

SUMMARY
The gut microbiome is the resident microbial community of the gastrointestinal tract. This community is highly diverse, but how microbial diversity confers resistance or susceptibility to intestinal pathogens is poorly understood. Using transplantation of human microbiomes into several animal models of infection, we show that key microbiome species shape the chemical environment of the gut through the activity of the enzyme bile salt hydrolase. The activity of this enzyme reduced colonization by the major human diarrheal pathogen Vibrio cholerae by degrading the bile salt taurocholate that activates the expression of virulence genes. The absence of these functions and species permits increased infection loads on a personal microbiome-specific basis. These findings suggest new targets for individualized preventative strategies of V. cholerae infection through modulating the structure and function of the gut microbiome.
Keywords: microbiome, cholera, pathogenesis, colonization resistance, interpersonal variation
Graphical Abstract
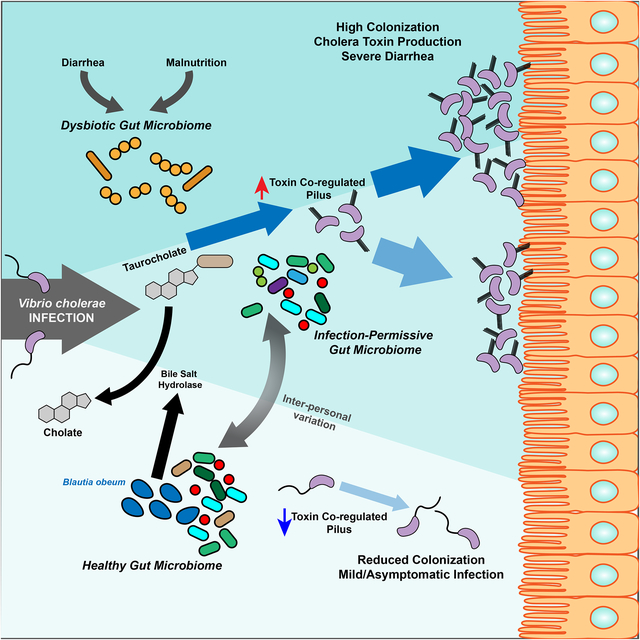
Differences in the gut microbiome between individuals determines resistance to cholera infection through the effects on the activity of a bile salt enzyme
INTRODUCTION
Gastrointestinal infections represent a major global health concern. One major human diarrheal pathogen is Vibrio cholerae, the etiologic agent of the severe disease cholera that affects millions of people annually (Clemens et al., 2017). V. cholerae cycles between aquatic reservoirs and the small intestine, requiring the coordinated regulation of environmental fitness genes versus virulence genes including the attachment factor Toxin Co-regulated Pilus (TCP) and Cholera Toxin (CT) (Herrington et al., 1988; Miller et al., 1987). In this and other pathogens, regulation depends on the chemical state of the gut, shaped by the gut microbiome, the dense resident gut microbial community (Eckburg et al., 2005) that varies dramatically across host species and across individuals as a function of diet, geography, and environmental insults (Yatsunenko et al., 2012). In cholera-endemic areas, the gut microbiome is subject to mutually-reinforcing pressures: malnutrition, leading to reduced host infection resistance, repeated diarrhea, and poorly-controlled antimicrobial usage in an attempt to mitigate the resulting sequelae. Previous 16S ribosomal gene studies of the gut microbiome in these areas demonstrate that these factors are able to drive the gut microbiome into a characteristic dysbiotic state, dominated by Streptococci such as Streptococcus salivarius, Enterococci, and Enterobacteriaceae. This configuration has been shown to be inducible by malnutrition (Subramanian et al., 2014), and diarrhea irrespective of etiology, including enterotoxigenic Escherichia coli, V. cholerae, and rotavirus infection (David et al., 2015; Hsiao et al., 2014; Kieser et al., 2018).
Generalized model “healthy” microbial communities of the human gut have been shown to be resistant to V. cholerae infection (Hsiao et al., 2014), and associative metagenomic studies have examined how the microbiome differs between cholera patients and household contacts that did not exhibit disease symptoms (Midani et al., 2018). Yet few studies have mechanistically explored how interpersonal microbiome variation can drive pathogen susceptibility. Here we show that the post-malnutrition/post-diarrheal dysbiotic community is highly vulnerable to subsequent infection. Moving beyond a dichotomous “normal” versus “dysbiotic” comparison, we show that microbiome differences among healthy humans drive striking differences in susceptibility. We show that fecal studies in animals and potentially humans may have limited utility for studies of community interactions with pathogens of the small intestine, and that microbiome-dependent infection susceptibility at the small intestine can be rescued by microbiome transplantation. In order to identify commensals that strongly interact with enteropathogens across many community contexts, we established an efficient unbiased experimental pipeline that revealed that the commensal species Blautia obeum can suppress virulence. We identify an enzymatic mechanism in B. obeum that degrades the host-produced virulence-inducing molecule taurocholate (TC), which B. obeum uses alongside other mechanisms (Hsiao et al., 2014) to suppress V. cholerae virulence gene activation and colonization.
RESULTS
Dysbiotic microbiomes are susceptible to V. cholerae colonization, and pathogen resistance can be rescued by microbiome transplantation
We took a two-pronged approach to study the effects of microbiome variation on pathogen resistance, both involving reconstituting human gut microbiomes in animal models of V. cholerae colonization and virulence. The first involved the construction of defined gut communities using cultured human isolates (Figure 1A), and the second involved studies with complete human fecal microbiomes (Figure 1B).
Figure 1. Model human gut microbiomes replicate structure of communities affected by diarrhea-induced dysbiosis.
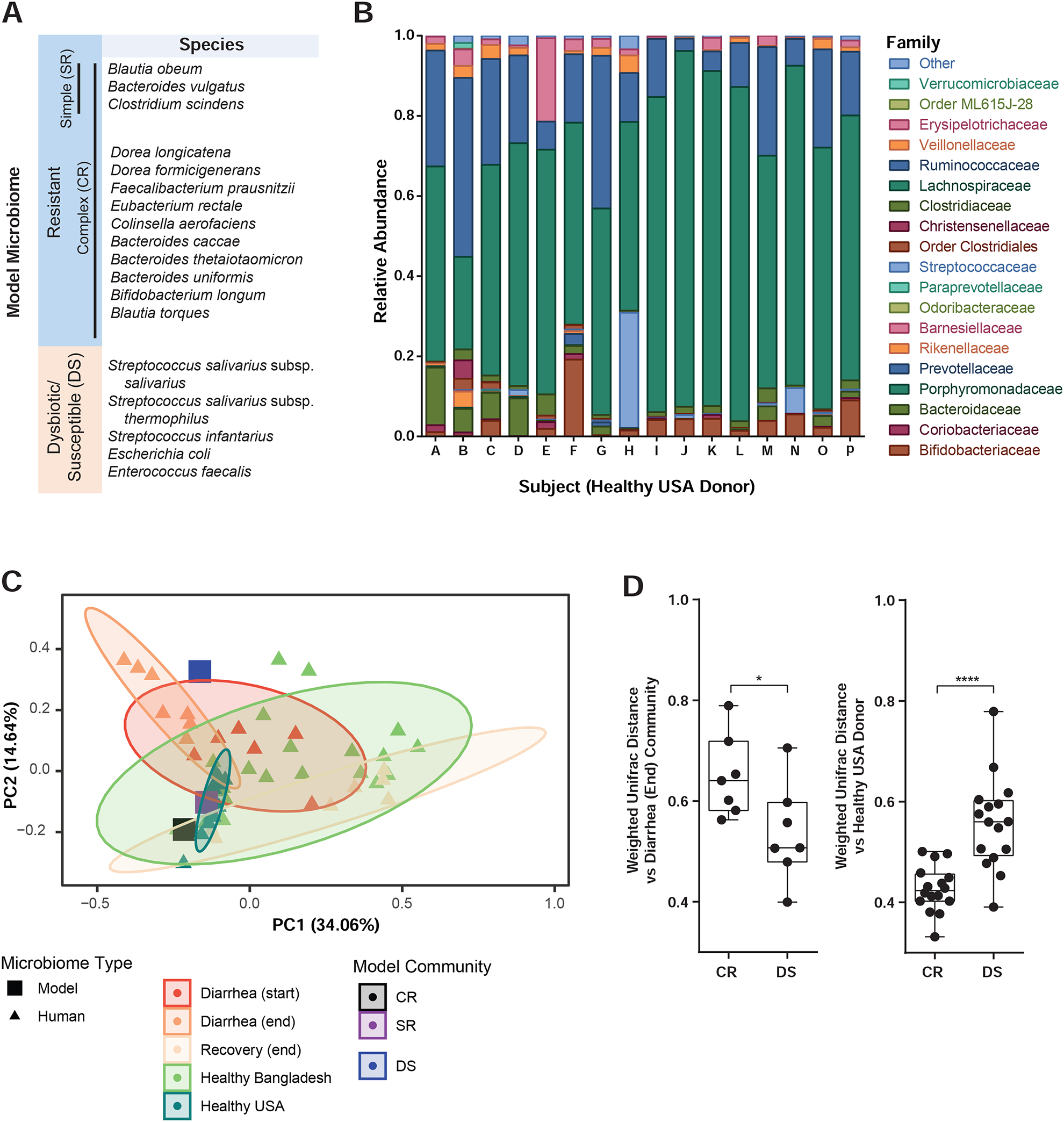
(A) Defined human gut communities. (B) Composition of healthy US human donor fecal microbiomes. (C) Principal coordinates analysis of defined and complete human gut microbiomes based on weighted UniFrac distance, % variance explained shown in parentheses. Ellipses show 95% confidence intervals. (D) Weighted UniFrac distance to indicated defined human model microbiomes of fecal samples from cholera patients at the end of diarrhea (left) and healthy human donors (right) *P<0.05, ****P<0.0001, Mann-Whitney U-test. Boxplots show inter-quartile range, whiskers minimum to maximum.
As the basis for designing defined model communities, we compared fecal microbiomes of a healthy adult volunteer cohort in the United States (Figure 1C, Table S2C) and previously published 16S ribosomal RNA gene sequencing of Bangladeshi adults (Hsiao et al., 2014; Subramanian et al., 2014). Previous studies in Bangladesh revealed that cholera drives the human gut microbial community to a highly dysbiotic, low-diversity state dominated by Streptococci, which recovers to a configuration similar to non-diarrheal individuals over the course of weeks after the cessation of acute disease (Hsiao et al., 2014). This has also been observed in other diarrheal infections such as enterotoxigenic E. coli and rotavirus (David et al., 2015; Kieser et al., 2018), and other gut pathologies such as severe malnutrition (Subramanian et al., 2014). Principal coordinates analysis (PCoA) of a human cohort from Bangladesh (Hsiao et al., 2014) displays the dysbiosis caused by cholera (Figure 1C, Diarrhea (start)), and community structure weeks after the cessation of diarrhea (Diarrhea (end) to Recovery (end)), when the microbiome becomes similar to that of individuals in the same area not suffering from acute malnutrition or diarrhea (Subramanian et al., 2014). Interpersonal microbiome variation in Bangladesh was far higher than among healthy US individuals sampled as part of this study; indeed, some Bangladeshi “healthy” microbiomes closely resemble cholera-diarrheal communities. As infectious diarrhea and malnutrition are frequent in cholera endemic areas, we hypothesized that the distinctive dysbiotic microbiome structure observed during recovery from multiple sources of environmental insult to the gut may be a recurring window of vulnerability to cholera.
We then used human-derived isolates to assemble defined gut communities (Figure 1A) based on these metagenomic analyses. One model microbiome (“CR”) was based on metagenomic surveys of healthy individuals, characterized by high taxonomic diversity but commonly including members of the genera Bacteroides, Clostridium, and Blautia (Arumugam et al., 2011; Qin et al., 2010; Yatsunenko et al., 2012). Another (“DS”) model microbiome is characteristic of the dysbiotic state found in cholera-endemic areas, comprising Streptococci, Enterococcus faecalis, and E. coli. 16S sequencing analysis confirmed that the CR community is more similar to healthy human microbiomes than dysbiotic diarrheal microbiomes, while the DS model community was more similar to microbiomes at the conclusion of cholera (Figure 1C–D).
We grew bacterial species from each defined group in pure culture, and used culture optical density (OD600) to introduce equivalent amounts of each member species with V. cholerae to germfree (GF) adult C57BL/6J mice by intra-gastric gavage. Overall microbial load during infection was equivalent as measured by quantitative PCR of 16S gene levels (Figure 2F). Mice that received the CR microbiome at infection were resistant to V. cholerae colonization, compared to animals receiving DS microbes (Figure 2A–B). We observed these colonization phenotypes in both feces and in the medial and distal thirds of the small intestine. In prior studies in adult mice, small intestinal colonization by V. cholerae required antibiotics (Freter, 1955, 1956) and ketamine anesthesia (Olivier et al., 2009). Our results with human, as opposed to murine, gut bacteria suggest that microbiome differences across host species and inter-individual variation within host species both play key roles in determining pathogen susceptibility. Significantly, we could restore colonization resistance by mixing the CR and DS bacteria, suggesting that susceptibility is reversible through microbiome modification (Figure 2A–B). In “Mix” groups, where mice were administered a 1:1 mixture of CR and DS, V. cholerae levels were significantly less compared to that in DS mice, in fact dropping below the level of CR mice 2 days post infection.
Figure 2. Gut microbiome composition contributes to V. cholerae infection resistance.
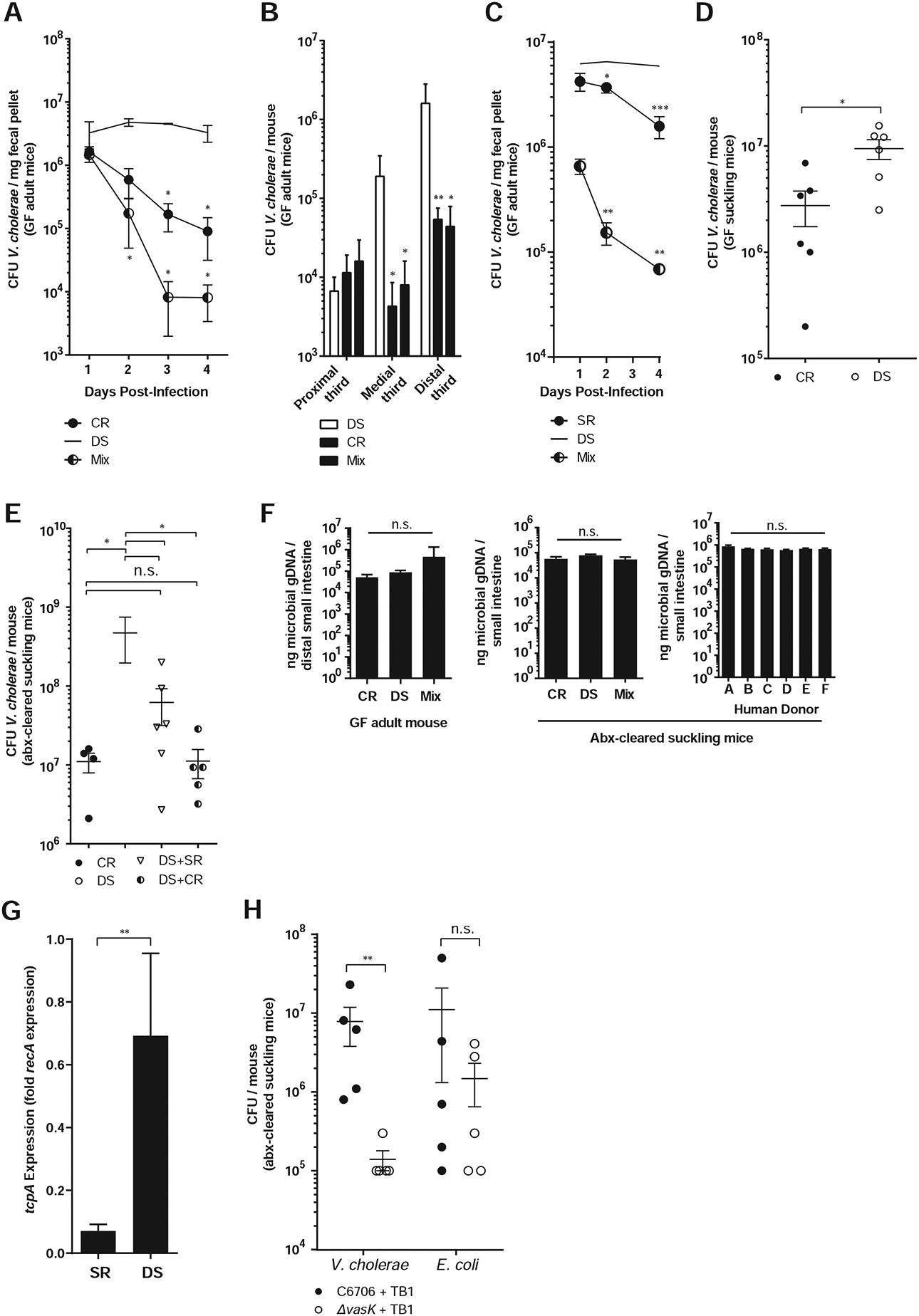
V. cholerae colonization in germfree adult mice harboring defined model communities co-gavaged with V. cholerae in (A) feces and (B) small intestines 4 days post infection. (C) Fecal V. cholerae colonization in GF adult mice harboring defined communities for 2 weeks and then gavaged with V. cholerae. Mix: 10 days DS colonization, followed by a SR microbes 4 days prior to V. cholerae infection. (D) Intestinal V. cholerae colonization of GF suckling mice co-gavaged with model communities and V. cholerae. (E) Intestinal V. cholerae colonization of antibiotic-treated CD-1 suckling mice co-gavaged with indicated communities. (F) 16S gene abundance in small intestine. (G) Expression of tcpA in infected mice with model human microbiomes. (H) T6SS effects on small intestine colonization in antibiotic-treated CD-1 pups. * P<0.05, ** P<0.01, *** P<0.001 (Mann-Whitney U-test); n.s. not significant. Error bars represent mean ± SEM. n=6–12 animals for all experiments.
We also observed increased colonization susceptibility of DS microbiomes when compared to a simplified model healthy microbiome (“SR”) when GF mice were colonized with defined communities for 2 weeks prior to introduction of V. cholerae (Figure 2C). The SR community consisted of three species representing major phylogenetic lineages commonly found in the healthy human gut. To model an attempted microbiome restoration of a fully established and dense gut community, we also introduced DS microbes for 10 days, followed by a gavage of SR microbes 4 days prior to infection with V. cholerae. In this Mix group, pathogen colonization was strongly inhibited compared to DS-colonized mice, suggesting that microbiome modification could be used to restore colonization resistance even to entrenched dysbiotic communities.
We then profiled gut microbiome structure during infection in feces and small intestines of gnotobiotic animals with different communities (Figures 3A–C, 3G). In these samples, the CR and DS communities were distinct and the CR community more similar to complete fecal microbiomes of healthy United States donors, while co-inoculation of CR and DS led to an intermediate final microbiome.
Figure 3. Addition of the CR model human microbiome to mice hosting DS microbes yields a community structure closer to complete fecal communities of healthy human volunteers.
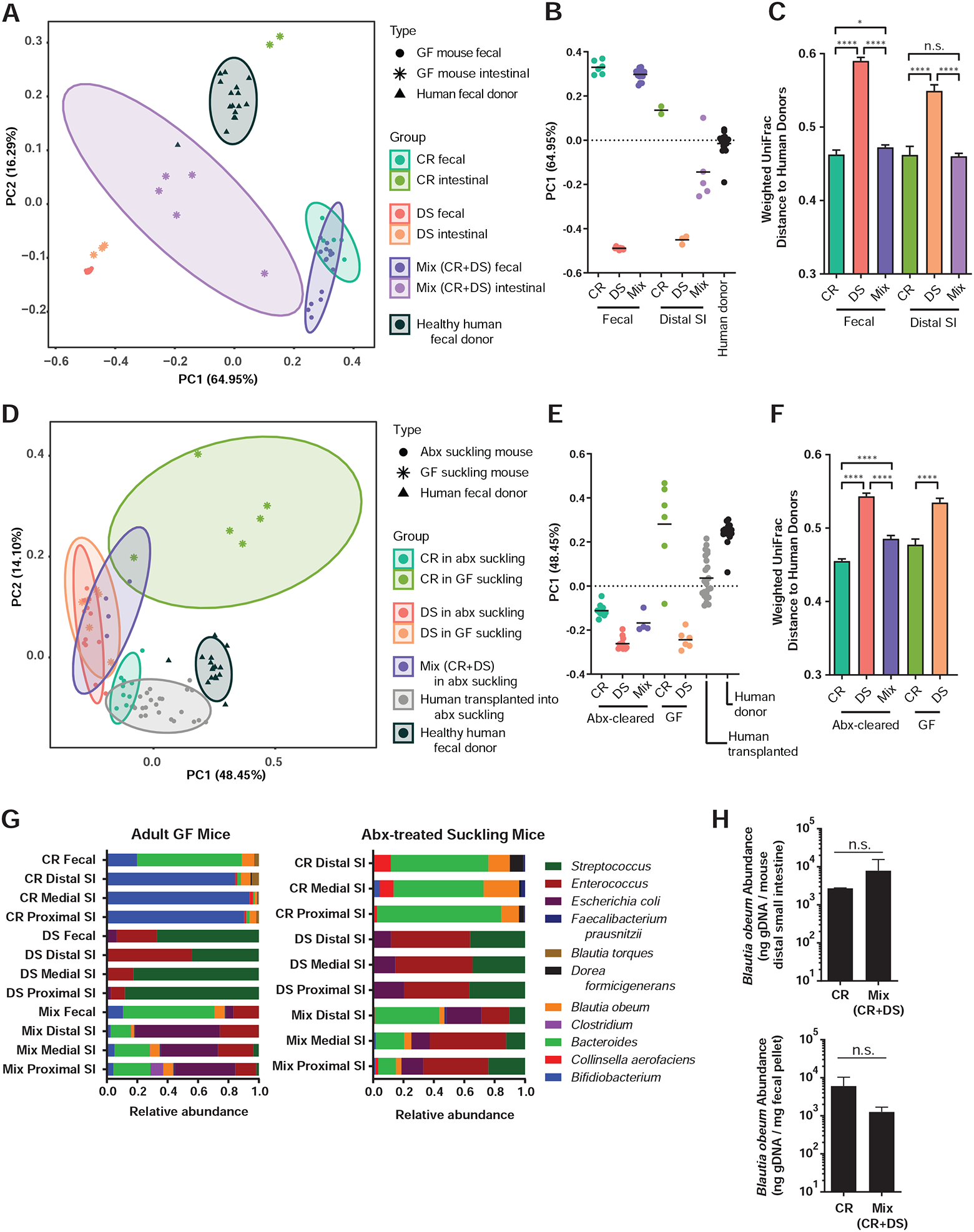
(A, D) Principal coordinates analysis (PCoA) of microbial community diversity based on weighted UniFrac distance, % variance explained shown in parentheses for each axis. Ellipses show 95% confidence intervals. (A) PCoA of fecal samples and distal third of small intestine of GF mice with model communities during V. cholerae infection compared to healthy US donor fecal samples, with (B) PC1 positions and (C) all pairwise weighted UniFrac distances to healthy US donor fecal samples. (D) PCoA of model communities and healthy human donor communities in suckling mice, with (E) PC1 positions and (F) all pairwise weighted UniFrac distances to healthy US donor fecal samples. (G) Microbiome structure during infection with V. cholerae and host reads filtered (left) and in antibiotic-treated suckling mice without V. cholerae (right). (H) B. obeum abundance in adult GF mice containing indicated microbiomes during V. cholerae infection (4d post infection). * P<0.05, **** P<0.0001; n.s. not significant (Mann-Whitney U-test). Error bars represent mean ± SEM.
Non-dysbiotic human microbiomes reduce virulence gene expression and colonization of V. cholerae in a suckling mouse model of infection
While gnotobiotic adult mice serve as a useful microbial-interaction model, we extended our studies to suckling mice, where the pathology and virulence gene expression observed during V. cholerae infection is closer to that of humans (Klose, 2000). First, we recapitulated the CR and DS resistance phenotypes in suckling GF C57BL/6J animals (Figure 2D). We then constructed a more accessible and scalable model of microbiome-pathogen interaction by clearing the native murine flora of CD-1 pups with streptomycin before introduction of human-associated species. Using this system, we observed similar microbiome-dependent infection outcomes; competitive CR/DS transplantation yielded a dominant CR-like phenotype (Figure 2E), while CR and DS colonization load did not differ in non-Vibrio-infected animals (Figure 2F). This pattern was reflected in 16S sequencing data (Figure 3D–F, Table S3). During infection, pups receiving CR microbes had very different community structure (with Vibrio reads filtered) compared to animals with DS microbes, and animals receiving a mixed inoculum (CR+DS) closely resembled CR mice.
Total microbial diversity was not a dominant factor in infection resistance, as we restored colonization resistance in DS mice to almost full CR levels by transplanting only a small subset of CR species (SR). Expression levels of the key colonization factor tcpA were reduced ~9.7 fold in SR compared to DS animals (Figure 2G). We did not observe significant microbiome-dependent differences in cholera toxin gene expression, diarrhea, or fluid accumulation in these animals (Figure S1).
Recent studies have shown Type VI secretion system (T6SS) killing of murine commensal E. coli acts to drive increased virulence in infection of suckling mice (Zhao et al., 2018). As our DS model community contains E. coli, albeit a different strain, we tested the effects of T6SS on V. cholerae colonization and E. coli levels in our experimental system. A T6SS ΔvasK mutant was deficient for colonization compared to wild type V. cholerae in antibiotic-cleared suckling mice (Figure 2H). However, T6SS activity did not significantly alter levels of a co-inoculated streptomycin-resistant K12 E. coli, and we observed E. coli at comparable levels in DS and Mix (DS+CR) communities in the small intestine (Figure 3G). These differences may be E. coli strain-specific, or due to the much higher levels of V. cholerae used in previous T6SS studies.
Together, our data suggested that the mechanism for improved colonization resistance of CR/SR microbes lay in T6SS-independent manipulation of V. cholerae virulence gene expression.
Inter-individual variation in pathogen colonization resistance of human gut microbiomes
Our microbiome transplantation system in suckling mice allowed us to screen numerous intact human fecal microbiomes collected from healthy adult volunteers without malnutrition or recent antibiotic usage or diarrhea for effects on V. cholerae (Figure 1B). These complete fecal communities were taxonomically similar to the CR, but not DS microbiomes in both original microbial content and community structure upon transplantation (Figures 3D–F). This was unsurprising, as the CR model community represented up to 73% of genus-level diversity by total relative abundance in these samples, while members of the DS community only represented <1% of the total (Table S4).
We normalized fecal slurries and transplanted these samples into antibiotic-treated suckling mice with V. cholerae (Figure 4), with dramatically different effects on V. cholerae colonization, even though these fecal communities colonized suckling animals at similar efficiencies and density (Figure 2F). We observed an approximately 1.5-log10 range of V. cholerae colonization depending on the human donor (Figure 4), suggesting wide variation in infection outcomes based on individual gut microbiome structure. The higher basal microbiome diversity in Bangladesh (Figure 1C) suggests that interpersonal variations in infection resistance in endemic areas could be substantially higher.
Figure 4. Gut microbiome composition contributes to inter-personal differences in V. cholerae infection resistance.
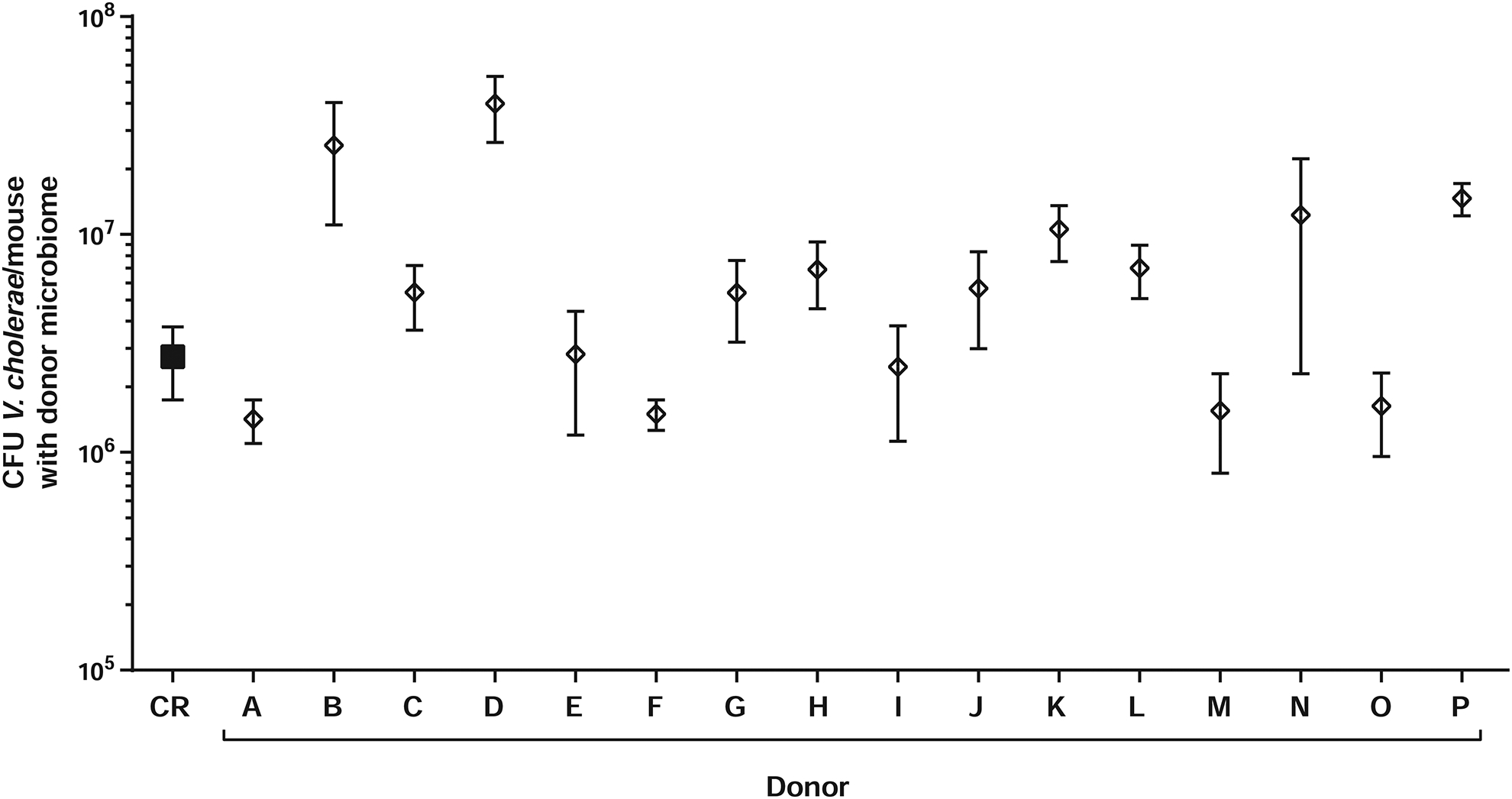
Intestinal V. cholerae colonization of antibiotic-treated CD-1 pups colonized with complete fecal microbiomes from healthy US human volunteers. n=5–7 animals for all experiments.
A pipeline for randomization of microbiome members identifies commensal species consistently correlated with V. cholerae infection outcome
We hypothesized that the CR species best able to colonize intestines with the DS community might be prophylactic for infection, since transplantation of CR microbes into DS communities reduced V. cholerae colonization. Therefore, we examined gut microbiome structure during V. cholerae infection in GF animals with a mixed CR+DS community (Figures 3A, 3G). The CR community in the small intestine was quite distinct from that in feces, but all animals with this community were consistently colonized by Blautia and Bacteroides species. The DS community was consistent in feces and small intestine, and dominated by Streptococci. The lack of generalizability of fecal data to other gut compartments suggested that fecal sampling studies may mask important differences for pathogenesis in the proximal gastrointestinal tract.
In the small intestine, B. obeum maintained its relative abundance in gnotobiotic CR+DS and CR-only animals (Figure 3H), suggesting that it may play a role in CR infection resistance and in transmitting this phenotype to DS animals. However, these findings had potentially limited translational applicability given the basal inter-personal diversity in humans; the ability to displace one dysbiotic microbiome and resist V. cholerae colonization may not be representative across diverse individuals and microbial communities. To identify Vibrio-antagonistic microbes across many different microbiome contexts, we generated random, unique, 5-member combinations drawn from CR and DS strains (Figure 5A–B) and established OD600-normalized mixtures of these bacteria in antibiotic-cleared suckling animals with and without Vibrio infection. We reasoned that species whose presence/absence or abundance consistently correlated against V. cholerae colonization in many different communities would be excellent putative targets for anti-Vibrio interventions.
Figure 5. An unbiased combinatorial strategy for identifying commensal correlates of protection against and susceptibility to V. cholerae colonization.
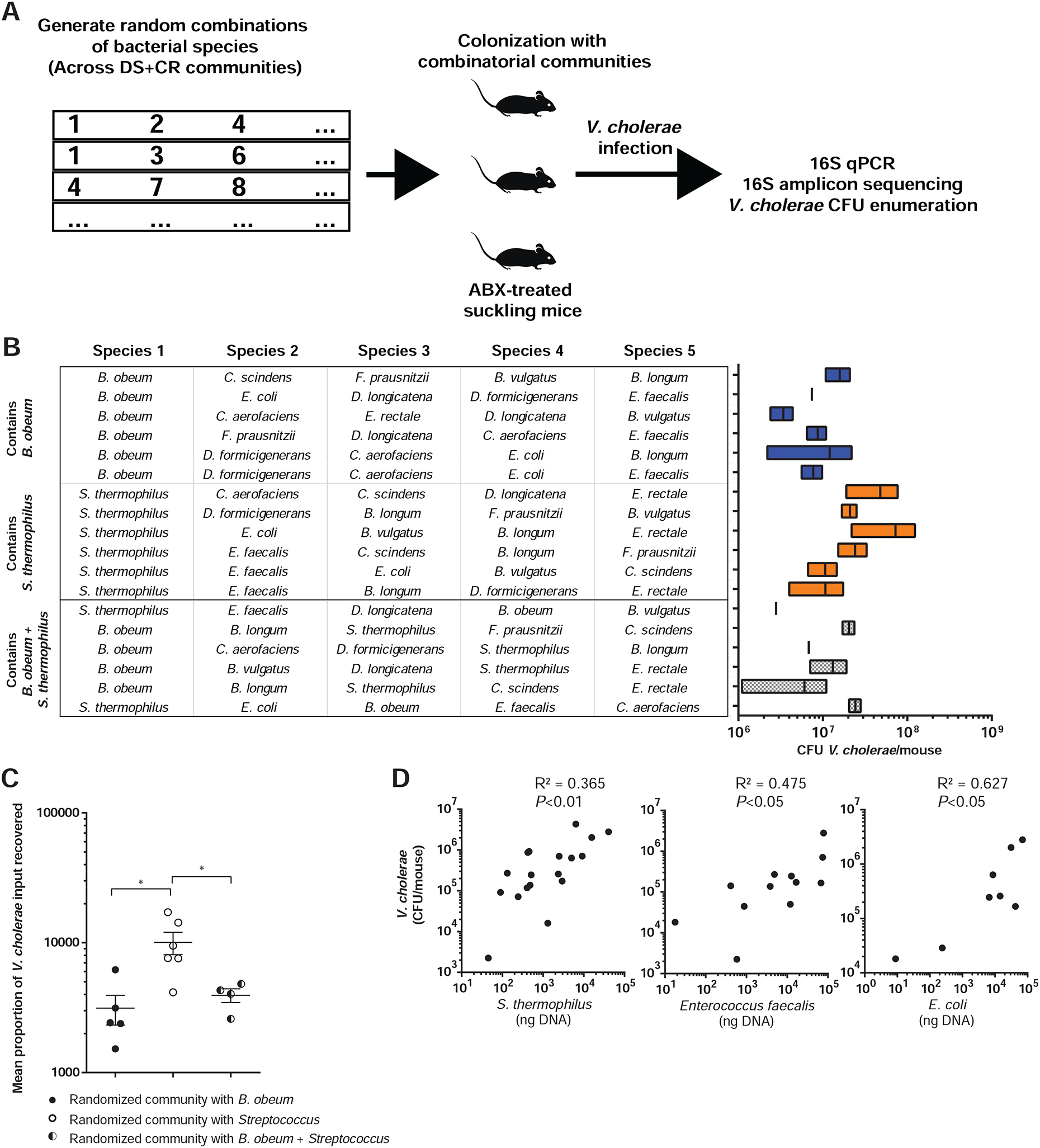
(A) Combinatorial strategy. (B) 5-member microbiome embodiments randomly generated using CR/DS members (left). Resulting V. cholerae infection of antibiotic-treated suckling mice containing defined microbiome embodiments are shown at right. (C) Mean V. cholerae colonization in suckling mice bearing communities containing B. obeum or Streptococcus species alone and in combination. Data normalized across experiments as fold CFU gavaged V. cholerae recovered after infection. (D) Abundance of DS member species in randomized microbiomes correlated to resulting V. cholerae abundance after infection. Points represent mice receiving different microbiome embodiments. * P<0.05, ** P<0.01 (Mann-Whitney U-test); n.s. not significant. Error bars represent mean ± SEM.
We identified several species consistently associated with pathogen levels across multiple microbiome combinations. Higher levels of B. obeum were significantly associated with reduced V. cholerae colonization (Figure 5C), but did not directly correlate with V. cholerae abundance. This is consistent with the effects of the mixed CR/DS microbiome on infection; B. obeum consistently established in the DS small intestine, but high levels were not required to affect V. cholerae. That this effect was seen across numerous randomized communities suggests that the inhibitory activity of B. obeum on V. cholerae infection may be broadly generalizable across many gut microbiomes. Except for B. obeum, we found no statistically significant effects on V. cholerae colonization based on the presence or absence of an SR or CR species. In contrast, levels during infection of DS microbes (Streptococcus, E. faecalis, and E. coli) all positively and significantly correlated with higher Vibrio levels (Figure 5D). In mice with combinations with both B. obeum and S. thermophilus, V. cholerae colonization was comparable to mice with combinations including B. obeum but not Streptococcus, again supporting the observation that B. obeum’s effects on pathogenesis are dominant across diverse microbiomes (Figure 5C).
Since SR microbes largely recapitulated the colonization resistance of the full CR microbiome, we performed similar analyses looking for whether combinations of different SR microbes with B. obeum yielded lower V. cholerae colonization than when those species were present in isolation. We observed no statistically significant additive effects on V. cholerae colonization of adding either Bacteroides vulgatus or Clostridium scindens to B. obeum in defined communities (Figure S2).
Intestinal signals that induce V. cholerae virulence gene expression are depleted by B. obeum
Having identified a candidate driver of V. cholerae infection resistance, we began to search for a molecular mechanism for these effects. Numerous host and some commensal microbial cues regulate V. cholerae gene expression in vivo (Gupta and Chowdhury, 1997; Kovacikova et al., 2010; Yang et al., 2013). Prior studies have identified a role for a B. obeum-produced AI-2 autoinducer in down-regulating the expression of TCP biogenesis genes during infection (Hsiao et al., 2014). Consistent with this finding, we observed reduced tcpA expression during infection of mice with microbiomes containing B. obeum (Figure 2G).
In vivo conditions for virulence gene regulation can be mimicked ex vivo using microaerophilic/anaerobic growth of V. cholerae with intestinal tissue from mice (Yang et al., 2013). We took homogenates of suckling mouse intestine and incubated them anaerobically with a V. cholerae lacZ:PtcpA –sh ble zeocin resistance reporter. As expected, intestinal homogenates induced tcpA expression, while pre-treatment of intestinal homogenates with B. obeum ablated tcpA induction (Figure 6A). As a control, we boiled intestinal homogenates that had been incubated with B. obeum cultures in order to remove AI-2, which is heat labile (Figure S3). Strikingly, homogenates incubated with B. obeum remained unable to induce tcpA even after boiling, in contrast to boiled homogenates alone or homogenates incubated with S. salivarius and then boiled (Figure 6A). These data suggested that B. obeum can deplete virulence-activating signals within the gut as well as produce virulence-suppressing signals.
Figure 6. B. obeum exerts effects on V. cholerae colonization through degradation of the in vivo virulence gene activating signal taurocholate (TC).
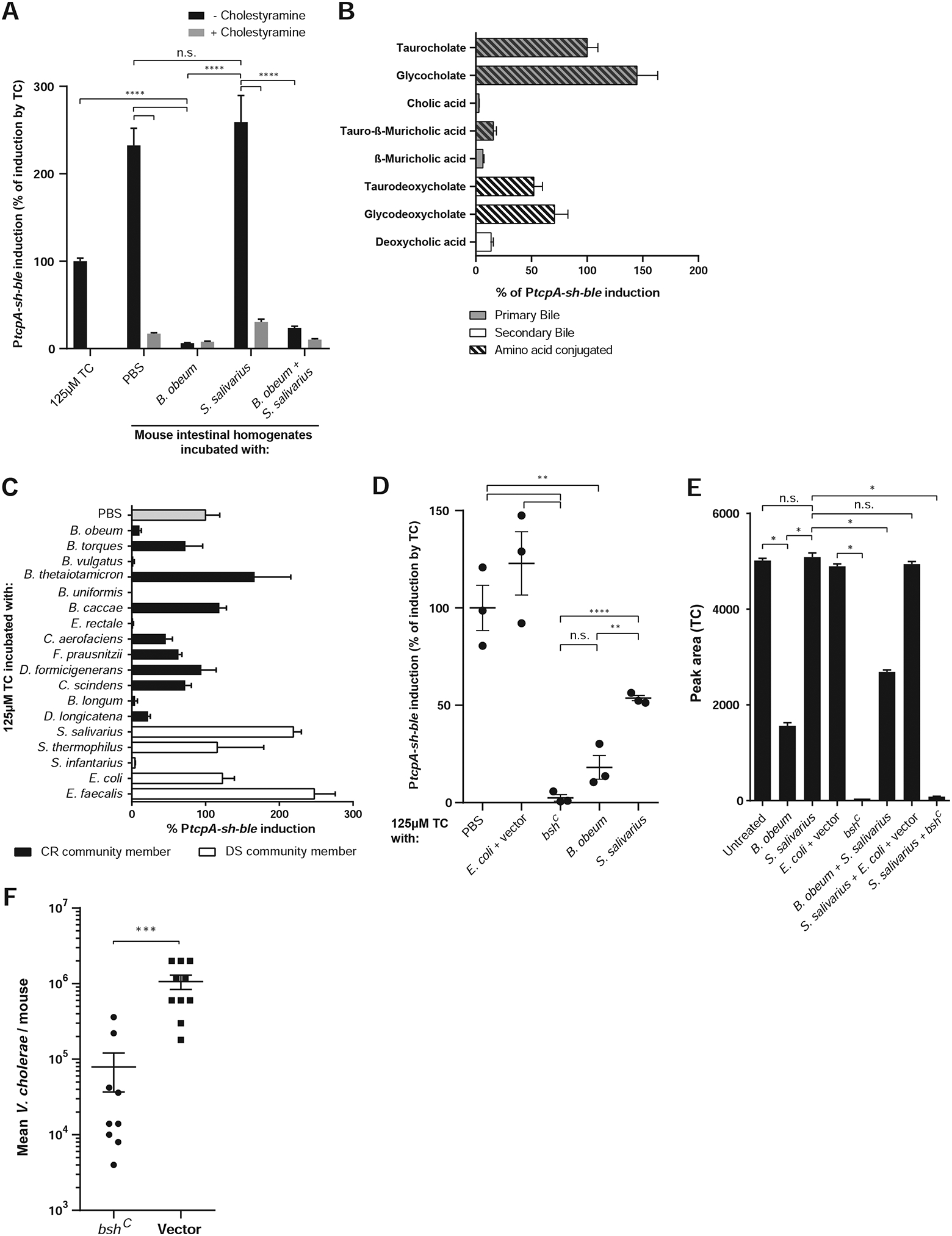
PtcpA activity normalized to tcpA induction by 125μM TC unless noted. (A) Modulation of tcpA-activating signals in suckling CD-1 mouse intestinal homogenates by pure cultures of B. obeum and S. salivarius, with heat treatment. (B) Bile effects on tcp gene expression in vitro. (C) Effects of CR and DS pure cultures on TC activation of virulence in vitro. (D) Effects of B. obeum bsh enzyme expression on TC-mediated tcp activation in vitro. (E) Mass spectrometry measurement of TC in suckling CD-1 mouse intestines after incubation with pure cultures of indicated strains. (F) V. cholerae infection of suckling CD-1 mice after 1-day of colonization with indicated E. coli strains. * P<0.05, ** P<0.01, *** P<0.001, **** P<0.0001 (unpaired Student’s t-test). Error bars represent mean ± SEM. n=3–10 for all experiments.
The bile salt taurocholate acts as a potent virulence gene activator, and is more efficiently degraded by commensal microbes in healthy but not dysbiotic communities
One abundant heat-resistant molecule present in the small intestine is bile. In humans and mice, bile acids are synthesized in the liver from cholesterol, and stored in the gallbladder. These primary bile acids are then secreted into the duodenum, where they, typically in their sodium salt form, act to aid in the emulsification and absorption of dietary fats. More than 95% of secreted bile acids are actively absorbed by the terminal ileum and sent back to the liver in a process known as enterohepatic circulation (Di Ciaula et al., 2017). Bile is a highly complex mixture, though bile acids dominate the dry weight of biliary bile (Muraca et al., 1991). Many prior studies have focused on crude extracts from varying sources, including ruminants, containing poorly defined mixtures of bile molecules. Human bile acids secreted into the small intestine are predominantly conjugated to taurine (taurocholic acid/sodium taurocholate) and glycine (glycocholic acid/sodium glycocholate), while taurine-conjugated forms predominate in mice (Li and Dawson, 2018; Sayin et al., 2013). Commensal microbial action is important for bile acid metabolism; bacterial enzymes (bsh, bile salt hydrolases) are able to remove the conjugated amino acids from secreted bile acids in the small intestine, for example converting glycocholate (GC) and taurocholate (TC) to cholate/cholic acid (CA) (Jones et al., 2008; Ridlon et al., 2006; Song et al., 2019). Indeed, in GF mice, the bile pool in the small intestine is almost exclusively taurine-conjugated (Sayin et al., 2013).
Previous studies have shown that TC, one of the most abundant bile molecules in humans and mice, can induce tcp expression under anaerobic conditions through modulating the structure and activity of the upstream virulence activator TcpP (Yang et al., 2013). Similarly, we saw that TC activated PtcpA, while CA was not an efficient inducer (Figure 6B). Intestinal homogenate effects on tcpA was bile-specific; pre-treatment of homogenates with the bile-sequestering resin cholestyramine ablated their ability to activate PtcpA (Figure 6A).
We next screened the DS and CR species for their effects on TC. We incubated TC solutions at a physiologically relevant concentration with pure cultures of these microbes, heat treated and filter-sterilized the resulting supernatants, and measured their ability to induce PtcpA (Figure 6C). We observed dramatic differences in the ability of these strains to affect TC virulence induction, with members of the CR/SR microbiomes in general being better able to prevent tcp activation. The ability to ablate TC-mediated induction of tcp expression varied at genus level, with Blautia torques unable to affect tcp induction by TC in comparison to B. obeum, and Streptococcus infantarius able to process TC in contrast to other DS Streptococci. Of SR species, B. vulgatus, but not C. scindens, showed efficient TC processing.
Since the SR community largely recapitulated the CR colonization resistance phenotype, and B. vulgatus demonstrated high activity against TC in vitro, we wanted to examine the relative contribution of B. obeum and B. vulgatus on TC levels in the distal small intestine. We colonized adult GF mice with CR members, or CR species without B. obeum, and measured TC and CA in the distal third of the small intestine 2 days post-colonization, compared to GF mice (Figure S4A). As expected given the absence of microbial bsh, GF distal small intestine showed a high TC/CA ratio, while the presence of CR microbes efficiently processed TC to CA, yielding low TC/CA ratios. Strikingly, the removal of B. obeum restored the TC/CA ratio in the distal small intestine to a level not statistically significantly different from GF animals, though there was a trend towards more TC in GF animals. This suggested that while other CR organisms can contribute to TC processing to CA, B. obeum is particularly well suited to manipulating the level of this bile acid in the distal small intestine. This agrees with our findings that B. obeum efficiently colonizes the small intestine (Figures 3, 5), and that the presence of B. vulgatus and B. obeum together does not significantly improve the ability of a microbiome to exclude V. cholerae (Figure S2).
A bile salt hydrolase enzyme encoded by B. obeum is able to degrade virulence-activating signals in the gut
To determine the molecular basis for B. obeum’s effect on TC-dependent virulence induction, we examined genetic determinants of bile acid processing. The B. obeum genome encodes for a hypothetical choloylglycine hydrolase (EC 3.5.1.24). Such bile salt hydrolase (bsh) enzymes catalyze the removal of the conjugated amino acids of bile salts, for example the removal of taurine from TC to form CA.
Putative bsh genes are broadly distributed across gut microbial species, as the ability to survive the inhibitory effects of bile are extremely important for enteric commensals (De Smet et al., 1995). A recent study classed bacterial bsh genes into several phylotypes based on sequence similarity and showed that bsh phylotypes have variable and substrate-dependent deconjugation activity (Song et al., 2019). We binned predicted bsh genes in the CR and DS genomes into these phylotypes (Table S5). By sequence alignment and predicted structure, B. obeum encodes for predicted type 1 bsh enzymes, which are highly effective at deconjugating TC, GC, glycodeoxycholate, and taurodeoxycholate (Song et al., 2019), the strongest activators of tcpA expression in V. cholerae (Figure S5). All DS members except S. infantarius lacked annotated bsh genes, while many CR species encoded bsh homologs. While S. infantarius showed high activity against TC in vitro, despite bearing bsh homologs to phylotypes with poor predicted TC activity, we observed no statistically significant difference in effects on V. cholerae colonization compared to S. salivarius (Figure S4B), suggesting that there may be differences in in vivo regulation of these enzymes in S. infantarius. Conversely, B. torques, despite encoding several putative bsh genes, was not able to prevent tcpA induction by TC. B. vulgatus was an efficient TC processor in vitro, but despite encoding 3 putative bsh genes could not drive significantly lower levels of TC to CA processing in the distal small intestine in the absence of B. obeum (Figure S4A), further suggesting that enzyme expression or function may diverge in in vitro vs in vivo.
V. cholerae also encodes a putative bsh enzyme (VCA0877). However, this is a predicted phylotype 6 bsh, which has poor activity against TC but higher activity against rarer bile acids that do not participate in regulation of virulence but may be bacteriostatic in vivo (Table S5). V. cholerae cannot prevent TC-mediated tcp activation in vitro, suggesting that V. cholerae has limited bsh activity against TC in comparison to B. obeum (Figure S4C).
We constitutively expressed the B. obeum bsh RUMOBE_000028 by cloning this locus downstream of a constitutive PLtet-O1 promoter in E. coli, generating strain bshC. This bshC strain efficiently reduced levels of TC and tcp activation compared to the isogenic vector control in both pure TC solutions (Figure 6D) and intestinal homogenates (Figure 6E). Significantly, given the dominant effect of B. obeum on Vibrio resistance, pure cultures of either B. obeum or bshC reduce tcp activation by TC (Figure 6D) and TC levels in intestinal homogenates (Figure 6E) in the presence of S. salivarius. The activity of this B. obeum enzyme is able to affect V. cholerae in vivo, as V. cholerae is unable to colonize mice gavaged with bshC as effectively as mice with the vector control (Figure 6F).
Bile salt hydrolase levels in human gut microbial communities are correlated to V. cholerae infection outcome
To determine the distribution of bsh phylotypes in human gut microbiomes predicted to be dysbiotic or healthy, we re-examined an existing deeply-sequenced shotgun metagenomic dataset of human cholera patients in Bangladesh (Table S2D) (David et al., 2015). Importantly, data was available from patients at presentation at clinic for cholera (“Diarrhea (d0)”) without any prior antibiotic usage, and from patients that received oral antibiotics as part of the standard of care for cholera (“Diarrhea + abx”). These data showed that bsh levels were already affected by diarrhea prior to any clinical intervention. We found that several bsh phylotypes followed diarrhea-dependent patterns in comparison to a healthy Bangladeshi cohort. Phylotypes 1, 3, 4, and 5 were significantly depleted in dysbiotic samples compared to healthy controls (Figure 7A, S6). Of these, phylotypes 1, 3, and 4 were shown to be highly active against TC ((Song et al., 2019), summarized in Table S5). These data suggested that a characteristic of healthy human microbiomes that may modulate V. cholerae susceptibility is their ability to deconjugate TC into non virulence-inducing forms.
Figure 7. Levels of B. obeum bsh enzymes in human samples correlate to infection outcome, and can independently modulate V. cholerae colonization.
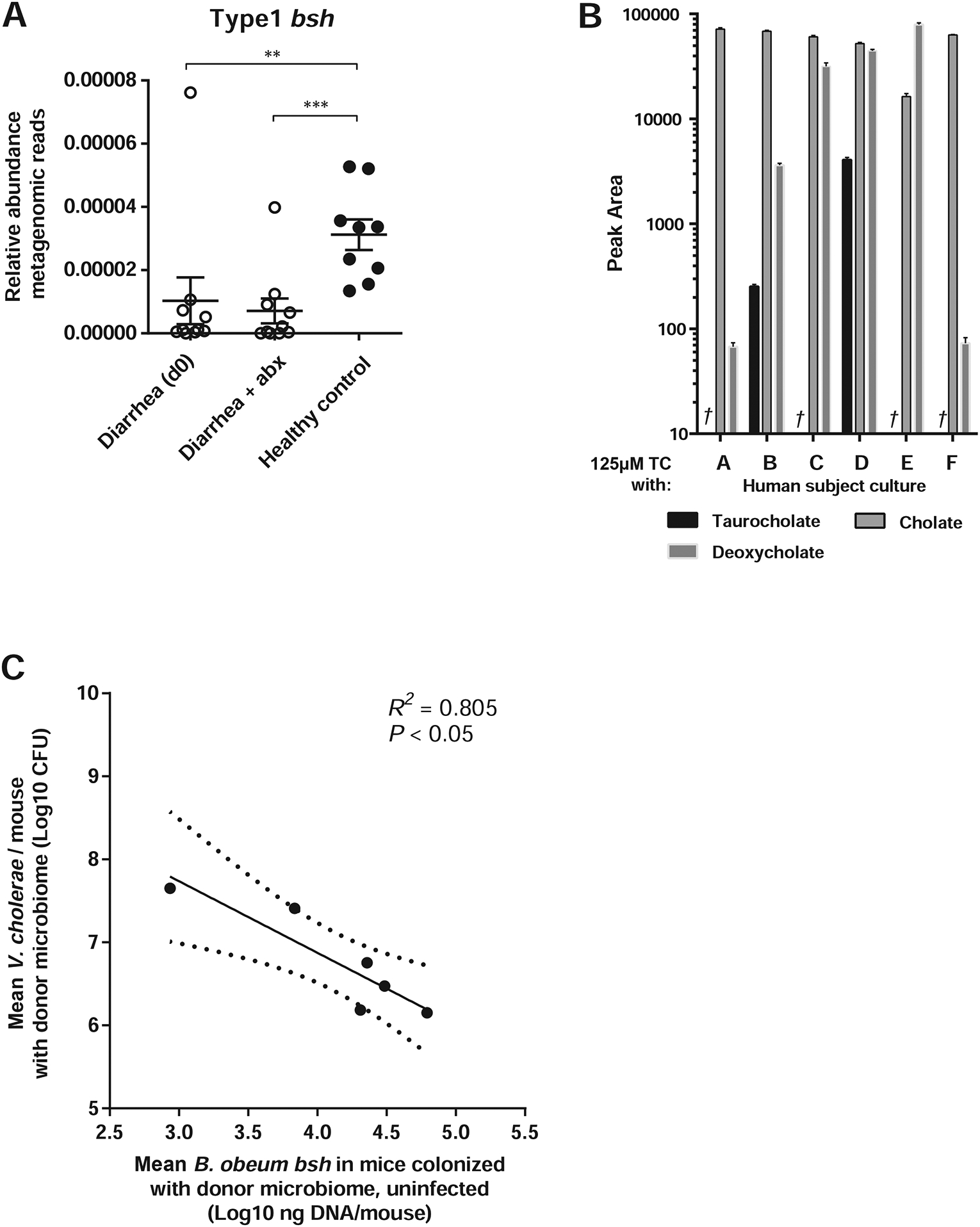
(A) Levels of phylotype 1 bsh enzymes in metagenomic sequencing of fecal microbiomes of cholera patients pre- (“Diarrhea (d0)”) and post-antibiotic (“Diarrhea + abx”) treatment, compared to healthy adults in Bangladesh. (B) Mass spectrometry measurement of bile levels in 125μM solutions of TC incubated with indicated cultured human fecal communities in vitro, †: TC not detected. (C) B. obeum bsh levels in intestines of antibiotic-cleared suckling CD-1 mice colonized with complex human fecal samples without V. cholerae, compared to V. cholerae colonization of antibiotic-cleared suckling animals bearing human donor microbiomes. * P<0.05, ** P<0.01, *** P<0.001 (Mann-Whitney U-test). Error bars represent mean ± SEM.
We then assayed whether complete healthy US fecal communities were differentially able to convert TC to non-tcp-activating forms. We took the first six healthy US donors and anaerobically cultured bacteria from their fecal samples in vitro, and were able to recover species representing 66–99% relative abundance of the original sample (Table S4C). We inoculated these complex fecal specimens in media and used standardized amounts of the resulting mixed cultures to treat TC solutions, which we then used for virulence reporter assays. Strikingly, we observed that microbiomes (subjects B, D) that allowed higher V. cholerae levels when transplanted in suckling animals were also unable to completely remove TC from solution after 24 hours, while communities exhibiting stronger colonization resistance (A, C and E) reduced TC to undetectable levels (Figure 7B).
Since species in genus Blautia demonstrated differences in bsh activity in vitro, as well as association with cholera patients and uninfected family members (Midani et al., 2018), we assayed for the level of the bsh gene of B. obeum specifically in total DNA extracted from human fecal samples by real-time PCR. This also served as a function-specific measure of B. obeum abundance in these complex fecal mixtures. We found that communities associated with higher V. cholerae colonization had lower levels of B. obeum bsh (Figure 7C), suggesting that B. obeum abundance and specifically the presence of the bsh activity is associated with resistance to V. cholerae infection.
DISCUSSION
A role for gastrointestinal microbes in resistance to enteropathogens such as V. cholerae has been recognized for some time (Freter, 1955, 1956). However, this colonization resistance has often been examined in dichotomous terms, either germfree or conventional, or “normal” and damaged by specific factors such as antibiotics. Our results suggest that beyond extreme fluctuations in structure, such as those due to diarrhea and severe malnutrition, diversity even within otherwise “healthy” human populations can serve as predictive markers for infection resistance.
A key difficulty in identifying taxa that can drive infection susceptibility is the complexity of animal gut microbiomes, compounded by dramatic differences in the taxonomic diversity across host species (Seedorf et al., 2014). The limited taxonomic and functional resolution of many commonly-employed metagenomic techniques is also a barrier for converting observations in large population studies to mechanistic insights on microbial effects on pathogenesis and other phenotypes. Findings in this study and others (Hsiao et al., 2014), in which single genes encoded by specific microbiome members are able to affect V. cholerae colonization in isolation from other functions, suggest that correlative studies have distinct limits in their abilities to provide mechanistic insights to microbiome-pathogen interactions. For instance, a recent sequencing-based study that sampled gut microbes of cholera patients and healthy household contacts identified microbes from the same genus as associated with both individuals with cholera and individuals with putative exposure but non-progression to disease (Midani et al., 2018). Thus, genus-, and possibly even species-, level data may be insufficient to identify clear targets for future mechanistic studies in the absence of experimental manipulations.
Recent developments in high-throughput sequencing, anaerobic microbiology, and gnotobiotic animal systems have allowed for mechanistic studies of the interaction of human commensals and human pathogens in animal models of colonization and disease. Specific target taxa identified by multi-omics approaches can be established in animals, with species and gene content defined before introduction of pathogens. These types of approaches allowed us to identify B. obeum as a key member of the human gut microbiome that drove infection resistance, and microbial interactions with bile acids as a driver of virulence gene regulation in V. cholerae and a mechanism of protection against infection.
We hypothesize that microbial bile metabolism most affects V. cholerae pathogenesis during early infection. V. cholerae tightly regulates gene expression in response to host environmental signals such as bile. Bile acids are thought to stabilize the structure of the key virulence regulator TcpP (Yang et al., 2013), which drives the activation of toxT transcription. ToxT then causes full induction of tcp and cholera toxin, with TCP-dependent colonization thought to begin prior to toxin expression (Lee et al., 1999). Following colonization, the activity of bile becomes less clear. Some studies have demonstrated that the unsaturated fatty acids in bile are able to modulate the binding of ToxT to target promoters, reducing CT and TCP expression (Plecha and Withey, 2015). The deconjugated bile salt sodium deoxycholate promotes interaction between the virulence activators ToxR and ToxS and subsequent activity (Midgett et al., 2017). However, ToxRS likely does not directly activate toxT, but rather acts to boost the activity of TcpP at the toxT promoter (Krukonis et al., 2000). Both conjugated and deconjugated bile acids are also able to induce ToxT-independent activation of cholera toxin dependent on ToxRS (Hung and Mekalanos, 2005).
Our results suggest a model where at the initial point of colonization in the small intestine, tcp gene expression and thus TCP biogenesis is determined by the balance of bile acids that is modulated by commensal microbes with differential bsh activity. Differences in early tcp gene expression and thus colonization may have disproportionate impact on the progression of V. cholerae infections; variation in microbiome bsh activity may help determine whether infection proceeds to fulminant diarrhea, or low or temporary colonization that leads to mild or asymptomatic infections that are common in cholera endemic areas (King et al., 2008). Once severe diarrhea has begun, the commensal community and existing lumenal bile is depleted, and any future bile secretion is predominantly conjugated primary species that stimulate virulence. While B. obeum bile modification is an important regulator of V. cholerae colonization, there may be additive effects on pathogen behavior of multiple community members, and through other mechanisms. Removal of B. obeum was sufficient to raise TC levels in the mouse distal small intestine, but while constitutive expression of B. obeum bsh yielded a 1 log drop in V. cholerae colonization, the full CR microbiome yielded almost 2 log fold differences in colonization compared to DS microbiomes. In V. cholerae, virulence gene expression is negatively regulated by several different quorum sensing systems involving the sensing of specific autoinducers (Jung et al., 2015). Prior studies identified B. obeum-produced autoinducer AI-2 as a suppressor of tcpA, functioning through a pathway bypassing the canonical AI-2 receptor LuxP and involving the regulator VqmA (Hsiao et al., 2014). VqmA has been shown to activate the master quorum sensing regulator HapR, which leads to repression of tcpP (Liu et al., 2006; Zhu et al., 2002). Thus, AI-2 expression and bsh activity by B. obeum may synergize to reduce the level and activity of TcpP during infection. Additional studies will be required to determine how these two inputs exert their effects on regulation of virulence determinants of V. cholerae in vivo. Non-B. obeum CR members may also impact V. cholerae colonization, both at the level of virulence regulation and possibly metabolic competition for colonization niches. Host diet may play a role in colonization; bile is secreted from the gallbladder in response to food ingestion, and this varies by the fat content of the meal (Marciani et al., 2013). Diet is also a potent driver of microbiome structure (Faith et al., 2011), but the effect of different dietary compositions on driving the microbiome to infection resistant or susceptible states has not been well studied. Ingestion of food has been shown to dramatically reduce the infectious dose of V. cholerae due to buffering of stomach pH, but also possibly by raising the levels of virulence-activating conjugated bile acids secreted into the small intestine.
Taken together, our results suggest that variation in human gut microbiomes can be a significant contributor to V. cholerae infection risk, and that this can be modulated through introduction of a human gut commensal with multiple molecular effects on V. cholerae, able to affect levels of both virulence-activating and virulence-suppressing signals at the site of infection. This suggests that targeted microbiome modification can be a promising prophylactic target against cholera.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Ansel Hsiao (ansel.hsiao@ucr.edu).
Materials Availability
Unique plasmids, strains, and reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Raw sequencing data has been deposited at the European Nucleotide Archive (ENA) under accession PRJEB31497.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human studies
All human samples were part of a study approved by the UCR Institutional Review Board and followed NIH guidelines. We collected intact fecal samples from a cohort of healthy adult volunteers at the University of California, Riverside. Inclusion criteria were: 1) age between 18 and 40 years, 2) must be able to provide signed and dated informed consent, 3) must be willing and able to provide stool specimen. Exclusion criteria were: 1) systemic antibiotic usage (oral, intramuscular, or intravenous) in the 2 weeks prior to sampling; 2) acute disease at time of enrollment (presence of moderate or severe illness with or without fever); 3) diarrhea (liquid or very loose stools not associated with a change in diet) in the 2 weeks prior to sampling; 4) active uncontrolled GI disorders or diseases including Inflammatory bowel disease (IBD), ulcerative colitis, Crohn’s disease, or indeterminate colitis, persistent, infectious gastroenteritis, colitis, or gastritis, and chronic constipation; 5) Major surgery of the GI tract, excluding cholecystectomy and appendectomy, but including major bowel resection at any time. Age inclusion criteria were chosen to avoid age-related microbiome differences, which are strongest in early life (Yatsunenko et al., 2012). Fecal samples were collected aseptically from each person at UCR and immediately preserved at −80°C until processing for DNA extraction, culturing, and animal colonization. Stocks of fecal slurries for subsequent experiments were prepared by resuspending samples at 1:3 weight/volume in sterile reduced PBS and adding sterile glycerol to a final concentration of 25% volume/volume.
Animal studies
All animal experiments used protocols approved by the Institutional Animal Care and Use Committee of the University of California, Riverside (UCR) and followed NIH guidelines. All CD-1 suckling animals were purchased from Charles River Laboratories. Suckling and adult germfree C57BL/6J mice were reared at the UCR gnotobiotic facility. No distinction was made between male and female animals for bacterial studies. Adult animals were used at >3 weeks of age. Germfree suckling mice used at 5–6 days of age. Animals were checked for signs of moribund condition prior to use in experiments, and used for one experimental procedure only. Adult animals were co-housed in cages without mixing sex. Male mice were separated except in cases of littermates.
For the antibiotic-cleared suckling mouse model, 4-day old suckling CD-1 animals were fasted for 1.5 hours, then orally dosed with ~1mg/g body weight streptomycin using 30-gauge plastic tubing, after which the animals were placed with a lactating dam for 1 day. After 24 hours, mice received microbial communities with V. cholerae in a maximum gavage volume of 50μl. At 18 hours post-infection, animals were sacrificed, and relevant sections of intestinal tissue dissected and homogenized for CFU numeration and nucleic acid extraction. Germ-free C57BL/6J mice were bred and maintained in plastic gnotobiotic isolators at University of California, Riverside. Mice were fed an autoclaved, low-fat plant polysaccharide-rich mouse chow (Lab Diet 5K52) and were 5–8 weeks old at time of gavage. Bacterial cultures were prepared as described above. Mice were fasted for 30 minutes prior to introduction of bacteria, and stomach pH was buffered by intra-gastric gavage of 100μL 1M NaHCO3, followed by gavage with 150uL of balanced defined microbial libraries. Fecal samples were collected across the course of the experiment. Mice were sacrificed 4 days post gavage and small intestine collected and cut to three equal (proximal, medial, distal) sections by length. Samples were homogenized and used for CFU enumeration of bacteria on LB agar containing 200 μg/mL streptomycin.
For establishing defined microbiomes prior to V. cholerae infection, germ-free C57BL/6J mice were maintained as mentioned above and used at 6–13 weeks of age. Mice were fasted and given NaHCO3 as previously described and either given 150 μL of the simple resistant (SR), or dysbiotic (DS) communities. For the Mix group, mice were initially given the DS microbiome embodiment. 10 days after microbiome introduction and 4 days prior to V. cholerae infection, the SR community was introduced into the Mix group animals by gavage. 2 weeks after human commensal colonization, each group was infected with ~5 × 109 CFU V. cholerae O1 El Tor C6706. Fecal samples were suspended in 500 μL of PBS and homogenized using a bead beater (BioSpec) at 1,400 RPM for 30 seconds. CFU enumeration of V. cholerae was done on LB agar containing 200 μg/mL streptomycin.
We used the antibiotic-treated infant mouse model described above to determine which members of the healthy human microbiome contribute most to resistance to V. cholerae. We made 18 random combinations of human gut microbiome strains and introduced them to suckling mice along with V. cholerae. Each combination included five unique strains, and each gavage contained the equivalent total microbial mass of 300μl of OD600=0.4 culture, divided evenly across all constituent strains. After introducing human microbiome and V. cholerae to suckling mice, V. cholerae levels in homogenized intestines were determined by plating on selective agar. The absolute abundance of each species was determined with a combination of 16S rRNA gene qPCR and 16S rRNA sequencing.
Bacterial strains and growth conditions
All human gut commensal strains used are listed in Table S1. Unless otherwise noted, human gut strains were propagated in LYHBHI liquid medium (BHI supplemented to 5g/L yeast extract, 5mg/L hemin, 1mg/mL cellobiose, 1mg/mL maltose and 0.5mg/mL cysteine-HCl). Cultures were then propagated in a Coy anaerobic chamber (5% H2, 20% CO2, balance N2) or aerobically at 37°C.
All V. cholerae strains were derived from the C6706 El Tor pandemic isolate, including the lacZ:PtcpA:sh-Ble zeocin resistance reporter strain (Liu et al., 2008), and propagated in LB media with appropriate antibiotics at 37°C. To construct a strain resistant to kanamycin, the plasmid pZE21 was cloned into V. cholerae C6706 (C6706-KMR) and propagated in LB with kanamycin sulfate (Fisher Scientific, 50μg/ml) and streptomycin sulfate salt (100μg/ml). Vibrio harveyi BB170 was propagated in LM medium (Bassler et al., 1994) aerobically at 37°C.
To construct bshC, a strain constitutively expressing a bile salt hydrolase found in B. obeum, the RUMOBE_00028 locus was amplified from B. obeum genomic DNA (primers: 5’-GTCGACGGTATCGATAATGCTTATGTGTACAGCTGC-3’ and 5’-GCAGGAATTCGATATCACTAATTCTGAAAATGAATCTGC-3’). All cloning and amplification primers are listed in Table S6. This amplicon was then cloned downstream of the constitutive PLtet-O1 promoter of plasmid pZE21 through digestion of the vector backbone with HindIII followed by Gibson assembly (New England Biolabs). The resulting plasmid was then introduced by electroporation into E. coli DH5αλpir to generate bshC. Strains were propagated aerobically in LB with kanamycin (50μg/ml) at 37°C.
A strain overexpressing the AI-2 signal of B. obeum (BW30045_RO_AI-2) was constructed by constitutively expressing the B. obeum luxS AI-2 synthase into E. coli BW30045 (ΔluxS). The B. obeum luxS coding region (from genome position 33,305–33,784) was codon-optimized for expression in E. coli, placed downstream of the PLtet-O-1 constitutive promoter sequence derived from the plasmid vector pZE21 vector, and the construct cloned into vector pMK using the GeneArt Subcloning & Express Cloning Service (ThermoFisher). This expression construct was then amplified and inserted into the endA gene of the E. coli genome using primers (forward: 5’-CCAAAACAGCTTTCGCTACGTTGCTGGCTCGTTTTAACACGGAGTAAGTGTTAGAAA AATTCATCCAGCA-3’, reverse: 5’-GGTTGTACGCGTGGGGTAGGGGTTAACAAAAAGAATCCCGCTAGTGTAGGCGGGCA GTGAAAGGAAGGCC-3’).
We used natural transformation (Dalia et al., 2014) to construct a ΔvasK V. cholerae. Fragments of flanking genomic DNA upstream (forward: 5’-GAACTTTCGTCACGTAAGTC-3’, reverse: 5’-GTCGACGGATCCCCGGAATCATGAATTGTGTCCTTGTTTAC-3’) and downstream (forward: 5’-GAACTTTCGTCACGTAAGTC-3’, reverse: 5’-GTCGACGGATCCCCGGAATCATGAATTGTGTCCTTGTTTAC-3’) of vasK and an antibiotic resistant gene cassette (forward: 5’-ATTCCGGGGATCCGTCGAC-3’, reverse: 5’-TGTAGGCTGGAGCTGCTTC-3’) were amplified from V. cholerae genomic DNA. Amplicons were then joined by overlapping PCR and introduced into C6706 via natural transformation. Resistant colonies were then selected on trimethoprim-containing agar (10ug/ml) and insertion confirmed via PCR.
METHOD DETAILS
16S library preparation
For DNA from human fecal samples, ~200 mg (average wet weight) fecal sample was suspended in 600μl PBS. For mouse intestinal samples, tissues were dissected, homogenized in 5mL PBS, and 500μl of the homogenate used for DNA extraction. ~500μl 0.1mm glass beads (BioSpec), 210μl SDS %20, and 500μl neutral phenol:chloroform:isoamyl-alcohol (24:24:1, Fisher Scientific) were added to each sample, and samples were lysed by bead-beating followed by ethanol precipitation (Hsiao et al., 2014).
The V4 variable region of bacterial 16S ribosomal RNA genes was amplified in 25μl total volume reactions comprising 1μl of extracted DNA as template, 10μl Platinum Hot Start PCR Master Mix (ThermoFisher), 13μl PCR-grade water and 0.5μl of forward and reverse primers (10μM). Cycling conditions were 94°C for 3 min, followed by 33 cycles (94°C for 45 sec, 50°C for 60 sec, 72°C for 90 sec), and 72°C for 10 min. An equal amount of each amplicon (~240ng) was pooled into libraries, which were then purified using QIAquick PCR purification columns (Qiagen) and subjected to sequencing using the Illumina MiSeq platform. Paired-end 150nt reads were assembled, de-multiplexed, rarefied to >900 reads per sample, and analyzed using the QIIME 1.9.1 software package (Caporaso et al., 2010). Sequencing run results are summarized in Table S2A and S3.
Human gut microbiome 16S meta-analysis
For the analysis of bacterial composition between the human gut microbial communities and our artificial communities, the sequencing data of the V4 region of the 16S rRNA gene from published studies and samples collected for this study. For the different phases of V. cholerae infection, we used the first and last time points of diarrhea, and the last time of recovery (Hsiao et al., 2014). The last time point of fecal samples collected from parents of malnourished Bangladesh children were selected as the healthy adult Bangladesh control (Subramanian et al., 2014). See Table S2C for sequence accession numbers. Defined community inputs were designed on the basis that all the strains in the specific community are evenly distributed (CR: 1000 reads/species; SR: 3000 reads/species; DS: 2000 reads/species). All of the sequencing data were collected together and analyzed using QIIME as described above.
Metagenomic analysis of bsh phylotypes
The protein sequences of the eight representative BSH were obtained from Song et al., 2019 (Song et al., 2019). Deep metagenomic sequencing data was obtained from David et al (David et al., 2015) (see Table S2D for sequence accession numbers). The microbial community DNA was aligned to the protein sequences using blastx. For queries that hit multiple protein sequences, the one that had the highest hit score was selected. The relative abundance of each type of the BSHs = the BSH reads count / (total reads count – human reads count). Host reads were determined by mapping metagenomic DNA to Homo sapiens reference genome (assembly GRCh38.p13) using HISAT2 (Kim et al., 2015).
Preparation of bacteria for animal studies
Each human gut bacterium was cultured from glycerol stocks in LYHBHI media for 48 hours at 37°C, and then diluted (1:50) in fresh LYHBHI media. After growth for an additional 48 hours, cultures were normalized for density by OD600. For inoculation into suckling mice, the equivalent of a total of 300μl of 0.4 OD600 culture divided evenly across strains by community was pooled, pelleted by centrifugation, and resuspended in fresh LYHBHI. Each mouse received this mass of bacterial cells in a maximum gavage volume of 50μl per pup. In mice containing multiple defined communities, normalized mixtures were prepared so that 300μl of OD600=0.4 equivalent of each community was represented in the final gavage. In mice receiving V. cholerae, the total resuspension volume of commensal strains was 25μl, with the remaining 25μl containing 10×104-1×105 CFU V. cholerae in PBS.
Microbial levels in human fecal slurries were estimated via real-time PCR using universal 16S primers as described below, and samples were normalized to so that each suckling animal received slurries containing the equivalent of ~20μg of microbial genomic DNA.
Measurement of fluid accumulation
Suckling mice were treated as described above. The fluid accumulation ratio percentage was determined as: [weight of intestines (Large and Small) / mouse body weight] × 100.
Assessment of T6SS killing by V. cholerae in vivo
CD-1 suckling animals were gavaged with antibiotics as described above. 3–5 day old infant mice were orally inoculated with total of ~1×109 CFU E. coli TB1 (lacZ-) and ~1×104 CFU V. cholerae (lacZ+) together in 50μl LB. Animals were sacrificed after 16 hr of infection. Mouse intestines were dissected and homogenized in 5 ml of PBS, and 10μl of the homogenate used for enumeration of bacteria via serial dilution on LB agar containing streptomycin and X-gal.
Quantitative real-time PCR
Total bacterial load in fecal samples and intestinal homogenates was determined by using real-time quantitative PCR. Reactions comprised 2 μL of extracted DNA (200ng/reaction) as template, 12.5 μL SYBRGreen Master Mix (BioRad), 10 μL PCR-grade water, and 0.25 μL of forward and reverse primers at 10μM (forward: 5’-CTCCTACGGGAGGCAGCAG-3’, reverse: 5’-TTACCGCGG CTGCTGGCAC-3’). Cycle conditions were 95°C for 3 min, followed by 39 cycles (95°C for 10 sec, 55°C for 30 sec, 95°C for 10 sec, 65°C for 5 sec, 95°C for 5 sec).
Levels of the B. obeum bsh enzyme (RUMOBE_00028) were determined by real-time PCR as described above, using the primers 5’-GCGATCAGATTACGATCACTC-3’ and 5’-GCCATGCCAACACCTTTTTC-3’. 200ng of purified DNA from intestinal homogenates of CD-1 mice colonized with complex human fecal samples were used as template for each reaction.
Levels of tcpA and ctxA expression in antibiotic-cleared CD-1 suckling animals containing SR and DS microbiomes were measured using real-time PCR (tcpA: primers 5’-GAAGAAGTTTGTAAAAGAAGAACACG-3’ and 5’-CGCTGAGACCACACCCATA-3’, ctxA: primers 5’-CACTAAGTGGGCACTTCTCA-3’ and 5’-TGATCATGCAAGAGGAACTCA-3’), with recA (primers 5’-ATTGAAGGCGAAATGGGCGATAG-3’ and 5’-TACACATACAGTTGGATTGCTTGAGG-3’) as a control. Templates were generated by Trizol (Ambion) extraction of total RNA from intestines of SR and DS-colonized mice infected with V. cholerae, followed by cDNA synthesis with the SuperScript IV First-Strand Synthesis System (Invitrogen) following manufacturers’ instructions. Real-time PCR was performed using conditions 95°C for 3 min, followed by 39 cycles (95°C for 10 sec, 55°C for 30 sec, 95°C for 10 sec, 65°C for 5 sec, 95°C for 5 sec).
Culturing of complex human fecal communities
Fecal slurries of complex human fecal samples were prepared as described above, and spread on LYH-BHI agar and incubated aerobically and anaerobically at 37°C. All colonies recovered were gathered by scraping, and DNA extracted and 16S rRNA genes amplified for sequencing as described.
AI-2 heat-stability assay
Cultures of BW31145_RO_AI-2, BB170 and C6706 were grown overnight. BW31145_RO_AI-2 was subcultured 1:100 into 12ml of LB and grown in a shaker at 37°C for until OD600 ≈ 0.22, centrifuged, and the supernatant filter sterilized. Aliquots of supernatant were then heated at 100°C for 30 minutes and cooled to room temperature. AI-2 activity was assessed using the
BB170 bioassay (Bassler et al., 1994). Briefly, overnight cultures of reporter strain BB170 in LM medium were diluted at 1:1000 in AB medium, and 10μl of cell-free supernatant or heat-treated cell-free supernatant were then added to 90μl of BB170 dilution. Luminescence and OD600 of each sample were measured immediately and after ~3.5hrs growth at 30°C with agitation.
In vitro bile-dependent tcp induction
PtcpA-sh-ble was grown as overnight culture, diluted 1:1000 in fresh LB, and grown for ~2 hours at 37°C. Each reaction was prepared in 40μl 0.5X pH 8.5 LB medium, with sodium taurocholate hydrate (TC, Sigma-Aldrich), sodium glycocholate hydrate (Sigma Aldrich), cholic acid (CA, Alfa Aesar), sodium taurodeoxycholate hydrate (Sigma-Aldrich), sodium glycodeoxycholate (Sigma Aldrich), deoxycholic acid (DCA, MB Biomedicals), tauro-β-muricholic acid (Steraloids Inc.), or β-muricholic acid (Steraloids Inc.) added to a final concentration of 125μM. 2μl of reporter strain subculture was then added, and samples incubated anaerobically at 37°C for 4hrs. 2μl of each reaction was then added to 200μl of 0.5X LB pH 8.5 +/−10μg/ml of zeocin (Sigma Aldrich), incubated for 30 minutes aerobically at 37°C with agitation, and then serially diluted and plated onto LB agar plates with streptomycin to determine survival rates. Induction represents percentage of PtcpA-sh-ble reporter cells surviving treatment with zeocin after incubation with indicated samples under anaerobic conditions, defined as (zeocin-treated sample survival/average of no-zeocin controls)*100. Where noted, survival rates were normalized to that induced by 125μM taurocholate.
BSH structure comparisons
The potential 3D models of unknown structure were produced by Phyre2 (Kelley et al., 2015), based on amino acid sequence alignment to known protein structure. The 3D structure comparison and root-mean-square distance (RMSD) per-column spatial variation among structures were calculated using UCSF developed Chimera (V1.14) (Pettersen et al., 2004).
Commensal effects on tcp-activating signals
Commensal isolate cultures were grown for 48 hrs, and then subcultured at 3:100 for 48hrs. Growth was measured by OD600 and cultures normalized to 1.5mL of OD600=0.4 culture. bshc and the corresponding vector strain were grown overnight in LB with kanamycin, subcultured at 1:100 for 24hr, and normalized as above. All cultures were clarified, the aqueous layer removed, and the pellets resuspended in sterile PBS with TC to a final concentration of 125uM. Cultures grown with antibiotics were washed one additional time with 1 volume of PBS prior to addition of TC. Samples were then incubated anaerobically for 24hr at 37°C followed by heat treatment at 100°C for 30 minutes, cooled to room temperature, centrifuged and the supernatant filter-sterilized with a 0.22μm filter. These samples were then used to induce PtcpA-sh-ble, and percent survival following zeocin treatment determined as described above.
Removal of tcp activating signals in the gut was assayed as above, replacing pure TC solution in PBS with homogenate. Tissues were collected from 5–6-day-old CD-1 suckling animals in 2.5ml sterile H2O, disrupted with a tissue homogenizer, pooled, and centrifuged to clear tissue debris. The resulting aqueous layer was heat-treated and filter sterilized as described above. The resulting sample was desiccated using a Savant Integrated speedvac system (Fisher Scientific) and resuspended in one-fifth volume of sterile water. Four volumes of acetonitrile (Sigma Aldrich) were then added and sample was incubated at room temperature for 20 minutes for deproteinization (Humbert et al., 2012). Samples were clarified, with the aqueous layer filter sterilized, desiccated and resuspended in one-fifth original volume sterile H2O as described above. To sequester bile salts, 12.5mg of cholestyramine (Sigma-Aldrich) was added to 0.5ml of de-proteinized sample, and the mixture incubated at 1 hour at 37°C with agitation followed by passage through a 3kDa protein concentrator (Pierce PES Protein Concentrators).
Human complex fecal sample TC processing was assayed in vitro. 100μl of fecal slurries in glycerol prepared as described above was inoculated into 5ml LYBHI and incubated anaerobically for 2 days at 37°C. Cells were then pelleted, normalized to ~OD600 =0.4 in 1.5ml sterile PBS with 125μM TC, and incubated anaerobically at 37°C for 24hrs. Supernatants were then collected via centrifugation, heat-treated and filter sterilized as described above, and submitted for mass spectroscopy (see below).
The effects of B. obeum bsh on V. cholerae colonization was assayed by introducing bshC and the isogenic vector control into suckling animals prior to infection with Vibrio. 4-day old CD-1 suckling mice were treated with 50μl of 75mg/mL kanamycin, then returned to lactating dams. Overnight cultures of bshC and vector strains were normalized to the equivalent 300μl of OD600=0.4 culture, and cells pelleted and resuspended in fresh LYHBHI. 50μl of this was then introduced via intra-gastric gavage into antibiotic-treated suckling mice that had been fasted for 1.5 hours. Pups were then returned to a lactating dam. After 1 day of pre-infection colonization with E. coli, animals were gavaged with V. cholerae as described above.
Quantification of bile salts
All standards (TC, CA, and DCA) were submitted as 10mM solutions. LC-MS analysis of bile acids was performed on a Synapt G2-Si quadrupole time-of-flight mass spectrometer (Waters) coupled to an I-class UPLC system (Waters). Separations were carried out on a CSH phenylhexyl column (2.1 × 100mm, 1.7μM) (Waters). The mobile phases were (A) water with 0.1% formic acid and (B) acetonitrile with 0.1% formic acid. The flow rate was 250 μL/min and the column was held at 40° C. The gradient was as follows: 0min, 1% B; 1min, 1% B; 8min, 40% B; 13min, 58.8% B; 13.5min, 100% B; 15.5min, 100% B; 16min, 1% B; 18min, 1% B. Flow rate was ramped to 600 μL/min at 13.5 min to speed up column flushing and re-equilibration.
The MS was operated in positive ion mode (50 to 1200 m/z) with a 100ms scan time. Source and desolvation temperatures were 150° C and 600° C, respectively. Desolvation gas was set to 1100L/hr and cone gas to 150L/hr. All gases were nitrogen except the collision gas, which was argon. Capillary voltage was 1kV. Injection volume was 1μl for all samples. The identity of bile acids in samples was confirmed by mass, retention time, and MS/MS as compared to authentic standards. Samples were analyzed in random order and injected in duplicate. Leucine enkephalin was infused and used for mass correction. Data processing (peak integration) was performed using QuanLynx software (Waters). Accuracy of peak integrations was checked manually.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests were performed in the GraphPad Prism software package. Results are representative of two independent experiments. Statistical parameters for studies are reported in relevant figure legends and tables.
Supplementary Material
Supplementary Table 1. Commensal strains, Related to Figure STAR Methods.
Supplementary Table 2. Sequencing statistics, Related to Figure STAR Methods.
Supplementary Table 3. Relative abundance of microbial taxa by sample. Related to Figures 1 and 3.
Supplementary Table 4. Recovery of microbial diversity from complex human fecal microbiomes by culturing and mouse colonization. Related to Figures 4 and 7.
Supplementary Table 5. Bile salt hydrolases found in genomes of defined community members. Related to Figure 6.
Supplementary Table 6. Oligonucleotides used in this study, Related to Figure STAR Methods.
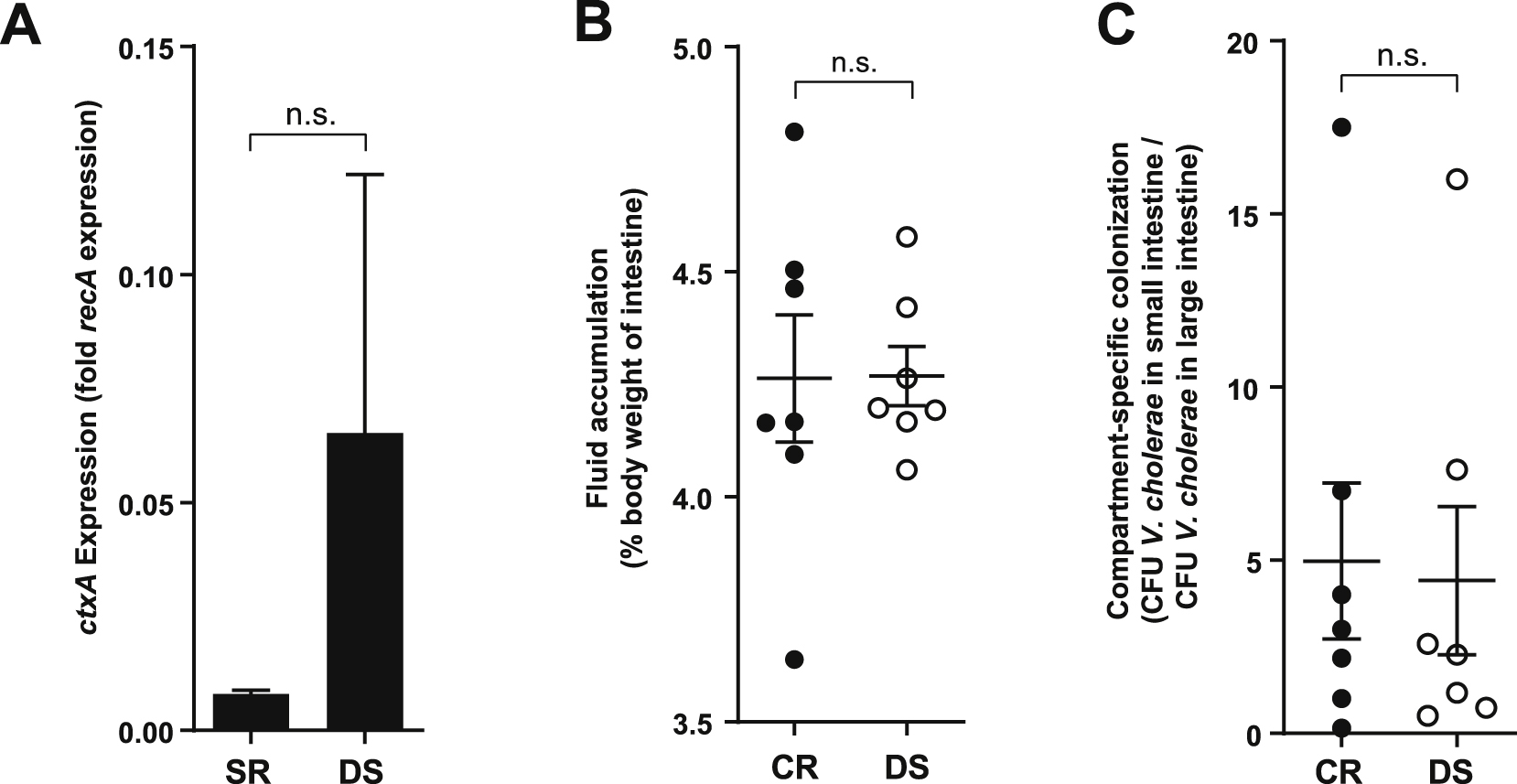
All experiments are in antibiotic-cleared suckling CD-1 mice. (A) Expression of ctxA in intestinal tissues of infected mice containing defined model human microbiomes. (B) Fluid accumulation in intestines of infected mice containing defined model human microbiomes. (C) Distribution of V. cholerae in infected mice containing defined model human microbiomes. n.s. not significant (Mann-Whitney U-test).
{kind=link}
Normalized colonization across experiments reported as fold CFU V. cholerae gavaged recovered after infection. n.s. not significant (Mann-Whitney U-test).
{kind=link}
** P<0.01, (Mann-Whitney U-test).
{kind=link}
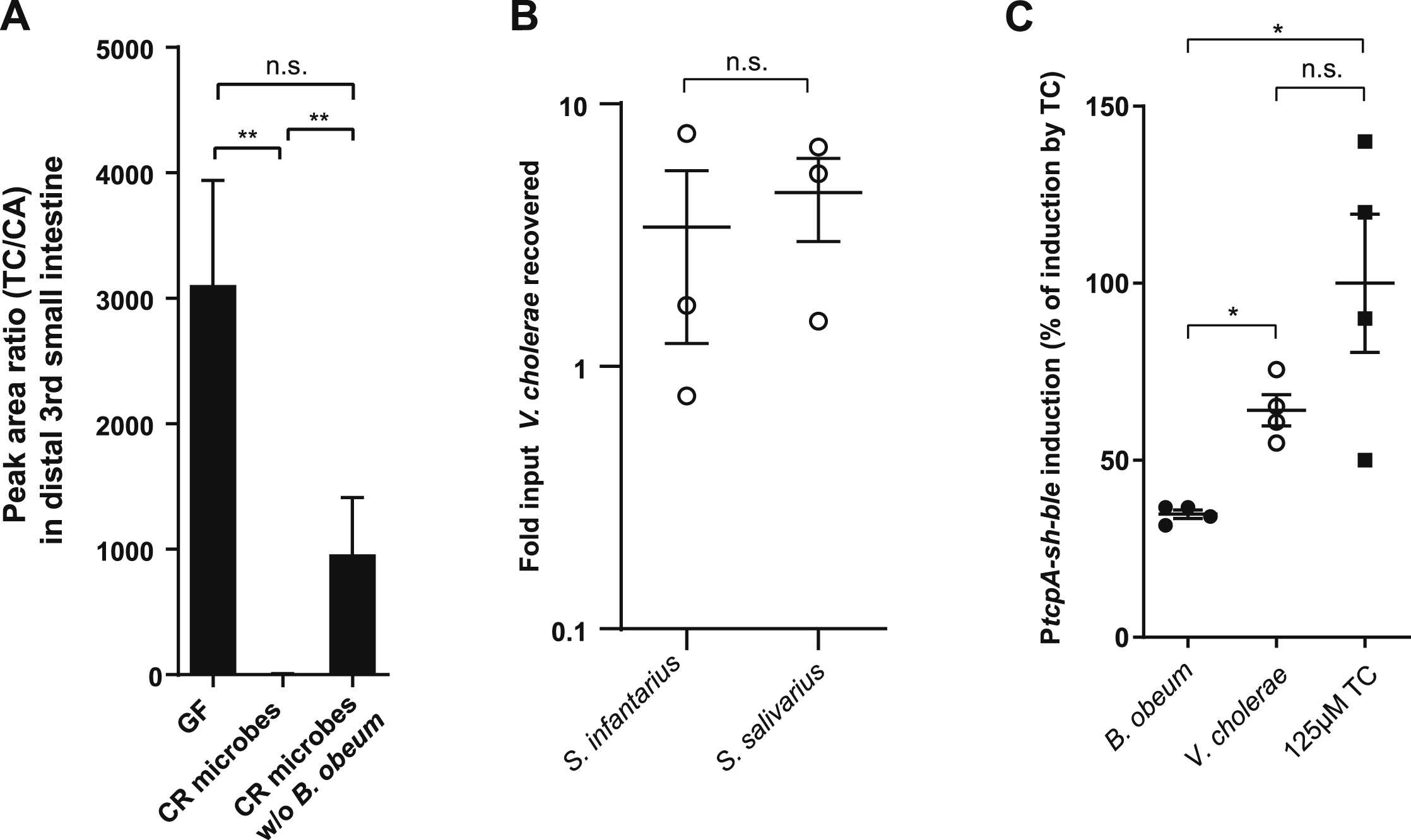
(A) Mass spectrometry measurement of taurocholate (TC) to cholic acid (CA) ratio in distal third of small intestine of adult germfree C57BL/6J mice 2 days after colonization with pure cultures of indicated strains. (B) Amount of V. cholerae recovered during co-infection of suckling CD-1 mice with either S. infantarius or S. salivarius, normalized to input CFU V. cholerae. (C) Ability of B. obeum and V. cholerae to interfere with TC activation of virulence in reporter V. cholerae in vitro after 5 hours incubation, normalized to induction by 125μM TC. * P<0.05, ** P<0.01 (Mann-Whitney U-test), n.s. not-significant.
{kind=link}
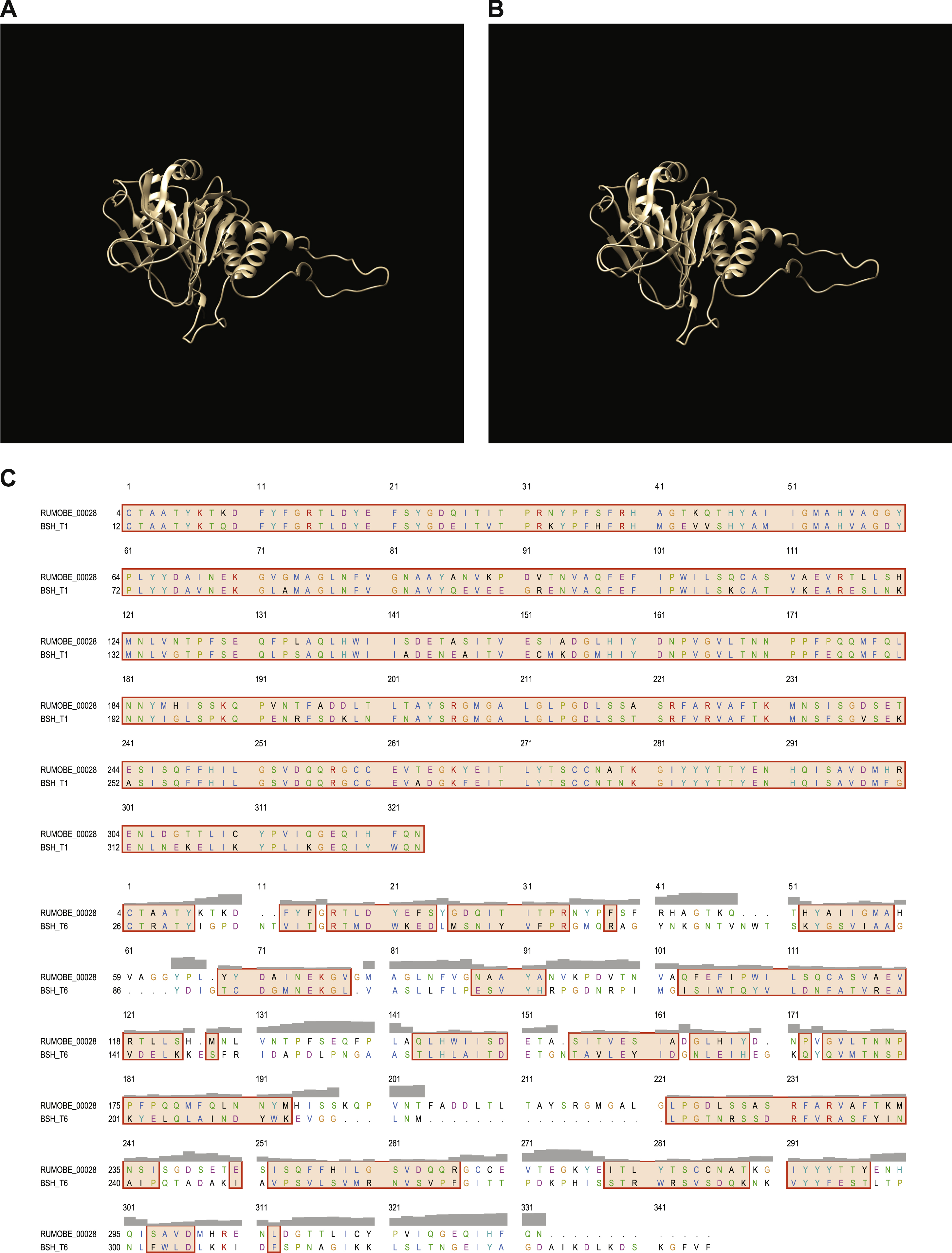
(A) Predicted structure of B. obeum bile salt hydrolase RUMOBE_00028. (B) Predicted structure of consensus phylotype 1 BSH. (C). Amino acid alignment of RUMOBE_00028, phylotype 1 BSH, and phylotype 6 BSH using Chimera. Header in gray shows the spatial variation per column. Colored boxes shows structural similarity between regions, with coloring of one-letter code amino acids using Clustal X, dependent on both residue type and the pattern of conservation across aligned sequences.
{kind=link}
* P<0.05, ** P<0.01, *** P<0.001, **** P<0.0001 (Mann-Whitney U-test), n.s. not-significant. Error bars represent mean ± SEM.
{kind=link}
Highlights.
Interpersonal human gut microbiome variation confers variable infection resistance
Microbiome-dependent infection resistance can be restored through co-transplantation
Colonization resistance is mediated through the bile salt hydrolase enzyme activity
Bile salt hydrolase abundance in human microbiomes correlates to final infection
Acknowledgments:
We thank Jun Zhu and Gary Dunny for the kind gift of V. cholerae and E. faecalis strains respectively. We thank the Metabolomics Core Facility at University of California, Riverside for mass spectrometry studies. This work was supported by National Institute of General Medical Sciences grant R35GM124724 (to A.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests:
The authors declare no competing interests.
REFERENCES
- Amin Marashi SM, Rajabnia R, Imani Fooladi AA, Hojati Z, Moghim S, and Nasr Esfahani B (2013). Determination of ctxAB expression in Vibrio cholerae Classical and El Tor strains using Real-Time PCR. Int J Mol Cell Med 2, 9–13. [PMC free article] [PubMed] [Google Scholar]
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto J-M, et al. (2011). Enterotypes of the human gut microbiome. Nature 473, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassler BL, Wright M, and Silverman MR (1994). Multiple signalling systems controlling expression of luminescence in Vibrio harveyi: sequence and function of genes encoding a second sensory pathway. Mol Microbiol 13, 273–286. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens JD, Nair GB, Ahmed T, Qadri F, and Holmgren J (2017). Cholera. Lancet 390, 1539–1549. [DOI] [PubMed] [Google Scholar]
- Dalia AB, McDonough E, and Camilli A (2014). Multiplex genome editing by natural transformation. Proceedings of the National Academy of Sciences of the United States of America 111, 8937–8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Weil A, Ryan ET, Calderwood SB, Harris JB, Chowdhury F, Begum Y, Qadri F, LaRocque RC, and Turnbaugh PJ (2015). Gut Microbial Succession Follows Acute Secretory Diarrhea in Humans. mBio 6, e00381–00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smet I, Van Hoorde L, Vande Woestyne M, Christiaens H, and Verstraete W (1995). Significance of bile salt hydrolytic activities of lactobacilli. J Appl Bacteriol 79, 292–301. [DOI] [PubMed] [Google Scholar]
- Di Ciaula A, Garruti G, Lunardi Baccetto R, Molina-Molina E, Bonfrate L, Wang DQ, and Portincasa P (2017). Bile Acid Physiology. Ann Hepatol 16 Suppl 1, S4–S14. [DOI] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, and Relman DA (2005). Diversity of the human intestinal microbial flora. Science 308, 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, McNulty NP, Rey FE, and Gordon JI (2011). Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 333, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freter R (1955). The fatal enteric cholera infection in the guinea pig, achieved by inhibition of normal enteric flora. J Infect Dis 97, 57–65. [DOI] [PubMed] [Google Scholar]
- Freter R (1956). Experimental enteric Shigella and Vibrio infections in mice and guinea pigs. The Journal of experimental medicine 104, 411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fykse EM, Skogan G, Davies W, Olsen JS, and Blatny JM (2007). Detection of Vibrio cholerae by real-time nucleic acid sequence-based amplification. Appl Environ Microbiol 73, 1457–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, and Chowdhury R (1997). Bile affects production of virulence factors and motility of Vibrio cholerae. Infect Immun 65, 1131–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington DA, Hall RH, Losonsky G, Mekalanos JJ, Taylor RK, and Levine MM (1988). Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. The Journal of experimental medicine 168, 1487–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao A, Ahmed AMS, Subramanian S, Griffin NW, Drewry LL, Petri WA, Haque R, Ahmed T, and Gordon JI (2014). Members of the human gut microbiota involved in recovery from Vibrio cholerae infection. Nature 515, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert L, Maubert MA, Wolf C, Duboc H, Mahe M, Farabos D, Seksik P, Mallet JM, Trugnan G, Masliah J, et al. (2012). Bile acid profiling in human biological samples: comparison of extraction procedures and application to normal and cholestatic patients. J Chromatogr B Analyt Technol Biomed Life Sci 899, 135–145. [DOI] [PubMed] [Google Scholar]
- Hung DT, and Mekalanos JJ (2005). Bile acids induce cholera toxin expression in Vibrio cholerae in a ToxT-independent manner. Proceedings of the National Academy of Sciences of the United States of America 102, 3028–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BV, Begley M, Hill C, Gahan CG, and Marchesi JR (2008). Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proceedings of the National Academy of Sciences of the United States of America 105, 13580–13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SA, Chapman CA, and Ng WL (2015). Quadruple quorum-sensing inputs control Vibrio cholerae virulence and maintain system robustness. PLoS Pathog 11, e1004837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJ (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieser S, Sarker SA, Sakwinska O, Foata F, Sultana S, Khan Z, Islam S, Porta N, Combremont S, Betrisey B, et al. (2018). Bangladeshi children with acute diarrhoea show faecal microbiomes with increased Streptococcus abundance, irrespective of diarrhoea aetiology. Environmental Microbiology 20, 2256–2269. [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AA, Ionides EL, Pascual M, and Bouma MJ (2008). Inapparent infections and cholera dynamics. Nature 454, 877–880. [DOI] [PubMed] [Google Scholar]
- Klose KE (2000). The suckling mouse model of cholera. Trends in microbiology 8, 189–191. [DOI] [PubMed] [Google Scholar]
- Kovacikova G, Lin W, and Skorupski K (2010). The LysR-type virulence activator AphB regulates the expression of genes in Vibrio cholerae in response to low pH and anaerobiosis. Journal of bacteriology 192, 4181–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krukonis ES, Yu RR, and Dirita VJ (2000). The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane-localized transcriptional activators on a single promoter. Mol Microbiol 38, 67–84. [DOI] [PubMed] [Google Scholar]
- Lee SH, Hava DL, Waldor MK, and Camilli A (1999). Regulation and temporal expression patterns of Vibrio cholerae virulence genes during infection. Cell 99, 625–634. [DOI] [PubMed] [Google Scholar]
- Li J, and Dawson PA (2018). Animal models to study bile acid metabolism. Biochimica et biophysica acta Molecular basis of disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Hsiao A, Joelsson A, and Zhu J (2006). The transcriptional regulator VqmA increases expression of the quorum-sensing activator HapR in Vibrio cholerae. Journal of bacteriology 188, 2446–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Miyashiro T, Tsou A, Hsiao A, Goulian M, and Zhu J (2008). Mucosal penetration primes Vibrio cholerae for host colonization by repressing quorum sensing. Proceedings of the National Academy of Sciences of the United States of America 105, 9769–9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciani L, Cox EF, Hoad CL, Totman JJ, Costigan C, Singh G, Shepherd V, Chalkley L, Robinson M, Ison R, et al. (2013). Effects of various food ingredients on gall bladder emptying. Eur J Clin Nutr 67, 1182–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midani FS, Weil AA, Chowdhury F, Begum YA, Khan AI, Debela MD, Durand HK, Reese AT, Nimmagadda SN, Silverman JD, et al. (2018). Human Gut Microbiota Predicts Susceptibility to Vibrio cholerae Infection. J Infect Dis 218, 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgett CR, Almagro-Moreno S, Pellegrini M, Taylor RK, Skorupski K, and Kull FJ (2017). Bile salts and alkaline pH reciprocally modulate the interaction between the periplasmic domains of Vibrio cholerae ToxR and ToxS. Mol Microbiol 105, 258–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller VL, Taylor RK, and Mekalanos JJ (1987). Cholera toxin transcriptional activator toxR is a transmembrane DNA binding protein. Cell 48, 271–279. [DOI] [PubMed] [Google Scholar]
- Muraca M, Vilei MT, Miconi L, Petrin P, Antoniutti M, and Pedrazzoli S (1991). A simple method for the determination of lipid composition of human bile. Journal of lipid research 32, 371–374. [PubMed] [Google Scholar]
- Olivier V, Queen J, and Satchell KJ (2009). Successful small intestine colonization of adult mice by Vibrio cholerae requires ketamine anesthesia and accessory toxins. PloS one 4, e7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Plecha SC, and Withey JH (2015). Mechanism for inhibition of Vibrio cholerae ToxT activity by the unsaturated fatty acid components of bile. Journal of bacteriology 197, 1716–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon JM, Kang DJ, and Hylemon PB (2006). Bile salt biotransformations by human intestinal bacteria. Journal of lipid research 47, 241–259. [DOI] [PubMed] [Google Scholar]
- Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, Angelin B, Hyotylainen T, Oresic M, and Backhed F (2013). Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell metabolism 17, 225–235. [DOI] [PubMed] [Google Scholar]
- Seedorf H, Griffin NW, Ridaura VK, Reyes A, Cheng J, Rey FE, Smith MI, Simon GM, Scheffrahn RH, Woebken D, et al. (2014). Bacteria from diverse habitats colonize and compete in the mouse gut. Cell 159, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Cai Y, Lao X, Wang X, Lin X, Cui Y, Kalavagunta PK, Liao J, Jin L, Shang J, et al. (2019). Taxonomic profiling and populational patterns of bacterial bile salt hydrolase (BSH) genes based on worldwide human gut microbiome. Microbiome 7, 9–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Huq S, Yatsunenko T, Haque R, Mahfuz M, Alam MA, Benezra A, DeStefano J, Meier MF, Muegge BD, et al. (2014). Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature 510, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Liu Z, Hughes C, Stern AM, Wang H, Zhong Z, Kan B, Fenical W, and Zhu J (2013). Bile salt-induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proceedings of the National Academy of Sciences of the United States of America 110, 2348–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Caro F, Robins W, and Mekalanos JJ (2018). Antagonism toward the intestinal microbiota and its effect on Vibrio cholerae virulence. Science 359, 210–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, and Mekalanos JJ (2002). Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proceedings of the National Academy of Sciences of the United States of America 99, 3129–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Commensal strains, Related to Figure STAR Methods.
Supplementary Table 2. Sequencing statistics, Related to Figure STAR Methods.
Supplementary Table 3. Relative abundance of microbial taxa by sample. Related to Figures 1 and 3.
Supplementary Table 4. Recovery of microbial diversity from complex human fecal microbiomes by culturing and mouse colonization. Related to Figures 4 and 7.
Supplementary Table 5. Bile salt hydrolases found in genomes of defined community members. Related to Figure 6.
Supplementary Table 6. Oligonucleotides used in this study, Related to Figure STAR Methods.
All experiments are in antibiotic-cleared suckling CD-1 mice. (A) Expression of ctxA in intestinal tissues of infected mice containing defined model human microbiomes. (B) Fluid accumulation in intestines of infected mice containing defined model human microbiomes. (C) Distribution of V. cholerae in infected mice containing defined model human microbiomes. n.s. not significant (Mann-Whitney U-test).
Normalized colonization across experiments reported as fold CFU V. cholerae gavaged recovered after infection. n.s. not significant (Mann-Whitney U-test).
** P<0.01, (Mann-Whitney U-test).
(A) Mass spectrometry measurement of taurocholate (TC) to cholic acid (CA) ratio in distal third of small intestine of adult germfree C57BL/6J mice 2 days after colonization with pure cultures of indicated strains. (B) Amount of V. cholerae recovered during co-infection of suckling CD-1 mice with either S. infantarius or S. salivarius, normalized to input CFU V. cholerae. (C) Ability of B. obeum and V. cholerae to interfere with TC activation of virulence in reporter V. cholerae in vitro after 5 hours incubation, normalized to induction by 125μM TC. * P<0.05, ** P<0.01 (Mann-Whitney U-test), n.s. not-significant.
(A) Predicted structure of B. obeum bile salt hydrolase RUMOBE_00028. (B) Predicted structure of consensus phylotype 1 BSH. (C). Amino acid alignment of RUMOBE_00028, phylotype 1 BSH, and phylotype 6 BSH using Chimera. Header in gray shows the spatial variation per column. Colored boxes shows structural similarity between regions, with coloring of one-letter code amino acids using Clustal X, dependent on both residue type and the pattern of conservation across aligned sequences.
* P<0.05, ** P<0.01, *** P<0.001, **** P<0.0001 (Mann-Whitney U-test), n.s. not-significant. Error bars represent mean ± SEM.
Data Availability Statement
Raw sequencing data has been deposited at the European Nucleotide Archive (ENA) under accession PRJEB31497.