions of the genome to coordinate gene expression [2]; however, the targeting mechanism of chromatin-remodeling complexes is largely undefined. Our studies showed a new targeting mechanism encoded within the ATPase subunit of chromatin-remodeling complexes [11]. We found that the helicase domain (also known as ATPase domain) of Brg1 is crucial for Brg1 to bind to its genomic target sites to control cardiac gene expression [11]. This evolutionarily conserved helicase domain belongs to the Deadbox RNA helicase subfamily of the superfamily 2 (SF2) helicase [2,19–21]. It consists of two homologous “RecA-like” domains (D1 and D2), which fold upon each other to form two clefts that bind ATP on one side and RNA nucleotides on the other. This structure couples ATP hydrolysis and RNA unwinding [22,23]. ATP hydrolysis by this ATPase/helicase domain is essential for driving chromatin remodeling activity of the BAF complex. However, how this same domain anchors Brg1 to the chromatin targets is previously unknown. Molecular and biochemical studies showed that this Brg1 helicase domain has dual nucleotide-binding specificities: it is capable of binding RNA and chromatinized DNA. In the hearts or cultured cells, Brg1 requires this domain to target to its downstream gene promoters to regulate gene expression. Disruption of the helicase domain prevents Brg1 from binding to its target sites to execute chromatin remodeling and gene regulation. Interestingly, both biochemical and cell-based as-says showed that Brg1 helicase domain can only bind to DNA that is chromatinized or histone-bound, but not to the naked DNA. The structural basis of how this domain distinguishes naked from chromatinized DNA is still unknown. In the studies of Brg1 and Myh expression regulation, we uncovered a cardiac-specific lncRNA (Mhrt) encoded within the Myh genomic loci. This lncRNA can disrupt the helicase-based targeting mechanism to inhibit Brg1’s chromatin activity and protect the heart from pathological hypertrophy (Fig. 2b) [11].
1.4. lncRNA and chromatin remodeling in stressed hearts
lncRNAs are known to be associated with human heart disease. These RNAs include MIAT (myocardial infarction associated transcript) [24], ANRIL (Antisense non-coding RNA of INK4/ARF locus) [25–27], KCNQ1OT1 (KCNQ1 opposite strand/antisense transcript 1) [28], LIPCAR (mitochondrially encoded long non-coding cardiac associated RNA) [29], and Myheart (Myosin heavy chain associated RNA transcript) [11]. lncRNA profiling from cardiac tissues of patients with left ventricular assistance device suggested that lncRNA signatures can predict the clinical outcome of these patients [30]. The mechanisms of these disease-associated lncRNAs are obscure.
The Myh-associated RNA transcripts or Myheart (Mhrt) represent a cluster of alternatively spliced nuclear lncRNA transcripts that are encoded within the Myh genomic loci [11]. These RNAs are transcribed from the promoter of Myh6 in an antisense direction into the genomic site of Myh7. In the adult mouse tissues, Mhrt is predominantly expressed in the heart with no or minimal expression in other tissues. In embryonic hearts Mhrt transcripts are expressed at low levels, and its abundance increases as the heart matures to adulthood. When the adult mouse heart is pathologically stressed, Mhrt expression is suppressed, and such suppression is essential for the development of heart failure. Transgenic restoration of Mhrt expression in stressed mouse cardiomyocytes prevents the heart from developing pathological hypertrophy and failure, indicating a cardioprotective role of Mhrt against environmental stress (Fig. 2b). In Mhrt-rescued hearts, Mhrt directly reverses Brg1-mediated pathological Myh switch and activation of the profibrotic Opn. Biochemical, cell-based, and genetic studies showed that Mhrt directly binds to the helicase domain of Brg1 to compete with chromatinized DNA for the binding of Brg1 (Fig. 2c). As a result, when Mhrt expression is transgenically restored in the stressed hearts, Brg1 is unable to find its target gene promoters, including Myh6, Myh7, and Opn [11]. Such removal of Brg1 from its gene targets alters gene expression in the stressed hearts. The helicase’s dual-binding specificity for nucleotides (RNA and chromatinized DNA) not only enables Brg1 to target the genome, but also allows such targeting by chromatin remodelers to be regulated by lncRNAs in response to environmental cues.
2. Discussion
The opposite regulations of Myh6 and Myh7 by the Brg1 complex can be caused at least by two different processes. The first one involves differential recruitment of chromatin regulators by Brg1 to Myh promoters. For example, Brg1 recruits the chromatin repressor G9a and Dnmt3 on Myh6 promoter, but not Myh7 promoter. This is associated with increased repressive chromatin marks on Myh6 promoter—H3K9me2 (catalyzed by G9a) and methylated CpG (catalyzed by Dnmt3)—causing its repression. These chromatin factor and histone mark changes provide a partial explanation for the differential regulation of Myh6 and Myh7. The epigenetic factor recruitment involves protein–protein interactions and may require additional information from local epigenetic landscape and/or DNA sequence. The second process is related to differential recruitment of transcription factors to Myh sites. YY1 is known to repress Myh6 expression, whereas NFAT1 and AP-1 are activators for Myh7 [31–34]. Brg1 may recruit different transcription repressors or activators to Myh sites to execute differential gene control. The transcription factor recruitment may require DNA sequence information. Further studies are needed to decipher how Brg1 interacts with epigenetic and transcription factors as well as with local DNA sequence to control the Myh promoters.
Brg1 and Mhrt form a feedback circuit in the heart, and such molecular circuit can be perturbed by environmental stress to cause cardiac hypertrophy and failure (Fig. 2b) [11,12]. Mhrt is capable of suppressing Brg1’s activity in normal hearts to protect the hearts from aberrant activities of Brg1. However, pathological stress activates the expression of Brg1, which then forms a repressive chromatin complex with HDAC and PARP to inhibit Mhrt expression, resulting in aberrant gene reprograming and heart failure. In normal hearts, Brg1 level is too low to overcome its suppression by the abundant Mhrt. Conversely, in stressed hearts, the level of Brg1 is much enhanced, allowing Brg1 to over-run Mhrt to gain its chromatin targeting to the Mhrt promoter and shut down Mhrt expression. The more Mhrt is shutdown, the further gain of Brg1 activity to turn off Mhrt, thus constituting a positive-feedback molecular competition that tips the balance toward Brg1 dominance in hearts that experience sustained environmental stress. The dose–dependent interactions among Brg1, Mhrt, and Mhrt promoter were demonstrated biochemically and in cell-based studies [11], providing a molecular basis for the pathogenesis of sustained environmental stress. Moreover, this positive Brg1–Mhrt feedback loop suggests the existence of a Brg1 expression threshold that converts a heart from a healthy into a disease state. Expression studies of Brg1 in hypertrophic human hearts are consistent with such threshold effect of Brg1 in triggering heart disease [12]. The chromatin–lncRNA interactions thus exemplify an epigenetic regulatory mechanism by which cardiomyocytes respond to environmental signals.
Transparency document.
The Transparency document associated with this article can be found, in online version.
b and c. Brg1, the ATPase subunit of BAF chromatin-remodeling complex, binds to its genomic targets through the helicase domain. Brg1 expression is activated by cardiac stress, and in stressed hearts Brg1 binds to Mhrt promoter to repress its expression (b). Conversely, Mhrt can bind to Brg1 helicase domain to prevent Brg1 from binding to its genomic targets (c). This reciprocal inhibition constitutes a molecular feedback circuit to maintain cardiac homeostasis.
Acknowledgments
C-P.C. is Charles Fisch Scholar of Cardiology and supported by American Heart Association (AHA, Established Investigator Award), National Institutes of Health (NIH, R01HL118087, R01HL121197), Indiana University (IU) School of Medicine-IU Health Strategic Research Initiative, and the IU Physician-Scientist Initiative, endowed by Lilly Endowment, Inc.
References
- [1].Han P, Hang CT, Yang J, Chang CP, Chromatin remodeling in cardiovascular development and physiology, Circ. Res 108 (2011) 378–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Clapier CR, Cairns BR, The biology of chromatin remodeling complexes, Annu. Rev. Biochem 78 (2009) 273–304. [DOI] [PubMed] [Google Scholar]
- [3].Wu JI, Lessard J, Crabtree GR, Understanding the words of chromatin regulation, Cell 136 (2009) 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ho L, Crabtree GR, Chromatin remodelling during development, Nature 463 (2010) 474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Saha A, Wittmeyer J, Cairns BR, Chromatin remodelling: the industrial revolution of DNA around histones, Nat. Rev. Mol. Cell Biol 7 (2006) 437–447. [DOI] [PubMed] [Google Scholar]
- [6].Iyer MK, et al. , The landscape of long noncoding RNAs in the human transcriptome, Nat. Genet (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Quinn JJ, et al. , Revealing long noncoding RNA architecture and functions using domain-specific chromatin isolation by RNA purification, Nat. Biotechnol 32 (2014) 933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rinn JL, lncRNAs: linking RNA to chromatin, Cold Spring Harb. Perspect. Biol 6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rinn JL, Chang HY, Genome regulation by long noncoding RNAs, Annu. Rev. Biochem 81 (2012) 145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Devaux Y, et al. , Long noncoding RNAs in cardiac development and ageing, Nat. Rev. Cardiol (2015). [DOI] [PubMed] [Google Scholar]
- [11].Han P, et al. , A long noncoding RNA protects the heart from pathological hypertrophy, Nature 514 (2014) 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hang CT, et al. , Chromatin regulation by Brg1 underlies heart muscle development and disease, Nature 466 (2010) 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chen H, et al. , BMP10 is essential for maintaining cardiac growth during murine cardiogenesis, Development 131 (2004) 2219–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Montgomery RL, et al. , Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure, Circulation 124 (2011) 1537–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Callis TE, et al. , MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice, J. Clin. Invest 119 (2009) 2772–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].van Rooij E, et al. , Control of stress-dependent cardiac growth and gene expression by a microRNA, Science 316 (2007) 575–579. [DOI] [PubMed] [Google Scholar]
- [18].van Rooij E, et al. , A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance, Dev. Cell 17 (2009) 662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jankowsky E, RNA helicases at work: binding and rearranging, Trends Biochem. Sci 36 (2011) 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Byrd AK, et al. , Dda helicase tightly couples translocation on single-stranded DNA to unwinding of duplex DNA: dda is an optimally active helicase, J. Mol. Biol 420 (2012) 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tang L, Nogales E, Ciferri C, Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription, Prog. Biophys. Mol. Biol 102 (2010) 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Linder P, Jankowsky E, From unwinding to clamping — the DEAD box RNA helicase family, Nat. Rev. Mol. Cell Biol 12 (2011) 505–516. [DOI] [PubMed] [Google Scholar]
- [23].Mallam AL, Del Campo M, Gilman B, Sidote DJ, Lambowitz AM, Structural basis for RNA-duplex recognition and unwinding by the DEAD-box helicase Mss116p, Nature 490 (2012) 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ishii N, et al. , Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction, J. Hum. Genet 51 (2006) 1087–1099. [DOI] [PubMed] [Google Scholar]
- [25].Pasmant E, et al. , Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF, Cancer Res. 67 (2007) 3963–3969. [DOI] [PubMed] [Google Scholar]
- [26].Jarinova O, et al. , Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus, Arterioscler. Thromb. Vasc. Biol 29 (2009) 1671–1677. [DOI] [PubMed] [Google Scholar]
- [27].Liu Y, et al. , INK4/ARF transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis, PLoS One 4 (2009), e5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Vausort M, Wagner DR, Devaux Y, Long noncoding RNAs in patients with acute myocardial infarction, Circ. Res 115 (2014) 668–677. [DOI] [PubMed] [Google Scholar]
- [29].Kumarswamy R, et al. , Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure, Circ. Res 114 (2014) 1569–1575. [DOI] [PubMed] [Google Scholar]
- [30].Yang KC, et al. , Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support, Circulation 129 (2014) 1009–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].von Harsdorf R, et al. , Identification of a cis-acting regulatory element conferring in-ducibility of the atrial natriuretic factor gene in acute pressure overload, J. Clin. Invest 100 (1997) 1294–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].McKinsey TA, Olson EN, Toward transcriptional therapies for the failing heart: chemical screens to modulate genes, J. Clin. Invest 115 (2005) 538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sucharov CC, Dockstader K, McKinsey TA, YY1 protects cardiac myocytes from pathologic hypertrophy by interacting with HDAC5, Mol. Biol. Cell 19 (2008) 4141–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mariner PD, Luckey SW, Long CS, Sucharov CC, Leinwand LA, Yin Yang 1 represses α-myosin heavy chain gene expression in pathologic cardiac hypertrophy, Biochem. Biophys. Res. Commun 326 (2004) 79–86. [DOI] [PubMed] [Google Scholar]