

Abstract
Malaria puts at risk nearly half the world’s population and causes high mortality in sub-Saharan Africa, while drug resistance threatens current therapies. The pyrimidine biosynthetic enzyme dihydroorotate dehydrogenase (DHODH) is a validated target for malaria treatment based on our finding that triazolopyrimidine DSM265 (1) showed efficacy in clinical studies. Herein we describe optimization of a pyrrole-based series identified by a target-based DHODH screen. Compounds with nanomolar potency versus Plasmodium DHODH and Plasmodium parasites were identified with good pharmacologic properties. X-ray studies showed the pyrroles bind an alternative enzyme conformation from 1 leading to improved species selectivity versus mammalian enzymes and equivalent activity on P. falciparum and P. vivax DHODH. The best lead DSM502 (37) showed in vivo efficacy at similar levels of blood exposure to 1, although metabolic stability was reduced. Overall the pyrrole-based DHODH inhibitors provide an attractive alternative scaffold for development of new antimalarial compounds.
Graphical Abstract
Introduction
Infectious diseases cause the majority of deaths in low income countries, primarily the result of respiratory infections, diarrheal diseases, HIV, malaria and tuberculosis (TB).1 Malaria alone is responsible for 0.4 million deaths per year, mostly amongst young children in sub-Saharan Africa.2 Most deaths are caused by Plasmodium falciparum malaria, however nearly 90 countries are endemic with malaria, and P. vivax, while less lethal, has the greater global reach.3–5 While a vaccine for the prevention of malaria has recently been recommended for large scale pilot implementation in Africa, its efficacy is sub-optimal resulting in <50% protection.6, 7 Therefore, both prevention and treatment programs continue to rely on chemotherapy. While a number of effective compounds have been employed, drug resistance has led to decreased efficacy in most cases (e.g chloroquine)8, a challenge that is shared with other infectious diseases like HIV and TB.9 The current standard of care treatments for malaria, artemisinin combination therapies (ACTs)10, 11, are safe, effective and credited with declining cases of malaria worldwide. However, parasites showing delayed clearance times to artemisinin derivatives have been identified in southeast Asia, and combined with increasing resistance to partner compounds, these issues underlie treatment failures in patients in this region.8, 10, 12
Concerns that ACTs may fail on a global scale are driving drug discovery efforts to find new therapies for the treatment of malaria.4, 13 A number of new chemical entities have reached clinical development, including three that are currently in Phase IIb human studies (the peroxide OZ43914, 15 combined with ferroquine, KAF15616, 17 combined with lumefantrine and spiroindolone KAE60918, 19 as a single agent for severe malaria). Combination therapy is considered key to reduce the potential for resistance to develop towards any new treatment, increasing the pressure on the malaria drug portfolio to identify a larger number of clinical candidates. While most of the current clinical candidates were identified by asexual blood stage screens, target based approaches have also been successful.4 Notably, dihydroorotate dehydrogenase (DHODH) was validated as a new target by our group through the identification of a clinical development candidate, the triazolopyrimidine DSM265 (1) (Fig. 1), targeting DHODH.20–25 A second triazolopyrimidine (DSM421; 2-(1,1-difluoroethyl)-5-methyl-N-(6-(trifluoromethyl)pyridin-3-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine) reached preclinical development but was not progressed due to unexpected off-target toxicity26, 27 and a thiophene analog 228 (Fig. 1) developed by Genzyme reached late lead development but was not advanced beyond this stage.
Fig. 1.
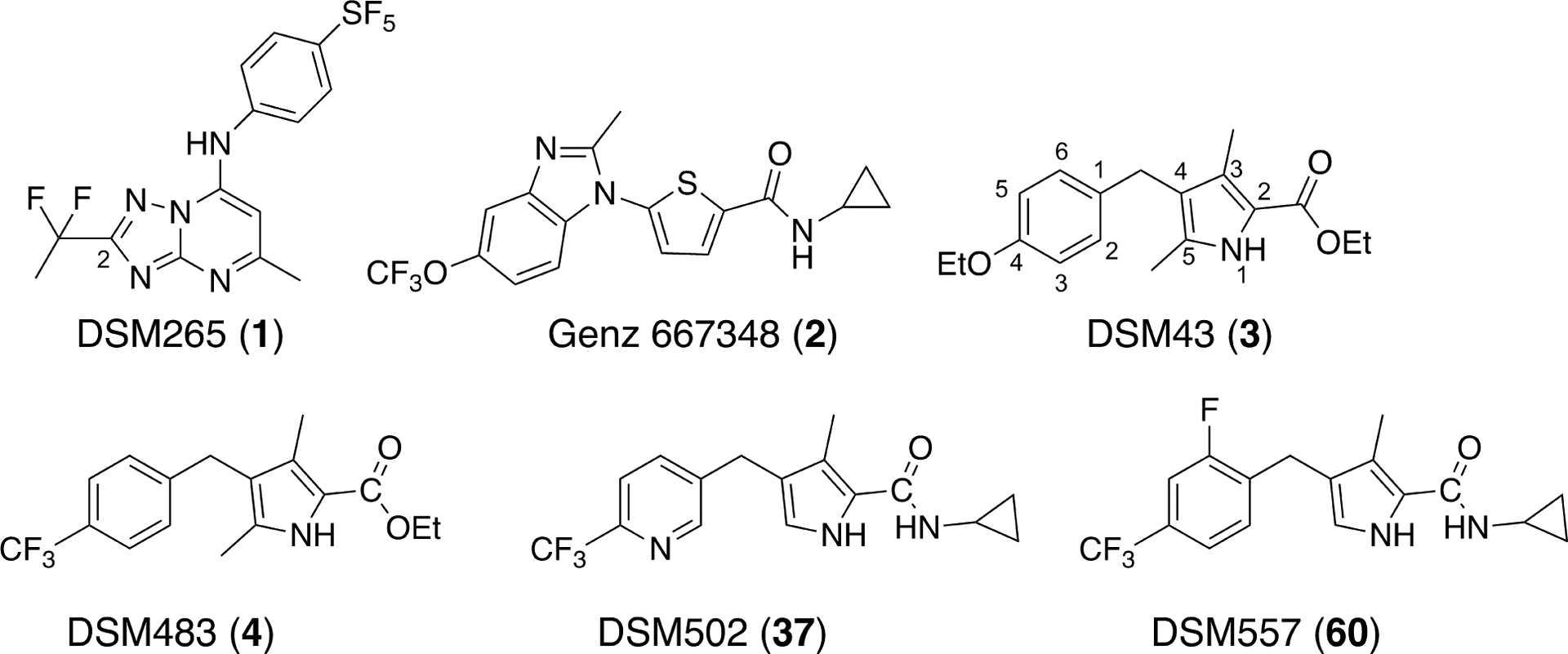
Structures of selected DHODH inhibitors.
DHODH is a mitochondrial enzyme that is localized to the inner mitochondrial membrane where it catalyzes a key step in the de novo synthesis of pyrimidines, the flavin-dependent conversion of dihydroorotate to orotic acid.29, 30 Ubiquinone serves as the final oxidant in the reaction. Because Plasmodium parasites lack salvage enzymes, the de novo pathway is essential to the formation of pyrimidines for DNA and RNA synthesis. As a consequence, inhibitors of P. falciparum DHODH (PfDHODH) block parasite replication at the early trophozoite stage prior to the burst of replication required to form the schizont.22 A key advantage of the target is that it is required for both blood and liver stage development, and thus DHODH inhibitors meet the product profile for either malaria treatment or chemoprevention.22, 31 1 has been clinically validated for the treatment of P. falciparum infections during Phase I and IIa studies to test safety and efficacy in volunteers and patients (Phase I24, 25 and Phase IIa23) and in human challenge studies to assess its potential for chemoprevention.32, 33 1 showed considerable strengths in the clinic, including good safety and a long human half-life (~100 hours) providing a single dose (400 mg dose) cure of P. falciparum malaria in human studies in patients in Peru.23, 25 Additionally a single dose (400 mg) given 1 day before human volunteers were challenged with the infectious mosquito stage of P. falciparum (sporozoites) prevented infection, a result that likely supports once weekly dosing for chemoprevention.32, 33 Two potential liabilities have been the identification of resistance mutations from both in vitro selections and in patients experiencing recrudescence23, 34 and the finding that in comparison to P. falciparum, 1 showed reduced activity on P. vivax in both ex vivo studies and in a human phase IIa clinical trial.23, 26
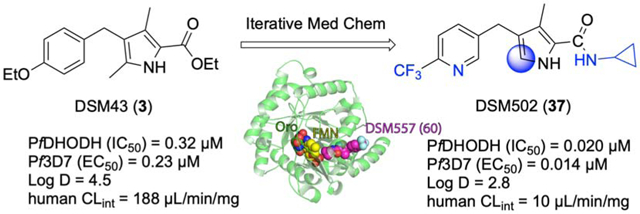
To position additional DHODH inhibitors from a chemical series distinct from 1, we undertook lead optimization of a pyrrole-based series that we identified in our original target-based high throughput screen35 but did not publish. Our identified hit (DSM43 (3); Fig. 1) was subsequently published by Genzyme36, but was not advanced into lead optimization. Herein we describe a structure-guided lead optimization program around the pyrrole scaffold leading to the identification of potent PfDHODH inhibitors with in vitro antimalarial activity and with good in vivo pharmacokinetic (PK) properties supporting in vivo efficacy in the SCID mouse model of P. falciparum malaria.
Results.
Chemistry and structure activity relationships (SAR) of the pyrrole series on DHODH and P. falciparum 3D7
The initial hit compound 3 was used as a starting point for a hit-to-lead optimization program to identify PfDHODH inhibitors that were differentiated from the current clinical candidate 1 (Fig. 1). Notably, our goals were to identify compounds that improved on the known liabilities of 1, including development of compounds with equivalent activity on P. falciparum and P. vivax parasites. We also sought to identify a series that maintained selectivity against human DHODH but also showed better selectivity versus the common toxicology species (e.g. rat, mouse and dog). While 1 is not a significant inhibitor of human DHODH, inhibition of the mouse and rat enzyme complicated its preclinical development.22 Finally, we also had the objective of identifying compounds with improved solubility to simplify formulation development. The initial hit 3 was a sub-micromolar inhibitor of both PfDHODH and P. falciparum 3D7 (Pf3D7) parasites in culture, it showed complete selectivity against human DHODH, and was more potent on P. vivax DHODH (PvDHODH) than PfDHODH (Table 1). These data suggested that the pyrrole series might meet our objective of improved P. vivax activity.
Table 1:
 | ||||||
|---|---|---|---|---|---|---|
| Cmpd | Cmpd ID | Ar | IC50 (μM) | EC50 (μM) | ||
| PfDHODH | PvDHODH | hDHODH | Pf3D7 cellsa | |||
| 1 | DSM265 | NA | 0.030 ±0.014 (12) | 0.072±0.028 (11) | >100 (4) | 0.0060 ± 0.0019 (9) |
| 2 | Genz-667348 * | NA | 0.022 | 0.042 | >30 | 0.007 |
| 3 | DSM43 |  |
0.27 ±0.092 (3) | 0.045 (0.04 – 0.050) | >100 (2) | 0.23, 0.25 |
| 4 | DSM483 | 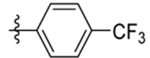 |
0.10±0.033 (3) | 0.014 (0.013–0.016) | >100 | 0.061, 0.071 |
| 5 | DSM484 | 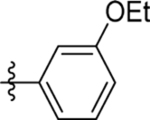 |
0.83 (0.74–0.92) | 0.021 (0.018–0.025) | >100 | 0.53 (0.28–0.78) |
| 6 | DSM485 | 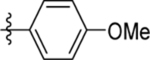 |
0.52 (0.46–0.60) | 0.055 (0.036–0.084) | >100 | 0.59 (0.47–0.75) |
| 7 | DSM486 | 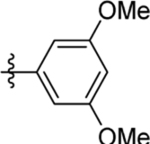 |
2.3 (2.0–2.7) | 0.47±0.065 (3) | >100 | 10 (6.7 – 15) |
| 8 | DSM487 | 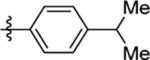 |
0.57 (0.46–0.71) | 0.040 (0.03–0.054) | >100 | 0.47 (0.2–1.1) |
| 9 | DSM491 | 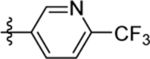 |
0.018 (0.016–0.020) | 0.0094 (0.0077–0.011) | >100 | 0.0092 (0.0076–0.011) |
| 10 | DSM520 | 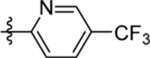 |
0.3 (0.25–0.37) | 0.06 (0.05–0.072) | >100 | 0.10 (0.090–0.11) |
| 11 | DSM490** | 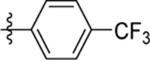 |
>100 | 16 (11–21) | nd | nd |
Pf, P. falciparum; Pv, P. vivax; h, human. For each experiment, triplicate data were collected at each inhibitor concentration. A 3-fold dilution series was used to determine IC50’s and either a 3-fold or 4-fold dilution series was used to determine the EC50’s. Values in parenthesis represent the 95% confidence interval. For key active compounds, multiple independent experiments were performed and for these compounds data represent mean ± standard deviation with the number of independent experiments in parenthesis. If only 2 biological replicates were collected than both values are shown.
data taken from28.
Compound has an N-methyl as shown in Scheme 1.
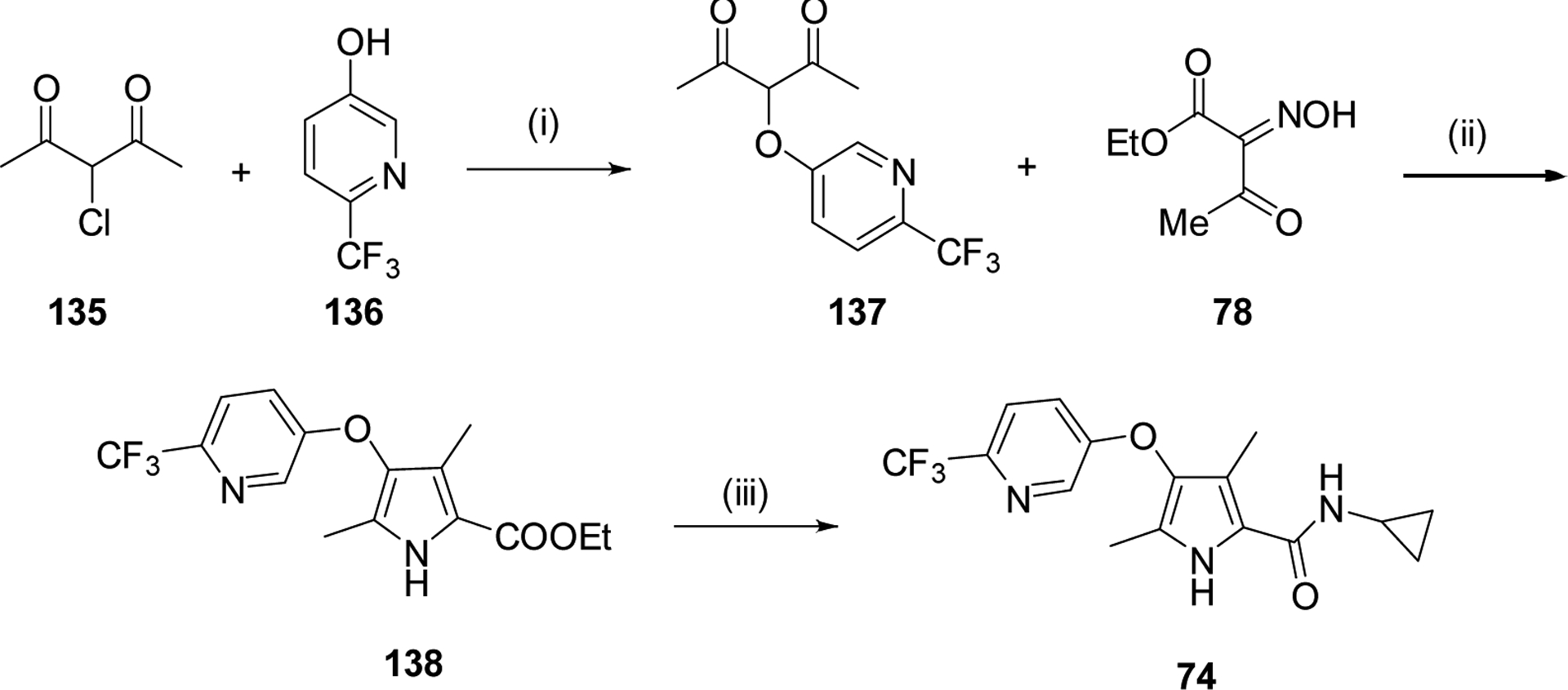
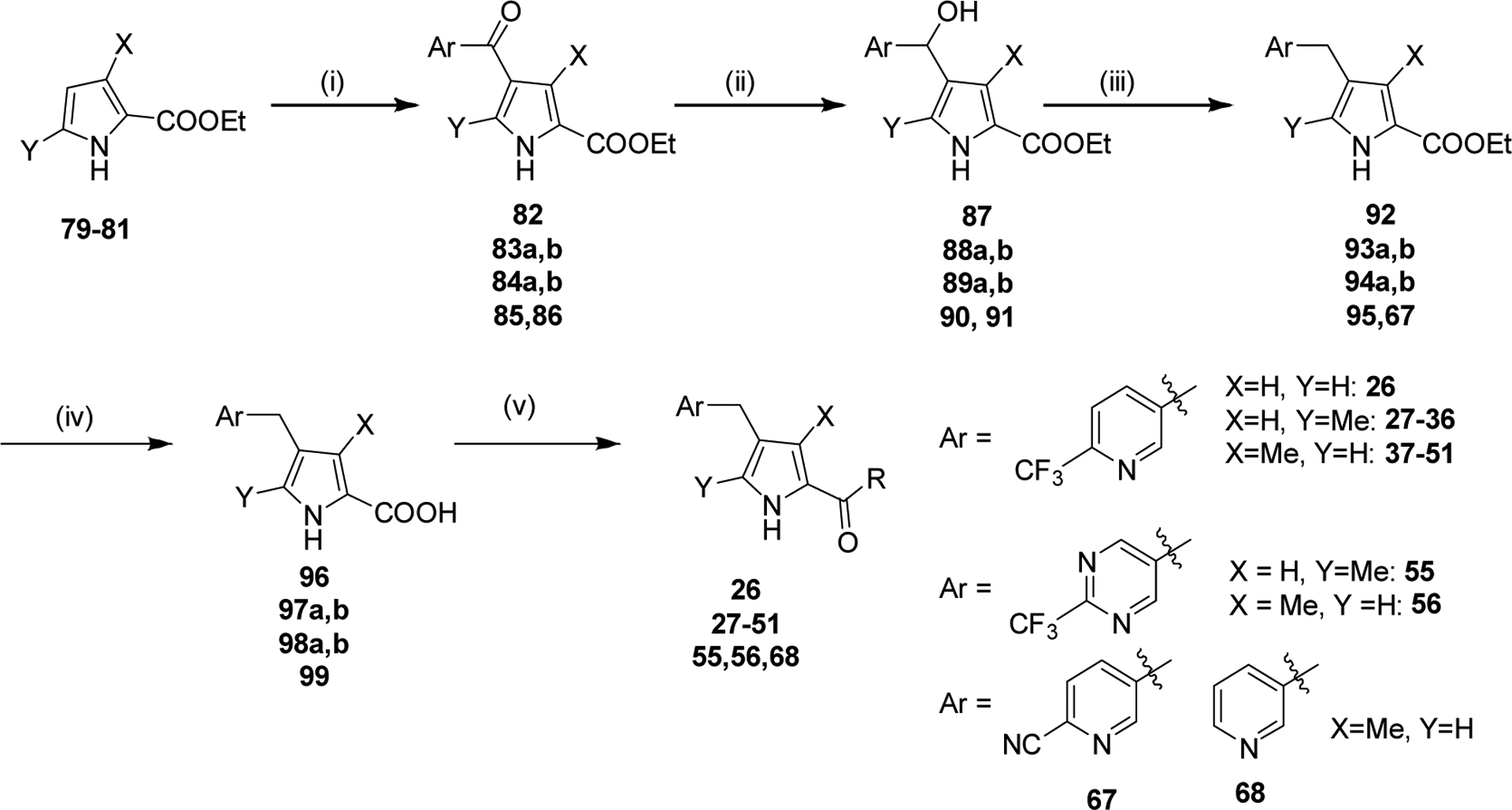
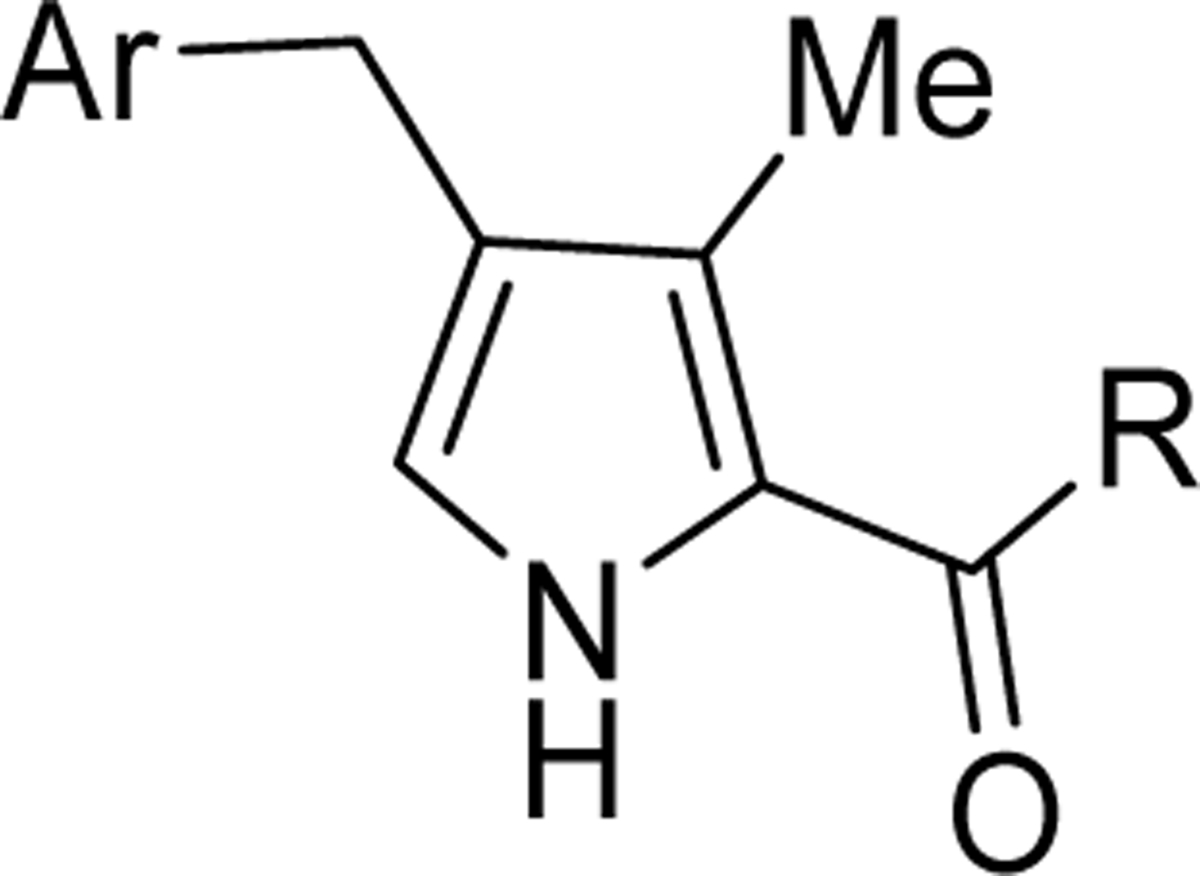
We systematically explored the SAR of the series by modifying each of the moieties attached to the pyrrole ring, including the aryl group attached at the 4 position, the two methyl groups attached at positions 3 and 5, and the ester group at position 2 (Table 1). The reported pyrrole derivatives 3–74 were synthesized as shown in schemes 1–6 and supplementary schemes 1–2. We utilized various protocols for the preparation of the pyrrole motif with different substitutions. Knorr condensation of α-amino β-keto ester with aryl substituted 2,4-pentanedione gave the aryl substituted dimethyl pyrroles (Scheme 1). Mono methyl substituted pyrroles were prepared by Friedel-Crafts acylation on the corresponding pyrrole followed by step wise reduction of the carbonyl group resulting in pyrrole ester intermediates (Scheme 2). 3-methoxymethyl and 3-hydroxymethyl substituted pyrroles were obtained via silver-catalyzed isocyanide-alkyne cycloaddition (Scheme 3).37 The 5-CF3 substituted pyrrole ester precursor 108 was prepared from the pentafluorophenyl ester 106, and subsequent iodination and trifluoromethylation38 afforded the precursor 108 leading to derivative 54 (Scheme 4). Friedel-Crafts arylation with some of the substituted aryl acid chlorides on the pyrrole proved difficult and gave poor yields. These phenyl substitutions were therefore prepared in a similar manner via the α,β alkynyl ketone as described in Scheme 3 starting from aryl aldehydes (Scheme 5).39 Similar reaction conditions were used to synthesize other pyrrole scaffolds as shown in Scheme 6 and supplementary schemes from the appropriate starting materials. Finally, hydrolysis of pyrrole esters obtained in the various protocols/methods followed by coupling with a variety of amines yielded pyrrole amides. Our initial analysis of the resultant compounds included determination of their inhibitory activity versus PfDHODH and PvDHODH, their selectivity versus human DHODH, and their potency against Pf3D7 parasites in whole cell assays (Tables 1–2, 4–7). Compounds that showed good potency and selectivity in these assays were then profiled further.
Scheme 1.
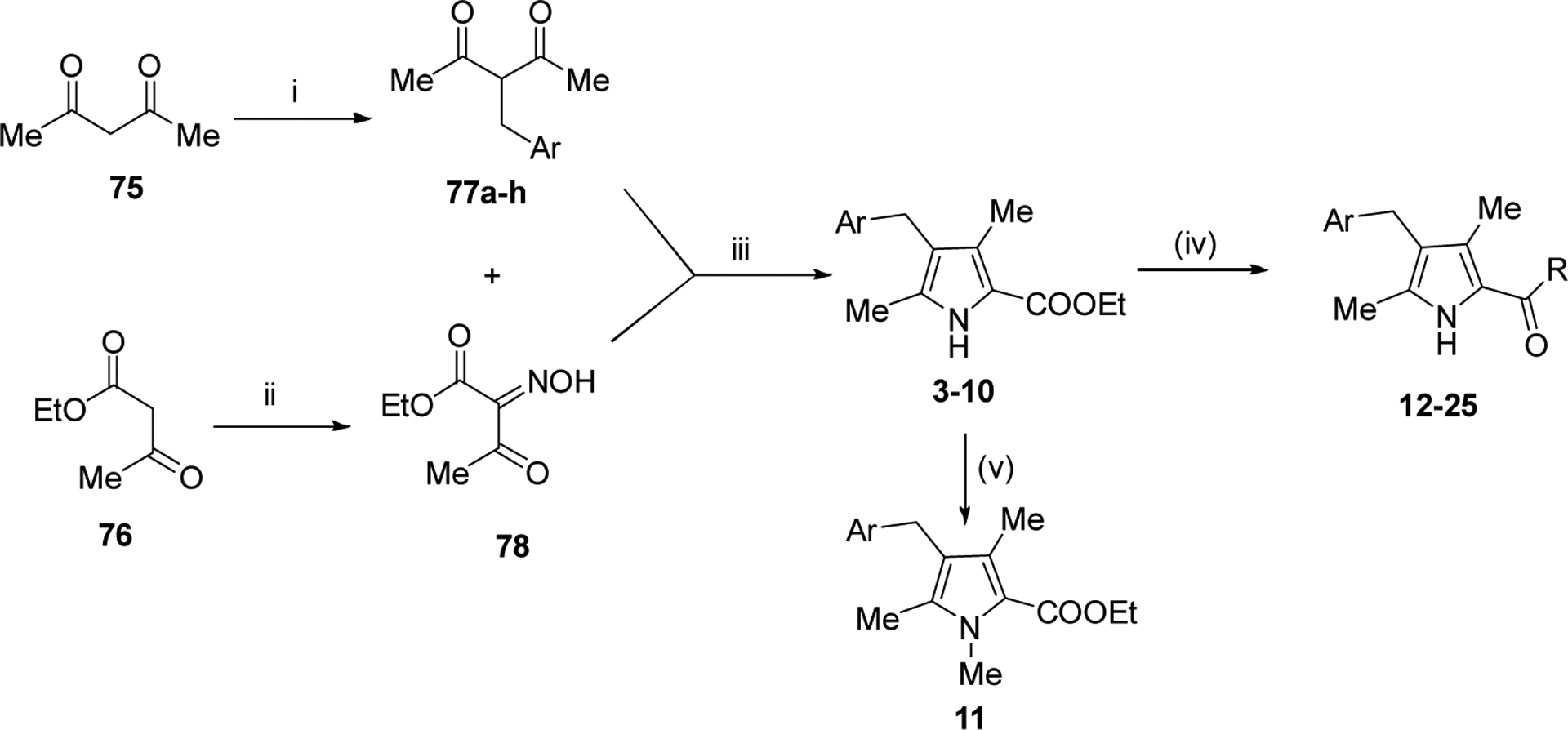
Reagents and conditions: (i) ArCH2Br, Acetone, K2CO3, 60 °C, 12h (ii) NaNO2 , AcOH, 0 °C- rt, 3h, (iii) Zn, AcOH, 80 °C, 6–8 h (iv) a) NaOH, EtOH, 80 °C, 2–4h b) EDC,TEA, DMF, RT, 4–6h (v) 4 -> 11, MeI, KOtBu, 18crown6, benzene, 6h
Scheme 6:
Reagents and conditions: (i) Cs2CO3, Acetone (ii) Zn, AcOH, reflux, 6h (iii) a) NaOH, EtOH-H2O, 80 °C; b) Amine, HATU, Et3N, CH2Cl2, 4h
Scheme 2.
Reagents and Conditions: (i) AlCl3, CH2Cl2, 0 °C-RT, 16 h (ii) NaBH4, EtOH, 1 h, RT (iii) TFA, triethylsilane, CH2Cl2, 85 °C, 1 h (iv) NaOH, H2O, MeOH, 80 °C, 2 h (vi) Cyclopropyl amine, HATU, TEA, CH2Cl2, RT, 4 h
Scheme 3.
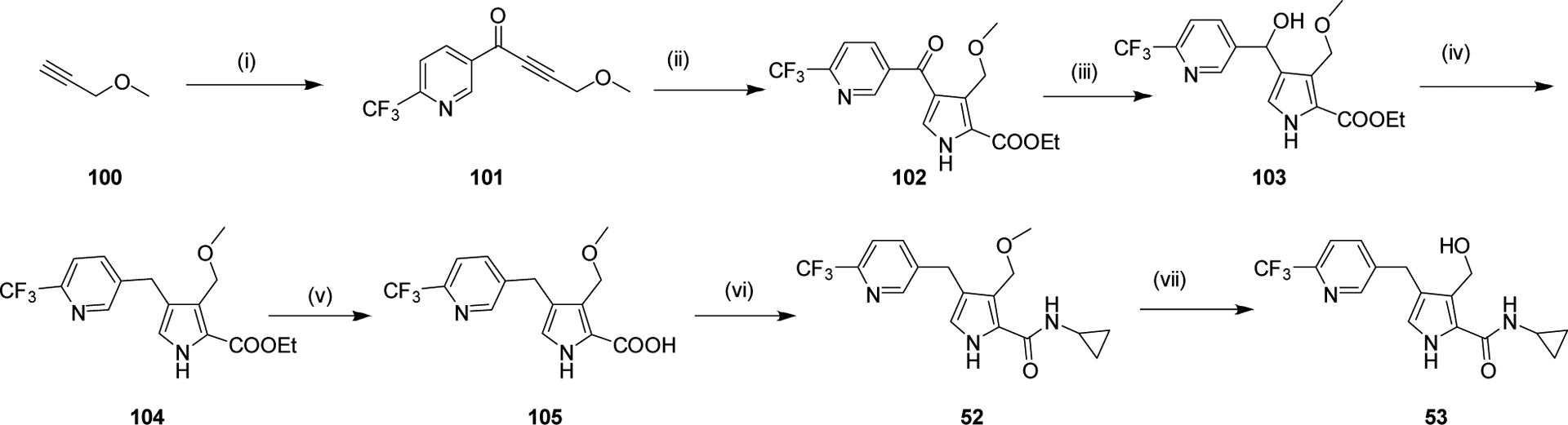
Reagents and Conditions: (i) N-methoxy-N-methyl-6-(trifluoromethyl)nicotinamide, n-BuLi, THF, −78 °C, 2 h (ii) Ethyl isocyanoacetate, Ag2CO3, NMP, 85 °C, 2 h (iii) NaBH4, EtOH, 0 °C-RT, 1 h (iv) TFA, triethylsilane, CH2Cl2, 85 °C, 1 h (v) NaOH, EtOH:H2O, 80 °C, 2 h (vi) Cyclopropyl amine, HATU, Et3N, CH2Cl2, 4–8 h. (vii) BBr3, CH2Cl2, 0 °C, 1h.
Scheme 4.
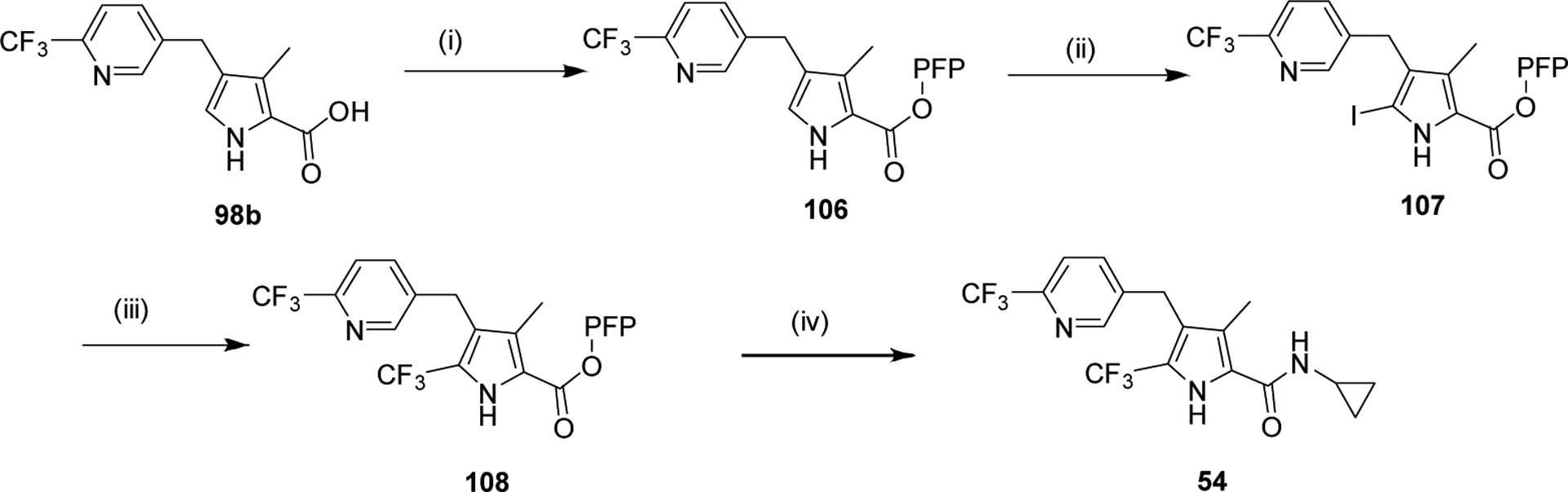
Reagents and Conditions: (i) Pentafluorophenol (PFP-OH), DCC, THF, RT, 1 h (ii) NIS, DMF, RT, 1 h (iii) Methyl 2,2-difluoro-2-(fluorosulfonyl)acetate, CuI, NMP, MW, 120 °C, 2 h (iv) THF, RT, 30 min
Scheme 5:
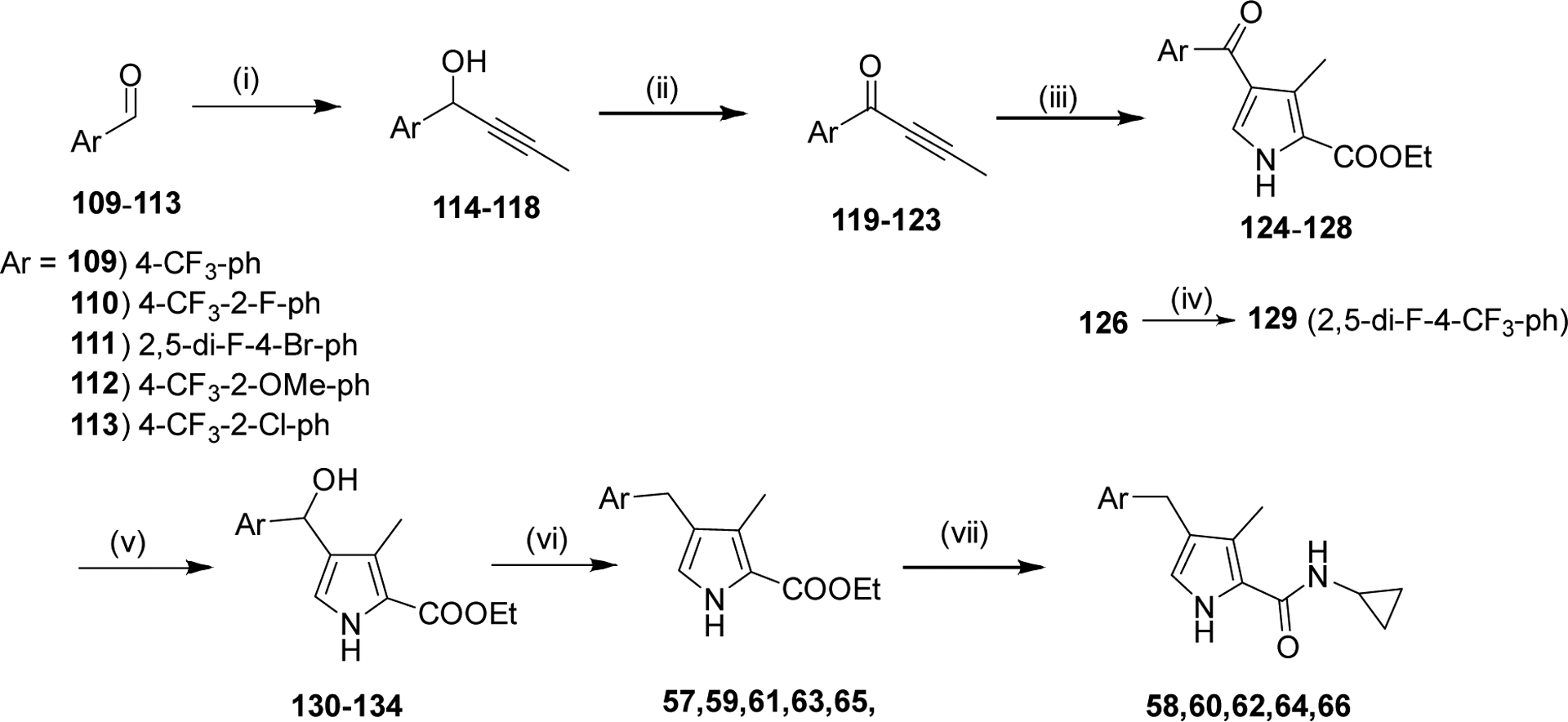
Reagents and Conditions: (i) 1-Propynylmagnesium bromide, THF, 0 °C-RT, 2 h (ii) Dess Martin, CH2Cl2, RT, 2 h (iii) Ethyl isocyanoacetate, Ag2CO3, NMP, 80 °C, 2 h (iv) methyl 2,2-difluoro-2-(fluorosulfonyl)acetate, KI, CuI, NMP, 120 °C, 16 h (v) NaBH4, EtOH, 0 °C-RT, 1 h (vi) TFA, triethylsilane, CH2Cl2, 85 °C, 1 h (vii) (a) NaOH, EtOH:H2O, 80 °C, 2 h (b) Amine, HATU, Et3N, CH2Cl2, 4–8 h.
Table 2.
Inhibitory activity on mammalian DHODHs
| Cmpd | hDHODH | mDHODH | rDHODH | dDHODH |
|---|---|---|---|---|
| Teriflunomide* | 0.30±0.052 (6) | 0.11, 0.14 | 0.017±0.0046 (3) | 0.20, 0.27 |
| 1 | >50 (10) | 2.1±0.62 (6) | 4.4±2.7 (8) | 14 ± 6.7 (5) |
| 4 | >100 (1) | >100 (1) | >100 (1) | >100 (1) |
| 6 | >100 (1) | >100 (1) | >100 (1) | >100 (1) |
| 9 | >100 (1) | >100 (1) | >100 (1) | >100 (1) |
| 37 | >100 (2) | >100 (2) | >100 (2) | >100 (2) |
| 45 | >100 (3) | >100 (1) | >100 (1) | >100 (1) |
| 60 | >100 (3) | nd | nd | nd |
h, human; m, mouse; r, rat; d, dog. Number of independent experiments in parenthesis. nd, not determined.
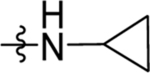
Human DHODH inhibitor teriflunomide ((Z)-2-cyano-3-hydroxy-N-(4-(trifluoromethyl)phenyl)but-2-enamide) is an approved drug for the treatment of rheumatoid arthritis and multiple sclerosis, data similar to previous reports40.
Table 4:
Replacement of the C2 pyrrole ester with amides
 | |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | Cmpd ID | Ar | R | IC50 (μM) | EC50 (μM) | ||
| PfDHODH | PvDHODH | hDHODH | Pf3D7 cellsa | ||||
| 12 | DSM489 |  |
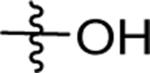 |
>100 | >100 | nd | >10 |
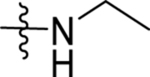
| 13 | DSM496 | 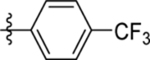 |
 |
0.14 (0.12–0.17) | 0.031 (0.023–0.041) | >100 | 0.051 (0.014–0.11) |
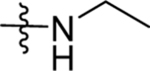
| 14 | DSM495 | 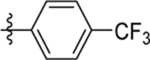 |
 |
0.3 (0.27–0.34) | 0.059 (0.05–0.069) | >100 | 0.11 (0.059–0.22) |
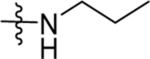
| 15 | DSM494 | 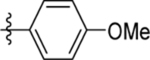 |
9.9±1.7 (3) | 1.8, 0.54 | >100 | 0.71 (0.57–0.90) | |
| 16 | DSM497 |  |
 |
0.054 (0.049–0.06) | 0.036 (0.03–0.043) | >100 | 0.033 (0.018–0.059) |
| 17 | DSM498 | 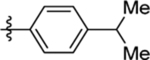 |
 |
0.072 (0.066–0.078) | 0.038 (0.034–0.044) | >100 | 0.047 (0.014–0.16) |
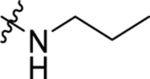
| 18 | DSM499 |  |
 |
0.050 ±0.013 (3) | 0.022 (0.019–0.026) | >100 | 0.036±0.0046 (3) |
| 19 | DSM500 | 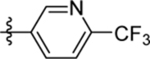 |
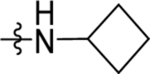 |
0.072, 0.042 | 0.036 (0.028–0.045) | >100 | 0.066 (0.057–0.077) |
| 20 | DSM508 | 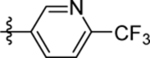 |
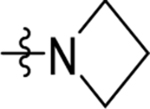 |
0.14±0.035 (3) | nd | nd | 0.13 ± 0.023 (3) |
| 21 | DSM509 | 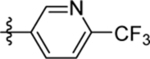 |
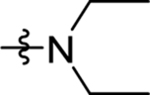 |
13 (11 – 15) | nd | nd | >10 |
| 22 | DSM517 | 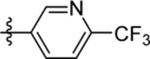 |
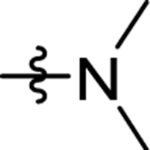 |
>30 | nd | nd | >10 |
| 23 | DSM519 | 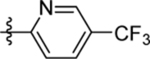 |
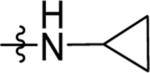 |
0.44 (0.38–0.49) | nd | nd | 0.34 (0.31–0.38) |
| 24 | DSM521 | 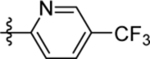 |
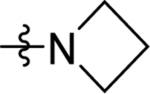 |
1.8 (1.5–2.0) | 0.6 (0.52–0.7) | >100 | 1.0 (0.99–1.1) |
| 25 | DSM522 | 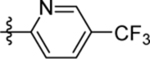 |
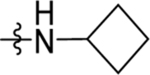 |
0.41 (0.26–0.65) | 0.22 (0.18–0.27) | >100 | 0.47, 0.68 |
See Table 1 footnote. nd, not determined.
Table 7:
Bridge modifications
| Cmpd | CmpdID | Structure | IC50 (μM) | EC50 (μM) | ||
|---|---|---|---|---|---|---|
| PfDHODH | PvDHODH | hDHODH | Pf3D7 cellsa | |||
| 69 | DSM504 | 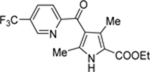 |
21 (17–25) | nd | nd | 6.5 (6.0–7.0) |
| 71 | DSM505 | 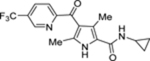 |
>100 (2) | nd | nd | >10, >20 |
| 73 | DSM558 | 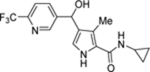 |
0.074 (0.059–0.092) | 0.23 (0.18–0.29) | >100 | 0.044 (0.038–0.052) |
| 74 | DSM537 | 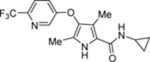 |
0.8 (0.68–0.94) | 0.16 (0.10–0.25) | >100 | 0.29 (0.24–0.36) |
See Table 1 footnote.
Optimization of the Aryl group.
To first further validate the series before beginning a full scale hit-to-lead program, a handful of additional ester analogs (4-11) were synthesized that explored different benzyl groups at the 4 position of the pyrrole with varied para, ortho and meta substituents (Scheme 1 and Tables 1 and 2). The data showed that para-substituted benzyl analogs yielded the most potent activity on both Plasmodium DHODHs and on Pf3D7. Substitution of the benzyl moiety with a para-CF3 benzyl (4) led to a 4-fold increase in potency against PfDHODH and Pf3D7 whereas replacement with para-CF3-pyridin-3-yl (9) led to a 15–25-fold improvement in enzyme and parasite activity over 3, and also to better activity than para-OMe (6) or para-iPr (8) (Table 1). Meta-substituted benzyl rings were less active (5, 7), as was the para-CF3-pyridin-2-yl analog (10). Methylation of the pyrrole nitrogen led to complete loss of activity (11). Both 4 and 9 retained good activity on PvDHODH while showing complete selectivity versus the human enzyme. We next tested a panel of mammalian DHODH enzymes from common species used in toxicologic studies (mouse, rat and dog), and again found that both 4 and 9 retained complete selectivity, while neither compound inhibited any of these mammalian enzymes up to the top concentration tested (100 μM) (Table 2). This contrasts with 1, which showed low to mid μM activity on the mouse, rat and dog enzymes while not significantly inhibiting hDHODH. Thus, these data confirmed that the pyrrole series could deliver both improved P. vivax DHODH activity and reduced mammalian DHODH inhibitory activity, meeting a key objective for a backup series for 1. Having met these objectives, we committed to a full scale hit-to-lead medicinal chemistry program on this series.
X-ray structure of 4 bound to PfDHODH.
To allow incorporation of a structure-based approach to optimize for good binding to PfDHODH, we solved the X-ray structure of the enzyme bound to 4 (Fig. 1) to a resolution of 1.78 A with an Rfree and Rwork of 0.194 and 0.151 respectively (Fig. 2 and Table 3). 4 bound adjacent to the flavin mononucleotide (FMN) cofactor in a binding pocket that overlaps the 1-binding pocket in the region between R265 and H185 (Fig. 2A), but which differs significantly in the position of the respective substituted phenyl rings (Fig. 2B). The different binding modes arise from the differing conformation of F188, which is rotated 180° to the “up” position in the 4 structure compared to the “down” position observed when bound to 1.22 The 4 binding configuration is similar to that observed for the thiophene-based inhibitor 228 (Fig. 1 and 2C). The pyrrole nitrogen of 4 forms a hydrogen bond with H185 (replacing the interaction with the bridging NH in 1), while the carbonyl forms an interaction with R265 (replacing the interaction with the pyridine nitrogen in 1). The ethyl ester linkage of 4 is bound in a hydrophobic pocket near the FMN cofactor formed by V532, I272, I263, E182 and G181, overlapping the binding site of the C2 -CF2CH3 of 1. The benzyl-CF3 of 4 forms interactions that are completely distinct from the aniline-SF5 of 1, as noted above. The 4 benzyl-binding pocket sits between the N-terminal α helices α1 and α2. These helices are composed of mostly hydrophobic amino acids that are thought to pack up against the mitochondrial membrane. The benzyl-CF3 of 4 instead makes close contacts (<5 Å) with L187, L191, C175, F171, Y168, F188 and M536.
Fig. 2.
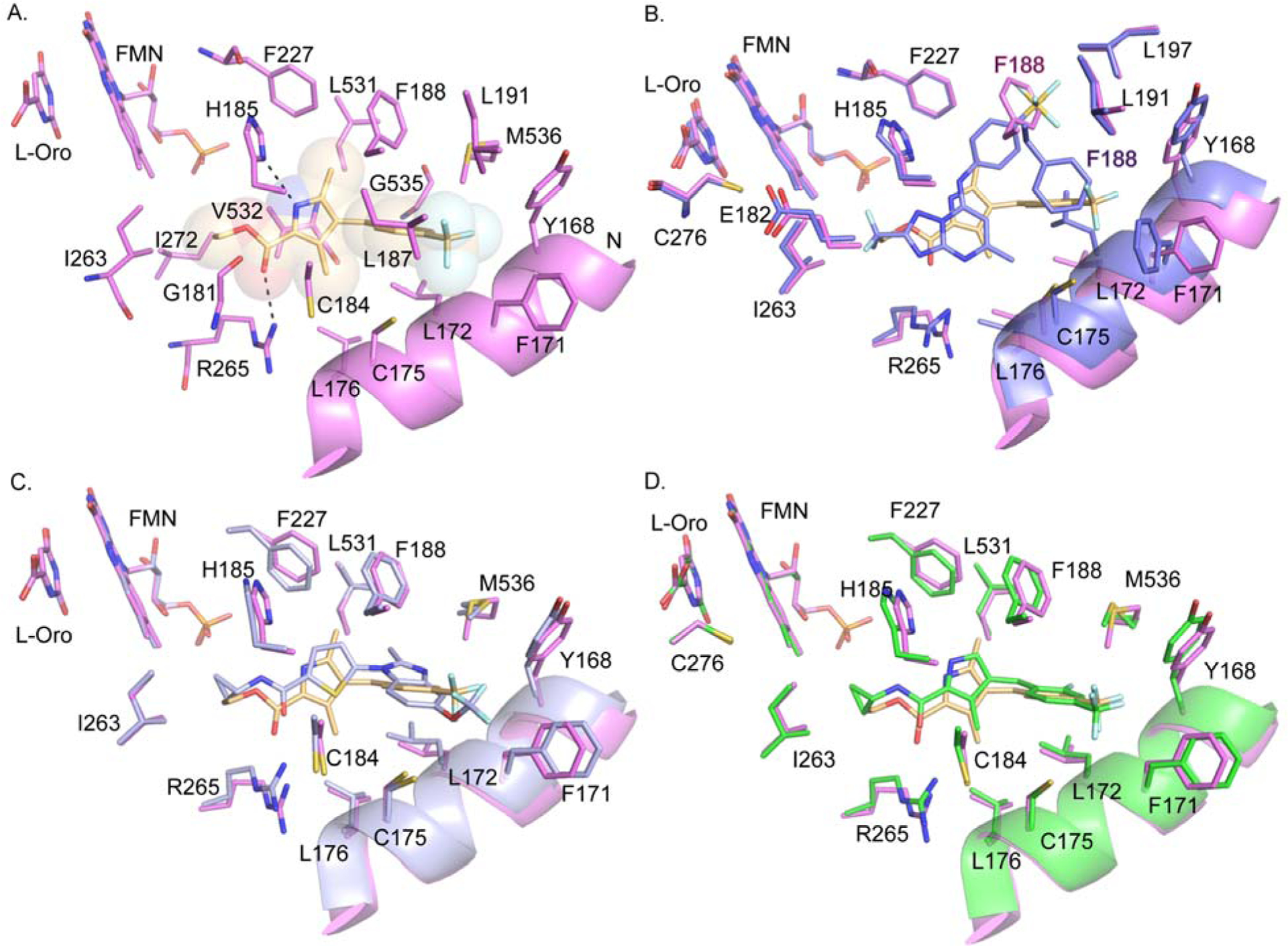
X-ray structures of PfDHODH bound to 4 and 60. A. 4 bound to PfDHODH showing amino acid residues within 4Å of the inhibitor. Dashed lines indicate H-bond interactions. Protein is displayed in pink and 4 is displayed in tan as sticks within shaded spheres. B. Comparison of the binding mode of 4 (colored as in A) with 1 (purple) (pdb 4RX0).22 C. Comparison of the binding mode of 4 (colored as in A) with 2 (light purple) (pdb 3O8A).28 D. Overlay of the binding modes of 4 (colored as in A) and 60 (green) showing residues within 4Å of the inhibitors. The coordinates for the structure of PfDHODH bound to 4 (pdb 6VTY) and 60 (pdb 6VTN) have been submitted to the Protein Data Bank.
Table 3.
PfDHODH co-inhibitor X-ray diffraction data collection and refinement statistics
| Data collection | ||
| Crystal | P/DHODHΔ384–413- 4 | P/DHODHΔ384–413- 60 |
| Space group | P212121 | P64 |
| Wavelength (Å) | 0.97924 | 0.97926 |
| Resolution (Å) | 42.03 – 1.78 (1.81–1.78) | 42.38 – 2.25 (2.29–2.25) |
| Unique reflections | 161,464 (8,016) | 26,640 (1,344) |
| Multiplicity | 9.8 (9.0) | 11.3 (10.2) |
| Data completeness (%) | 100.0 (100.0) | 100.0 (100.0) |
| Rmerge (%)a | 8.3 (100) | 5.1 (100) |
| Rpim (%)b | 3.0 (65.6) | 1.9 (71.9) |
| I/σ(I) | 26.2 (1.1) | 37.9 (1.0) |
| Wilson B-value (Å2) | 19.0 | 29.4 |
| Refinement | ||
| Resolution range (Å) | 42.02 – 1.78 (1.82–1.78) | 42.38 – 2.25 (2.33–2.25) |
| No. of reflections Rwork/Rfree | 148,953/2,002 (4,308/57) | 21,071/1,482 (453/31) |
| Data completeness (%) | 92.2 (38.3) | 79.2 (18.9) |
| No. of atoms/water | 12,086/897 | 2,958/72 |
| Rwork/Rfree(%) | 15.14/19.39 (23.85/30.16) | 20.24/21.87 (23.72/39.70) |
| R.m.s.d. bond length (Å) | 0.020 | 0.004 |
| R.m.s.d. bond angle (°) | 1.497 | 0.609 |
| Mean B (protein/ligand, Å2) | 28.9/20.4 | 45.4/23.3 |
| Ramachandran plot (%) (favored/additional)c | 97.96/2.04 | 98.12/1.82 |
Rmerge = 100 ΣhΣi|Ih,i— 〈Ih〉|/ΣhΣi 〈Ih,i〉, where the outer sum (h) is over the unique reflections and the inner sum (i) is over the set of independent observations of each unique reflection.
Rpim = 100 ΣhΣi [1/(nh - 1)]1/2|Ih,i— 〈Ih〉|/ΣhΣi 〈Ih,i〉, where nh is the number of observations of reflections h.
As defined by the validation suite MolProbity41
Replacement of ethyl ester with amides.
The ester linkage in compounds 3-11 is metabolically unstable and thus was replaced to advance the series towards in vivo activity. Based on the small size of the ethyl ester pocket (Fig. 2), as well as by analogy to previously identified moieties that bound well to this site in either the triazolopyrimidine (e.g. 1)21 or in the thiophene series (e.g. 3)28, a library of small cyclic and linear chain amides (12-25) were synthesized to replace the ester as described in Scheme 1 and Table 4. These modifications were made in the context of 4-CF3-benzyl, 4-CF3-3-pyridinyl and 4-CF3-2-pyridinyl groups at C4 of the pyrrole ring. The acid 12 was inactive, but several amide replacements were well tolerated. Within a series, PfDHODH inhibitory activity was best for the smaller substituents with ethyl amines 13 and 16 showing better activity than propyl amines 14 and 17 and with activity dropping off significantly for butyl amine 15. Cyclopropyl amine 18 showed the best potency of the tested derivatives with cyclobutyl amine 19 a close second. Azetidines 20 and 24 were tolerated with a 5-fold reduction in activity compared to the cyclopropyl amine 18, but dimethyl 22 or diethyl 21 amines showed poor to no activity. Activity was best for 4-CF3-3-pyridinyl, with the order of preference 4-CF3-3-pyridinyl 16–19 > 4-CF3-2-pyridinyl 23–25> 4-CF3-benzyl 13–14. Activity against PvDHODH and against Pf3D7 parasites paralleled the activity observed for PfDHODH, with the best compounds 16 and 18 showing sub-50 nM activity on all three (Table 4). Complete species selectivity was also maintained in the amide series for all tested compounds, with no observed inhibition of mammalian enzymes (Table 4).
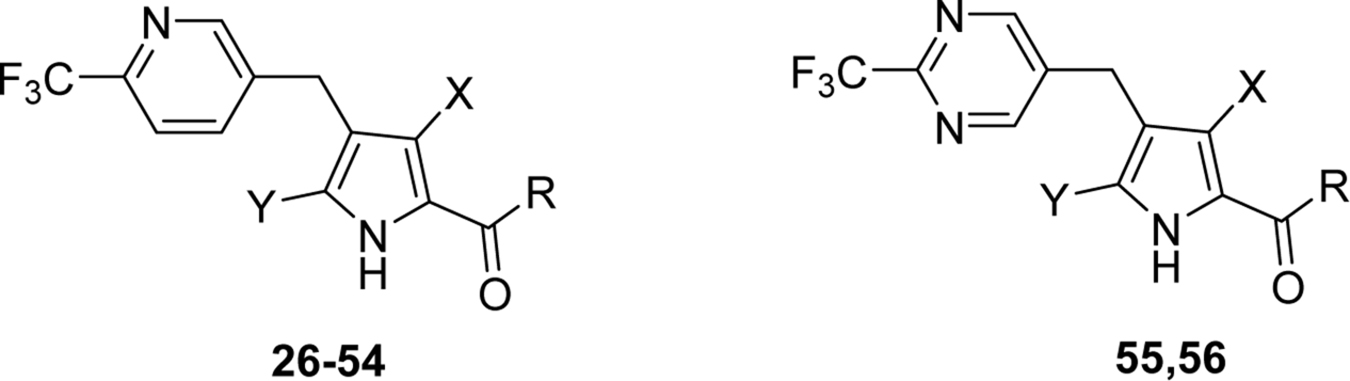
Replacement and modification of the pyrrole ring methyl substituents.
The importance of the methyl substituents on the pyrrole ring was next assessed by either removal or by replacement with alcohols, methoxy ether, or CF3 and these variations were made within the context of a wider range of amides compared to those in Table 4. Pyrrole derivatives (26–55) were synthesized by Friedel-Crafts acetylation of the corresponding pyrrole with aryl acid chloride in the presence of Lewis acid (AlCl3) or via the alkynyl ketone (Schemes 2–4, Supporting Information Scheme S1 and Table 5). Removal of both pyrrole methyl groups 26 led to a 50-fold drop in potency against the Plasmodium enzymes and Pf3D7 when compared to the matched cyclopropyl compound 18 containing both methyls (Tables 4 and 5). Methyl at C5 was detrimental to activity, leading to a further 4-fold drop in PfDHODH activity relative to the compound lacking both methyls (27 vs 26). The best activity was observed for compounds containing methyl at C3 (37–41, 43–45, 47, 49, 51) in the absence of the C5 methyl, with those linked to cyclopropyl 37 or cyclobutyl amine 51 showing the highest potency on Plasmodium DHODH and Pf3D7 (Table 5). Amides linked to carbon rings with >4 atoms showed poor to no activity (28, 29, 32, 33, 34, 42) regardless of the position of the pyrrole methyl. Branched chain alkyl groups including dimethyl 40, tert-butyl 41, -CH2CF3 39 and azetidine 38, including rings with fluorine substitutions (45 and 47) all provided sub-100 nM activity, with 45 showing similar activity to the cyclobutyl analog 51. Fluorination of cyclobutyl amine led to reduced activity versus 51 with the difluoro analog 48 showing less activity than the monofluoro 49 analog. Substitution of the C3 methyl with an alcohol 53 or ether 52 led to a 200-fold and 20-fold reduction in potency, respectively, against PfDHODH relative to 37. Within the context of Me at C3, replacement of the C5 methyl with CF3 54 reduced activity 80-fold, as did analogs of 27 and 37 with pyrimidinyl-CF3 (55 and 56) capping the pyrrole 4 position. A strong correlation between potency on PfDHODH and Pf3D7 was observed for this set, and equivalent to better potency was maintained for these compounds on PvDHODH. No inhibition of the human enzyme was observed for any of the tested compounds.
Table 5:
Replacement or modification of the pyrrole ring methyl groups
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | Cmpd ID | X | Y | R | IC50 (μM) | EC50 (μM) | ||
| PfDHODH | PvDHODH | hDHODH | Pf3D7 cellsa | |||||
| 26 | DSM501 | H | H | 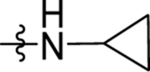 |
2.5 (1.9–3.2) | 1.2 (0.94–1.5) | >100 | 1.7, 0.50 |
| 27 | DSM503 | H | Me | 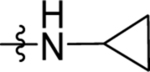 |
10 ± 0.96 (3) | 2.3 (1.5–3.6) | >100 | 3.6 (3.1–4.2) |
| 28 | DSM506 | H | Me | 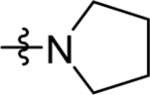 |
>100 (2) | nd | nd | >10 |
| 29 | DSM507 | H | Me | 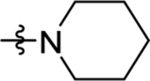 |
>100 | nd | nd | >20 |
| 30 | DSM511 | H | Me | 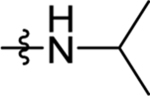 |
2.8 (2.1–3.8) | nd | nd | 1.9 (1.2–3.0) |
| 31 | DSM512 | H | Me | 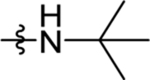 |
5.7 (4.4–7.4) | nd | nd | 3.5 (2.5 – 4.9) |
| 32 | DSM513 | H | Me | 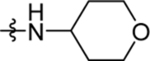 |
>100 | nd | nd | >10 |
| 33 | DSM514 | H | Me | >100 | nd | nd | >10 | |
| 34 | DSM515 | H | Me | >100 | nd | nd | >10 | |
| 35 | DSM518 | H | Me | 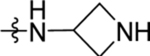 |
>100 | nd | nd | >20 |
| 36 | DSM524 | H | Me | >100 | nd | nd | >20 | |
| 37 | DSM502 | Me | H | 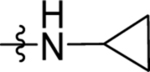 |
0.020 ± 0.0054 (7) | 0.014 ± 0.0046 (3) | >100 (3) | 0.014 ± 0.0055 (6) |
| 38 | DSM525 | Me | H | 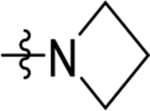 |
0.17 (0.15–0.2) | 0.21 (0.17–0.26) | >100 | 0.082 (0.073–0.093) |
| 39 | DSM528 | Me | H | 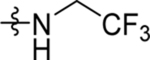 |
0.21 (0.16–0.27) | 0.051 (0.043–0.061) | >100 | 0.044, 0.059 |
| 40 | DSM529 | Me | H | 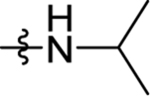 |
0.080 (0.062–0.1) | 0.018 (0.015–0.021) | >100 | 0.013 (0.011–0.016) |
| 41 | DSM530 | Me | H | 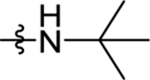 |
0.11 (0.084–0.15) | 0.064 (0.051–0.082) | >100 | 0.051 (0.029–0.090) |
| 42 | DSM531 | Me | H | 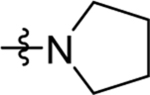 |
9.8 (7.4–12.8) | nd | nd | 4.8 (3.5 – 6.5) |
| 43 | DSM532 | Me | H | 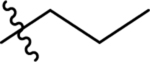 |
0.098 (0.077–0.12) | 0.041 (0.034–0.049) | >100 | 0.065 ± 0.029 (4) |
| 44 | DSM536 | Me | H | 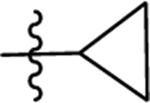 |
0.018±0.0021 (3) | 0.0075± 0.0031 (3) | >100 (3) | 0.0099 ±0.0020 (3) |
| 45 | DSM538 | Me | H | 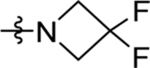 |
0.033±0.0079 (3) | 0.018 ±0.0025 (3) | >100 (3) | 0.029 ± 0.0079 (4) |
| 46 | DSM539 | Me | H | 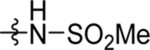 |
>100 | nd | nd | >100 |
| 47 | DSM546 | Me | H | 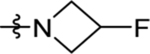 |
0.12 (0.11–0.14) | 0.2 (0.17–0.25) | >100 | 0.051 (0.036–0.072) |
| 48 | DSM552 | Me | H | 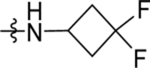 |
3.6 (4.0–5.4) | nd | nd | 1.3 (0.86–2.0) |
| 49 | DSM553 | Me | H | 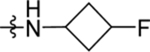 |
0.32 (0.40 – 0.90) | nd | nd | 0.17 (0.16–0.19) |
| 50 | DSM559 | Me | H | 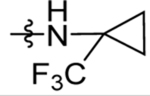 |
9.3 (6.3–12) | nd | nd | 3.9 (3.3 – 6.9) |
| 51 | DSM560 | Me | H | 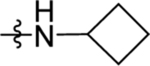 |
0.044 ±0.19 (3) | 0.019 ± 0.0058 (3) | >100 (3) | 0.027 (0.023–0.032) |
| 52 | DSM534 | H | 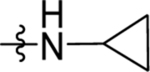 |
0.45 (0.34–0.6) | 0.07 (0.05–0.096) | >100 | 0.24±0.046 (3) | |
| 53 | DSM535 | 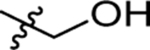 |
H | 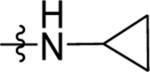 |
4.3 (2.5–7.4) | nd | nd | 1.2±0.64 (3) |
| 54 | DSM526 | Me | CF3 | 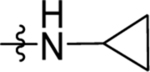 |
1.6 (1.4–1.9) | nd | nd | 0.77 (0.60 – 1.0) |
| 55 | DSM527 | H | Me | 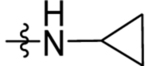 |
>100 (2) | nd | nd | >10 |
| 56 | DSM533 | Me | H | 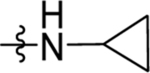 |
0.88 (0.78–1.0) | 0.21 (0.17–0.26) | >100 | 0.39±0.085 (3) |
See Table 1 footnote. nd, not determined
Modification of the 4-position benzylic substitutions.
To further explore the potential for the benzyl group to yield potent compounds we synthesized additional analogs (57 – 68) in the context of either cyclopropyl amide or ethyl ester (Scheme 5 and Table 6). 4-CF3-benzyl (57, 58), or this ring with a single ortho fluoro (59, 60) or ortho chloro (65, 66) all showed similar activity to 4-CF3-pyridinyl 37, with the cyclopropyl amide showing better activity than the ethyl ester. There was a significant drop in activity upon adding a second ortho-fluoro to the ring (61, 62), with this substitution leading to complete loss of activity for the ethyl ester. Addition of a single ortho O-methyl to the 4-CF3 benzyl ring led to ~10-fold drop in potency (63, 64). Replacement of the 4-CF3 with CN in the context of the pyridinyl ring 67 led to a potency drop of 30-fold, whereas removal of the 4-CF3 led to complete loss in activity (68).
Table 6:
Modification of the 4-position benzylic substitutions
 | |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | Cmpd ID | Ar | R | IC50 (μM) | EC50 (μM) | ||
| PfDHODH | PvDHODH | hDHODH | Pf3D7 cellsa | ||||
| 57 | DSM561 | 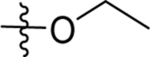 |
0.13 (0.11–0.15) | 0.019 (0.014–0.027) | >100 | 0.060 (0.05–0.07) | |
| 58 | DSM562 | 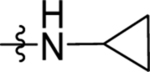 |
0.048 ±0.019 (3) | 0.0067 ±0.017 (3) | >100 (3) | 0.018 ±0.0038 (3) | |
| 59 | DSM556 | 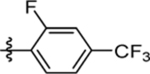 |
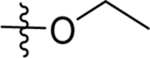 |
0.035 (0.029–0.043) | 0.024 (0.018–0.03) | >100 | 0.028 (0.024–0.031) |
| 60 | DSM557 | 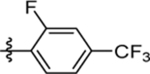 |
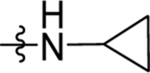 |
0.044 ±0.19 (3) | 0.015 ± 0.0080 (3) | >100 (3) | 0.016 ±0.0056 (5) |
| 61 | DSM563 | 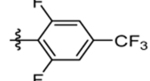 |
 |
>30 | nd | nd | >10 |
| 62 | DSM564 | 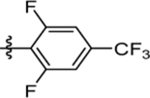 |
 |
1.2 (1.1–1.4) | nd | nd | 0.42 (0.26–0.67) |
| 63 | DSM566 | 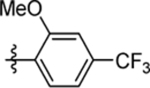 |
 |
1.1 (0.66–1.8) | nd | nd | 0.81 (0.4–1.6) |
| 64 | DSM567 | 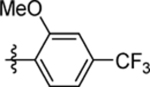 |
 |
0.11 (0.093–0.14) | 0.092 (0.064–0.13) | >100 | 0.26 (0.18–0.39) |
| 65 | DSM570 | 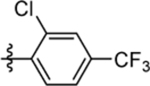 |
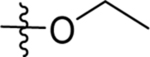 |
0.042 (0.036–0.048) | 0.059 (0.049–0.072) | >100 | 0.029 (0.025–0.033) |
| 66 | DSM571 | 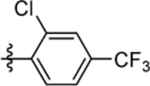 |
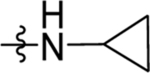 |
0.017 ±0.0061 (3) | 0.014 ±0.0050 (3) | >100 (3) | 0.020 ± 0.00086 (5) |
| 67 | DSM584 | 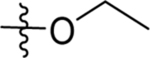 |
0.63 (0.51–0.77) | nd | nd | 0.20± 0.017 (3) | |
| 68 | DSM585 | 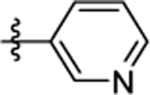 |
 |
>100 | nd | nd | >10 |
See Table 1 footnote. nd, not determined.
Comparison of the X-ray structure of PfDHODH bound to cyclopropyl amide 60 versus ethylester 4.
Having identified several potent pyrrole analogs containing amide replacements for the original ester, we solved the X-ray structure of PfDHODH bound to cyclopropyl amide derivative 60 (Fig. 1 and Table 6). The structure of PfDHODH bound to 60 was solved to 2.3 Å in a space group of P64 with the cell dimension of a=b=84.8, c=138.0 and one molecule of PfDHODH in the asymmetric unit binding modes (Table 3 and Fig. 2D). The structures of PfDHODH bound to 60 superimposed with the 4-bound structure with an RMSD = 0.26 over 1199 atoms. The positions of 60 and 4 in the inhibitor binding site were similar, although 60 formed an additional H-bond between the amide NH and H185 and as a consequence 60 was shifted closer to H185 than 4 (Fig. 2D). H-bonds between the pyrrole NH and H185 and the carbonyl oxygen and R265 were observed for both 60 and 4. Thus interestingly for 60, H185 is involved in a bifurcated H-bonding interaction. Differences in the binding mode between the ester and the amide may explain why benzyl modifications had differential impact depending on which was present (Table 6).
Replacement or modification of the bridging methyl.
Finally, the impact of replacing or modifying the bridging methyl was explored (Schemes 6 and S2; Table 7). Oxidation to a ketone (69 and 71) led to almost complete loss of activity, whereas replacement with oxygen (74) led to a ~40-fold drop in potency against PfDHODH. In contrast, modification of the bridging carbon with hydroxyl (73) caused only a 4-fold drop in potency, demonstrating that hydroxyl modification of this position is tolerated. Additionally since 73 is racemic, it is likely that the active enantiomer will be 2-fold more potent than what is observed for the racemic mixture. Attempts to replace the bridging carbon with NH were made but this compound was not synthetically feasible in our hands.
SAR summary.
We identified a number of compounds (37, 45, 51, 60 and 66) with sub-30 nM potency on PfDHODH and Pf3D7 parasites that were of interest for potential follow up. Overall, we observed a strong correlation between PfDHODH and activity on Pf3D7 throughout the series and across the full dose response range of measured values (Fig. 3). Good activity on PvDHODH was also observed, with a tendency for greater potency on PvDHODH than PfDHODH. No inhibition of the mammalian enzymes was observed for compounds in the series.
Fig. 3.
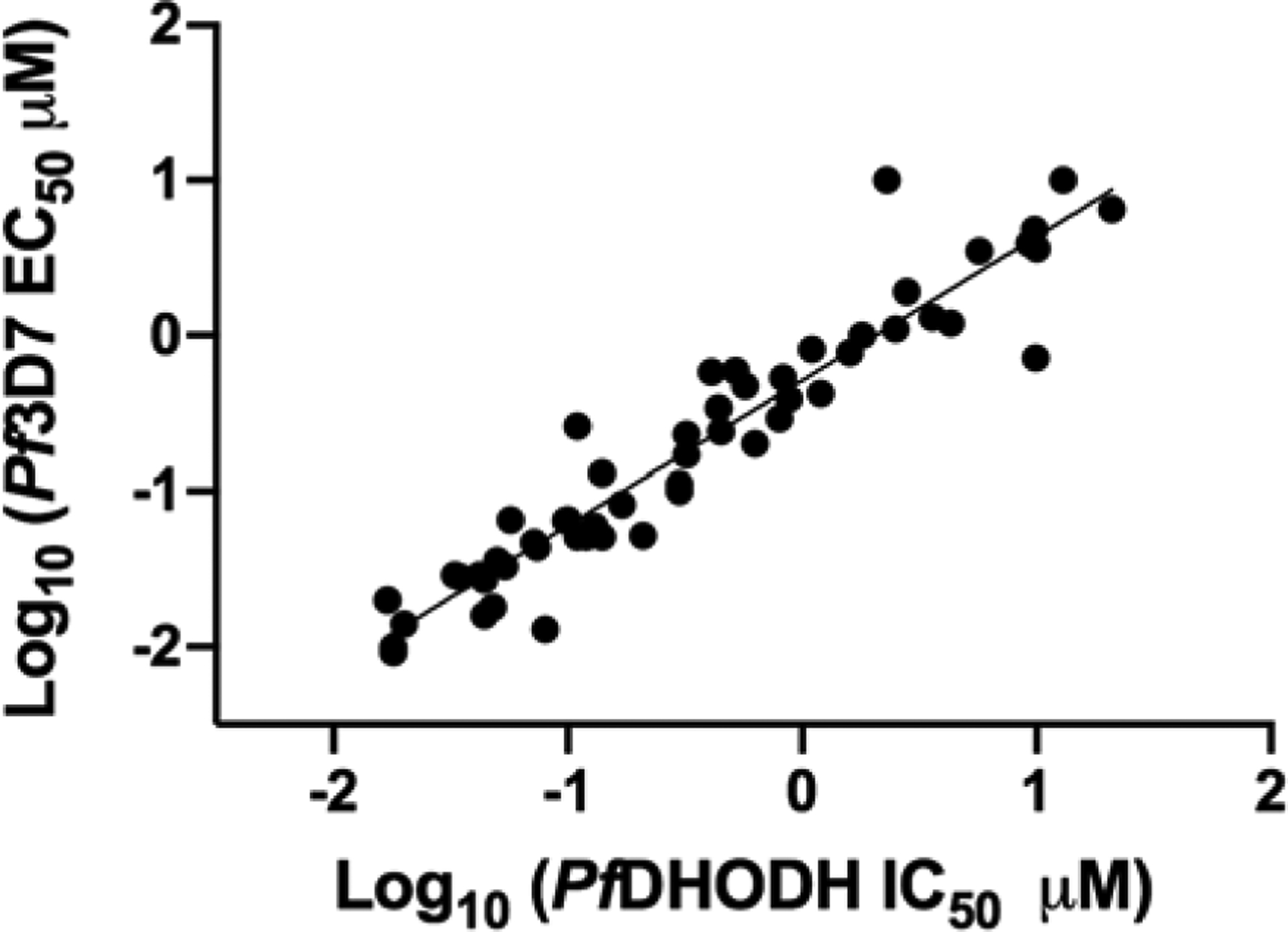
SAR correlation between inhibitory activity on PfDHODH and Pf3D7 parasites. Data are taken from Tables 1, 3–6. For compounds where either the PfDHODH IC50 or Pf3D7 EC50 was greater than the top concentration tested in the study (e.g. > 100 μM) were not included.
In vitro ADME.
A subset of compounds was evaluated to assess their physicochemical properties and to determine their propensity to be metabolized using in vitro human and mouse liver microsome assays (Table 8). Compounds were typically selected based on having demonstrated good potency in the PfDHODH and Pf3D7 assays, however some compounds were chosen to explore the SAR around this subset of ADME properties. The Log D7.4 for compounds containing an amide was generally in good physicochemical space (range 1.7 to 4.1). Kinetic solubility (phosphate buffered saline pH 6.5) ranged from poor to quite good, with compounds that contained an azetidine attached to the carbonyl bond showing the best solubility (20, 38, 45 and 47). The azetidine contains a tertiary nitrogen that is unable to act as a H-bond donor, in contrast to the amide secondary nitrogen observed in most of the other non-ester compounds. Thus, the poorer solubility of the amides may potentially result from intramolecular H-bonding within the crystal lattice. Within the amide series, compounds containing a cyclopropyl amide (27, 37, 55 and 71) were more soluble than those with other amide substituents, while replacement of the pyridinyl-CF3 with a pyrimidinyl-CF3 improved solubility (55 vs 27). Compounds with benzyl-CF3 were predictably less soluble (58 vs 37), and addition of an ortho-chloro led to further reductions (66). Variation of the number of methyl groups on the pyrrole ring (0–2), had little impact on solubility although those with a single methyl were improved over the others (27 and 37 vs 18 and 26).
Table 8:
Physicochemical properties, in vitro metabolism.
| Compd | Log DpH 7.4 | Kinetic solubility range in pH 6.5 PBS (μg/mL) | cPPBa (% bound) | CLint (H/M)b (μL/min/mg protein) |
|---|---|---|---|---|
| 1 | 3.6 | 12.5–25 | 97.4 | <7/8 |
| 3 | 4.5 | <1.6 | nd | 188c/470c |
| 9 | 3.7 | 1.6–3.1 | 93.6 | 286/779 |
| 16 | 2.8 | 12.5–25 | 77.1 | 70/54 |
| 17 | 3.2 | 6.3–12.5 | 84.5 | 172/64 |
| 18 | 3 | 12.5–25 | 82.8 | 15/33 |
| 20 | 2.9 | >50 | 77.2 | 31/53 |
| 26 | 2.6 | 12.5–25 | 66.7 | 31/96 |
| 27 | 2.8 | 25–50 | 72.6 | 36/41 |
| 28 | 3.2 | 6.3–12.5 | 81.5 | 226/138 |
| 29 | 3.5 | 12.5–25 | 87.5 | 173/93 |
| 37 | 2.8 | 25–50 | 76.2 | 10/24 |
| 38 | 2.7 | >100 | 65.9 | <7/192 |
| 39 | 3.3 | 12.5–25 | 81.9 | 15/57 |
| 45 | 3.3 | 25–50 | 74.9 | 21/94 |
| 47 | cnd | 25–50 | cnd | 7/145 |
| 51 | 3.2 | 12.5–25 | 83.5 | 29/204 |
| 55 | 2.5 | 50–100 | 44.5 | <7/8 |
| 56 | 2.5 | 12.5–25 | nd | <7/nd |
| 58 | 3.7 | 3.1–6.3 | 97.1 | 19/173 |
| 60 | 3.8 | 1.6–3.1 | 97 | 15/184 |
| 66 | 4.1 | <1.6 | 98.1 | 14/264 |
| 68 | 2.6 | 12.5–25 | 73.2 | 19/138 |
| 71 | 2.4 | 25–50 | 50.4 | 8/24 |
| 73 | 3.2 | 6.3–12.5 | 89.5 | 20/40 |
| 74 | 1.7 | 12.5–25 | 42.7 | 8/22 |
Binding estimated using a chromatographic method with an albumin-based column as described.21
Human (H) or mouse (M) liver microsomes
Measurable loss of 3 in control incubations without cofactor suggesting the contribution of non-NADPH mediated metabolism to overall degradation rate.
cnd, could not determine; nd, not determined
Metabolic stability ranged from good to poor across the series, but for many compounds species differences were also observed between human and mouse microsomes (Table 8). Cyclopropyl amides with either para-CF3-pyridine or para-CF3-pyrimidine (18, 27, 37, 55, 56, 71, 73 and 74) showed the best stability with compounds containing pyrimidine performing best (55 and 56). The difluoroazetidine 38 showed excellent stability in human liver microsomes but was much more rapidly metabolized in mouse microsomes, and this was likewise a problem for three potentially interesting compounds with good potency 51, 60 and 66. This issue would likely make development of these compounds difficult as it suggests that there will be poor plasma exposure in the mouse efficacy model. Replacement or modification of the bridging carbon did not significantly alter stability, e.g. hydroxylation (73 vs 37), oxidation to a carbonyl (71 vs 18), or replacement with oxygen (74 vs 18). Not surprisingly esters (3 and 9) showed poor solubility and were very rapidly metabolized.
The estimated extent of protein binding (using an albumin-based column and chromatographic method) ranged from ~43% (74) to 98% (66) and the extent of binding strongly correlated with Log D across the series.
Identification of 37 as a lead compound.
Taking into consideration both the potency data against Pf3D7 parasites and key parasite enzymes (PfDHODH and PvDHODH), and the in vitro pharmacology, we selected 37 for additional biological, ADME and pharmacokinetic profiling. It was the only compound with sufficiently strong properties across these criteria to suggest it could meet development criteria.
Additional Parasitology Profiling of 37.
As 37 showed good potency against both Pf3D7 and the Plasmodium enzymes (Table 5), we undertook additional parasitology studies to further define its profile. Good activity was also observed for P. falciparum Dd2, which is an isolate that is resistant to multiple clinically used drugs (EC50 0.016 vs 0.014 μM on Dd2 vs 3D7, respectively) (Tables 5 and 9), thus demonstrating that like other DHODH inhibitors (e.g. 1), 37 is not cross resistant with 8-aminoquinolines or DHFR inhibitors. To provide further demonstration that cell killing occurs through DHODH inhibition we tested 37 against a P. falciparum strain that has been transformed with yeast DHODH and is resistant to both P. falciparum DHODH and cytochrome bc1 inhibitors.42, 43 Inclusion of proguanil in this study further allowed us to distinguish between bc1 and DHODH inhibitors, as resistance of the yeast DHODH strain to bc1 inhibitors is rescued in the presence of proguanil. As expected for a DHODH inhibitor, and like 1, the yeast DHODH strain was resistant to 37, and this resistance was not rescued by proguanil consistent with DHODH inhibition as the mechanism of parasite killing (Table 9).
Table 9.
Activity of 37 on drug resistant P. falciparum and mammalian cell lines
| P. falciparum Strain | Resistance Profile | Selection Agent | EC50 37 (μM) | Fold change | EC50 1 (μM) | Fold- change |
|---|---|---|---|---|---|---|
| Dd2 (parental) | CQ, Py, SD, MF, CG | na | 0.016±0.0070(6) | NA | 0.0048, 0.0046# | NA |
| D10 w/yeast DHODH | Genetic insertion | na | >20 ±PG* | >100 | >20 ±PG* | >100 |
| 1D3 | 3 copies DHODH | 1 | 0.083 ± 0.028 (3) | 5.2 | 0.023 ± 0.0036 (3) | 4.8 |
| R10C1B | G181C DHODH | 1 | 0.024±0.018 (3) | 1.5 | 0.12, 0.15# | 25 |
| R1BC1A | L531F DHODH | 1 | 0.013 ± 0.0069 (3) | 0.81 | 0.080, 0.11# | 17 |
| R1AC1B | C276F DHODH | 1 | 0.089±0.071 (3) | 5.6 | 0.17, 0.17# | 35 |
| R2B | R265G DHODH | 1 | 0.078 ±0.024 (3) | 4.9 | 0.074, 0.15# | 15 |
| Mammalian Cell line (CC50 μM) | ||||||
| Human HepG2 | na | na | >50 | na | >50 | na |
| Mouse L1210 | na | na | >50 | na | >50 | na |
| Human Ion Channel activity | ||||||
| hERG | na | na | >33 | na | 1.6##, 7.0## | na |
| NaV1.5 | na | na | >33 | na | nd | na |
| Cav1.2 | na | na | >33 | na | nd | na |
| KV1.5 K+ | na | na | >11 | na | nd | na |
See Table 1 footnote for description of error analysis.
Study was conducted ± 3 and 10 μM proguanil (PG) to distinguish between DHODH and bc1 inhibition. CQ, chloroquine; Py, pyrimethamine, SD, sulphadoxine, MF, mefloquine, CG, cycloguanil. DSM265 resistant clones were previously reported.22, 34, 42 For DSM265 an additional set of EC50 data were collected in parallel to the 37 data and that value is reported first followed by
data taken from34, which was the average of triplicate biological repeats.
DSM265 hERG data were taken from22. Ion channel data; hERG was performed by IonWorks patch clamp electrophysiology under contract by AZ UK. Sodium and potassium channel assays were performed by Essen Bioscience using IonWorks patch clamp electrophysiology and calcium channel (cardiac L-type) data were collected using a Flexstation fluorescence assay by Essen Bioscience. na, not applicable. nd, not determined.
To assess cross-resistance with 1, we tested 37 against a panel of 1 resistant cell lines that we previously described22, 34 (Table 9). The cell line containing a DHODH gene amplification (1D3) showed a similar 5-fold resistance to both 1 and 37, consistent with their common mechanism of action. Cell lines harboring DHODH point mutations caused a 15–40-fold shift in EC50 for 1, but the effects on 37 were less (range 0.8–6-fold), with the L531F mutation actually leading to slightly improved binding of 37. The additional data set collected for 1 in this study agreed closely with the previously published results34, data are included in Table 9 for comparison. The largest effect (6-fold) for 37 was caused by the C276F mutation, which is the mutation that was observed clinically.23 However, this mutation caused a significantly smaller shift for 37 than for 1. Overall, the greatly reduced effects of the 1 selected mutations on 37 efficacy are consistent with the different binding modes of the triazolopyrimidine and pyrrole series compounds (Fig. 2).
Finally, 37 did not show any evidence of cytotoxicity when tested against both human HepG2 cells and mouse L1210 cells (CC50 > 50 μM) (Table 9). Preliminary safety pharmacology studies showed that 37 did not inhibit the hERG channel, nor a set of additional ion channels that were tested (Table 9).
Advanced ADME studies on 37.
Additional studies were conducted to further profile the physicochemical and ADME properties of 37. First, solubility studies were conducted in fasted state simulated gastric fluid (FaSSGF), fasted and fed state simulated intestinal fluid (FaSSIF and FeSSIF, respectively) and pH 7.4 phosphate buffer (PBS). Across these media, solubility was moderate and largely unaffected by differences in pH in FaSSIF, FaSSGF and PBS (Table 10). The somewhat improved solubility under fed state intestinal conditions (FeSSIF) most likely occurs through solubilization in mixed micellar colloidal species present under these conditions.
Table 10.
Physicochemical properties and in vitro binding and metabolism
| Cmpd | 37 | 1a |
| MW | 323 | 415 |
| Log D 7.4 | 2.8b | 4.0c |
| Solubility (μg/mL) | ||
| FaSSGF (pH 1.6) | 46.4 | 6.8 |
| FaSSIF (pH 6.5) | 60.1 | 5.1 |
| FeSSIF (pH 5.8) | 115 | 27.6 |
| PBS (pH 7.4) | 45.6 | 2.0 |
| H/R/M plasma protein binding (% bound) | 90.0 / 88.2 / 88.9 | 99.9 / 97.0 / 99.7 |
| Albumax binding (% bound) | 51.8 | 83.1 |
| Microsome binding (% bound) | 29.9 | 71.7 |
| DMEM/10% FCS binding (% bound) | 31.5 | 78.5 |
| H/R/M CLint (μL/min/mg) | 10 / 14 / 24 | <7 / <7 / <7 |
Plasma protein binding and in vitro metabolism were also confirmed and extended to additional mammalian species. Protein binding (determined by ultracentrifugation) for 37 ranged from 90% in human plasma to 88% in mouse plasma (Table 10). Binding was also assessed in human liver microsomes, Albumax medium, and DMEM containing 10% fetal calf serum (Table 10) where binding was relatively low in each medium (30–50% bound). Metabolic stability was greatest for human liver microsomes (intrinsic clearance (CLint) of 10 μL/min/mg protein), while the CLint was 1.4 and 2.4-fold higher for rats and mice, respectively (Table 10). Studies using cryopreserved hepatocytes from humans, rats, and mice suggested that there was no additional metabolism by enzymes other than those present in microsomes (e.g. CYPs and FMO).
In vitro metabolite identification studies were also conducted with 37. Following incubation of 37 (m/z 324.13, Fig. 4, panel A) with human liver microsomes, four oxygenated metabolites were detected by high resolution MS (eluting at 3.08, 3.45, 3.69 and 3.77 min, Fig. 4 panel B and C). Metabolites M+16 (I) to M+16 (III) had molecular ions at m/z 340.13 (Panel B) and M+16 (IV) was detected as an in-source fragment (Panel C). The peak at 3.77 min (Panel C) is an in-source fragment of M+16 (III) (Panel B). We hypothesized that the likely sites of oxygenation could include the cyclopropyl ring, the methyl carbon, the methylene carbon and the pyrrole ring. Based on relative peak areas, metabolite M+16 (I) appeared to be the predominant product (20% of the total area following a 2-h incubation with 1 mg/mL microsomal protein and 10 μM 37) followed by M+16 (III) (6% of the total area) with other metabolites representing minor products only (<3% of the total area).
Fig. 4.
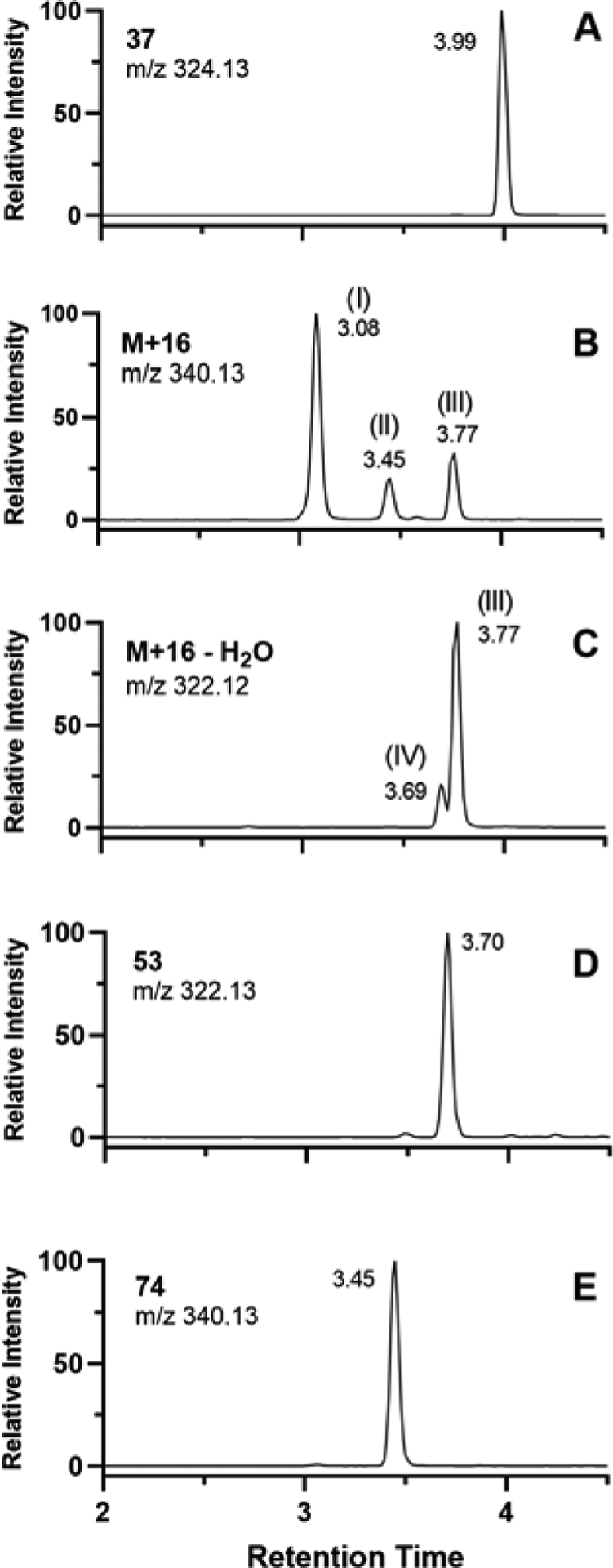
Extracted ion chromatograms obtained by high resolution MS. A. Authentic standard for 37; B and C. M+16 metabolites and an in-source fragment of a M+16 metabolite, respectively, formed by incubation of 37 with human liver microsomes in the presence of NADPH. D and E represent authentic standards for 53 and 74, respectively.
Authentic metabolites corresponding to oxygenation of the methylene carbon (74, Table 7) and oxygenation of the methyl carbon (53, Table 5) were then synthesized. Metabolite M+16 (II) was confirmed to be 74 by comparison of the retention times and CID fragmentation patterns (Fig. S1). Metabolite 53 was found to be highly susceptible to in-source fragmentation and instead of the expected molecular ion of m/z 340.13, its dehydrated fragment ion was observed at m/z 322.13 (Fig. S2). The CID spectrum of M+16 (IV) was too weak to be used for structure assignment. Instead, the structure of M+16 (IV) was proposed based the on similarity of the retention time and MS/MS transition at 322.05 > 264.93 compared to the data for 53 (Fig. S3).
The identity of the peaks corresponding to M+16 (I) and M+16 (III) were proposed based on comparison of their CID fragmentation patterns to that of 37 (Fig. 5). For M+16 (III), the molecular ion at m/z 340.14 was not detected and the peak at m/z 322.13 represents an in-source fragment resulting from loss of water. Below m/z 280, the fragmentation pattern for M+16 (III) was identical to that of 37suggesting that oxygenation had occurred on the cyclopropyl ring. The fragmentation pattern of M+16 (I) (Fig. 5) suggested that it was likely the product of oxygenation on the pyrrole ring. The fragment at m/z 255 likely results from loss of the carboxamide side chain in the absence of dehydration. This is then followed by loss of HF to produce the fragment at m/z 235. Further loss of 28 to produce the fragment at m/z 227 suggests loss of CO from the 2-oxopyrrole, supporting the proposal for oxygenation at the 5 position. The presence of the fragments at m/z 161 and 256, suggest that fragmentation possibly occurred via radical cleavage pathways as a result of the modified pyrrole. Further structural confirmation of both metabolites M+16 (I) and (III) would require the synthesis of authentic standards. It is worth noting that each of metabolites M+16 (I), (II) and (III) were resistant to reduction by sodium metabisulphite suggesting the absence of N-oxidation products.
Fig. 5.
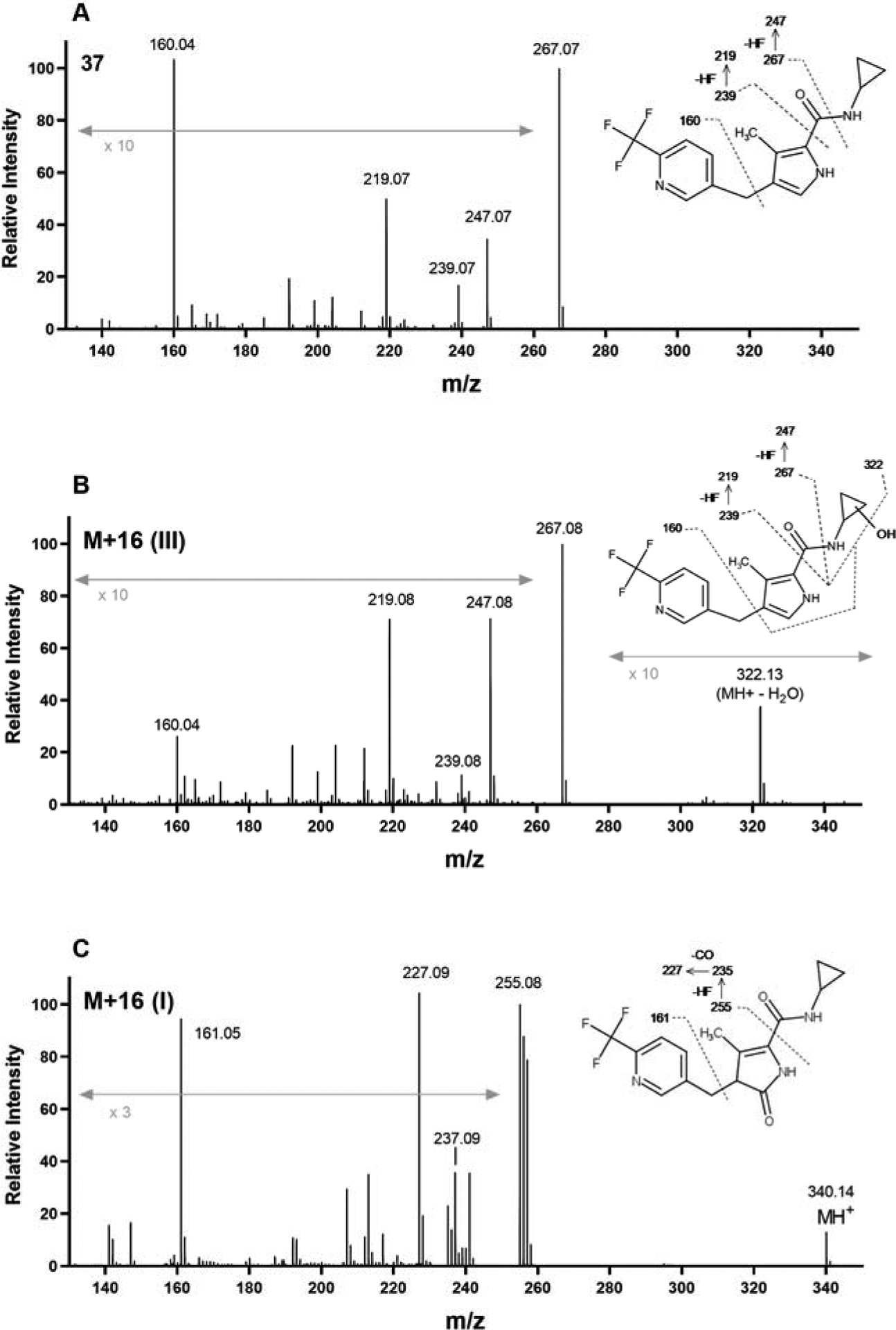
High resolution MS spectra and proposed fragmentation sites for 37 (A), and the oxygenation metabolites M+16 (III) (B) and M+16 (I) (C) formed upon incubation of 37 with human liver microsomes in the presence of NADPH. The molecular ion (m/z 324.13) for 37 was only observed under low energy conditions. For M+16 (III), the molecular ion (m/z 340.14) was observed as an in-source fragment resulting from loss of water.
Cytochrome P450 inhibition studies were conducted using a substrate specific interaction approach and suggested that 37 does not directly inhibit the major CYP isoforms 1A2, 2C9, 2C19, 2D6 or 3A4/5 with IC50 values all exceeding 20 μM. Additional time-dependent inhibition studies with 37 were conducted using an “IC50 shift” protocol as described previously.44 There was no evidence of time-dependent inhibition of any isoform except for CYP3A4/5 where the IC50 shifted from > 20 μM when a 30 min preincubation was conducted in the absence of NADPH, to ~5.5 μM when the preincubation step included NADPH suggestive of time-dependent inhibition of this isoform.
Pharmacokinetic properties of 37 and analogs in mice and rats.
Compound 37 was dosed to mice and rats to assess its in vivo pharmacokinetic properties (Tables 11 and 12 and Figs. 6 and 7). After intravenous (IV) administration to mice, clearance and volume of distribution were both moderate. Clearance of 37 was approximately 3-fold higher than that seen for 1 in mice.22 Following oral (PO) administration to mice, plasma exposure was similar to that observed previously for 122 but bioavailability was greater than 100% suggesting dose-dependent kinetics. Cmax and AUC increased to a greater extent than the increase in dose from 20 and 50 mg/kg (4–5-fold increase versus a 2.5-fold increase in dose) (Table 11). The observed exposure of 37 in mice after oral dosing was significantly better than that for 18 and 45 (Fig. 6) consistent with the higher CLint observed in vitro for these latter compounds (Table 8).
Table 11.
Pharmacokinetic properties of 37 in mice after single IV and PO doses.
| Cmpd | 37 | 37 | 37 | 37 | 1 |
|---|---|---|---|---|---|
| Dose and route | 2.8 mg/kg IV | 10.5 mg/kg PO | 18.3 mg/kg PO | 50 mg/kg PO | 10 mg/kg POa |
| Apparent t1/2 (h) | 2.8 | cnd | 2.6 | 3.6 | 2.4 |
| IV CLplasma (mL/min/kg) | 26.1 | --- | --- | --- | --- |
| IV Vss (L/kg) | 1.2 | --- | --- | --- | --- |
| B/P ratio | 0.8 | 0.8 | 0.8 | 0.8 | 0.7 |
| Tmax (h) | --- | 2 | 0.5 | 1 | 1–2 |
| Cmax (μM) | --- | 4.6 (3.7) | 8.4 (6.7) | 42.3 (33.8) | 3.94 |
| AUC0−∞h (μM.h) | 5.6 | cnd | 55.6 (44.5) | 220 (176) | 29.7 |
| Bioavailability (%) | --- | cnd | >100 | >100 | 61 |
Data taken from22. Values in parenthesis are blood values obtained by multiplying the plasma data by the blood to plasma ratio. cnd, could not determine
Table 12.
Rat plasma pharmacokinetic parameters after a single PO dose
| Cmpd | 37a | 1b |
|---|---|---|
| IV dose (mg/kg) | 3.4 | 2.6 |
| Apparent t1/2 (h) | 14.6 | 12.6 |
| CLplasma (mL/min/kg) | 28.6 | 6.5 |
| Vss (L/kg) | 4.7 | 5.9 |
| PO dose (mg/kg) | 3.0 | 2.1 |
| PO Tmax(h) | 1 | 3.3 |
| Apparent t1/2 (h) | 3.9 | 11.6 |
| Cmax (μM) | 1.7 | 0.75 |
| AUC∞ (μM.h) | 5.1 | 15.6 |
| Bioavailability (%) | 91 | 66 |
| PO dose (mg/kg) | 33 | 18.8 |
| Tmax(h) | 4 | 7 |
| Apparent t1/2 (h) | 2.8 | 15.5 |
| Cmax (μM) | 27 | 3.95 |
| AUC∞ (μM.h) | 204 | 122 |
| Bioavailability (%) | >100 | 57 |
Mean of two animals per dose group.
Data taken from22
Fig. 6.
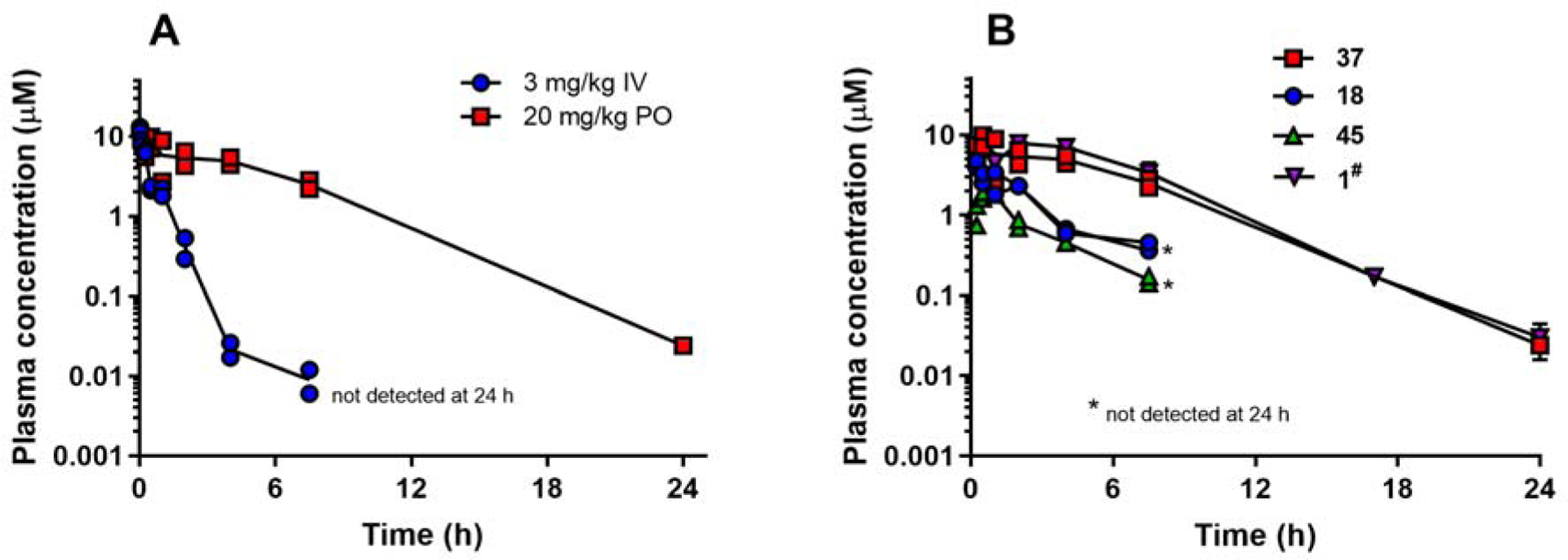
Plasma concentration versus time profiles in mice (n=2 animals per time point). A. Data for 37 after IV and PO dosing. B. Comparative profiles after PO dosing at 20 mg/kg for compounds as indicated in the graph legend. #Data for 1 are based on results obtained at an oral dose of 10 mg/kg22 which were then scaled to 20 mg/kg for comparison purposes with the assumption of linear kinetics.
Fig. 7.
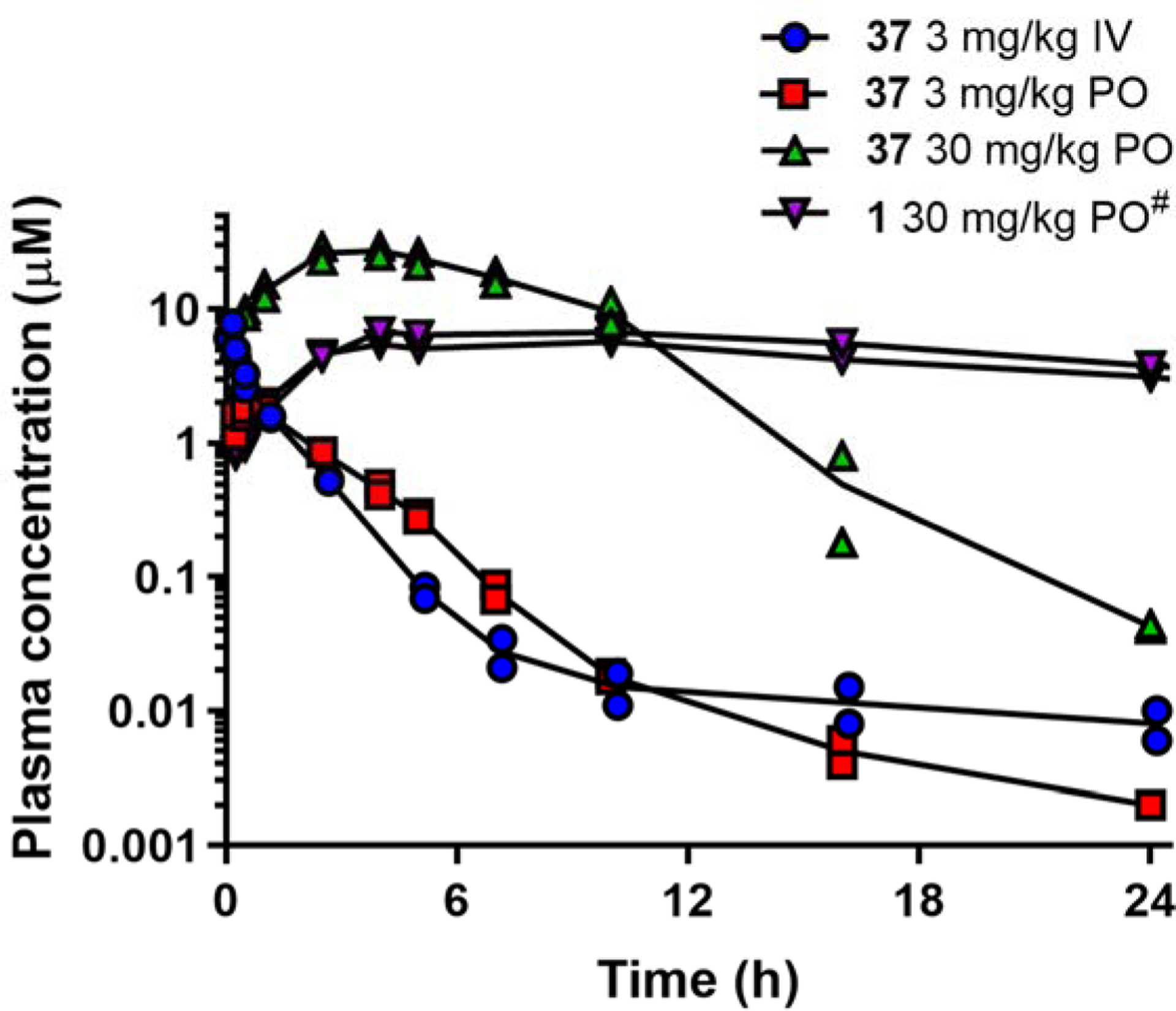
Plasma concentration versus time profiles for 37 in rats (n=2 animals per dose group). #Data for 1 are based on results obtained at a PO dose of 20 mg/kg22 which were then scaled to 30 mg/kg for comparison purposes and with the assumption of linear kinetics over this dose range.
After IV dosing of 37 to rats, plasma clearance was moderate (~50% of hepatic blood flow in rats) and volume of distribution was high (~4.7 L/kg) (Fig. 7 and Table 12). Compared to 1, clearance of 37 was nearly 5-fold higher in rats. At an oral dose of 3 mg/kg, bioavailability was approximately 90%. Increasing the oral dose to 30 mg/kg resulted in a bioavailability of >100% suggesting saturation of clearance processes. There was minimal excretion of 37 in urine after either IV or PO administration of 37 suggesting that metabolism was likely the major route of clearance. At the higher oral dose, the time to reach the maximum plasma concentration was prolonged, possibly suggesting slow dissolution and absorption within the gastrointestinal tract.
SCID mouse efficacy studies.
Compound 37 was tested for in vivo efficacy in the SCID mouse P. falciparum model in two separate studies, the first of which was accompanied by PK analysis of drug concentrations in blood (Fig. 8). Oral doses were administered once daily for 4 days starting on day 3 after mice had been inoculated with parasites. Parasitemia was monitored daily and final parameters were determined 24 h after the last dose. In the first study (conducted by GSK), mice were dosed at both 10 and 50 mg/kg. The 10 mg/kg mouse died on day 5 so a full set of parasitemia data could not be collected. The death was assessed as unlikely to be drug related, and the compound was well tolerated at the higher 50 mg/kg dose in both studies. In the GSK study the ED90 (dose that provides 90% reduction in parasitemia 24 h after the last dose) was ~50 mg/kg corresponding to an AUCED90 of 9 μg.h/mL (28 μM.h), which compares favorably to the AUCED90 observed previously for 1 in a similar study (Table 13 and 14). The dose required for the ED90 was significantly higher for 37 than for 1 (Table 14). Interestingly, the exposure levels for 37 in the SCID mouse were lower than observed in the normal mice (Figs. 6 and 8) where the SCID mouse Cmax was 4–6-fold lower (10 and 50 mg/kg) and AUC was 7-fold lower (50 mg/kg) (Tables 11 and 13). The basis for these differences in exposure are not yet known. In the second confirmatory SCID study (conducted at Swiss TPH) only a single dose of 50 mg/kg was administered and PK data were not collected. In this study, higher parasitemia levels were observed for the vehicle control, but 37 showed similar to slightly improved efficacy compared to the first study (50 mg/kg dose resulted in 97% parasite clearance compared to 85% clearance in the GSK study) (Fig. 8).
Fig. 8.
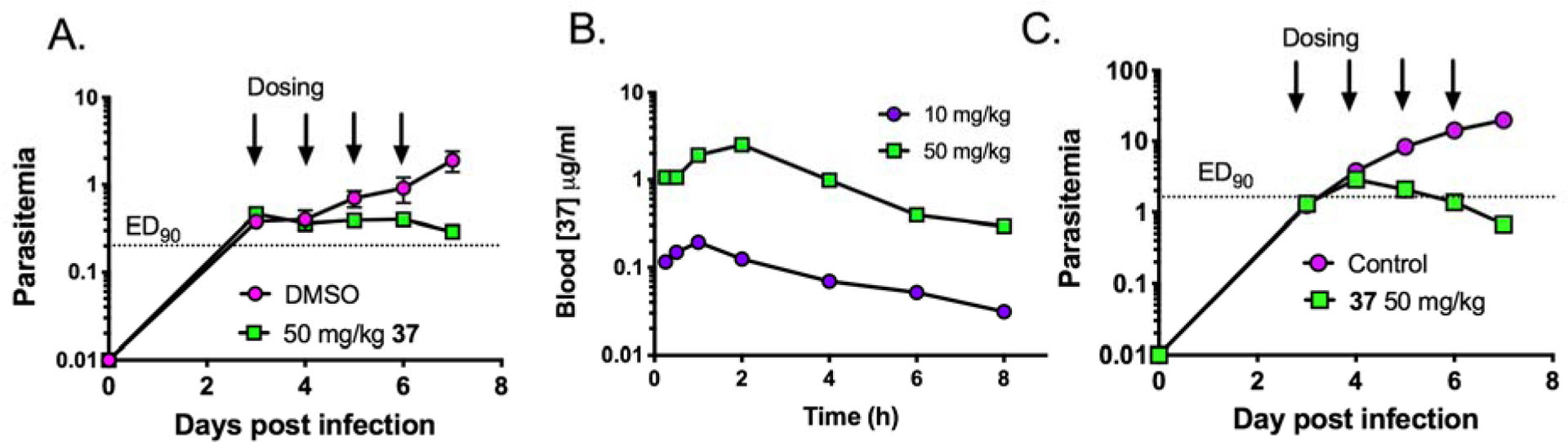
SCID mouse efficacy data. Two separate studies were conducted either at GSK (A and B) or at Swiss TPH (C). A and C. Parasitemia versus days post infection for the DMSO control (pink circles) and the 37 treated mice (green squares). B. Blood levels of 37 for the mice treated in A. PK data were not collected in the Swiss TPH study (C).
Table 13.
Blood exposure parameters for 37 following oral dosing to P. falciparum-infected SCID mice.
| Dose mg/kg | Blood Cmax μg/mL(μM) | Blood AUC23 h μg.h/mL (μM.h) |
|---|---|---|
| 10 | 0.194 (0.6) | 0.680 (2.10) |
| 50 | 2.52 (7.81) | 8.60 (26.7) |
Table 14.
Comparison of in vitro potency (3D7 parasites) and SCID mouse efficacy data for PfDHODH inhibitors.
| Cmpd | 3D7 EC50 (μM) | Unbound 3D7 EC50 (μM)a | ED90 (mg/kg/day) | Blood AUCED90 (μg.h/ml) | Unbound blood AUC (μg.h/ml)b |
|---|---|---|---|---|---|
| 37 | 0.016 | 0.0077 | ~50* (QD) | ~9 | ~1.1 |
| 1# | 0.0060 | 0.0010 | 8.1 (QD) | 16.7 | 0.023 |
| 1# | --- | --- | 3.0 (BID) | 10.8 | 0.015 |
| DSM421# | 0.014 | 0.0070 | 2.6 (QD) | 12.9 | 0.25 |
QD=once daily dosing; BID=twice daily dosing. Studies compared use the standard Peters test with 4 days of consecutive dosing. Parasite levels one day after the final dose were used to determine the ED90.
For 37 the ED90 reflects the average from the two studies where 24 h after the last dose 85% reduction in parasitemia was observed in the GSK study and 97% reduction was observed in the Swiss TPH study, both at the 50 mg/kg dose.
obtained by multiplying the EC50 by the fraction unbound in Albumax medium
approximated by multiplying the blood AUCED90 by the fraction unbound in blood (fu(blood)) where fu(blood) = fu(plasma)/B:P
Discussion
Malaria remains a serious public health issue to 50% of the world’s population, and drug resistance threatens to derail existing therapies. Our group has previously validated DHODH as a drug target for the treatment of malaria through the identification of 1, which reached clinical development.23 Herein we sought to identify a potential backup candidate from a distinct chemical scaffold. The pyrrole scaffold was identified as a hit in our original HTS program to discover PfDHODH inhibitors. We progressed the series to the point of identifying a lead candidate 37 that showed potent and selective Plasmodium DHODH inhibition, equivalent activity on P. falciparum parasites in vitro, and efficacy in the SCID mouse P. falciparum mouse model. The pyrrole lead 37 was ~4-fold less potent than 1 in parasite assays but showed efficacy at similar blood exposure as 1 in the SCID mouse model, likely due to the significantly higher unbound concentrations compared to 1. Compound 37 showed improved species selectivity over 1; it was not an inhibitor of any of the tested mammalian enzymes and showed equivalent activity on both P. falciparum and P. vivax DHODH. Additionally, while some cross-resistance was observed for 37 versus some of the 1-selected resistant P. falciparum cell lines, the magnitude of the EC50 shifts was significantly lower, and not all 1-selected point mutations led to 37 resistance. Thus 37 achieved three of our key milestones for a 1 backup: improved species selectivity, equivalent P. falciparum and P. vivax activity, and a different resistance profile
X-ray structure analysis of PfDHODH bound to both an ester (4) and amide (60) analog in the series demonstrated that the pyrroles bound to the enzyme in an overlapping but distinct binding mode when compared to the triazolopyrimidines (e.g. 1). The pyrrole ring bound between the conserved R265 and H185 making H-bond interactions similar to the triazolopyrimidine ring of 1. The cyclopropyl amide of 60 also bound in a similar position to the CF2CH3 in 1, but the polar amide NH donates a second H-bond to the side chain N of H185. In contrast, the benzyl group binds in an alternative pocket compared to the aniline of 1 due to a 180° rotation of F188. This different binding mode also likely explains the equivalent activity on Pf and PvDHODH and the differing resistance profiles for the pyrrole series compared to the triazolopyrimidines. None of the tested pyrrole analogs bound to the mammalian enzymes, leading to better over all species selectivity compared to 1. In contrast, prior crystallographic studies demonstrated that rodent and human DHODH have a binding pocket that can accommodate compounds from the triazoloypyrimidine series with binding affinity dictated by aniline hydrophobicity and facilitated by the addition of meta-fluorines on the aniline ring.40
Comparison of the cross-resistance of 37 on multiple 1-selected P. falciparum Dd2 mutant clones demonstrated that the differing binding modes did lead to a different resistance profile. As expected the strain that harbors a DHODH gene amplification showed ~5-fold higher EC50 for both compounds, however two of the 1-selected resistance mutants G181C and L531F showed wild-type to slightly improved activity for L531F towards 37. Parasites harboring C276F or R256G mutations showed some cross-resistance but had a less pronounced EC50 shift when compared to 1 (5-fold for 37 vs 15–25-fold for 1). These two mutations are of note as C276F was observed in a single patient receiving the 400 mg dose in the Phase IIb clinical study23 and both the C276F and R265G mutations have been observed after in vivo selections in SCID mice45. The ability of DHODH point mutations to show differential affinity towards inhibitors from different scaffolds has been previously noted and our data further support the hypothesis that DHODH inhibitors from different structural classes might be able to protect each other from the generation of resistance if dosed together.46
During the medicinal chemistry effort to identify 37 we explored variation of the substituents at all positions of the pyrrole scaffold. The ester linkage of the original HTS hit 3 could be readily replaced with an ethyl amide that yielded good activity and this substitution was also key to improving metabolic stability. Exploration around the amide showed that small linear or small cyclic alkanes provided the best activity with a cyclopropyl amine as in 37 showing the best activity. The order of preference for linear alkanes was ethyl>propyl>butyl amide and for the cyclic alkanes cyclopropyl>cyclobutyl>azetidine, although di-fluoro-azetidine retained good activity. Larger rings (n = 5 or greater) were inactive. Dimethyl and diethyl amide had poor to no activity. We determined that only a single methyl on the pyrrole at the 3-position contributed to binding, whereas the methyl at the 5-position was detrimental to good binding affinity. The benzyl groups substituted with 4-CF3 and 3-pyridinyl-4-CF3 yielded both the best potency and most favorable ADME properties (e.g. solubility and metabolic stability). Addition of fluoro or chloro at the 2-position of the benzyl ring when combined with 4-CF3 also yielded good activity. The bridging carbon could be hydroxylated with only a 2-fold reduction in activity or replaced with oxygen with a 10-fold reduction.
In vitro ADME data suggested that 37 is marginally more soluble than 1, it has lower plasma protein binding but a higher intrinsic clearance in liver microsomes from various species. In vitro metabolite identification studies suggested the formation of multiple oxygenation products. Whereas there was no evidence for direct inhibition of CYP enzymes by 37, IC50 shift experiments highlighted the potential for time-dependent CYP inhibition, possibly as a result of reactive metabolite formation. While further mechanistic and kinetic studies could shed light on these questions, the observed time-dependent CYP inhibition represents a potential risk for the advancement of 37 and a point of inferiority when compared to triazolopyrimidines including 1. In vivo PK studies in mice and rats indicated that, similar to the in vitro results, 37 has a higher clearance compared to 1 (3–5-fold). Volume of distribution was found to be similar or marginally higher for 1 than for 37, however in general, the data suggested that 37 was unlikely to have a sufficiently long half-life in humans to support a single dose regimen.
Interestingly, the efficacy studies conducted in SCID mice indicated that the exposure of 37 after oral dosing was considerably lower (by ~ 5–7-fold) than that seen in normal mice (similar formulations and dose levels were used for normal and SCID mice) (Table 11 and 13). This result may possibly reflect increased hepatic metabolic activity in the SCID mice versus normal mice resulting in higher clearance, however data are currently not available to confirm this hypothesis. The lower than expected exposure in SCID mice meant that the dose required to achieve a 90% reduction in parasitemia (ED90 ~50 mg/kg/day for 37) was considerably higher than for 1 in the QD study (ED90 ~8 mg/kg/day21 ) however the blood exposure levels (AUCED90, Table 14) were similar. Correcting blood exposure for the fraction unbound in blood suggests a 50-fold higher unbound exposure is required for 37 to reach a 90% reduction in parasitemia compared to 1. In contrast, the difference in unbound in vitro potency (3D7 parasites, Table 14) differed by only 8-fold (EC50 of 0.0077 μM for 37, 0.0010 μM for 1). These results suggest that the in vivo efficacy of 37 is below what would be expected if it performed as well as the triazolopyrimidines. Conducting a similar analysis for DSM42126 compared to 1 yielded consistent differences between the unbound in vitro 3D7 EC50 and the unbound AUCED90 (which were 10-fold lower for 1 for both parameters, Table 14). The basis for the discrepancy in efficacy for 37 compared to the triazolopyrimidines is not currently understood.
Conclusion.
While 37 has many attractive features and improves on some aspects of 1, it is less metabolically stable and does not show the sustained exposure needed to fulfill the criteria for single dose cure or once weekly chemoprevention, which is the current objective of new antimalarials.47 Additional improvements in metabolic stability, and modifications that would eliminate the time-dependent CYP inhibition could be undertaken to improve the series further. However, 37 may have sufficient potency and plasma exposure to be able to support a therapeutic use if dosed more frequently than once for treatment or once weekly for chemoprevention. Thus, we have demonstrated herein that the pyrrole-based series has the potential to deliver an anti-malarial candidate that could advance to preclinical development.
Experimental Section.
Materials.
Routine chemicals were sourced from Sigma or Aldrich unless otherwise stated.
Protein Expression and Purification.
BL21-DE3 E coli phage resistant cells containing His6-tagged DHODH-pRSETb (N-terminal tag)(Pf and PvDHODH), pET22b C-terminal tag (human) or pET-28b C-terminal tag (rat, mouse and dog) constructs were grown and harvested as described using appropriate antibiotics.21, 22, 34 Cells in lysis buffer A (50 mM Tris pH 8.5, 20 mM imidazole pH 8.0, 5 mM 2-mercaptoethanol, 2% Triton X-100, 0.5 mM FMN, 10 % glycerol, and 1X Protease Inhibitor Cocktail (Sigma P8849)) were passed through an Emulsi-Flex-C5 high pressure homogenizer (3x) and the lysate clarified by centrifugation (20,000 rpm at 4°C). Lysate was loaded onto a 5 ml HisTrap HP column (20 mM Tris pH 8.5, 300 mM NaCl, 10% glycerol, 20 mM imidazole, pH 8.0, 0.05% Triton X-100), DHODH was eluted using a linear imidazole gradient (20 to 400 mM) and then further purified by gel filtration chromatography on a Pharmacia HiLoad 16/600 Superdex 200 column (100 mM HEPES, pH 8.0, 300 mM NaCl, 0.05% Triton X-100 reduced, 1 mM DTT) to a purity >95% based on SDS-PAGE analysis. The purified DHODH concentration was determined based on absorbance at 280 nm with the molar extinction coefficient calculated by ProtParam in ExPASy (PfDHODH - 30,830 M−1cm−1, PvDHODH - 29,340 M−1cm−1, human DHODH – 15930 M−1cm−1, rat DHODH – 11920 M−1cm−1, mouse DHODH – 11920 M−1cm−1, dog DHODH – 11920 M−1cm−1).48
DHODH Kinetic Analysis.
Steady-state DHODH assays to determine the 50% inhibitory concentration (IC50) of the described compounds were performed using the 2,6-dichloroindophenol (DCIP) assay to monitor the reaction rate at 25°C in assay buffer (100 mM HEPES, pH 8.0, 150 mM NaCl, 10% glycerol, 0.1% Triton X-100 reduced, 20 μM CoQD, 200 μM L-DHO, 120 μM DCIP and 5–20 nM enzyme) as described.48 Inhibitor stocks (100 mM) were prepared in DMSO in amber bottles. Stocks were diluted in DMSO to generate a 3-fold dilution series and then dispensed into assay buffer via a 1/100 dilution to generate a final concentration range of (0.001–100 μM). Triplicate rate data were collected at each inhibitor concentration. To determine the IC50, data were fitted to log (inhibitor) vs. response equation Y=Bottom + (Top-Bottom)/(1+10^((X-LogIC50))) in GraphPad Prism.
P. falciparum growth and inhibition assays.
P. falciparum 3D7 cells were grown in RPMI media supplemented Albumax (2.5g/L) and hypoxanthine (50 mg/L) with human red blood cells and plated to achieve a 0.5% hematocrit, 0.5% parasitemia solution. Inhibitor stocks (as above) were serially diluted in DMSO in 2 or 3 -fold steps (final inhibitor concentrations 0.001 – 30 μM depending on the cell line and a final DMSO concentration of 0.2%). Parasites in the presence of either DMSO controls or DHODH inhibitors were grown at 37°C for 72 h before growth was assessed using the SYBR Green I method (as described49 with minor modifications40), which measures fluorescence (ex./em. 485/535nm) as the output. Data were collected in triplicate and to determine the 50% effective concentration (EC50) by fitting data to log (inhibitor) vs. response equation Y=Bottom + (Top-Bottom)/(1+10^((X-LogIC50))) equation in GraphPad Prism.
X ray Crystallography. Protein preparation.
The pET28b-TEV- PfDHODΔ384–413 expression construct used for crystallography was previously described.28, 50 In this construct, the N-terminal non-enzymatic domain and membrane spanning domain (residues 1–157) were deleted to allow expression of the soluble enzyme, and the surface loop 384–413 was deleted to improve crystallization, such that the expressed protein contains PfDHODH residues 158–383 contiguous with residues 414 – 569. PfDHODHΔ384–413 was expressed in E.coli BL21 phage-resistant cells (NEB, C252H) and protein was purified with Nickel and Gel-filtration chromatography as previously described.50 Protein was concentrated to 20 mg/ml in buffer with 20 mM Hepes pH 7.8, 20 mM NaCl, and 2 mM n-Dodecyl-N,N-Dimethylamine-N-Oxide (LDAO, Anatrace), and 10 mM DTT, and stored at −80°C.
Crystallization and data collection of PfDHODHΔ384–413 −4 and −60.
Preliminary crystallization conditions were found using the random crystallization screen Cryos suite (Nextal) followed by variation of pH, precipitant, and protein concentrations to find optimal conditions. Crystals of PfDHODHΔ384–413 in complex with 4 or 60 grew from the same condition (reservoir solution = 0.17 M Ammonium Acetate (Fluka, Ultra), 0.1 M tri-Sodium citrate (Sigma, BioUltra), pH 5.6, 25% PEG4000 (Sigma), 16% Glycerol (Sigma), and 10 mM DTT (Sigma)). Crystallizations were setup using hanging drop vapor diffusion at 20°C from an equal volume mixture of reservoir solution and PfDHODHΔ384–413 (20 mg/ml) pre-equilibrated with inhibitor (1 mM) and dihydroorotate (DHO) (2 mM). Crystals of PfDHODHΔ384–413- 4 typically grew in 1 week whereas crystals of PfDHODHΔ384–413- 60 grew slowly (~5 weeks).
Diffraction data were collected at 100K on beamline 19ID at Advanced Photon Source (APS) using an ADSC Q315 detector with 240 degree of data collected for PfDHODHΔ384–413- 4 and 200 degree of data collected for PfDHODHΔ384–413- 60. The crystal of PfDHODHΔ384–413- 4 diffracted to 1.8 Å in a space group of P212121 with the cell dimension of a=92.1, b=97.5, c=186.3 and four molecules were found PfDHODH in the asymmetric unit. PfDHODHΔ384–413- 60 diffracted to 2.3 Å in a space group of P64 with the cell dimension of a=b=84.8, c=138.0 and one molecule of PfDHODH was found in the asymmetric unit. Diffraction data were integrated and intensities were scaled with HKL2000 package.51
Structure determination and refinement of PfDHODH −4 and −60 co-crystal structures.
Crystallographic phases for PfDHODH inhibitor complexes were solved by molecular replacement with Phaser52 using the structure of PfDHODHΔ384–413 bound to a triazolopyrimidine (PDB ID 3I65)50 (with inhibitor removed) as the search model. Structures were rebuilt with COOT53 and refined with phenix.refine.54 PfDHODHΔ384–413- 4 was refined to R and Rfree of 0.151 and 0.194, respectively. Electron density was observed for the following residues (Chain A: 158 −377, including 3 amino acids (Ala, Asp, and Pro) from the N-terminal-His tag linker, and 414–566. Chain B: 160–378, 414–566. Chain C: 166–378, 414–566. Chain D: 163–377, 414–566). All residues were within the allowed section of the Ramachandran plot (Table 3). Additionally water molecules (926) were modeled into the structure using the default setting in phenix.refine. PfDHODHΔ384–413- 60 was refined with twin operator (h, -h-k, -l) to R and Rfree of 0.202 and 0.219, respectively. The refined structure contains amino acid residues 162–383, and 414–566, and 75 waters (Table 3). All amino acid conformations were within the allowed section of the Ramachandran plot. Data collection and refinement statistics are provided in Table 3.
Structure display and analysis.
Structures were rendered in in MacPyMOL: PyMOL v1.8.2.3 Enhanced for Mac OS X (Schrödinger LLC). Structural alignments were conducted using the Align command.
Physicochemical and binding properties and in vitro metabolism.
Methods for assessing solubility in biorelevant media (FaSSIF, FeSSIF and FaSSGF), binding to plasma proteins, microsomes and in vitro assay media, and whole blood-to-plasma partitioning ratio determination have been described previously.22
In vitro Metabolism.
Compound 37 (1 μM) was incubated at 37°C for up to 60 minutes with human, rat or mouse liver microsomes or cryopreserved hepatocytes (all Xenotech LLC, Kansas City, KS) and samples were analysed by LC-MS (Waters Xevo G2QTOF coupled to a Waters Acquity UPLC operating in positive electrospray ionization under MSE mode). Microsome incubations were conducted at a protein concentration of 0.4 mg/mL and hepatocyte studies at a cell concentration of 7 × 105 cells/mL. The degradation half-life and in vitro intrinsic clearance in microsomes were calculated from the apparent first-order degradation rate constant.
Metabolite identification and reactive metabolite trapping.
Compound 37 was incubated at 37°C with human liver microsomes. To maximise the metabolite yield, a high substrate concentration (10 μM) and a high microsomal protein concentration (1 mg/mL) were used. Reactions were initiated by the addition of an NADPH-regenerating system and the samples were incubated for up to 60 min. Controls containing no NADPH were included in the incubation. The reaction was quenched by protein precipitation with an equal volume of ice-cold acetonitrile (containing 0.15 μg/mL diazepam as internal standard). Following protein precipitation, samples were vortexed and then centrifuged for 3 minutes at 10,000 rpm. The supernatant was removed and analysed by LC/MS. Chromatography and mass spectrometry were conducted using a Waters Acquity UPLC coupled to a Waters Xevo G2 QTOF. The column was an Asscentis Express Amide (50 × 2.1 mm, 2.7 μm) and the mobile phase comprised water and acetonitrile with 0.05% formic acid delivered with a gradient program and 6 min cycle time. The flow rate was 0.4 mL/min and the injection volume was 5 μL. Mass spectrometry was conducted in positive electrospray ionisation under MSE mode with metabolites confirmed using accurate mass and MS/MS fragmentation.
Cytochrome P450 inhibition and time-dependent inhibition studies.
The CYP inhibition assay was performed utilising a substrate-specific interaction approach which relies on the formation of a metabolite that is mediated by a specific CYP isoform using human liver microsomes. The specific CYP-mediated metabolic pathways included phenacetin O-deethylation (CYP1A2), tolbutamide methylhydroxylation (CYP2C9), (S)-mephenytoin 4’-hydroxylation (CYP2C19), dextromethorphan O-demethylation (CYP2D6), midazolam 1’-hydroxylation (CYP3A4/5) and testosterone 6β-hydroxylation (CYP3A4/5). Multiple concentrations of 37 (0.25 to 20 μM) and positive control inhibitors were incubated at 37°C concomitantly with each probe substrate in a suspension of human liver microsomes with the total organic solvent concentration being 0.47% (v/v). The microsomal protein concentration was 0.1 mg/mL for all pathways except for CYP2C9 and 2C19 (both 0.2 mg/mL microsomal protein). The reactions were initiated by the addition of an NADPH-regenerating system and the samples were quenched by the addition of ice-cold acetonitrile containing analytical internal standard (diazepam). Metabolite formation was monitored over variable periods from 10 to 30 min depending on the pathway. Concentrations of the substrate-specific metabolites in quenched samples were determined by UPLC-MS (Waters Xevo TQD triple-quadrupole mass spectrometer) with an Ascentis Express RP Amide column and acetonitrile/water (both containing 0.05% formic acid) gradient. Calibration standards were prepared in quenched microsomal matrix and analysed along with the samples. Control samples confirmed that the UPLC-MS assay of the specific metabolites was not affected by the presence of each test compound (and potential metabolites).
Assessment of time-dependent inhibition was conducted using human liver microsomes and the “IC50 shift“-method. Multiple concentrations of inhibitor were incubated in the absence or presence of NADPH at 37°C with human liver microsomes suspended in 0.1 M phosphate buffer (pH 7.4) for 30 min at 10-fold the final target concentration. The microsomal protein concentration during this pre-incubation period was 1 mg/mL (for CYPs 1A2, 2D6 and 3A4) and 2 mg/mL (for CYPs 2C9 and 2C19), and the total concentration of organic solvent in the pre-incubation mixture was 1% (v/v). Following the 30 min pre-incubation, aliquots of the mixture were diluted 10-fold in 0.1 M phosphate buffer (pH 7.4) containing the probe substrate for an individual CYP isoform (same as for the CYP inhibition assay). An NADPH-regenerating cofactor system was added to all samples which were then incubated at 37°C for 10 – 40 min. The total concentration of organic solvent in this incubation mixture was 0.3% (v/v). The incubation was quenched by the addition of ice-cold acetonitrile (containing 0.15 μg/mL of diazepam as an internal standard), samples were centrifuged and an aliquot of the supernatant transferred to a fresh 96-well plate for analysis. Concentrations of the probe metabolites were determined by UPLC-MS as described above. IC50 values were determined by curve fitting to a four-parameter logistic function following log transformation of the concentration data (GraphPad Prism software, ver 7.01)
Animal Studies All animal studies were ethically reviewed and carried out in accordance with either the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (mouse and rat PK studies) or European Directive 2010/63/EEC and the GSK Policy on the Care, Welfare and Treatment of Animals (SCID mouse studies). The human biological samples for the SCID mouse studies were sourced ethically and their research use was in accord with the terms of the informed consents under an IRB/EC approved protocol. Animal experiments described at the Swiss Tropical and Public Health Institute (Basel, Switzerland) were adhering to local and national regulations of laboratory animal welfare in Switzerland (awarded permission no. 2303). Protocols are regularly reviewed and revised following approval by the local authority (Veterinäramt Basel Stadt).
Mouse and Rat Pharmacokinetic Studies.
Protocols for the in vivo assessment of 37 pharmacokinetics in male Sprague Dawley rats and male Swiss outbred mice were reviewed and approved by the Monash University of Pharmaceutical Sciences Animal Ethics Committee. 37 was administered intravenously (IV) to rats in a saline vehicle containing 10% (v/v) DMSO and 4% (v/v) Solutol HS 15 (total volume of 1 mL) as a 10 min infusion into the jugular vein via a surgically implanted catheter. In mice, the IV dose was administered as a bolus injection into the tail vein in a vehicle comprised of 40% (v/v) propylene glycol, 10% (v/v) ethanol and 0.5 % (v/v) Tween 80 in water (2 mL/kg dose volume). Oral doses in both rats and mice were administered by oral gavage in an aqueous suspension vehicle containing 0.5% (w/v) carboxymethyl cellulose, 0.4% (v/v) Tween 80 and 0.5 % (v/v) benzyl alcohol (10 mL/kg volume). Blood samples from rats and mice were collected up to 24 h post-dose into tubes containing heparin as an anticoagulant and the plasma fraction transferred to clean tubes following centrifugation. Proteins were precipitated with the addition of two volumes of acetonitrile and the supernatant was analysed by LC-MS against calibration standards prepared in blank rat or mouse plasma and extracted in the same way. Diazepam was added to all plasma samples and calibration standards (prior to protein precipitation) as an internal standard. Plasma samples were analysed on a Waters Xevo TQ mass spectrometer coupled to a Waters Acquity UPLC operating in positive electrospray ionization multiple-reaction monitoring mode. The column was a Sulpeco Ascentis Express RP amide column (50 × 2.1 mm, 2.7 μm). The mobile phase consisted of an acetonitrile-water gradient with 0.05% formic acid, a gradient cycle of 4 min and a flow rate of 0.4 mL/min.
SCID Mouse Efficacy studies.
In vivo antimalarial efficacy studies in P. falciparum conducted at GSK.
The in vivo efficacy of 37 was measured against P. falciparum 3D70087/N9 growing in the peripheral blood of NOD-scidIL2Rγnull mice (Jackson Laboratory, USA) (23–36 g) engrafted with human erythrocytes as described.55 The parasites (20 × 106) were inoculated by intravenous injection, and antimalarial efficacy was assessed using a standard “4-day test”. Blood parasitemia was measured by FACS analysis. 37 was administered in vehicle (0,5% hydroxypropylmethylcellulose, 0,4% tween 80 0,5% Benzoyl) by oral gavage for four consecutive days starting on the third day after infection. With the goal to determine PD endpoints the pharmacokinetics of 37 were evaluated in whole blood in the same animals as part of the efficacy study after dosing 10 and 50 mg/kg. With this goal, peripheral blood samples (25 μl) were taken at different times (0.25, 0.5, 1.0, 2.0, 4.0, 6.0, 8.0 and 23 h) at day 1, after first administration of parent compound. Samples were mixed with 25 μL of Milli-Q water and immediately frozen on dry ice. The frozen samples were stored at −80 °C until analysis. Vehicle-treated mice experienced the same blood-sampling regimen. Blood samples were analysed for 37 using a method based upon protein precipitation followed by LC-MS/MS analysis (Waters Acquity coupled to a Sciex API4000) (LLOQ= 1 ng/mL). Reversed-phase HPLC separation was achieved with a UPLC HSS T3 column (50×2.1mm 1.8μm). MS/MS detection was set at mass transitions of 324.193→266.2887 m/z for 37 and 326→291 m/z for Midazolam, (IS) in positive mode. Pharmacokinetic analysis was performed by noncompartmental methods by using WinNonLin Phoenix Version 6.3 (Pharsight Corporation).
In vivo antimalarial efficacy studies in P. falciparum conducted at Swiss TPH.
Reduction of existing parasitemia in vivo was tested in the murine P. falciparum SCID model essentially as described.55 Briefly, 37 was formulated in vehicle (0,5% hydroxypropylmethylcellulose, 0,4% tween 80, 0,5% Benzoyl) and administered to a cohort of age-matched female immunodeficient NOD-scidIL2Rγnull mice (The Jackson Laboratory, Bar Harbor, ME) (20–22 g) previously engrafted for 11 days with human erythrocytes (generously provided by the Blood Bank in Zürich, Switzerland). The mice were intravenously infected with 2×107 P. falciparum Pf3D70087/N9-infected erythrocytes (day 0). On day 3 after infection, mice (n=2) were randomly allocated to treatments that were administered once a day for 4 consecutive days by oral gavage at 10 ml/kg. Parasitemia was measured by microscopy and flow cytometry using anti-murine erythrocyte TER119 monoclonal antibody (Pharmingen, San Diego, CA) in serial 2 μl blood samples taken every 24 h until assay completion.
Chemistry.
All reagents and starting materials were obtained from commercial suppliers and used without further purification unless otherwise stated. Reaction progress was monitored by thin layer chromatography (TLC) on preloaded silica gel 60 F254 plates. Visualization was achieved with UV light and iodine vapor. Flash chromatography was carried out using prepacked Teledyne Isco Redisep Rf silica-gel columns as the stationary phase and analytical grade solvents as the eluent unless otherwise stated. Yields were of purified product and were not optimized. 1H NMR spectra were recorded on Bruker Avance 300 and 500 MHz spectrometer at ambient temperature. Chemical shifts are reported in parts per million (δ) and coupling constants in Hz. 1H NMR spectra were referenced to the residual solvent peaks as internal standards (7.26 ppm for CDCl3, 2.50 ppm for DMSO-d6, and 3.34 ppm for MeOD). Spin multiplicities are described as s (singlet), brs (broad singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), dt (doublet of triplets) and m (multiplet). Total ion current traces were obtained for electrospray positive and negative ionization (ES+/ES-) on a Bruker Esquire Liquid Chromatograph-Ion trap mass spectrometer. Analytical chromatographic conditions used for the LC/MS analysis: Column, Zorbax Extend C18 from Agilent technologies, 2.1×100 mm. The stationary phase particle size is 3.5 μM. Solvents were A, aqueous solvent = water +5% acetonitrile+1% acetic acid; B, organic solvent = acetonitrile + 1% acetic acid; Methods, 22 min run time (0–10 min 10–60% B, flow rate-0.275 mL/min; 10–20 min 60–100% B, flow rate-0.350 mL/min; 20–22 min 100–10% B, flow rate-0.350 mL/min). The following additional parameters were used: injection volume (10 μL), column temperature (30 °C), UV wavelength range (254–330 nm). Analytical HPLC analyses were performed on a SupelcoSIL LC18 column (5 μm, 4.6mm x 25cm) with a linear elution gradient ranging from 0–100% ACN over 27 min, using a SupelcoSIL LC18 column (5 μm, 4.6mm x 25cm) at a flow rate of 1 mL/min. The purity of all tested compounds was ≥95% using the analytical method (LC/MS) described above unless stated otherwise.
Chemistry Synthetic Methods.
The reported pyrrole analogs were synthesized as shown in Schemes 1–5 using the following methods.
General procedure for intermediate (77a-h):
To a solution of acetylacetone (75) (10 mmol) in acetone (8 mL) was added solid potassium carbonate (10 mmol), followed by corresponding aryl bromide (12 mmol). The mixture was heated to 60 °C for 12 h. The solution was allowed to cool to room temperature (RT) and acidified with aq. HCl (10%, 20 mL). The mixture was extracted with Ethyl acetate (3 × 25 mL) and the combined organic fractions were washed with brine, dried over Na2SO4 and concentrated in vacuo. Flash column chromatography [hexane:ethyl acetate 9:1 v/v] afforded 77a-h (35–40% yield). Compounds 77c, e, f and h were purified as described then used in the next step without characterization. The NMR data for the remaining intermediates is provided below.
3-(4-ethoxy-benzyl)-pentane-2,4-dione (77a):
1H NMR (300 MHz, CDCl3): δ 7.15 – 6.94 (m, 2H), 6.93 – 6.71 (m, 2H), 4.09 – 3.89 (m, 2.6H), 3.61 (s, 1.6H), 3.09 (d, J = 7.6 Hz, 0.8 H), 2.14 (s, 1.6 H), 2.10 (s, 4.4H), 1.47 – 1.33 (m, 3H). (mixture of keto-enol forms) MS (ESI+) m/z: 235.2
3-(4-trifluoromethyl)-pentane-2,4-dione (77b):
1H NMR (300 MHz, CDCl3): δ7.52 −7.48 (m, 2H), 7.33 – 7.29 (m, 2H), 4.0 (t, J = 7.6 Hz, 0.5H) 3.8 (s, 1H), 3.2 (d, J = 7.6 Hz, 1H) 2.10 (s, 2.7H), 2.05 (s, 3.3H) (mixture of keto-enol forms) MS (ESI+) m/z: 259.1
3-(4-methoxy-benzyl)-pentane-2,4-dione (77d):
1H NMR (300 MHz, CDCl3): δ 7.19 – 7.10 (m, 2H), 6.94 – 6.73 (m, 2H), 3.99 (t, J = 7.6 Hz, 0.25 H), 3.81 (s, 3H), 3.61 (s, 1.5H), 3.1 (d, J = 7.6Hz, 0.5 H) 2.14 (s, 1.6 H), 2.10 (s, 4.4 H). (mixture of keto-enol forms) MS (ESI+) m/z: 221.2
3-(6-(trifluoromethyl)pyridin-3-yl)-pentane-2,4-dione (77g):
1H NMR (300 MHz, CDCl3): δ8.60 (d, J = 7.7 Hz, 1H), 7.77 – 7.51 (m, 2H), 4.02 (t, J = 7.4 Hz, 0.23 H), 3.78 (s, 1.66 H), 3.24 (d, J = 7.4 Hz, 0.64 H), 2.21 (s, 1.69H), 2.10 (s, 4.31H). (mixture of keto-enol forms)MS (ESI+) m/z: 260.2
Ethyl-2-(hydroxyimino)-3-oxobutanoate 78:
To a solution of ethyl acetoacetate (76) (5g, 38 mmol) in 6 ml of acetic acid was added a solution of sodium nitrate (3.4g, 49 mmol) in 6 ml of cold water at −10 °C. The reaction mixture was stirred for 3 h. A saturated aqueous solution of sodium bicarbonate was added to adjust the pH to 7–8, washed with brine and the mixture was extracted with diethyl ether. Then dried over anhydrous sodium sulphate, filtered and concentrated and then purified by flash chromatography on silica gel [hexane:ethyl acetate 10:1 v/v] to give ethyl-2-hydroxyimino-3-oxobutanoate (78) as pale yellow oil (4g, 80% yield).
1H NMR (300 MHz, CDCl3): δ 4.15 (q, J = 7.0 Hz, 2 H), 2.30 (s, 3H), 1.92 (brs, 1H), 1.33 (t, J = 7.0 Hz, 3H)
General procedure for Knorr pyrrole synthesis of pyrroles (3–10):
Freshly prepared ethyl-2-(hydroxyimino)-3-oxobutanoate (78) (4 mmol) and corresponding acetylacetone (77a-h) (4 mmol) was dissolved in glacial acetic acid (25 mL) at 15 °C. Zn dust (10 mmol) was then slowly added to the mixture and allowed to react at RT with vigorous stirring for 15–20 min. The resulting mixture was heated to 60 °C for 2 h. The mixture was allowed to attain RT and poured into ice water. The aqueous phase was extracted with ethyl acetate (3×25ml), and the combined organic phases were washed with brine, dried (Na2SO4), filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (eluent: gradient, hexane/EtOAc = 95:5 to 70:30) to give the corresponding pyrroles 3–10 (60–70% yield)
Ethyl 4-(4-ethoxybenzyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (3)
(DSM43, 65% yield, white solid, mp: 82 – 84 °C): 1H NMR (500 MHz, CDCl3): δ 8.61 (brs, 1H), 7.01 (d, J = 8.7 Hz, 2H), 6.80 (d, J = 8.7 Hz, 2H), 4.32 (q, J = 7.1Hz, 2H), 4.01 (q, J = 7.0 Hz, 2H), 3.70 (s, 2H), 2.23 (s, 3H), 2.19 (s, 3H), 1.39 (dt, J =12.8 Hz, 7.1 Hz, 6H). MS (ESI+) m/z: 302.3
Ethyl 3,5-dimethyl-4-(4-(trifluoromethyl)benzyl)-1H-pyrrole-2-carboxylate (4)
(DSM483, 60% yield, off white solid): 1H NMR (500 MHz, CDCl3): δ 8.63 (brs, 1H), 7.51 (d, J = 8.1 Hz, 2H), 7.22 (d, J = 8.0 Hz, 2H), 4.32 (q, J = 7.1 Hz, 2H), 3.83 (s, 2H), 2.22 (s, 3H), 2.20 (s, 3H), 1.37 (t, J = 7.1 Hz, 3H). MS (ESI+) m/z: 326.3
Ethyl 4-(3-ethoxybenzyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (5)
(DSM484, 60% yield, white solid, mp: 114 – 116 °C): 1H NMR (500 MHz, CDCl3): δ 8.60 (brs, 1H), 7.18 (t, J = 7.9 Hz, 1H), 6.72 (dd, J = 7.9 Hz, 3Hz, 2H), 6.66 (s, 1H), 4.33 (q, J = 7.1 Hz, 2H), 3.75 (q, J = 7.0 Hz, 2H), 3.75 (s, 2H), 2.25 (s, 3H), 2.21 (s, 3H), 1.40 (dt, J =19.0, 7.1 Hz, 6H). MS (ESI+) m/z: 302.5
Ethyl 4-(4-methoxybenzyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (6)
(DSM485, 70% yield, white solid, mp: 129 – 131 °C): 1H NMR (300 MHz, CDCl3): δ 8.63 (brs, 1H), 7.05 (d, J = 8.7 Hz, 2H), 6.82 (d, J = J = 8.8 Hz, 2H), 4.33 (q, J = 7.1 Hz, 2H), 3.80 (s, 3H), 3.72 (s, 2H), 2.24 (s, 3H), 2.19 (s, 3H), 1.35 (t, J = 7.3 Hz, 3H). MS (ESI+) m/z: 288.4
Ethyl 4-(3,5-dimethoxybenzyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (7)
(DSM486, 60% yield, off white solid): 1H NMR (300 MHz, CDCl3): δ 8.50 (brs, 1H), 7.20 (s, 2H), 6.20 (s, 1H), 4.43 (q, J = 7.1 Hz, 2H), 3.68 (s, 6H), 3.62 (s, 2H), 2.15 (s, 3H), 2.11 (s, 3H), 1.38 (t, J = 7.1 Hz, 3H) MS (ESI−) m/z: 316.7
Ethyl 4-(4-isopropylbenzyl)-3,5-dimethyl-1H-pyrrole-2-carboxylate (8)
(DSM487, 70% yield, light yellow solid, mp:113 – 115 °C): 1H NMR (300 MHz, CDCl3): δ 8.67 (brs, 1H), 7.13 (d, J = 8.0 Hz, 2H), 7.04 (d, J = 8.0 Hz, 2H), 4.33 (q, J = 7.1 Hz, 2H), 3.76 (s, 2H), 2.96–2.79 (m, 1H), 2.25 (s, 3H), 2.21 (s, 3H), 1.37 (t, J = 7.1 Hz, 3H), 1.25 (d, J = 6.9 Hz, 6H). MS (ESI+) m/z: 300.3
Ethyl 3,5-dimethyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxylate (9)
(DSM491, 60% yield, off white solid): 1H NMR (300 MHz, CDCl3): δ 8.75 (brs, 1H), 8.56 (s, 1H), 7.68–7.48 (m, 2H), 4.31 (q, J =7.1 Hz, 2H), 3.84 (s, 2H), 2.81 (s, 3H), 2.21 (s, 3H), 1.37 (t, J = 7.1 Hz, 3H). MS (ESI+) m/z: 327.3
Ethyl 3,5-dimethyl-4-((5-(trifluoromethyl)pyridin-2-yl)methyl)-1H-pyrrole-2-carboxylate (10)
(DSM520, 60% yield, yellow solid): 1H NMR (300 MHz, CDCl3): δ 8.95 (s, 1H), 8.83 (brs, 1H), 8.00 (d, J = 8.2 Hz, 1H), 7.24 (d, J = 8.3 Hz, 1H), 4.34 (q, J = 7.1 Hz, 2H), 4.15 (s, 2H), 2.22(s, 3H), 2.21 (s, 3H), 1.38 (t, J = 7.1 Hz, 3H). MS (ESI+) m/z: 327.1
Ethyl 1,3,5-trimethyl-4-(4-(trifluoromethyl)benzyl)-1H-pyrrole-2-carboxylate (11)
(DSM490): To a solution of 4 mg (0.015 mmol) of 18-crown-6 in 10 mL of benzene was added 17 mg (0.153 mmol) of potassium tert-butoxide and 50 mg (0.15 mmol) of 4. The reaction mixture was stirred for 15 min. Iodomethane 0.01 mL (0.169 mmol) dissolved in 1 mL of benzene was added dropwise to the reaction mixture. Stirring was continued for 6 h. The reaction was quenched with water (20 mL) and the mixture was extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous Na2SO4 and evaporated to give 11 as a white solid (25mg, 48% yield). 1H NMR (300 MHz, CDCl3): δ 7.51 (d, J = 8.1 Hz, 2H), 7.21 (d, J = 7.9 Hz, 2H), 4.31 (q, J = 7.1 Hz, 2H), 3.83 (s, 2H), 3.82 (s, 3H), 2.22 (s, 3H), 2.14 (s, 3H), 1.38 (t, J = 7.1 Hz, 3H). MS (ESI+) m/z: 340.4
General Procedure for the Preparation of 12–25.
To a solution of pyrrole ester (3–10) (1 equiv) and NaOH (4equiv) in EtOH: water (8:2 mL) were heated to 80 °C for 2–4 h. When the reaction was complete, as determined by TLC, water (10 mL) was added to the residue and neutralized with 1 M aqueous HCl. The precipitate was extracted with EtOAc (3×100 mL), the organic extracts were washed with water, brine and dried with Na2SO4 and concentrated in vacuo. To this residue (acid, 1equiv) was added DMF (2.5 to 5 mL), trimethylamine (4 equiv), EDC (1.25 equiv) and HOBt (1 equiv) and the mixture was stirred for 10 min. To this solution, appropriate amine (1 equiv) was added and stirring continued at RT for 4–6 h. The reaction mixture was poured into sat. aq NaHCO3 and extracted with EtOAc. The organic layer was separated, washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (eluted with 20−100% EtOAc in hexane) to afford the title compounds (12–25) typically a white or light yellow solid.
3,5-Dimethyl-4-(4-(trifluoromethyl)benzyl)-1H-pyrrole-2-carboxylic acid (12)
(DSM489, 60% yield, red solid). 1H NMR (300 MHz, CDCl3): δ 8.78 (brs, 1H), 7.53 (d, J = 8.1 Hz, 2H), 7.23 (d, J = 8.0 Hz, 2H), 3.83 (s, 2H) 2.26 (s, 3H), 2.21 (s, 3H). MS (ESI−) m/z: 296.4
N-Ethyl-3,5-dimethyl-4-(4-(trifluoromethyl)benzyl)-1H-pyrrole-2-carboxamide (13)
(DSM496, 50% yieldd, yellow solid, mp: 188 – 190 °C). 1H NMR (300 MHz, CDCl3): δ 9.5 (brs, 1H), 7.49 (d, J = 8.19, Hz, 2H), 7.18 (d, J = 8.19 Hz, 2H), 5.60 (brs, 1H), 3.82 (s, 2H), 3.46 (qd, J = 7.2, 5.8 Hz, 2H), 2.18 (s, 3H), 2.13 (s, 3H), 1.22 (t, J = 7.2 Hz, 3H). MS (ESI+) m/z: 325.2
3,5-Dimethyl-N-propyl-4-(4-(trifluoromethyl)benzyl)-1H-pyrrole-2-carboxamide (14)
(DSM495, 50% yield, off white solid). 1H NMR (300 MHz, CDCl3): δ 9.5 (brs, 1H), 7.52 (d, J = 8.1 Hz, 2H), 7.22 (d, J = 7.9 Hz, 2H), 5.68 (t, J = 5.2 Hz, 1H), 3.83 (s, 2H), 3.43 (dd, J = 13.0, 6.9 Hz, 2H), 2.21 (s, 3H), 2.16 (s, 3H), 1.70–1.55 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H). MS (ESI+) m/z: 339.2
N-Butyl-3,5-dimethyl-4-(4-(trifluoromethyl)benzyl)-1H-pyrrole-2-carboxamide (15)
(DSM494, 50% yield, off white solid, mp: 134 – 136 °C). 1H NMR (300 MHz, CDCl3): δ 8.96 (brs, 1H), 7.50 (d, J = 8.1, 2H), 7.20 (d, J = 7.6, 2H), 5.63 (t, J = 5.6 Hz, 1H), 3.81 (s, 2H), 3.49–3.38 (m, 2H), 2.18 (s, 3H), 2.12 (s, 3H), 1.63–1.31 (m, 4H), 0.95 (t, J = 7.1, 3H). MS (ESI+) m/z: 353.4
N-Ethyl-3,5-dimethyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (16)
(DSM497, 60% yield, off white solid, mp: 171 – 173 °C). 1H NMR (300 MHz, CDCl3): δ 9.67 (brs, 1H), 8.58 (s, 1H), 7.71 – 7.41 (m, 2H), 5.64 (brs, 1H), 3.86 (s, 2H), 3.50 (td, J = 12.8, 7.2 Hz, 2H), 2.23 (s, 3H), 2.15 (s, 3H), 1.25 (t, J = 7.2 Hz, 3H). MS (ESI+) m/z: 326.3
3,5-Dimethyl-N-propyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (17)
(DSM498, 55% yield, off white solid, mp: 155 – 157 °C). 1H NMR (300 MHz, CDCl3): δ 9.83 (brs, 1H), 8.58 (s, 1H), 7.55 (dd, J = 17.1, 7.8 Hz, 2H), 5.70 (brs, 1H), 3.85 (s, 2H), 3.43 (dd, J = 13.1, 6.7 Hz, 2H), 2.22 (s, 3H), 2.15 (s, 3H), 1.76 – 1.53 (m, 2H), 0.98 (t, J = 7.4 Hz, 3H). MS (ESI+) m/z: 340.3
N-Cyclopropyl-3,5-dimethyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (18)
(DSM499, 50% yield, off white solid, mp: 176 – 178 °C). 1H NMR (300 MHz, CDCl3): δ 9.03 (brs, 1H), 8.55 (s, 1H), 7.55 (d, J = 8.1 Hz, 1H), 7.48 (d, J = 8.2 Hz, 1H), 5.72 (brs, 1H), 3.82 (s, 2H), 2.84 (td, J = 7.0, 3.0 Hz, 1H), 2.19 (s, 3H), 2.07 (s, 3H), 0.90 – 0.75 (m, 2H), 0.62 – 0.52 (m, 2H). MS (ESI+) m/z: 338.2
N-Cyclobutyl-3,5-dimethyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (19)
(DSM500, 50% yield, off white solid, mp: 177 – 179 °C). 1H NMR (300 MHz, CDCl3): δ 9.89 (brs, 1H), 8.56 (s, 1H), 7.62 – 7.44 (m, 2H), 5.82 (d, J = 7.7 Hz, 1H), 4.74 – 4.44 (m, 1H), 3.85 (s, 2H), 2.52 – 2.33 (m, 2H), 2.21 (s, 3H), 2.17 (s, 3H), 2.02 – 1.84 (m, 2H), 1.83 – 1.66 (m, 2H). MS (ESI+) m/z: 352.3
Azetidin-1-yl(3,5-dimethyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrol-2-yl)methanone (20)
(DSM508, 35% yield, off white solid). 1H NMR (300 MHz, CDCl3): δ 8.87 (brs, 1H), 8.56 (s, 1H), 7.62 – 7.49 (m, 2H), 4.23 (t, J = 7.7 Hz, 4H), 3.83 (s, 2H), 2.52 – 2.27 (m, 2H), 2.20 (s, 3H), 2.06 (s, 3H). MS (ESI+) m/z: 338.4
N,N-Diethyl-3,5-dimethyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (21)
(DSM509, 45% yield, red solid). 1H NMR (300 MHz, CDCl3): δ 8.82 (brs, 1H), 8.56 (s, 1H), 7.61 – 7.50 (m, 2H), 3.82 (s, 2H), 3.52 (q, J = 7.32 Hz, 4H), 2.18 (s, 3H), 2.05 (s, 3H), 1.18 (t, J = 7.32, 6H). MS (ESI+) m/z: 354.2
N,N,3,5-Tetramethyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (22)
(DSM517, 40% yield, red solid, mp: 150 −152 °C). 1H NMR (500 MHz, CDCl3): δ 9.23 (brs, 1H), 8.55 (s, 1H), 7.60 – 7.50 (m, 2H), 3.82 (s, 2H), 3.09 (s, 6H), 2.18 (s, 3H), 1.94 (s, 3H). MS (ESI+) m/z: 326.2
N-Cyclopropyl-3,5-dimethyl-4-((5-(trifluoromethyl)pyridin-2-yl)methyl)-1H-pyrrole-2-carboxamide (23)
(DSM519, 50% yield, light yellow solid, mp: 182 – 184 °C). 1H NMR (300 MHz, CDCl3): δ 9.49 (brs, 1H), 8.80 (s, 1H), 7.80– 7.75 (m, 1H), 7.11 – 7.0 (m, 1H), 5.81 (brs, 1H), 4.02 (s, 2H), 2.88 – 2.82 (m, 1H), 2.24 (s, 3H), 2.13 (s, 3H), 0.88 – 0.81 (m, 2H), 0.74 – 0.69 (m 2H). MS (ESI+) m/z: 338.2
Azetidin-1-yl(3,5-dimethyl-4-((5-(trifluoromethyl)pyridin-2-yl)methyl)-1H-pyrrol-2-yl)methanone (24)
(DSM521, 40% yield, yellow solid, mp: 114–116 °C). 1H NMR (300 MHz, CDCl3): δ 8.83 (brs, 1H), 8.80 (s, 1H), 7.78 (dd, J = 8.2, 1.9 Hz, 1H), 7.08 (d, J = 8.2 Hz, 1H), 4.21 (t, J = 7.45 Hz, 4H), 4.0 (s, 2H), 2.44 – 2.27 (m, 2H), 2.20 (s, 3H), 2.07 (s, 3H). MS (ESI+) m/z: 338.3
N-Cyclobutyl-3,5-dimethyl-4-((5-(trifluoromethyl)pyridin-2-yl)methyl)-1H-pyrrole-2-carboxamide (25)
(DSM522, 50% yield, light red solid, mp: 78 – 80 °C). 1H NMR (300 MHz, CDCl3): δ 9.60 (brs, 1H), 8.82 (s, 1H), 7.79 (dd, J = 8.2, 1.8 Hz, 1H), 7.08 (d, J = 8.2 Hz, 1H), 5.81 (d, J = 7.5 Hz, 1H), 4.68 – 4.49 (m, 1H), 4.04 (s, 2H), 2.53 – 2.32 (m, 2H), 2.23 (s, 3H), 2.21 (s, 3H), 2.02 – 1.84 (m, 2H), 1.83 – 1.67 (m, 2H). MS (ESI+) m/z: 352.4
Friedel-Crafts acetylation of pyrroles: 82, 83a,b, 84a,b, 85, 86.
Appropriately substituted ethyl pyrrole-2-carboxylate (79–81) (1 equiv) and aroyl chloride (1.5 equiv) were dissolved in anhydrous CH2Cl2 (50 mL) under argon and cooled to 0 °C in an ice/water bath. AlCl3 (3 equiv) was added portion wise to the reaction mixture and stirred at RT for 16 h. The completion of reaction was determined by UPLC. Then, reaction mixture was poured into ice water (50 mL) slowly. The resulting reaction mixture was extracted with dichloromethane (2×150 mL). The combined organic layer was washed with saturated sodium bicarbonate solution, dried (Na2SO4) and concentrated. The resulting concentrated product was purified by column chromatography using 10–50% ethyl acetate in petroleum ether to afford 82, 83a,b, 84a,b, 85 and 86 (40 – 50% yield).
Partial reduction of ketone: 87, 88a,b, 89a,b, 90, 91:
Sodium borohydride (1.5 equiv) was added slowly to a solution of appropriate ketone 82, 83a,b, 84a,b, 85, 86 (1 equiv) in ethanol (6 mL) at 0 °C. The reaction mixture was stirred 1 h at RT. Then, the reaction mixture was concentrated under reduced pressure, dissolved in water and extracted with ethyl acetate (2×30 mL). The combined organic layers were washed with brine solution, dried over Na2SO4 and concentrated to afford compounds 87, 88a,b, 89a,b, 90 and 91 respectively (80 – 90% yield). Product was taken for next step without any purification.
Silane reduction in acidic media 92, 93a,b, 94a,b, 95 and 67.
To a stirred solution of compound 87, 88a,b, 89a,b, 90 and 91 (1 equiv) in TFA (4 equiv) was added triethylsilane (2 equiv). The reaction mixture was stirred at 85 °C for 1 h. Then, saturated NaHCO3 (10 mL) was added dropwise to the reaction mixture and was extracted with ethyl acetate (3×20 mL, with careful venting). The resulting ethyl acetate was dried (Na2SO4), filtered, and concentrated to dryness on a rotary evaporator. The resulting concentrated product was triturated with pet ether to afford compounds 92, 93a,b, 94a,b, 95 and 67 as white solid. (36 – 50% yield)
Ester hydrolysis: preparation of 96, 97a,b, 98a,b, and 99.
Compound 92, 93a,b, 94a,b and 95 (300 mg, 1.0 mmol) and NaOH (80 mg, 2.01 mmol) in EtOH: water (8:2 mL) were heated to 80 °C for 2 h. After concentration, water (10 mL) was added to the residue and acidified with 10% citric acid solution. The solid was collected on a filter, washed with water and dried to afford compounds 96, 97a,b, 98a,b, and 99 as off white solid. (60 – 80% yield)
General synthetic method for amides (26–51, 55, 56, and 68).
Pyrrole acid 96, 97a,b, 98a,b, and 99 (1 equiv) in dichloromethane (2 mL) was added to appropriate amine (1.1 equiv), triethylamine (2 equiv) and stirred for 5 min. Then, HATU (1.5 equiv) was added to the reaction mixture and stirred for 4 h at RT. After completion of starting material (monitored by TLC), water was added to reaction mixture and extracted with dichloromethane (2×30 mL). Combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The resulting concentrated product was purified by column chromatography using 10–50% ethyl acetate in pet ether to afford 26–51, 55, 56 and 68.
N-cyclopropyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (26)
(DSM501). (42% yield, off white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.26 (s, 1H), 8.65 (s, 1H), 7.81 – 7.87 (m, 3H), 6.75 (s, 1H), 6.53 (s, 1H), 3.87 (s, 2H), 2.69 – 2.74 (m, 1H), 0.62 – 0.65 (m, 2H), 0.46 –0.48 (m, 2H). MS (ESI+) m/z: 310.2.
N-Cyclopropyl-5-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (27)
(DSM503). (41%, off white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.13 (s, 1H), 8.62 (s, 1H), 7.79 –7.83 (m, 2H), 7.73 (d, J = 3.8Hz, 1H), 6.44 (d, J = 2.2 Hz, 1H), 3.80 (s, 2H), 2.68 – 2.74 (m, 1H), 2.16 (s, 3H), 0.59 – 0.63 (m, 2H), 0.44 – 0.47 (m, 2H). MS (ESI+) m/z: 324.2.
(5-Methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrol-2-yl)(pyrrolidin-1-yl) methanone (28)
(DSM506). (85% yield, off white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.15 (s, 1H), 8.64 (s, 1H), 8.78 –7.84 (m, 2H), 6.44(d, J = 2.4 Hz, 1H), 3.84 (s, 2H), 3.45 –3.59 (m, 4H), 2.15 (s, 3H), 1.80 –1.90 (m, 4H); MS (ESI+) m/z: 338.2.
(5-Methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrol-2-yl)(piperidin-1-yl) methanone (29)
(DSM507). (61% yield, off white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.11 (s, 1H), 8.62 (s, 1H), 7.78 –7.83 (m, 2H), 6.27 (d, J = 2.7 Hz, 1H), 3.81 (s, 2H), 3.58 (t, J = 5.3 Hz, 4H), 2.12 (s, 3H), 1.57 – 1.62 (m, 2H), 1.45 – 1.51 (m, 4H): MS (ESI+) m/z: 352.2.
N-Isopropyl-5-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (30)
(DSM511). (39% yield, off white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.10 (s, 1H), 8.64 (s, 1H), 7.82 (s, 2H), 7.50 (d, J = 8.0 Hz, 1H), 6.49 (s, 1H), 3.97 – 4.02 (m, 1H), 3.80 (s, 2H), 2.16 (s, 3H), 1.08 (d, J = 6.4 Hz, 6H): MS (ESI+) m/z: 326.2.
N-(tert-Butyl)-5-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (31)
(DSM512). (50% yield, off white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.03 (s, 1H), 8.63 (s, 1H), 7.81 (s, 2H), 7.05 (s, 1H), 6.51 (s, 1H), 3.80 (s, 2H), 2.15 (s, 3H), 1.32 (s, 9H): MS (ESI+) m/z: 340.2.
5-Methyl-N-(tetrahydro-2H-pyran-4-yl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (32)
(DSM513). (39% yield, off white solid). 1H NMR (400 MHz, DMSO d6) δ (ppm): 11.14 (s, 1H), 8.64 (s, 1H), 7.82 (s, 2H), 7.61 (d, J = 8.0 Hz, 1H), 6.51 (s, 1H), 3.86 – 3.90 (m, 7H), 2.16 (s, 3H), 1.67 – 1.70 (m, 2H), 1.45 – 1.49 (m, 2H): MS (ESI+) m/z: 368.2.
5-Methyl-N-(piperidin-4-yl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (33)
(DSM514). 1-Boc-4-(Amino)piperidine (127 mg, 0.63 mmol) and triethylamine (0.15 mL, 1.05 mmol) were added to compound 97a (150 mg, 0.52 mmol) in dichloromethane (4 mL) was added and stirred for 5 min. Then, HATU (301 mg, 0.79 mmol) was added to the reaction mixture and stirred at room temperature for 4 h. After completion of reaction (monitored by TLC), water was added to reaction mixture and extracted with dichloromethane (2×40 mL). The combined organic layer was dried over Na2SO4 and concentrated. The resulting concentrated product was purified by column chromatography using 10–90% ethyl acetate in petroleum ether to afford intermediate (100 mg, 41% yield). The obtained intermediate was dissolved in 1, 4-dioxane and added HCl in 1, 4-dioxane solution (4.5 M) at 0 °C for 2 h. Then, it was concentrated under reduced pressure and washed with acetonitrile and dried to afford 5-methyl-N-(piperidin-4-yl)-4-((6-(trifluoromethyl) pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (33) (69%, yield) as off white solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.28 (s, 1H), 8.89 (brs, 2H), 8.63 (s, 1H), 7.87 (d, J = 7.7 Hz, 1H), 7.82 (s, 2H), 6.55 (s, 1H), 3.95–3.97 (m, 1H), 3.81 (s, 2H), 3.25 – 3.28 (m, 2H), 2.95 – 2.97 (m, 2H), 2.08 (s, 3H), 1.87 – 1.90 (m, 2H), 1.70 – 1.72 (m, 2H); MS (ESI+) m/z: 367.2
5-Methyl-N-(1-methylpiperidin-4-yl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (34)
(DSM515). (45% yield, off white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.11 (s, 1H), 8.62 (s, 1H), 7.81 (brs, 2H), 7.54 (d, J = 8.0 Hz, 1H), 6.49 (s, 1H), 3.80 (s, 2H), 3.60 – 3.70 (m, 1H), 2.75 – 2.85 (m, 2H), 2.15 – 2.30 (m, 6H), 1.90 – 2.05 (m, 2H), 1.68 – 1.71 (m, 2H), 1.47 – 1.49 (m, 2H); MS (ESI+) m/z: 381.1.
N-(Azetidin-3-yl)-5-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (35)
(DSM518). tert-Butyl 3-aminoazetidine-1-carboxylate (109 mg, 0.63 mmol) and triethylamine (0.15 mL, 1.05 mmol) were added to Compound 97a (150 mg, 0.53 mmol) in dichloromethane (4 mL) and stirred for 5 min. Then HATU (301 mg, 0.79 mmol) was added to the reaction mixture and stirred at same temperature for 4 h. After completion of reaction (monitored by TLC), water was added to reaction mixture and extracted with dichloromethane (2×40 mL). The combined organic layer was dried over Na2SO4 and concentrated. The resulting concentrated product was purified by column chromatography using 10–60% ethyl acetate in petroleum ether to afford intermediate (120 mg, 51.9%). Then, the resulting obtained Intermediate was dissolved in 1, 4-dioxane and added HCl in dioxane solution (4.5 M) at 0 °C and stirred for 2 h. Then, it was concentrated under reduced pressure and washed with acetonitrile and dried to afford N-(azetidin-3-yl)-5-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (35) . (52% yield, off white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.32 (s, 1H), 8.79 – 8.81 (m, 2H), 8.62 (s, 1H), 8.44 (d, J = 7.1 Hz, 1H), 7.83 (s, 1H), 7.81 (d, J = 7.1 Hz, 1H), 6.49 (s, 1H), 4.77 – 4.71 (m, 1H), 3.95 – 4.08 (m, 4H), 3.82 (s, 2H), 2.16 (s, 3H); MS (ESI+) m/z: 339.2;
5-Methyl-N-(1-methylazetidin-3-yl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (36)
(DSM524). (16% yield, off white solid) 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.16 (s, 1H), 8.64 (s, 1H), 8.04 (d, J = 6.1 Hz, 1H), 7.83 (s, 2H), 6.54 (s, 1H), 4.36 – 4.38 (m, 1H), 3.81 (s, 2H), 3.51 (brs, 2H), 2.88 (brs, 2H), 2.23 (s, 3H), 2.16 (s, 3H); MS (ESI+) m/z : 353.4.
cyclopropyl-3-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (37)
(DSM502). (44% yield, off white solid) 1H NMR (400 MHz, DMSO-d6) δ 10.85 (s, 1H), 8.61 (s, 1H), 7.79 (s, 2H), 7.42 (d, J = 7.2 Hz, 1H), 6.66 – 6.67 (m, 1H), 3.83 (s, 2H), 2.69 – 2.74 (m, 1H), 2.10 (s, 3H), 0.63 – 0.68 (m, 2H), 0.43–0.47 (m, 2H); MS (ESI+) m/z: 324.2. See Fig. S4 for the LC/MS data demonstrating purity and mass.
Azetidin-1-yl(3-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrol-2-yl)methanone (38)
(DSM525). (25% yield, off white solid). 1H NMR (300 MHz, CDCl3): δ 8.90 (brs, 1H), 8.60 (s, 1H), 7.75 – 7.51 (m, 2H), 6.64 (d, J = 2.6 Hz, 1H), 4.25 (t, J = 7.45 Hz, 4H), 3.87 (s, 2H), 2.51 – 2.27 (m, 2H), 2.10 (s, 3H). MS (ESI+) m/z: 324.8.
3-Methyl-N-(2,2,2-trifluoroethyl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (39)
(DSM528). (30% yield, off white solid). 1H NMR (500 MHz, CDCl3): δ 9.30 (brs, 1H), 8.62 (s, 1H), 7.68 – 7.59 (m, 2H), 6.73 (d, J = 2.93 Hz 1H), 5.92 (t, J = 5.86 Hz, 1H), 4.20 – 4.09 (m, 2H), 3.92 (s, 2H), 2.24 (s, 3H). MS (ESI+) m/z: 366.4.
N-Isopropyl-3-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (40)
(DSM529). (61% yield, off white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.93 (s, 1H), 8.62 (s, 1H), 7.79 (s, 2H), 7.13 (d, 1H, J = 7.6 Hz), 6.67 (brs, 1H), 3.96–4.01 (m, 1H), 3.83 (s, 2H), 2.11 (s, 3H), 1.11 (d, 6H, J = 6.4 Hz); MS (ESI+) m/z: 326.2.
N-(tert-Butyl)-3-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (41)
(DSM530). (34% yield, white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.01 (s, 1H), 8.62 (s, 1H), 7.79 (s, 2H), 6.78 (brs, 1H), 6.67 (s, 1H), 3.83 (s, 2H), 2.10 (s, 3H), 1.33 (s, 9H); MS (ESI+) m/z: 340.2.
(3-Methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrol-2-yl)(pyrrolidin-1-yl) methanone (42)
(DSM531). (64% yield, off white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.82 (s, 1H), 8.64 (s, 1H), 7.82 (brs, 2H), 6.62 (brs, 1H), 3.84 (s, 2H), 3.42 (t, J = 6.8 Hz, 4H), 1.97 (s, 3H), 1.79 – 1.82 (m, 4H); MS (ESI+) m/z: 338.2.
1-(3-Methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrol-2-yl)butan-1-one (43)
(DSM532):(supplementary scheme 1) Step1: A mixture of pyrrole ester (93b, 200 mg, 0.64 mmol) and sodiumhydroxide (50mg, 1.28 mmol) in 10 mL of ethylene glycol was slowly heated to 200 °C over a 2-h period, while the distillate was collected. The mixture was then cooled to 20°C, 10 mL of water was added, and the aqueous mixture was extracted with 3×20 mL of chloroform. The pooled extracts were dried over Na2S04. Evaporation of the solvent under reduced pressure afforded 111 mg (73%) of 3-methylpyrrole as a colorless oil that was directly used in the next step without further purification.
Step 2: To a stirred suspension of anhydrous AlC13 (1.5 eq) in 20 mL of 1,2-dichloroethane at 0 °C was added slowly propionyl chloride (1 eq). The resulting solution was stirred at 25 °C for 10 min, a solution of 3-methylpyrrole (0.8 eq) in 10 mL of 1,2-dichloroethane was added slowly, and the mixture was stirred at 25 °C for 4 h. The reaction mixture was poured into ice-water, and the aqueous layer was extracted with dichloromethane (3×50 mL). The combined organic layer was washed with brine, dried over anhydrous Na2SO4 and filtered, and the solvent was removed under reduced pressure. The crude product was chromatographed on silica gel using 20% ethyl acetate in hexane to afford 1-(3-Methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrol-2-yl)butan-1-one (43). (30 % yield, off white solid)
1H NMR (300 MHz, CDCl3): δ 9.17 (brs, 1H), 8.62 (s, 1H), 7.69 – 7.55 (m, 2H), 6.73 (d, J = 3.0 Hz, 1H), 3.91 (s, 2H), 2.75 (t, J = 7.3 Hz, 2H), 2.28 (s, 3H), 1.87 – 1.70 (m, 2H), 1.02 (t, J = 7.45 Hz, 3H). MS (ESI+) m/z: 311.3
Cyclopropyl(3-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrol-2-yl)methanone 44
(DSM536). (20 % yield, off white solid). 44 was prepared according to the procedure reported for compound 43, treating 3-methyl pyrrole intermediate with cyclopropane carbonylchloride and AlCl3, afforded 44. 1H NMR (300 MHz, CDCl3): δ 9.14 (brs, 1H), 8.63 (s, 1H), 7.73 – 7.54 (m, 2H), 6.72 (s, 1H), 3.92 (s, 2H), 2.51 – 2.39 (m, 1H), 2.37 (s, 3H), 1.32 – 1.15 (m, 2H), 1.08 – 0.94 (m, 2H). MS (ESI+) m/z: 309.3
(3,3-Difluoroazetidin-1-yl)(3-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrol-2-yl)methanone (45)
(DSM538). (35 % yield, off white solid, mp: 130 – 132 °C). 1H NMR (300 MHz, CDCl3): δ 9.21 (brs, 1H), 8.59 (s, 1H), 7.68 – 7.58 (m, 2H), 6.68 (d, J = 3.06 Hz, 1H), 4.48 (t, J = 12.27 Hz, 4H), 3.87 (s, 2H), 2.12 (s, 3H). MS (ESI+) m/z: 360.3
3-Methyl-N-(methylsulfonyl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (46)
(DSM539). (30% yield, off white solid). 1H NMR (500 MHz, CDCl3): δ 9.53 (brs, 1H), 8.61 (s, 1H), 8.28 (brs, 1H), 7.65 – 7.60 (m, 2H), 6.85 (s, 1H), 3.92 (s, 2H), 3.44 (s, 3H), 2.32 (s, 3H). MS (ESI+) m/z: 362.5
(3-Fluoroazetidin-1-yl)(3-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrol-2-yl)methanone (47)
(DSM546). (40% yield, off white solid, mp: 108 – 110 °C). 1H NMR (300 MHz, CDCl3): δ 9.71 (brs, 1H), 8.57 (s, 1H), 7.67 – 7.53 (m, 2H), 6.63 (d, J = 2.9 Hz, 1H), 5.54 – 5.14 (m, 1H), 4.60 – 4.17 (m, 4H), 3.85 (s, 2H), 2.09 (s, 3H). MS (ESI+) m/z: 342.2
N-(3,3-Difluorocyclobutyl)-3-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (48)
(DSM552). (40% yield, off white solid). 1H NMR (300 MHz, CDCl3): δ 9.64 (brs, 1H), 9.19 (s, 1H), 7.71 – 7.56 (m, 2H), 6.69 (d, J = 2.9 Hz, 1H), 3.89 (s, 2H), 3.17 – 3.01 (m, 2H), 2.73 – 2.48 (m, 2H), 2.21 (s, 3H). MS (ESI+) m/z: 374.0
N-(3-Fluorocyclobutyl)-3-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (49)
(DSM553). (60% yield, off white solid, mp: 174 – 176 °C). 1H NMR (500 MHz, CDCl3): δ 9.49 (brs, 1H), 8.60 (s, 1H), 7.75 – 7.47 (m, 2H), 6.67 (d, J = 2.8 Hz, 1H), 5.84 (d, J = 5.8 Hz, 1H), 5.46 – 5.07 (m, 1H), 4.90 – 4.55 (m, 1H), 3.90 (s, 2H), 2.96 – 2.61 (m, 2H), 2.50 – 2.32 (m, 2H), 2.20 (s, 3H). MS (ESI+) m/z: 356.2
3-Methyl-N-(1-(trifluoromethyl)cyclopropyl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (50)
(DSM559). (40% yield, off white solid). 1H NMR (500 MHz, CDCl3): δ 9.20 (brs, 1H), 8.62 (s, 1H), 7.65 – 7.58 (m, 2H), 6.72 (d, J = 2.8 Hz, 1H), 6.11 (brs, 1H), 3.91 (s, 2H), 2.23 (s, 3H), 1.51 – 1.38 (m, 2H), 1.28 – 1.20 (m, 2H). MS (ESI+) m/z: 392.2
N-Cyclobutyl-3-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (51)
(DSM560). (60% yield, off white solid). 1H NMR (500 MHz, CDCl3): δ 9.82 (brs, 1H), 8.59 (s, 1H), 7.68 – 7.49 (m, 2H), 6.62 (d, J = 2.9 Hz, 1H), 6.01 (d, J = 7.6 Hz, 1H), 4.67 – 4.44 (m, 1H), 3.88 (s, 2H), 2.51 – 2.35 (m, 2H), 2.20 (s, 3H), 1.99 – 1.85 (m, 2H), 1.80 – 1.69 (m, 2H). MS (ESI+) m/z: 338.3
4-methoxy-1-(6-(trifluoromethyl)pyridin-3-yl)but-2-yn-1-one (101).
To a solution of methyl propargyl ether 100 (4 g, 57.14 mmol) in THF (60 mL), was added n-BuLi (1.6 M in hexane) (25 mL, 62.85 mmol) at −78 °C and stirred for 40 min at −78 °C. Then, N-methoxy-N-methyl-6-(trifluoromethyl)nicotinamide (10.7 g, 45.7 mmol) was added to the reaction mixture at −78°C and was stirred for 30 min at the same temperature. The reaction mixture was quenched with saturated NH4Cl solution and extracted with ethyl acetate (2×300 mL) and washed with brine solution, dried over Na2SO4 and concentrated. The resulting concentrated product was purified by column chromatography using 20–50% ethyl acetate in pet ether to afford 4-methoxy-1-(6-(trifluoromethyl)pyridin-3-yl)but-2-yn-1-one (101; 6 g, 43% yield) as brown oil.
3-(methoxymethyl)-4-(6-(trifluoromethyl)nicotinoyl)-1H-pyrrole-2-carboxylate 102.
To a stirred solution of compound 101 (6 g, 24.69 mmol) in NMP (30 mL) were added silver carbonate (0.68 g, 2.47 mmol) and ethyl isocyanoacetate (4.2 g, 37.04 mmol) at RT. It was stirred 2 h at 85 °C. The reaction mixture was taken in water and extracted with ethyl acetate (2×200 mL). The resulting organic layer was washed with brine solution, dried over Na2SO4 and concentrated under reduced pressure. The resulting concentrated product was purified by column chromatography using 5–40% ethyl acetate in pet ether to afford ethyl 3-(methoxymethyl)-4-(6-(trifluoromethyl)nicotinoyl)-1H-pyrrole-2-carboxylate (102; 4.7 g, 54% yield).
Ethyl 4-(hydroxy(6-(trifluoromethyl)pyridin-3-yl)methyl)-3-(methoxymethyl)-1H-pyrrole-2-carboxylate (103).
Sodium borohydride (0.76 g, 19.8 mmol) was added to a stirred solution of compound 102 (4.7 g, 13.20 mmol) in ethanol (25 mL) and stirred 1 h at RT. The reaction mixture was concentrated under reduced pressure, taken in water and extracted with ethyl acetate (2×30 mL). The resulting organic layer was washed with brine solution, dried over Na2SO4 and concentrated to afford ethyl 4-(hydroxy(6-(trifluoromethyl)pyridin-3-yl)methyl)-3-(methoxymethyl)-1H-pyrrole-2-carboxylate (103; 4.5 g, 95% yield).
Ethyl 3-(methoxymethyl)-4-((6-(trifluoromethyl)pyrimidi-3-yl)methyl)-1H-pyrrole-2-carboxylate (104).
To a stirred solution of 103 (4.5 g, 12.57 mmol) in TFA (9 mL, 75.4 mmol) was added triethylsilane (3.2 mL, 25.13 mmol). The reaction mixture was stirred at 85 °C for 1 h. The reaction mixture was quenched with saturated NaHCO3 solution and extracted with ethyl acetate. The combined organic layers were dried (Na2SO4), filtered and concentrated to dryness on a rotary evaporator. The resulting concentrated product was purified by column chromatography using 5–20% ethyl acetate in pet ether to afford 104 (3.6 g, 84% yield).
3-(methoxymethyl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxylic acid (105).
NaOH (0.84 g, 11.04 mmol) was added to a stirred solution of compound 104 (3.6 g, 10.52 mmol) and in EtOH: water (18:2 mL) was heated to 80 °C for 2 h. After concentration, water (10 mL) was added and acidified with 10% citric acid solution. The solid obtained was filtered, washed with water and dried to afford 3-(methoxymethyl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxylic acid, (105; 2.5 g, 75% yield) as off white solid.
N-Cyclopropyl-3-(methoxymethyl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (52)
(DSM534). Cyclopropyl amine (0.5 g, 9.55 mmol) was added to a solution of 105 (2.5 g, 7.96 mmol) and triethylamine (3 mL, 15.92 mmol) in dichloromethane (20 mL). Then, HATU (4.5 g, 11.94 mmol) was added to the reaction mixture and stirred for 4h at RT. After completion of starting (monitored by TLC), water was added to reaction mixture and extracted with dichloromethane (2×30 mL). Combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The resulting concentrated product was purified by column chromatography using 20–50% ethyl acetate in pet ether to afford N-cyclopropyl-3-(methoxymethyl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (52, 2.1 g, 73% yield) as off white solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.33 (s, 1H), 8.62 (s, 1H), 7.75–7.84 (m, 3H), 6.70 (d, 1H, J = 3.0 Hz), 4.39 (s, 2H), 3.90 (s, 2H), 3.14 (s, 3H), 2.73–2.77 (m, 1H), 0.67–0.72 (m, 2H), 0.40–0.44 (m, 2H); MS (ESI+) m/z: 354.2
N-cyclopropyl-3-(hydroxymethyl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide (53)
(DSM535). 1M BBr3 solution (0.5 g, 9.55 mmol) was added to a stirred solution of 52 (2.0 g, 7.96 mmol) in dichloromethane (20 mL) at 0 °C and it was stirred for 1h at same temperature. After completion of stating material (monitored by TLC), water was added to reaction mixture and extracted with dichloromethane (2×30 mL). Combined organic layers were dried over Na2SO4 and concentrated under reduced pressure to afford N-cyclopropyl-3-(hydroxymethyl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxamide. (53, 150 mg, 8%). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.26 (s, 1H), 8.26 (s, 1H), 8.43 (d, 1H, J= 3.6 Hz), 7.79 (s, 2H), 6.65 (s, 1H), 5.62 (t, 1H, J=4.4 Hz), 4.43 (d, 2H, J=4.4 Hz), 3.89 (s, 2H), 2.76–2.67 (m, 1H), 0.71–0.69 (m, 2H), 0.44–0.42 (m, 2H). MS (ESI+) m/z: 340.2
Perfluorophenyl 3-methyl-4-((6-(trifluoromethyl) pyridin-3-yl)methyl)-1H-pyrrole-2-carboxylate (106) :
Pentafluorophenol (0.15 mg, 0.845 mmol) was added to a stirred solution of 3-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxylic acid 98b (0.2 g, 1.53 mmol) and DCC (0.17 g, 0.84) in THF (5 mL) at RT. It was stirred for 1 h. The reaction mixture was filtered through and filtrate was concentrated under reduced pressure. The concentrated product was purified by flash chromatography (silica gel, eluting with hexane: EtOAc mixtures from 100% to 80:20%) to afford perfluorophenyl 3-methyl-4-((6-(trifluoromethyl) pyridin-3-yl)methyl)-1H-pyrrole-2-carboxylate (106; 0.16 mg, 48% yield) as white solid.
Perfluorophenyl 5-iodo-3-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxylate (107).
N-Iodosuccinimide (82 mg, 0.366 mmol) was added to a stirred solution of 106 (0.15 mg, 0.33 mmol) in DMF (1 mL) at 0 °C. The reaction mixture was stirred at RT for 1 h. After stirring for 1 h, the reaction mixture was poured into water and filtered and dried to afford perfluorophenyl 5-iodo-3-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxylate (107; 0.15 g, 79% yield) as off white solid.
Perfluorophenyl 3-methyl-5-(trifluoromethyl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxylate (108).
Methyl 2,2-difluoro-2-(fluorosulfonyl)acetate (100 mg, 0.52 mmol) was added to a solution of 107 (0.15 g, 0.26 mmol) and CuI (15 mg, 0.08 mmol) in NMP (2 mL) at RT and it was heated to 120 °C for 16 h. The reaction mixture was cooled to RT and water (10 mL) was added. It was extracted with ethyl acetate (2×30 mL), washed with brine solution, dried over Na2SO4 and concentrated. The resulting concentrated product was purified by column chromatography using 0–5% ethyl acetate in pet ether to afford perfluorophenyl 3-methyl-5-(trifluoromethyl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxylate (108); 15 mg, 11% yield) as white solid.
N-Cyclopropyl-3-methyl-5-(trifluoromethyl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2carboxamide (54)
(DSM526). Cyclopropyl amine (13 mg, 0.23 mmol) was added to a solution of 108 (60 mg, 0.12 mmol) in THF (1 mL) and stirred for 30 min. After 30 min. reaction mixture was concentrated and triturated with pet ether to afford N-cyclopropyl-3-methyl-5-(trifluoromethyl)-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2carboxamide (54, 6 mg, 55% yield) as white solid. 1H NMR (400 MHz, CD3OD) δ (ppm); 8.50 (s, 1H), 7.73 (s, 2H), 4.09 (s, 2H), 2.79 – 2.85 (m, 1H), 2.17 (s, 3H), 0.79 – 0.89 (m, 2H), 0.62 – 0.63 (m, 2H). MS (ESI+) m/z: 392.4
N-Cyclopropyl-5-methyl-4-((2-(trifluoromethyl)pyrimidin-5-yl)methyl)-1H-pyrrole-2-carboxamide (55)
(DSM527). (50% yield, off white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.16 (s, 1H), 8.88 (s, 2H), 7.72 (s, 1H), 6.41 (brs, 1H), 3.81 (s, 2H), 2.66 – 2.72 (m, 1H), 2.17 (s, 3H), 0.58 – 0.63 (m, 2H), 0.41 – 0.45 (m, 2H). MS (ESI+) m/z: 325.2;
N-Cyclopropyl-3-methyl-4-((2-(trifluoromethyl)pyrimidin-5-yl)methyl)-1H-pyrrole-2-carboxamide (56)
(DSM533). (50% yield, off white solid). 1HNMR (400 MHz, DMSO-d6) δ (ppm): 10.90 (s, 1H), 8.89 (s, 2H), 7.44 (s, 1H), 6.68 (s, 1H), 3.86 (s, 2H), 2.68 – 2.73 (m, 1H), 2.13 (s, 3H), 0.64 – 0.68 (m, 2H), 0.44 – 0.48 (m, 2H). MS (ESI+) m/z: 325.2
Compounds (114–118): 1-Propynylmagnesium bromide (0.5 M in THF) (1.1 equiv) was added to corresponding aryl aldehyde 109–113 (1 equiv) in THF (150 mL) at 0 °C and stirred for 2 h at RT. Reaction mixture was quenched with 1.5 N HCl solution and extracted with ethyl acetate (2×30 mL). The resulting organic layer was washed with brine, dried over Na2SO4 and concentrated to afford compounds 114–118 (11.2 g, 80 – 94% yield) as colorless liquid.
Compounds (119–123): Dess Martin (1.5 equiv) was added to stirred solution of compound 114–118 (1 equiv) in CH2Cl2 (15 mL) at RT and continued for 2 h. The reaction mixture was quenched with saturated sodium bicarbonate solution, extracted with CH2Cl2. The combined organic layers were dried (Na2SO4), filtered and concentrated. The concentrated product was purified by flash chromatography (silica gel, eluting with hexane: EtOAc mixtures from 100% to 80:20% to afford the title compounds 119–123 (72 – 82% yield) as an off white solid.
Compounds (124–128): compounds (119–123) (1 equiv) were added to stirred solution of silver carbonate (0.1 equiv) in NMP (10 mL) at RT. Ethyl isocyanoacetate (1.5 equiv) was added at RT and stirred for 2 h at 80 °C. Reaction mixture was cooled to RT, quenched with water (800 mL), and extracted with ethyl acetate (2×40 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated. The concentrated product was purified by flash chromatography (silica gel, eluting with hexane: EtOAc mixtures from 100% to 70:30%) to afford compounds 124–128; (35 – 41%, as an off white solid).
Ethyl 4-(2,6-difluoro-4-(trifluoromethyl)benzoyl)-3-methyl-1H-pyrrole-2-carboxylate (129).
Methyl 2, 2-difluoro-2-(fluorosulfonyl)acetate (1.3 g, 6.44 mmol) was added to a solution of 126 (1.2 g, 3.22 mmol), KI (53 mg, 0.32 mmol) and CuI (29 mg, 0.32 mmol) in NMP (2 mL) at RT. It was heated to 120 °C for 16 h. Reaction mixture was cooled to RT and water (20 mL) was added and extracted with ethyl acetate (2×30 mL). The resulting organic layer was washed with brine solution, dried over Na2SO4 and concentrated. The resulting concentrated product was purified by preparative HPLC to afford ethyl 4-(2,6-difluoro-4-(trifluoromethyl)benzoyl)-3-methyl-1H-pyrrole-2-carboxylate (129) (200 mg, 17% yield) as white solid. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.51 (s, 1H), 7.79 (s, 1H), 7.78 (s, 1H), 7.49 (s, 1H), 4.30 (q, J =7.1 Hz, 2H), 3.78 (s, 2H), 2.67 (s, 3H), 1.26 (t, J = 7.1 Hz, 3H).
Compounds 130–134 were prepared according to the reported procedure for 87, reducing 124, 125, 127, 128, 129 with sodium borohydride (60 – 75% yield).
Compounds 57, 59, 61, 63, 65 were prepared according to the procedure reported for compound 92, treating with triethylsilane-TFA.
Ethyl 3-methyl-4-((6-(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrole-2-carboxylate (57)
(DSM561) (0.1 g, 70%, white solid). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 11.36 (s, 1H), 7.63 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 6.77 (s, 1H), 4.21 (q, J = 7.2 Hz, 2H), 3.87 (s, 2H), 2.13 (s, 3H), 1.27 (t, J = 7.2 Hz, 3H); MS (ESI+) m/z: 312.2.
N-cyclopropyl-3-methyl-4-(4-(trifluoromethyl)benzyl)-1H-pyrrole-2-carboxamide (58)
(DSM562) (53% yield, off white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.80 (s, 1H), 7.61 (d, J = 8.0 Hz, 2H), 7.35 – 7.41 (m, 3H), 6.64 (s, 1H), 3.78 (s, 2H), 2.70 – 2.73 (m, 1H), 2.08 (s, 3H), 0.63 –0.67 (m, 2H), 0.45 –0.47 (m, 2H); MS (ESI+) m/z: 323.2;
Ethyl 4-(2-fluoro-4-(trifluoromethyl)benzyl)-3-methyl-1H-pyrrole-2-carboxylate (59)
(DSM556) (47% yield, white solid). 1H NMR (400 MHz, CDCl3) δ 8.85 (s, 1H), 7.30 – 7.33 (m, 2H), 7.19 – 7.23 (m, 1H), 6.68 (s, 1H), 4.33 (q, J = 7.1 Hz, 2H), 3.86 (s, 2H), 2.27 (s, 3H), 1.38 (t, J = 7.1 Hz, 3H). MS (ESI+) m/z: 330.2
N-Cyclopropyl-4-(2-fluoro-4-(trifluoromethyl)benzyl)-3-methyl-1H-pyrrole-2-carboxamide (60)
(DSM557) (36% yield, off white solid). 1H NMR (400 MHz, CDCl3) δ 9.13 (s, 1H), 7.30 –7.32 (m, 2H), 7.16 – 7.20 (m, 1H), 6.68 (s, 1H), 5.84 (s, 1H), 3.85 (s, 2H), 2.85 – 2.87 (m, 1H), 2.16 (s, 3H), 0.86 – 0.90 (m, 2H), 0.60 – 0.62 (m, 2H). MS (ESI+) m/z: 341.0;
Ethyl 4-(2,6-difluoro-4-(trifluoromethyl)benzyl)-3-methyl-1H-pyrrole-2-carboxylate 61
(DSM563) (77% yield, white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.37 (s, 1H), 7.61 (s, 1H), 7.60 (s, 1H), 6.57 (s, 1H), 4.20 (q, J = 7.2 Hz, 2H), 3.78 (s, 2H), 2.24 (s, 3H), 1.26 (t, J = 7.2 Hz, 3H); MS (ESI+) m/z: 348.2
N-cyclopropyl-4-(2,6-difluoro-4-(trifluoromethyl)benzyl)-3-methyl-1H-pyrrole-2-carboxamide (62)
(DSM564) (51% yield, off white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.82 (s, 1H), 7.62 (s, 1H), 7.60 (s, 1H), 7.45 (s, 1H), 6.47 (s, 1H), 3.75 (s, 2H), 2.71 – 2.72 (m, 1H), 2.21 (s, 3H), 0.64 – 0.66 (m, 2H), 0.44 – 0.46 (m, 2H); MS (ESI+) m/z: 359.0
Ethyl 4-(2-methoxy-4-(trifluoromethyl)benzyl)-3-methyl-1H-pyrrole-2-carboxylate (63)
(DSM566) (73% yield, white solid). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.79 (s, 1H), 7.07 – 7.14 (m, 3H), 6.64 (s, 1H), 4.34 (q, J = 7.2 Hz, 2H), 3.91 (s, 3H), 3.79 (s, 2H), 2.27 (s, 3H), 1.38 (t, J = 7.2 Hz, 3H); MS (ESI+) m/z: 342.2
N-cyclopropyl-4-(2-methoxy-4-(trifluoromethyl)benzyl)-3-methyl-1H-pyrrole-2-carboxamide (64)
(DSM567) (64% yield, off white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.79 (s, 1H), 7.42 (d, J = 3.6 Hz, 1H), 7.19 – 7.21 (m, 2H), 7.10 –7.12 (d, J = 7.7 Hz, 1H), 6.57 (s, 1H), 3.89 (s, 3H), 3.68 (s, 2H), 2.68 – 2.74 (m, 1H), 2.09 (s, 3H), 0.64 – 0.69 (m, 2H), 0.45 – 0.48 (m, 2H); MS (ESI+) m/z: 353.0;
Ethyl 4-(2-chloro-4-(trifluoromethyl)benzyl)-3-methyl-1H-pyrrole-2-carboxylate (65)
(DSM570) (75% yield, white solid). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.85 (s, 1H), 7.66 (s, 1H), 7.42 (d, J = 8.0 Hz, 1H), 7.20 (d, J = 8.0 Hz, 1H), 6.64 (s, 1H), 4.35 (q, J = 7.2 Hz, 2H), 3.93 (s, 2H), 2.26 (s, 3H), 1.38 (t, J = 7.2 Hz, 3H): MS (ESI+) m/z: 346.2
4-(2-chloro-4-(trifluoromethyl)benzyl)-N-cyclopropyl-3-methyl-1H-pyrrole-2-carboxamide (66)
(DSM571) (55% yield, off white solid). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.86 (s, 1H), 7.82 (s, 1H), 7.63 (d, 1H, J=7.2 Hz), 7.45 (s, 1H), 7.32 (d, 1H, J=7.2 Hz), 6.58 (s, 1H), 3.84 (s, 2H), 2.66–2.74 (m, 1H), 2.10 (s, 3H), 0.64–0.68 (m, 2H), 0.44–0.48 (m, 2H); ESIMS m/z: 357.0
Ethyl 4-((6-cyanopyridin-3-yl)methyl)-3-methyl-1H-pyrrole-2-carboxylate (67)
(DSM584) (50% yield, off white solid). 1H NMR (500 MHz, CDCl3): δ 9.06 (s, 1H), 8.60 (s, 1H), 7.68 – 7.48 (m, 2H), 6.68 (d, J = 3.0 Hz, 1H), 4.34 (q, J = 7.1 Hz, 2H), 3.89 (s, 2H), 2.22 (s, 3H), 1.38 (t, J = 7.1 Hz, 3H). MS (ESI+) m/z: 270.2
N-Cyclopropyl-3-methyl-4-(pyridin-3-ylmethyl)-1H-pyrrole-2-carboxamide (68)
(DSM585) (45% yield, light brown solid). 1H NMR (500 MHz, CDCl3): δ 8.40 (s, 1H), 8.13 (m, 3H), 7.64 (d, J = 8.2 Hz, 1H), 6.57 (s, 1H), 6.43 (s, 1H), 3.85 (s, 2H), 2.99 – 2.90 (m, 1H), 1.99 (s, 3H), 0.92 – 0.80 (m, 2H), 0.69 – 0.51 (m, 2H). MS (ESI+) m/z: 256.3
3-(6-(trifluoromethyl)pyridin-3-yloxy)pentane-2,4-dione (137):
A mixture of commercially available 3-Chloroacetylacetone (135) (0.2 ml, 1.84 mmol), 5-hydroxy-2-trifluoromethyl pyridine (136) (250 mg, 1.5 mmol), caesium carbonate (599 mg, 1.84 mmol) and acetone (10 ml) was heated under reflux for 5 h. After cooling, a solid was removed by filtration and it was washed with dichloromethane (25 ml), the combined filtrates were concentrated under reduced pressure. The remaining oil was diluted with dichloromethane (25 ml), washed with 1M hydrochloric acid (20 ml), then brine (20 ml), dried over sodium sulphate, filtered and finally concentrated under reduced pressure. The crude product was purified by flash chromatography on silica gel eluting with 0–10% ethyl acetate in hexane to provide the titled compound (180 mg, 45%) as a white solid (137).
Ethyl 3,5-dimethyl-4-(6-(trifluoromethyl)pyridine-3yloxy)-1H-pyrrole-2-carboxylate (138):
Freshly prepared ethyl-2-(hydroxyimino)-3-oxobutanoate (78) (4mmol) and corresponding acetylacetone (137) (4mmol) was dissolved in glacial acetic acid (25 ml) at 15 °C. Zn dust (10 mmol) was then slowly added to the mixture and allowed to react at RT with vigorous stirring for 15–20 min. The resulting mixture was heated to 60 °C for 2 h. The mixture was allowed to attain RT and was then poured into ice water. The aqueous phase was extracted with ethylacetate (3×25ml), and the combined organic phases were washed with brine, dried (Na2SO4), filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (eluent: gradient, hexane/EtOAc = 95:5 to 70:30) to give the pyrrole ester 138 (60% yield)
N-Cyclopropyl-3,5-dimethyl-4-(5-(trifluoromethyl)pyridin-2-yloxy)-1H-pyrrole-2-carboxamide (74)
(DSM537): NaOH (24mg, 0.6 mmol) was added to a stirred solution of compound 138 (200mg, 0.3 mmol) and in EtOH: water (18:2 mL) was heated to 80 °C for 2 h. After concentration, water (10 mL) was added and acidified with 10% citric acid solution. The solid obtained was filtered, washed with water and dried to afford carboxylic acid intermediate which was directly used in the next step without further purification. (80% yield)
Cyclopropyl amine (20mg, 0.36 mmol) was added to a solution of carboxylicacid intermediate (100mg, 0.33 mmol) and triethylamine (66μL, 0.66 mmol) in dichloromethane (5 mL). Then, HATU (188mg, 0.5 mmol) was added to the reaction mixture and stirred for 4h at RT. After completion of starting (monitored by TLC), water was added to reaction mixture and extracted with dichloromethane (2×30 mL). Combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The resulting concentrated product was purified by column chromatography using 20–50% ethyl acetate in pet ether to afford compound 74. (60% yield, off white solid, mp: 124 – 126 °C). 1H NMR (300 MHz, CDCl3): δ 10.32 (brs, 1H), 8.47 (s, 1H), 7.59 (d, J = 8.7 Hz, 1H), 7.22 (dd, J = 8.7, 2.8 Hz, 1H), 5.82 (s, 1H), 2.94 – 2.81 (m, 1H), 2.18 (s, 3H), 2.07 (s, 3H), 0.92 – 0.81 (m, 2H), 0.68 – 0.55 (m, 2H). MS (ESI+) m/z: 340.3.
Compounds 69, 71 and 73 were synthesized as shown in Supplementary Scheme2. Friedel-crafts acetylation of appropriate aryl acid chloride on corresponding pyrrole afforded compounds 69 and 70. Further hydrolysis of ester and amide coupling with cyclopropylamine gave aryl-pyrroleamides 71 and 72. Partial reduction of 72 with NaBH4 in ethanol afforded the hydroxy derivative 73.
Ethyl 3,5-dimethyl-4-(5-(trifluoromethyl)picolinoyl)-1H-pyrrole-2-carboxylate (69)
(DSM504, 45% yield, off white solid): 1H NMR (300 MHz, CDCl3): δ 9.12 (brs, 1H), 8.94 (s, 1H), 8.14 (d, J = 8.2 Hz, 1H), 8.00 (d, J = 8.2 Hz, 1H), 4.34 (q, J = 7.1 Hz, 2H), 2.31 (s, 3H), 2.21 (s, 3H), 1.37 (t, J = 7.1 Hz, 3H). MS (ESI+): 341.2.
N-Cyclopropyl-3,5-dimethyl-4-(5-(trifluoromethyl)picolinoyl)-1H-pyrrole-2-carboxamide (71)
(DSM505) (55% yield, light yellow solid). 1H NMR (300 MHz, CDCl3): δ 10.53 (brs, 1H), 8.92 (s, 1H), 8.13 (dd, J = 8.2, 1.7 Hz, 1H), 7.99 (d, J = 8.1 Hz, 1H), 5.94 (brs, 1H), 3.02 – 2.71 (m, 1H), 2.24 (s, 3H), 2.20 (s, 3H), 0.96 – 0.77 (m, 2H), 0.72 – 0.48 (m, 2H). MS(ESI+): 352.4
N-Cyclopropyl-4-(hydroxy(6-(trifluoromethyl)pyridin-3-yl)methyl)-3-methyl-1H-pyrrole-2-carboxamide (73)
(DSM558) (70% yield, light red liquid). 1H NMR (300 MHz, CDCl3): δ 9.44 (brs, 1H), 8.74 (s, 1H), 7.95 (d, J = 8.1 Hz, 1H), 7.69 (d, J = 8.1 Hz, 1H), 6.61 (d, J = 3.0 Hz, 1H), 5.95 (s, 1H), 5.89 (brs, 1H), 2.86 (qd, J = 7.1, 3.9 Hz, 1H), 2.23 (s, 3H), 0.96 – 0.81 (m, 2H), 0.66 – 0.54 (m, 2H). MS (ESI+) m/z: 340.1
Supplementary Material
Acknowledgments.
The authors thank Sibylle Sax (Swiss TPH) for technical assistance with the SCID mouse model. Results shown in this report are derived from work performed at the Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. This work was supported by funds from the United States National Institutes of Health grant, R01AI103947 (to MAP and PKR) and from Medicines for Malaria Venture (MMV). MAP acknowledges the support of the Welch Foundation (I-1257). MAP holds the Sam G. Winstead and F. Andrew Bell Distinguished Chair in Biochemistry.
Abbreviations.
- DHODH
dihydroorotate dehydrogenase
- Pf
Plasmodium falciparum
- Pv
Plasmodium vivax
- hDHODH
human DHODH
- mDHODH
mouse DHODH
- rDHODH
rat DHODH
- dDHODH
dog DHODH
- Pf3D7
P. falciparum 3D7 parasites
- PfDd2
P. falciparum Dd2 parasites
- PG
proguanil
- CQ
chloroquine
- Py
pyrimethamine
- SD
sulphadoxine
- MF
mefloquine
- CG
cycloguanil
Footnotes
PDB coordinates.
The coordinates for the structure of PfDHODH bound to 4 and 60 have been submitted to the Protein Data Bank (PDB 6VTY and 6VTN, respectively). Authors will release the atomic coordinates and experimental data upon article publication
References.
- 1.World Health Organization. Leading Causes of Death. https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death. (Accessed Feb. 27, 2020)
- 2.World Health Organization. World Malaria Report 2019. ISBN: 978-92-4-156572-1, Geneva, Switzerland. [Google Scholar]
- 3.Miller LH; Ackerman HC; Su XZ; Wellems TE, Malaria biology and disease pathogenesis: insights for new treatments. Nat Med 2013, 19 (2), 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phillips MA; Burrows JN; Manyando C; van Huijsduijnen RH; Van Voorhis WC; Wells TNC, Malaria. Nat Rev Dis Primers 2017, 3, 17050. [DOI] [PubMed] [Google Scholar]
- 5.White NJ; Pukrittayakamee S; Hien TT; Faiz MA; Mokuolu OA; Dondorp AM, Malaria. Lancet 2014, 383 (9918), 723–735. [DOI] [PubMed] [Google Scholar]
- 6.Schuerman L, RTS S malaria vaccine could provide major public health benefits. Lancet 2019, 394 (10200), 735–736. [DOI] [PubMed] [Google Scholar]
- 7.Adepoju P, RTS S malaria vaccine pilots in three African countries. Lancet 2019, 393 (10182), 1685. [DOI] [PubMed] [Google Scholar]
- 8.Haldar K; Bhattacharjee S; Safeukui I, Drug resistance in Plasmodium. Nat Rev Microbiol 2018, 16 (3), 156–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldberg DE; Siliciano RF; Jacobs WR Jr., Outwitting evolution: fighting drug-resistant TB, malaria, and HIV. Cell 2012, 148 (6), 1271–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tilley L; Straimer J; Gnadig NF; Ralph SA; Fidock DA, Artemisinin action and resistance in Plasmodium falciparum. Trends Parasitol 2016, 32 (9), 682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plewes K; Leopold SJ; Kingston HWF; Dondorp AM, Malaria: What’s new in the management of malaria? Infect Dis Clin North Am 2019, 33 (1), 39–60. [DOI] [PubMed] [Google Scholar]
- 12.Menard D; Dondorp A, Antimalarial drug resistance: a threat to malaria elimination. Cold Spring Harb Perspect Med 2017, 7 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hooft van Huijsduijnen R; Wells TN, The antimalarial pipeline. Curr Opin Pharmacol 2018, 42, 1–6. [DOI] [PubMed] [Google Scholar]
- 14.Charman SA; Arbe-Barnes S; Bathurst IC; Brun R; Campbell M; Charman WN; Chiu FC; Chollet J; Craft JC; Creek DJ; Dong Y; Matile H; Maurer M; Morizzi J; Nguyen T; Papastogiannidis P; Scheurer C; Shackleford DM; Sriraghavan K; Stingelin L; Tang Y; Urwyler H; Wang X; White KL; Wittlin S; Zhou L; Vennerstrom JL, Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc Natl Acad Sci U S A 2011, 108 (11), 4400–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moehrle JJ; Duparc S; Siethoff C; van Giersbergen PL; Craft JC; Arbe-Barnes S; Charman SA; Gutierrez M; Wittlin S; Vennerstrom JL, First-in-man safety and pharmacokinetics of synthetic ozonide OZ439 demonstrates an improved exposure profile relative to other peroxide antimalarials. Br J Clin Pharmacol 2013, 75 (2), 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuhen KL; Chatterjee AK; Rottmann M; Gagaring K; Borboa R; Buenviaje J; Chen Z; Francek C; Wu T; Nagle A; Barnes SW; Plouffe D; Lee MC; Fidock DA; Graumans W; van de Vegte-Bolmer M; van Gemert GJ; Wirjanata G; Sebayang B; Marfurt J; Russell B; Suwanarusk R; Price RN; Nosten F; Tungtaeng A; Gettayacamin M; Sattabongkot J; Taylor J; Walker JR; Tully D; Patra KP; Flannery EL; Vinetz JM; Renia L; Sauerwein RW; Winzeler EA; Glynne RJ; Diagana TT, KAF156 is an antimalarial clinical candidate with potential for use in prophylaxis, treatment, and prevention of disease transmission. Antimicrob Agents Chemother 2014, 58 (9), 5060–5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.White NJ; Duong TT; Uthaisin C; Nosten F; Phyo AP; Hanboonkunupakarn B; Pukrittayakamee S; Jittamala P; Chuthasmit K; Cheung MS; Feng Y; Li R; Magnusson B; Sultan M; Wieser D; Xun X; Zhao R; Diagana TT; Pertel P; Leong FJ, Antimalarial activity of KAF156 in falciparum and vivax malaria. N Engl J Med 2016, 375 (12), 1152–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rottmann M; McNamara C; Yeung BK; Lee MC; Zou B; Russell B; Seitz P; Plouffe DM; Dharia NV; Tan J; Cohen SB; Spencer KR; Gonzalez-Paez GE; Lakshminarayana SB; Goh A; Suwanarusk R; Jegla T; Schmitt EK; Beck HP; Brun R; Nosten F; Renia L; Dartois V; Keller TH; Fidock DA; Winzeler EA; Diagana TT, Spiroindolones, a potent compound class for the treatment of malaria. Science 2010, 329 (5996), 1175–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White NJ; Pukrittayakamee S; Phyo AP; Rueangweerayut R; Nosten F; Jittamala P; Jeeyapant A; Jain JP; Lefevre G; Li R; Magnusson B; Diagana TT; Leong FJ, Spiroindolone KAE609 for falciparum and vivax malaria. N Engl J Med 2014, 371 (5), 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phillips MA; Gujjar R; Malmquist NA; White J; El Mazouni F; Baldwin J; Rathod PK, Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite, Plasmodium falciparum. J Med Chem 2008, 51, 3649–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coteron JM; Marco M; Esquivias J; Deng X; White KL; White J; Koltun M; El Mazouni F; Kokkonda S; Katneni K; Bhamidipati R; Shackleford DM; Angulo-Barturen I; Ferrer SB; Jimenez-Diaz MB; Gamo FJ; Goldsmith EJ; Charman WN; Bathurst I; Floyd D; Matthews D; Burrows JN; Rathod PK; Charman SA; Phillips MA, Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J Med Chem 2011, 54 (15), 5540–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phillips MA; Lotharius J; Marsh K; White J; Dayan A; White KL; Njoroge JW; El Mazouni F; Lao Y; Kokkonda S; Tomchick DR; Deng X; Laird T; Bhatia SN; March S; Ng CL; Fidock DA; Wittlin S; Lafuente-Monasterio M; Benito FJ; Alonso LM; Martinez MS; Jimenez-Diaz MB; Bazaga SF; Angulo-Barturen I; Haselden JN; Louttit J; Cui Y; Sridhar A; Zeeman AM; Kocken C; Sauerwein R; Dechering K; Avery VM; Duffy S; Delves M; Sinden R; Ruecker A; Wickham KS; Rochford R; Gahagen J; Iyer L; Riccio E; Mirsalis J; Bathhurst I; Rueckle T; Ding X; Campo B; Leroy D; Rogers MJ; Rathod PK; Burrows JN; Charman SA, A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci Transl Med 2015, 7 (296), 296ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Llanos-Cuentas A; Casapia M; Chuquiyauri R; Hinojosa J-C; Kerr N; Rosario M; Toovey S; Arch RH; Phillips MA; Rozenberg FD; Bath J; Ng CL; Cowell AN; Winzeler EA; Fidock DA; Baker M; Möhrle JJ; Hooft van Huijsduijnen R; Gobeau N; Araeipour N; Andenmatten N; Rückle T; Duparc S, A Phase IIa study of antimalarial activity of single dose DSM265, a novel Plasmodium dihydroorotate dehydrogenase inhibitor, in patients with Plasmodium falciparum or P. vivax infection observed over 35 days in the Peruvian Amazon Lancet Infect Dis 2018, 18, 874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCarthy JS; Lotharius J; Rückle T; Chalon S; Phillips MA; Elliott S; Sekuloski S; Griffin P; Ng CL; Fidock DA; Marquart L; Williams NS; Gobeau N; Bebrevska L; Rosario M; Marsh K; Möhrle JJ, Safety, tolerability, pharmacokinetics, and activity of the novel long-acting antimalarial DSM265: a two-part first-in-human phase 1a/1b randomised study. Lancet Infect Dis 2017, 17 (6), 626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collins KA; Ruckle T; Elliott S; Marquart L; Ballard E; Chalon S; Griffin P; Mohrle JJ; McCarthy JS, DSM265 at 400 milligrams clears asexual stage parasites but not mature gametocytes from the blood of healthy subjects experimentally infected with Plasmodium falciparum. Antimicrob Agents Chemother 2019, 63 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phillips MA; White KL; Kokkonda S; Deng X; White J; El Mazouni F; Marsh K; Tomchick DR; Manjalanagara K; Rudra KR; Wirjanata G; Noviyanti R; Price RN; Marfurt J; Shackleford DM; Chiu FC; Campbell M; Jimenez-Diaz MB; Bazaga SF; Angulo-Barturen I; Martinez MS; Lafuente-Monasterio M; Kaminsky W; Silue K; Zeeman AM; Kocken C; Leroy D; Blasco B; Rossignol E; Rueckle T; Matthews D; Burrows JN; Waterson D; Palmer MJ; Rathod PK; Charman SA, A Triazolopyrimidine-Based Dihydroorotate Dehydrogenase Inhibitor with Improved Drug-like Properties for Treatment and Prevention of Malaria. ACS Infect Dis 2016, 2 (12), 945–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lynch JJ 3rd; Rossignol E; Moehrle JJ; Van Vleet TR; Marsh KC; Parman T; Mirsalis J; Ottinger SE; Segreti JA; Rao M; Mittelstadt SW, Increased stress associated with head-out plethysmography testing can exacerbate respiratory effects and lead to mortality in rats. J Pharmacol Toxicol Methods 2019, 106580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Booker ML; Bastos CM; Kramer ML; Barker RH Jr.; Skerlj R; Sidhu AB; Deng X; Celatka C; Cortese JF; Guerrero Bravo JE; Crespo Llado KN; Serrano AE; Angulo-Barturen I; Jimenez-Diaz MB; Viera S; Garuti H; Wittlin S; Papastogiannidis P; Lin JW; Janse CJ; Khan SM; Duraisingh M; Coleman B; Goldsmith EJ; Phillips MA; Munoz B; Wirth DF; Klinger JD; Wiegand R; Sybertz E, Novel inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in the mouse model. J Biol Chem 2010, 285 (43), 33054–33064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phillips MA, Antimalarial agents targeting nucleotide synthesis and electron transport: insight from structural biology In Neglected Diseases and Drug Discovery; Palmer M and Wells TNC, Eds; RCS Publishing: Cambridge, UK, 2011; pp 65.-. [Google Scholar]
- 30.Phillips MA; Rathod PK, Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect Disord Drug Targets 2010, 10 (3), 226–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flannery EL; Foquet L; Chuenchob V; Fishbaugher M; Billman Z; Navarro MJ; Betz W; Olsen TM; Lee J; Camargo N; Nguyen T; Schafer C; Sack BK; Wilson EM; Saunders J; Bial J; Campo B; Charman SA; Murphy SC; Phillips MA; Kappe SH; Mikolajczak SA, Assessing drug efficacy against Plasmodium falciparum liver stages in vivo. JCI Insight 2018, 3 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy SC; Duke ER; Shipman KJ; Jensen RL; Fong Y; Ferguson S; Janes HE; Gillespie K; Seilie AM; Hanron AE; Rinn L; Fishbaugher M; VonGoedert T; Fritzen E; Kappe SH; Chang M; Sousa JC; Marcsisin SR; Chalon S; Duparc S; Kerr N; Mohrle JJ; Andenmatten N; Rueckle T; Kublin JG, A Randomized trial evaluating the prophylactic activity of dsm265 against preerythrocytic Plasmodium falciparum infection during controlled human malarial infection by mosquito bites and direct venous inoculation. J Infect Dis 2018, 217 (5), 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sulyok M; Ruckle T; Roth A; Murbeth RE; Chalon S; Kerr N; Samec SS; Gobeau N; Calle CL; Ibanez J; Sulyok Z; Held J; Gebru T; Granados P; Bruckner S; Nguetse C; Mengue J; Lalremruata A; Sim BKL; Hoffman SL; Mohrle JJ; Kremsner PG; Mordmuller B, DSM265 for Plasmodium falciparum chemoprophylaxis: a randomised, double blinded, phase 1 trial with controlled human malaria infection. Lancet Infect Dis 2017, 17 (6), 636–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White J; Dhingra SK; Deng X; El Mazouni F; Lee MCS; Afanador GA; Lawong A; Tomchick DR; Ng CL; Bath J; Rathod PK; Fidock DA; Phillips MA, Identification and mechanistic Understanding of dihydroorotate dehydrogenase point mutations in plasmodium falciparum that confer in vitro resistance to the clinical candidate DSM265. ACS Infect Dis 2019, 5 (1), 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baldwin J; Michnoff CH; Malmquist NA; White J; Roth MG; Rathod PK; Phillips MA, High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J Biol Chem 2005, 280 (23), 21847–21853. [DOI] [PubMed] [Google Scholar]
- 36.Patel V; Booker M; Kramer M; Ross L; Celatka CA; Kennedy LM; Dvorin JD; Duraisingh MT; Sliz P; Wirth DF; Clardy J, Identification and characterization of small molecule inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J Biol Chem 2008, 283 (50), 35078–35085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu J; Fang Z; Zhang Q; Liu Q; Bi X, Silver-catalyzed isocyanide-alkyne cycloaddition: a general and practical method to oligosubstituted pyrroles. Angew Chem Int Ed Engl 2013, 52 (27), 6953–6957. [DOI] [PubMed] [Google Scholar]
- 38.Eusterwiemann S; Martinez H; Dolbier WR Jr., Methyl 2,2-difluoro-2-(fluorosulfonyl)acetate, a difluorocarbene reagent with reactivity comparable to that of trimethylsilyl 2,2-difluoro-2-(fluorosulfonyl)acetate (TFDA). J Org Chem 2012, 77 (12), 5461–5464. [DOI] [PubMed] [Google Scholar]
- 39.Falck J; Barma D; Mioskowski C; Schlama T, α-chlorovinylation: Synthesis of 2-chloropropenyl and propargyl alcohols. Tetrahedron Lett 1999, 40 (11), 2091–2094. [Google Scholar]
- 40.Deng X; Kokkonda S; El Mazouni F; White J; Burrows JN; Kaminsky W; Charman SA; Matthews D; Rathod PK; Phillips MA, Fluorine modulates species selectivity in the triazolopyrimidine class of Plasmodium falciparum dihydroorotate dehydrogenase inhibitors. J Med Chem 2014, 57 (12), 5381–5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen VB; Arendall WB; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ganesan SM; Morrisey JM; Ke H; Painter HJ; Laroiya K; Phillips MA; Rathod PK; Mather MW; Vaidya AB, Yeast dihydroorotate dehydrogenase as a new selectable marker for Plasmodium falciparum transfection. Mol Biochem Parasitol 2011, 177 (1), 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Painter HJ; Morrisey JM; Mather MW; Vaidya AB, Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 2007, 446 (7131), 88–91. [DOI] [PubMed] [Google Scholar]
- 44.Obach RS; Walsky RL; Venkatakrishnan K, Mechanism-based inactivation of human cytochrome p450 enzymes and the prediction of drug-drug interactions. Drug Metab Dispos 2007, 35 (2), 246–255. [DOI] [PubMed] [Google Scholar]
- 45.Mandt REK; Lafuente-Monasterio MJ; Sakata-Kato T; Luth MR; Segura D; Pablos-Tanarro A; Viera S; Magan N; Ottilie S; Winzeler EA; Lukens AK; Gamo FJ; Wirth DF, In vitro selection predicts malaria parasite resistance to dihydroorotate dehydrogenase inhibitors in a mouse infection model. Sci Transl Med 2019, 11 (521). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ross LS; Lafuente-Monasterio MJ; Sakata-Kato T; Mandt REK; Gamo FJ; Wirth DF; Lukens AK, Identification of collateral sensitivity to dihydroorotate dehydrogenase inhibitors in Plasmodium falciparum. ACS Infect Dis 2018, 4 (4), 508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burrows JN; Duparc S; Gutteridge WE; Hooft van Huijsduijnen R; Kaszubska W; Macintyre F; Mazzuri S; Mohrle JJ; Wells TNC, New developments in anti-malarial target candidate and product profiles. Malar J 2017, 16 (1), 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Malmquist NA; Gujjar R; Rathod PK; Phillips MA, Analysis of flavin oxidation and electron-transfer inhibition in Plasmodium falciparum dihydroorotate dehydrogenase. Biochemistry 2008, 47 (8), 2466–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bennett TN; Paguio M; Gligorijevic B; Seudieu C; Kosar AD; Davidson E; Roepe PD, Novel, rapid, and inexpensive cell-based quantification of antimalarial drug efficacy. Antimicrob Agents Chemother 2004, 48 (5), 1807–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deng X; Gujjar R; El Mazouni F; Kaminsky W; Malmquist NA; Goldsmith EJ; Rathod PK; Phillips MA, Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J Biol Chem 2009, 284 (39), 26999–27009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Otwinowski Z; Minor W, Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 52.McCoy A; Grosse-Kunstleve R; Adams P; Winn M; Storoni L; Read R, Phaser crystallographic software. J Appl Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and development of Coot. Acta Crystallogr D 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adams P; Afonine P; Bunkóczi G; Chen V; Davis I; Echols N; Headd J; Hung L; Kapral G; Grosse-Kunstleve R; McCoy A; Moriarty N; Oeffner R; Read R; Richardson D; Richardson J; Terwilliger T; Zwart P, PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D Biological Crystallography 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jimenez-Diaz MB; Mulet T; Viera S; Gomez V; Garuti H; Ibanez J; Alvarez-Doval A; Shultz LD; Martinez A; Gargallo-Viola D; Angulo-Barturen I, Improved murine model of malaria using Plasmodium falciparum competent strains and non-myelodepleted NOD-scid IL2Rgammanull mice engrafted with human erythrocytes. Antimicrob Agents Chemother 2009, 53 (10), 4533–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.