

Abstract
PHARC (polyneuropathy, hearing loss, cerebellar ataxia, retinitis pigmentosa, early-onset cataracts) is a human neurological disorder caused by deleterious mutations in the ABHD12 gene, which encodes an integral membrane lyso-phosphatidylserine (lyso-PS) lipase. Pharmacological or genetic disruption of ABHD12 leads to heightened lyso-PS lipids in human cells and the central nervous system of mice. ABHD12 loss also causes rapid rewiring of PS content, resulting in selective elevations in arachidonoyl (C20:4) PS and reductions in other PS species. The biochemical basis for ABHD12-dependent PS remodeling and its pathophysiological significance remain unknown. Here, we show that genetic deletion of the lysophospholipid acyltransferase LPCAT3 blocks accumulation of brain C20:4 PS in mice lacking ABHD12 and concurrently produces hyper-elevations in lyso-PS in these animals. These lipids changes correlate with exacerbated auditory dysfunction and brain microgliosis in mice lacking both ABHD12 and LPCAT3. Taken together, our findings reveal that ABHD12 and LPCAT3 coordinately regulate lyso-PS and C20:4 PS content in the CNS and point to lyso-PS lipids as the likely bioactive metabolites contributing to PHARC-related neuropathologies.
Lipid metabolic enzymes play critical roles in the nervous system, regulating not only the composition and structure of specialized membrane-delineated compartments (e.g., myelinated axons, dendritic spines, synaptic vesicles), but also the magnitude and duration of signaling of lipid transmitters, such as endocannabinoids1, 2 and lysophospholipids3, 4. Accordingly, several human neurological diseases are caused by genetic perturbations in lipid metabolic enzymes5, 6. Polyneuropathy, hearing loss, ataxia, retinitis pigmentosa and cataract (PHARC; OMIM 612674) is a rare, autosomal recessive monogenic disease that manifests as a slowly progressive sensorimotor disorder and is caused by deleterious mutations in the ABHD12 gene7. ABHD12 is an integral membrane serine hydrolase with high expression in the central nervous system (CNS) and innate immune cells, including microglia and macrophages. ABHD12 has been shown to hydrolyze several lipid substrates in vitro, including lyso-8 and oxidized-9 phosphatidylserines (PS) and monoacylglycerols, such as the endocannabinoid 2-arachidonoylglycerol (2-AG)10–12.
ABHD12(–/–) mice display a subset of PHARC-related phenotypes, including age-dependent hearing loss and motor impairments8. These behavioral phenotypes are accompanied by heightened microgliosis in the CNS8, pointing to a potential immunomodulatory function for ABHD12. Consistent with this premise, ABHD12(–/–) mice and mice treated with the selective ABHD12 inhibitor DO26413 show greater immunopathological responses in a viral infection model13, and macrophages from ABHD12(–/–) mice display heightened cytokine production9, 14.
The (neuro)immunological phenotypes caused by ABHD12 disruption are accompanied by discrete lipid changes in the CNS9, 13, 15, in particular, elevations in several lyso-PS lipids, as well as remodeled PS content reflecting a shift toward arachidonoyl (C20:4) PS with reduction in other PS species8, 13. The extent to which these different lipid changes contribute to PHARC-related neuropathologies remains an open and mechanistically important question. Lyso-PS lipids can activate immune-restricted GPCRs16–18 and Toll-like receptors (TLRs)19 and have been found to produce diverse immunomodulatory effects16, 20, 21. C20:4 PS, on the other hand, is susceptible to oxidization to produce ligands for the class B scavenger receptor CD36, which mediates phagocytosis of apoptotic cells by innate immune cells22. Acknowledging the independent biological activities of lyso-PS and C20:4 PS lipids, we aimed to investigate both the mechanistic basis for their regulation by ABHD12 in the CNS and their respective contributions to PHARC-related neuropathologies.
Considering that ABHD12 lacks PS lipase activity8, 9, we hypothesized that the remodeling of brain PS lipids caused by disruption of this enzyme reflected a secondary metabolic consequence of elevations in lyso-PS. A strong candidate enzyme responsible for converting lyso-PS to C20:4 PS is lysophosphatidylcholine acyltransferase 3 (LPCAT3; or membrane-bound O-acyltransferase 5 (MBOAT5)), which accepts lysophospholipid substrates, including lyso-PS, and shows high specificity for polyunsaturated fatty acyl coenzyme A (CoA) substrates, specifically C18:2 and C20:4 CoA23, 24. The role of LPCAT3 in PC and PE remodeling has been well-documented in various tissues from mice25–27. However, a potential contribution of LPCAT3 to modulating PS lipid content in the brain, or in human cells, has not, to our knowledge, been investigated.
We first evaluated ABHD12-LPCAT3 crosstalk in the human monocytic THP-1 cell line, where we have previously shown that the ABHD12 inhibitor DO264 increases both lyso-PS and C20:4 PS in a concentration-dependent manner13. We generated LPCAT3-deficient THP-1 cells using CRISPR/Cas9 (clustered regularly interspaced short palindromic repeat/Cas9) methods (see Detailed Experimental Procedures) and confirmed disruption of the LPCAT3 gene by DNA sequencing (Figure S1). Three independent populations of LPCAT3-deficient THP-1 cells were generated (sgLPCAT3–1-3) and each showed a >90% loss of LPCAT3 activity compared to a cell population treated with a control single-guide RNA (sgCtrl), as measured using cell lysates and an LC-MS assay that quantified conversion of 17:1 lyso-PS and C20:4 CoA to 17:1/20:4 PS (Figure 1A). We then treated sgLPCAT3 and sgCtrl THP-1 cells with DO264 (1 μM, 4 h) and analyzed phospholipids by LC-MS using targeted multiple reaction monitoring (MRM) methods. Consistent with previous results13, DO264 treatment resulted in elevations in several lyso-PS and C20:4 PS lipids in control (sgCtrl) THP-1 cells (Figure 1B, C and Figure S2 and Table S1). Notably, DO264-induced elevations in lyso-PS were substantially higher in sgLPCAT3 cells, despite these cells showing unaltered basal concentrations of lyso-PS (DMSO-treated groups) (Figure 1B and Figure S2 and Table S1), and, conversely, the increases in C20:4 PS caused by DO264 were blocked in sgLPCAT3 cells (Figure 1C). Other (lyso-)phospholipids showed more limited changes, with subsets of lyso-PCs and lyso-PEs either decreasing or being unaltered in DO264-treated sgCtrl cells and showing no further alteration in sgLPCAT3 cells (Figures S3 and S4 and Table S1). None of the measured PC or PE lipids were altered by DO264 in either sgCtrl or sgLPCAT3 cells (Figures S3 and S4 and Table S1). These data, taken together, support a model where, in ABHD12-disrupted cells, LPCAT3 converts heightened lyso-PS to C20:4 PS, creating a shunt pathway that both increases C20:4 PS and, interestingly, blunts the magnitude of elevation in lyso-PS caused by ABHD12 loss.
Figure 1.
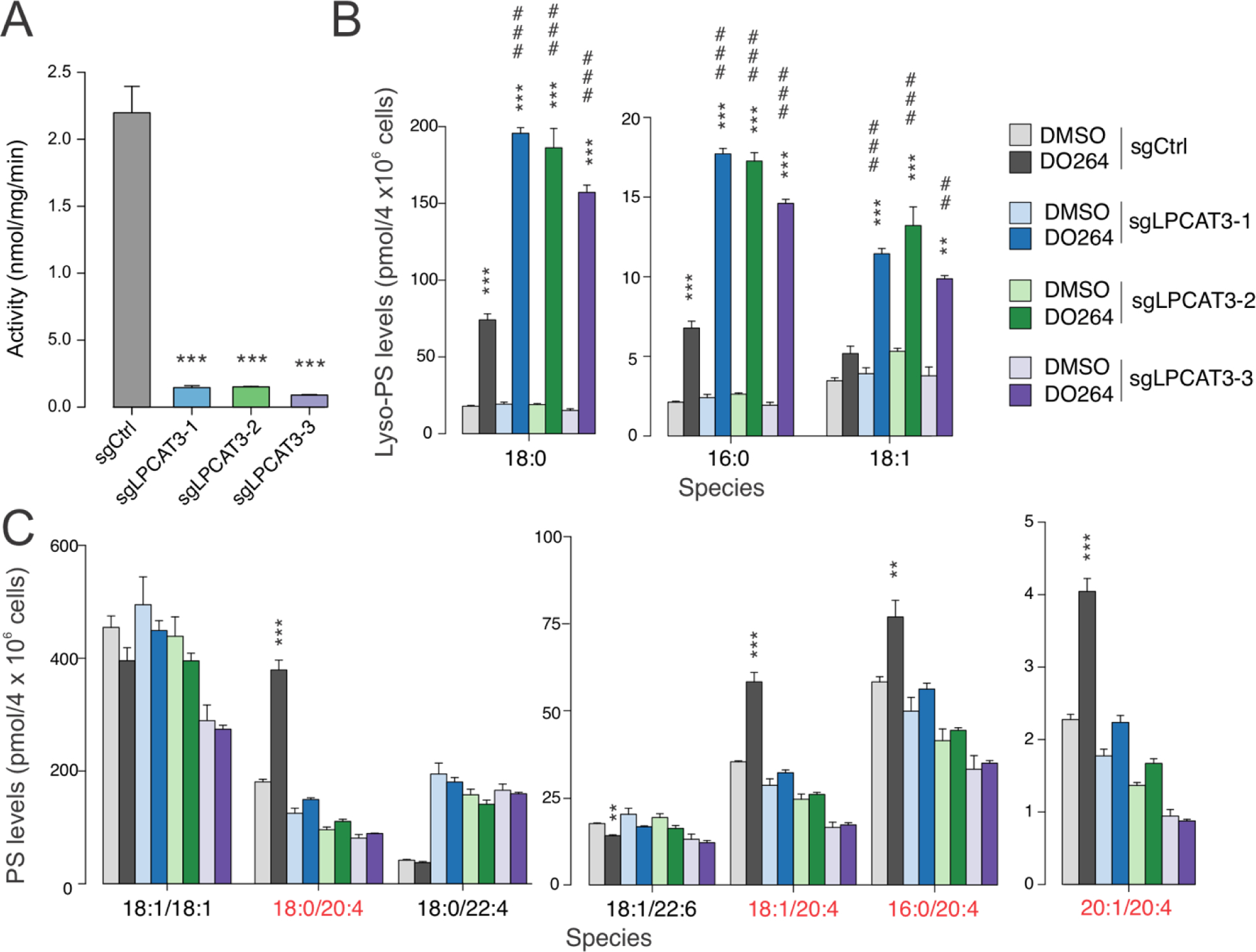
Lyso-PS and PS lipid profiles of LPCAT3-deficient THP-1 cells treated with the ABHD12 inhibitor DO264. A) Lyso-PS acyltransferase activity of membrane lysates (0.05 mg/mL) from LPCAT3-wild type (sgCtrl) and LPCAT3-null (sgLPCAT3–1-3) THP-1 cells generated by the CRISPR/Cas9 system were tested using 17:1 lyso-PS and C20:4 CoA as substrates. Data represent mean ± SE values. n = 3 per group. B, C) Lyso-PS (B) and PS (C) lipid profiles of LPCAT3-wild type and LPCAT3-null THP-1 cells treated with DMOSO or DO264 (1 μM, 4 h). C20:4 PS lipids are highlighted in red. Data represent mean ± SE values. n = 3 per group. *P < 0.05; ** or ## P < 0.01; *** or ### P < 0.001. (asterisk (*) used for two-sided Student’s t-test performed relative to sgCtrl for (A) and to respective DMSO controls for (B) and (C). Pound (#) used for two-sided Student’s t-test performed for DO264-treated sgLPCAT3 cell populations relative to DO264-treated sgCtrl cells.
We next set out to investigate LPCAT3-ABHD12 crosstalk in the CNS of mice. Global genetic deletion of LPCAT3 is neonatally lethal in mice25, 28, so we generated nervous system-specific LPCAT3-knockout mice (designated hereafter as “N-LPCAT3(–/–) mice”) by crossing floxed-LPCAT3(fl/fl) mice28 with nestin-Cre transgenic mice. N-LPCAT3(–/–) mice were born at the expected Mendelian frequency (Table S2), survived to adulthood, and were not distinguishable from LPCAT3(fl/fl) mice in their appearance and general cage behavior. The lyso-PS acyltransferase activity of brain membrane lysates from N-LPCAT3(–/–) mice was ~80% decreased compared to LPCAT3(fl/fl) mice (Figure 2A). It is unclear whether the residual lyso-PS acyltransferase activity reflects contributions from other LPCAT enzymes or the retention of LPCAT3 in brain cell types that do not express nestin-Cre mice. Attempts to assess LPCAT3 protein content in brain tissue from LPCAT3(fl/fl) mice and N-LPCAT3(–/–) mice by western blotting were thwarted due to the lack of commercial antibodies showing specificity for LPCAT328. Nonetheless, our results confirm that LPCAT3 is a major source of bulk lyso-PS acyltransferase activity in mouse brain.
Figure 2.
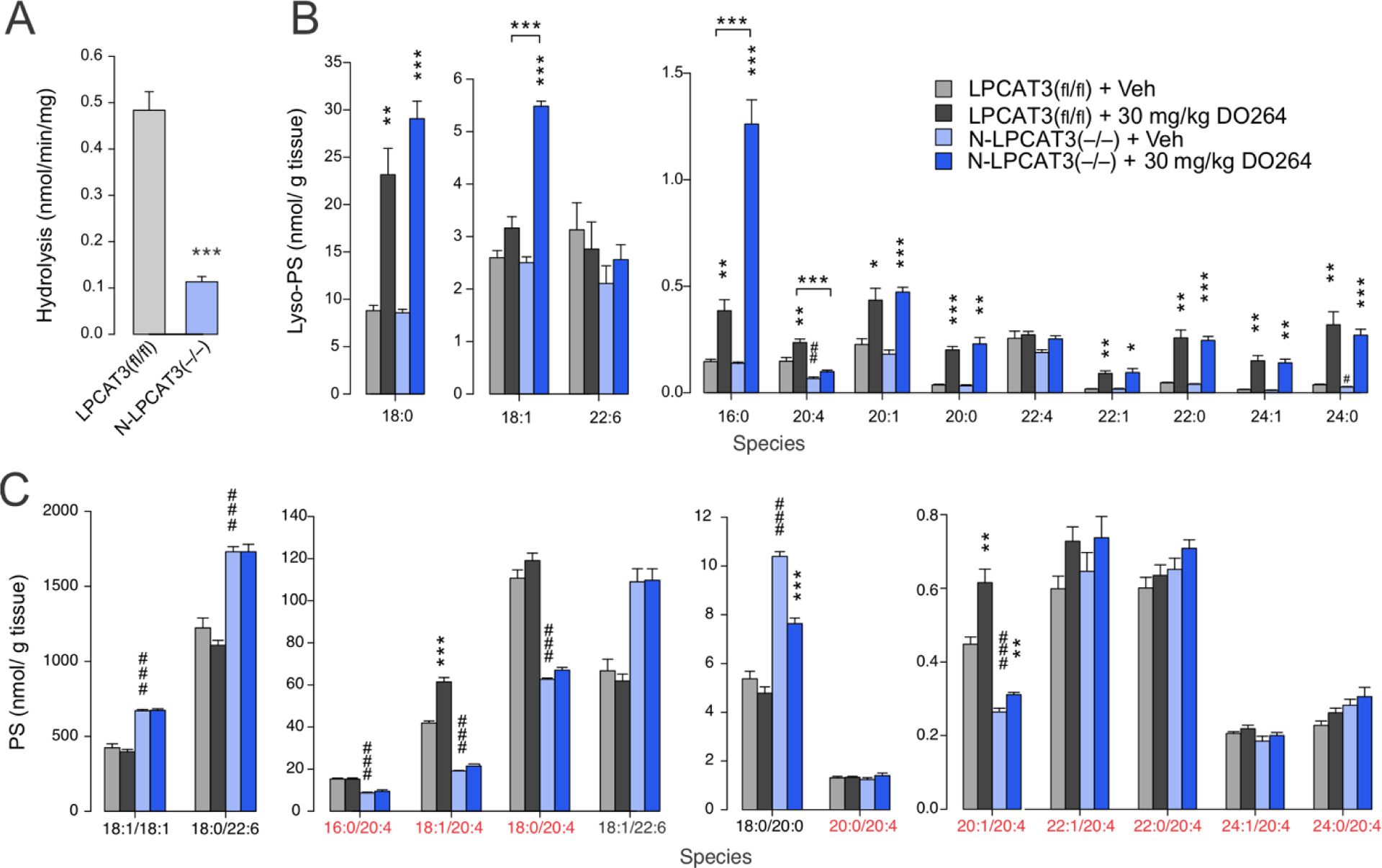
Lyso-PS and PS lipid profiles of N-LPCAT3(–/–) mice treated with the ABHD12 inhibitor DO264. A) Lyso-PS acyltransferase activity from brain membrane lysates (0.05 mg/mL) of N-LPCAT3(–/–) mice. B, C) Brain lyso-PS (B) and PS (C) lipid profiles of wild type (WT; LPCAT3(fl/fl)) and N-LPCAT3(–/–) mice treated with vehicle (Veh) or DO264 (30 mg/kg, 4h, p.o.). Data represent mean ± SE values. n = 5/group. C20:4 PS species are highlighted in red. * or # P < 0.05; ** or ## P < 0.01; *** or ### P < 0.001 (two-sided Student’s t-test performed relative to LPCAT3(fl/fl) for (A). For (B) and (C), asterisks (*) were used for LPCAT3(fl/fl) + Veh vs LPCAT3(fl/fl) + 30 mg/kg DO264 and N-LPCAT3(–/–) + Veh vs N-LPCAT3(–/–) + 30 mg/kg DO264 comparisons. Sharps (#) were used for LPCAT3(fl/fl) + Veh vs N-LPCAT3(–/–) + Veh comparisons.
N-LPCAT3(–/–) and LPCAT3(fl/fl) control mice were treated with vehicle or DO264 (30 mg/kg, 4 h), after which animals were sacrificed and brain tissue harvested and analyzed for phospholipid content. The lyso-PS concentrations in vehicle-treated LPCAT3(fl/fl) and N-LPCAT3(–/–) mice were similar except for a reduction in C20:4 lyso-PS in N-LPCAT3(–/–) mice (Figure 2B and Table S1). DO264 caused the expected elevations in several lyso-PS lipids in LPCAT3(fl/fl) mice, and, interestingly, these increases were even greater in N-LPCAT3(–/–) mice for some of the most abundant lyso-PS species (16:0, 18:0, 18:1) (Figure 2B and Table S1). Notably, however, the elevation in C20:4 lyso-PS observed in DO264-treated LPCAT3(fl/fl) mice was completely blocked in N-LPCAT3(–/–) mice. The PS content of N-LPCAT3(–/–) mice showed substantial basal rewiring, with vehicle-treated animals displaying decreased concentrations of C20:4 PS lipids and increased concentrations of other PS species (e.g. 18:1, 22:6, and 22:4 PS) (Figure 2C and Figure S5 and Table S1). Consistent with previous studies 13, acute inhibition of ABHD12 by DO264 produces a more limited set of PS changes in control mice compared to chronic disruption of this enzyme (e.g., ABHD12(–/–) mice; see below), but these changes – elevations in 18:1/20:4 and 20:1/20:4 PS – did not occur in N-LPCAT3(–/–) mice (Figure 2C and Figure S5 and Table S1).
We next assessed the impact of chronic loss of both ABHD12 and LPCAT3 in the nervous system by crossing ABHD12(+/−)/LPCAT3(fl/fl) and ABHD12(+/−)/LPCAT3(fl/fl) nestin-Cre mice to generate ABHD12/N-LPCAT3 double-knockout (double-KO) mice. The double-KO mice were born at the expected Mendelian ratio (Table S2) and indistinguishable from LPCAT3(fl/fl), ABHD12(–/–), or N-LPCAT3(–/–) mice in their appearance and general cage behavior. The expected reductions in both brain lyso-PS lipase (ABHD12) and lyso-PS acyltransferase (LPCAT3) activity were observed across the genotypes (Figure 3A). As we found for DO264-treated N-LPCAT3(–/–) mice, the double-KO mice, evaluated at either 11 weeks or nine-months of age, showed hyper-elevations in several brain lyso-PS species compared to ABHD12(–/–) mice (Figure 3B and Table S1). The principal exception was again C20:4 lyso-PS, for which the elevation in ABHD12(–/–) mice was blocked in double-KO mice. The widespread increases in C20:4 PS lipids and decreases in saturated/monounsaturated PS lipids in ABHD12(–/–) brain tissue were also mostly ablated in double-KO mice (Figure 3C and Table S1). In general, the PS profile of double-KO mice resembled that of N-LPCAT3(–/–) mice (Figure 3C and Figure S6 and Table S1). These results demonstrate that ABHD12 and LPCAT3 coordinately regulate lyso-PS and C20:4 PS content in the mammalian brain and enable construction of a metabolic pathway diagram to explain the flux of specific lyso-PS and PS lipids (Figure 3D).
Figure 3.
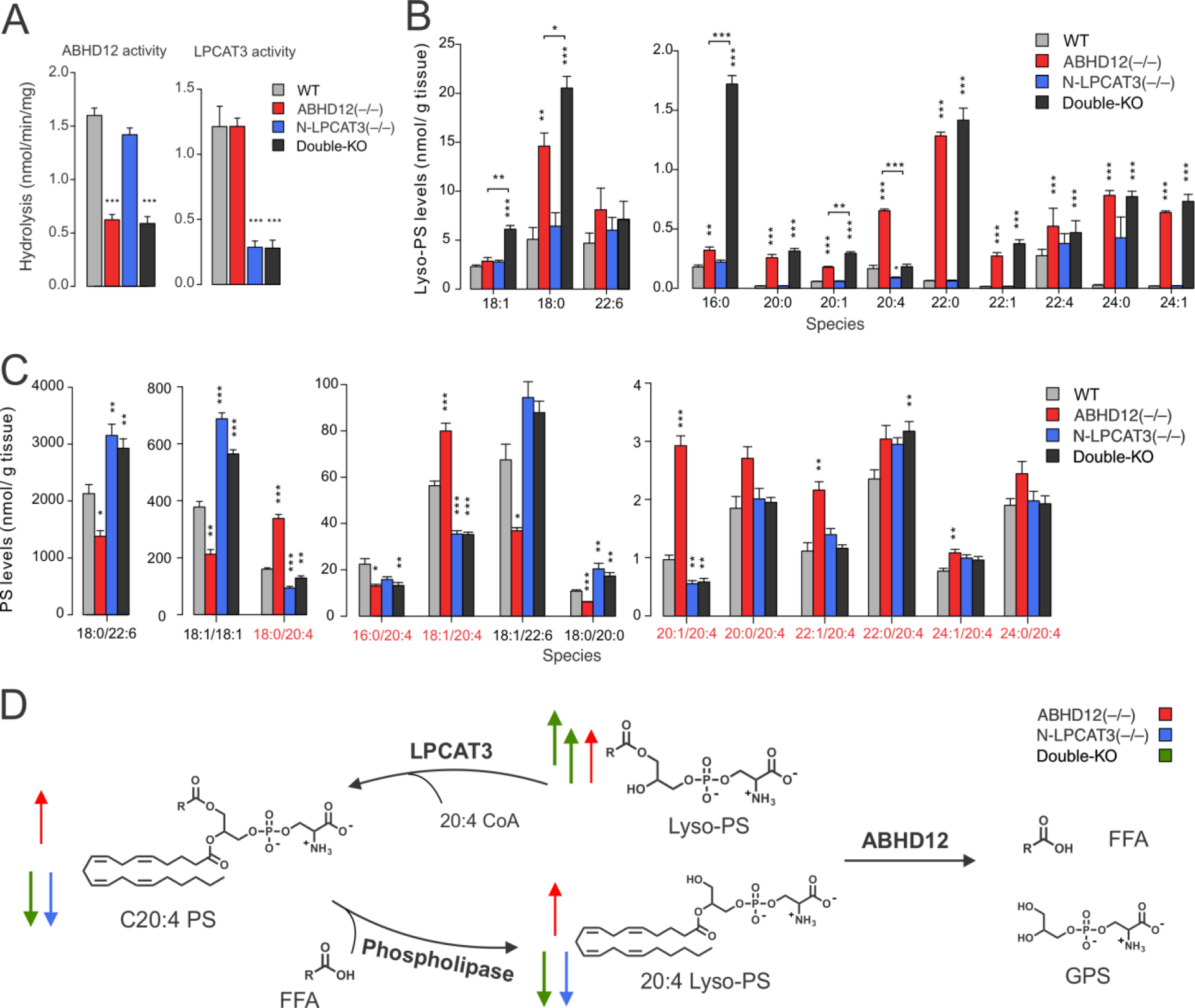
Lyso-PS and PS lipid profiles of ABHD12(–/–), N-LPCAT3(–/–), and ABHD12/N-LPCAT3 double-KO mice. A) Lyso-PS hydrolysis (ABHD12) and lyso-PS acyltransferase (LPCAT3) activities from brain membrane lysates (0.05 mg/mL) of WT (LPCAT3(fl/fl)), ABHD12(–/–), N-LPCAT3(–/–), and double-KO mice. B, C) Brain lyso-PS (B) and PS (C) lipid profiles of WT, ABHD12(–/–), N-LPCAT3(–/–), and double-KO mice. Data represent mean ± SE values and are shown for eleven week-old mice. n = 5/group. Similar profiles were observed in nine month-old mice (Table S1). D) Metabolic pathway diagram delineating the coordinated regulation of lyso-PS and C20:4 PS content in the mammalian brain by ABHD12 and LPCAT3. Arrows indicate abundance changes of indicated metabolites in ABHD12(–/–) (red), N-LPCAT3(–/–) (blue), and double-KO (green) mice. *P < 0.05; **P < 0.01; ***P < 0.001 (two-sided Student’s t-test performed relative to WT.
Additional, generally more restricted changes in other (lyso-)phospholipids were observed in ABHD12- and LPCAT3-disrupted mice, including: 1) increases in 18:0 lyso-PC and 18:0 lyso-PE (Figures S7 and S8 and Table S1) in ABHD12(–/–) and double-KO mice; and 2) reductions in C20:4 (lyso)-PC and C20:4 (lyso)-PE (Figures S7 and S8 and Table S1) in N-LPCAT3(–/–) and double-KO mice. Unlike PS, however, substantia acyl chain remodeling, as reflected in elevations in other non-C20:4 lipids, was not observed for PC or PE in N-LPCAT3(–/–) and double-KO mice (Figures S7 and S8 and Table S1). We also do not know whether the C18:0 lyso-PC/PE changes observed in ABHD12-disrupted mice reflect direct utilization of these lipids as substrates by ABHD12 or are an indirect consequence of remodeling of the brain lyso-PS-PS network in these animals.
The bidirectional changes in lyso-PS (hyper-elevated) and C20:4 PS (suppressed) observed in double-KO mice compared to ABHD12(–/–) mice afforded an opportunity to investigate the respective contributions of these lipid classes to ABHD12-dependent neuropathologies. We hypothesized that, if lyso-PS lipids were contributing to ABHD12-dependent neurological phenotypes, then the double-KO mice should show equivalent or even greater defects compared to ABHD12(–/–) mice, while, if C20:4 PS lipids were a principle source of pathogenicity, then the double-KO mice should be protected from ABHD12-dependent neurological impairments. We compared the auditory startle responses of nine-month old LPCAT3(fl/fl), ABHD12(–/–) mice, N-LPCAT3(–/–), and double-KO mice and found, as reported previously8, 13, that ABHD12(–/–) mice show a substantial impairment (Figure 4A). The auditory function of N-LPCAT3(–/–) mice, on the other hand, was generally similar, albeit modestly reduced, compared to LPCAT3(fl/fl) control mice (Figure 4A). The double-KO mice, however, displayed the greatest loss of auditory function, significantly exceeding in magnitude the impairment observed in ABHD12(–/–) mice (Figure 4A). A similar trend was observed for the degree of cerebellar microgliosis as determined by immunostaining for ionized calcium binding adaptor molecule 1 (Iba1), a marker of activated microglia and macrophages. As reported previously8, the number of enlarged, activated microglia was higher in ABHD12(–/–) mice compared to wild type control (LPCAT3(fl/fl)) mice (Figure 4B, C). N-LPCAT3(–/–) mice also showed a higher number of activated microglia, and the microgliosis was greatest in the double-KO mice. We interpret the exacerbated auditory dysfunction and microgliosis state of the double-KO mice as being more supportive of the hypothesis that elevations in lyso-PS lipids, rather than C20:4 PS lipids, contribute to the neuropathological phenotypes observed in ABHD12(–/–) mice. While we do not yet understand why the N-LPCAT3(–/–) mice independently show a microgliosis phenotype, it is possible that the broader reductions in C20:4 phospholipids (e.g., PC, PE) occurring in these animals also produce a neuroimmunological effect.
Figure 4.
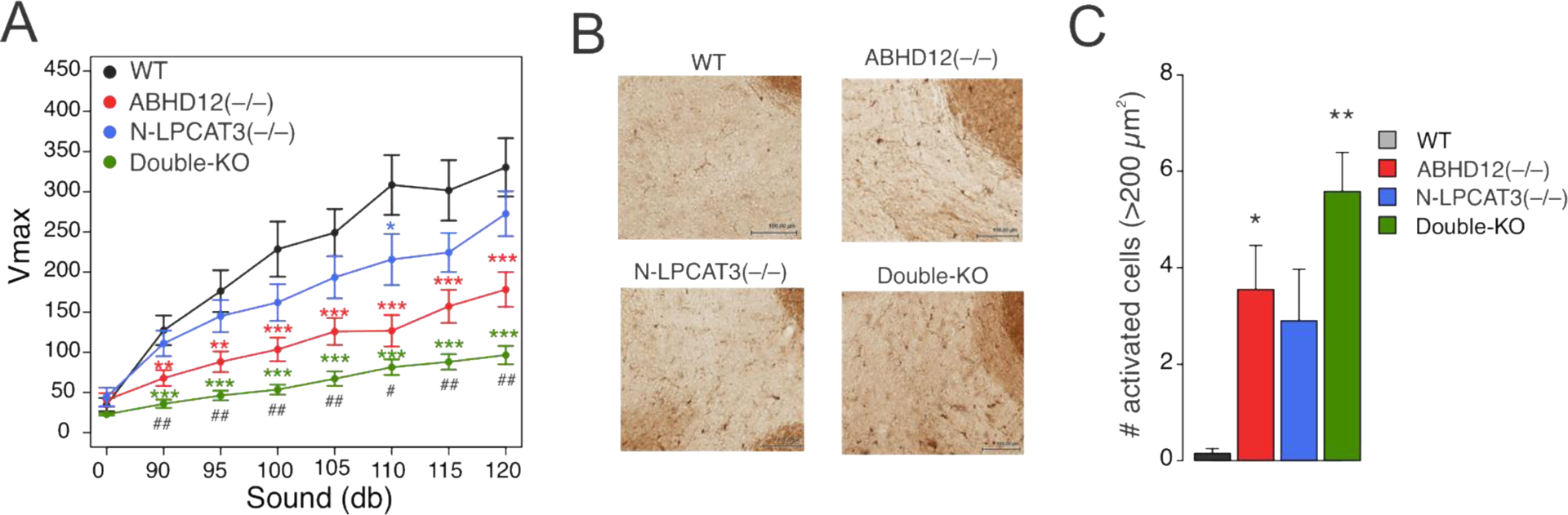
Auditory function and microgliosis of ABHD12(–/–), N-LPCAT3(–/–), and ABHD12/N-LPCAT3 double-KO mice. A) Auditory startle response test performed on nine-month-old WT, ABHD12(–/–), N-LPCAT3(–/–), and double-KO mice. Data represent mean ± SE values. n = 23–30/group. B) Representative images of Iba-1 immunostaining for activated microglia in the cerebellum of 12-month-old WT, ABHD12(–/–), N-LPCAT3(–/–), and double-KO mice. Scale bar, 100 μm. C) Quantification of activated microglia (Iba-1+ cells with area greater than 200 μm2) in the cerebellum of 12-month-old WT, ABHD12(–/–), N-LPCAT3(–/–), and double-KO mice. Data represent mean ± SE values. n = 5/group. * or # P < 0.05; ** or ## P < 0.01; ***P < 0.001 (asterisk (*) used for two-sided Student’s t-test performed relative to WT for (A) and (C); pound (#) used for two-sided Student’s t-test performed between ABHD12(–/–) and double-KO data in (A).
In summary, our findings provide compelling insights into both the metabolic regulation of (lyso-)PS lipids in the nervous system and how specific perturbations in this lipid network relate to neurological disease. We are struck by the tight coupling of ABHD12 and LPCAT3, as even acute pharmacological blockade of ABHD12 resulted in rapid LPCAT3-dependent remodeling of PS lipids. Additionally, while LPCAT3 disruption did not, on its own, alter the lyso-PS content of human cells or mouse brain, with the notable exception of C20:4 lyso-PS (see below), the loss of this enzyme had a major impact on lyso-PS lipids in ABHD12-disrupted systems, leading, in many cases, to the hyper-elevation of these lipids. We interpret this result to indicate that, when ABHD12 is blocked, LPCAT3 becomes a rate-limiting enzyme for controlling lyso-PS. The curious case of C20:4 lyso-PS suggests an alternative metabolic pathway for this lipid – instead of serving as a direct substrate for ABHD12, we posit that the elevations in C20:4 lyso-PS in ABHD12-disrupted systems reflect the action of a distinct, as-of-yet unidentified, sn-1 PS lipase acting on elevated C20:4 PS lipids (Figure 3D). Further advances in our understanding of the dynamic interplay between ABHD12 and LPCAT3, as well as possibly other lipid enzymes (e.g. sn-1 or sn-2 PS lipases that generate lyso-PS14, 29; or additional LPCATs, such as MBOAT1, that can also accept lyso-PS lipids as substrates23, 24) would benefit from the development of selective and in vivo-active LPCAT3 inhibitors, as these compounds would overcome some of the technical challenges associated with the lethality caused by the global, genetic loss of LPCAT3.
The integration of our lipidomic and neuropathology data point to lyso-PS lipids, rather than C20:4 PS lipids, as the more likely source of neurological defects caused by ABHD12 disruption. The in vivo signaling mechanisms of lyso-PS lipids, however, remain poorly understood. Candidate lyso-PS receptors, such as GPR34 and GPR174, are implicated in (neuro)immunological processes30, but we do not know yet which, if any of the established lyso-PS receptors, contribute to the neuropathological phenotypes caused by ABHD12 loss. We also recently identified ABHD12 as a modulator of ferroptosis in human cancer cells31, and this non-apoptotic form of cell death is also regulated by LPCAT332. PHARC has been suggested to have a neurodegenerative basis7, and it will be interesting, in future studies, to determine if neurons in ABHD12-disrupted mice show altered ferroptotic potential. Finally, our findings provide some directionality to the pursuit of treatment strategies for PHARC and related neuroimmunological disorders, as they point to enzymes involved in lyso-PS biosynthesis, such as PLA1 and/or PLA2 PS lipases14, 29, as potentially attractive targets. Even more ambitious would be the development of small-molecule activators of LPCAT3, which would be predicted, based our findings, to counter the rise in lyso-PS caused by ABHD12 loss.11,17,26
Supplementary Material
ACKNOWLEDGMENT
We thank Hui Jing for experimental guidance and helpful discussions and Michele Martes for maintenance of mouse colonies.
Funding Sources
This work was supported by the NIH (DA033760, HL136618) and Lundbeck.
Footnotes
REFERENCES
- [1].Ahn K, McKinney MK, and Cravatt BF (2008) Enzymatic pathways that regulate endocannabinoid signaling in the nervous system, Chem Rev 108, 1687–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tsuboi K, Uyama T, Okamoto Y, and Ueda N (2018) Endocannabinoids and related N-acylethanolamines: biological activities and metabolism, Inflamm Regen 38, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rosen H, Germana Sanna M, Gonzalez-Cabrera PJ, and Roberts E (2014) The organization of the sphingosine 1-phosphate signaling system, Curr Top Microbiol Immunol 378, 1–21. [DOI] [PubMed] [Google Scholar]
- [4].Yung YC, Stoddard NC, and Chun J (2014) LPA receptor signaling: pharmacology, physiology, and pathophysiology, J Lipid Res 55, 1192–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Long JZ, and Cravatt BF (2011) The metabolic serine hydrolases and their functions in mammalian physiology and disease, Chem Rev 111, 6022–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Darios F, Mochel F, and Stevanin G (2020) Lipids in the Physiopathology of Hereditary Spastic Paraplegias, Front Neurosci 14, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fiskerstrand T, H’Mida-Ben Brahim D, Johansson S, M’Zahem A, Haukanes BI, Drouot N, Zimmermann J, Cole AJ, Vedeler C, Bredrup C, Assoum M, Tazir M, Klockgether T, Hamri A, Steen VM, Boman H, Bindoff LA, Koenig M, and Knappskog PM (2010) Mutations in ABHD12 cause the neurodegenerative disease PHARC: An inborn error of endocannabinoid metabolism, Am J Hum Genet 87, 410–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Blankman JL, Long JZ, Trauger SA, Siuzdak G, and Cravatt BF (2013) ABHD12 controls brain lysophosphatidylserine pathways that are deregulated in a murine model of the neurodegenerative disease PHARC, Proc Natl Acad Sci U S A 110, 1500–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kelkar DS, Ravikumar G, Mehendale N, Singh S, Joshi A, Sharma AK, Mhetre A, Rajendran A, Chakrapani H, and Kamat SS (2019) A chemical-genetic screen identifies ABHD12 as an oxidized-phosphatidylserine lipase, Nat Chem Biol 15, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Blankman JL, Simon GM, and Cravatt BF (2007) A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol, Chem Biol 14, 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Joshi A, Shaikh M, Singh S, Rajendran A, Mhetre A, and Kamat SS (2018) Biochemical characterization of the PHARC-associated serine hydrolase ABHD12 reveals its preference for very-long-chain lipids, J Biol Chem 293, 16953–16963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Navia-Paldanius D, Savinainen JR, and Laitinen JT (2012) Biochemical and pharmacological characterization of human alpha/beta-hydrolase domain containing 6 (ABHD6) and 12 (ABHD12), J Lipid Res 53, 2413–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ogasawara D, Ichu TA, Vartabedian VF, Benthuysen J, Jing H, Reed A, Ulanovskaya OA, Hulce JJ, Roberts A, Brown S, Rosen H, Teijaro JR, and Cravatt BF (2018) Selective blockade of the lyso-PS lipase ABHD12 stimulates immune responses in vivo, Nat Chem Biol 14, 1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kamat SS, Camara K, Parsons WH, Chen DH, Dix MM, Bird TD, Howell AR, and Cravatt BF (2015) Immunomodulatory lysophosphatidylserines are regulated by ABHD16A and ABHD12 interplay, Nat Chem Biol 11, 164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ogasawara D, Ichu TA, Jing H, Hulce JJ, Reed A, Ulanovskaya OA, and Cravatt BF (2019) Discovery and Optimization of Selective and in Vivo Active Inhibitors of the Lysophosphatidylserine Lipase alpha/beta-Hydrolase Domain-Containing 12 (ABHD12), J Med Chem 62, 1643–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Barnes MJ, Li CM, Xu Y, An J, Huang Y, and Cyster JG (2015) The lysophosphatidylserine receptor GPR174 constrains regulatory T cell development and function, J Exp Med 212, 1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kitamura H, Makide K, Shuto A, Ikubo M, Inoue A, Suzuki K, Sato Y, Nakamura S, Otani Y, Ohwada T, and Aoki J (2012) GPR34 is a receptor for lysophosphatidylserine with a fatty acid at the sn-2 position, J Biochem 151, 511–518. [DOI] [PubMed] [Google Scholar]
- [18].Inoue A, Ishiguro J, Kitamura H, Arima N, Okutani M, Shuto A, Higashiyama S, Ohwada T, Arai H, Makide K, and Aoki J (2012) TGFalpha shedding assay: an accurate and versatile method for detecting GPCR activation, Nat Methods 9, 1021–1029. [DOI] [PubMed] [Google Scholar]
- [19].van der Kleij D, Latz E, Brouwers JF, Kruize YC, Schmitz M, Kurt-Jones EA, Espevik T, de Jong EC, Kapsenberg ML, Golenbock DT, Tielens AG, and Yazdanbakhsh M (2002) A novel host-parasite lipid cross-talk. Schistosomal lyso-phosphatidylserine activates toll-like receptor 2 and affects immune polarization, J Biol Chem 277, 48122–48129. [DOI] [PubMed] [Google Scholar]
- [20].Sugo T, Tachimoto H, Chikatsu T, Murakami Y, Kikukawa Y, Sato S, Kikuchi K, Nagi T, Harada M, Ogi K, Ebisawa M, and Mori M (2006) Identification of a lysophosphatidylserine receptor on mast cells, Biochem Biophys Res Commun 341, 1078–1087. [DOI] [PubMed] [Google Scholar]
- [21].Shinjo Y, Makide K, Satoh K, Fukami F, Inoue A, Kano K, Otani Y, Ohwada T, and Aoki J (2017) Lysophosphatidylserine suppresses IL-2 production in CD4 T cells through LPS3/GPR174, Biochem Biophys Res Commun 494, 332–338. [DOI] [PubMed] [Google Scholar]
- [22].Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, and Hazen SL (2006) Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells, J Exp Med 203, 2613–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gijon MA, Riekhof WR, Zarini S, Murphy RC, and Voelker DR (2008) Lysophospholipid acyltransferases and arachidonate recycling in human neutrophils, J Biol Chem 283, 30235–30245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hishikawa D, Shindou H, Kobayashi S, Nakanishi H, Taguchi R, and Shimizu T (2008) Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity, Proc Natl Acad Sci U S A 105, 2830–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hashidate-Yoshida T, Harayama T, Hishikawa D, Morimoto R, Hamano F, Tokuoka SM, Eto M, Tamura-Nakano M, Yanobu-Takanashi R, Mukumoto Y, Kiyonari H, Okamura T, Kita Y, Shindou H, and Shimizu T (2015) Fatty acid remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport, Elife 4, e06328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rong X, Albert CJ, Hong C, Duerr MA, Chamberlain BT, Tarling EJ, Ito A, Gao J, Wang B, Edwards PA, Jung ME, Ford DA, and Tontonoz P (2013) LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition, Cell Metab 18, 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang B, Rong X, Duerr MA, Hermanson DJ, Hedde PN, Wong JS, Vallim TQ, Cravatt BF, Gratton E, Ford DA, and Tontonoz P (2016) Intestinal Phospholipid Remodeling Is Required for Dietary-Lipid Uptake and Survival on a High-Fat Diet, Cell Metab 23, 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rong X, Wang B, Dunham MM, Hedde PN, Wong JS, Gratton E, Young SG, Ford DA, and Tontonoz P (2015) Lpcat3-dependent production of arachidonoyl phospholipids is a key determinant of triglyceride secretion, Elife 4, e06557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sato T, Aoki J, Nagai Y, Dohmae N, Takio K, Doi T, Arai H, and Inoue K (1997) Serine phospholipid-specific phospholipase A that is secreted from activated platelets. A new member of the lipase family, J Biol Chem 272, 2192–2198. [DOI] [PubMed] [Google Scholar]
- [30].Sayo A, Konishi H, Kobayashi M, Kano K, Kobayashi H, Hibi H, Aoki J, and Kiyama H (2019) GPR34 in spinal microglia exacerbates neuropathic pain in mice, J Neuroinflammation 16, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kathman SG, Boshart J, Jing H, and Cravatt BF (2020) Blockade of the Lysophosphatidylserine Lipase ABHD12 Potentiates Ferroptosis in Cancer Cells, ACS Chem Biol. 15, 871–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, and Stockwell BR (2015) Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death, ACS Chem Biol 10, 1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.