

Abstract
Background
In the current scenario, designing of world-wide effective malaria vaccine against Plasmodium falciparum remain challenging despite the significant progress has been made in last few decades. Conventional vaccinology (isolate, inactivate and inject) approaches are time consuming, laborious and expensive; therefore, the use of computational vaccinology tools are imperative, which can facilitate the design of new and promising vaccine candidates.
Results
In current investigation, initially 5548 proteins of P. falciparum genome were carefully chosen for the incidence of signal peptide/ anchor using SignalP4.0 tool that resulted into 640 surface linked proteins (SLP). Out of these SLP, only 17 were predicted to contain GPI-anchors using PredGPI tool in which further 5 proteins were considered as malarial antigenic adhesins by MAAP and VaxiJen programs, respectively. In the subsequent step, T cell epitopes of 5 genome derived predicted antigenic adhesins (GDPAA) and 5 randomly selected known malarial adhesins (RSKMA) were analysed employing MHC class I and II tools of IEDB analysis resource. Finally, VaxiJen scored T cell epitopes from each antigen were considered for prediction of population coverage (PPC) analysis in the world-wide population including malaria endemic regions. The validation of the present in silico strategy was carried out by comparing the PPC of combined (MHC class I and II) predicted epitope ensemble among GDPAA (99.97%), RSKMA (99.90%) and experimentally known epitopes (EKE) of P. falciparum (97.72%) pertaining to world-wide human population.
Conclusions
The present study systematically screened 5 potential protective antigens from P. falciparum genome using bioinformatics tools. Interestingly, these GDPAA, RSKMA and EKE of P. falciparum epitope ensembles forecasted to contain highly promiscuous T cell epitopes, which are potentially effective for most of the world-wide human population with malaria endemic regions. Therefore, these epitope ensembles could be considered in near future for novel and significantly effective vaccine candidate against malaria.
Electronic supplementary material
The online version of this article (10.1186/s12859-018-2482-x) contains supplementary material, which is available to authorized users.
Keywords: Malaria, Antigen, Epitopes, Population coverage, Vaccine, Plasmodium falciparum
Background
The human malaria is triggered as a result of 5 species of Plasmodium protozoan parasite (P. falciparum, P. vivax, P. ovale, P. malariae and P. knowlesi). However, P. falciparum is one of the most deadly species that responsible towards > 90% of overall malaria mediated deaths. On the basis of 2016 data, WHO evaluated that almost 445,000 deaths were due to malaria with a total of 216 million cases from 91 countries. The African region (15 countries), continues to account for about 91% of malaria deaths worldwide including 80% by sub-Saharan Africa as well as virtually all of the remaining incidence reported from South-East Asia and the Indian sub-continent with South America. Thus, if we want to get the global response of malaria vaccine, these heavily affected countries in African region must be our primary focus [1].
Human infection with the malaria parasite is developed following the inoculation of the sporozoite stage of the protozoan parasite by female anopheles mosquitoes. Infants and young children in malaria-endemic countries of African region naturally come across numerous clinical episodes of malaria before they build up partial immunity that defends towards severe disease as well as malaria mediated death. The mechanisms related to naturally acquired immunity are not completely explored; nevertheless, there are two foremost theories. First is the gradual attainment of strain-specific immunity, while second is the recurring antigenic exposure, possibly in combination with an age-linked immune maturation [2]. Although, important roles of both humoral as well as cell-mediated immune responses were demonstrated in animal models and humans subsequently natural malaria infection and exposure to experimental malaria vaccines. However, no clear correlation for protection have been documented for existing vaccine candidates except antibodies against circumsporozoite protein (CSP), which depict some correlation for protection towards the pre-erythrocytic stages of the parasite [3, 4].
Although, there are more than 30 vaccine candidates of malaria that were involved in different clinical phase trail, but they did not make a globally effective vaccine [5, 6]. The RTS,S is latest leading vaccine candidate with partial protection efficacy (39.0% in clinical malaria and 20.5% in severe malaria case) and restricted in limited regions of African countries [7]. The RTS,S is a recombinant vaccine of CSP and Hepatitis B surface antigen with liposomal adjuvant [5, 8], which induces the anti-CSP antibodies and CD4+ T cells during phase-III clinical trials [3, 4]. However, CD8+ T cell response remain missing from the RTS,S vaccine. Moreover, the attainment of naturally acquired partial immunity against malaria infection and some correlation to protection against experimental malaria vaccines, provide a positive perception towards the design of globally efficient malaria vaccines. Nonetheless, the large size of the Plasmodium genome (23.3 Mb) with more than 5000 genes pose significant challenges in experimental identification of immunodominant epitopes for activating both CD4+ and CD8+ T cells.
In past two decades, as a result of development of reverse vaccinology and immunoinformatics, identification of antigen-specific CD4+ and CD8+ T cell epitopes turn out to be more straightforward approach along with less laborious and low cost [9–14]. Concisely, this approach is based on the screening of antigenic features from the genome sequence of pathogen and further prediction of peptide ligands, which establish stable complexes (high affinity) with major histocompatibility complex (MHC) molecules. These MHC-peptide complexes can be used to monitor antigen-specific CD4+ and CD8+ T cell responses [15, 16]. Hence, the finding of targets for protective immunity has been the sole utmost significant objective of the overall immunologist involved towards the design and improvement of anti-malarial vaccines [17]. Critical review of literature depicted that there is little research work has been carried related to population coverage analysis of predicted and experimentally known epitopes (EKE) of P. falciparum [18].
In the present work, we hypothesized that application of reverse vaccinology along with immunological bioinformatics tools might uncover P. falciparum-resulting epitopes specific for CD4+ and CD8+ T cells involved in world-wide protection towards malaria. This study focus on genome-wide in silico screening of putative antigens of P. falciparum 3D7 genome and further prediction of human leukocyte antigen (HLA) class I and II binding epitopes covered by major world-wide population including malaria endemic regions through immune epitope database (IEDB) based prediction of population coverage (PPC). Ultimately, the predicted epitope ensemble of genome derived predicted antigenic adhesins (GDPAA) showed considerably higher world-wide population coverage (99.97%) compared to randomly selected known malarial adhesins (RSKMA) (99.90%) and EKE of P. falciparum (97.72%). These predicted epitope ensembles could be considered as promising candidate for effective nano vaccine design against malaria.
Methods
Data resources
The whole protein sequences (5548) of P. falciparum 3D7 genome were retrieved from PlasmoDB database (http://plasmodb.org) into the FASTA format, accessed on October 3, 2017 together with 5 RSKMA. Similarly, 151 non redundant EKE of P. falciparum 3D7 including 121 MHC class I and 30 MHC class II were retrieved from IEDB database (www.iedb.org) accessed on February 20, 2018 using MHC biding assay as search criteria (Additional file 1).
Methodology of the work
This investigation facilitates the screening of vaccine candidates from entire protein sequences (5548) of P. falciparum genome involved three major steps: i) prediction of probable antigens ii) prediction of T cell epitopes iii) screening of minimal epitope ensemble for maximum human population coverage. The methodology flow chart is given in Fig. 1.
Fig. 1.

Methodology flow chart used in the present study for genome-wide screening of effective malaria vaccine candidates
Prediction of antigens and physico-chemical characterization
The first step involved hypothetical confirmation for occurrence of surface protein and antigenic features assessed from protein primary sequences employing well-known tools: SignalP4.0, PredGPI, MAAP, VaxiJen and TMHMM in sequential manner to identify probable antigens [19, 20]. The proteins of P. falciparum were reflected as high-grade antigen candidates if they have forecasted features of a surface protein, i.e., a signal peptide, a glycophosphatidylinositol (GPI) anchor and transmembrane domain(s). Currently, SignalP4.0 is the most advanced and widely used tool for prediction of signal peptides from amino acid sequences of protein. The protein with signal peptide is targeted to the secretory route, nevertheless it is not necessarily to be secreted [21]. Numerous eukaryotic proteins are accompanying with the extracellular leaflet of the plasma, which carries a GPI-anchor associated with the C-terminal residue after a proteolytic cleavage occurring at omega-site [22]. The position of the GPI omega site within the secretary route proteins was predicted using PredGPI tool [23]. Surface proteins having functions in immune evasion and/ or cytoadhesion, are of importance also as promising vaccine candidates. Adhesion of Plasmodium parasites is mediated by proteins called adhesins. The parasite can be inhibited by immunizing the host with adhesins. Therefore, these GPI-anchored surface proteins were screened as malarial adhesins and adhesin-like proteins by MAAP tool [24–26]. The VaxiJen2.0 program was used for the evaluation of antigenicity of the malarial adhesins protein sequence [27, 28]. Furthermore, to assess the potential impact of membrane proximity, transmembrane regions of potential antigens (pathogenesis-related secretome of P. falciparum) were predicted using TMHMM2.0 tool [29–31]. The physico-chemical characterization [(grand average of hydropathicity (Gravy), theoretical isoelectric point (pI)] and water solubility analysis of all the 5 GDPAA and 5 RSKMA were studied by using tools ExPASy-ProtParam and INNOVAGEN, respectively [32–34]. Moreover, the clustering tree analysis of all the 5 GDPAA was carried out by MAFFT7.0 web server. In this context, the detail web addresses and threshold criteria of in silico tools exploited in the current investigation are provided in Table 1.
Table 1.
Details of the computational tools used for the prediction of antigens, epitopes and their population coverage analysis
| S.No. | Database/tool name | Description | URL | Threshold criteria |
|---|---|---|---|---|
| 1 | SignalP4.0 | Presence and location of signal peptide cleavage sites | http://www.cbs.dtu.dk/services/SignalP/ | Default |
| 2 | PredGPI | Prediction system for GPI-anchored proteins | http://gpcr.biocomp.unibo.it/predgpi/pred.htm | Default |
| 3 | MAAP | Prediction of Malarial adhesins and adhesins-like proteins. | http://maap.igib.res.in/ | ≥ 0 |
| 4 | TMHMM2.0 | Transmembrane helices in the integral membrane proteins | http://www.cbs.dtu.dk/services/TMHMM/ | ≤ 1 |
| 5 | VaxiJen2.0 | Prediction of protective antigens | http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html | ≥ 0.5 |
| 6 | ExPASy-ProtParam | Calculation of physicochemical parameters | https://web.expasy.org/protparam/ | Default |
| 7 | MAFFT7.0 | Multiple sequence alignment | http://mafft.cbrc.jp/alignment/server/large.html. | Default |
| 8 | INNOVAGEN | Prediction of water solubility of proteins | https://pepcalc.com/ | Default |
| 9 | IEDB-MHC class I-Consensus | Prediction of MHC class I binding epitopes | http://tools.iedb.org/mhci/ | IC50 ≤ 500 nM |
| 10 | IEDB-MHC class II-Consensus | Prediction of MHC class II binding epitopes | http://tools.iedb.org/mhcii/ | ≤ 3 percentile rank |
| 11 | IEDB-Population Coverage | Population coverage analysis of selected epitopes | http://tools.iedb.org/population/ | Default |
| 12 | CTLPred | Prediction of cytotoxic T cell epitopes | http://crdd.osdd.net/raghava/ctlpred/ | Default |
| 13 | IL-10Pred | Prediction of IL-10 inducer epitopes | http://webs.iiitd.edu.in/raghava/il10pred/predict3.php | Default |
| 14 | TAPPred | Prediction of TAP binding affinity of epitopes | http://crdd.osdd.net/raghava/tappred/ | Default |
| 15 | IFNepitope | Prediction of IFN-γ inducer epitopes | http://crdd.osdd.net/raghava/ifnepitope/predict.php | Default |
| 16 | PEP-FOLD 3 | Peptide and miniprotein structure prediction | http://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/ | Default |
| 17 | PatchDock | Molecular docking algorithm based on shape complementarity principles | https://bioinfo3d.cs.tau.ac.il/PatchDock/ | Default |
| 18 | FireDock | Refinement and re-scoring of rigid-body protein-protein docking | http://bioinfo3d.cs.tau.ac.il/FireDock/ | Default |
| 19 | ClusPro | Molecular docking | https://cluspro.bu.edu/login.php | Default |
Prediction of T cell epitopes
The subsequent step predicts the presence of immunogenic T cell epitopes in all the 5 GDPAA and 5 RSKMA by employing consensus technique of MHC class I and II tools accessible at IEDB web server. The prediction of T cell epitopes were performed for 51 HLA class I (cut off threshold IC50 ≤ 500 nM) and 54 HLA class II alleles (cut off threshold ≤ 3 percentile rank) [35]. These T cell epitopes were further screened by VaxiJen2.0 tool that predicted the antigenic epitopes with threshold ≥ 0.5 [32].
Population coverage analysis of predicted T cell epitope ensemble
In the following step, PPC analysis of aforesaid combined (MHC class I and II) epitopes for all 5 GDPAA were conducted by utilizing IEDB based PPC tool against world-wide population. Furthermore, the minimal combined epitope set for each antigen was formed along with the inclusion of only those epitope which have highest PPC value and restricted by different set of MHC alleles. In case, PPC value of epitopes are equal then epitope with highest VaxiJen score was included in the minimal epitope set. Then, after employing the same protocol of minimal epitope selection, an ultimate epitope ensemble was designed with the joint screening of minimal epitope set of all the 5 GDPAA. Finally, the PPC of 5 GDPAA minimal epitope set and epitope ensemble were executed for the selected malaria endemic regions in the present study (India, South America, South-east Asia, Central Africa, East Africa, North Africa, South Africa and West Africa). So as to compare the predictive efficiency of PPC analysis for GDPAA epitope ensemble selection, the same protocol was also applied to predict epitope ensemble of 5 RSKMA and 151 EKE of P. falciparum [19, 36].
Prediction of immunogenic induction
The immunogenicity of the MHC class I epitope ensemble designed from GDPAA was estimated by CTLPred and TAPPred [34, 37, 38] while induction of IFN-γ and IL-10 by MHC class II epitope ensemble from GDPAA were predicted using tools IFNepitope and IL-10Pred, respectively [39, 40].
Prediction of epitope structure and docking studies
A structure-based docking approach was further carried out so as to improve the predictive capability of peptide-MHC binding. The combination of sequence and structure-based approaches not merely enhances the probability of MHC binding prediction but also calculates the docked epitope orientation. The complex crystal structures of HLA–A*02:01 (PDB ID: 1I4F) and HLA–DRB1*01:01 (PDB ID: 1AQD) were retrieved from the protein data bank (PDB) for MHC class I and class II, respectively. We used peptides NQMIFVSSI (C1) and LKELIKVGLPSFENL (C2) from EKE of P. falciparum as positive controls in docking studies. The structural information about the test peptides of GDPAA epitope ensemble (P1, P3 and P11) and positive control peptides were modelled using the PEP-FOLD 3 web server. The test peptides of GDPAA epitope ensemble were then docked by employing PatchDock web server to HLA–A*0201 and HLA–DRB1*0101 interacting residues as input. The best 10 HLA-peptide complexes were further refined by the FireDock web server. In order to compare the study of molecular docking, ClusPro tool was also used for the same test and control peptides along with target MHC molecule of interest [41]. The details of the docking web servers are given in the Table 1.
Results and discussion
Prediction and characterization of antigens
Primarily, 5548 proteins of P. falciparum genome were screened for the presence of signal peptide/ anchor using SignalP4.0 and found 640 surface accompanying/screatory proteins. Out of 640, only 17 are predicted to contain GPI-anchors using PredGPI tool and further 5 proteins are predicted as malarial adhesins and adhesin-like proteins. These 5 malarial adhesin proteins viz. CSP, surface protein P113 (P113), merozoite surface protein 1 (MSP1), 28 kDa ookinete surface protein (P28) and 25 kDa ookinete surface antigen precursor (P25), were also predicted as antigenic using VaxiJen, which contain ≤ 1 transmembrane helix as predicted by TMHMM2.0 server (Table 2). Among these malarial adhesin proteins, CSP is recognized as multifunctional protein that needed towards the sporozoite formation so as to mediate the parasite passage from vector mosquito midgut into the hepatocyte cells [42]. On the other hand, P113 is surface protein that expressed in almost every stage of parasite pertaining to its life-cycle. Such surface protein not only facilitates effective transformation of sporozoite in liver stage but also serve as a molecular link between the parasite and an erythrocyte [43, 44]. Likewise, MSP1 is an extremely abundant protein that covers the merozoite surface in almost all the species of malaria parasites and important with reference to invasion of erythrocyte [45]. In addition, P25 as well as P28 that behave as post-fertilization proteins are expressed in large quantities upon the surfaces of zygotes including the maturing ookinete stage of the parasite, which can likely to facilitate parasite clustering [46]. Interestingly, these GPI-anchored surface linked antigens are projected as promising vaccine targets against various developmental stage of the P. falciparum owing to their significant roles not only in the invasion of host cell but also in the completion of life-cycle of parasite as reviewed by Draper group [47]. These antigens are extremely conserved amongst Plasmodium species as well as in various isolates of different geographical region. Such important characteristics lead to the development of simpler malaria vaccine as single target gene sequence will be efficient against various parasite species like P. falciparum, P. vivax and P. berghai. Furthermore, a similar analysis data are also stored in MalVac database, which presently accessible as a scientific community resource for only 161 adhesin proteins belonging to P. falciparum [48].
Table 2.
The predicted physicochemical properties of 5 genome derived predicted antigenic adhesins (GDPAA) and 5 randomly selected known malarial adhesins (RSKMA)
| S.No. | PlasmoDB ID | Name of protein | No. of amino acids | GRAVY | pI (pH) | Water Solubility |
|---|---|---|---|---|---|---|
| GDPAA | ||||||
| 1 | PF3D7_0304600 | CSP | 397 | −1.249 | 5.36 | Good |
| 2 | PF3D7_0930300 | MSP1 | 1720 | −0.655 | 6.11 | Good |
| 3 | PF3D7_1030900 | P28 | 218 | −0.046 | 5.94 | Poor |
| 4 | PF3D7_1031000 | P25 | 217 | −0.001 | 6.55 | Good |
| 5 | PF3D7_1420700 | P113 | 969 | −1.063 | 4.48 | Good |
| RSKMA | ||||||
| 1 | PF3D7_0617400 | Erythrocyte membrane protein 1, PfEMP1 | 2394 | −1.013 | 6.10 | Good |
| 2 | PF3D7_0616500 | TRAP-like protein | 1371 | −1.183 | 5.12 | Good |
| 3 | PF3D7_1133400 | Apical membrane antigen 1 | 622 | −0.812 | 5.36 | Good |
| 4 | PF3D7_0731500 | Erythrocyte binding antigen-175 | 1502 | −1.093 | 5.52 | Good |
| 5 | PF3D7_0202000 | Knob-associated histidine-rich protein | 654 | −1.432 | 9.17 | Good |
The appropriate physico-chemical properties as well as stable structure of proteins are needed to trigger immune response [33, 49]. The negative Gravy values of 5 GDPAA clearly indicate their hydrophilic nature and good water solubility except P28 (Table 2). The clustering analysis of these antigens also revealed a close similarity between CSP and P113 as well as P28 and P25 inferred by MAFFT7.0 web server (Fig. 2). A vaccine involving a little number of alleles may be enough for coverage towards naturally-circulating strains, which also supports the concept of multi-allele methodology for designing polymorphic antigens as malaria vaccines [50, 51]. The study conducted by Soulama group revealed that AMA-1 antigen of P. falciparum 3D7 showed lower allelic diversity in Central Africa compared to West Africa [52]. Thus, the limited allelic diversity of antigens screened in the present study is suitable vaccine candidates for further immunological experiment.
Fig. 2.
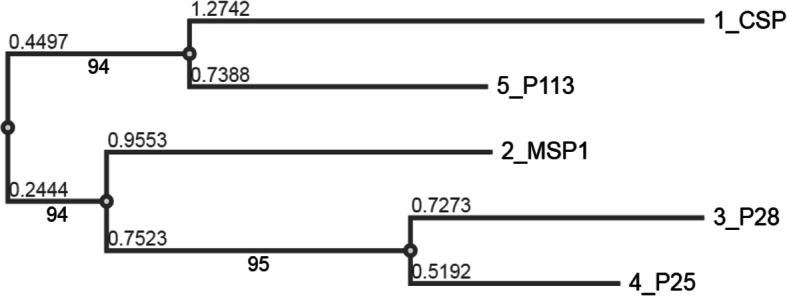
The clustering tree of 5 genome derived predicted antigenic adhesions (GDPAA) of P. falciparum generated by web server MAFFT version 7 with 100 times bootstrapping. The numbers above the branch showing branch length while below the branch showing bootstrap values
Prediction of antigenic T cell epitopes
It would be a noticeable option to provoke sufficient T cell response that may enhance the protective efficacy as a result of additive/ synergistic effects [53, 54]. Considering this, the ultimate goal of the current investigation focused on integrating the epitopes belonging to aforementioned antigens presented by common human MHC class I and II molecules. Apart from this, the task of epitope discovery with vaccine designing is gradually dependent on bioinformatics tools and access to data relevant to immune reactions of specific pathogens. The tools involve validated and benchmarked methods to predict T cell epitopes of MHC class I and II binding peptides [36]. Furthermore, the protective efficacy of the vaccine is dependent on the polymorphic human MHC alleles and malaria antigens [55]. A total of 2647 T cell epitopes (755 HLA class I and 1892 HLA class II) were predicted for all the 5 GDPAA which were further screened by VaxiJen tool with threshold ≥ 0.5 resulted into a manageable 1270 antigenic epitopes (Table 3, Additional file 2) [32]. A similar data of T cell epitopes prediction for RSKMA are also presented in Table 3 and Additional file 3.
Table 3.
Predicted number of T cell epitopes against 5 genome derived predicted antigenic adhesins (GDPAA) and 5 randomly selected known malarial adhesins (RSKMA)
| S.No. | PlasmoDB ID | Total no. of predicted epitopes | No. of epitopes with VaxiJen score ≥ 0.5 | ||||
|---|---|---|---|---|---|---|---|
| MHC class I | MHC class II | Combined | MHC class I | MHC class II | Combined | ||
| GDPAA | |||||||
| 1 | PF3D7_0304600 | 55 | 110 | 165 | 28 | 46 | 74 |
| 2 | PF3D7_0930300 | 426 | 1005 | 1431 | 197 | 466 | 663 |
| 3 | PF3D7_1030900 | 62 | 122 | 184 | 28 | 65 | 93 |
| 4 | PF3D7_1031000 | 48 | 122 | 170 | 32 | 66 | 98 |
| 5 | PF3D7_1420700 | 164 | 533 | 697 | 82 | 260 | 342 |
| RSKMA | |||||||
| 1 | PF3D7_0617400 | 485 | 1167 | 1652 | 238 | 427 | 665 |
| 2 | PF3D7_0616500 | 299 | 796 | 1095 | 137 | 408 | 545 |
| 3 | PF3D7_1133400 | 183 | 364 | 547 | 94 | 188 | 282 |
| 4 | PF3D7_0731500 | 326 | 714 | 1040 | 175 | 427 | 602 |
| 5 | PF3D7_0202000 | 110 | 167 | 277 | 58 | 99 | 157 |
Population coverage analysis and selection of minimal epitope ensemble
The immunogens based on population coverage analysis enable us towards the screening of best possible minimal epitope combination, i.e., epitope ensemble so as to maximize vaccine effectiveness. In this background, the PPC strategy was followed for developing the epitope ensemble of chosen antigens towards improving the vaccine efficacy in the world-wide as well as endemic population of malaria. Primarily, the PPC of predicted T cell epitopes (MHC class I, II and combined) from 5 GDPAA with VaxiJen score ≥ 0.5 were performed for the world-wide, South East Asia, India, South-America and Central Africa, East Africa, North Africa and West Africa populations (Additional file 4). The minimum PPC value of combined epitopes (94.84%) for P25 is reported for South-America population, whereas the maximum PPC (99.99%) is forecasted for MSP1 antigen for world-wide population (Fig. 3) [36]. However, the PPC values of EKE of P. falciparum for 121 MHC class I, 30 MHC class II and 151 combined epitopes were found 96.97, 65.35 and 99.25%, correspondingly. In order to minimize the epitopes in the vaccine construct design, we have restricted epitopes number on the basis of considering minimal epitopes binding with maximum number of different MHC alleles for the entire 5 GDPAA (Additional file 5). This resulted into 5 minimal set of epitopes covering populations world-wide, South East Asia, India, South America, Central Africa, East Africa, North Africa and West Africa (Additional file 6). The PPC value (86.18%) of combined minimal epitopes is minimum for CSP antigen for South-East Asian population while the maximum coverage (99.95%) for MSP1 antigen is reported for world-wide population. Further, employing the above protocol of minimal epitope selection, an epitope ensemble was designed for each of the combined minimal epitopes of 5 GDPAA and 5 RSKMA as well as 151 EKE. Here, as detailed in Table 4, the PPC value of GDPAA epitope ensemble (99.97%) (Additional file 6) is significantly higher over the epitope ensemble of RSKMA (99.90%) (Additional file 3) and EKE of P. falciparum epitope ensemble (97.72%) for the world-wide population (Table 4). Also, it is interesting to note that the PPC value of predicted MHC class II epitope ensemble of GDPAA (93.71%) is much higher than the MHC class II epitope ensemble of EKE of P. falciparum (75.31%). However, an exception was recorded for South African population (0.43%) that could be due to limited HLA typing data as detailed in Table 5.
Fig. 3.

The predicted population coverage (PPC) of combined (MHC class I and II) T cell epitopes for 5 genome derived predicted antigenic adhesins (GDPAA) with VaxiJen score ≥ 0.5
Table 4.
The world-wide PPC coverage of epitope ensemble predicted from all 5 genome derived predicted antigenic adhesins (GDPAA) and 5 randomly selected known malarial adhesins (RSKMA) as well as 151 experimentally known epitopes (EKE) of P. falciparum
| S.No. | Epitope ensemble | PPC for world-wide population (%) | ||
|---|---|---|---|---|
| MHC class I | MHC class II | Combined | ||
| 1. |
GDPAA MHC class I: FAMSNALLV, ILMLILYSF, YTLTAGVCV, FSSSNNSVY, YEMKFNNNF, LIVCSIFIK, YFNDDIKQF, LLKSYKYIK, SRLKKRKYF, KGMSSSQEM MHC class II: IPFFILHILLLQFLL, FIQLYITLNKARVTE, TCGNGIQVRIKPGSA |
99.53 | 93.71 | 99.97 |
| 2. |
RSKMA MHC class I: YFFASFFVL, FTYDSEEYY, FAFPPTEPL, FMPPRRQHF, FRDEWWKVI, RIYDKNLLM, KLYFPTPAL, YAFSEECPY, ISFQNYTYL MHC class II: DKMKIIIASSAAVAV, YKYAASFTLAAILFL |
98.67 | 92.31 | 99.90 |
| 3. |
EKE of P. falciparum MHC class I: RPRGDNFAV, TPYAGEPAPF, YLINKHWQR, NQMIFVSSI, KVSDEIWNY MHC class II: DAEVAGTQYRLPSGKCPVFG, LKELIKVGLPSFENL, ALLIIPPKIHISIEL |
90.78 | 75.31 | 97.72 |
Table 5.
The PPC value of epitope ensemble vaccine candidate of 5 genome derived predicted antigenic adhesins (GDPAA) and 5 randomly selected known malarial adhesins (RSKMA) as well as 151 experimentally known epitopes (EKE) of P. falciparum for malaria endemic regions
| Population | GDPAA (%) | RSKMA (%) | EKE of P. falciparum (%) |
|---|---|---|---|
| North Africa | 99.93 | 99.84 | 95.34 |
| India | 99.54 | 99.17 | 93.01 |
| East Africa | 99.68 | 99.41 | 92.17 |
| Southeast Asia | 99.39 | 99.07 | 81.71 |
| South America | 98.94 | 97.96 | 87.66 |
| Central Africa | 99.31 | 98.78 | 90.47 |
| West Africa | 99.84 | 99.19 | 92.88 |
| South Africa | 0.43 | 0.43 | 0.40 |
The GDPAA epitope ensemble as a vaccine candidate includes 10 MHC class I epitopes (P1:FAMSNALLV, P2:ILMLILYSF, P3:YTLTAGVCV, P4:FSSSNNSVY, P5:YEMKFNNNF, P6:LIVCSIFIK, P7:YFNDDIKQF, P8:LLKSYKYIK, P9:SRLKKRKYF, P10:KGMSSSQEM) and 3 MHC class II epitopes (P11:IPFFILHILLLQFLL, P12:FIQLYITLNKARVTE, P13:TCGNGIQVRIKPGSA) as presented in Table 4 (Additional file 7). Out of 10 MHC class I epitopes, 4 epitopes (P1, P2, P5 and P9) were confirmed as cytotoxic T-lymphocyte (CTL) epitopes by using CTLPred, while 7 epitopes (P1, P2, P3, P5, P7, P8, and P9) were confirmed as TAP binders by TAPPred. Also, the epitope P11 was found to be inducer of both IFN-γ and IL-10 cytokines predicted by IFN-epitope and IL-10Pred, respectively while epitope P13 induced only IFN-γ. The GDPAA epitope ensemble showed 99.54% of PPC value for Indian population (Table 5). In the West African population, HLA–B*5301 and HLA–DRB1*1302 alleles are linked with the reduction in life-threatening malaria [56]. In our study, the GDPAA epitope ensemble P1: FAMSNALLV binds to the HLA–B*5301 and P12: FIQLYITLNKARVTE also binds to the HLA–DRB1*1302. Moreover, the epitope P4: FSSSNNSVY and P8: LLKSYKYIK of GDPAA ensemble binds to HLA–A*30:02 and HLA–A*30:01, correspondingly, which are also linked with cerebral malaria [57]. In addition, the MHC class II epitopes P12: FIQLYITLNKARVTE and P11: IPFFILHILLLQFLL binds to the HLA–DRB1*04:01, which is accompanying with severe malaria in Northern Ghana [58]. There are 5 most frequently HLA alleles (HLA–B*15:03, HLA–B*42:01, HLA–B*53:01, HLA–B*58:02 and HLA–B*57:03) found in African regions [59]. The peptide P10: KGMSSSQEM found in the GDPAA epitope ensemble also binds to HLA–B*5801, while peptide P1: FAMSNALLV binds to HLA–B*5301.
The interaction between the peptide and MHC is an important characteristic of T cell epitope and the accessibility of the crystal structures facilitates us to determine the mode of interaction by computational methodology [60]. The docking data obtained from the freely available online tools PatchDock and ClusPro depicted that the anchor residues of majority of the epitope ensemble associate with the active site residues of HLA–A*0201 and HLA–DRB1*0101 that specify the superior mode of binding of screened epitopes with the T cell receptor. For HLA–A*0201, the geometric shape complementarities scores for test peptides P1 and P3 were 7284 and 7586, respectively, which were very close to the score of the positive control peptide C1 (7604) and global energy for test peptides P1and P3 were − 11.93 and − 15.72, correspondingly that were less than to the positive control peptide C1 (− 7.99). Similarly for HLA–DRB1*0101, the geometric shape complementarity scores for P11 was 11,870, which was very close to the score of the positive control C2 peptide (10806) and global energy for P11 was − 61.82, which was less than the positive control C2 peptide (− 55.11). The ClusPro docking tool was also used to predict the HLA–epitope complex molecular model and calculate the clustering score. The test epitopes (P1, P3 and P11), target MHC proteins and control peptides were same as selected in the PatchDock tool to compare the docking results. For HLA–A*0201, the lowest clustering scores for P1 and P3 peptides were − 694.4 and − 758.3, respectively, which were lower to the score of the positive control C1 peptide (− 647.1). Similarly, for HLA-DRB1*0101, the lowest clustering score for P11 peptides was − 1159.8 which was very much lower than the score of the positive control C2 peptide (− 715.7). The docking models for HLA–A*0201-C1, HLA–A*0201-P1, HLA–A*0201-P3, HLA–DRB1*0101-C2 and HLA–DRB1*0101-P11 are shown in the Fig. 4.
Fig. 4.
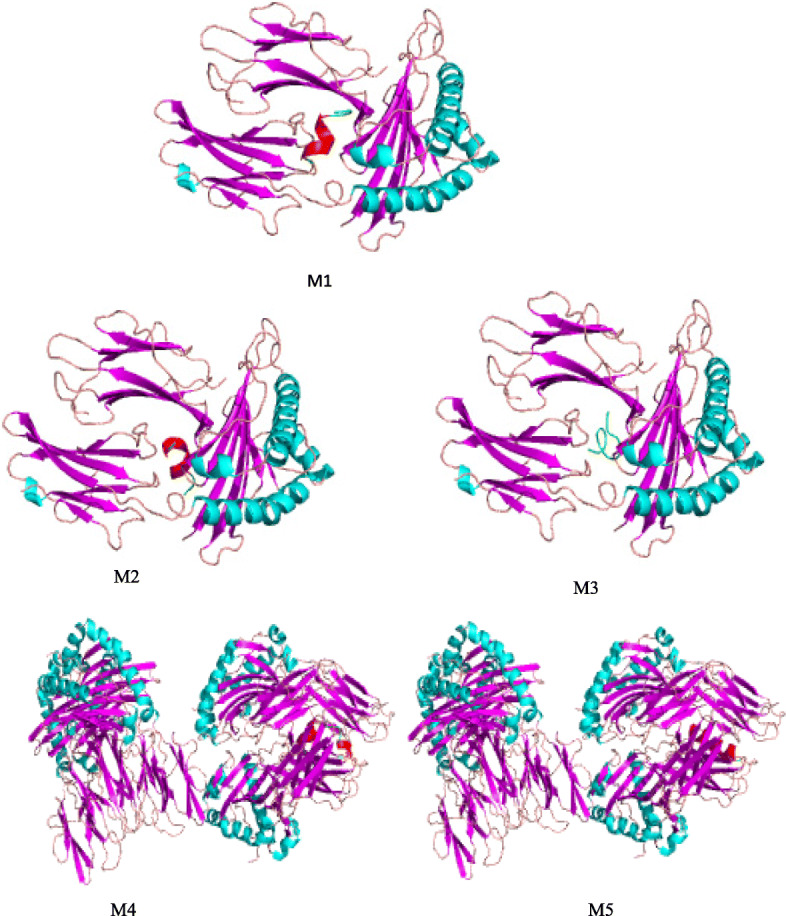
The visual models (M1-M5) of the best docking orientation as predicted by ClusPro for the HLA–A*0201 and HLA–DRB1*0101 molecules complexed with test epitopes (P1 and P3) and control epitopes (C1 and C2). M1: complex of epitope C1 with HLA–A*0201, M2: complex of epitope P1 with HLA–A*0201, M3: complex of epitope P3 with HLA–A*0201 M4: complex of epitope C2 with HLA–DRB1*0101 and (e) M5: complex of epitope P11 with HLA–DRB1*0101. The epitope in models are shown in red and green colour
The reverse vaccinology and immunoinformatics strategies are still under progressive phase, however reasonable triumphs may result in substantial advancement on epitope-based ensemble vaccine efficacy against malaria pathogens for example, by enhancing coverage in the target populations through reasonably bearing in mind the specificity as well as occurrence of the HLA molecules [61, 62]. Therefore, epitope ensemble provided in the present study provides the basis for effective malaria vaccine design. A well-known drawback of epitope ensemble vaccine is poor immunogenicity, usually necessitating the use of suitable adjuvants [63]. Therefore, these predicted and experimentally validated epitopes ensemble could be tested for further studies like nanovaccine formulation and evaluation in the experimental animal model for actual efficacy of nano sized malaria vaccine. Numerous investigations found to depict that malarial antigens showed more immunogenicity and superior correlated with protection when presented on nanoparticles based carrier systems like self-assembling protein nanoparticle (SAPN). The SAPN depend on coiled-coil domains of proteins to form stable nanoparticles [64]. Recently, Burkhard and Lanar developed protein based nano vaccines, which provide robust immunity against malaria [65]. This SAPN contains HLA-supertypes-restricted CD8+ T cell epitopes (separated by N/KAAA spacers and optimized for proteasomal cleavage) from antigens expressed during malaria pathogen life-cycle, the universal CD4+ T cell epitope, and flagellin as a scaffold and TLR5 agonist. On the other hand, used de novo designed amino acid domains to fuel the development of the coiled-coil scaffolds that present the antigenic epitopes on the nanoparticles surface [66]. As the surface area of the nanoparticles increases with the reduction of particles size, therefore there is a great need to develop more in silico strategy for effective nanovaccine designs that fulfil the vaccine requirement of needy human being [67].
Conclusions
The design of epitope ensemble using computational vaccinology is one of the promising alternatives, which enables finding of new epitope based vaccine candidate in a cost-effective manner for global as well as P. falciparum malaria endemic population. The present study screened 5 GDPAA as potential vaccine targets due to their extreme conservancy amongst Plasmodium species including isolates of different geographical region. The PPC analysis with respect to epitope ensemble of 5 GDPAA and 5 RSKMA as well as 151 EKE of P. falciparum showed more than 81% population coverage in the world-wide along with malarial endemic regions except South Africa. These promiscuous T cell epitope ensembles will significantly aid towards the fast development of more efficacious malaria vaccine against P. falciparum. Therefore, this promising strategy could be extended to other infectious diseases as well. Overall, the computational tools used here are not yet ready to substitute the wet laboratory experimentation, rather they are assisting in experimental design and reducing the time and cost of the vaccine development process.
Additional files
Experimentally known epitopes of P. falciparum 3D7. (XLSX 11 kb)
(a) The PPC of predicted MHC class I binding T cell epitopes from 5 genome derived predicted antigenic adhesions (GDPAA) with VaxiJen score ≥ 0.5, (b) The PPC of predicted MHC class II binding T cell epitopes from 5 genome derived predicted antigenic adhesions (GDPAA) with VaxiJen score ≥ 0.5. (XLSX 88 kb)
(a) The PPC of predicted T cell epitopes (MHC class I, MHC class II and combined) from 5 randomly selected known malarial adhesions (RSKMA) with VaxiJen score ≥ 0.5, (b) The PPC of predicted T cell epitopes (MHC class I, MHC class II and combined) from 5 RSKMA, (c) Predicted set of T cell epitopes of 5 RSKMA, (d) The PPC of minimal T cell epitopes (MHC class I, MHC class II and combined) from 5 RKSMA, (e) The predicted ensemble epitope (MHC class I and MHC class II) from 5 RKSMA. (XLSX 145 kb)
The PPC of predicted T cell epitopes for 5 genome derived predicted antigenic adhesions (GDPAA). (XLSX 9 kb)
The PPC of individual epitope found in minimal epitope set of 5 genome derived predicted antigenic adhesions (GDPAA). (XLSX 13 kb)
The PPC of minimal combined epitope set for 5 GDPAA and jointly screened epitope ensemble. (XLSX 10 kb)
The PPC value of individual epitope of epitope ensemble designed from GDPAA and experimentally known epitope (EKE) of P. falciparum. (XLSX 11 kb)
Acknowledgements
The authors are thankful to member of doctoral research committee at Amity University Uttar Pradesh, Lucknow Campus for providing necessary support and guidance.
Funding
Publication costs were self-financed by the authors.
Availability of data and materials
The data analysed but not presented in the main text are available as supplementary materials provided as Additional files 1, 2, 3, 4, 5, 6 and 7.
About this supplement
This article has been published as part of BMC Bioinformatics Volume 19 Supplement 13, 2018: 17th International Conference on Bioinformatics (InCoB 2018): bioinformatics. The full contents of the supplement are available online at https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-19-supplement-13.
Abbreviations
- AMA-1
Apical membrane antigen-1
- CSP
Circumsporozoite protein
- CTL
Cytotoxic T-lymphocyte
- GPI-anchors
Glycophosphatidylinositol-anchors
- GRAVY
Grand average of hydropathicity
- HLA
Human leukocyte antigen
- IEDB
Immune epitope database
- IFN
Interferon
- IL-10
Interleukin-10
- MHC
Major histocompatibilty complex
- MSP
Merozoite surface protein
- PDB
Protein data bank
- PPC
Predicted population coverage
- SAPN
Self-assembling protein nanoparticle
- TAP
Transporter associated with antigen processing
- WHO
World Health Organisation
Authors’ contributions
SPS conceived and designed the study and experiments and wrote the manuscript. MP performed the experiment and analyses. GS, SS and AKS contributed by providing suggestions in experimental design. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Manisha Pritam, Email: manishapritam12@gmail.com.
Garima Singh, Email: garima.singh1107@gmail.com.
Suchit Swaroop, Email: drsswaroop@gmail.com.
Akhilesh Kumar Singh, Email: aksingh2@lko.amity.edu.
Satarudra Prakash Singh, Phone: +91-522-2399553, Email: spsingh5@amity.edu, Email: sprakashsingh@gmail.com.
References
- 1.WHO. Global response to malaria at crossroads. Geneva: World Health Organization; 2017. http://www.who.int/news-room/detail/29-11-2017-global-response-to-malaria-at-crossroads. Accessed 11 June 2018.
- 2.Neafsey DE, Juraska M, Bedford T, Benkeser D, Valim C, Griggs A, et al. Genetic Diversity and Protective Efficacy of the RTS,S/AS01 Malaria Vaccine. N Engl J Med. 2015;373(21):2025–2037. doi: 10.1056/NEJMoa1505819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moorthy VS, Good MF, Hill AV. Malaria vaccine developments. Lancet. 2004;363(9403):150–156. doi: 10.1016/S0140-6736(03)15267-1. [DOI] [PubMed] [Google Scholar]
- 4.Moorthy VS, Ballou WR. Immunological mechanisms underlying protection mediated by RTS,S: a review of the available data. Malar J. 2009;8:312. doi: 10.1186/1475-2875-8-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies DH, Duffy P, Bodmer JL, Felgner PL, Doolan DL. Large screen approaches to identify novel malaria vaccine candidates. Vaccine. 2015;33(52):7496–7505. doi: 10.1016/j.vaccine.2015.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh SP, Verma V, Mishra BN. Characterization of Plasmodium falciparum proteome at asexual blood stages for screening of effective vaccine candidates: an Immunoinformatics approach. Immunol Immunogenet Insights. 2015;7:21–30. doi: 10.4137/III.S24755. [DOI] [Google Scholar]
- 7.WHO. Malaria position paper.World Health organization; 2016. http://www.who.int/wer/2016/wer9104.pdf?ua=1. Accessed 11 June 2018.
- 8.Birkett AJ, Moorthy VS, Loucq C, Chitnis CE, Kaslow DC. Malaria vaccine R&D in the decade of vaccines: breakthroughs, challenges and opportunities. Vaccine. 2013;31(Suppl 2):B233–B243. doi: 10.1016/j.vaccine.2013.02.040. [DOI] [PubMed] [Google Scholar]
- 9.Rappuoli R, Pizza M, Del Giudice G, De Gregorio E. Vaccines, new opportunities for a new society. Proc Natl AcadSci U S A. 2014;111(34):12288–12293. doi: 10.1073/pnas.1402981111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rappuoli R. Vaccines, emerging viruses, and how to avoid disaster. BMC Biol. 2014;12:100. doi: 10.1186/s12915-014-0100-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Centlivre M, Combadière B. New challenges in modern vaccinology. BMC Immunol. 2015;16:18. doi: 10.1186/s12865-015-0075-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh SP, Mishra BN. Major histocompatibility complex linked databases and prediction tools for designing vaccines. Hum Immunol. 2016;77(3):295–306. doi: 10.1016/j.humimm.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 13.Delany I, Rappuoli R, Seib KL. Vaccines, reverse vaccinology, and bacterial pathogenesis. Cold Spring Harb Perspect Med. 2013;3(5):a012476. doi: 10.1101/cshperspect.a012476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rizwan M, Naz A, Ahmad J, Naz K, Obaid A, Parveen T, et al. VacSol: a high throughput in silico pipeline to predict potential therapeutic targets in prokaryotic pathogens using subtractive reverse vaccinology. BMC Bioinformatics. 2017;18(1):106. doi: 10.1186/s12859-017-1540-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodswen SJ, Kennedy PJ, Ellis JT. A novel strategy for classifying the output from an in silico vaccine discovery pipeline for eukaryotic pathogens using machine learning algorithms. BMC Bioinformatics. 2013;14:315. doi: 10.1186/1471-2105-14-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meza B, Ascencio F, Sierra-Beltrán AP, Torres J, Angulo C. A novel design of a multi-antigenic, multistage and multi-epitope vaccine against helicobacter pylori: an in silico approach. Infect Genet Evol. 2017;49:309–317. doi: 10.1016/j.meegid.2017.02.007. [DOI] [PubMed] [Google Scholar]
- 17.Pichugin A, Zarling S, Perazzo L, Duffy PE, Ploegh HL, Krzych U. Identification of a novel CD8 T cell epitope derived from Plasmodium berghei protective liver-stage antigen. Front Immunol. 2018;9:91. doi: 10.3389/fimmu.2018.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh SP, Srivastava D, Mishra BN. Genome-wide identification of novel vaccine candidates for Plasmodium falciparum malaria using integrative bioinformatics approaches. 3 Biotech. 2017;7(5):318. doi: 10.1007/s13205-017-0947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalyanaraman N. In silico prediction of potential vaccine candidates on capsid protein of human bocavirus 1. MolImmunol. 2018;93:193–205. doi: 10.1016/j.molimm.2017.11.024. [DOI] [PubMed] [Google Scholar]
- 20.Kahsay RY, Gao G, Liao L. An improved hidden Markov model for transmembrane protein detection and topology prediction and its applications to complete genomes. Bioinformatics. 2005;21(9):1853–1858. doi: 10.1093/bioinformatics/bti303. [DOI] [PubMed] [Google Scholar]
- 21.Nielsen H. Predicting secretory proteins with SignalP. Methods Mol Biol. 2017;1611:59–73. doi: 10.1007/978-1-4939-7015-5_6. [DOI] [PubMed] [Google Scholar]
- 22.Guy AJ, Irani V, Beeson JG, Webb B, Sali A, Richards JS, Ramsland PA. Proteome-wide mapping of immune features onto Plasmodium protein three-dimensional structures. Sci Rep. 2018;8(1):4355. doi: 10.1038/s41598-018-22592-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swearingen KE, Lindner SE, Flannery EL, Vaughan AM, Morrison RD, Patrapuvich R, et al. Proteogenomic analysis of the total and surface-exposed proteomes of Plasmodium vivax salivary gland sporozoites. PLoSNegl Trop Dis. 2017;11(7):e0005791. doi: 10.1371/journal.pntd.0005791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen JH, Chen SB, Wang Y, Ju C, Zhang T, Xu B, et al. An immunomics approach for the analysis of natural antibody responses to Plasmodium vivax infection. MolBiosyst. 2015;11(8):2354–2363. doi: 10.1039/c5mb00330j. [DOI] [PubMed] [Google Scholar]
- 25.Anderson DC, Lapp SA, Barnwell JW, Galinski MR. A large scale Plasmodium vivax- Saimiriboliviensistrophozoite-schizont transition proteome. PLoS One. 2017;12(8):e0182561. doi: 10.1371/journal.pone.0182561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ansari FA, Kumar N, BalaSubramanyam M, Gnanamani M, Ramachandran S. MAAP: malarial adhesins and adhesin-like proteins predictor. Proteins. 2008;70(3):659–666. doi: 10.1002/prot.21568. [DOI] [PubMed] [Google Scholar]
- 27.Delfani S, Imani Fooladi AA, Mobarez AM, Emaneini M, Amani J, Sedighian H. In silico analysis for identifying potential vaccine candidates against Staphylococcus aureus. ClinExp Vaccine Res. 2015;4(1):99–106. doi: 10.7774/cevr.2015.4.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cravo P, Machado RB, Leite JA, Leda T, Suwanarusk R, Bittencourt N, et al. In silico epitope mapping and experimental evaluation of the Merozoite adhesive Erythrocytic binding protein (MAEBL) as a malaria vaccine candidate. Malar J. 2018;17(1):20. doi: 10.1186/s12936-017-2144-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehla K, Ramana J. Surface proteome mining for identification of potential vaccine candidates against campylobacter jejuni: an in silico approach. Funct Integr Genomics. 2017;17(1):27–37. doi: 10.1007/s10142-016-0530-z. [DOI] [PubMed] [Google Scholar]
- 30.Jing L, Guo D, Hu W, Niu X. The prediction of a pathogenesis-related secretome of Pucciniahelianthi through high-throughput transcriptome analysis. BMC Bioinformatics. 2017;18(1):166. doi: 10.1186/s12859-017-1577-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lopez D, Ribeiro S, Label P, Fumanal B, Venisse JS, Kohler A, et al. Genome-wide analysis of Corynesporacassiicola leaf fall disease putative effectors. Front Microbiol. 2018;9:276. doi: 10.3389/fmicb.2018.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oany AR, Emran AA, Jyoti TP. Design of an epitope-based peptide vaccine against spike protein of human coronavirus: an in silico approach. Drug Des Devel Ther. 2014;8:1139–1149. doi: 10.2147/DDDT.S67861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Negahdaripour M, Nezafat N, Eslami M, Ghoshoon MB, Shoolian E, Najafipour S, et al. Structural vaccinology considerations for in silico designing of a multi-epitope vaccine. Infect Genet Evol. 2018;58:96–109. doi: 10.1016/j.meegid.2017.12.008. [DOI] [PubMed] [Google Scholar]
- 34.Dikhit MR, Ansari MY, Vijaymahantesh K, Mansuri R, Sahoo BR, et al. Computational prediction and analysis of potential antigenic CTL epitopes in Zika virus: a first step towards vaccine development. Infect Genet Evol. 2016;45:187–197. doi: 10.1016/j.meegid.2016.08.037. [DOI] [PubMed] [Google Scholar]
- 35.Singh SP, Khan F, Mishra BN. Computational characterization of Plasmodium falciparum proteomic data for screening of potential vaccine candidates. Hum Immunol. 2010;71(2):136–143. doi: 10.1016/j.humimm.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 36.Fleri W, Paul S, Dhanda SK, Mahajan S, Xu X, Peters B, et al. The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front Immunol. 2017;8:278. doi: 10.3389/fimmu.2017.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhasin M, Raghava GP. Prediction of CTL epitopes using QM, SVM and ANN techniques. Vaccine. 2004;22(23–24):3195–3204. doi: 10.1016/j.vaccine.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 38.Bhasin M. Analysis and prediction of affinity of TAP binding peptides using cascade SVM. Protein Science. 2004;13(3):596–607. doi: 10.1110/ps.03373104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct. 2013;8:30. doi: 10.1186/1745-6150-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagpal G, Usmani SS, Dhanda SK, Kaur H, Singh S, Sharma M, et al. Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci Rep. 2017;7:42851. doi: 10.1038/srep42851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, et al. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12(2):255–278. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coppi A, Natarajan R, Pradel G, Bennett BL, James ER, Roggero MA, et al. The malaria circumsporozoite protein has two functional domains, each with distinct roles as sporozoites journey from mosquito to mammalian host. J Exp Med. 2011;208(2):341–356. doi: 10.1084/jem.20101488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Offeddu V, Rauch M, Silvie O, Matuschewski K. The Plasmodium protein P113 supports efficient sporozoite to liver stage conversion in vivo. Mol Biochem Parasitol. 2014;193(2):101–109. doi: 10.1016/j.molbiopara.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 44.Galaway F, Drought LG, Fala M, Cross N, Kemp AC, Rayner JC, et al. P113 is a merozoite surface protein that binds the N terminus of Plasmodium falciparum RH5. Nat Commun. 2017;8:14333. doi: 10.1038/ncomms14333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beeson JG, Drew DR, Boyle MJ, Feng G, Fowkes FJ, Richards JS. Merozoite surface proteins in red blood cell invasion, immunity and vaccines against malaria. FEMS Microbiol Rev. 2016;40(3):343–372. doi: 10.1093/femsre/fuw001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saxena AK, Wu Y, Garboczi DN. Plasmodium p25 and p28 surface proteins: potential transmission-blocking vaccines. Eukaryot Cell. 2007;6(8):1260–1265. doi: 10.1128/EC.00060-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Draper SJ, Sack BK, King CR, Nielsen CM, Rayner JC, Higgins MK, et al. Malaria vaccines: recent advances and new horizons. Cell Host Microbe. 2018;24(1):43–56. doi: 10.1016/j.chom.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chaudhuri R, Ahmed S, Ansari FA, Singh HV, Ramachandran S. MalVac: database of malarial vaccine candidates. Malar J. 2008;7:184. doi: 10.1186/1475-2875-7-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh SP, Mishra BN. Identification and characterization of merozoite surface protein 1 epitope. Bioinformation. 2009;4(1):1. doi: 10.6026/97320630004001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ulrich A, Partridge JR, Schwartz TU. The stoichiometry of the nucleoporin 62 subcomplex of the nuclear pore in solution. MolBiol Cell. 2014;25(9):1484–1492. doi: 10.1091/mbc.E13-12-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Terheggen U, Drew DR, Hodder AN, Cross NJ, Mugyenyi CK, Barry AE, et al. Limited antigenic diversity of Plasmodium falciparum apical membrane antigen 1 supports the development of effective multi-allele vaccines. BMC Med. 2014;12:183. doi: 10.1186/s12916-014-0183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soulama I, Bigoga JD, Ndiaye M, Bougouma EC, Quagraine J, Casimiro PN, et al. Genetic diversity of polymorphic vaccine candidate antigens (apical membrane antigen-1, merozoite surface protein-3, and erythrocyte binding antigen-175) in Plasmodium falciparum isolates from western and Central Africa. Am J Trop Med Hyg. 2011;84(2):276–284. doi: 10.4269/ajtmh.2011.10-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stone WJR, Campo JJ, Ouédraogo AL, Meerstein-Kessel L, Morlais I, Da D, et al. Unravelling the immune signature of Plasmodium falciparum transmission-reducing immunity. Nat Commun. 2018;9(1):558. doi: 10.1038/s41467-017-02646-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sherrard-Smith E, Sala KA, Betancourt M, Upton LM, Angrisano F, Morin MJ, et al. Synergy in anti-malarial pre-erythrocytic and transmission-blocking antibodies is achieved by reducing parasite density. Elife. 2018;7:e35213. doi: 10.7554/eLife.35213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patel P, Bharti PK, Bansal D, Raman RK, Mohapatra PK, Sehgal R, et al. Genetic diversity and antibody responses against Plasmodium falciparum vaccine candidate genes from Chhattisgarh, Central India: implication for vaccine development. PLoS One. 2017;12(8):e0182674. doi: 10.1371/journal.pone.0182674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hill AV, Allsopp CE, Kwiatkowski D, Anstey NM, Twumasi P, Rowe PA, et al. Common west African HLA antigens are associated with protection from severe malaria. Nature. 1991;352(6336):595–600. doi: 10.1038/352595a0. [DOI] [PubMed] [Google Scholar]
- 57.Lyke KE, Fernández-Viňa MA, Cao K, Hollenbach J, Coulibaly D, Kone AK, et al. Association of HLA alleles with Plasmodium falciparum severity in Malian children. Tissue Antigens. 2011;77(6):562–571. doi: 10.1111/j.1399-0039.2011.01661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Osafo-Addo AD, Koram KA, Oduro AR, Wilson M, Hodgson A, Rogers WO. HLA-DRB1*04 allele is associated with severe malaria in northern Ghana. Am J Trop Med Hyg. 2008;78(2):251–255. doi: 10.4269/ajtmh.2008.78.251. [DOI] [PubMed] [Google Scholar]
- 59.Cao K, Moormann AM, Lyke KE, Masaberg C, Sumba OP, Doumbo OK, et al. Differentiation between African populations is evidenced by the diversity of alleles and haplotypes of HLA class I loci. Tissue Antigens. 2004;63(4):293–325. doi: 10.1111/j.0001-2815.2004.00192.x. [DOI] [PubMed] [Google Scholar]
- 60.Bertholet S, Goldszmid R, Morrot A, Debrabant A, Afrin F, Collazo-Custodio C, et al. Leishmania antigens are presented to CD8+T cells by a transporter associated with antigen processing-independent pathway in vitro and in vivo. J Immunol. 2006;177(6):3525–3533. doi: 10.4049/jimmunol.177.6.3525. [DOI] [PubMed] [Google Scholar]
- 61.Oyarzun P, Ellis JJ, Gonzalez-Galarza FF, Jones AR, Middleton D, Boden M, et al. A bioinformatics tool for epitope-based vaccine design that accounts for human ethnic diversity: application to emerging infectious diseases. Vaccine. 2015;33(10):1267–1273. doi: 10.1016/j.vaccine.2015.01.040. [DOI] [PubMed] [Google Scholar]
- 62.Oyarzún P, Kobe B. Recombinant and epitope-based vaccines on the road to the market and implications for vaccine design and production. Hum Vaccin Immunother. 2016;12(3):763–767. doi: 10.1080/21645515.2015.1094595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Damfo SA, Reche P, Gatherer D, Flower DR. In silico design of knowledge-based Plasmodium falciparum epitope ensemble vaccines. J Mol Graph Model. 2017;78:195–205. doi: 10.1016/j.jmgm.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 64.Kaba SA, McCoy ME, Doll TA, Brando C, Guo Q, Dasgupta D, et al. Protective antibody and CD8+ T-cell responses to the Plasmodium falciparum circumsporozoite protein induced by a nanoparticle vaccine. PLoS One. 2012;7(10):e48304. doi: 10.1371/journal.pone.0048304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Burkhard P, Lanar DE. Malaria vaccine based on self-assembling protein nanoparticles. Expert Rev Vaccines. 2015;14(12):1525–1527. doi: 10.1586/14760584.2015.1096781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Karch CP, TAPF D, Paulillo SM, Nebie I, Lanar DE, Corradin G, Burkhard P. The use of a P. falciparum specific coiled-coil domain to construct a self-assembling protein nanoparticle vaccine to prevent malaria. J Nanobiotechnology. 2017;15(1):62. doi: 10.1186/s12951-017-0295-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Singh AK, Pal P, Gupta V, Yadav TP, Gupta V, Singh SP. Green synthesis, characterization and antimicrobial activity of zinc oxide quantum dots using Eclipta alba. Mater Chem Phys. 2018;203:40–48. doi: 10.1016/j.matchemphys.2017.09.049. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimentally known epitopes of P. falciparum 3D7. (XLSX 11 kb)
(a) The PPC of predicted MHC class I binding T cell epitopes from 5 genome derived predicted antigenic adhesions (GDPAA) with VaxiJen score ≥ 0.5, (b) The PPC of predicted MHC class II binding T cell epitopes from 5 genome derived predicted antigenic adhesions (GDPAA) with VaxiJen score ≥ 0.5. (XLSX 88 kb)
(a) The PPC of predicted T cell epitopes (MHC class I, MHC class II and combined) from 5 randomly selected known malarial adhesions (RSKMA) with VaxiJen score ≥ 0.5, (b) The PPC of predicted T cell epitopes (MHC class I, MHC class II and combined) from 5 RSKMA, (c) Predicted set of T cell epitopes of 5 RSKMA, (d) The PPC of minimal T cell epitopes (MHC class I, MHC class II and combined) from 5 RKSMA, (e) The predicted ensemble epitope (MHC class I and MHC class II) from 5 RKSMA. (XLSX 145 kb)
The PPC of predicted T cell epitopes for 5 genome derived predicted antigenic adhesions (GDPAA). (XLSX 9 kb)
The PPC of individual epitope found in minimal epitope set of 5 genome derived predicted antigenic adhesions (GDPAA). (XLSX 13 kb)
The PPC of minimal combined epitope set for 5 GDPAA and jointly screened epitope ensemble. (XLSX 10 kb)
The PPC value of individual epitope of epitope ensemble designed from GDPAA and experimentally known epitope (EKE) of P. falciparum. (XLSX 11 kb)
Data Availability Statement
The data analysed but not presented in the main text are available as supplementary materials provided as Additional files 1, 2, 3, 4, 5, 6 and 7.