

Abstract
Objective:
Abdominal aortic aneurysm (AAA) is a life-threatening vascular disease without an effective pharmaceutical treatment. Genetic studies have proved the involvement of smooth muscle phenotype switch in the development of AAA. The alpha subunit of the heterotrimeric G stimulatory protein (Gsα) mediates receptor-stimulated production of cyclic adenosine monophosphate (cAMP). However, the role of smooth muscle Gsα in AAA formation remains unknown.
Approach and results:
In this study, mice with knockout of smooth muscle-specific Gsα (GsαSMKO) were generated by cross-breeding Gsαflox/flox mice with SM22-CreERT2 transgenic mice, induced in adult mice by tamoxifen treatment. Gsα deficiency induced a smooth muscle phenotype switch from a contractile to a synthetic state. Mechanically, Gsα deletion reduced cAMP level and increased the level of human antigen R (HuR), which binds with the adenylate uridylate–rich elements of the 3′ untranslated region of Krüppel-like factor 4 (KLF4) mRNA, thereby increasing the stability of KLF4. Moreover, genetic knockdown of HuR or KLF4 rescued the phenotype switch in Gsα-deficient smooth muscle cells. Furthermore, with acute infusion of angiotensin II, the incidence of AAA was markedly higher in ApoE−/−/GsαSMKO than ApoE−/−/Gsαflox/flox mice and induced increased elastic lamina degradation and aortic expansion. Finally, the levels of Gsα and SM α-actin were significantly lower while those of HuR and KLF4 were higher in human AAA samples than adjacent nonaneurysmal aortic sections.
Conclusions:
Gsα may play a protective role in AAA formation by regulating the smooth muscle phenotype switch and could be a potential therapeutic target for AAA disease.
Keywords: Abdominal aortic aneurysm, Gsα, Phenotype switch, HuR, KLF4
1. Introduction
Abdominal aortic aneurysm (AAA) represents the top 20th cause of mortality in the United States and results in about 15,000 deaths per year [1]. In humans > 65 years old, the incidence of AAA is up to 9% [2]. The pathological characteristics of AAA are localized structural deterioration of the aortic wall and progressive aortic dilation [3]. AAA is often accompanied by downregulation of multiple contractile proteins in aortic smooth muscle cells (SMCs) [4,5]. Furthermore, aortic intimal layer injury and inflammatory cell infiltration participate in the aneurysm progression via complicated mechanisms such as cytokine secretion and increased reactive oxygen species levels [3,6]. Finally, vascular collagen and elastin extracellular matrix is thought to undergo degradation by matrix metalloproteinases and contribute to AAA formation [4,7]. However, the detailed mechanisms underlying AAA formation remain unknown.
SMCs have remarkable phenotypic plasticity different from some terminally differentiated cells [8]. The SMC phenotype switch was defined as transformation from a quiescent contractile state to a synthetic state [9]. This process is characterized by the coordinate downregulation of differentiated SMC markers such as SM α-actin (α-SMA), SM22α and SM myosin heavy chain (SMMHC) required for contraction [10]. The SMC phenotype switch is considered to play an important role in the development of cardiovascular diseases such as atherosclerosis and AAA [11–13]. Many studies have identified proteins such as Krüppel-like factor 4 (KLF4) regulating the SMC phenotype switch [14]. Smooth muscle-specific deletion of KLF4 attenuated AAA formation via phenotypic modulation [15]. However, the mechanism that regulates KLF4 expression and the SMC phenotype switch remains unclear.
The alpha-subunit of the stimulatory G protein (Gsα) is expressed in many cell types and is responsible for receptor-stimulated cyclic adenosine monophosphate (cAMP) generation and activation of the downstream signaling pathways [16]. For example, in rat aortic SMCs, cAMP could decrease the expression of human antigen R (HuR), which is an mRNA binding protein and increase the stability of target mRNAs [17]. Gsα dysfunction is involved in many diseases. The endothelium-specific Gsα knockout mice were embryonic lethal due to abnormal vessel structure and serious bleeding [18]. However, mice overexpressing a dominant negative Gsα-mutant in heart leads to decreased β-adrenergic responsiveness and is protective against ISO-induced hypertrophy [19]. In our recent report, we found that Gsα could regulate intestinal smooth muscle contraction in mice [20]. However, the roles of Gsα in the SMC phenotype switch and AAA formation are undefined.
In the present study, we used mice with SM22-CreERT2–mediated Gsα knockout to demonstrate that Gsα deficiency in smooth muscle exacerbates angiotensin II (AngII)-induced AAA formation by regulating the SMC phenotype switch.
2. Materials and methods
2.1. Animals
Gsαflox/flox mice were kindly provided by Dr. Lee S. Weinstein (generation /characterization of this mouse strain described previously [21]). SM22-CreERT2 mice expressing a tamoxifen-inducible Cre recombinase under control of the SM22 promoter [22] were kindly provided by Dr. Robert Feil. ApoE−/− mice were from Vital River Laboratories (Distributor of Jackson Laboratory, Beijing, China). All mice were C57BL/6 J background. Mice with smooth muscle-specific Gsα knockout (GsαSMKO) were generated as follows. Gsαflox/flox mice were cross-bred with SM22-CreERT2 mice to produce Gsαflox/flox/Cre+ mice, which were injected with tamoxifen (1 mg/day) intraperitoneally for 5 consecutive days at age 8 weeks to generate GsαSMKO mice. ApoE−/−/GsαSMKO double knockout mice were generated by crossing Gsαflox/flox/Cre+ mice with ApoE−/− mice, induced in adult mice by tamoxifen treatment. For AngII-induced AAA, after 2 weeks of tamoxifen injection, male ApoE−/−/GsαSMKO and ApoE−/−/Gsαflox/flox mice were infused with AngII (1000 ng/kg/min) via mini-osmotic pumps (Alzet, Model 2004, DURECT Corp., Cupertino, CA). Blood pressure was monitored to confirm effective AngII. The status of mice was observed and Kaplan-Meier curves were plotted during the experiment. Mice were sacrificed and aortic aneurysmal segments were harvested after 28 days. All experiments were conducted in accordance with the guidelines of the Animal Care and Use Committee of Shandong University.
2.2. Statistical analysis
All statistical analyses involved using GraphPad Prism 7.0 (GraphPad Software, San Diego, CA). Two groups were compared by Student t-test and > 2 groups by 2-way ANOVA and Bonferroni post-tests. Data are presented as mean ± SEM. P < .05 was considered statistically significant.
Detailed descriptions of other unmentioned materials, methods, and experimental procedures are available in Supplemental Materials and Methods.
3. Results
3.1. Generation and analysis of GsαSMKO mice
To elucidate the biological significance of Gsα expression in vascular SMCs in vivo, Gsαflox/flox mice were cross-bred with SM22-CreERT2 mice expressing a tamoxifen-activated Cre recombinase to generate Gsαflox/+/Cre+ mice. These were further intercrossed to obtain Gsαflox/flox/Cre+ mice, which were injected with tamoxifen at age 8 weeks to generate GsαSMKO mice. Littermate Gsαflox/flox/Cre− mice with the same dose of tamoxifen were controls.
To confirm smooth muscle-specific Gsα deletion in GsαSMKO mice, immunofluorescence was used to detect Gsα expression in aortas. SMCs were detected by α-SMA staining. Gsα expression was significantly lower in the smooth muscle layer from GsαSMKO than control aortas (Fig. 1A). The protein and mRNA levels of Gsα were significantly decreased in aortas from GsαSMKO mice (Fig. 1B and C). Gsα plays its physiological roles mainly via generating receptor-stimulated cAMP, so we measured cAMP level in aortas. As expected, cAMP level was significantly reduced after Gsα knockout (Fig. 1D), which further confirmed effective Gsα deletion in GsαSMKO mice. However, there was no significant difference for the Gsα expression in the macrophage and vascular adventitial fibroblasts between CTR and GsαSMKO mice (Supplementary Fig. 1E).
Fig. 1.
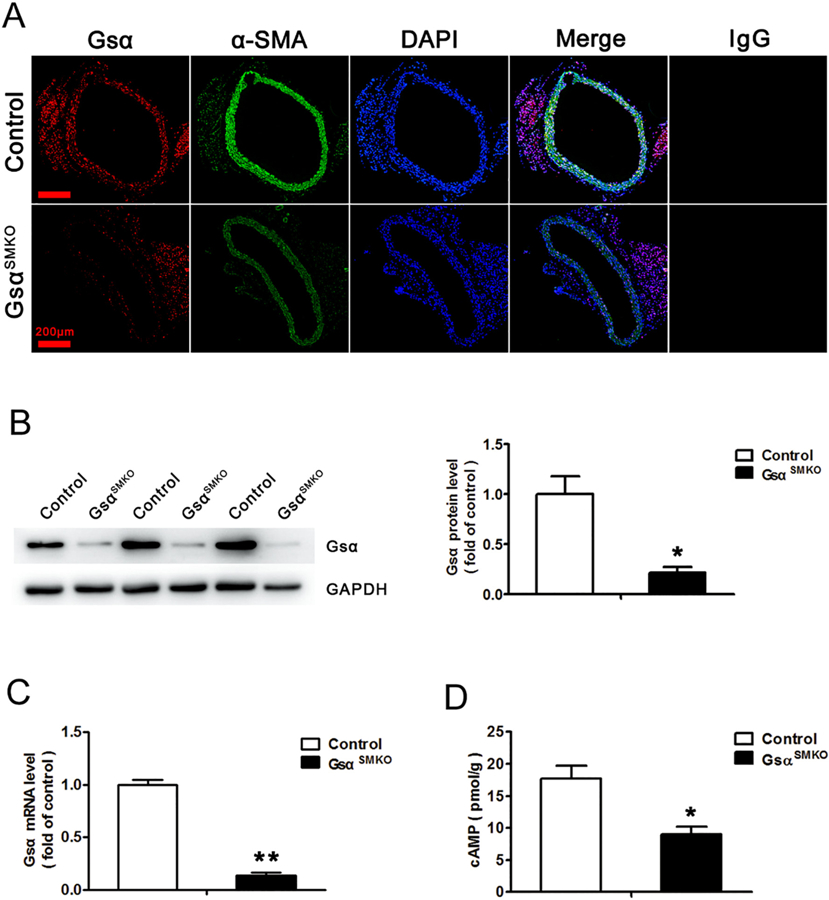
Identification of Gsα deletion in aortic smooth muscle from GsαSMKO mice. A, Representative immunofluorescence staining of Gsα, α-SMA and 4′,6-diamidino-2-phenylindole (DAPI) for nuclei in abdominal aortas from control and GsαSMKO mice. IgG was labeled as the negative control. Scale bar, 200 μm. B, Western blot analysis of aortic Gsα protein expression from control and GsαSMKO mice and quantification (n = 5). *P < 0.05 vs control. C, Quantitative RT-PCR analysis of aortic Gsα mRNA level in control and GsαSMKO mice (n = 5). **P < 0.01 vs control. D, Aortic cAMP level in control and GsαSMKO mice (n = 6). *P < 0.05 vs control. Data are mean ± SEM.
Gsα deletion did not affect the blood pressure. However, the heart rate, the body weight and ejection function were lower in the GsαSMKO mice (Supplementary Table 1). Besides, there was no significant difference in the aortic media thickness and SMC size between control and GsαSMKO mice (Supplementary Fig. 1G–I). Thus, VSMC-specific Gsα knockout didn’t alter aortic medial SMC content in mice. In addition, we detected the mRNA levels of some cytokines in heart tissues from the GsαSMKO and control mice. The mRNA levels of IL-17C, ICAM-1, IL-1β and TGF-β were increased significantly in GsαSMKO mice compared with control mice (Supplementary Fig. 5E).
3.2. Gsα deficiency induces the SMC phenotype switch
We next explored whether Gsα modulates the expression of smooth-muscle marker genes. Abdominal aortas from control and GsαSMKO mice were isolated; immunohistochemistry revealed significantly reduced level of α-SMA and SMMHC (marker of SMC contractile phenotype) in GsαSMKO than control aortas with higher level of Vimentin and Osteopontin (marker of SMC synthetic phenotype) (Fig. 2A, B); Gsα deletion in SMCs decreased the protein and mRNA levels of α-SMA and SMMHC; in the meanwhile, increased those of Vimentin and Osteopontin (Fig. 2C–E). Besides, Gsα knockout also reduced SM22 and Calponin expression (Supplementary Fig. 1A). Thus, Gsα deficiency induced the SMC phenotype switch from a contractile to a synthetic state. Moreover, Gsα deletion increased the phosphorylation of P65 (the marker of NF-κB activation) (Supplementary Fig. 1F) and the levels of SMC derived cytokines such as TGF-β1, IL-6, E-selectin, IL-17C, ICAM-1, TNF-α and CCL-2 (Supplementary Fig. 1B). Gsα deficiency also increased the expression of MMP2, fibronectin, collagen I and collagen III (Supplementary Fig. 1A).
Fig. 2.
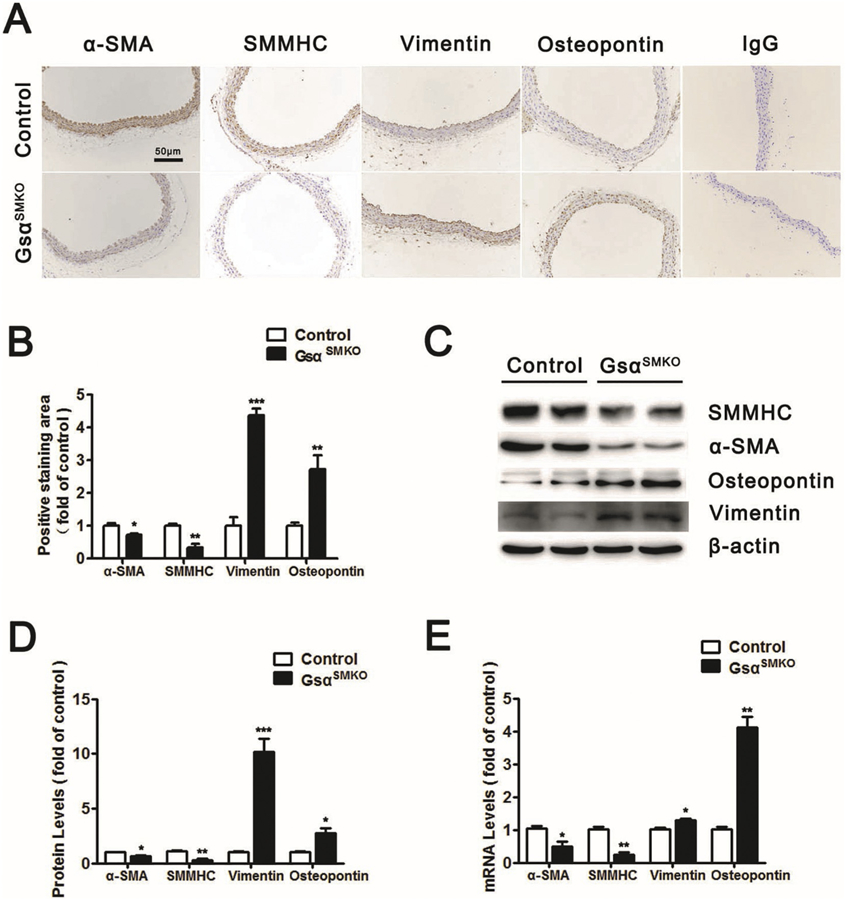
Gsα deficiency induces SMC phenotype switch. A, Representative immunochemical staining of α-SMA, SMMHC, Vimentin and Osteopontin in abdominal aortas from control and GsαSMKO mice. IgG was labeled as the negative control. Scale bar, 50 μm. B, Quantification of IHC (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001 vs control. C, Western blot analysis of α-SMA, SMMHC, Osteopontin and Vimentin protein expression in control and GsαSMKO mice. D, Quantification of Western blot (n = 5). *P < 0.05, ***P < 0.001 vs control. E, Quantitative RT-PCR analysis of aortic α-SMA, SMMHC, Osteopontin and Vimentin mRNA levels in control and GsαSMKO mice (n = 5). *P < 0.05 vs control. Data are mean ± SEM.
3.3. Gsα deficiency increases KLF4 and HuR expression
KLF4 is known as a key factor required for the SMC phenotype switch, and its deletion attenuated AAA formation [13]. Our results also suggested that Gsα could regulate the SMC phenotype switch, so we next determined whether KLF4 level was changed by Gsα deficiency. KLF4 protein level was significantly elevated in GsαSMKO aortas (Fig. 3A and B), which was further confirmed by RT-PCR and immunohistochemistry (Fig. 3C–E). Thus, Gsα deficiency increased KLF4 expression.
Fig. 3.
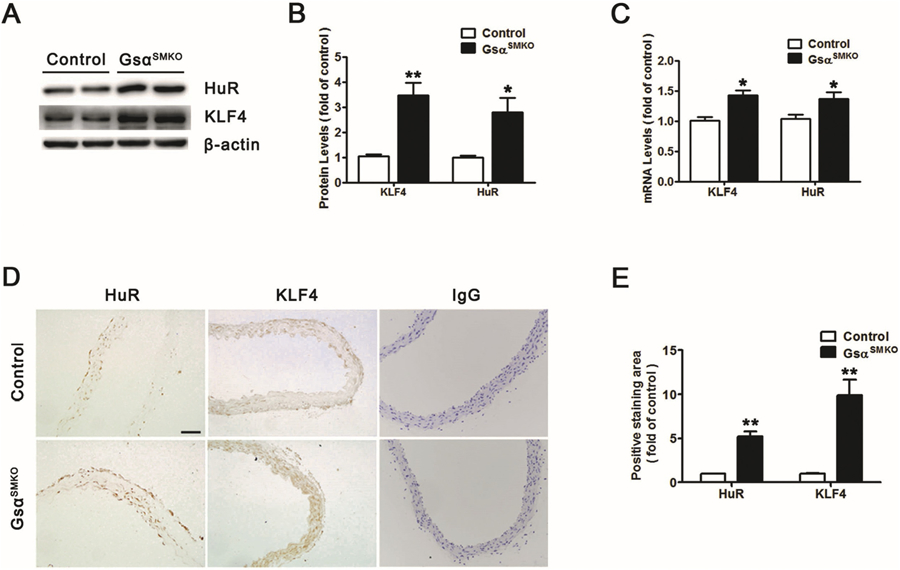
Gsα deficiency increases KLF4 and HuR expression. A, Western blot analysis of protein levels of KLF4 and HuR in primary control and Gsα-deficient SMCs and B, quantification (n = 5). *P < 0.05, **P < 0.01 vs control. C, Quantitative analysis of KLF4 and HuR mRNA levels in primary control and Gsα-deficient SMCs (n = 5). *P < 0.05, **P < 0.01 vs control. D, Representative immunochemical staining for KLF4 and HuR in abdominal aortas from control and GsαSMKO mice. IgG was labeled as the negative control. E, Quantification of IHC (n = 5). **P < 0.01 vs control. Data are mean ± SEM.
Previous study showed that cAMP could inhibit HuR expression [17]. In the SMCs, both 8-Br-cAMP (the cAMP analog) and Forskolin (adenylate cyclase activator) could decrease HuR expression (Supplementary Fig. 5A), suggesting that cAMP is required for HuR downregulation. Moreover, H-89 (PKA inhibitor) increased the HuR expression (Supplementary Fig. 5B), suggesting that PKA is required for HuR downregulation. Thus, cAMP/PKA could reduce HuR expression in SMCs. In addition, Gsα deletion in smooth muscle reduced cAMP level (Fig. 1D) and HuR protein level was increased after Gsα deletion (Fig. 3A and B), which was also confirmed by RT-PCR and immunohistochemistry (Fig. 3C–E). Hence, Gsα deficiency could increase KLF4 and HuR expression in mouse aortic smooth muscle.
3.4. KLF4 is the HuR target gene
HuR is an RNA-binding protein that regulates the stability of adenylate uridylate (AU)-rich element (ARE)-containing transcripts. We examined the 3′ untranslated region (UTR) of KLF4 mRNA to assess whether KLF4 may be a target of HuR and found 3 conserved AREs in the 3′ UTR (Fig. 4A), which suggested that HuR may bind with KLF4 mRNA and regulate its expression.
Fig. 4.
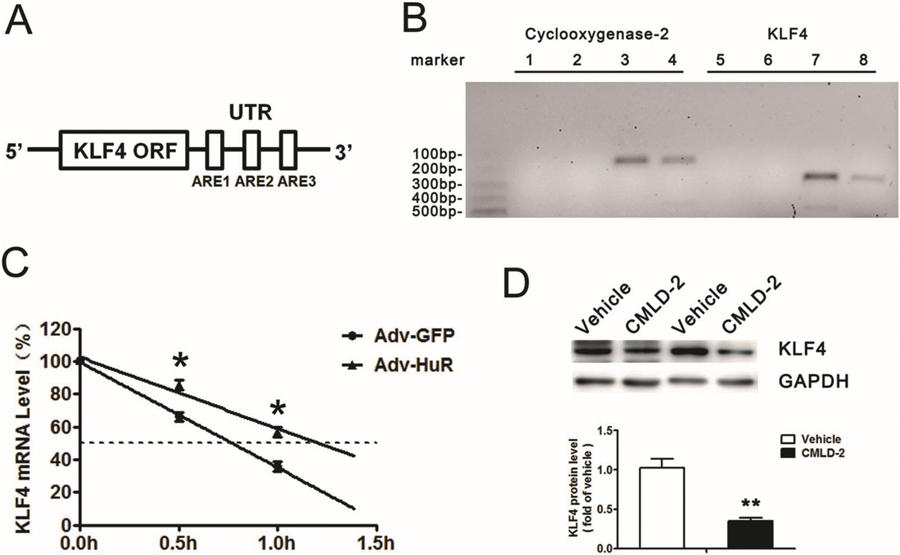
KLF4 is the HuR target gene. A, Schematic representations of predicted adenylate uridylate (AU)-rich elements (AREs) in the 3′ untranslated region (UTR) of KLF4 mRNA. ORF: Open Reading Frame. B, RNA immunoprecipitation (IP) with anti-HuR antibody or control IgG. Cyclooxygenase-2 was the positive control. 1&5: No template PCR control. 2&6: IgG RNA IP. 3&7: Anti-HuR RNA IP. 4&8: 10% Input. C, Primary SMCs were infected with adenovirus expressing GFP or HuR for 24 h, then treated with actinomycin D (5 μg/mL). Quantified RT-PCR analysis of KLF4 mRNA level (n = 5). D, SMCs were treated with Vehicle (DMSO) or HuR inhibitor, CMLD-2 (20 μM) for 24 h, then underwent western blot analysis to detect KLF4 protein level (n = 4). **P < 0.01 vs Vehicle. Data are mean ± SEM.
RNA immunoprecipitation with anti-HuR antibody or control IgG showed that HuR could bind with KLF4 mRNA (Fig. 4B). Cyclooxygenase-2, a known target of HuR, was used as a positive control in this assay. To examine the effect of HuR on the stability of endogenous KLF4 mRNA, primary aortic SMCs were infected with adenovirus expressing GFP or HuR and then treated with actinomycin D, a transcriptional inhibitor. The half-life of KLF4 mRNA was increased from 0.75 to 1.25 h after HuR overexpression (Fig. 4C), so HuR enhanced KLF4 mRNA stability. Furthermore, CYLD2, an HuR inhibitor, significantly reduced KLF4 expression (Fig. 4D). Thus, KLF4 is the target of HuR, which could bind to and stabilize endogenous KLF4 mRNA transcripts, thus elevating KLF4 protein level.
3.5. Gsα deletion induces the SMC phenotype switch via the HuR/KLF4 pathway
To detect whether Gsα deletion induces the SMC phenotype switch via the HuR/KLF4 pathway, primary aortic SMCs from control or GsαSMKO mice were transfected with HuR siRNA to knock down HuR expression. HuR knockdown suppressed the levels of KLF4 and Vimentin and increased that of α-SMA in control and Gsα-deficient SMCs (Fig. 5A and B). Hence, HuR-mediated KLF4 expression may participate in the Gsα deletion–induced SMC phenotype switch. To further confirm the role of KLF4 in the process, KLF4 siRNA was used to transfect SMCs. As expected, KLF4 knockdown increased α-SMA expression and decreased Vimentin level in control and Gsα-deficient SMCs (Fig. 5C and D), so KLF4 inhibition could rescue the SMC phenotype switch caused by Gsα knockout. Hence, Gsα deletion induced the SMC phenotype switch via the HuR/KLF4 pathway.
Fig. 5.
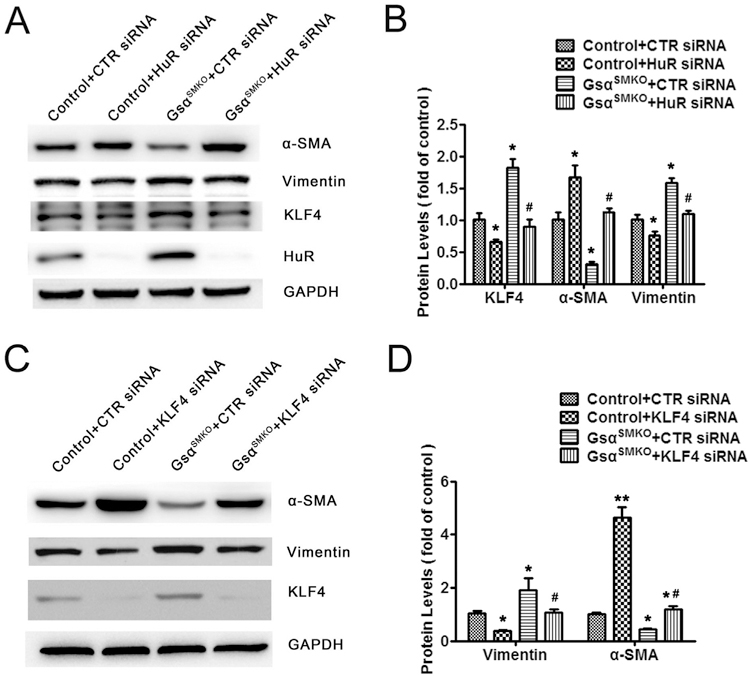
Gsα deletion induces SMC phenotype switch via the HuR/KLF4 pathway. A, Primary control and Gsα-deficient SMCs were transfected with control siRNA or HuR siRNA for 48 h followed by western blot analysis and B, quantified (n = 5). *P < 0.05 vs Control+CTR siRNA. #P < 0.05 vs GsαSMKO + CTR siRNA. C, Primary control and Gsα-deficient SMCs were transfected with control siRNA or KLF4 siRNA for 48 h followed by western blot analysis and D, quantified (n = 5). *P < 0.05, **P < 0.01 vs Control+CTR siRNA. #P < 0.05 vs GsαSMKO + CTR siRNA. Data are mean ± SEM.
3.6. Smooth muscle-specific Gsα deletion exaggerates AngII-induced AAA formation
Because Gsα regulates the SMC phenotype switch, which plays an important role in the development of AAA [13], we sought to determine whether Gsα expression changed during AAA formation. ApoE−/− mice were infused with saline or AngII for 28 days to induce AAA; Gsα expression was significantly reduced in abdominal aortas with AngII infusion as compared with saline infusion (Fig. 6A and Supplementary Fig. 2A). Ang II infusion also decreased cAMP levels (Supplementary Fig. 2C).
Fig. 6.
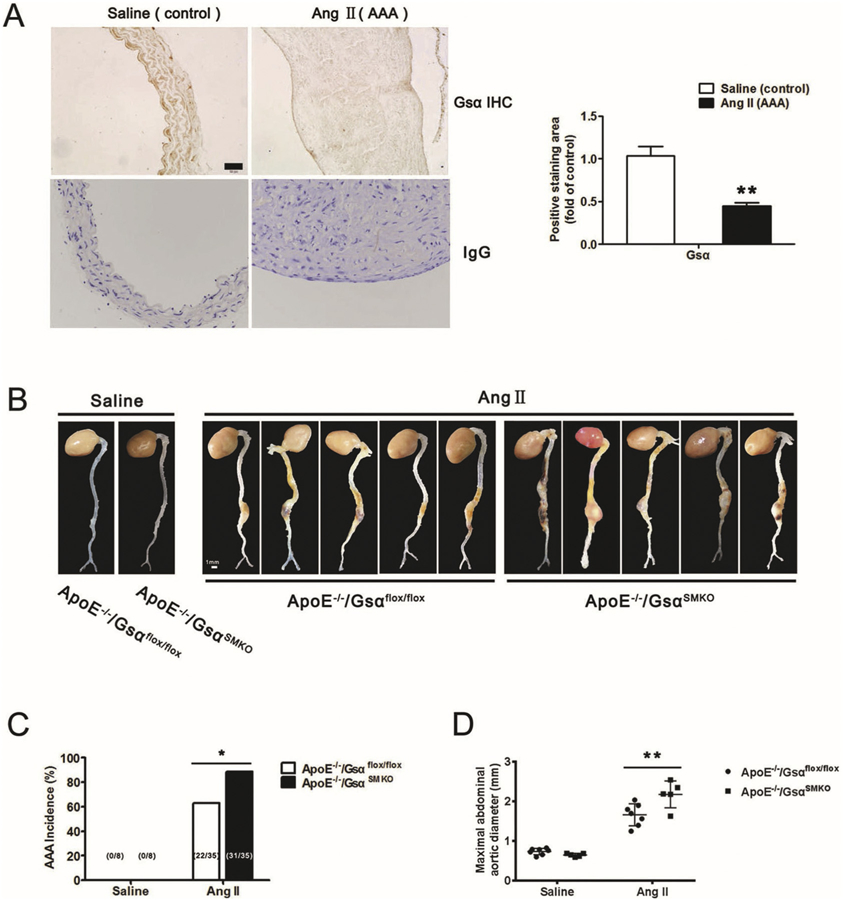
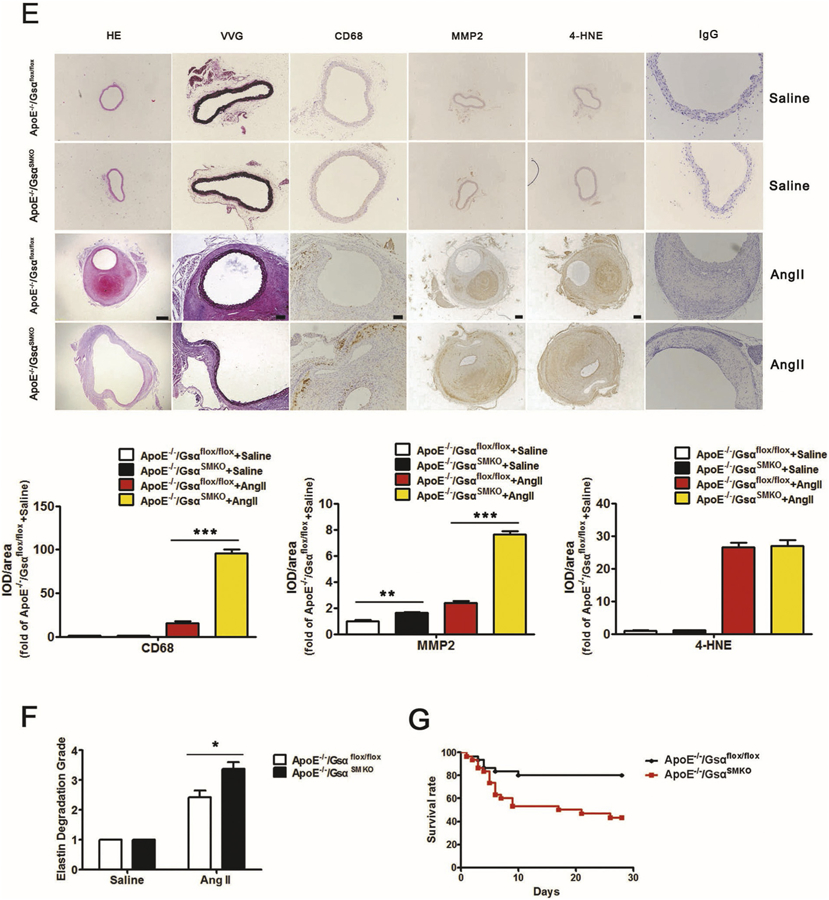
Smooth muscle-specific Gsα deletion exaggerates AngII-induced AAA formation. A, Representative immunochemical staining of Gsα in abdominal aortas from ApoE−/− mice infused with Saline or AngII (1000 ng/kg/min) for 28 days and quantification (n = 5). IgG was labeled as the negative control. **P < 0.01 vs Saline. B, Male adult ApoE−/−/Gsαflox/flox and ApoE−/−/GsαSMKO mice were infused with saline or AngII (1000 ng/kg/min) for 28 days. Representative photographs showed macroscopic features of aneurysms induced by AngII. C, Data represent percentage incidence of AAAs in ApoE−/−/Gsαflox/flox (saline n = 8, AngII n = 35) and ApoE−/−/GsαSMKO (saline n = 8, AngII n = 35) mice. *P < 0.05 for AngII-infused ApoE−/−/GsαSMKO compared with ApoE−/−/Gsαflox/flox mice. D, Maximal abdominal aortic diameters in mice with different treatment (n = 7). **P < 0.01 vs ApoE−/−/Gsαflox/flox + AngII. E, Representative HE, VVG staining, and immunochemical staining of CD68, MMP2 and 4-HNE in AAA and their quantification (n = 4). **P < 0.01, ***P < 0.001. Scale bar, 200 μm for HE, MMP2 and 4-HNE, 100 μm for VVG and CD68. IgG was labeled as the negative control. F, Grade of elastin degradation in aortic wall (n = 7). *P < 0.05 for AngII-infused ApoE−/−/GsαSMKO compared with ApoE−/−/Gsαflox/flox mice. G. Kaplan-Meier curves represent percentage survival of AngII-infused ApoE−/−/Gsαflox/flox and ApoE−/−/GsαSMKO mice (n = 30). Data are mean ± SEM.
To determine the role of smooth muscle-specific Gsα deletion in the development of AAA, ApoE−/−/Gsαflox/flox and ApoE−/−/GsαSMKO mice were generated, the levels of Gsα protein and cAMP were decreased in ApoE−/−/GsαSMKO mice (Supplementary Fig. 2B and D). Consistent with data in C57BL/6 J background, the levels of HuR, KLF4 and Vimentin were increased, while the levels of α-SMA and SM22 were decreased in ApoE−/−/GsαSMKO mice compared with ApoE−/−/Gsαflox/flox mice (Supplementary Fig. 5D). In addition, the mRNA levels of TGF-β1, IL-6, E-selectin, IL-17C, ICAM-1, TNF-α and CCL-2 were elevated in aortas from ApoE−/−/GsαSMKO mice compared with ApoE−/−/Gsαflox/flox mice (Supplementary Fig. 5G). With AngII or saline infusion, the mRNA levels of CCL-2, E-selectin, TNF-α and ICAM-1 were increased in ApoE−/−/GsαSMKO mice compared with ApoE−/−/Gsαflox/flox mice (Supplementary Fig. 6B). Thus Gsα knockout could also increase the proinflammatory phenotype in ApoE−/− background.
Male adult ApoE−/−/Gsαflox/flox and ApoE−/−/GsαSMKO mice were infused with saline or AngII for 28 days, AngII infusion could reduce the cAMP levels (Supplementary Fig. 5F) and PKA activity (Supplementary Fig. 5C) in both ApoE−/−/Gsαflox/flox and ApoE−/−/GsαSMKO mice. AAA was defined as ≥50% enlargement of the external diameter of the suprarenal aorta compared to aortas from saline-infused control mice. Gsα knockout in smooth muscle did not affect the blood pressure in saline and AngII-infused groups (Supplementary Table 2). Gross morphology of aortas did not differ between saline-infused ApoE−/−/Gsαflox/flox and ApoE−/−/GsαSMKO mice (Fig. 6B); however, with AngII infusion, the incidence of AAA in ApoE−/−/GsαSMKO mice was increased compared with in ApoE−/−/Gsαflox/flox mice (31 [88.6%] of 35 ApoE−/−/GsαSMKO mice vs 22 [62.8%] of 35 ApoE−/−/Gsαflox/flox mice, Fig. 6C). Kaplan-Meier curves demonstrated that smooth muscle-specific Gsα deletion increased mortality rate due to aneurysm rupture in both AngII-induced models of aneurysm formation (Fig. 6G). Additionally, the maximal diameter of the abdominal aorta was significantly higher in AngII-infused ApoE−/−/GsαSMKO than ApoE−/−/Gsαflox/flox mice (Fig. 6B and D). Gsα deficiency also increased the incidence of thoracic aortic aneurysm (TAA) with AngII infusion (13 [37.1%] of 35 ApoE−/−/GsαSMKO mice vs 5 [14.3%] of 35 ApoE−/−/Gsαflox/flox mice). However, Gsα deficiency did not affect the atherosclerotic plaque area in AngII-induced ApoE−/−Gsαflox/flox and ApoE−/−GsαSMKO mice (Supplementary Fig. 1C–D).
Hematoxylin and eosin (HE) and Verhoff-Van Gieson (VVG) staining revealed that AngII infusion caused positive remodeling of the abdominal aorta, breakdown of the aortic media and adventitia and destruction, and discontinuity of elastin fibers in ApoE−/−/Gsαflox/flox mice (Fig. 6E and Supplementary Fig. 2E). These pathological changes induced by AngII were largely exaggerated in ApoE−/−/GsαSMKO mice (Fig. 6E and F). Additionally, the matrix metalloproteinase 2 (MMP2) level and inflammatory cell infiltration as determined by CD68 of the AAA were elevated in AngII-infused ApoE−/−/GsαSMKO than ApoE−/−/Gsαflox/flox mice (Fig. 6E and Supplementary Fig. 2E). However, Gsα deficiency did not affect the level of reactive oxygen species (ROS) as determined by 4-hydroxynonenal (4-HNE) immunostaining in the AAA (Fig. 6E and Supplementary Fig. 2E). Moreover, Gsα deficiency increased Vimentin and Osteopontin expression with AngII infusion compared with control groups (Supplementary Fig. 6A), which confirms the important role of SMC phenotype switch in the AAA progression. Taken together, Gsα deficiency in SMCs aggravated the AngII-induced AAA formation.
3.7. The levels of Gsα and α-SMA are reduced while those of HuR and KLF4 are increased in human AAA samples
To establish the clinical relevance of Gsα/HuR/KLF4 signaling and AAA formation, we further examined the levels of Gsα, HuR, KLF4 and α-SMA in human AAA samples. Human AAA tissues and their control adjacent aortic sections without aneurysm were obtained from patients undergoing open surgery (Supplementary Fig. 5H). As expected, the levels of Gsα and α-SMA were significantly lower in human AAA sections than adjacent nonaneurysmal aortic sections (Fig. 7A–B, Supplementary Fig. 3 and Fig. 4). Meanwhile, both HuR and KLF4 were significantly upregulated in human AAA samples (Fig. 7A–B, Supplementary Fig. 3 and Fig. 4), which indicates the important role of Gsα/HuR/KLF4 signaling in the development of AAA.
Fig. 7.
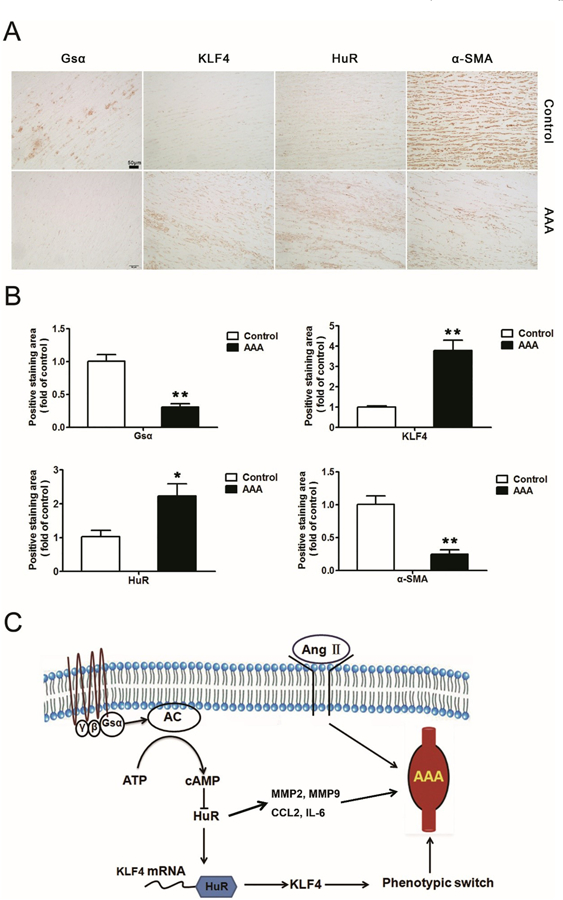
The levels of Gsα and α-SMA are reduced while those of HuR and KLF4 are increased in human AAA samples. A, Representative immunohistochemical staining of Gsα, HuR, KLF4 and α-SMA in human AAA tissues and adjacent normal aortas and B, quantification (n = 7). *P < 0.05, **P < 0.01 vs Control. Data are mean ± SEM. C, Diagram for the protective role of Gsα in AngII-induced AAA formation via HuR/KLF4. AC, adenylyl cyclase.
4. Discussion
In this study, to explore the functions of Gsα in SMCs in vivo, we generated tamoxifen-inducible Gsαflox/flox/SM22-CreERT2 mice, which are smooth muscle-specific Gsα knockout mice, by using a tamoxifen-activated Cre recombinase method. Deletion of Gsα in aortic SMCs resulted in an SMC phenotype switch, with decreased expression of contractile marker α-SMA and increased expression of the synthetic marker Vimentin in aortas. Mechanically, Gsα deficiency reduced cAMP level and increased the level of HuR, which bound with KLF4 mRNA and enhanced its stability (Fig. 7C). Moreover, Gsα was downregulated in mouse and human AAA samples; smooth muscle-specific Gsα deletion exaggerated AngII-induced AAA formation in vivo. Taken together, our results reveal that Gsα plays a protective role in the development of AAA by inhibiting the HuR/KLF4-mediated SMC phenotype switch.
Increasing evidence indicates that Gsα plays important roles in cell proliferation, apoptosis, differentiation and metabolism [23,24]. To detect the functions of Gsα in vivo, many tissue-specific Gsα knockout mice have been generated. For example, adipose-specific Gsα knockout mice showed a lean phenotype with impaired cold tolerance and increased diet-induced thermogenesis [25]. Pancreas-specific Gsα deficiency led to increased pancreatic alpha-cell proliferation and decreased pancreatic beta-cell proliferation [26]. Recent research showed that multiple human diseases are related to somatic GNAS mutations encoding the Gsα. Constitutively activating Gsα mutations were extensively detected in McCune-Albright syndrome and parosteal osteosarcoma, whereas heterozygous GNAS mutations causing decreased expression of Gsα resulted in Albright hereditary osteodystrophy [23]. Transgenic mice overexpressing cardiac Gsα exhibited increased cardiac contractility in response to β-adrenergic receptor stimulation. However, these mice developed cardiomyopathy with aging [27–29], suggesting that overexpression of Gsα has a detrimental effect on cardiac function. In this study, we demonstrated that Gsα has a protective role on vascular structure, which indicates that Gsα plays different roles in the different tissues. Moreover, consideration about the synthetic phenotype of Gsα-deficient smooth muscle cells, Gsα deletion might increase the angiogenesis and reduce the vascular resistance in the small vessels. In our previous study, Gsα deficiency impaired intestinal smooth muscle contraction [20], which affect their food intake. Thus, GsaSMKO mice exhibited the phenotype of lower body weight. Moreover, the increased inflammation in the heart of GsαSMKO mice may contribute to the phenotype of lower heart rate and ejection fraction. However, the reason of the cardiac dysfunction needs to be explored in the further study.
A phenotype switch is the characteristic reaction of SMCs in response to adverse stimuli such as biological factors, extracellular matrix components and chemico-physical factors [30]. The SMC phenotype switch affects the progression of many diseases such as atherosclerosis and AAA [12,14,15]. The mechanism of the phenotype switch is complicated and involves multiple factors. Myocardin and caveolin-3 were both identified as the guardians of a vascular smooth muscle contractile phenotype [31,32], whereas miR-143/145 and KLF4 promoted the SMC phenotype switch from a contractile to a synthetic state [12,14,33]. Although various data demonstrated that KLF4 is a key molecule regulating the SMC phenotype switch, the regulation of KLF4 expression is still unclear. Recent study showed that Sp1 could bind to the KLF4 gene promoter to increase its expression [34]. In this study, we demonstrated that HuR could bind with KLF4 mRNA and increase its mRNA stability, so KLF4 mRNA stability is regulated by HuR. This discovery uncovers a novel mechanism of KLF4 gene regulation and also broadens the biological functions of HuR. Our study demonstrated that Gsα and HuR participate in regulating the aortic SMC phenotype switch. Because HuR has a wide range of target mRNAs, the HuR/KLF4 may not be the single mechanism in AAA. Previous study showed that HuR could increase the stability of MMP2 and MMP9 mRNAs [35]. Moreover, some inflammatory cytokines such as CCL-2 and IL-6 are the targets of HuR [36,37]. Thus, HuR may participate in the AAA progression through regulating some different pathways. Additionally, there are many PKA substrates and PKA regulated genes, many of which might modulate the phenotype of Gsα-deficient mice. Future study will be performed to explore their roles.
Gsα deficiency in SMCs also caused increased inflammation in aortas. Inflammatory factors such as IL-6, ICAM-1, CCL-2 and TNF-α were up-regulated significantly in GsαSMKO compared with control mice. However, the possible mechanisms were still unclear. Previous studies show that HuR regulates the expression of many inflammatory cytokines including IL-6, ICAM-1, CCL-2, IL-17C and TNF-α through binding to their mRNAs [36–39]. Thus, the increased inflammation caused by Gsα deficiency may be resulted from increased HuR expression. Moreover, Gsα deletion increased the phosphorylation of p65, the aberrant activation of NF-κB pathway is another potential mechanism of increased inflammation.
The mechanism underlying AAA is complicated; many kinds of cells (SMCs, macrophages, endothelial cells, etc.) and various elements (inflammation, cell apoptosis, extracellular matrix, etc.) participate in this process. Here, we show that smooth muscle-specific Gsα deletion increased AAA formation by promoting the SMC phenotype switch, which was consistent with the mechanism found in smooth muscle-specific KLF4-knockout mice [14]. Another study directly demonstrated that loss of α-SMA increased the sensitivity to AngII by increasing Agtr1a level, which was established to drive AAA formation [40]. Thus, the SMC phenotype switch may be one of the most important mechanisms of AAA formation. Moreover, there is no effective clinical pharmacological therapy to prevent or suppress AAA development. Some studies explored the effect of cAMP agonist on the aneurysm. Cilostazol, a selective inhibitor of cAMP phosphodiesterase 3, could increase cellular cAMP and suppress the development of AAA in the mouse and rat models [41,42], which was consistent with the conclusion in this study.
In conclusion, we have established a causative relation between smooth-muscle Gsα deficiency and AAA formation in vivo and identified the protective role of Gsα in AngII-triggered AAA. The identification of Gsα expression in AngII-triggered AAA and in human patients with AAA suggests that Gsα downregulation might be a reason for AAA formation, and the agents that activate the Gsα signaling pathway may have therapeutic potential for AAA and other cardiovascular diseases.
Supplementary Material
Acknowledgements
We thank Dr. Robert Feil of Interfakultäres Institut für Biochemie (IFIB) Signaltransduktion - Transgene Modelle for providing the SM22-CreERT2 mice. This study was supported by grants from the National Natural Science Foundation of China (No. 81770473, 81570393, 81770436, 81570324, 81425004, 81770442, 31770977, 31400771), the Taishan Scholar Project of Shandong Province of China (No. tsqn20161066) and the Key Research and Development Plan of Shandong Province (No. 2015GSF118118).
Abbreviations:
- AAA
Abdominal aortic aneurysm
- AngII
angiotensin II
- ARE
adenylate uridylate-rich element
- α-SMA
SM α-actin
- cAMP
cyclic adenosine monophosphate
- Gsα
alpha-subunit of the stimulatory G protein
- GsαSMKO
smooth muscle-specific Gsα knockout
- HuR
human antigen R
- KLF4
Krüppel-like factor 4
- SMCs
Smooth muscle cells
- UTR
untranslated region
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.yjmcc.2019.05.002.
Disclosures
None.
References
- [1].Zhou HF, Yan H, Stover CM, Fernandez TM, Rodriguez de Cordoba S, Song WC, Wu X, Thompson RW, Schwaeble WJ, Atkinson JP, Hourcade DE, Pham CT, Antibody directs properdin-dependent activation of the complement alternative pathway in a mouse model of abdominal aortic aneurysm, Proc. Natl. Acad. Sci. U. S. A 109 (2012) 415–422, 10.1073/pnas.1119000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Thompson RW, Detection and management of small aortic aneurysms, N. Engl. J. Med 346 (2002) 1484–1486, 10.1056/NEJM200205093461910. [DOI] [PubMed] [Google Scholar]
- [3].McCormick ML, Gavrila D, Weintraub NL, Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms, Arterioscler. Thromb. Vasc. Biol 27 (2007) 461–469, 10.1161/01.ATV.0000257552.94483.14. [DOI] [PubMed] [Google Scholar]
- [4].Moehle CW, Bhamidipati CM, Alexander MR, Mehta GS, Irvine JN, Salmon M, Upchurch GR Jr., Kron IL, Owens GK, Ailawadi G, Bone marrow-derived MCP1 required for experimental aortic aneurysm formation and smooth-muscle phenotypic modulation, J. Thorac. Cardiovasc. Surg 142 (2011) 1567–1574, 10.1016/j.jtcvs.2011.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ailawadi G, Moehle CW, Pei H, Walton SP, Yang Z, Kron IL, Lau CL, Owens GK, Smooth muscle phenotypic modulation is an early event in aortic aneurysms, J. Thorac. Cardiovasc. Surg 138 (2009) 1392–1399, 10.1016/j.jtcvs.2009.07.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Emeto TI, Moxon JV, Au M, Golledge J, Oxidative stress and abdominal aortic aneurysm: potential treatment targets, Clin. Sci. (Lond.) 130 (2016) 301–315, 10.1042/CS20150547. [DOI] [PubMed] [Google Scholar]
- [7].Nosoudi N, Nahar-Gohad P, Sinha A, Chowdhury A, Gerard P, Carsten CG, Gray BH, Vyavahare NR, Prevention of abdominal aortic aneurysm progression by targeted inhibition of matrix metalloproteinase activity with batimastat-loaded nanoparticles, Circ. Res 117 (2015) e80–e89, 10.1161/CIRCRESAHA.115.307207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang YN, Xie BD, Sun L, Chen W, Jiang SL, Liu W, Bian F, Tian H, Li RK, Phenotypic switching of vascular smooth muscle cells in the ‘normal region’ of aorta from atherosclerosis patients is regulated by miR-145, J. Cell. Mol. Med 20 (2016) 1049–1061, 10.1111/jcmm.12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chamley-Campbell J, Campbell GR, Ross R, The smooth muscle cell in culture, Physiol. Rev 59 (1979) 1–61, 10.1152/physrev.1979.59.1.1. [DOI] [PubMed] [Google Scholar]
- [10].Rensen SS, Doevendans PA, van Eys GJ, Regulation and characteristics of vascular smooth muscle cell phenotypic diversity, Neth. Hear. J 15 (2007) 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kawai-Kowase K, Owens GK, Multiple repressor pathways contribute to phenotypic switching of vascular smooth muscle cells, Am. J. Phys. Cell Physiol 292 (2007) C59–C69, 10.1152/ajpcell.00394.2006. [DOI] [PubMed] [Google Scholar]
- [12].Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK, KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis, Nat. Med 21 (2015) 628–637, 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Busch A, Hartmann E, Grimm C, Ergün S, Kickuth R, Otto C, Kellersmann R, Lorenz U, Heterogeneous histomorphology, yet homogeneous vascular smooth muscle cell dedifferentiation, characterize human aneurysm disease, J. Vasc. Surg 66 (2017) 1553–1564, 10.1016/j.jvs.2016.07.129. [DOI] [PubMed] [Google Scholar]
- [14].Yoshida T, Kaestner KH, Owens GK, Conditional deletion of Krüppel-like factor 4 delays downregulation of smooth muscle cell differentiation markers but accelerates neointimal formation following vascular injury, Circ. Res 102 (2008) 1548–1557, 10.1161/CIRCRESAHA.108.176974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Salmon M, Johnston WF, Woo A, Pope NH, Su G, Upchurch GR Jr., G.K. Owens, G. Ailawadi, KLF4 regulates abdominal aortic aneurysm morphology and deletion attenuates aneurysm formation, Circulation 128 (2013) S163–S174, 10.1161/CIRCULATIONAHA.112.000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lei R, Zhang K, Wei Y, Chen M, Weinstein LS, Hong Y, Zhu M, Li H, Li H, G-protein α-subunit Gsα is required for craniofacial morphogenesis, PLoS ONE 11 (2016) e0147535, 10.1371/journal.pone.0147535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Klöss S, Srivastava R, Mülsch A, Down-regulation of soluble guanylyl cyclase expression by cyclic AMP is mediated by mRNA-stabilizing protein HuR, Mol. Pharmacol 65 (2004) 1440–1451, 10.1124/mol.65.6.1440. [DOI] [PubMed] [Google Scholar]
- [18].Shao X, Liu K, Fan Y, Ding Z, Chen M, Zhu M, Weinstein LS, Li H, Li H, Gαs relays Sphingosine-1-phosphate receptor 1 signaling to stabilize vascular endothelial-cadherin at endothelial junctions to control mouse embryonic vascular integrity, J. Genet. Genomics 42 (2015) 613–624, 10.1016/j.jgg.2015.08.006. [DOI] [PubMed] [Google Scholar]
- [19].Streit MR, Weiss CS, Meyer S, Ochs MM, Hagenmueller M, Riffel JH, Buss SJ, Heger T, Katus HA, Hardt SE, Cardiac effects of attenuating Gsα-dependent signaling, PLoS ONE 11 (2016) e0146988, 10.1371/journal.pone.0146988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Qin X, Liu S, Lu Q, et al. , Heterotrimeric G stimulatory protein α subunit is required for intestinal smooth muscle contraction in mice, Gastroenterology 152 (2017) 1114–1125, 10.1053/j.gastro.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chen M, Gavrilova O, Zhao WQ, Nguyen A, Lorenzo J, Shen L, Nackers L, Pack S, Jou W, Weinstein LS, Increased glucose tolerance and reduced adiposity in the absence of fasting hypoglycemia in mice with liver-specific Gs alpha deficiency, J. Clin. Invest 115 (2005) 3217–3227, 10.1172/JCI24196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kühbandner S, Brummer S, Metzger D, Chambon P, Hofmann F, Feil R, Temporally controlled somatic mutagenesis in smooth muscle, Genesis 28 (2000) 15–22. [DOI] [PubMed] [Google Scholar]
- [23].Turan S, Bastepe M, GNAS Spectrum of disorders, Curr. Osteoporos. Rep 13 (2015) 146–158, 10.1007/s11914-015-0268-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lu C, Xia J, Zhou Y, et al. , Loss of Gsα impairs liver regeneration through a defect in the crosstalk between cAMP and growth factor signaling, J. Hepatol 64 (2016) 342–351, 10.1016/j.jhep.2015.08.036. [DOI] [PubMed] [Google Scholar]
- [25].Chen M, Chen H, Nguyen A, Gupta D, Wang J, Lai EW, Pacak K, Gavrilova O, Quon MJ, Weinstein LS, G(s)alpha deficiency in adipose tissue leads to a lean phenotype with divergent effects on cold tolerance and diet-induced thermogenesis, Cell Metab 11 (2010) 320–330, 10.1016/j.cmet.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Xie T, Chen M, Weinstein LS, Pancreas-specific Gsalpha deficiency has divergent effects on pancreatic alpha- and beta-cellproliferation, J. Endocrinol 206 (2010) 261–269, 10.1677/JOE-10-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Geng YJ, Ishikawa Y, Vatner DE, Wagner TE, Bishop SP, Vatner SF, Homcy CJ, Apoptosis of cardiac myocytes in Gsalpha transgenic mice, Circ. Res 84 (1) (1999) 34–42. [DOI] [PubMed] [Google Scholar]
- [28].Vatner DE, Yang GP, Geng YJ, Asai K, Yun JS, Wagner TE, Ishikawa Y, Bishop SP, Homcy CJ, Vatner SF, Determinants of the cardiomyopathic phenotype in chimeric mice overexpressing cardiac Gsalpha, Circ. Res 86 (7) (2000) 802–806. [DOI] [PubMed] [Google Scholar]
- [29].Shen W, Vatner DE, Vatner SF, Ingwall JS, Progressive loss of creatine maintains a near normal DeltaG approximately (ATP) in transgenic mouse hearts with cardiomyopathy caused by overexpressing Gsalpha, J. Mol. Cell. Cardiol 48 (4) (2010) 591–599, 10.1016/j.yjmcc.2009.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Scirocco A, Matarrese P, Carabotti M, Ascione B, Malorni W, Severi C, Cellular and molecular mechanisms of phenotypic switch in gastrointestinal smooth muscle, J. Cell. Physiol 231 (2016) 295–302, 10.1002/jcp.25105. [DOI] [PubMed] [Google Scholar]
- [31].Gutierrez-Pajares JL, Iturrieta J, Dulam V, Wang Y, Pavlides S, Malacari G, Lisanti MP, Frank PG, Caveolin-3 promotes a vascular smooth muscle contractile phenotype, Front Cardiovasc. Med 2 (2015) 27, 10.3389/fcvm.2015.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ackers-Johnson M, Talasila A, Sage AP, Long X, Bot I, Morrell NW, Bennett MR, Miano JM, Sinha S, Myocardin regulates vascular smooth muscle cell inflammatory activation and disease, Arterioscler. Thromb. Vasc. Biol 35 (2015) 817–828, 10.1161/ATVBAHA.114.305218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sala F, Aranda JF, Rotllan N, Ramírez CM, Aryal B, Elia L, Condorelli G, Catapano AL, Fernández-Hernando C, Norata GD, MiR-143/145 deficiency attenuates the progression of atherosclerosis in Ldlr−/−mice, Thromb. Haemost 112 (2014) 796–802, 10.1160/TH13-11-0905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Deaton RA, Gan Q, Owens GK, Sp1-dependent activation of KLF4 is required for PDGF-BB-induced phenotypic modulation of smooth muscle, Am. J. Physiol. Heart Circ. Physiol 296 (2009) H1027–H1037, 10.1152/ajpheart.01230.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kong J, Zhang Y, Liu S, Li H, Liu S, Wang J, Qin X, Jiang X, Yang J, Zhang C, Zhang W, Melatonin attenuates angiotensin II-induced abdominal aortic aneurysm through the down-regulation of matrix metalloproteinases, Oncotarget 8 (2017) 14283–14293, 10.18632/oncotarget.15093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fan J, Ishmael FT, Fang X, Myers A, Cheadle C, Huang SK, Atasoy U, Gorospe M, Stellato C, Chemokine transcripts as targets of the RNA-binding protein HuR in human airway epithelium, J. Immunol 186 (2011) 2482–2494, 10.4049/jimmunol.0903634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ouhara K, Munenaga S, Kajiya M, Takeda K, Matsuda S, Sato Y, Hamamoto Y, Iwata T, Yamasaki S, Akutagawa K, Mizuno N, Fujita T, Sugiyama E, Kurihara H, The induced RNA-binding protein, HuR, targets 3’-UTR region of IL-6 mRNA and enhances its stabilization in periodontitis, Clin. Exp. Immunol 192 (2018) 325–336, 10.1111/cei.13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rhee WJ, Ni CW, Zheng Z, Chang K, Jo H, Bao G, HuR regulates the expression of stress-sensitive genes and mediates inflammatory response in human umbilical vein endothelial cells, Proc. Natl. Acad. Sci 107 (2010) 6858–6863, 10.1073/pnas.1000444107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dean JL, Wait R, Mahtani KR, Sully G, Clark AR, Saklatvala J, The 3′ untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA-stabilizing factor HuR, Mol. Cell. Biol 21 (2001) 721–730, 10.1128/MCB.21.3.721-730.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chen J, Peters A, Papke CL, et al. , Loss of smooth muscle α-actin leads to NF-κB-dependent increased sensitivity to angiotensin II in smooth muscle cells and aortic enlargement, Circ. Res 120 (2017) 1903–1915, 10.1161/CIRCRESAHA.117.310563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Umebayashi R, Uchida HA, Kakio Y, Subramanian V, Daugherty A, Wada J, Cilostazol attenuates angiotensin II-induced abdominal aortic aneurysms but not atherosclerosis in apolipoprotein E-deficient mice, Arterioscler. Thromb. Vasc. Biol 38 (2018) 903–912, 10.1161/ATVBAHA.117.309707. [DOI] [PubMed] [Google Scholar]
- [42].Zhang Q, Huang JH, Xia RP, Duan XH, Jiang YB, Jiang Q, Sun WJ, Suppression of experimental abdominal aortic aneurysm in a rat model by the phosphodiesterase 3 inhibitor cilostazol, J. Surg. Res 167 (2011) e385–e393, 10.1016/j.jss.2011.01.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.