

The study describes a synthetic hemostatic agent that can be intravenously injected to reduce internal hemorrhage.
Abstract
Uncontrolled noncompressible hemorrhage is a major cause of mortality following traumatic injuries in civilian and military populations. An injectable hemostat for point-of-care treatment of noncompressible hemorrhage represents an urgent medical need. Here, we describe an injectable hemostatic agent via polymer peptide interfusion (HAPPI), a hyaluronic acid conjugate with a collagen-binding peptide and a von Willebrand factor–binding peptide. HAPPI exhibited selective binding to activated platelets and promoted their accumulation at the wound site in vitro. In vivo studies in mouse tail vein laceration model demonstrated a reduction of >97% in both bleeding time and blood loss. A 284% improvement in the survival time was observed in the rat inferior vena cava traumatic model. Lyophilized HAPPI could be stably stored at room temperature for several months and reconstituted during therapeutic intervention. HAPPI provides a potentially clinically translatable intravenous hemostat.
INTRODUCTION
For decades, uncontrollable hemorrhage has been the leading cause of death in populations aged 1 to 46 years, and 30 to 40% of these deaths are associated with major blood loss in both civilians and military populations (1–5). Because most deaths occur during the initial hour after a traumatic injury, hemorrhage control is extremely time sensitive, as illustrated by the notion of the “golden hour” for trauma care; the hemostatic intervention within the first hour will determine the survival in trauma patients (6, 7). However, currently available on-site interventions such as tourniquets and topical hemostatic dressings can only cater to externally accessible and potentially compressible wounds. In recent years, some advancements have been made in the development of intratissue or intracavitary injectable, self-expandable hemostatic polymer foam technologies, e.g., XSTAT and ResQFoam, and embolic catheter technologies, e.g., resuscitative endovascular balloon occlusion of the aorta, that may enable higher efficacy of torso hemorrhage control (8). However, for internal, diffuse, and noncompressible bleeding, besides few hemostatic drugs (9), transfusion of blood products (i.e., packed red blood cells and fresh frozen plasma and platelets) and coagulation factor concentrates remains the primary clinical treatment to stanch bleeding (6, 10). Although highly effective, there are several logistical challenges associated with point-of-care administration of biological products. Taking the platelet as an example, its application is limited by (i) short shelf life (5 to 7 days) with stringent storage requirements, (ii) extensive prescreening to avoid immunogenicity issues, and (iii) high risk of biological contamination (11–13). Even in sterile clinical settings, shortage of platelet supply from donors adds to the challenges of maintaining platelet inventory for the treatment of bleeding complications in patients with surgery-associated hemorrhage, oncologic clotting disorders, and chemo/radiotherapy-induced myelosuppression, besides trauma (12, 14). Thus, there is an unmet need to develop intravenous hemostats that can overcome the aforementioned challenges and promote rapid clot formation at inaccessible injury sites.
Several attempts have been made to develop platelet substitutes that either initiate or supplement the natural hemostasis process (14). These include the use of Arg-Gly-Asp (RGD) peptide for targeting activated platelets or the use of collagen-binding peptide (CBP) and von Willebrand factor (vWF)–binding peptide (VBP) for targeting vascular injury sites. These peptides have been typically delivered using micro- or nanoparticles, including albumin microspheres (15), liposomes (16), poly(l-lactic-co-glycolic acid) nanoparticles (17), layer-by-layer–assembled discoidal nanoparticles (18), and poly(N-isopropylacrylamide-co-acrylic acid) microgels (19). Polymer constructs offer an appealing platform for therapeutics owing to their high aqueous solubility, flexibility, and multivalent affinity toward their targeted moieties at a molecular level (20). Using hyaluronic acid (HA) as a model polymeric backbone, we report that HA conjugates with VBP (TRYLRIHPQSQVHQI) and CBP ([GPO]7H) (16, 18), termed as hemostatic agents via polymer peptide interfusion (HAPPI), which are highly effective in promoting hemostasis without systemic toxicity. CBP mediates binding of HAPPI to collagen exposed at the site of vascular injury, whereas VBP, a short sequence from factor VIII protein that is responsible for its binding to vWF, mediates binding of HAPPI to vWF immobilized on activated platelets and damaged endothelium at the injury site.
RESULTS
Design and synthesis of HAPPI
As one of the key components of extracellular matrix and endothelial glycocalyx (21), HA provides an excellent framework for designing macromolecular therapeutics. Along with fibrin, it is also the primary macromolecular regulator during the wound healing process following vascular injury. Shortly after tissue injury with vascular denudation, there is an increase in the HA content of the wound, following which, it binds to the fibrin clot and reorganizes the clot into an HA-fibrin matrix (22). From the synthesis perspective, HA provides an excellent building block because of the presence of multiple functional groups on its backbone (23). On the basis of this rationale, CBP and VBP were conjugated to HA using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)/sulfo-NHS (N-hydroxysuccinimide)–mediated coupling reactions (Fig. 1A). The carboxylic acid group of HA was first activated by EDC and sulfo-NHS and further reacted with primary amines of the peptides, yielding the HA-CBP-VBP conjugates, i.e., HAPPI. The orientation of the peptides is thereby adopted as C terminus of peptide is directed outward and N terminus toward the HA backbone. A fluorescent dye [Alexa Fluor 647 (AF 647)] was also conjugated to the HA backbone to facilitate in vivo biodistribution and pharmacokinetic studies. Conjugation was confirmed by gel permeation chromatography (GPC) (fig. S1A).
Fig. 1. Synthesis and characterization of AF 647–labeled HA-CBP-VBP conjugates (HAPPI).

(A) Reaction scheme for conjugation of fluorescent dye and peptides on HA backbone using EDC/sulfo-NHS chemistry. (B) 1H nuclear magnetic resonance (NMR) spectrum shows the presence of characteristic peaks b, c, d, and e from the peptides, indicating their successful conjugation to HA. (C) GPC profile of HAPPI shows the appearance of ultraviolet (UV) absorbance at the elution time of conjugates [refractive index (RI) signals], in contrast to the profile of fluorescent HA alone, further validating the chemical conjugation. (D) Image of freeze-dried HAPPI as the bluish material in the vial. Topographic air tapping mode atomic force micrographs for HA (E), HA-VBP (F), HA-CBP (G), and HAPPI (H) (scale bars, 500 nm). Photo credit: (D) Y. Gao, Harvard University.
Chemical and structural characterization of HAPPI
HAPPI was characterized by nuclear magnetic resonance (NMR) spectroscopy, GPC, and atomic force microscopy (AFM) to confirm the successful conjugation and to probe their morphology. Characteristic 1H NMR peaks from VBP, CBP, and HA were detected in the final, purified products, demonstrating the successful conjugation (Fig. 1B). Specifically, the peaks at 6.8 and 7.1 parts per million (ppm) correspond to aromatic rings in tyrosine from VBP. The peaks at 8.6 and 7.3 ppm belong to histidine from VBP and CBP, respectively. In addition, the peak at 1.95 to 2.05 ppm corresponds to methyl groups in the acetamido moiety of HA. For a quantitative analysis, the methyl resonance of acetamido moiety of HA at (δ 1.95 to 2.05 ppm) was used as an internal standard (a in Fig. 1B). The degree of VBP modification was estimated as ca. 7.8 mole percent (mol %) by comparing the peak areas of tyrosine and HA (d and e in Fig. 1B). Similarly, the CBP modification was found to be ~5.6 mol %. On the basis of this, the average numbers of conjugated VBP and CBP molecules per single HA chain were estimated to be 51 and 37, respectively. VBP and CBP were also conjugated to HA individually in a similar manner, yielding HA-VBP and HA-CBP conjugates with comparable degree of substitution achieved (fig. S2).
Successful conjugation to HA was further confirmed by GPC (Fig. 1C, fig. S1, B and C). Unmodified HA does not absorb at 280 nm, and therefore, no peak was observed in ultraviolet (UV) detection. On the other hand, HAPPI absorbs at the same location and exhibits the refractive index (RI) peak, indicating that the peptides were covalently attached to the HA molecules and are not just physically mixed with the polymer. A similar retention volume of AF 647–HA and AF 647–HAPPI demonstrated that HA was intact, with no detectable degradation occurring during the conjugation reaction and purification process. The increase in molecular weights after conjugation (HA-VBP, HA-CBP, and HAPPI versus HA), as determined from their nonfluorescent analogs, is further manifested by the parallel shift of molecular weight distribution peaks (fig. S1, D and E). To assess the storage stability of HAPPI, they were stored at room temperature in lyophilized solids (Fig. 1D) for 3 months and assessed by GPC. The elution profile of the conjugates was comparable to the starting materials, demonstrating the outstanding stability of these formulations.
To probe their morphology, HA and HAPPI, along with conjugates of HA with individual peptides, were deposited on a highly ordered pyrolytic graphite (HOPG) substrate from dilute aqueous solutions and imaged in air by tapping mode AFM (Fig. 1, E to H). HA molecules exhibited both condensed globular forms and extended coil structures, which are known to be due to the intramolecular and intermolecular aggregation (24). While both globular and coiled forms were also observed for HA-VBP, HA-CBP, and HAPPI, HA-VBP tends to form extended conformation, whereas HA-CBP forms more isolated globular forms. This conformational difference could be attributed to the difference in hydrophobicity and isoelectric points of VBP and CBP, which can regulate their molecular conformations both in the solution and on the surface (24). While it would be ideal to characterize the morphology of HAPPI in physiologically relevant condition, such as in saline solution, the hydrophilic nature of HA backbone makes it practically challenging, as HA has weaker affinity toward the substrate than to aqueous solution (24). After modifying the mica substrate with positively charged polylysine, we obtained the morphology of HAPPI in saline (fig. S3), which is similar with those observed in air.
In vivo hemostatic efficacy in mouse tail bleeding model
HAPPI was next investigated in vivo for the ability to halt bleeding in mouse tail vein incision (laceration) model, a well-established model for determination of hemostatic capability (Fig. 2A) (25, 26). HA alone (4.6 mg/kg; without peptides) and free peptides alone did not exhibit any decrease in bleeding time compared to the untreated group (Fig. 2B). This is reasonable as, to the best of our knowledge, HA molecules do not have any preferable binding capacity toward platelets, vWF, or collagen. The lack of hemostatic efficacy for free peptides could presumably be attributed to the absence of multivalent binding ability, resulting in the incapability of accelerating the clot formation process. However, HAPPI, administered with equivalent HA dose of 4.6 mg/kg, exhibited an outstanding hemostatic performance (Fig. 2, B and C). In particular, HA-VBP and HAPPI significantly lowered the bleeding time and bleeding volume compared to the untreated group. About 99% reduction in bleeding time and 97% reduction in blood loss were achieved with HAPPI. While the saline group exhibited an unexpected reduction of bleeding time and blood loss, compared to the HA group, HAPPI still led to a remarkable reduction in bleeding time and volume. Among the two peptides, VBP clearly dominated the hemostatic effect since HA-CBP did not exhibit a significant reduction of bleeding time or volume. On the other hand, equivalent dosage of HA-VBP exhibited a strong reduction of both. To eliminate the contributions arising from different degrees of peptide conjugation in HA-VBP, HA-CBP, and HAPPI, the dosage of the formulation was further calibrated to the total amount of peptides (0.68 μmol/kg) by tuning the conjugate concentration. A similar trend in the reduction of bleeding time and blood loss was observed (Fig. 2, D and E), further substantiating that the hemostatic effect arises primarily from the two peptides, in particular, VBP. When mice were administered with a mixture of HA-VBP and HA-CBP (HA-VBP/HA-CBP; Fig. 2, B and C), their bleeding time and blood loss were comparable to those treated with HA-VBP and slightly higher than those from the HAPPI group, indicating a synergistic effect of conjugating two types of peptides onto a single HA molecule.
Fig. 2. Hemostatic action of HA-peptide conjugates in mouse tail laceration model.
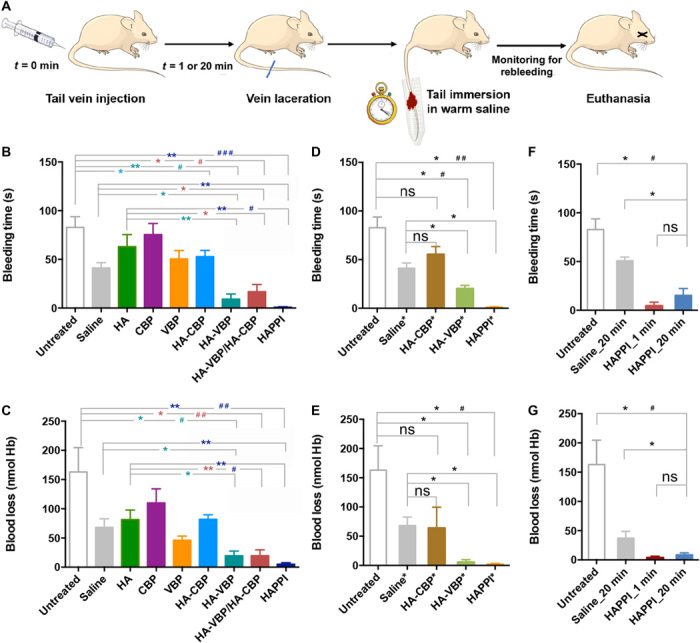
(A) Schematic representation of the hemostatic efficacy studies in BALB/c mice. Mice were intravenously administered with saline, HA, or HA-peptide conjugates, and a tail vein laceration was induced following 1- or 20-min circulation time. Bleeding times (B, D, and F) and total blood loss (C, E, and G) were quantified. (B and C) Hemostatic efficacy of HA-CBP, HA-VBP, HA-VBP/HA-CBP, and HAPPI with untreated, saline, native HA, and free peptides (CBP and VBP) as controls. In HA-CBP, HA-VBP, and HAPPI groups, mice were administered with 4.6 mg/kg of equivalent HA dose. HA-VBP/HA-CBP was formulated by mixing the desired amounts of HA-VBP and HA-CBP conjugates to obtain final individual concentrations equal to the HA-VBP group and HA-CBP group, respectively (n = 5 mice). (D and E) HA-CBP*, HA-VBP*, and HAPPI* were dosed with the same amount of peptides as detailed in Materials and Methods (n ≥ 3 mice). (F and G) Efficacy of HAPPI with 20 min circulation time was compared with untreated mice and mice treated with saline of 20 min circulation and HAPPI of 1 min circulation time (n ≥ 4 mice). All data are means ± SEM; statistics by two-tailed, nonparametric Mann-Whitney test (*P < 0.05 and **P < 0.01) and Kruskal-Wallis test followed by Dunn’s multiple comparisons test (#P < 0.05, ##P < 0.01, and ###P < 0.01). ns, not significant; Hb, hemoglobin.
To assess the effect of delay between the injection time and injury, a three-arm experiment was designed, where HAPPI and saline were injected 20 min before the tail vein laceration and compared to HAPPI with 1-min circulation time. The hemostatic efficacy was observed in both time groups compared to the untreated and saline-treated groups (Fig. 2, F and G).
Biodistribution, pharmacokinetics, and systemic toxicity of HAPPI
The pharmacokinetics and biodistribution of HAPPI were determined in injured mice. The circulation half-life of HAPPI was found to be almost identical to that of native HA at ~1 hour (Fig. 3A). Following the injection, HAPPI was primarily concentrated in liver and spleen by 6 hours (Fig. 3B), thus reducing the risk of prolonged systemic exposure. Circulation and biodistribution of the conjugates are in agreement with the literature reports on clearance of intravenously injected HA (27, 28). The liver is the major site of elimination by virtue of its greater mass, where specific receptor-mediated endocytosis of HA brings about its degradation (29). Little accumulation was found in the brain, kidney, and heart. While some accumulation was observed in the lungs within 30 min of injection, it was cleared relatively quickly so that no detectable signal was observed in the lungs after 1 hour. Some accumulation was also detected at the site of the tail injury. To assess the role of CBP and VBP in the observed biodistribution of the conjugates, we measured biodistribution of HA alone (fig. S4). Overall, the differences between the two were modest. Accumulation in the liver and spleen was comparable in both cases. Accumulation of HAPPI at the injury site was not statistically different from that of the native HA.
Fig. 3. Pharmacokinetic, biodistribution, and histology analyses of HAPPI in BALB/c mice.
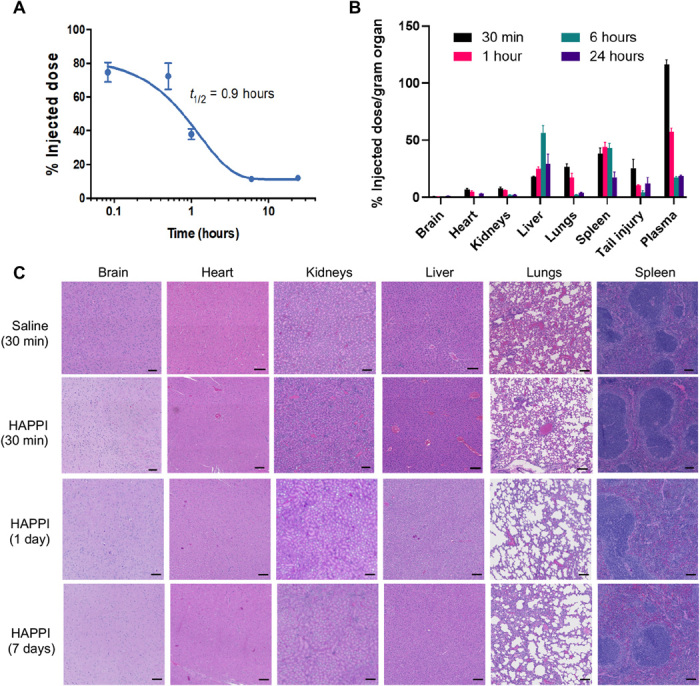
(A) Pharmacokinetic studies for HAPPI indicating plasma circulation half-life (t1/2) of ~1 hour. Data were fitted using a one-phase exponential decay model with half-life values that are extracted as shown. (B) Percent injected dose normalized per organ mass at 30-min, 1-hour, 6-hour, and 24-hour time points after injection (n = 5 per group). (C) Representative micrographs of H&E staining of six vital organs, after 30 min, 1 day, and 7 days of treatment administration (scale bars, 100 μm).
Histopathological analysis of hematoxylin and eosin (H&E)–stained sections of various organs harvested after 30 min, 1 day, and 7 days of treatment did not show any inflammation or toxicity in either treatment group (Fig. 3C and fig. S4B). No microthrombi were found in any of the vital organs. A slight increase in the blood content in the liver and spleen was noted for both HA- and HAPPI-treated mice compared to saline. We further confirmed the safety of HAPPI through the hematological and biochemistry analyses. Blood collected after 1 or 7 days of treatment showed that the composition of the blood was unaffected by the treatment, as none of the measured parameters showed statistically significant differences between saline and HAPPI groups (fig. S5). Similarly, no significant difference was observed for liver and kidney parameter levels between HAPPI and saline. In particular, the concentrations of enzymes alanine aminotransferase and aspartate aminotransferase and blood urea nitrogen in HAPPI-treated groups were found to be in the normal range (fig. S5) (30–32), indicating the lack of detectable systemic toxicity. HAPPI, at the dosage equivalent to in vivo efficacy studies, did not affect systemic coagulopathy as verified through ex vivo testing of mouse whole blood using thromboelastography (TEG). No abnormalities were observed in clot formation kinetics or clot strength in the presence of HA or HA-peptide conjugates (table S1).
Mechanism of action
To understand the mechanism of improved hemostatic efficacy of HAPPI, the interaction between HAPPI and murine platelets was first studied by fluorescence-activated cell sorting (FACS). CD41 and CD62P receptors were used as pan markers and activation markers for the platelets, respectively (33). Neither HA nor HAPPI induced significant activation of platelets, as judged by up-regulation of CD62P (Fig. 4A). Activation by adenosine diphosphate (ADP) was used as a positive control. The lack of platelet activation by HAPPI has significant positive implications for systemic safety by avoiding inadvertent emboli formation, which can be potentially fatal in the absence of an injury site. HAPPI, however, exhibited increased binding to activated platelets, as compared to respective controls (Fig. 4, B and C). HA by itself exhibited no binding to activated or nonactivated platelets. However, HAPPI exhibited 5.4-fold higher binding to activated platelets compared to nonactivated platelets. This could possibly be attributed to the binding of vWF on ADP-activated platelet surface (34).
Fig. 4. Interaction of HAPPI with platelets and their preferable binding ability.

(A) FACS analysis of HAPPI-mediated platelet activation as measured by coexpression of CD62P/CD41 after 10 min of incubation. Data are presented as percentage of platelets positive for both epitopes. (B) FACS analysis of attachment of AF 647–HA and HAPPI to platelets in their nonactivated and activated states [n = 3, one-way analysis of variance (ANOVA) test and Tukey’s multiple comparisons test, ****P < 0.0001]. (C) Representative dot plots for expression of AF 647–HA and HAPPI to indicate binding of platelets to HA and HAPPI following 10 min of incubation. (D) Human platelet aggregation assay of HAPPI, HA-VBP, and HA-CBP with saline as control. (E) Schematic of experimental setup for dynamic binding studies using PPFC to validate preferable binding ability of HAPPI to wound-specific proteins, collagen, and vWF, under flow. (F) Representative fluorescence images (×20 magnification) of (F1) DiOC6-stained platelets (green), (F2) AF 647–HAPPI (red), and (F3) overlay of F1 and F2 in the same field of view, demonstrating colocalization of platelets with HAPPI at the collagen + vWF surface (scale bars, 100 μm). ns, not significant; ADP, adenosine diphosphate; FACS, fluorescence-activated cell sorting.
We next tested the potential activity of HAPPI in human platelet aggregometry assay. Light transmission aggregometry has shown that HAPPI, HA-VBP, and HA-CBP did not cause platelet aggregation, but the addition of HAPPI and HA-VBP enhanced the ADP-induced aggregation as shown by the increase in light transmission compared to HA-CBP and saline solvent controls (Fig. 4D). HAPPI itself did not cause aggregation of inactive human platelets, which is consistent with the findings in the above mouse platelet activation study. Still, the attachment of HAPPI to activated platelets, presumably through the mediation of platelet granule–derived vWF (34), may be responsible for platelet aggregation. Note that the ~10% increase in light transmission following the addition of conjugates or saline solution is most likely caused by the ~16% dilution of platelet-rich plasma (PRP) with hemostatic agents and/or solvent (saline) effect. We also confirmed that HAPPI did not cause an undesired aggregation of solubilized vWF, analogous to the circulating vWF in plasma. vWF protein size in saline, as determined by dynamic light scattering (DLS), was unaffected by the addition of HAPPI for at least 20 hours (fig. S6, A to C). No precipitation was observed when HAPPI was mixed with plasma for at least 7 days (fig. S6D).
The preferable binding of HAPPI to vWF and collagen was further validated using parallel-plate flow chamber (PPFC) with casein as a negative control (Fig. 4E). At a physiologically relevant shear stress of 25 dynes/cm2, HAPPI, by virtue of VBP and CBP, showed relatively higher binding to collagen and vWF surface as compared to HA alone (fig. S7). When HAPPI was perfused through the PPFC along with fluorescently labeled resting platelets, there was distinct binding of both HAPPI and platelets on the collagen and vWF surface (Fig. 4F). Adhesion of HAPPI and platelets was minimal on the control casein surface. The selective binding of HAPPI, in the presence of platelets, toward collagen and vWF proteins over control protein is of high physiological relevance, as the adhesion of the platelets toward collagen and collagen-bound vWF at injury site and their subsequent activation are the initial yet critical steps to halt the bleeding. Adhesion of HAPPI to this surface, either through directly binding to collagen and vWF or through the mediation of activated platelets (fig. S7, C and D), as verified above in the FACS study, could indicate their active involvement in vivo during the clot formation process at the site of vascular injury.
To assess cooperativity between HAPPI and natural platelets, hemostatic efficacy of HAPPI was assessed in thrombocytopenic mice. Following platelet-depleting antibody administration, the natural platelet count in these mice was ~95% lower than that of normal mice (as per the manufacturer’s specifications). When these mice were injected with HAPPI, there was a modest decrease in the bleeding time compared to the untreated group (Fig. 5A). A closer inspection revealed that the bleeding was nearly halted for ~4 min upon injection of conjugates (Fig. 5B). However, the bleeding resumed thereafter. These results indicate that, while HAPPI themselves do not form a firm physical plug to stop bleeding, they are highly effective in cooperation with natural platelets.
Fig. 5. Hemostatic action of HAPPI on the tail laceration models in thrombocytopenic BALB/c mice (A,B) and the IVC traumatic models in Sprague-Dawley rat (C,D).

Blood loss in injured, thrombocytopenic mice treated with HAPPI (equivalent HA dose of 4.6 mg/kg): total blood loss over 20-min observation time (A) and graph representing significant difference in the bleeding kinetics (B). Survival time (C) and blood loss (D) in IVC injured rats following treatment with HAPPI (12 mg/kg) versus equal volume of saline (***P < 0.001 and ****P < 0.0001).
Hemostatic efficacy in internal, noncompressible traumatic model
On the basis of the understandings of the hemostatic efficacy of HAPPI, we further evaluated its capability to improve the hemostasis and survival rate in a lethal traumatic bleeding model in rat, induced by the puncture of inferior vena cava (IVC) (fig. S8). In this model, the efficacy of HAPPI was evaluated as a rescue treatment strategy, where HAPPI infusion was performed at the onset of IVC hemorrhage. Rats treated with HAPPI experienced significantly longer survival time compared to those treated with saline (P = 0.0003). The averaged survival time increased from 25 (±4.3) to 71 (±8.1) min (Fig. 5C). This outcome is of clinical significance, as therapies that can extend the window of survival beyond the golden hour after such noncompressible traumatic hemorrhage are critically needed to prevent prehospital deaths (7). No statistical significance was observed in the blood loss between HAPPI and saline (Fig. 5D). Nevertheless, the IVC harvested after euthanasia showed accumulation of HAPPI at the injury site (fig. S8B).
DISCUSSION
The studies presented here establish a unique polymer-based, hemostatic construct, HAPPI, that can be injected systemically to accumulate at the injury sites to enable hemostatic amplification and halt bleeding. This design is of clinical significance, especially for internal, noncompressible bleeding. Hemorrhage from traumatic injury sites is responsible for 90% of survivable prehospital mortalities in military battlefield and 64% of civilian trauma-related deaths (4, 5). Immediate intervention at the point of injury to stop the bleeding or extend the window of survival is imperative to preventing death. Thus, therapies that can treat such hemorrhage in the prehospital environment are urgently needed. In contrast to the prevalent designs of topical hemostats for accessible bleeding, agents that can be systemically injected to halt the bleeding endovascularly (Fig. 6) are tremendously beneficial for emergency procedures.
Fig. 6. Graphical schematic of the proposed hemostatic mechanism of HAPPI following vascular injury.
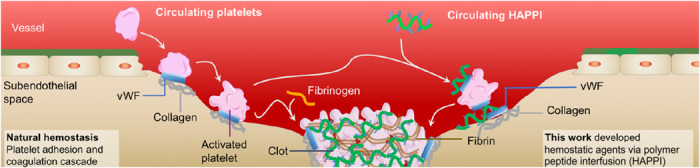
HAPPI binds to vWF and collagen exposed at the site of vascular injury and vWF immobilized on the activated platelet surface, facilitating the recruitment and aggregation of platelets at the injury site to further augment the clotting process.
Successful intravenous hemostats must simultaneously target the injury sites and exhibit limited off-target procoagulant effects. Excellent blood compatibility and stability are also essential requirements for a synthetic hemostatic agent. Our results reported here confirmed that this newly designed HAPPI meets all these requirements, highlighting their translational feasibility.
To achieve the injury-binding effect, we chose vWF and collagen as our target molecules, which are indeed the trigger molecules for platelet-mediated natural hemostasis. Physiologically, following vascular injury, subendothelial collagen is exposed to the blood stream, and vWF, the primary adhesion molecule for platelets, binds to the exposed collagen and changes its conformation under shear forces of the flow, thereby facilitating platelet adhesion to both vWF and collagen (Fig. 6, left) (35). Previous research efforts have identified targeting peptides toward collagen and vWF and achieved selective accumulation of particulate carriers at the wound site (16, 18). Through dynamic binding assay in PPFC, we verified that HAPPI, together with platelets, can bind to these injury-specific proteins under flow (Fig. 4, E and F). From the FACS study, we found that HAPPI can also selectively bind to activated platelets at about 5.4-fold more than the binding to nonactivated platelets (Fig. 4B), which could potentially originate from the binding of HAPPI to the vWF bound to GPIIb/IIIa receptors on activated platelet surface (34). This additional selectivity toward activated platelets can further promote the recruitment and adhesion of activated platelets to the injury sites, augmenting the clot formation process (Fig. 6, right).
Studies were also designed to verify off-target coagulation effects of HAPPI. HAPPI is stable in plasma for several days without precipitating out, and HAPPI did not cause unnecessary aggregation of vWF solubilized in saline. FACS and platelet aggregometry studies demonstrated that HAPPI themselves neither activate quiescent platelets nor cause their aggregation, thus building an inherent margin of safety against nonspecific aggregation of circulating resting platelets. TEG studies further confirm that HAPPI neither initiates the coagulation reactions nor changes the viscoelasticity of mouse whole blood and the subsequent clot. In vivo experiments show similar pharmacokinetics and biodistribution profiles of HAPPI and native HA, unchanged blood compositions and serum biochemistry, and lack of inflammation and toxicity in vital organs, indicating that HAPPI carrying two peptides do not cause systematic toxicity or thrombi formation. This is an important consideration for intravenous hemostats to maintain the delicate balance between physiological hemostasis and pathological thrombosis since unregulated platelet activation or coagulation may pose a severe risk of thromboembolism.
Use of the biomacromolecule HA to carry peptides ensures the blood compatibility of HAPPI. HA is one of the key components of endothelial glycocalyx regulating the vascular integrity (21). HA is also a key molecule in the regulation of many cellular and biological processes, and in particular, it is the primary macromolecular regulator for wound healing following vascular injury (22). Given its specific biological and bioactive properties, HA-based therapeutic formulations have been approved and used clinically for the treatment of arthritis and skin diseases (23). The degree of modification of HA is within 15%, less than the thresholds for affecting biological functions such as the interaction with CD44 receptors (36). Given that the biological properties of HA are highly sensitive to its molecular weight (37), we investigated the molecular size of HAPPI and found that HA backbone is still intact after the conjugation and purification process (Fig. 1C). Histological, hematological, and serum chemistry analyses further confirm the safety of HAPPI for short (1 day) and long term (7 days).
Collectively, HAPPI shows excellent promise as fast-acting intravenous hemostats. We validated its hemostatic efficacy in two animal models: the mouse tail vein laceration model and rat IVC traumatic injury (Figs. 2 and 5, C and D). In the mouse tail vein laceration model, the substantial reduction (>97%) in blood loss by injecting HAPPI, compared with the untreated, or HA and free peptids–treated groups, highlights the superior hemostatic efficacy of multivalent presentation of peptides on HA polymer. The prominent effect of VBP-containing conjugates (e.g., HAPPI and HA-VBP) can be attributed to the presence of vWF on the activated platelets (34). The temporary hemostasis achieved in thrombocytopenic mouse model with low level of platelets (Fig. 5, A and B) is also consistent with this hypothesis. These results indicate that HAPPI by itself does not form a physical plug at the injury sites but rather selectively accelerates the hemostatic ability of natural platelets to induce hemostasis. In the lethal traumatic hemorrhage model in rat induced by IVC rupture, HAPPI was administered after injury as such to mimic its actual application as a rescue therapy. HAPPI can significantly extend the survival time up to 71 min. This is remarkable, given that for such internal, noncompressible traumatic hemorrhage, it is critical to extend the window of survival beyond the golden hour to facilitate the trauma patients to reach definitive surgical care and prevent the prehospital death (7, 38). Note that, in the mouse model, as much as 50% of injected dose remains circulating in plasma for about an hour. Long circulation time combined with rapid hemostatic activity is advantageous for resolving bleeding issues during transport of trauma patients to the clinics. The facile synthesis and long-term ambient storage stability of these polymeric agents add advantages over blood or other biological-based hemostats for point-of-care administration.
Although HAPPI showed hemostatic efficacy in two distinct bleeding models, several aspects of the study need further exploration. The binding of HAPPI to activated platelets was presumably due to the affinity of VBP toward vWF, which is bound to activate platelet surface after secretion from α-granules (34), but the exact molecular binding mechanism remains unclear. In the mouse tail vein laceration bleeding model, reduction in bleeding time and blood loss were observed in saline-treated groups, comparing to untreated group, which differs from previous observations in mouse tail transection model (16). The reason for this phenomenon needs further investigation. In the rat IVC trauma model, HAPPI extended the survival time but did not reduce the blood loss, as expected, which could be attributed to the rapid bleeding and hemorrhagic shock right after the injury, and the challenges of detecting the subtle change in the kinetics of clot formation in our experimental settings. A detailed real-time hemodynamic monitoring should be included in the future to monitor the subtle hemodynamic change during the hemostasis process. Moreover, the hemostatic efficacy of HAPPI should be verified in a traumatic bleeding model in large animals, such as swine, with complete vital signs/urine output and arterial blood gas monitoring. While HAPPI has been demonstrated as a highly effective hemostatic agent, the dependence on natural platelets potentially limits its applications for patients with thrombocytopenia or platelet dysfunction or patients under antiplatelet medication. Nevertheless, HAPPI can be further applied to halt bleeding episodes in combination with other therapies by virtue of its synthetic ease of attaching multiple moieties for added functionality. As a versatile drug delivery platform, polymer conjugates provide numerous degrees of freedom to include additional drug molecules for diagnostic or therapeutic action in other vascular disorders such as stroke and atherosclerosis.
MATERIALS AND METHODS
Synthesis of HA-peptide conjugates
HA (250 kDa) purchased from Creative PEGWorks (Chapel Hill, NC) was fluorescently labeled with AF 647 Hydrazide (Thermo Fisher Scientific, Waltham, MA) for all studies to facilitate detection in subsequent assays. Cross-linking reagents EDC hydrochloride (EDC·HCl) and N-hydroxysulfosuccinimide sodium salt (sulfo-NHS), as well as dimethyl sulfoxide (DMSO) solvent, were purchased from Sigma-Aldrich (St. Louis, MO). Peptides including CBP ([GPO]7H) and VBP (TRYLRIHPQSQVHQI) were commercially obtained from GenScript USA Inc. (Piscataway, NJ). All materials were used from their indicated commercial sources without further purification. Unless otherwise stated, deionized (DI) water from a Milli-Q Plus system (Millipore, Schwalbach, Germany) was used for all experiments.
For fluorescent labeling of HA, the polymer was dissolved in DI water at 10 mg/ml under constant stirring for 1 hour at room temperature. EDC·HCl (2× molar excess of fluorescent dye) was dissolved in 1:1 DMSO:water at 50 mg/ml and added to the HA solution immediately. AF 647 (0.3 mol %) relative to HA disaccharide units was dissolved in DI water (5 mg/ml) and added to the reaction mixture. After an overnight reaction as shown in Fig. 1A, the fluorescent HA was purified by precipitation in ethanol. On redissolution of the precipitated polymer in DI water, the solution was transferred to Spectrum Spectra/Por 3 regenerated cellulose (RC) dialysis membrane tubing [molecular weight cutoff (MWCO), 10 kDa; Fisher Scientific, Pittsburgh, PA] and dialyzed against a large excess amount of DI water for 24 hours (three times water changes), followed by lyophilization.
Resultant AF 647–HA was used for further conjugation with CBP and VBP using EDC/sulfo-NHS chemistry. The polymer was dissolved in 1:1 mixture of DI water and DMSO at 7.5 mg/ml under constant stirring for 1 hour at room temperature. Sulfo-NHS (2× molar excess of peptides) was dissolved in DI water at 150 mg/ml, and EDC·HCl (2× molar excess of peptides) was dissolved in DMSO at 50 mg/ml. Both solutions were freshly prepared and added to the HA solution and stirred for 1 hour at room temperature to activate the carboxyl groups. On the basis of desired conjugates, 10 mol % of CBP (2026 Da, 50 mg/ml) and/or 10 mol % of VBP (1876.2 Da, 50 mg/ml) in DMSO solution were added to the activated polymer solution. The reaction was allowed to proceed overnight at room temperature. The products were purified by precipitation in ethanol and dialyzed (Spectrum Spectra/Por 3 RC dialysis membrane tubing; MWCO, 10 kDa; Fisher Scientific, Pittsburgh, PA) against a large excess amount of DMSO/DI water solution for 3 days (50, 30, and 10% DMSO solution, each for 1 day) and pure DI water for another 4 days (solvent renewal: twice daily). No purification procedure was performed to separate the unconjugated polymers from polymer-peptide conjugates. The fluorescent HA-peptide conjugates were then freeze-dried and stored at −20°C until further use. The same procedure was adopted to synthesize nonfluorescent HA-peptide conjugates for the molecular weight analysis by GPC.
Chemical characterization using NMR and GPC
The HA-peptide conjugates were characterized by proton NMR (1H NMR). Polymer conjugates were dissolved in deuterated water (D2O), and 1H NMR spectra were obtained on Agilent DD2 600-MHz NMR Spectrometer (Santa Clara, CA) with MNova 10.0.1 (Mestrelab Research, Spain) processing software. The chemical shifts were represented in parts per million, with reference to the signal for D2O at 4.79 ppm. The amount of conjugated peptides in the reaction products was quantified by 1H NMR through comparing the characteristic peaks from peptides and HA.
GPC analysis was carried out on a Viscotek 270max GPC system (Malvern Instruments Ltd., UK), equipped with GPCmax solvent and sample delivery module, a TDA 305 triple detector, a UV detector 2600, and OmniSEC software. A set of two GPC columns (TSKgel G5000SWXL or G4000SWXL and TSKgel G3000SWXL) were eluted with triple filtered 0.2 M NaNO3 and 0.01 M monosodium phosphate aqueous solution (pH 6.8) at a flow rate of 0.6 ml/min at 35°C. Polymer conjugates were dissolved in water or saline and filtered through a 0.22-μm filter. Polymer solutions (100 μl) were injected each time. Light scattering detector with 670-nm laser was used to confirm the conjugation of AF 647 to HA after separation. UV absorbance at 280 nm was used to confirm the conjugation of peptides to the separated polymers. Pullulan with Mw of 200 kDa and polydispersity index of 1.09 (Sigma-Aldrich, 01615) was used for triple detection calibration.
Structural characterization using AFM
Polymer conjugates were dissolved in saline at 1 mg/ml overnight and diluted with DI water to 100 μg/ml with overnight rotation and then diluted to 5 μg/ml. The dilute solutions were stirred using a tube revolver overnight at room temperature, with intermittent vortexing to ensure mixing at the molecular scale. For imaging in air, sample preparation is based on a previous procedure from Spagnoli et al. (24) with slight modification. Briefly, 10 μl of the solution was dropped on a freshly cleaved HOPG, incubated for 30 min at room temperature under controlled humidity, rinsed twice with 100 μl of DI water, and dried under nitrogen. Morphology of the polymer conjugates was scanned in air with Cypher microscope (Asylum Research, Santa Barbara, CA), operated in tapping mode. Silicon cantilevers having chromium/gold (Cr/Au) coating, with resonance frequencies between 44 and 95 kHz and spring constant in the range of 0.3 to 4.8 N/m, and 9 ± 2 mm uncoated silicon tip (AC240TSA-R3, Asylum Research, Santa Barbara, CA) were used. For imaging in liquid, freshly cleaved mica was firstly incubated with 20 μl of polylysine aqueous solution (1 mg/ml) for 30 s, rinsed with 4 ml of DI water, and dried under nitrogen stream. Immediately following this, 20 μl of conjugates/saline solution (5 μg/ml) was dropped on the surface, incubated for 2 min, rinsed with 400 μl of saline, and covered with ~100 μl of saline. Subsequently, an AC240TSA-R3 cantilever (Asylum Research) was mounted on the droplet holder, wet with ~50 μl of saline, and placed in the instrument. Depending on the frame size, the scan rate was set in the 0.7- to 2.4-Hz range. The images were processed and analyzed using Gwyddion 2.47 software.
In vivo hemostasis studies
All in vivo experiments were performed as per protocols approved by the Institutional Animal Care and Use Committee of Harvard University. BALB/c mice (9 to 10 weeks old, female) and Sprague-Dawley rats (8 to 10 weeks old, male) were purchased from Charles River Laboratories (Wilmington, MA). Animals were housed under standard 12-hour light/12-hour dark conditions with ad libitum access to both food and water until the time of the procedure.
For the mouse tail vein laceration bleeding model, the procedure shown in Fig. 2A was followed to evaluate the hemostasis efficacy. To mimic the actual application, it would be ideal to induce the injury first, followed by the treatment of intravenous administration of the hemostatic agents. Here, however, because of the difficulty in handling, a reverse procedure was followed (16). Briefly, healthy female BALB/c mice were randomized into different control and treatment groups, including untreated, saline, HA, CBP, VBP, HA-CBP, HA-VBP, HA-VBP/HA-CBP, and HAPPI. Control or experimental formulations were intravenously administered via tail vein injection using 27-gauge disposable needles. As detailed in the caption of Fig 2, the dosage of experimental formulations (HA-CBP, HA-VBP, and HAPPI) were firstly normalized to HA (4.6 mg/kg; determined from a pilot study; fig. S9), with equivalent and peptide amounts for peptide controls, and later normalized to peptides (0.68 μmol/kg) based on the peptide conjugation degree, as per one of the reviewers’ suggestion. For the HA-VBP/HA-CBP–treated group, equal volumes of HA-VBP and HA-CBP solutions were mixed to give final concentrations that are the same as those used in the HA-VBP group and HA-CBP group, respectively. After 1- or 20-min (as specified in Fig. 2) circulation time, a 0.5- to 1-mm incision was made on the opposite lateral vein at the site, with tail diameter of ~2.5 mm, using sterile scalpel #10 and a previously reported laceration template (25). The lacerated tail was immediately immersed in 14 ml of sterile saline at 37°C, and bleeding time was recorded using a stopwatch. Even after the bleeding stopped, the tail was kept immersed in warm saline for 4 min after injury to observe any further bleeding episodes. Care was taken to not disturb or touch the injured vessel throughout the observation period. Given that the majority of bleeding time data points were within 2 min and no rebleeding was observed, animals are euthanized 5 min after injection. The volume of the blood from the bleeding site was quantified using Drabkin’s reagent (39). Blood collected in warm saline was centrifuged at 1000g for 5 min at 4°C. After discarding the supernatant, the pelleted red blood cells were lysed by resuspension in 10 ml of Drabkin’s reagent (Sigma-Aldrich, St. Louis, MO) to quantify the released hemoglobin (Hb) by absorbance measurements in Corning UV-transparent 96-well microplate at 540 nm using SpectraMax i3 microplate reader (Molecular Devices LLC., San Jose, CA). Bleeding volume was calculated in terms of Hb amount (nanomole) using Eq. 1
| (1) |
For thrombocytopenia induction, antibodies against mouse GPIbα platelet receptors (R300, Emfret Analytics, Germany) were intravenously injected at a dosage of 2 μg/g bodyweight to cause platelet depletion (40). Following 12 hours of antibody administration, tail vein bleeding assays were performed in the thrombocytopenic mice, as described previously. The bleeding phenomenon was observed until experimental end point of 20 min after injury was reached.
For the rat trauma model of IVC, 18 rats were randomized into two different groups (nine rats each): saline and HAPPI. The dosage of HAPPI was set to 12 mg/kg, as determined from the mice study. The same volume of saline was used for the control group. Following anesthesia by inhalation of a combination of oxygen and isoflurane (2 to 3%), left inguinal was incised and left femoral vein is identified, separated from the connected tissue, and cannulated using a saline-prefilled 22-gauge catheter. To avoid clotting inside the catheter, 0.05 ml was then flushed. Subsequently, a long midline incision was performed, and the intestine was gently placed out of the abdominal cavity and kept hydrated by warm saline–wet gauze. IVC was exposed through a four-armed retractor and separated from the aorta and connecting fatty tissues below the renal veins. Size 3 suture was used to gently isolate the IVC. If the IVC was ruptured during the dissection, then rats were not used further. An 18-gauge catheter was used to completely puncture through the IVC. Immediately following the removal of the catheter (starting bleeding, time 0), saline (1.25 ml/kg) or HAPPI (1.25 ml/kg) was infused intravenously through the femoral vein catheter. The animal was observed for 2 hours or until mortality as determined by the stop of the pulmonary function. Survival time was recorded, and blood loss was quantified using preweighted 4-inch by 4-inch gauzes. IVC was harvested and imaged by In Vivo Imaging System (PerkinElmer, Waltham, MA).
Biodistribution, pharmacokinetic, and systemic toxicity studies in mice
Fluorescent HA conjugates (equivalent to HA dose of 4.6 mg/kg) with or without CBP and VBP were reconstituted in saline for intravenous injection followed by tail vein incision in healthy female BALB/c mice weighing 18 to 21 g (n = 3 per group). At indicated time points (5 min, 30 min, 1 hour, 6 hours, and 24 hours) after injection, mice were euthanized with anesthetic overdose of carbon dioxide inhalation. Blood was extracted by cardiac puncture, and the organs were perfused with phosphate-buffered saline (PBS). For pharmacokinetic and biodistribution studies, plasma was obtained by centrifugal separation of blood cells at 1250g for 10 min at 4°C. Organs of interest (brain, heart, kidneys, liver, lungs, spleen, and injured tail section) were harvested, weighed, and digested in 1 to 2 ml of Thermo Scientific radioimmunoprecipitation assay lysis and extraction buffer using a T10 basic ULTRA TURRAX homogenizer (IKA Works, Wilmington, NC) to obtain tissue lysates. The homogenate was then centrifuged at 1000g for 5 min, and the supernatant was used to quantify the amount of HA conjugates in each organ using fluorescence spectroscopy (λex/λem: 640/665). Similar sample preparation was used to measure fluorescence of standard solutions to calculate the percent injected dose per gram of tissue (% ID/g).
To determine systemic toxicity, mice (n = 3) were administered with saline, HA, or HAPPI and euthanized after 30 min, 1 day, and 7 days of injury infliction. Vital organs including the brain, heart, kidneys, liver, lungs, and spleen were collected, formalin-fixed, and paraffin-embedded for further histological analysis. Organ sections were stained with H&E. For hematological and biochemistry analyses, blood samples were collected at days 1 and 7 after injury, submitted to IDEXX Laboratories (North Grafton, MA), and analyzed within 4 hours after collection. Blood samples were collected into a 0.5-ml EDTA anticoagulant tube (LTT-MINI Top tube) and a 0.8-ml serum gel tube (SST-MINI Gold Top Tube), both provided by IDEXX Laboratories, for hematological and biochemistry analyses, respectively. Whole blood was mixed by gently inverting the tube to prevent clotting and hemolysis and stored on ice. Serum was separated by centrifugation at 2000g for 10 min after 20 min of clotting at room temperature.
TEG, platelet aggregometry, and plasma stability test of HAPPI
TEG using TEG 5000 Thrombelastograph Hemostasis Analyzer system (Haemonetics Corporation, Braintree, MA) was performed on citrated BALB/c mouse whole blood, commercially obtained from BioIVT. Briefly, blood was mixed with saline or HA conjugates at doses equivalent to the in vivo efficacy studies, and the clot formation kinetics was studied in clear disposable cups and pins.
Platelet aggregometry test using a four-channel platelet ionized calcium aggregometer (model 660, Chrono-log Corp, PA) was done on human PRP. Briefly, whole-blood samples were drawn from healthy donors in the presence of 10% sodium citrate and spun at ~200g for 20 min. The top PRP layer was collected and rested at a 37°C water bath over 30 min. The platelet concentration was determined and adjusted to ~2 × 105/μl with prewarmed Hepes-Tyrode-glucose buffer using Hemavet 850FS (Drew Scientific, FL). Under stirring conditions at 37°C, 39 μl of HAPPI, HA-VBP, HA-CBP, or saline control was added to the cuvette containing 211 μl of PRP at doses equivalent to the in vivo study. Six minutes later, 3 μl of ADP (2.5 mM) was added as the agonist, resulting in platelet aggregation and, thus, increase in light transmission. Data were analyzed using AGGRO/LINK software package version 5.2.5 and Microsoft Office Professional Plus 2013.
To test the stability of HAPPI in plasma and PBS, 100 μl of HAPPI was mixed with 566.7 μl of BALB/c mouse plasma (3.2% NaCit pooled female, BioIVT) and incubated at 37°C for checking of the formation of precipitates over 1 week. In addition, the size of vWF protein (Sigma-Aldrich, 681300-100UG) was measured using DLS (Zetasizer Zen 3600, Malvern Instruments) before and after adding an equal amount HAPPI (40 μg/ml). Three replicates were measured for each time point.
Dynamic binding assays
Fisherbrand plain glass microscope slides (2 inches by 3 inches) were degreased with 2 M HCl in 50% ethanol and rinsed with DI water. The cleaned slides were marked with two distinct circular spots and coated with 10 μl of casein blocking buffer (B6429, Sigma-Aldrich, St. Louis, MO) and collagen and vWF mixture, respectively. Equal volumes of collagen type 1 (100 μg/ml; CC050, Sigma-Aldrich, St. Louis, MO) and of vWF (50 μg/ml; RP-43132, Thermo Fisher Scientific, Waltham, MA) in carbonate-bicarbonate buffer (pH 9.6) were mixed together to obtain the collagen and vWF mixture. Microspot-coated glass slides were incubated in a humid chamber for 2 hours at 37°C and washed with PBS containing 0.1% Tween 20. Mouse platelets were obtained from PRP (BioIVT), washed thrice with PBS, and stained with 3,3′-dihexyloxacarbocyanine iodide (DiOC6; Sigma-Aldrich, St. Louis, MO) for fluorescence detection (λex: 485 nm; λem: 501 nm). For dynamic binding assays, a PPFC device with rectangular flow channel (flow width: 0.25 cm; height: 0.0254 cm) was obtained from GlycoTech (Gaithersburg, MD, USA). The coated glass slides were vacuum-sealed onto the transparent PPFC, and fluid flow was maintained via silicone tubing and fixtures using a programmable pump (VWR Variable-Speed Peristaltic Pumps 70730-062, VWR International). AF 647–labeled HA conjugates, normalized for HA amount and, thus, fluorescence, with or without DiOC6-stained mouse platelets were perfused through the chamber at 25 dynes/cm2 for 5 min in a recirculating loop. After rinsing with PBS, the glass slides were imaged under an epifluorescence microscope to observe the binding or platelets and/or conjugates at the nonspecific casein as well as target collagen and vWF–coated microspots. Images were processed and analyzed using ImageJ (National Institutes of Health) to quantify the fluorescence intensity of bound conjugates and platelets.
FACS studies
Anti-mouse CD62P (catalog no. 148303, clone: RMP-1) and anti-mouse CD41 (catalog no. 133917, clone: MWReg30) antibodies were purchased from BioLegend (San Diego, CA). They were diluted appropriately in eBioscience Flow Cytometry Staining Buffer from Thermo Fisher Scientific (Waltham, MA) before use. Murine platelets were identified with CD41 surface antigen, and CD62P (P-selectin) was used as an exclusive marker to determine their activation (33) on incubation with AF 647–HA and AF 647–HA-CBP-VBP. To study the effect on activation, murine plasma was incubated for 10 min with equivalent concentrations of HA and its conjugates, respectively. ADP solution was used as a positive control for activation. Briefly, platelets in murine PRP obtained from BioIVT (Westbury, NY) were activated using 2 × 10−5 M ADP in PBS for 10 min. For activation and binding studies with conjugates, activated platelets were incubated with appropriate conjugate concentrations for another 10 min. Following centrifugal washing with staining buffer at 1250g for 10 min at 22°C, platelet samples were stained with antibodies for 20 min at room temperature in the dark, and flow cytometry analysis was carried out using BD LSR II Analyzer (Franklin Lakes, NJ). Experimental data were analyzed using FCS Express 6 (De Novo Software, Glendale, CA).
Statistical analysis
Statistical analysis for all studies was performed using Prism 8.2.0 (GraphPad Software, San Diego, CA) to calculate the mean and SEM for each experimental and control group. Statistical significance of normally distributed flow cytometry data was determined using one-way analysis of variance (ANOVA) test and Tukey’s multiple comparisons test. For in vivo studies, bleeding time and blood loss (nanomole Hb) were analyzed with two-tailed, nonparametric Mann-Whitney test (*P < 0.05 and **P < 0.01) and nonparametric Kruskal-Wallis test followed by post hoc comparisons using Dunn’s test at a 95% confidence interval (P < 0.05).
Supplementary Material
Acknowledgments
We thank J. Vieira for analysis of histology images. We also thank J. Kim, D. Vogus, and Q. M. Qi for training on in vivo procedures and for helpful suggestions regarding experiment design and data analysis. Free images available at Servier Medical Art (http://smart.servier.com) were used in preparation of graphical schemes. Funding: S.M. and A.S.G. acknowledge financial support from the NIH (R01HL129179). S.M. acknowledges support from Blavatnik Biomedical Accelerator of Harvard University. This work was performed, in part, at the Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Coordinated Infrastructure Network (NNCI), which is supported by the NSF under NSF award no. 1541959. CNS is part of Harvard University. S.G. is a visitor from Minjiang University, Fujian, China visiting Beth Israel Deaconess Medical Center with support from China Scholarship Council. Author contributions: S.M. conceived the idea. A.S., Y.G., A.U., Z.Z., N.K., and S.G. performed experiments. All authors designed, analyzed, and discussed data, with guidance from N.S., R.F., A.S.G., and S.M. A.S., Y.G., and S.M. wrote the paper, with contributions and critical revisions from all co-authors. Competing interests: A.S., Y.G., and S.M. are inventors on a patent application US2018/0311378A1 that covers some aspects of the technology reported in this manuscript (owned and managed by Harvard University). Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper as source data (Figs. 1 to 6) and/or in the Supplementary Materials (figs. S1 to S9 and table S1). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eaba0588/DC1
REFERENCES AND NOTES
- 1.D. Berwick, A. Downey, Elizabeth Cornett, A National Trauma Care System: Integrating Military and Civilian Trauma Systems to Achieve Zero Preventable Deaths After Injury (National Academies Press, 2016). [PubMed] [Google Scholar]
- 2.S. Shackelford, B. J. Eastridge, in Damage Control Resuscitation (Springer, 2020), pp. 31–40. [Google Scholar]
- 3.Rhee P., Joseph B., Pandit V., Aziz H., Vercruysse G., Kulvatunyou N., Friese R. S., Increasing trauma deaths in the United States. Ann. Surg. 260, 13–21 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Davis J. S., Satahoo S. S., Butler F. K., Dermer H., Naranjo D., Julien K., Van Haren R. M., Namias N., Blackbourne L. H., Schulman C. I., An analysis of prehospital deaths: Who can we save? J. Trauma Acute Care Surg. 77, 213–218 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Spinella P. C., Zero preventable deaths after traumatic injury: An achievable goal. J. Trauma Acute Care Surg. 82, S2–S8 (2017). [DOI] [PubMed] [Google Scholar]
- 6.King D. R., Initial care of the severely injured patient. N. Engl. J. Med. 380, 763–770 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Alarhayem A. Q., Myers J. G., Dent D., Liao L., Muir M., Mueller D., Nicholson S., Cestero R., Johnson M. C., Stewart R., O’Keefe G., Eastridge B. J., Time is the enemy: Mortality in trauma patients with hemorrhage from torso injury occurs long before the “golden hour”. Am. J. Surg. 212, 1101–1105 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Van Oostendorp S. E., Tan E. C. T. H., Geeraedts L. M. G. Jr., Prehospital control of life-threatening truncal and junctional haemorrhage is the ultimate challenge in optimizing trauma care; a review of treatment options and their applicability in the civilian trauma setting. Scand. J. Trauma Resusc. Emerg. Med. 24, 110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mannucci P. M., Hemostatic drugs. N. Engl. J. Med. 339, 245–253 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Peng H. T., Hemostatic agents for prehospital hemorrhage control: A narrative review. Mil. Med. Res. 7, 13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kruskall M. S., The perils of platelet transfusions. N. Engl. J. Med. 337, 1914–1915 (1997). [DOI] [PubMed] [Google Scholar]
- 12.Slichter S. J., Platelet transfusion therapy. Hematol. Oncol. Clin. North Am. 21, 697–729 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Prodger C. F., Rampotas A., Estcourt L. J., Stanworth S. J., Murphy M. F., Platelet transfusion: Alloimmunization and refractoriness. Semin. Hematol. 10.1053/j.seminhematol.2019.10.001 , (2019). [DOI] [PubMed] [Google Scholar]
- 14.Modery-Pawlowski C. L., Tian L. L., Pan V., McCrae K. R., Mitragotri S., Sen Gupta A., Approaches to synthetic platelet analogs. Biomaterials 34, 526–541 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Levi M., Friederich P. W., Middleton S., de Groot P. G., Wu Y. P., Harris R., Biemond B. J., Heijnen H. F., Levin J., ten Cate J. W., Fibrinogen-coated albumin microcapsules reduce bleeding in severely thrombocytopenic rabbits. Nat. Med. 5, 107–111 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Modery-Pawlowski C. L., Tian L. L., Ravikumar M., Wong T. L., Sen Gupta A., In vitro and in vivo hemostatic capabilities of a functionally integrated platelet-mimetic liposomal nanoconstruct. Biomaterials 34, 3031–3041 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Hubbard W. B., Lashof-Sullivan M., Greenberg S., Norris C., Eck J., Lavik E., VandeVord P., Hemostatic nanoparticles increase survival, mitigate neuropathology and alleviate anxiety in a rodent blast trauma model. Sci. Rep. 8, 10622 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anselmo A. C., Modery-Pawlowski C. L., Menegatti S., Kumar S., Vogus D. R., Tian L. L., Chen M., Squires T. M., Sen Gupta A., Mitragotri S., Platelet-like nanoparticles: Mimicking shape, flexibility, and surface biology of platelets to target vascular injuries. ACS Nano 8, 11243–11253 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown A. C., Stabenfeldt S. E., Ahn B., Hannan R. T., Dhada K. S., Herman E. S., Stefanelli V., Guzzetta N., Alexeev A., Lam W. A., Lyon L. A., Barker T. H., Ultrasoft microgels displaying emergent platelet-like behaviours. Nat. Mater. 13, 1108–1114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan L. W., Wang X., Wei H., Pozzo L. D., White N. J., Pun S. H., A synthetic fibrin cross-linking polymer for modulating clot properties and inducing hemostasis. Sci. Transl. Med. 7, 277ra29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lennon F. E., Singleton P. A., Hyaluronan regulation of vascular integrity. Am. J. Cardiovasc. Dis. 1, 200–213 (2011). [PMC free article] [PubMed] [Google Scholar]
- 22.Weigel P. H., Fuller G. M., LeBoeuf R. D., A model for the role of hyaluronic acid and fibrin in the early events during the inflammatory response and wound healing. J. Theor. Biol. 119, 219–234 (1986). [DOI] [PubMed] [Google Scholar]
- 23.Kim H., Shin M., Han S., Kwon W., Hahn S. K., Hyaluronic acid derivatives for translational medicines. Biomacromolecules 20, 2889–2903 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Spagnoli C., Korniakov A., Ulman A., Balazs E. A., Lyubchenko Y. L., Cowman M. K., Hyaluronan conformations on surfaces: Effect of surface charge and hydrophobicity. Carbohydr. Res. 340, 929–941 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Johansen P. B., Tranholm M., Haaning J., Knudsen T., Development of a tail vein transection bleeding model in fully anaesthetized haemophilia A mice—Characterization of two novel FVIII molecules. Haemophilia 22, 625–631 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Stella L., Donati M. B., de Gaetano G., Bleeding time in laboratory animals. I. Aspirin does not prolong bleeding time in rats. Thromb. Res. 7, 709–716 (1975). [DOI] [PubMed] [Google Scholar]
- 27.Fraser J. R., Laurent T. C., Pertoft H., Baxter E., Plasma clearance, tissue distribution and metabolism of hyaluronic acid injected intravenously in the rabbit. Biochem. J. 200, 415–424 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fraser J. R., Alcorn D., Laurent T. C., Robinson A. D., Ryan G. B., Uptake of circulating hyaluronic acid by the rat liver. Cellular localization in situ. Cell Tissue Res. 242, 505–510 (1985). [DOI] [PubMed] [Google Scholar]
- 29.Smedsrød B., Cellular events in the uptake and degradation of hyaluronan. Adv. Drug Deliv. Rev. 7, 265–278 (1991). [Google Scholar]
- 30.Santos E. W., de Oliveira D. C., Hastreiter A., da Silva G. B., de O’Beltran J. S., Tsujita M., Crisma A. R., Neves S. M. P., Fock R. A., Borelli P., Hematological and biochemical reference values for C57BL/6, Swiss Webster and BALB/c mice. Brazilian J. Vet. Res. Anim. Sci. 53, 138–145 (2016). [Google Scholar]
- 31.Park C. G., Hartl C. A., Schmid D., Carmona E. M., Kim H.-J., Goldberg M. S., Extended release of perioperative immunotherapy prevents tumor recurrence and eliminates metastases. Sci. Transl. Med. 10, eaar1916 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Zaias J., Mineau M., Cray C., Yoon D., Altman N. H., Reference values for serum proteins of common laboratory rodent strains. J. Am. Assoc. Lab. Anim. Sci. 48, 387–390 (2009). [PMC free article] [PubMed] [Google Scholar]
- 33.Vardon Bounes F., Mémier V., Marcaud M., Jacquemin A., Hamzeh-Cognasse H., Garcia C., Series J., Sié P., Minville V., Gratacap M.-P., Payrastre B., Platelet activation and prothrombotic properties in a mouse model of peritoneal sepsis. Sci. Rep. 8, 13536 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adelman B., Carlson P., Powers P., Von Willebrand factor is present on the surface of platelets stimulated in plasma by ADP. Blood 70, 1362–1366 (1987). [PubMed] [Google Scholar]
- 35.Leebeek F. W. G., Eikenboom J. C. J., Von Willebrand’s disease. N. Engl. J. Med. 375, 2067–2080 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Kwon M. Y., Wang C., Galarraga J. H., Puré E., Han L., Burdick J. A., Influence of hyaluronic acid modification on CD44 binding towards the design of hydrogel biomaterials. Biomaterials 222, 119451 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang L., Liu G., Liu H., Han J., Liu Z., Ma H., Molecular weight impact on the mechanical forces between hyaluronan and its receptor. Carbohydr. Polym. 197, 326–336 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Swaroop M., Straus D. C., Agubuzu O., Esposito T. J., Schermer C. R., Crandall M. L., Pre-hospital transport times and survival for hypotensive patients with penetrating thoracic trauma. J. Emerg. Trauma Shock 6, 16–20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Acker J. P., Croteau I. M., Yi Q.-L., An analysis of the bias in red blood cell hemolysis measurement using several analytical approaches. Clin. Chim. Acta 413, 1746–1752 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Bergmeier W., Rackebrandt K., Schröder W., Zirngibl H., Nieswandt B., Structural and functional characterization of the mouse von Willebrand factor receptor GPIb-IX with novel monoclonal antibodies. Blood 95, 886–893 (2000). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eaba0588/DC1