

Abstract
Sex is a key modifier of neurological disease outcomes. Microglia are implicated in neurological diseases and modulated by miRNAs, but it is unknown whether microglial miRNAs have sex-specific influences on disease. We show that microglial miRNA expression differs in males and females and that loss of miRNAs leads to sex-specific changes in the microglial transcriptome and tau pathology. These findings suggest microglial miRNAs influence tau pathogenesis in a sex-specific manner.
Sex has heterogeneous effects on human disease pathogenesis, including in Alzheimer’s disease (AD) and other neurological diseases1,2. Dysfunction of microglia, the resident innate immune cells of the central nervous system, has been linked to many neurological diseases3–5. Sex differences in microglial gene expression and functions are seen in young adult mice6,7, and may be especially pronounced in the aging brain8,9. MicroRNAs (miRNAs) regulate immune networks in microglia10,11 and exhibit sex-specific expression in some cell types12. However, whether microglial miRNAs are expressed and function in a sex-specific manner is unknown. A better understanding of microglial miRNA function could identify novel molecular networks that contribute to neurological diseases.
We performed miRNA sequencing (miRNA-seq) on microglia isolated from brains of adult mice (B6C3F1/J). Unsupervised clustering grouped samples by sex (Fig. 1a). Sixty-one miRNAs were enriched in males and 26 in females (Fig. 1b, Supplementary Table 1). Thirteen of these (8 in males, 5 in females) were encoded by the X chromosome (Supplementary Table 2). qPCR analyses validated several differentially expressed (DE) miRNAs (Fig. 1c). Despite differing miRNA profiles, male and female cortical microglia had similar branching complexities (Fig. 1d–f).
Figure 1. Adult microglia have sex-dependent microRNA expression.

(a) Heatmap of 50 miRNAs measured by miRNA-seq of microglia isolated from 6-month-old mice. Counts were log-transformed, normalized, and centered. n = 4 biologically independent samples/sex, 2 animals/sample.
(b) Volcano plot of data from (a). Turquoise, enriched in males (61 miRNAs; P ≤ 0.05 by Benjamini-Hochberg correction and log2FC ≥ 1); pink, enriched in females (26 miRNAs; P ≤ 0.05 and log2FC ≤ –1); grey, not significantly different. Top 10 miRNAs in each sex with highest log2FC values and normalized counts over 100 are labeled. Vertical dashed lines indicate log2FC ± 1. Horizontal dashed line indicates -log10(0.05). Full list of DE miRNAs in Supplementary Table 1. Wald test used.
(c) qPCR validation of differentially expressed miRNAs from (b). Each dot represents one mouse. Bar denotes mean. n = 3 biologically independent samples/sex, 2 animals/sample. * t = 2.784, df = 4, P = 0.0496 (miR-200c-3p), * t = 4.119, df = 4, P = 0.0146 (miR-1298–5p), * t = 2.864, df = 4, P = 0.05 (miR-365–3p), unpaired, two-tailed t test.
(d) Representative 3D Imaris morphology reconstruction (top) and corresponding Iba1 immunostaining (bottom) of cortical microglia from 9-month-old mice. Image acquired and analysis repeated for every mouse quantified in (e,f). Scale bar, 15 μm.
(e,f) Number of branch points (e) and total length of processes (f) per cell, determined by Imaris-based automatic quantification. Error bars represent SEM. Bar denotes mean. Each dot represents one mouse. ~60 cells/sex.
To evaluate the transcriptional role of microglial miRNAs, we selectively depleted the miRNA-processing enzyme Dicer in adult microglia by crossing Cx3cr1CreERT2/+ with Dicerfl/fl mice (Dicer KO). Dicer mRNA and Dicer protein levels were significantly reduced in CD11b+ cells, but not in CD11b− cells (Fig. 2a–d), accompanied by depletion of several Dicer-dependent miRNAs (Fig. 2e). Interestingly, loss of mature miRNAs resulted in far greater changes in the transcriptome of male microglia than female microglia (Fig. 2f, Supplementary Tables 3,4). Pathway analysis of male DE genes showed enrichment in immune system pathways, including genes involved in TNFα signaling through NFκB activation, such as Il1b, Cd69, Tlr2, and Vegfa (Fig. 2g,h). Upstream activators of this expression pattern included TNFα, NFκB, and interleukins (Il2, Il1b) (Fig. 2i), as predicted by Ingenuity Pathway Analysis, suggesting an overall enhanced inflammatory state. Thus, removing miRNAs alters the expression of immune-related genes to a greater extent in male compared to female microglia.
Figure 2. Loss of mature microRNAs affects microglia in a sex-dependent manner.

(a,b) qPCR of Dicer mRNA in CD11b+ (a) and CD11b– (b) cells isolated from female and male 9-month-old Dicer WT and KO mice. Error bars represent SEM. Bar denotes mean. Each dot represents one mouse. n = 3 biologically independent animals/genotype for females, 4 for males (a). n = 3 biologically independent animals for Dicer WT, 5 for Dicer KO (b). * t = 3.464, df = 4, P = 0.025, ** t = 4.855, df = 6, P = 0.0028, unpaired, two-tailed t test.
(c) Representative images from 9-month-old Dicer WT and KO mice. Iba1+ cells outlined in white. Image acquired and analysis repeated for every mouse quantified in (d). Scale bar, 10 μm.
(d) Relative fluorescence intensity of cortical Dicer immunostaining. Error bars represent SEM. Bar denotes mean. Each dot represents one mouse. n = 8 biologically independent animals/genotype, ~40 cells/genotype. ** t = 3.740, df = 14, P = 0.0022, unpaired, two-tailed t test.
(e) qPCR of miRNAs in CD11b+ cells isolated from 9-month-old Dicer WT and KO mice. Each dot represents one mouse. Bar denotes mean. n = 3 biologically independent samples/genotype, 2 animals/sample. ** t = 4.597, df = 4, P = 0.0059 (miR-16–5p), * t = 4.328, df = 4, P = 0.0124 (miR-23a-3p), * t = 2.961, df = 4, P = 0.0415 (miR-342–3p), unpaired, two-tailed t test.
(f) Venn diagram of DE mRNAs comparing microglia isolated from Dicer WT and KO females and males. Purple, genes up-regulated in Dicer KO; green, genes down-regulated in Dicer KO. n = 3 Dicer WT female samples, 2 Dicer KO female samples, 5 Dicer WT male samples, 4 Dicer KO male samples with 2 mice/sample. Full list of DEGs in Supplementary Tables 3,4.
(g) Pathway analysis with GSEA Hallmark gene set on DE genes uniquely changed in male Dicer KO compared to Dicer WT microglia identified in (f). Significance calculated by a phenotypic-based permutation test, P ≤ 0.05 by Benjamini-Hochberg correction.
(h) STRING network analysis of genes overlapping with the “TNFɑ signaling via NFκB” pathway identified in (g). Purple, log2FC ≥ 1; green, log2FC ≤ −1.
(i) Ingenuity Pathway Analysis for predicted upstream regulators of genes overlapping with the “TNFɑ signaling via NFκB” pathway identified in (g). One-sided Fisher’s exact test based on a Hypergeometric distribution.
Microglia have been implicated in the spread of tau burden in the brain4,5,13, but it is not known whether male and female microglia respond to tau pathology differently. To probe this, we used the P301S (PS19) tauopathy mouse model14 and labeled pathogenic forms of tau with the MC1 antibody15. The densities of MC1-positive tau inclusions were similar in male and female animals (Extended Data Fig. 1). However, the miRNA and mRNA profiles were altered by tau pathology to a far greater extent in male microglia (Extended Data Fig. 2a–c, Supplementary Tables 5–7). We then identified a subset of the DE mRNAs predicted to be targets of DE miRNAs in male microglia, focusing only on miRNA and mRNA targets that have anti-correlated expression patterns (Extended Data Fig. 2d). Out of these, downregulation of miR-1249–5p explained the greatest number of upregulated targets in male PS19 microglia. These data suggest sex-specific microglial responses to similar levels of tau pathology.
Depleting Dicer in neurons results in tau hyperphosphorylation16, but it is unknown how loss of microglial miRNAs alters tau pathology. We found that the density of MC1-positive tau inclusions was higher in male PS19 Dicer KO mice than in females, reflecting differences in accumulation in the cortex, amygdala, and piriform region (Fig. 3a–d). Male PS19 Dicer KO mice also had more amoeboid-like microglia, consistent with increased pathology (Fig. 3e–g). Bulk sequencing of isolated microglia from these mice showed that aside from differential expression of sex-chromosome genes, such as Xist, Ddx3y, Uty, and Eif2s3y, male microglia had enrichment of genes involved in inflammation and phagocytosis, including Spp1, Ccl6, Lpl, Il1b, and Cst7 (Fig. 3h, Supplementary Table 8), characteristic of disease-associated microglia (DAMs)17,18. These differences may be exacerbated by age and tau pathology, as DAM genes were not upregulated in younger mice (Extended Data Fig. 3a). Transcriptional sex differences, however, were still observed in younger mice, highlighting the dynamic role of miRNAs in the sex-dependent adaptations to tau pathology at different stages of the disease.
Figure 3. Loss of microglial microRNAs increases DAMs and tau pathology in male PS19 mice.

(a-c) Representative images of MC1 immunostaining of hemibrains from 9-month-old Dicer KO PS19 female (a) and male (b) mice. Scale bar, 600 μm. Yellow dashed box is magnified in (c). 2 independent experimental cohorts were used. Scale bar, 300 μm.
(d) MC1 density of hemibrain from Dicer KO nontransgenic (--) and transgenic (+) PS19 mice. n = 8 Dicer KO females, 6 Dicer KO males, 12 Dicer KO PS19 females, 10 Dicer KO PS19 males. * t = 2.578, df = 20, P = 0.02, unpaired, two-tailed t test. Boxplot elements: center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range.
(e) Representative 3D Imaris morphology reconstruction of cortical microglia from 9-month-old Dicer KO PS19 mice. Scale bar, 10 μm. Image acquired and analysis repeated for every mouse quantified in (f,g).
(f,g) Number of branch points (f) and total length of processes (g) per cell in Dicer KO PS19 mice, determined by Imaris-based automatic quantification. Error bars represent SEM. Bar denotes mean. Each dot represents one mouse. n = 7 biologically independent animals/sex, ~70 cells/sex. * t = 2.200, df = 12, P = 0.0481 (f), * t = 2.319, df = 12, P = 0.0388 (g), unpaired, two-tailed t test.
(h) Volcano plot of RNA-seq data from Dicer KO microglia from 9-month-old male and female PS19 mice. Pink, enriched in females; turquoise, enriched in males; grey, not significantly different. Vertical dashed lines indicate log2FC ± 1. Horizontal dashed line indicates -log10(0.05). n = 4 biologically independent samples/sex, 2 animals/sample. Wald test used.
(i) t-SNE plots of cells from female or male mice plotted based on clustering from Supplementary Fig. 3h. 2 biologically independent animals/sex.
(j) Percent of cells in each microglial cluster for each sex from (i). ** P = 0.0088 (cluster 1), ** P = 0.0021 (cluster 2), **** P < 0.0001 (cluster 3), ns P = 0.6139 (cluster 4), ns P = 0.1145 (cluster 5), two-sided Fisher’s exact test.
(k-m) Volcano plot of DEGs for cluster 1 (k), cluster 2 (l), and cluster 3 (m) as defined in (i). Purple, up-regulated genes; green, down-regulated genes; grey, not significantly different in each cluster compared to all other clusters. Wilcoxon rank sum test. DEGs defined as P ≤ 0.05 by Benjamini-Hochberg correction. Full list of DEGs in Supplementary Tables 9–11.
(n) Representative images of Lpl immunostaining of 9-month-old PS19 female and male cortical Dicer KO microglia. Scale bar, 15 μm. Iba1+ cells outlined. Image acquired and analysis repeated for every mouse quantified in (o).
(o) Average Lpl fluorescence intensity per Iba1+ cell in each sex. n = 43 female cells from 3 mice, 78 male cells from 4 mice. **** t = 7.413, df = 118, P = 2.04*10−11, unpaired, two-tailed t test. Boxplot elements: center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range.
To further probe these sex-specific effects in tauopathy, we did single-cell RNA-seq on PS19 Dicer KO microglia, gating on CD45+CD11b+ cells (Extended Data Fig. 3b–d). Of the 721 cells sequenced, 676 cells passed quality control and clustered into 5 distinct groups (Extended Data Fig. 3e–j). Female and male cells differentially distributed, such that females had more cluster 1 and 2 cells while males had more cluster 3 cells (Fig. 3i,j). While the clusters were distinguishable by distinct markers (Extended Data Fig. 3k), expression of resident microglial markers was relatively uniform, except in cluster 5, which likely comprised of CD45hi (Ptprc) peripheral macrophages (Extended Data Fig. 3l). Cluster 1 cells expressed higher levels of homeostatic genes such as P2ry12, Tmem119, Hexb, and Sall1 compared to all other clusters (Fig. 3k, Supplementary Table 9). Cluster 2 cells expressed higher levels of genes associated with microglia in human AD and experimental autoimmune encephalomyelitis mouse models19,20, such as Apoe, Ms4a7, Klra2, Clec12a, and Mrc1, with a downregulation of homeostatic genes, such as Sall1, P2ry12, and Gpr56 (Fig. 3l, Supplementary Table 10). The male-enriched Cluster 3 cells expressed higher levels of genes involved in phagocytosis, exosome biology, and immune modulation seen in DAMs17, such as Lpl, Cst7, Spp1, Tyrobp, Cd9, and Cd63 (Fig. 3m, Supplementary Table 11), consistent with the bulk RNA-seq (Fig. 3h). IHC confirmed upregulation of lipoprotein lipase (Lpl) in male microglia (Fig. 3n,o). This sex-dependent cluster enrichment supports the differential microglial response to tau pathology, where male PS19 mice exhibited increased DAMs and decreased homeostatic microglia compared with females.
In summary, male and female microglia expressed different miRNAs, both at baseline and in tauopathy. Loss of these miRNAs had sex-dependent consequences on the microglial transcriptome and tau pathogenesis. Therefore, microglial miRNAs are key contributors to sex-specific phenotypes and should be further studied to understand microglial biology in the context of neurodegenerative diseases.
Online Methods
Animals
For RNA-seq and histological analyses, Cx3cr1CreERT2/+ (https://www.jax.org/strain/021160) were crossed with Dicerfl/fl (https://www.jax.org/strain/006366) and P301S transgenic (https://www.jax.org/strain/008169) mice to obtain Cx3cr1+/+;Dicerfl/fl, Cx3cr1+/+;Dicerfl/fl;P301S+, Cx3cr1CreERT2/+;Dicerfl/fl, and Cx3cr1CreERT2/+;Dicerfl/fl;P301S+ mice. For miRNA-seq, P301S transgenic mice (https://www.jax.org/strain/008169) and their background control mice (https://www.jax.org/strain/100010) were used. Mice of both sexes were studied. For animal studies, sample size was determined by using power analysis with type 1 error = 5%, power = 80%, and effect size and standard deviation based on previous studies using the P301S (PS19) transgenic mouse line. Randomization of groups was not relevant for this study as all animals in the study were treated similarly. Mice had free access to food and water and were housed in a pathogen-free barrier facility with a 12-h light/12-h dark cycle. All mouse experiments were approved by the Institutional Animal Care and Use Committee, University of California, San Francisco and complied with all relevant ethical regulations.
Drug administration
Tamoxifen (Sigma-Aldrich, T5648) was prepared in corn oil at 20 mg/ml and given by intraperitoneal injection (2 mg/day) for 10 consecutive days, starting when mice were 3–3.5 months of age.
Brain tissue harvest
Mice were anesthetized with avertin and transcardially perfused with phosphate-buffered saline (PBS). Whole brains were fixed in 4% paraformaldehyde for 48 h and transferred to 30% sucrose for 48 h. 30–μm thick coronal sections were prepared on a sliding microtome (Leica, SM2010R), immersed in cyroprotectant, and stored at –20 °C before staining.
Immunohistochemistry
Free-floating coronal sections were washed in PBS, permeabilized in PBST buffer (0.5% Triton X-100 diluted in PBS), and immersed in 5% normal donkey serum at room-temperature for 1 h. For anti-Dicer and anti-Lpl primary antibodies, antigen retrieval was performed using Reveal Decloaker (BioCare, RV1000M) for 10 min at 90 °C then cooled to room temperature before blocking. Primary antibodies were diluted in PBST containing 5% normal donkey serum. Brain sections were incubated in primary antibodies at 4 °C overnight and then with secondary antibodies at room temperature for 1 h. Tissues were mounted on glass slides with antifade mounting medium (Life Technologies, P36935). Primary antibodies and dilutions used for staining were the following: Anti-Iba1 (Abcam, ab5076, 1:500 and Wako, 019–19741, 1:500), anti-Dicer (Abcam, ab167444, N167/7, 1:100), anti-Lpl (Abcam, ab21356, LPL.A4, 1:50) and anti-MC1 (a kind gift from P. Davies, 1:500)15. Secondary antibodies used were the following at 1:500 dilution: Cy3 AffiniPure Donkey Anti-Goat (Jackson ImmunoResearch, 705–165-147), Alexa Fluor 488 Donkey Anti-Mouse (Jackson ImmunoResearch, 715–545-150), Alexa Fluor 488 Donkey Anti-Mouse (Thermo Fisher Scientific, R37114). Validation information for primary antibodies included in “Life Sciences Reporting Summary”.
Epifluorescence fluorescence microscopy
Each coronal hemibrain slice was scanned at 10x magnification with an epifluorescence microscope (BZ-9000, Keyence). The images were stitched together with Keyence BZ-X Analyzer software (v1.3.0.3).
Confocal fluorescence microscopy
Images of hemibrain slices were acquired using an LSM880 confocal microscope (Carl Zeiss Microscopy, Thornwood, NY) at 1 μm focal plane intervals at 40x magnification. Images were examined by maximum intensity Z-projection.
Image analyses
All images were analyzed with ImageJ 1.5121. For MC1 density quantification, tiff images were processed with the auto local threshold Phansalkar plugin22, regions of interest were hand-traced, and MC1+ puncta were counted with the “Analyze Particles” function. For morphological analysis, confocal images of the lower cortical region were analyzed with Imaris software (v9.0.2, Bitplane) using the “Filament Tracer” function for microglial processes. Investigators were blinded during image acquisition and analysis.
Adult microglia isolation
Adult microglia were isolated by magnetic activated cell sorting as previously described23. Briefly, mice were anesthetized with avertin and transcardially perfused with PBS to remove circulating blood cells in the central nervous system. Dissected brains were digested with 3% collagenase type 3 (Worthington, LS004182) and 3 U/ml dispase (Worthington, LS02104) and incubated at 37 °C for 45 min. Digestion was stopped with inactivation buffer containing 2.5mM EDTA (Thermofisher, 15575020) and 1% fetal bovine serum (Invitrogen, 10082147). Tissue was then triturated in a serological pipette several times and passed through a 70-μm filter. Myelin in the homogenate was depleted with myelin removal beads (Miltenyi Biotec, 130–096-733) and a magnetic LD column (Miltenyi Biotec, 130–042-901). Microglia were isolated from the elutant with CD11b magnetic beads (Miltenyi Biotec, 130–049-601) and a magnetic MS column (Miltenyi Biotec, 130–042-201).
High-throughput miRNA-sequencing
Freshly isolated microglia from 2 mice were pooled, and RNA enriched in miRNAs was extracted with the miRvana miRNA Isolation Kit and phenol (Invitrogen, AM1560) as recommended by the manufacturer. Total RNA (100 ng) was used for adapter ligation with the Total RNAseq Kit v2 (Life Technologies, 4475936) and subsequently run on a 1% Tris-borate-EDTA acrylamide gel followed by gel extraction of small RNA (45–60 bp). The small RNA fraction was used for downstream reactions to build a small RNA library. Libraries were sequenced in multiplex on an Ion Proton (ThermoFisher Scientific, 4476610) and reads were mapped to mature miRNA from mirBase with Bowtie2 (v2.3.4)24.
miRNA-seq data analyses
Batch effects were removed with the R package RUVSeq25 (v1.16.1) and normalized to in silico control genes. 1 male and 1 female sample were excluded from the miRNA-seq analysis based on principle component analysis. Differences in gene expression were calculated with the R package DESeq226. Counts were normalized with the trimmed mean normalization method27. Genes with <15 counts across all samples were excluded from analysis. The false discovery rate (FDR) was calculated by the Benjamini-Hochberg method28. Predicted miRNA targeting of mRNA transcripts was done using Ingenuity Pathway Analysis (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity- pathway-analysis)29. Results were filtered for those with opposing log fold change values between mRNA and miRNAs and were either experimentally observed or highly predicted to be miRNA-mRNA target interactions.
High-throughput bulk RNA-sequencing
Freshly isolated microglia from 2 mice were pooled, and total RNA was extracted with the RNeasy mini kit (Qiagen, 74104). RNA quality was examined with a Bioanalyzer 2100 (Agilent Genomics). RNA samples with RNA integrity numbers >8 were used to construct a cDNA library. Oligo(dT) beads were used to enrich for mRNA. cDNA library generation was done using the QuantSeq 3´ mRNA-Seq Library Prep Kit for Illumina (Lexogen, 015.96). The quality of the cDNA library was assessed by using a Nanodrop spectrophotometer to determine concentration and a Bioanalyzer 2100 to determine insert size. cDNA library samples were then sequenced with the HiSeq 4000 system (Illumina).
RNA-seq data analyses of bulk RNA-seq
RNA-seq reads were mapped using the Bluebee Genomics Platform and using the GENCODE mouse genome GRCm38 (Lexogen QuantSeq 2.2.3). Differential gene expression was calculated with the R package DESeq226, using the Wald test. Counts were normalized with the trimmed mean normalization method27. Genes with <15 counts across all samples were excluded from analysis. The FDR was calculated with the Benjamini-Hochberg method28.
Real-time quantitative PCR
For miRNA qPCR, 10 ng of total RNA was reverse transcribed with the miRCURY LNA RT Kit (Qiagen). RT-qPCR was done with Power SYBR Green PCR master mix (Thermo Scientific) and miR-specific LNA primers (Qiagen) according to the Q5 Quantstudio system protocol. A small ribosomal RNA, 5S, was used for normalization across all samples, and relative differences were calculated with the 2-ΔΔCt method. To validate Dicer mRNA reduction, RNA was converted to cDNA with iScript Reverse Transcription Supermix (BioRad). Quantitative RT-PCR was done with an ABI 7900 HT sequence detector (Applied Biosystems) and SYBR Green PCR master mix (Applied Biosystems). GAPDH was used for normalization, and relative differences were calculated with the 2-ΔΔCt method. The following primers were used for quantitative RT-PCR. Dicer: forward, CCTCGGGCCTTGAGGATTTT; reverse, CGACGCCACAGTTCTCTTCT. GAPDH: forward, GGGAAGCCCATCACCATCTT; reverse, GCCTTCTCCATGGTGGTGAA.
Gene network and functional analyses
Gene network analyses of RNA-seq data were done with gene set enrichment analysis (GSEA)30 and the Hallmark gene dataset31. Networks were visualized with Cytoscape (v3.6.1)32, the STRING database33, and perfuse force directed layout.
Preparation of tissue for scRNA-seq
Brain tissue was prepared as described34. Briefly, 9-month-old mice were anesthetized with avertin and transcardially perfused with phosphate-buffered saline (PBS). The brain without the cerebellum was harvested and collected into cold medium containing 15mM HEPES, 0.5% glucose in Hank’s balanced salt solution without Phenol Red; the entire procedure was done on ice. Brains were minced with a razor blade and homogenized with a 2ml douncer containing 2ml of medium A with 80 μl DNase (12500 units/ml) and 5 μl of recombinant RNase inhibitor (Takara Bio 2313B). Homogenized tissue was filtered through a 70-μm strainers to obtain a single cell suspension. Cells were washed with medium A and resuspended in 850 μl of MACS buffer containing 1.8 μl of RNase inhibitor (sterile-filtered 0.5% bovine serum albumin, 2 mM EDTA in PBS). Cells were incubated with 100 μl of myelin removal beads (MACS Miltenyl Biotec) for 10 min, and loaded onto LD columns (Miltenyi Biotec). Cells were collected and washed for FACS staining.
Single cell sorting for scRNA-seq
Cells were blocked in 5 μl of mouse Fc block for 5 min on ice then incubated with primary antibodies for 10 min and washed with FACS buffer (sterile-filtered 1% fetal calf serum, 2mM EDTA, 25 mM HEPES in 1XPBS). Cells were incubated with secondary antibodies for 10min then washed with FACS buffer. Cells were resuspended in 500 μl of FACS buffer with RNase inhibitor (Takara Bio 2313B, 1:500) and 0.5 μl of propidium iodide (Thermo Fisher Scientific P3566, 1:1000) for single cell index sorting. Cell sorting/flow cytometry analysis was done on the cell sorter (BD InFlux) at the Stanford FACS Facility. The following gates were used for sorting microglia: (1) forward scatter-area (FSC-A)/side scatter-area (SSC-A) (2) Trigger Pulse Width/ FSC (3) Live- Dead negative using propidium iodide (4) CD45lowCD11b+ and CD45hiCD11b+. Single cells were sorted into 96-well plates containing 4 μl of lysis buffer (4U Recombinant RNase Inhibitor (Takara Bio 2313B), 0.05% Triton X-100, 2.5mM dNTP mix (Thermo Fisher Scientific R0192), 2.5 μM Oligo-dT30VN (50-AAGCAGTGGTATCAACGCAGAGTACT30VN-30, ERCC Spike-ins (Thermo Fisher Scientific 4456740) diluted at 1:2.4×107). Plates were vortexed, spun down and frozen on dry ice, and plates were stored at −80°C freezer. Antibodies used for FACS: rabbit anti-mouse Tmem119 (Abcam ab210405, ~200 μg/μl, 106–6, 1:400 dilution), CD45-PE-Cy7 (Thermo Fisher Scientific 25–0451-82, 30-F11, 1:300), CD11b-BV421 (BioLegend 101236, M1/70, 1:300), goat anti-rabbit Alexa 488 (Thermal Fisher Scientific 11034, 1:300).
ScRNA-seq library preparation
Sequencing libraries were prepared following the Smart-seq2 published protocol35. Briefly, plates were thawed and incubated at 72°C for 3 min in order to anneal RNAs to the Oligo-dT30VN primer. The 6 μl of the reverse transcription mixture was added to each well: 95U SMARTScribe Reverse Transcriptase (100U/μl, Clontech 639538), 10U RNase inhibitor (40U/μl), 1XFirst-Strand buffer, 5 mM DTT, 1M Betaine, 6mM MgCl2, 1 μM TSO (Exiqon, Rnase free HPLC purified). RT was performed at 42°C for 90 min, followed by 70°C, 5 min. 15 μl of PCR amplification mix containing the following reagents was added to each well: 1X KAPA HIFI Hotstart Master Mix (Kapa Biosciences KK2602), 0.1 μM ISPCR Oligo (AAGCAGTGGTAT CAACGCAGAGT), 0.56U Lambda Exonuclease (5U/μl, New England BioLabs M0262S). cDNA was amplified using the following PCR program: (1) 37°C 30 min; (2) 95°C 3 min; (3) 23 cycles of 98°C 20 s, 67°C 15 s, 72°C 4 min; (4) 72°C 5 min. cDNA samples were purified using PCRClean DX beads (0.7:1 ratio, Aline C-1003–50), and resuspended in 20 μl EB buffer. cDNA quality was examined with a Fragment Analyzer (AATI, High Sensitivity NGS Fragment Analysis Kit:1 bp - 6000 bp). To make libraries, all samples were diluted down to 0.15 ng/μl in 384-well plates using Mantis Liquid Handler (Formulatrix) and Mosquito X1 (TTP Labtech) with customized scripts. Nextera XT DNA Sample Prep Kit (Illumina FC-131–1096) was used at 1/10 of recommendation volume, with the help of a Mosquito HTS robot for liquid transfer. Tagmentation was done in 1.6 μl (1.2 μl Tagment enzyme mix, 0.4 μl diluted cDNA) at 55°C, 10 min. 0.4 μl Neutralization buffer was added to each well and incubated at room temperature for 5 min. 0.8 μl Illumina Nextera XT 384 Indexes (0.4 μl each, 5 μM from 4 sets of 96 indexes) and 1.2 μl PCR master mix were added to amplify whole transcriptomes using the following PCR program: (1) 72°C 3 min; (2) 95°C 30 s; (3) 10 cycles of 95°C 10 s, 55°C 30 s, 72°C 1 min; (4) 72°C 5 min. Libraries from one 384 plate were pooled into an Eppendorf tube and purified twice using PCRClean DX beads. Quality and concentrations of the final libraries were measured with Bioanalyzer and Qubit, respectively. Libraries were sequences on the Illumina HiSeq 4000 at the Weill Cornell Medicine Genomics and Epigenomics Core Facility.
Processing of scRNA-seq raw data
Prinseq36 v0.20.4 was first used to filter sequencing reads shorter than 30 bp (-min_len 30), to trim the first 10 bp at the 50 end (-trim_left 10) of the reads, to trim low-quality reads from the 30 end (-trim_qual_right 25) and to remove low-complexity reads (-lc_method entropy, -lc_threshold 65). Then, Trim Galore v0.4.3 (https://github.com/FelixKrueger/TrimGalore) was applied to trim the Nextera adapters (–stringency 1). The remaining reads were aligned to the mm10 genome by calling STAR37 v2.5.3a with the following options: –outFilterType BySJout,–outFilterMultimapNmax 20,–alignSJoverhangMin 8,–alignSJDBoverhangMin 1,– outFilterMismatchNmax 999,–outFilterMismatchNoverLmax 0.04,–alignIntronMin 20,–alignIntronMax 1000000,– alignMatesGapMax 1000000,–outSAMstrandField intronMotif. Picard was then used to remove the duplicate reads (VALIDATION_ STRINGENCY = LENIENT, REMOVE_DUPLICATES = true). Finally, the aligned reads were converted to counts for each gene by using HTSeq (-m intersection-nonempty, -s no)38.
Quality control for scRNA-seq data
The following criteria were used to filter out cells with low-quality sequencing. The distribution of total reads (in logarithmic scale) was fitted by a truncated Cauchy distribution, and data points in two tails of the estimated distribution were considered to be outliers and eliminated. Fitting and elimination were then applied to the remaining data. This process was run iteratively until the estimated distribution became stable. The threshold was set to the value at which the cumulative distribution function of the estimated distribution reaches 0.05. Cells with small numbers of detected genes and poor correlation coefficients for ERCC (low sequencing accuracy) were dropped. 676 cells were retained for downstream analysis after filtering from 721 cells.
Clustering analysis of scRNA-seq data
The Seurat v3.0.1 R package was used to perform unsupervised clustering analysis on the filtered scRNA-seq data39,40. Gene counts were normalized to the total expression and log-transformed. Principal component analysis was performed on the scaled data using highly variable genes as input. The JackStrawPlot function was used to determine the statistically significant principal components. These principal components were used to compute the distance metric and generate cell clusters. Non-linear dimensional reduction (t-SNE) was used to visualize clustering results. Differentially expressed genes were found using the FindAllMarkers function that ran Wilcoxon rank sum tests.
Statistics
All experiments were done with at least two biological replicates. Mean values from each mouse were used for computing statistical differences. Data distribution was assumed to be normal but this was not formally tested. Individual data points are shown when possible. Statistical analyses were done with Prism 7.0e (Graphpad, San Diego, CA) and R v3.5.1 (F Foundation for Statistical Computing, Vienna, Austria). Data were visualized with Prism or the R package ggplot241. Two-tailed t tests were used to compare two groups and two-sided Fisher’s exact tests were used to analyze contingency tables. Differential expression analysis for bulk-sequencing and single-cell sequencing was done using the Wald test and the Wilcoxon rank sum test, respectively, with P values corrected by Benjamini-Hochberg. P < 0.05 was considered statistically significant.
Reporting Summary
Further information on research design is available in the “Life Sciences Reporting Summary”.
Data Availability:
The bulk RNA-seq and miRNA-seq data that support the findings of this study were deposited in the Gene Expression Omnibus repository (https://www.ncbi.nlm.nih.gov/geo/). Bulk RNA-seq data was deposited under accession number GSE122663, and single-cell RNA-seq data was deposited under accession number GSE135330.
Extended Data
Extended Data Fig. 1. Male and female PS19 mice have similar tau pathology.
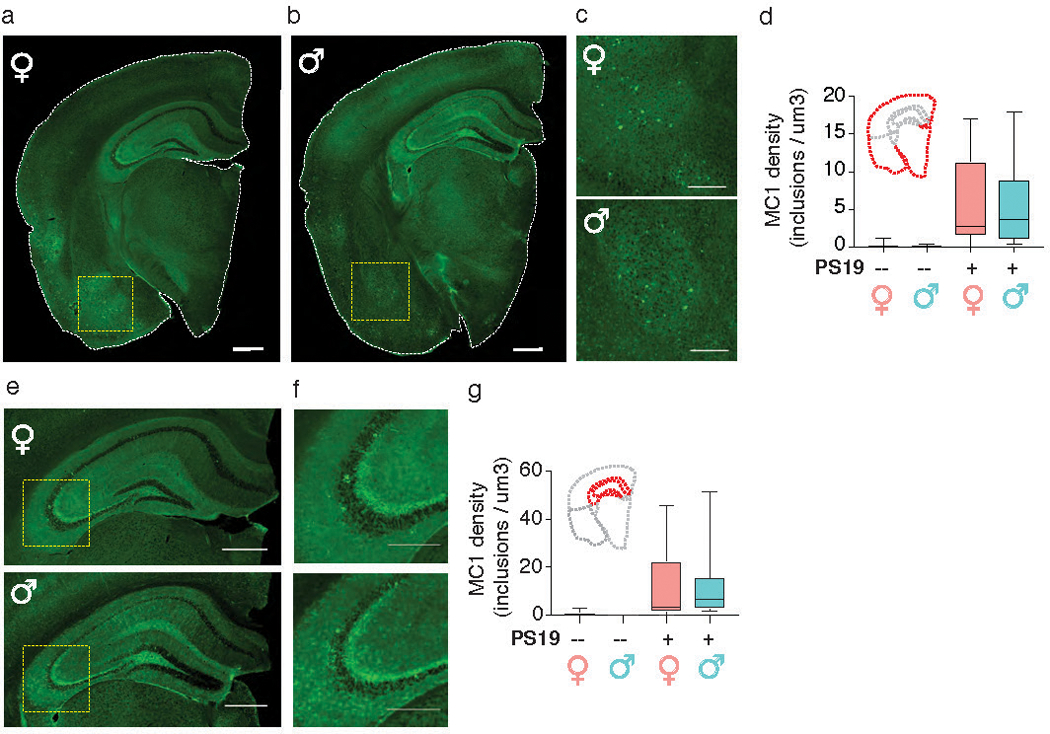
(a–c,e,f) Representative images of MC1 immunostaining of 9-month-old PS19 female (a) and male (b) hemibrains. Scale bar, 600 μm. Yellow dashed boxes magnified in (c). Scale bar, 300 μm. (e) Representative image of hippocampus. Scale bar, 600 μm. Yellow dashed boxes magnified in (f). Scale bar, 150 μm. 2 independent experimental cohorts were used.
(d,g) MC1 density of entire hemibrain (d) and hippocampus (g) of nontransgenic (–) and transgenic (+) male and female mice. n = 11 nontransgenic females, 5 nontransgenic males, 10 PS19 females, and 9 PS19 males. Boxplot elements: center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range.
Extended Data Fig. 2. Male and female microglia have differential transcriptional responses to tau pathology.

(a,c) Venn diagram of differentially expressed (DE) miRNAs (a) and mRNAs (c) comparing microglia from PS19 vs nontransgenic control (Ctrl) mice. DE genes defined as those with log2FC ≥ 1 or ≤ −1 and FDR < 0.05. Purple numbers, up-regulated DE genes; green numbers, down-regulated DE genes. n = 4 Ctrl samples/sex, 5 PS19 samples/sex, 2 mice/sample (a). n = 5 male and 3 female Ctrl samples, 3 male and 4 female PS19 samples, 2 mice/sample (c). * P = 0.05 (a), * P = 0.0146 (c), two-sided Fisher’s exact test. Full list of DEGs in Supplementary Tables 5–7.
(b) Volcano plot of male miRNA-seq data from (a). Purple and green dots represent miRNAs upregulated in PS19 samples (11 miRNAs; P ≤ 0.05 by Benjamini-Hochberg correction and log2FC ≥ 1) and downregulated in PS19 samples (43 miRNAs; P ≤ 0.05 and log2FC ≤ –1), respectively. Grey dots are miRNAs not significantly different. Dots with black circles represent those that were analyzed in (d). Vertical dashed lines indicate log2FC ± 1. Horizontal dashed line indicates -log10(0.05). Wald test was used.
(d) Bar graph showing 9 Ingenuity Pathway Analysis predicted target coverage of DE mRNAs from male PS19 vs Ctrl microglia (c) by DE miRNAs from male PS19 vs Ctrl microglia (a). Results were filtered for those with opposing miRNA and mRNA log fold changes (i.e. focusing on miRNA and mRNA targets that have anti-correlated expression patterns) and were either experimentally observed or highly predicted to be miRNA-mRNA target interactions.
Extended Data Fig. 3. RNA sequencing of Dicer KO microglia from PS19 mice.

(a) Volcano plot of RNA-seq data from Dicer KO microglia from 3-month-old male and female PS19 mice. Pink, female-enriched; turquoise, male-enriched; grey, not significantly different. Vertical dashed lines indicate log2FC ± 1. Horizontal dashed line indicates -log10(0.05). n = 4 male samples, 2 female samples, 2 mice/sample. Wald test was used.
(b) Schematic of the single-cell isolation method. Brains without the cerebellum were harvested from 9-month-old Dicer KO PS19 female and male mice and homogenized. After myelin depletion, cells were sorted using flow cytometry and gated by CD45+;CD11b+ expression.
(c) Representative FACS plots showing gating strategy and the cells sequenced. Similar gating strategy was used for all samples sequenced (n = 2 biologically independent animals/sex).
(d) Number of cells, proportion and statistics for FACS plots from (c).
(e-g) Quality control criteria for the single-cell sequencing data. Fitted curves for histograms of mapped reads (e), numbers of detected genes (f) and ERCC correlation coefficient (g) are labeled in red. Dashed lines are statistical cutoffs. Cells that passed all three criteria were retained for analysis.
(h) Scatter plot highlighting cells that passed QC (red) among all cells sequenced. Each dot is a cell.
(i) Summary of the numbers of cells sequenced and cells that passed QC (red).
(j) t-SNE plot of microglia clusters from 9-month-old Dicer KO PS19 female and male mice. n = 2 biologically independent animals/sex.
(k) Heatmap of top genes defining each microglial cluster.
(l) Ridge plots of microglial marker expression levels by each microglial cluster.
Supplementary Material
Supplementary Table 2: DE miRNAs encoded by the X chromosome in nontransgenic mice (Related to Fig. 1)w
Acknowledgements
We thank the Gladstone Histology & Light Microscopy Core and the Weill Cornell Medicine CLC Microscopy and Image Analysis Core Facility for help with imaging, and the Gladstone Bioinformatics Core for help with sequencing data analysis. Bulk RNA-seq was done at the Center for Advanced Technology, University of California, San Francisco. Single-cell RNA-seq was done at the Weill Cornell Medicine Genomics and Epigenomics Core Facility. FACS was done at the Stanford FACS Facility. We thank S. Ordway and K. Claiborn for editing the manuscript, K. M. Ansel and his lab members for comments on the manuscript and discussions on miRNA biology, and Y. Fu (University of California, San Francisco) for the Dicerfl/fl mice. We apologize that many relevant publications are not cited due to space limitations. This work was supported by NIH grants 1R01AG054214–01A1, U54NS100717, R01AG051390, and Tau Consortium grant (to L.G.), NIH grants 1F30AG062043–02 and T32GM007618 (to L.K.), NIH grant F31AG058505 (to F.A.S), NIH grant U54NS100717, Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, Larry L. Hillblom Foundation, and the Edward N. & Della L. Thome Memorial Foundation (to K.S.K.), Alzheimer’s Association AACSF 17–531484, NIH grant R25 R25NS070680, and UCSF Clinical and Translational Science Institute 5TL1TR0018 (to C.D.C), IRACDA Postdoctoral fellowship (to J.C.U.), and R01MH110504 (to G.Y.). Gladstone Institutes received support from the National Center for Research Resources Grant RR18928.
Footnotes
Competing Interests:
The authors declare no competing interests.
Accession codes:
Gene Expression Omnibus repository (https://www.ncbi.nlm.nih.gov/geo/):
References for main text:
- 1.Ferretti MT et al. Sex differences in Alzheimer disease - the gateway to precision medicine. Nature reviews. Neurology 14, 457–469, doi: 10.1038/s41582-018-0032-9 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Mazure CM & Swendsen J Sex differences in Alzheimer’s disease and other dementias. The Lancet Neurology 15, 451–452, doi: 10.1016/s1474-4422(16)00067-3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salter MW & Stevens B Microglia emerge as central players in brain disease. Nature medicine 23, 1018–1027, doi: 10.1038/nm.4397 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Guerreiro R et al. TREM2 variants in Alzheimer’s disease. The New England journal of medicine 368, 117–127, doi: 10.1056/NEJMoa1211851 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jonsson T et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. The New England journal of medicine 368, 107–116, doi: 10.1056/NEJMoa1211103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Villa A et al. Sex-Specific Features of Microglia from Adult Mice. Cell Reports 23, 3501–3511, doi: 10.1016/j.celrep.2018.05.048 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guneykaya D et al. Transcriptional and Translational Differences of Microglia from Male and Female Brains. Cell Rep 24, 2773–2783 e2776, doi: 10.1016/j.celrep.2018.08.001 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Mangold CA et al. Sexually divergent induction of microglial-associated neuroinflammation with hippocampal aging. J Neuroinflammation 14, 141, doi: 10.1186/s12974-017-0920-8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang SS et al. Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. The Journal of experimental medicine 215, 2235–2245, doi: 10.1084/jem.20180653 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM & Weiner HL MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-α–PU.1 pathway. Nature medicine 17, 64, doi:doi: 10.1038/nm.2266 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varol D et al. Dicer Deficiency Differentially Impacts Microglia of the Developing and Adult Brain. Immunity 46, 1030–1044 e1038, doi: 10.1016/j.immuni.2017.05.003 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Sharma S & Eghbali M Influence of sex differences on microRNA gene regulation in disease. Biology of sex differences 5, 3, doi: 10.1186/2042-6410-5-3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asai H et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nature neuroscience 18, 1584–1593, doi: 10.1038/nn.4132 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshiyama Y et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351, doi: 10.1016/j.neuron.2007.01.010 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Jicha GA, Bowser R, Kazam IG & Davies P Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. Journal of neuroscience research 48, 128–132 (1997). [DOI] [PubMed] [Google Scholar]
- 16.Hebert SS et al. Genetic ablation of Dicer in adult forebrain neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum Mol Genet 19, 3959–3969, doi: 10.1093/hmg/ddq311 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Keren-Shaul H et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e1217, doi: 10.1016/j.cell.2017.05.018 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Krasemann S et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581.e569, doi: 10.1016/j.immuni.2017.08.008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett FC et al. A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 98, 1170–1183.e1178, doi: 10.1016/j.neuron.2018.05.014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jordao MJC et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science (New York, N.Y.) 363, doi: 10.1126/science.aat7554 (2019). [DOI] [PubMed] [Google Scholar]
Methods-only References:
- 21.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nature methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phansalkar N, More S, Sabale A & Joshi M Adaptive local thresholding for detection of nuclei in diversity stained cytology images | Engineering360, <https://reference.globalspec.com/ref/3065459/adaptive-local-thresholding-for-detection-of-nuclei-in-diversity-stained-cytology-images> (1 February 2011).
- 23.Minami SS et al. Progranulin protects against amyloid beta deposition and toxicity in Alzheimer’s disease mouse models. Nature medicine 20, 1157–1164, doi: 10.1038/nm.3672 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359, doi: 10.1038/nmeth.1923 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Risso D, Ngai J, Speed TP & Dudoit S Normalization of RNA-seq data using factor analysis of control genes or samples. Nature Biotechnology 32, 896, doi:doi: 10.1038/nbt.2931 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550, doi:doi: 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robinson MD & Oshlack A A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biology 11, doi:doi: 10.1186/gb-2010-11-3-r25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benjamini Y & Hochberg Y Vol. 57 289–300 (Royal Statistical Society, Journal of the Royal Statistical Society. Series B (Methodological), 1995). [Google Scholar]
- 29.Kramer A, Green J, Pollard J Jr. & Tugendreich S Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530, doi: 10.1093/bioinformatics/btt703 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian A et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. doi: 10.1073/pnas.0506580102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liberzon A et al. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Systems 1, 417–425, doi: 10.1016/j.cels.2015.12.004 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shannon P et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research 13, 2498–2504, doi: 10.1101/gr.1239303 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szklarczyk D et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45, D362–d368, doi: 10.1093/nar/gkw937 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Q et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 101, 207–223.e210, doi: 10.1016/j.neuron.2018.12.006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Picelli S et al. Full-length RNA-seq from single cells using Smart-seq2. Nature Protocols 9, 171, doi:doi: 10.1038/nprot.2014.006 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Schmieder R & Edwards R Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864, doi: 10.1093/bioinformatics/btr026 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anders S, Pyl PT & Huber W in Bioinformatics Vol. 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology 36, 411, doi:doi: 10.1038/nbt.4096 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821, doi: 10.1016/j.cell.2019.05.031 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wickham H ggplot2 - Elegant Graphics for Data Analysis. (Springer, 2009). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 2: DE miRNAs encoded by the X chromosome in nontransgenic mice (Related to Fig. 1)w
Data Availability Statement
The bulk RNA-seq and miRNA-seq data that support the findings of this study were deposited in the Gene Expression Omnibus repository (https://www.ncbi.nlm.nih.gov/geo/). Bulk RNA-seq data was deposited under accession number GSE122663, and single-cell RNA-seq data was deposited under accession number GSE135330.