

Abstract
Objective
Inflammation is closely linked to angiogenesis, and Toll-like receptors (TLRs) are the key mediators of inflammatory responses. However, the impact of TLRs on angiogenesis is incompletely understood. In this study, we determined the involvement of TLRs in angiogenesis.
Methods and Results
In a mouse model of alkali-induced corneal neovascularization (CNV), we found that CNV was attenuated in TLR4−/− but not TLR2−/− mice. Further study revealed that the absence of TLR4 led to decreased production of proangiogenic factors in association with reduced accumulation of macrophages at the site of wounds, which was associated with reduced expression of high-mobility group box-1 (HMGB1) protein, an endogenous ligand for TLR4. Topical application of HMGB1 to the injured cornea promoted CNV with increased macrophage accumulation in wild-type mice but not in TLR4−/− mice. HMGB1 treatment in vitro also promoted the production of proangiogenic factors by mouse macrophages in a TLR4-dependent manner. Furthermore, antagonists of HMGB1 and TLR4 reduced CNV and macrophage recruitment in the injured cornea of wild-type mice.
Conclusion
Our results suggest that the release of HMGB1 in the wounds initiates TLR4-dependent responses that contribute to neovascularization. Thus, targeting HMGB1-TLR4 signaling cascade may constitute a novel therapeutic approach to angiogenesis-related diseases.
Keywords: angiogenesis, macrophages, HMGB1, inflammation, Toll-like receptor
Angiogenesis is a normal process in growth, development, and wound healing. It also participates in a variety of pathogenesis, such as cancer, atherosclerosis, diabetic retinopathy, rheumatoid arthritis, and corneal neovascularization (CNV).1 Pathological angiogenesis is linked to inflammatory responses through the production of proangiogenic mediators by innate and adaptive immune cells that express Toll-like receptors (TLRs).2
TLRs are a family of pattern recognition receptors that recognize distinct molecular patterns associated with microbial pathogens, including viral or bacterial nucleic acids, lipopolysaccharide (LPS) or lipoteichoic acids, and flagellin.3 Thus far, 11 TLRs in human and 13 TLRs in mouse have been identified, and some of their endogenous ligands, such as heat shock proteins,4 surfactant protein A18,5 extracellular matrix components,6 and high-mobility group box-1 (HMGB1),7 are known. Recent studies showed that TLRs might be involved in angiogenesis. It is reported that TLR4 expressed on liver endothelial cells regulates the invasive capacity of liver endothelial cells by promoting extracellular protease production.8 Activation of TLR4 by endotoxin could induce the proliferation and differentiation of endothelial progenitor cells.9 Previous study also showed that activation of TLR4 by LPS induced the production of proangiogenic factor vascular endothelial growth factor (VEGF) by macrophages.10 However, the role of endogenous TLR4 ligands released from damaged tissues in angiogenesis remains to investigate.
HMGB1 is one of the endogenous ligands for TLR4.7 It is either passively released by injured or necrotic cells or actively secreted by monocyte/macrophages.11,12 HMGB1 has not only been demonstrated as a late mediator of sepsis but also been implicated as a putative danger signal involved in the pathogenesis of a variety of noninfectious inflammatory conditions, including autoimmunity, cancer, trauma, hemorrhagic shock, and ischemia-reperfusion injury.13–15 HMGB1 has been reported to signal mainly through 3 putative receptors, including the receptor for advanced glycation end products (RAGE),16 TLR2, and TLR4.17 Recent studies have indicated that HMGB1 might be a proangiogenic factor in tissue injury by stimulating the sprouting, proliferation, and chemotaxis of endothelial cells.18 HMGB1 also attracts endothelial progenitor cells and hematopoietic stem cells to the sites of tissue injury and tumors to promote neovascularization.19,20 HMGB1 enhanced vessel density in skin wound or promoted collateral blood flow in ischemic hindlimbs of diabetic mice and increased the release angiogenic factors, such as VEGF and basic fibroblast growth factor (b-FGF), by macrophages or cultured human cardiac fibroblasts.21–24
These proangiogenic functions of HMGB1 were reported to be mediated by the RAGE signaling pathway.19,25 Mice deficient in TLR4 or RAGE but not TLR2 had a drastically reduced production of inflammatory cytokines in response to HMGB1 administration. In contrast, TLR2-deficient mice displayed an increased production of inflammatory cytokines.26 These results suggest that TLR4 but not TLR2 is a receptor involved in the induction of inflammatory processes by HMGB1 in vivo.
Mouse cornea and cultured human corneal epithelial cells express TLR2, TLR3, TLR4, TLR5, and TLR9.27 Animal model studies indicate that development of inflammation and injurious responses in cornea depends on TLR4 signaling.28,29 Because inflammation is a key process linked to angiogenesis,2 in this study, we investigated the role of HMGB1-TLR4 signaling in angiogenesis using a model of CNV in mice. We found that TLR4 but not TLR2 was involved in the development of CNV. HMGB1 from injured tissues triggered TLR4-dependent responses, including the recruitment of macrophages and the generation of proangiogenic factors. Our results addressed an essential role for HMGB1-TLR4 signaling in promoting neovascularization.
Methods
A detailed description of the methods is given in the Supplemental Materials, available online at http://atvb.ahajournals.org.
Animals
TLR2−/− and TLR4−/− mice were backcrossed onto the C57BL/6 background and then intercrossed to obtain the knockout genotypes and wild-type (WT) mice (control littermates). Littermates of both sexes between 8 and 12 weeks old were used in all experiments. Animals were kept in a specific pathogen-free facility. Animal care and use were in compliance with institutional guidelines.
Recombinant Full-Length HMGB1, HMGB1 Box A, and Anti-HMGB1 Rabbit Serum
The cloning, expression, and purification of recombinant full-length HMGB1 and Box A and preparation of anti-HMGB1 rabbit serum were carried out as previously described.30
Alkali-Induced Corneal Injury Model
Mice were anesthetized. A 2-mm disc of filter paper saturated with 1 N NaOH was placed onto the right cornea of each mouse for 40 seconds, followed by rinsing extensively with 25 mL of PBS. In some experiments, recombinant HMGB1, Box A, LPS, or Rhodobacter sphaeroides LPS (LPS-RS) was applied topically to the alkali-treated eye twice a day for 7 days.
Histology
Immunohistochemical analyses were performed using anti-HMGB1 or anti-CD31 antibodies (Abs). As the immunofluorescence analysis for macrophage infiltration, the cryostat sections of isolated eyes were immunostained with fluorescein isothiocyanate–conjugated anti-F4/80 Ab. For 2-color analyses, sections were incubated with the combinations of anti-F4/80 and anti-TLR4, anti-F4/80 and anti-HMGB1, anti-CD31 and anti-TLR4, or anti-CD31 and anti-HMGB1 Abs. For an immunocytochemical analysis of VEGF expression, murine peritoneal macrophages were harvested and stimulated with recombinant HMGB1 for 24 hours and then subjected to immunocytochemical study.
Biomicroscopic Examination
Eyes were examined with a slit lamp (Zeiss, Germany) 7 and 14 days after alkali injury, and microscopic assessment was done by observers without prior knowledge of the procedures.
Enumeration of CNV
The sections were stained using anti-CD31 polyclonal Abs, and the numbers and sizes of the CNV were determined.
Real-Time Quantitative Reverse Transcription–Polymerase Chain Reaction
Total RNAs were extracted from the corneas or cultured peritoneal macrophages, and real-time polymerase chain reaction (PCR) was performed. The tested mRNA expression was finally determined after correction with GAPDH expression. Each measurement of a sample was conducted in duplicate.
Reproducibility and Statistical Analysis
Experiments were repeated at least 3 times. Results were highly reproducible. Representative results are shown in the figures. The means and SEM were calculated on all parameters determined in the study. Data were analyzed statistically using 1-way ANOVA or 2-tailed Student t test. A value of P<0.05 was accepted as statistically significant.
Results
TLR4 Is Involved in Mouse Alkali Injury–Induced CNV
To test the involvement of TLR4 in angiogenic response, we first examined neovascularization in TLR4-deficient mice with alkali-induced corneal injury. In control mice, alkali burn caused a rapid neovascularization, with limbal vessels sprouted into corneas 7 days after burn (Figure 1A). However, in TLR4-deficient mice, few new blood vessels grew into the cornea on day 7 (Figure 1A). The macroscopic CNV in WT mice reached a maximal level at 14 days after injury, whereas only a small number of vessels appeared near the limbal area in TLR4-deficient mice (Figure 1A). Immunohistochemical analysis using anti-CD31 Ab showed that vascular areas were increased to a greater extent in WT mice than in TLR4−/− mice (Figure 1B to 1D). These observations indicate that TLR4 is involved in the development of new vessels in alkali-induced injured cornea.
Figure 1.
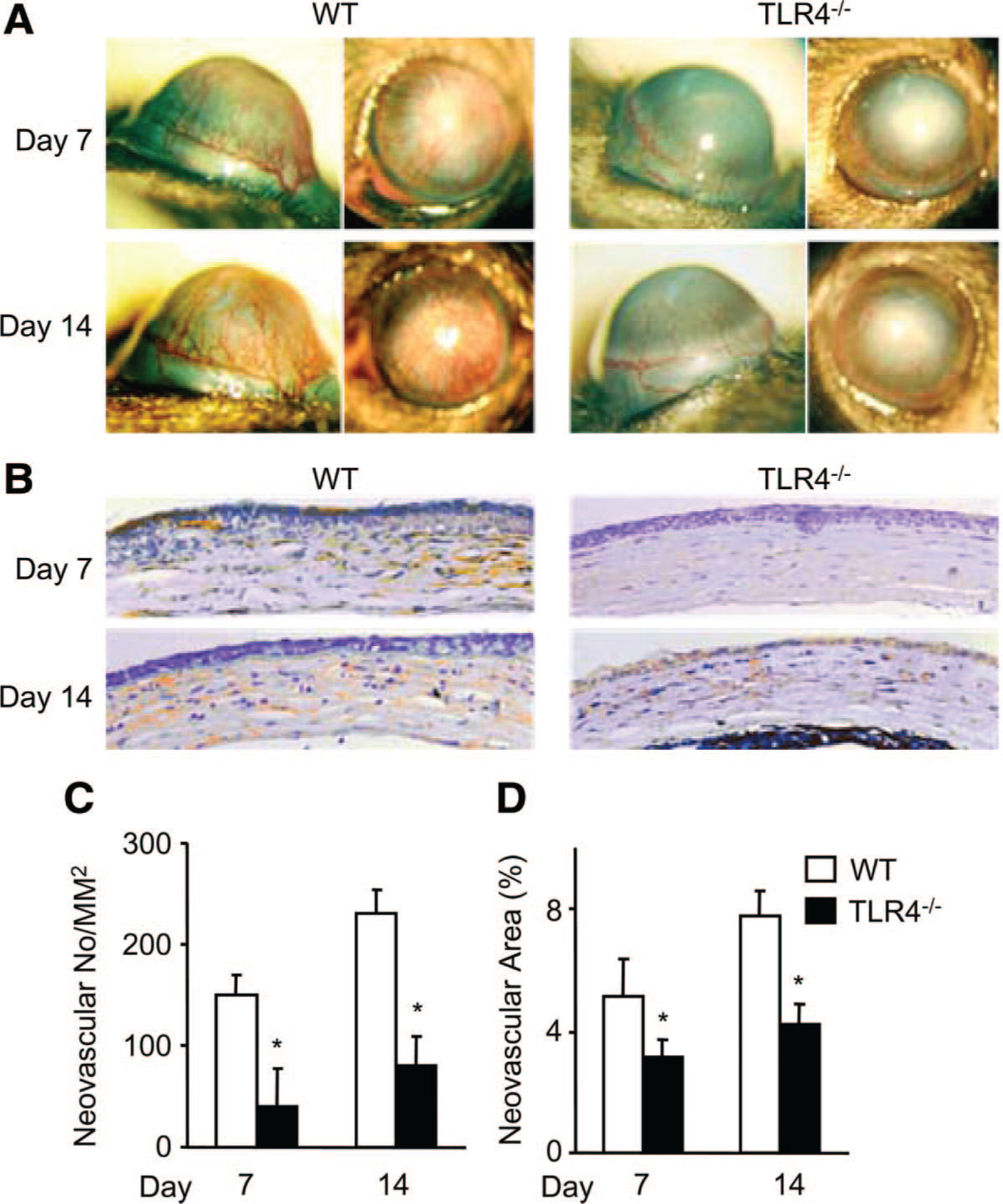
Alkali injury-induced CNV in WT and TLR4-deficient mice. A, Macroscopic appearance of WT (left panels) and TLR4-deficient (right panels) mouse eyes 7 days (upper panels) and 14 days (lower panels) after alkali injury. Images were taken with a slit lamp to show the frontal and lateral view of each eye. B, Corneal tissues were immunostained with anti-CD31 Abs. Magnification ×400. C and D, Quantitative analysis of data presented in B. CNV numbers per mm2 in hot spots (C) or percentage of CNV areas in hot spots (D) were determined on corneas. Data represent means±SEM (n=6 animals). *P<0.05 vs WT mice.
TLR4 Is Required for the Recruitment of Macrophages in CNV
Macrophages have been known to participate in the development of neovascularization in various tissues, including cornea.31–35 Given the fact that TLR4 is expressed on macrophages to mediate inflammatory responses,28 we examined the effects of TLR4 deficiency on macrophage accumulation in injured corneas. There was no obvious accumulation of macrophages in the uninjured corneas of WT and TLR4−/− mice (day 0) (Figure 2A). F4/80+ macrophages started to accumulate on day 1 and reached a peak on day 4 after injury (Figure 2A and 2B). Although the kinetics of macrophage infiltration in the wounds were similar in control and TLR4−/− animals (Figure 2B), the infiltration of macrophages in TLR4−/− wounded corneas was significantly attenuated (Figure 2B). Decreased infiltration of macrophages may imply that chemokine expression is downregulated in TLR4−/− wounds. To test this hypothesis, quantitative PCRs were used to examine changes in mRNA expression of chemokines MIP-2/CXCL2, MIP-1α/CCL3, and MCP-1/CCL2, which have been shown to play a critical role in macrophage recruitment.36 All 3 chemokine genes tested were upregulated in the wounded corneas. However, these expressions were significant reduced in TLR4−/− mice (Supplemental Figure I). These observations indicate that in damaged corneas, TLR4 is critical for macrophage accumulation, which may be attributed to the TLR4-induced production of chemokines.
Figure 2.
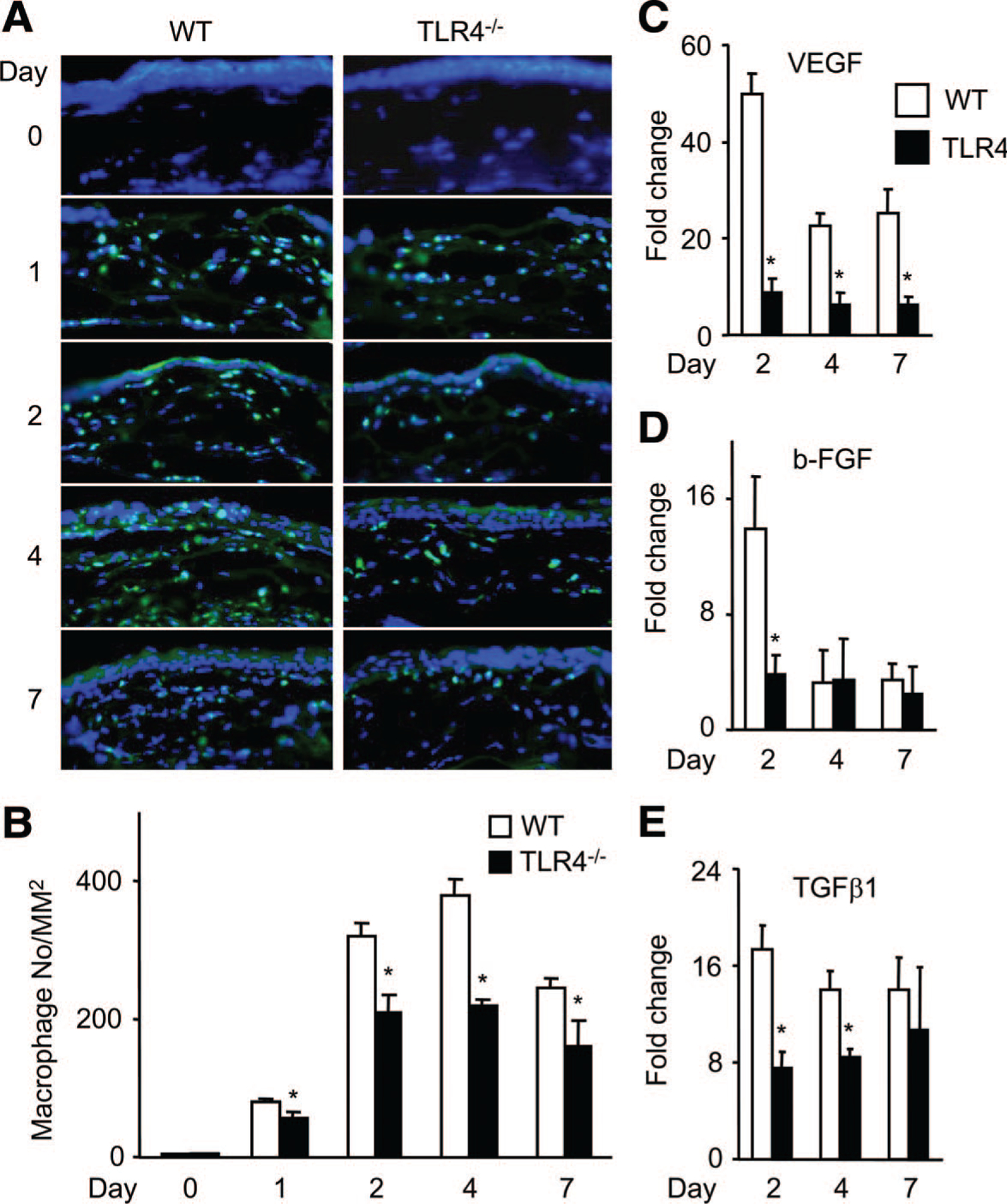
Macrophage accumulation and angiogenic factor expression in injured corneas. A, Corneal tissues from WT mice (left panels) or TLR4−/− (right panels) mice were stained with fluorescein isothiocyanate–conjugated anti-F4/80 monoclonal Abs. Magnification ×400. B, The numbers of infiltrating F4/80-positive macrophages were determined, and the means±SEM are shown (n=6). *P<0.05 vs WT mice. C to E, Angiogenic factor expression in injured corneas. The mRNA expression of VEGF (C), b-FGF (D), and TGFβ1 (E) in wound sites was determined by quantitative reverse transcription–PCR. Results are expressed as mean±SEM of fold increase over control. *P<0.05 vs WT mice.
TLR4 Is Required for Alkali Injury–Induced Proangiogenic Factor Gene Expression
Macrophages are a rich source of growth factors.31 Markedly decreased infiltration of macrophages also implied that proangiogenic factor expression might be downregulated in wounded TLR4−/− corneas. To test this hypothesis, quantitative PCRs were used to examine the changes in mRNA expression of 3 angiogenic factors, VEGF, transforming growth factor (TGF) β1, and b-FGF, in injured corneas. Alkali injury markedly increased intracorneal mRNA expression of these 3 proangiogenic factors in WT and TLR4−/− mice (Figure 2C to 2E). The level of enhancement was significantly lower in TLR4−/− corneas, with the expression of VEGF increased up to 49-fold on day 2 in WT mice but only 10-fold in the TLR4−/− mice (Figure 2C). These results suggest that TLR4 plays a critical role in proangiogenic factor production by infiltrating macrophages in the wounded corneas.
The Expression of HMGB1 and TLR4 in Injured Cornea
We examined TLR4 expression in corneas after alkali-induced injury and found that the mRNA of TLR4 was significantly upregulated in wounded corneas (Figure 3A). Large numbers of TLR4-positive cells were found in the limbus, epithelium, and stroma of the wounded corneas on day 4, and the number remained elevated 7 days after alkali treatment (Figure 3B).
Figure 3.
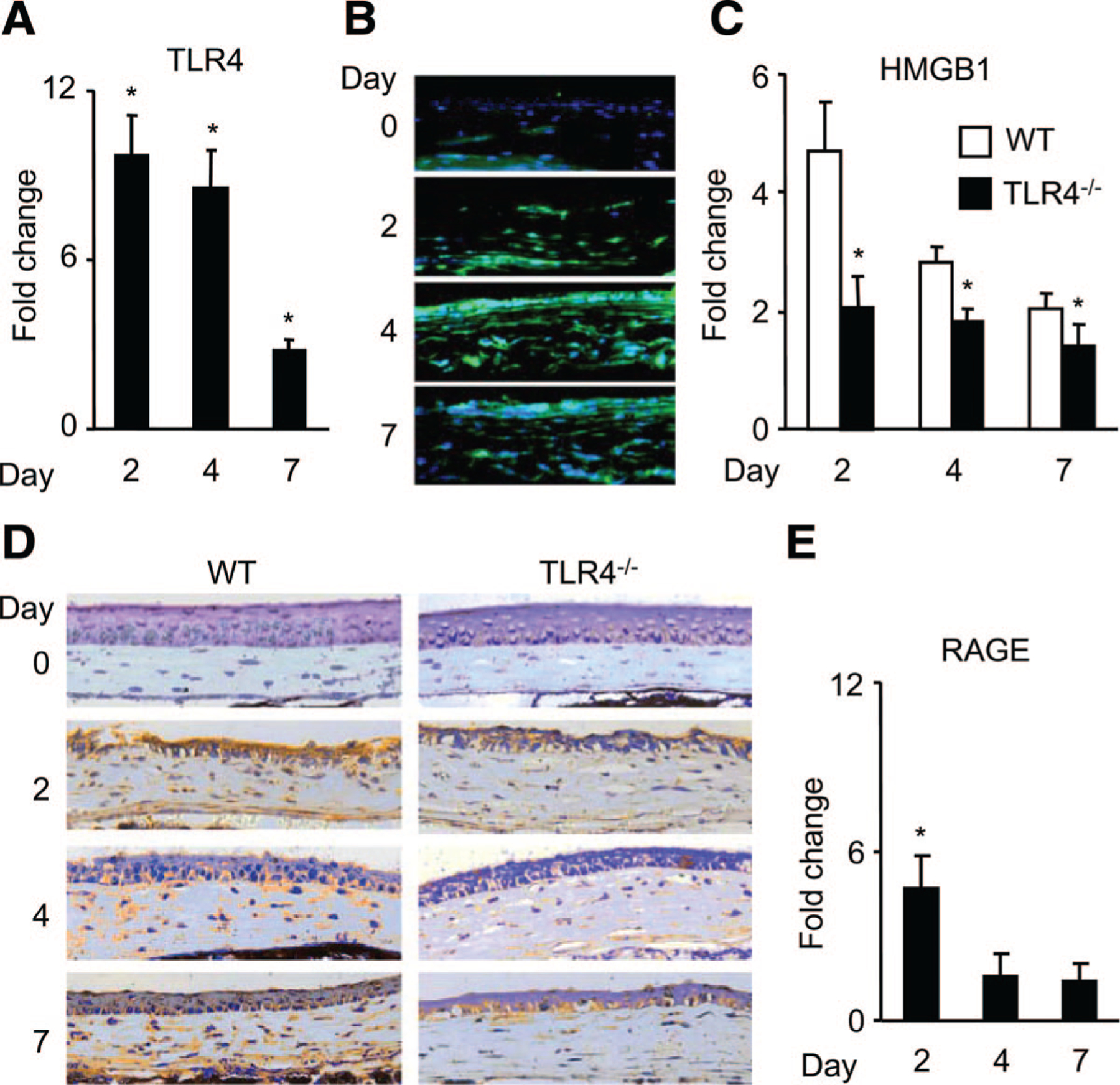
The expression of TLR4, HMGB1, and RAGE in corneas after alkali injury. A, Quantitative reverse transcription–PCR to assess TLR4 gene expression in corneas of WT mice. Data represent means±SEM (n=6). *P<0.05 vs normal corneas. B, Immunofluorescence analysis of TLR4 protein expression in intact or wounded cornea from WT mice. Magnification ×400. C, Real-time PCR analysis of HMGB1 expression in wounded corneas at the indicated time points. Results were expressed as mean±SEM (n=6) of fold increase over control. *P<0.05 vs WT mice. D, HMGB1 protein expression in injured corneas. Whole eyes were obtained and processed for immunohistochemical analysis using an anti-HMGBI Ab. Magnification ×400. E, Real-time PCR analysis of RAGE expression in wounded corneas. All values represent mean±SEM (n=6). *P<0.05 vs intact corneas.
To test the involvement of HMGB1, an endogenous ligand for TLR4, in CNV, we evaluated HMGB1 expression in the corneas before and after injury. Wounding significantly enhanced the intracorneal HMGB1 mRNA expression in both WT and TLR4−/− mice (Figure 3C). However, the magnitudes were markedly lower in TLR4−/− mice throughout a 7-day period of observation (Figure 3C). Concomitantly, HMGB1 protein was detected in epithelial cells and infiltrating leukocytes after injury (Figure 3D), with marked reduction in TLR4−/− mice (Figure 3D). These results indicate that loss of TLR4 results in impaired injury-induced HMGB1 production. The induction of both TLR4 and HMGB1 expression in cornea by wounding suggests that their interaction may regulate CNV through an autocrine mechanism, a paracrine mechanism, or both. We also examined the expression of RAGE, another putative receptor for HMGB1 in CNV. Wounding induced a transient and moderate upregulation in the RAGE mRNA expression in the corneas on day 2 (Figure 3E). These data indicate that TLR4 plays a major role in angiogenesis after the corneal injury.
TLR4-Positive Macrophages and Endothelial Cells Express HMGB1
In line with the previous observation that TLR4 is expressed on intracorneally infiltrating macrophages during fungal keratitis,28 we observed that a substantial proportion of F4/80-positive macrophages expressed TLR4 in injured cornea on day 2 postinjury (Supplemental Figure IIA). HMGB1 protein was detected in macrophages at the same time (Supplemental Figure IIA). The expression of HMGB1 by TLR4-positive macrophages in cornea suggests that triggering HMGB1-TLR4 signaling pathway may create a positive feedback loop to regulate macrophage activation. Moreover, TLR4 protein was also detected on most CD31-positive endothelial cells at the wound sites 7 days after injury, and HMGB1 was coexpressed on a minority of endothelial cells (Supplemental Figure IIB). The HMGB1-TLR4 intensity in macrophages was stronger than that in endothelial cells. These observations suggest that the recruitment of macrophages may be a key step in CNV formation.
HMGB1 Promotes CNV in a TLR4-Dependent Manner
We next used pharmacological and genetic approaches to examine the effects of the TLR4 endogenous agonist HMGB1 on angiogenesis in vivo. Recombinant HMGB1 was topically administered to wounded corneas after alkali injury. Because CNV reaches a peak on day 14 after injury (Figure 1A), we chose this time point to compare the effect of HMGB1. HMGB1 showed a notable effect on the enhancement of CNV in WT mice at 2 weeks after the injury (Figure 4A and Supplemental Figure IIIA). Similar results were observed in the corneal cryosections with anti-CD31 staining (Figure 4A, 4C, and 4D). Concomitantly, topical administration of HMGB1 augmented the number of intracorneal macrophages (Figure 4E and 4F) at the early stage of alkali-induced wounds. However, HMGB1 treatment failed to enhance CNV and macrophage accumulation in the wounds of TLR4−/− animals (Figure 4B to 4F and Supplemental Figure IIIA). Similar to HMGB1, treatment with LPS, the exogenous ligand for TLR4, also enhanced CNV and macrophage accumulation in WT mice but not in TLR4−/− mice (Figure 4A to 4F and Supplemental Figure IIIA). These data indicate that activation of TLR4 by HMGB1 enhanced the recruitment macrophages, which in turn may produce angiogenic factors and promote CNV.
Figure 4.
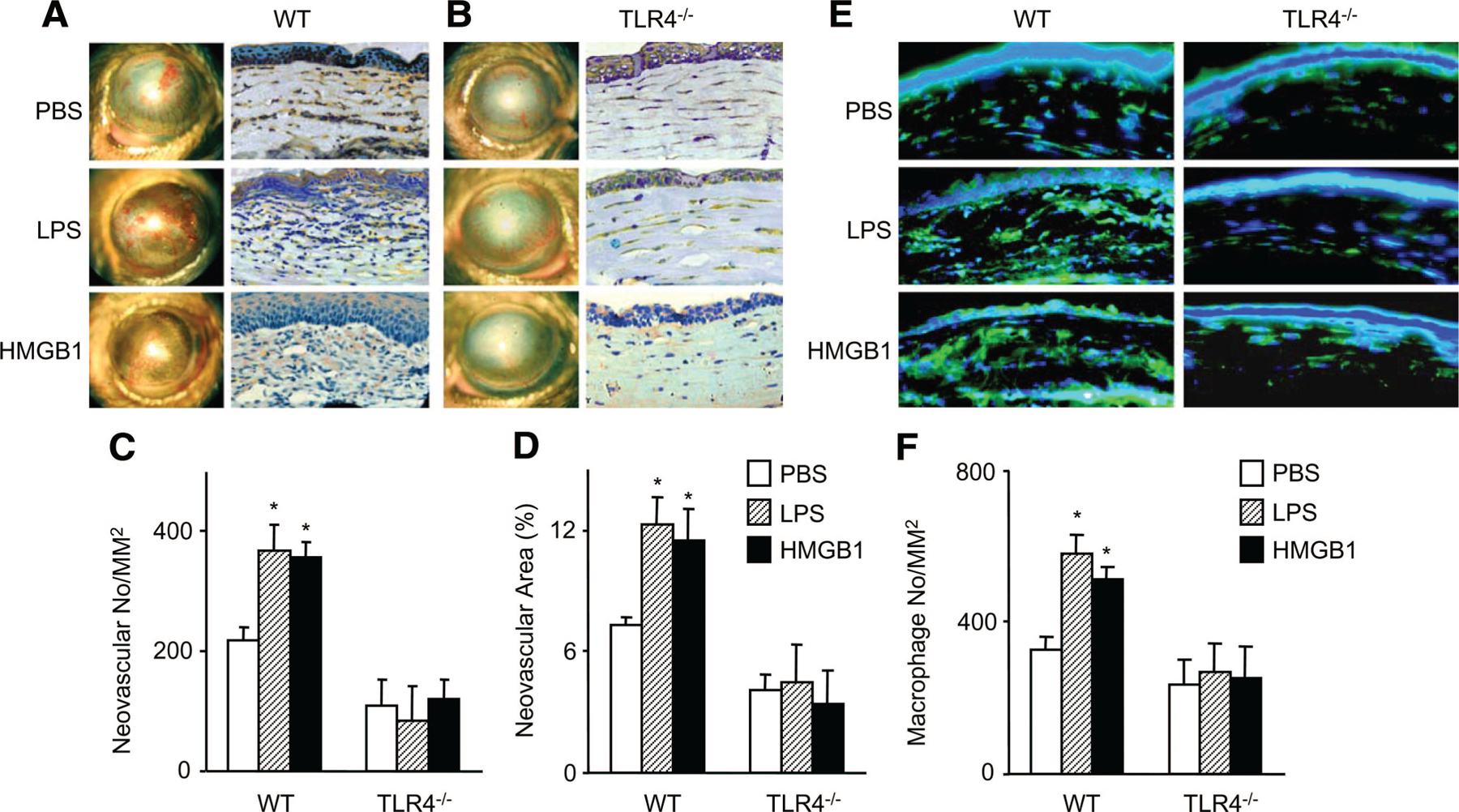
The effects of topical HMGB1 application on CNV. A and B, Macroscopic appearances of WT (A) and TLR4−/− (B) mice topically applied with HMGB1 2 weeks after alkali injury are shown. LPS treatment was used as a positive control. Images were taken with a slit lamp (left panels), and the corresponding cryosections from treated corneal tissues were immunostained with anti-CD31 Ab (right panels). Magnification ×400. C and D, Quantitative analysis of data presented in A and B. The CNV numbers per mm2 in hot spots (C) and percentage of CNV areas in hot spots (D) were determined. Data represent means±SEM (n=6). *P<0.05 vs PBS-treated groups. E, Corneal tissues removed 4 days after injury from WT (left panel) and TLR4−/− (right panel) mice were stained with anti-F4/80 monoclonal Ab. Magnification ×400. F, The numbers of F4/80-positive macrophages were determined, and the mean±SEM are shown (n=6). *P<0.05 vs PBS-treated groups.
HMGB1-Induced Angiogenic Factor Expression by Murine Peritoneal Macrophages Is TLR4 Dependent
Because TLR4-mediated signaling can enhance the functions of macrophages,28 we next examined the effects of exogenous HMGB1 on angiogenic factor expression by primary peritoneal murine macrophage. HMGB1 markedly increased the expression of the genes for VEGF, b-FGF, and TGFβl in peritoneal macrophages, with an increase in the expression of the VEGF gene of 40.5-fold as compared with untreated cells (Figure 5A). Immunofluorescence analysis showed that HMGB1-stimulated macrophages exhibited increased VEGF protein expression as compared with untreated cells (Figure 5B). In contrast, macrophage from TLR4−/− mice showed markedly reduced expression of the genes for VEGF, b-FGF, and TGFβ1; in particular, the magnitudes in VEGF expression were approximately 80% lower in the TLR4−/− macrophage compared with the WT macrophage (Figure 5A). This is associated with a decrease of VEGF protein expression in TLR4−/− macrophages (Figure 5B). These results confirmed the observations in vivo, indicating that HMGB1 induces the production of angiogenic factors by macrophage via TLR4-dependent mechanisms.
Figure 5.
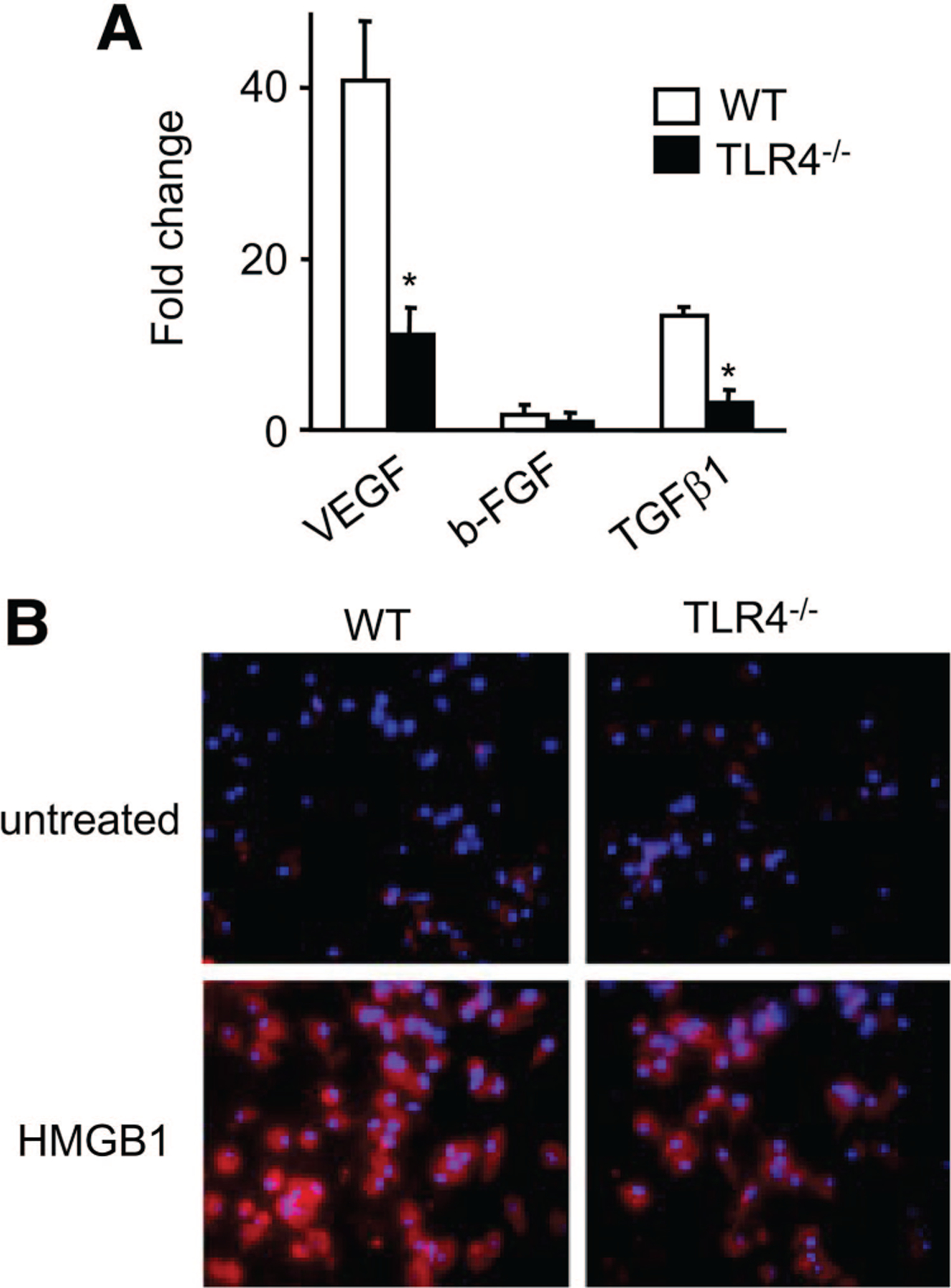
The effects of HMGB1 on angiogenic factor expression by murine peritoneal macrophages. A, Peritoneal macrophages from wT and TLR4−/− mice were incubated with 1.0 μg/mL recombinant HMGB1 for 12 hours. The levels of VEGF, b-FGF, and TGFβ1 mRNA were determined by quantitative reverse transcription–PCR, and data represent means±SEM. *P<0.05 vs WT mice. B, Peritoneal macrophages from WT mice were stimulated in the absence (upper panels) or presence of HMGB1 (lower panels) for 24 hours. The cells were stained with anti-VEGF Ab and then Cy3-conjugated secondary Ab and Hoechst 33342. Original magnification, ×400.
Alkali-Induced CNV Is Not Dependent on TLR2
Because TLR2 has also been reported to recognize HMGB1,17 we analyzed alkali-induced corneal angiogenesis in TLR2−/− mice. We found that CNV developed in TLR2 mutant mice was not different from that in WT animals (Supplemental Figure IV). These data suggest that CNV induction is primarily dependent on TLR4 but not on TLR2.
Reduction of CNV and Macrophage Recruitment by Antagonists of HMGB1 and TLR4
We then attempted to block the activity of endogenous HMGB1 by topical administration of Box A, a fragment of HMGB1 with antagonist activity. Box A topical instillation reduced alkali-induced CNV at 2 weeks after the injury (Figure 6A to 6C and Supplemental Figure IIIB). We then tested the effects of the TLR4 antagonist LPS-RS on CNV. LPS-RS also reduced the degree of CNV in WT mice (Figure 6A to 6C and Supplemental Figure IIIB). Box A and LPS-RS were also highly effective in inhibiting alkali injury-induced cornea macrophage infiltration in WT mice (Figure 6D and 6E). These results confirm macrophages as the central effector cell type in HMGB1/TLR4-mediated angiogenesis in CNV. Furthermore, LPS-RS pretreatment significantly suppressed HMGB1-enhanced CNV, whereas Box A pretreatment reduced LPS-aggravated CNV in WT mice (Supplemental Figure V), suggesting that the effect of HMGB1 on promoting CNV is TLR4 dependent.
Figure 6.
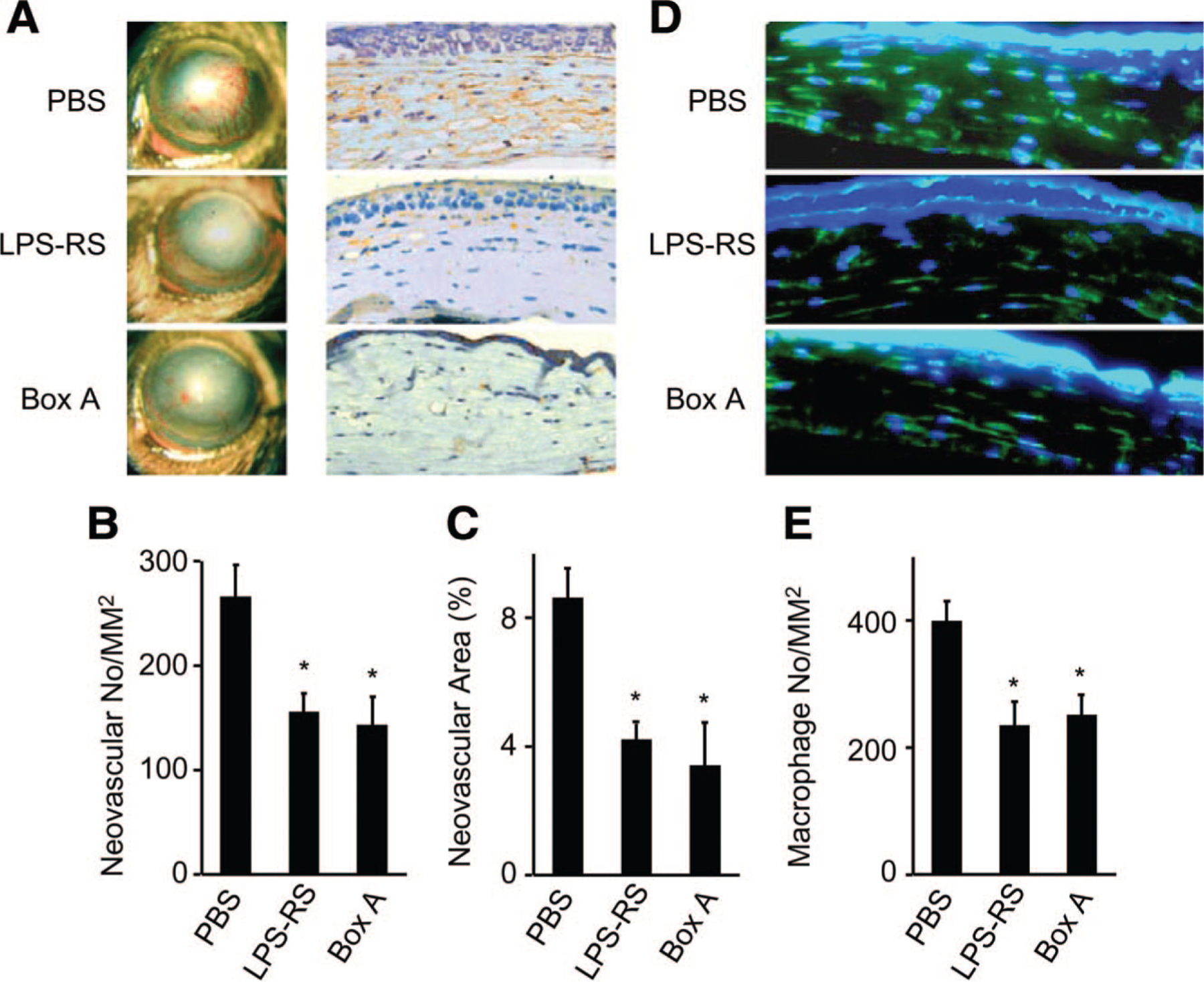
Antiangiogenic effects of Box A and LPS-RS. A, Macroscopic observations of CNV in WT mice instilled with the antagonists 2 weeks after alkali injury are shown. Images were taken with a slit lamp (left panels), and the corresponding cryosections from corneal tissues were immunostained with anti-CD31 Ab (right panels). Magnification ×400. B and C, Quantitative analysis of data presented in A. The CNV numbers per mm2 in hot spots (B) and percentage of CNV areas in hot spots (C) were determined. Each value represents the mean±SEM (n=6). *P<0.05 vs PBS-treated groups. D, Corneal tissues removed 4 days after the injury were stained with anti-F4/80 monoclonal Ab. Original magnification, ×400. E, The numbers of F4/80-positive macrophages were determined, and the mean±SEM are shown (n=6). *P<0.05 vs PBS-treated groups.
Discussion
To our knowledge, this study was the first demonstration of an essential role of the HMGB1-TLR4 signal pathway in angiogenesis in vivo.
In the vasculature, TLR4 is expressed in different cell types, such as endothelial cells, smooth muscle cells, adventitial fibroblasts, dendritic cells, macrophages, and endothelial progenitor cells.9,37 TLR4 signaling has been shown to play an important role in the pathogenesis of atherosclerosis and the models of hepatic, pulmonary, brain myocardial, and renal ischemia/reperfusion injury.15,37,38 In ocular surface tissues, activation of TLR4 by LPS mediates inflammatory responses in keratitis.29 TLR4 is also involved in corneal injury, including corneal incision, corneal epithelium scraping, and corneal suture.39 In vitro, LPS-induced TLR4 signaling in primary cultures of the epithelial cells and fibroblasts from cornea or conjunctiva also induces the production of inflammatory cytokines, chemokines, growth factors, and adhesion molecules.40,41 Our findings showed that topical treatment with LPS promotes alkali-induced CNV in WT mice but not in TLR4−/− mice. In specific pathogen-free conditions without any exogenous agonist treatment, mice deficient in TLR4 showed reduced CNV, suggesting that under sterile conditions without pathogen-associated molecular patterns, TLR4 could be activated by endogenous agonists produced by damaged tissues or infiltrating immune cells in the injured cornea, such as HMGB1.
HMGB1 is a nuclear protein that acts as a cytokine when released into the extracellular milieu by necrotic and inflammatory cells.11,12 A large body of evidence indicates that HMGB1 is required for the development or progression of inflammation in the absence of infection, as occurs during experimental autoimmune arthritis, cerebral ischemia, hemorrhagic shock, pancreatitis, sterile hepatic necrosis, and other conditions that lead to inflammation and tissue injury.42 Recently, HMGB1 has been recognized as a putative proangiogenic factor mediated by RAGE based on the ability of HMGB1 to induce endothelial cell sprouting, proliferation, and chemotaxis.18 HMGB1 may also attract endothelial progenitor cells and hematopoietic stem cells to the sites of injured tissues and tumors.19,20 Blockade of HMGB1 binding to RAGE with soluble RAGE or anti-RAGE Ab inhibited angiogenesis.19,25 HMGB1 has been implicated in several vasculature-related diseases, including tumor, and diabetes, arthritis, and atherosclerosis.43 In our study, we found that injury induced the expression of HMGB1 in mouse cornea. We revealed that exogenous HMGB1 enhanced CNV and macrophage recruitment. Treatment with Box A inhibited CNV with reduced macrophage accumulation in WT mice but not in TLR4-deficient mice, indicating that HMGB1-enhanced CNV was TLR4 dependent. Furthermore, LPS-RS inhibited HMGB1-enhanced CNV and Box A inhibited LPS-promoted CNV in WT mice (Supplemental Figure V). Because Box A functions by competitive binding to the receptors for HMGB1, including TLR4,44,45 and LPS-RS is known as a specific antagonist for TLR4,46,47 these findings confirmed the involvement of HMGB1 and TLR4 in CNV formation.
Macrophages, the main source of HMGB1 under inflammatory conditions,12 are proposed to play central roles in tissue repair based on the observations that these cells produce various growth and angiogenic factors.31,32,48 It has also been reported that macrophages are essential in corneal and choroidal neovascularization because they produce angiogenic mediators.35,49,50 Previous reports have shown that systemic or local macrophage depletion inhibited pathological neovascularization in several ocular models.33,34,51–53 In our study, the majority of TLR4- and HMGB1-positive cells were F4/80 positive cells in the injured corneas. Moreover, in vitro, the main HMGB1-induced angiogenic factor from murine peritoneal macrophages of WT mice but not of TLR4−/− mice was VEGF. These observations support the hypothesis that HMGB1 activates macrophages in a positive feedback mechanism to sustain inflammation and angiogenesis cascade under pathological conditions.43 Recent studies showed that the receptor binding of HMGB1 is TLR4 dependent and the cysteine in position 106 within the HMGB1 B box is required for the binding.54–56 Consistently, our results suggest that the corneal injury induced the extracellular release of HMGB1, which may act as an autocrine or paracrine mediator to stimulate the infiltrating macrophages to produce more chemokines, proangiogenic factors, and HMGB1 itself in a TLR4-dependent manner, which contributes to the neovascularization.
To date, 3 putative HMGB1 receptors, RAGE, TLR2, and TLR4, have been reported. Although TLR2 was expressed in normal human and mouse corneal epithelial cells,27,29 our data indicate that there is no effect of deleting TLR2 on induction of CNV. One possibility, as suggested, is that TLR2 acts as a receptor for nucleosome-bound HMGB1 but not for free HMGB1.57 Our data also showed that in murine cornea, RAGE mRNA is inducible after injury. Although the kinetics of alkali injury-induced RAGE expression were similar to those of TLR4, the magnitudes were markedly lower, and TLR4 is sufficient to promote CNV, suggesting that TLR4 may play a major role in this process. However, the possibility that HMGB1 may interact with the other purported cellular receptors in the process of CNV development requires further investigation.
Recently, HMGB1-TLR4 signaling has been implicated in the inflammatory process of a vast array of disease models.38,54,58,59 HMGB1-TLR4 interactions also regulate the processing and presentation of tumor antigens by dendritic cells, which is considered as important for the death-driven immunoadjuvant effects of chemotherapy.60 More recently, the proinflammatory role of TLR4-HMGB1 axis in the model of acute and chronic seizures or skin tumor development has also been established.46,61 A 2-step inflammation-induced skin tumorigenesis model showed that carcinogen-induced inflammation was dependent on endogenous HMGB1 but not on skin bacteria–associated LPS. In addition, we found that TLR4 was involved in the aberrant angiogenesis in an oxygen-induced retinopathy model (data not shown). Because TLR4 and HMGB1 are detected in the retina,62,63 our data suggest that the HMGB1-TLR4 signaling cascade may also contribute to the retinal neovascularization induced by ischemic injury.
In conclusion, targeting the HMGB1-TLR4 signaling pathway may be a novel therapeutic approach for a variety of vasculature-related disorders, such as atherosclerosis, diabetic retinopathy, rheumatoid arthritis, psoriasis, cancer, and as CNV.
Supplementary Material
Sources of Funding
This project was supported by the grants from the National Basic Research Program of China (2007CB512206, 2010CB529400 and 2011CB966200) and the National Natural Science Foundation of China (81072483).
Footnotes
Disclosures
None.
References
- 1.Folkman J Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. [DOI] [PubMed] [Google Scholar]
- 2.Frantz S, Vincent KA, Feron O, Kelly RA. Innate immunity and angiogenesis. Circ Res. 2005;96:15–26. [DOI] [PubMed] [Google Scholar]
- 3.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. [DOI] [PubMed] [Google Scholar]
- 4.Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107–15112. [DOI] [PubMed] [Google Scholar]
- 5.Guillot L, Balloy V, McCormack FX, Golenbock DT, Chignard M, Si-Tahar M. Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J Immunol. 2002;168:5989–5992. [DOI] [PubMed] [Google Scholar]
- 6.Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. 2009;9:57–63. [DOI] [PubMed] [Google Scholar]
- 7.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. [DOI] [PubMed] [Google Scholar]
- 8.Jagavelu K, Routray C, Shergill U, O’Hara SP, Faubion W, Shah VH. Endothelial cell toll-like receptor 4 regulates fibrosis-associated angiogenesis in the liver. Hepatology. 2010;52:590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He J, Xiao Z, Chen X, Chen M, Fang L, Yang M, Lv Q, Li Y, Li G, Hu J, Xie X. The expression of functional Toll-like receptor 4 is associated with proliferation and maintenance of stem cell phenotype in endothelial progenitor cells (EPCs). J Cell Biochem. 2010;111:179–186. [DOI] [PubMed] [Google Scholar]
- 10.Leibovich SJ, Chen JF, Pinhal-Enfield G, Belem PC, Elson G, Rosania A, Ramanathan M, Montesinos C, Jacobson M, Schwarzschild MA, Fink JS, Cronstein B. Synergistic up-regulation of vascular endothelial growth factor expression in murine macrophages by adenosine A(2A) receptor agonists and endotoxin. Am J Pathol. 2002;160:2231–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. [DOI] [PubMed] [Google Scholar]
- 12.Jiang W, Bell CW, Pisetsky DS. The relationship between apoptosis and high-mobility group protein 1 release from murine macrophages stimulated with lipopolysaccharide or polyinosinic-polycytidylic acid. J Immunol. 2007;178:6495–6503. [DOI] [PubMed] [Google Scholar]
- 13.Peltz ED, Moore EE, Eckels PC, Damle SS, Tsuruta Y, Johnson JL, Sauaia A, Silliman CC, Banerjee A, Abraham E. HMGB1 is markedly elevated within 6 hours of mechanical trauma in humans. Shock. 2009;32:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brezniceanu ML, Volp K, Bosser S, Solbach C, Lichter P, Joos S, Zornig M. HMGB1 inhibits cell death in yeast and mammalian cells and is abundantly expressed in human breast carcinoma. FASEB J. 2003;17:1295–1297. [DOI] [PubMed] [Google Scholar]
- 15.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201:1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin: mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–25761. [DOI] [PubMed] [Google Scholar]
- 17.Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–179. [DOI] [PubMed] [Google Scholar]
- 18.Schlueter C, Weber H, Meyer B, Rogalla P, Roser K, Hauke S, Bullerdiek J. Angiogenetic signaling through hypoxia: HMGB1: an angiogenetic switch molecule. Am J Pathol. 2005;166:1259–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chavakis E, Hain A, Vinci M, Carmona G, Bianchi ME, Vajkoczy P, Zeiher AM, Chavakis T, Dimmeler S. High-mobility group box 1 activates integrin-dependent homing of endothelial progenitor cells. Circ Res. 2007;100:204–212. [DOI] [PubMed] [Google Scholar]
- 20.Palumbo R, Sampaolesi M, De Marchis F, Tonlorenzi R, Colombetti S, Mondino A, Cossu G, Bianchi ME. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol. 2004;164:441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biscetti F, Straface G, De Cristofaro R, Lancellotti S, Rizzo P, Arena V, Stigliano E, Pecorini G, Egashira K, De Angelis G, Ghirlanda G, Flex A. High-mobility group box-1 protein promotes angiogenesis after peripheral ischemia in diabetic mice through a VEGF-dependent mechanism. Diabetes. 2010;59:1496–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Straino S, Di Carlo A, Mangoni A, De Mori R, Guerra L, Maurelli R, Panacchia L, Di Giacomo F, Palumbo R, Di Campli C, Uccioli L, Biglioli P, Bianchi ME, Capogrossi MC, Germani A. High-mobility group box 1 protein in human and murine skin: involvement in wound healing. J Invest Dermatol. 2008;128:1545–1553. [DOI] [PubMed] [Google Scholar]
- 23.Andersson U, Erlandsson-Harris H, Yang H, Tracey KJ. HMGB1 as a DNA-binding cytokine. J Leukoc Biol. 2002;72:1084–1091. [PubMed] [Google Scholar]
- 24.Rossini A, Zacheo A, Mocini D, Totta P, Facchiano A, Castoldi R, Sordini P, Pompilio G, Abeni D, Capogrossi MC, Germani A. HMGB1-stimulated human primary cardiac fibroblasts exert a paracrine action on human and murine cardiac stem cells. J Mol Cell Cardiol. 2008;44:683–693. [DOI] [PubMed] [Google Scholar]
- 25.Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, Hofmann MA, Kislinger T, Ingram M, Lu A, Tanaka H, Hori O, Ogawa S, Stern DM, Schmidt AM. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405:354–360. [DOI] [PubMed] [Google Scholar]
- 26.van Zoelen MA, Yang H, Florquin S, Meijers JC, Akira S, Arnold B, Nawroth PP, Bierhaus A, Tracey KJ, van der Poll T. Role of toll-like receptors 2 and 4, and the receptor for advanced glycation end products in high-mobility group box 1-induced inflammation in vivo. Shock. 2009;31:280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar A, Yu FS. Toll-like receptors and corneal innate immunity. Curr Mol Med. 2006;6:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu J, Wang Y, Xie L. Potential role of macrophages in experimental keratomycosis. Invest Ophthalmol Vis Sci. 2009;50:2087–2094. [DOI] [PubMed] [Google Scholar]
- 29.Johnson AC, Heinzel FP, Diaconu E, Sun Y, Hise AG, Golenbock D, Lass JH, Pearlman E. Activation of toll-like receptor (TLR)2, TLR4, and TLR9 in the mammalian cornea induces MyD88-dependent corneal inflammation. Invest Ophthalmol Vis Sci. 2005;46:589–595. [DOI] [PubMed] [Google Scholar]
- 30.Lin Q, Fang J, Fang D, Li B, Zhou H, Su SB. Production of recombinant human HMGB1 and anti-HMGB1 rabbit serum [epub ahead of print Jan. 19, 2011.]. Int Immunopharmacol. [DOI] [PubMed] [Google Scholar]
- 31.Sunderkotter C, Steinbrink K, Goebeler M, Bhardwaj R, Sorg C. Macrophages and angiogenesis. J Leukoc Biol. 1994;55:410–422. [DOI] [PubMed] [Google Scholar]
- 32.Crowther M, Brown NJ, Bishop ET, Lewis CE. Microenvironmental influence on macrophage regulation of angiogenesis in wounds and malignant tumors. J Leukoc Biol. 2001;70:478–490. [PubMed] [Google Scholar]
- 33.Cursiefen C, Chen L, Borges LP, Jackson D, Cao J, Radziejewski C, D’Amore PA, Dana MR, Wiegand SJ, Streilein JW. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J Clin Invest. 2004;113:1040–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakurai E, Anand A, Ambati BK, van Rooijen N, Ambati J. Macrophage depletion inhibits experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3578–3585. [DOI] [PubMed] [Google Scholar]
- 35.Tsutsumi C, Sonoda KH, Egashira K, Qiao H, Hisatomi T, Nakao S, Ishibashi M, Charo IF, Sakamoto T, Murata T, Ishibashi T. The critical role of ocular-infiltrating macrophages in the development of choroidal neovascularization. J Leukoc Biol. 2003;74:25–32. [DOI] [PubMed] [Google Scholar]
- 36.Gillitzer R, Goebeler M. Chemokines in cutaneous wound healing. J Leukoc Biol. 2001;69:513–521. [PubMed] [Google Scholar]
- 37.Li H, Sun B. Toll-like receptor 4 in atherosclerosis. J Cell Mol Med. 2007;11:88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaczorowski DJ, Nakao A, Vallabhaneni R, Mollen KP, Sugimoto R, Kohmoto J, Zuckerbraun BS, McCurry KR, Billiar TR. Mechanisms of Toll-like receptor 4 (TLR4)-mediated inflammation after cold ischemia/reperfusion in the heart. Transplantation. 2009;87:1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang H, Brignole-Baudouin F, Labbe A, Pauly A, Warnet JM, Baudouin C. LPS-stimulated inflammation and apoptosis in corneal injury models. Mol Vis. 2007;13:1169–1180. [PubMed] [Google Scholar]
- 40.Rodriguez-Martinez S, Cancino-Diaz ME, Miguel PS, Cancino-Diaz JC. Lipopolysaccharide from Escherichia coli induces the expression of vascular endothelial growth factor via toll-like receptor 4 in human limbal fibroblasts. Exp Eye Res. 2006;83:1373–1377. [DOI] [PubMed] [Google Scholar]
- 41.Chung SH, Kweon MN, Lee HK, Choi SI, Yang JY, Kim EK. Toll-like receptor 4 initiates an innate immune response to lipopolysaccharide in human conjunctival epithelial cells. Exp Eye Res. 2009;88:49–56. [DOI] [PubMed] [Google Scholar]
- 42.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. [DOI] [PubMed] [Google Scholar]
- 43.van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis. 2008;11:91–99. [DOI] [PubMed] [Google Scholar]
- 44.Huang Y, Yin H, Han J, Huang B, Xu J, Zheng F, Tan Z, Fang M, Rui L, Chen D, Wang S, Zheng X, Wang CY, Gong F. Extracellular hmgb1 functions as an innate immune-mediator implicated in murine cardiac allograft acute rejection. Am J Transplant. 2007;7:799–808. [DOI] [PubMed] [Google Scholar]
- 45.Gong Q, Xu JF, Yin H, Liu SF, Duan LH, Bian ZL. Protective effect of antagonist of high-mobility group box 1 on lipopolysaccharide-induced acute lung injury in mice. Scand J Immunol. 2009;69:29–35. [DOI] [PubMed] [Google Scholar]
- 46.Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi AA, Bianchi ME, Vezzani A. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16:413–419. [DOI] [PubMed] [Google Scholar]
- 47.Coats SR, Pham TT, Bainbridge BW, Reife RA, Darveau RP. MD-2 mediates the ability of tetra-acylated and penta-acylated lipopolysaccharides to antagonize Escherichia coli lipopolysaccharide at the TLR4 signaling complex. J Immunol. 2005;175:4490–4498. [DOI] [PubMed] [Google Scholar]
- 48.Sunderkotter C, Goebeler M, Schulze-Osthoff K, Bhardwaj R, Sorg C. Macrophage-derived angiogenesis factors. Pharmacol Ther. 1991;51:195–216. [DOI] [PubMed] [Google Scholar]
- 49.Lu P, Li L, Liu G, Zhang X, Mukaida N. Enhanced experimental corneal neovascularization along with aberrant angiogenic factor expression in the absence of IL-1 receptor antagonist. Invest Ophthalmol Vis Sci. 2009;50:4761–4768. [DOI] [PubMed] [Google Scholar]
- 50.Lu P, Li L, Wu Y, Mukaida N, Zhang X. Essential contribution of CCL3 to alkali-induced corneal neovascularization by regulating vascular endothelial growth factor production by macrophages. Mol Vis. 2008;14:1614–1622. [PMC free article] [PubMed] [Google Scholar]
- 51.Chung ES, Chauhan SK, Jin Y, Nakao S, Hafezi-Moghadam A, van Rooijen N, Zhang Q, Chen L, Dana R. Contribution of macrophages to angiogenesis induced by vascular endothelial growth factor receptor-3-specific ligands. Am J Pathol. 2009;175:1984–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Espinosa-Heidmann DG, Suner IJ, Hernandez EP, Monroy D, Csaky KG, Cousins SW. Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3586–3592. [DOI] [PubMed] [Google Scholar]
- 53.Ishida S, Usui T, Yamashiro K, Kaji Y, Amano S, Ogura Y, Hida T, Oguchi Y, Ambati J, Miller JW, Gragoudas ES, Ng YS, D’Amore PA, Shima DT, Adamis AP. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J Exp Med. 2003;198:483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, Stolz DB, Geller DA, Rosengart MR, Billiar TR. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204:2913–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang F, Lu Z, Hawkes M, Yang H, Kain KC, Liles WC. Fas (CD95) induces rapid, TLR4/IRAK4-dependent release of pro-inflammatory HMGB1 from macrophages. J Inflamm (Lond). 2010;7:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y, Akira S, Bierhaus A, Erlandsson-Harris H, Andersson U, Tracey KJ. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107:11942–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Urbonaviciute V, Furnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, Bianchi ME, Kirschning C, Wagner H, Manfredi AA, Kalden JR, Schett G, Rovere-Querini P, Herrmann M, Voll RE. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205:3007–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qiu J, Xu J, Zheng Y, Wei Y, Zhu X, Lo EH, Moskowitz MA, Sims JR. High-mobility group box 1 promotes metalloproteinase-9 upregulation through Toll-like receptor 4 after cerebral ischemia. Stroke. 2010;41:2077–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dai S, Sodhi C, Cetin S, Richardson W, Branca M, Neal MD, Prindle T, Ma C, Shapiro RA, Li B, Wang JH, Hackam DJ. Extracellular high mobility group box-1 (HMGB1) inhibits enterocyte migration via activation of Toll-like receptor-4 and increased cell-matrix adhesiveness. J Biol Chem. 2010;285:4995–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer G, Zitvogel L. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. [DOI] [PubMed] [Google Scholar]
- 61.Mittal D, Saccheri F, Venereau E, Pusterla T, Bianchi ME, Rescigno M. TLR4-mediated skin carcinogenesis is dependent on immune and radioresistant cells. EMBO J. 2010;29:2242–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ross RJ, Zhou M, Shen D, Fariss RN, Ding X, Bojanowski CM, Tuo J, Chan CC. Immunological protein expression profile in Ccl2/Cx3cr1 deficient mice with lesions similar to age-related macular degeneration. Exp Eye Res. 2008;86:675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Watanabe T, Keino H, Sato Y, Kudo A, Kawakami H, Okada AA. High mobility group box protein-1 in experimental autoimmune uveoretinitis. Invest Ophthalmol Vis Sci. 2009;50:2283–2290. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.