

Abstract
Oncolytic virotherapy represents a promising approach for treating recurrent and/or drug-resistant ovarian cancer. However, its successful application in the clinic has been hampered by rapid immune-mediated clearance, which reduces viral delivery to the tumor. Patient-derived mesenchymal stem cells that home to tumors have been used as viral delivery tools, but variability associated with autologous cell isolations limits the clinical applicability of this approach. We previously developed an allogeneic, clonal neural stem cell (NSC) line (HB1.F3.CD21) that can be used to deliver viral cargo. Here, we demonstrate that this NSC line can improve the delivery of a thymidine kinase gene-deficient conditionally replication-competent orthopoxvirus, CF33, in a preclinical cisplatin-resistant peritoneal ovarian metastases model. Overall, our findings provide the basis for using off-the-shelf allogeneic cell-based delivery platforms for oncolytic viruses, thus providing a more efficient delivery alternative compared with the free virus administration approach.
Keywords: oncolytic virotherapy, CF33, ovarian cancer, neural stem cells, cellular therapy, Cancer, Oncolysis, Oncolytic virus, Immunity, Immunotherapy
Graphical Abstract
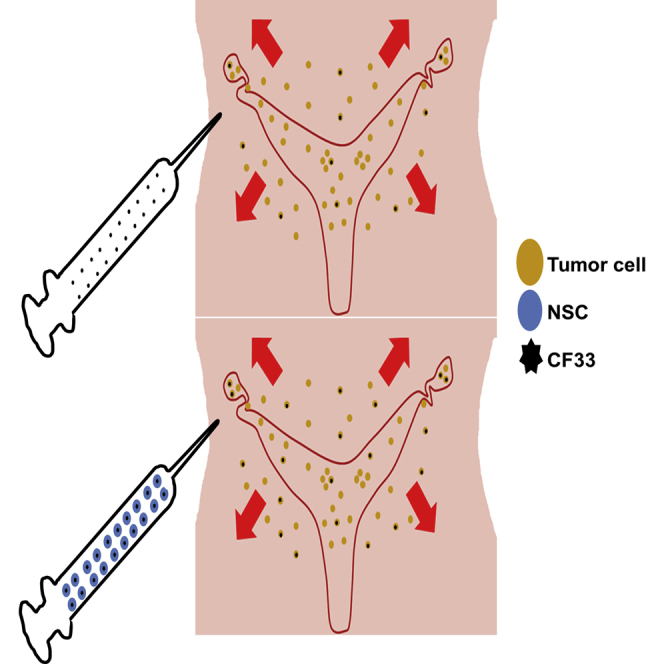
More effective and less toxic therapeutic approaches for ovarian cancer are urgently needed. Our data suggest that packaging CF33 oncolytic virus in our neural stem cell line improves distribution, and thus might reduce toxicities, improve clinical outcome, and improve quality of life for patients with advanced cancer.
Introduction
Epithelial ovarian cancer accounts for 140,000 annual deaths worldwide and is the leading cause of gynecologic cancer-related mortality in the United States.1, 2, 3 Unfortunately, it is usually not diagnosed until patients have abdominal metastases, at which point their 5-year survival rate is only 34% after standard surgical debulking and chemotherapy.4 Hence there is an urgent need for the development of safer and more efficacious therapeutic approaches. Advances in oncolytic virotherapy, in which oncolytic viruses (OVs) are used to selectively target, reproduce in, and kill tumor cells while sparing normal cells, have led to successful clinical applications.5,6 Interestingly, OVs can instate cancer cell killing7 regardless of chemoresistance8 and can expose tumor antigens, and consequently fuel immune recognition of cancer cells. So far, out of 11 OVs that have been tested in pre-clinical human ovarian cancer models, four moved to phase I/II clinical trials that established the safety of this approach.5 Despite having an excellent safety profile in clinical settings, the anti-tumor efficacy of most OVs has been modest so far mainly due to inefficient vector distribution.9, 10, 11, 12 Specifically, OVs injected systemically are subject to rapid clearance by the host’s immune system because of their small size13 and inactivation by complement and antibodies (Abs).14
To overcome these limitations, there is a growing interest in developing cellular vehicles that are cytogenetically stable, amenable to viral infection, and capable of shielding encapsulated viruses from the host immune system.13 However, it is important that such carriers are able to localize to tumors and enhance viral infection and distribution at these sites.5 To address these limits, we previously developed an immortalized HB1.F3.CD21 neural stem cell (NSC) line that was shown to be tumor tropic, minimally immunogenic,15,16 non-tumorigenic, and clinically safe.17 We showed that this cell line enhanced tumor-confined production of the oncolytic adenovirus, CRAd-S-pk7, in glioma18, 19, 20, 21, 22 and ovarian cancer models.19 Consequently, HB1.F3.CD21 NSCs loaded with CRAd-S-pk7 (NSC.CRAd-S-pk7) are currently in clinical trial as an adjunct oncolytic virotherapy for newly diagnosed glioma patients (ClinicalTrials.gov: NCT03072134). Good manufacturing practices (GMPs) for the viral loading and release kinetics and freeze-thaw standard operating procedures (SOPs) are established. These SOPs could provide standard “off-the-shelf” allogeneic clinical-grade banks for other OVs, facilitating an expedited translation to the clinic for ovarian cancer. One such OV is CF33, a chimeric poxvirus created by chimerization of nine species of orthopoxvirus, including multiple strains of vaccinia virus selected for enhanced anti-tumor activity.23, 24, 25 CF33 has a mutation in the J2R (thymidine kinase [TK]) gene, and hence it depends on the cellular pool of thymidine triphosphate. Because cellular TK is highly expressed in many tumor cells,26 it is expected that CF33 replicates preferentially in these cells. We hypothesized that using the NSC line to deliver CF33 (NSC.CF33) solely to tumor locations could minimize off-target distribution, particularly to adult mesenchymal stem cells (MSCs) that express high levels of TK1.27 In the current study, we performed in vitro and in vivo studies to evaluate the pre-clinical utility of CF33 delivered via NSCs in the context of ovarian cancer metastases within the peritoneal cavity. Our results show that CF33 replicates within ovarian cancer cells causing oncolysis, and NSC.CF33 selectively targets and penetrates tumor metastases sites, effectively delivering CF33. Overall, this study further demonstrates the potential of our off-the-shelf allogeneic NSC line as a delivery vehicle for oncolytic virotherapy in ovarian cancer.
Results
TK1 Expression Is High and Correlates with Poor Survival in Ovarian Cancer
We searched The Cancer Genome Atlas (TCGA) RNA sequencing (RNA-seq) database to evaluate expression of TK1 in 37 cancers and normal tissues. We observed that there was a broad spectrum of TK1 mRNA expression, and it was highly expressed in all TCGA cancer types compared with TCGA normal samples (Figure 1A). These results agree with previous reports that showed that TK1 is overexpressed in many human cancers.28 Furthermore, based on the interquartile range, the spread of TK1 expression was more varied within some cancer types than others. For example, glioblastoma multiforme low-grade glioma (GBMLGG) had a wider spread compared with uterine carcinosarcoma (UCS) (Figure 1A), possibly because some cancers have more than one clearly defined subtype and hence more genetic diversity. We assessed the frequency of TK1 in ovarian cancer as compared with normal tissues to ensure that our approach of targeting TK1 could potentially be applied for treatment of this cancer. We used the publicly available GEO Affymetrix human U133A microarray datasets (GEO: GSE14407, GSE1926, GSE9891, GSE102073, and GSE102085) and identified that the TK1 gene (Gene ID#7083) is highly expressed in ovarian cancer. This query dataset included gene expression data for an extensive set of samples: 606 ovarian cancer samples from 594 patients, 307 samples from 304 patients for ovarian mRNA sequencing (mRNA-seq) expression, and 12 samples representing normal ovarian surface epithelium.29 We observed that ovarian cancer samples showed the presence of 44.09 transcripts per million (TPMs) of TK1 as compared with only 0.83 in normal ovarian tissue (Figure 1A). Similarly, results using the Affymetrix Human Genome U133 Plus 2.0 Array [HG-U133_Plus_2]29 showed that TK1 mRNA expression was remarkably higher by more than 1 log in ovarian cancer compared with normal tissue (Figure 1B). Results using the GEO: GSE9891 Relapse-Free-Survival Affymetrix HG-133 Plus 2 (285 samples)30 showed that TK1 expression had a high negative correlation to relapse-free survival for ovarian cancer (Figure 1C), suggesting that TK1 expression might improve early prognosis in individual patients.
Figure 1.

Pan-Cancer TK1 mRNA-Seq Expression Is High in Ovarian Cancer and Correlates with Poor Survivability
(A) TK1 mRNA-seq expression TCGA pan-cancer. (B) GEO: GSE14407 matched tumor versus normal29 (Affymetrix HG-133 Plus 2). (C) GEO: GSE9891 relapse-free survival30 (Affymetrix HG-133 Plus 2; 285 samples).
We used a specificity index probability (PSI) to analyze TK1 expression in an mRNA-seq dataset of 54 normal human cell lines. It was observed that expression of TK1 was low in all cell lines except cultured fibroblasts and Epstein-Barr virus (EBV)-transformed lymphocytes (Figure 2A).
Figure 2.

Ovarian Cancer Cell Lines Exhibit High TK1 RNA-Seq Values Compared with Normal Ones
(A) TK1 mRNA-seq expression in normal human tissues. (B) NCI-60 cancer cell lines RNA-seq. (C) BROAD CCLE cancer cell lines RNA-seq.
We further analyzed the TK1 mRNA-seq dataset for NCI-60 cancer cell lines from nine different cancers and observed that ovarian cancer had the highest TK1 mRNA expression compared with other cancers (Figure 2B). Furthermore, we analyzed the TK1 mRNA-seq dataset for 36 cancer cell lines from the BROAD CCLE cancer cell lines RNA-seq dataset and observed that TK1-mRNA expression was highest in prostate, esophagus, and ovarian cancer cell lines (Figure 2C). Overall, these results suggest that TK1 is highly expressed in ovarian cancer, and its expression correlates with poor survival in these patients.
CF33 Infects and Replicates in Human and Mouse Ovarian Cancer Cell Lines In Vitro
Because CF33 has a mutation in the gene encoding TK1 it is expected to preferentially infect and replicate in ovarian cancer cell lines that overexpress TK1. To confirm this hypothesis, we infected murine (ID8) and human (OVCAR8) ovarian cancer cells with CF33 at various MOIs. We performed an 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) assay to analyze cell viability after 3 days postinfection of these cells (Figures 3A and 3B). In both tested cell types, non-infected cells were used as the negative control. We observed that infection with CF33 significantly reduced the viability of these cells at lower MOIs and completely eliminated both ID8 and OVCAR8 cell lines at MOIs of 1 and 10 (p < 0.004 and p < 0.007; Figures 3A and 3B, respectively). These results indicate that CF33 can infect, replicate in, and kill ovarian cancer cell lines in vitro.
Figure 3.

Validation of CF33 Ovarian Anticancer Activity
(A) ID8 cells were completely eliminated at MOI of 10 at 3 days after exposure. (B) OVCAR8 cells exposed to CF33 of MOI of 1 have been completely eradicated. Cytotoxicity was measured with MTS assay. Error bars indicate ± SEM. n = 10.
NSC.CF33 Remains Viable and Retains Tumor Tropism to Orthotopic Ovarian Cancer
We have previously demonstrated that NSCs home to ovarian tumors after intraperitoneal (i.p.) injection into mice bearing either SKOV3 or OVCAR8 i.p. xenografts.19,31 Accordingly, using this immortalized cell line as a carrier vehicle might allow for predictable and reproducible viral loading and release kinetics.20 Therefore, we analyzed the effect of CF33 infection on the viability of NSCs and observed virus-induced cell lysis after infection with CF33. At 4 h following infection, 90%, 80%, and 70% of infected cells were still viable at MOIs of 1, 3, and 5, respectively (Figure 4A). Next, to test the effect of CF33 infection on the tumor-tropic properties of NSCs, we performed an in vitro Boyden chamber migration assay using media conditioned with cancer-derived cytokines from the OVCAR8 cell line. It was observed that NSC.CF33 effectively migrated toward the conditioned media for up to 5 h following transduction (Figure 4B). Importantly, there was no significant difference in viability between transduced and non-transduced cells (Figure 4C). Overall, these results suggest that NSCs transduced with CF33 remain viable and retain their tumor-tropic abilities.
Figure 4.

NSCs Are Permissive to CF33, and NSCs.CF33 Migrate to Ovarian Cancer In Vitro
(A) NSCs 4 h after infection with CF33 at MOI of 3 and 5 were not significantly different in viability from each other, with 80% of NSCs viable at MOI of 3. (B) Migration of NSCs toward OVCAR8 conditioned media through Boyden chamber assay. These cells were kept with CF33 at MOI of 1 and 5 for different time periods. (C) Viability of migrated cells. Noninfected NSCs in FBS and noninfected NSCs in bovine serum albumin are the positive and negative controls, respectively. Cytotoxicity was measured with MTS assay. Error bars indicate ± SEM. n = 10.
Using a NSC Line as a Cell Carrier for CF33 Improves Delivery of CF33 in Orthotopic Tumor Models
Next, we used orthotopic mouse models to observe the effect of using NSCs as a carrier vehicle for CF33 and to test whether this improves CF33 delivery in vivo compared with free virus administration. First, B6 mice (n = 3 per group) were injected i.p. with EGFP-expressing ID8 cells. One week after tumor cell inoculation, the mice were injected i.p. with either free CF33.FFLUC (2 weeks of 3 × 106 plaque-forming units [PFUs]/week), NSC.CF33.FFLUC (2 weeks of 2 × 106 cells [3 × 106 PFUs]/week), or PBS (control). Following this, we performed bioluminescence imaging to quantify viral infection and replication in the infected tissues, to assess the relative infectivity of CF33.FFLUC when delivered either as the free virus or with NSCs.
We observed increased CF33 infection in tumor tissue in NSC.CF33-treated mice compared with free CF33-treated mice in an OVCAR8 orthotopic mouse model (Figure 5A). We investigated whether packaging CF33 in NSCs would improve retention of the virus in the tumor tissue by protecting it against a potential antiviral immune response. Therefore, we compared bioluminescence imaging (BLI) signal from the second to the first treatment round and observed that NSC maintains CF33 retention, while free CF33 lost 75% of the BLI signal (Figure 5B).
Figure 5.

NSC.CF33 Improves CF33 Distribution In Vivo
(A) Seven days after i.p. treatment, NSCs remarkably increase infection and replication of CF33.FFLUC in the ovcar8 NSG orthotopic mouse model. (B) NSCs maintain CF33.FFLUC BLI after the second round of treatment compared with the free CF33.FFLUC-treated group that lost 2/3 of their signal. BLI signal was acquired 1 h after the first and second treatment rounds. Error bars represent the SEM. (C) Tumors with adjacent tissue were harvested 2 days after the first treatment, and a representative micrograph of a tumor mass (black dotted line) stained with FFLUC antibody-DAB (brown) on the surface of normal omentum. A sister serial section of the section shown was stained with an anti-pox antibody-DAB (brown). This staining pattern suggests that CF33 delivered by NSCs spreads through tumors from the periphery inward (black arrows). Scale bars: 200 μm. (D) C57BL/6 mice with established i.p. ID8 tumors with NSC.CF33 (2 × 106), free CF33 with matched viral load 3 × 106 plaque-forming units (PFUs), or PBS control. Immunofluorescence staining with anti-vaccinia Ab (red) confirmed transfer of CF33 (red) to tumor (green) in the NSC.CF33 group and free CF33 group with matched viral loads (3 × 106 PFUs/mouse), compared with the untreated group, 4 days after the first treatment. Scale bar: 100 μm.
Next, we investigated whether NSCs enhance delivery of CF33 to human tumors in an OVCAR8 xenograft ovarian tumor model. Mice were treated with either PBS or NSC.CF33, and tumors with adjacent tissue were harvested 2 days later and stained for tumor and CF33. The sections were stained with FFLUC Ab-3,3′-diaminobenzidine (DAB) (brown). Sister serial sections of the section shown were stained with an anti-pox Ab-DAB (brown). This staining pattern suggests that CF33 delivered by NSCs spreads through tumors from the periphery inward (black arrows) (Figure 5C).
Finally, we looked at immunofluorescence staining of tumors in an ID8-immunocompetent syngeneic murine ovarian cancer model harvested 4 days after the last treatment. Immunofluorescence staining with anti-vaccinia Ab (red) confirmed transfer of CF33 (red) to tumor (green) in the NSC.CF33 group and free CF33 group with matched viral loads (3 × 106 PFUs/mouse), compared with the untreated group, 4 days after the first treatment. The results suggested that packaging CF33 in NSCs remarkably improved seeding of the virus within the tumor compared with the free CF33-treated group (Figure 5D). Taken together, these data suggest that using a NSC carrier for CF33 improves the delivery and efficacy of the virus within the peritoneal setting.
Discussion
Several studies have demonstrated that oncolytic virotherapy is safe and nontoxic.4,5,32 Despite the high safety profile of OVs in clinical trials, oncolytic virotherapy has shown a limited efficacy specifically for ovarian cancer, mainly because of the host-antiviral immune responses and insufficient viral delivery to tumors.33 Hence it is crucial to use a well-characterized delivery system to enhance viral distribution to tumors and subsequently their clinical applicability. Many groups have used autologous cells for viral delivery, for example, adipose tissue-derived MSCs,5,34,35 irradiated tumor cells, interleukin-2 (IL-2) expanded T cells,36 and CD14+-derived monocytes.34 However, there are potential variabilities in ex vivo virus loading capacities, cytogenetic stabilities, cell expansion potential, and tumor homing associated with such approaches. For instance, Mader et al.35 reported that it took 2 weeks to produce sufficient amounts of autologous MSCs for patient treatment, and 20% of these displayed abnormal karyotypes. Hence we believe that using an “off-the-shelf” allogeneic cell line is a more viable approach for long-term scale-up and clinical translation. Consequently, delivering OVs through a cellular vehicle might significantly improve therapeutic outcome with the need to give multiple administrations and the potential succeeding buildup of anti-OV neutralizing Abs.
Given the potential utility of the NSC delivery platform, we are interested in exploring oncolytic viral cargo with potent anti-tumor efficacy. Given its deficiency in TK1, CF33 seems a promising OV because its replication is dependent on the overexpression of TK1 that is common in many cancer cells and encapsulating it in a cell carrier might significantly improve its therapeutic potential. For example, it was recently shown that a low dose of 104 PFUs of CF33 in a preclinical flank setting slowed pancreatic tumor progression without measurable toxicities associated with virotherapy.25,37
We have previously shown that the HB1.F3.CD cell line enhances adenoviral delivery to brain and peritoneal cancers; however, HB1.F3.CD-encapsulated pox virus delivery was not tested. In the current study, we utilized a research lot of HB1.F3.CD, equivalent to the clinical lot used in the glioma clinical trial (ClinicalTrials.gov: NCT03072134) to package CF33. NSC.CF33 demonstrated greater tropism toward ovarian cancer-conditioned media as compared with uninfected NSCs. In addition, NSC.CF33-treated mice showed a remarkably higher CF33 PFUs per milligram of tumor tissue compared with free CF33 in an OVCAR8 orthotopic murine model. We also demonstrate enhanced CF33 delivery in an orthotopic ovarian cancer mouse model. This result is in agreement with another study showing that MSC-encapsulated measles virus has a better therapeutic outcome compared with virus alone in an orthotopic ovarian tumor model.38 However, our results need to be tested in preimmunized syngeneic murine models to verify the potential of HB1.F3.CD as an improved delivery mechanism for OVs in peritoneal carcinomatosis.
A potential mechanism, based on overexpression of three chemoattractant receptors (CXCR4, c-met, VEGF-R2) on HB1.F3.CD NSCs upon infection with OVs, was proposed in a malignant glioma model.39 In addition to chemotaxis, other factors such as adhesion molecules in the peritoneal cavity play a role in homing of cell carriers to tumors.35 Additional studies are required to confirm whether this mechanism holds true for NSC.CF33 in ovarian cancer. Further studies characterizing the temporal and spatial distribution and intratumoral retainment of the virus by NSCs before being neutralized by the immune system especially in repeated administration settings are required.40 Additionally, studies are warranted to understand whether the improvement of OV distribution through NSCs delivery is due to in situ amplification of the virus and/or improvements in viral delivery. It will also be interesting to identify whether CF33 provokes anti-tumor immunogenicity potentially by uncovering neoantigens through tumor cell lysis, as well as to manipulate it to express therapeutic transgenes.41 Overall, our data suggest that encapsulating CF33 in NSCs enhanced its distribution in a peritoneal ovarian cancer setting and therefore call for investigating the therapeutic potential of NSC.CF33. In conclusion, we demonstrate the utility of an off-the-shelf allogeneic cell line as a delivery vehicle for oncolytic virotherapy.
Materials and Methods
Microarray Analysis of TK1 Expression in Patient Cohorts
Two ovarian cancer microarray datasets from TCGA42 and GEO: GSE989130 composed of clinical samples were analyzed. All patients were classified into two distinct groups: high and low TK1 expressers. The log rank test was utilized to evaluate whether significant differences existed in the recurrence-free survival (RFS). Tumor versus normal TK1 gene expression was calculated using 12 normal and 12 tumor patient samples.29 Primary versus metastatic ovarian cancer genome-wide transcriptional analysis of carboplatin response was calculated using 18 human cell cultures (drug resistance and drug sensitive) from six patients.43 TK1 expression intensity was calculated using 12 matched ovary serous adenocarcinoma versus 12 normal ovarian tissues.29
Chimerization of Orthopoxviruses, High-Throughput Screening, and Luciferase Insertion
Generation of CF33 through chimerization of different poxviruses has been previously reported with CF33 acquiring significantly improved oncolytic characteristics compared with its parental viruses.23 CF33 caused significant cell death in the majority of the NCI-60 solid cancer cell lines even at a low MOI of 0.01. This virus was engineered to express Firefly luciferase (FFLUC) (CF33-FFLUC) as described in O’Leary et al.23
Cell Culture
NSC lines, including the human v-myc immortalized HB1.F3.CD NSC line, were obtained from Dr. Seung Kim (University of British Columbia, Canada).44 To generate NSC.CF33 lines, we infected them with CF33 at MOI of 3 for 4 h in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Gemini Bio), 1% L-glutamine (Invitrogen), and 1% penicillin-streptomycin (Invitrogen) and maintained at 37°C in a humidified incubator (Thermo Electron Corporation) containing 6% CO2 and then harvested them. Ovarian cancer cell lines were cultured in RPMI basal media with the same supplements. For all cell lines, when cells reached 80% confluency, they were passaged using 0.25% trypsin and EDTA solution (Invitrogen); media were changed every 2–3 days. Ovarian cancer cell lines including FFLUC, and/or EGFP-expressing OVCAR8 (OVCAR8.EGFP. and/or FFLUC) cell lines were generously provided by Dr. Carlotta Glackin. The ID8 murine ovarian cancer cell line was obtained from Dr. Katherine Roby (University of Kansas). All tumor lines were used to generate tumor-conditioned media by replacing culture media with serum-free media when cells were 80%–100% confluent followed by a 48-h incubation period.
In Vitro Boyden Migration Assay
We used a classic Boyden chamber assay to evaluate cell migration. In a 24-well tissue culture plate, 500 mL target media (either containing only BSA as a negative control or derived from the culture of ovarian cancer cells) was added to each well. At a density of 1 × 105 cells/well, unmodified NSCs or NSC.CF33 in DMEM and 5% w/v BSA were placed in transwell polycarbonate membrane cell culture inserts (Fisher) and incubated at 37°C for 4 h. After the incubation period, the transwell inserts were placed in a new 24-well tissue culture plate containing Accutase and incubated for 10 min at 37°C. Detached cells were then transferred to a 96-well V-bottom plate, centrifuged at 1,500 rpm for 5 min, and resuspended in 1:1 PBS to Guava ViaCount Reagent (EMD Millipore). NSC migration to tumor-conditioned media was assessed using a Guava EasyCyte flow cytometer (EMD Millipore).
In Vitro Cell Viability Assay
In a 96-well tissue culture plate, a density of 5 × 103 cells/well of either OVCAR8 or ID8 cells was added to each well in 100 μL and allowed to grow overnight in the incubator. CF33 at different MOIs (0.001, 0.01, 0.1, 1, and 10) in 100 μL was added to their respective wells and was allowed to replicate for 72 h. Each sample was done in triplicate. After 72 h, an MTS assay was performed by adding 20 μL MTS reagent to each well. The plate was covered with aluminum foil and placed in the incubator for 1 h. The samples were then read using SpectraMax M3 (Molecular Devices) at a wavelength of 490 nm.
In Vivo NSC.CF33 Tropism in Orthotopic Ovarian Cancer Model
All animal experiments were conducted following NIH guidelines for housing and care of laboratory animals and in accordance with City of Hope regulations after review and approval by the City of Hope Institutional Animal Care and Use Committee (protocol number 18002).
Female B6 mice (National Cancer Institute [NCI]) that were 6–8 weeks old were inoculated with 5 × 106 ID8.EGFP.FFLUC cells via i.p. injection. At 1 and 2 weeks, mice (n = 3) were administered i.p. 2e6 NSC.CF33.FFLUC cells. One hour after each treatment, BLI was performed using Lago (Spectral Instruments Imaging, Tucson, AZ, USA). Mice were injected i.p. with D-luciferin (Xenogen; 150 μL/mouse). Anesthesia was induced with 2% isoflurane (Abbott Laboratories, Chicago, IL, USA) inhalation in a special airtight transparent anesthesia box for 5–7 min before the mice were moved to the light-tight chamber of the charge-coupled device (CCD) camera in the imaging position. The images were then analyzed with Aura version 2.2.0 software (Spectral Instruments Imaging, Tucson, AZ, USA). Two days after NSC injection, tumors were harvested. Three tumors per mouse were frozen in Tissue Tek OCT (Sakura Finetek USA) and sectioned on a Leica CM1510 S cryostat (Leica Biosystems). Sections (10 mm thick) were collected on positively charged slides (Thermo Fisher Scientific), stained with either anti-ffLuc goat polyclonal Ab (Promega) or rabbit anti-vaccinia Ab (YVS8101; Accurate Chemical and Scientific, Westbury, NY, USA) and then with biotinylated secondary Ab for 1 h as described previously (1:250 dilution, Vector BA-2001).45 Sections were washed in PBS, incubated in avidin-biotin complex (ABC) solution in DAB substrate solution, followed by bright-field imaging (Richard-Allan Scientific). Other sections were stained with DAPI (1 mg/mL; Sigma) and then imaged using the Zeiss Axio Observer Z1 fluorescence microscope (ZEISS Microscopy).
Statistical Analysis
Data are presented as mean ± SEM unless otherwise stated. Statistical significance for virus flux at each time point between two groups was examined using a two-sample t test with a two-sided alternative (∗p < 0.05 deemed to be significant).
Author Contributions
Conceptualization, K.S.A., M.H., T.H.D., N.G.C.; Methodology, M.H., Y.R.C., J.B.-C., A.A.M., C.B., Y.-C.Y., Z.L.; Formal Analysis, M.H.; Investigation, M.H., Y.R.C., J.B.-C., A.A.M., C.B., J.L.; Writing – Original Draft, M.H.; Writing – Review & Editing, M.H., K.S.A., Y.F., R.M.; Visualization: M.H., Y.R.C., A.A.M., J.B.-C., K.S.A., R.M.; Funding Acquisition, M.H., Y.R.C., K.S.A.; Resources, Y.F., K.S.A.; Supervision, R.M., K.S.A., Y.F.
Conflicts of Interest
K.S.A. and Y.F. disclose patent holder and research funding. K.S.A. discloses employment, an advisory role, and stock ownership with CSO and TheraBiologics, Inc., and intellectual property with City of Hope and Harvard Patents. All other authors declare no competing interests.
Acknowledgments
This work was funded by Stop Cancer, The Rosalinde and Arthur Gilbert Foundation, California Institute of Regenerative Medicine, the Alvarez Family Foundation, the Anthony F. & Susan M. Markel Foundation, the Ben and Catherine Ivy Foundation, City of Hope, and National Cancer Institute grants (R43CA86768, R44CA8678, and R01CA197359). Research reported in this publication included work performed by X.L. in the City of Hope Biostatistics Core, which is supported by the National Cancer Institute of the NIH under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Materials Transfer Information is available from the City of Hope Office of Technology Licensing. Special thanks for COH scientific writers Kerin Higa, PhD, and Supriya Deshpande, PhD, for editing this manuscript.
Contributor Information
Mohamed Hammad, Email: mhammad@coh.org.
Rachael Mooney, Email: rmooney@coh.org.
References
- 1.Siegel R., Naishadham D., Jemal A. Cancer statistics, 2013. CA Cancer J. Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Znaor A., van den Hurk C., Primic-Zakelj M., Agius D., Coza D., Demetriou A., Dimitrova N., Eser S., Karakilinc H., Zivkovic S. Cancer incidence and mortality patterns in South Eastern Europe in the last decade: gaps persist compared with the rest of Europe. Eur. J. Cancer. 2013;49:1683–1691. doi: 10.1016/j.ejca.2012.11.030. [DOI] [PubMed] [Google Scholar]
- 3.Bray F., Ren J.S., Masuyer E., Ferlay J. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int. J. Cancer. 2013;132:1133–1145. doi: 10.1002/ijc.27711. [DOI] [PubMed] [Google Scholar]
- 4.Cannistra S.A. Cancer of the ovary. N. Engl. J. Med. 2004;351:2519–2529. doi: 10.1056/NEJMra041842. [DOI] [PubMed] [Google Scholar]
- 5.Li S., Tong J., Rahman M.M., Shepherd T.G., McFadden G. Oncolytic virotherapy for ovarian cancer. Oncolytic Virother. 2012;1:1–21. doi: 10.2147/OV.S31626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim J., Hall R.R., Lesniak M.S., Ahmed A.U. Stem Cell-Based Cell Carrier for Targeted Oncolytic Virotherapy: Translational Opportunity and Open Questions. Viruses. 2015;7:6200–6217. doi: 10.3390/v7122921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakamori M., Fu X., Meng F., Jin A., Tao L., Bast R.C., Jr., Zhang X. Effective therapy of metastatic ovarian cancer with an oncolytic herpes simplex virus incorporating two membrane fusion mechanisms. Clin. Cancer Res. 2003;9:2727–2733. [PubMed] [Google Scholar]
- 8.Ding J. Oncolytic virus as a cancer stem cell killer: progress and challenges. Stem Cell Investig. 2014;1:22. doi: 10.3978/j.issn.2306-9759.2014.12.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun L., Funchain P., Song J.M., Rayman P., Tannenbaum C., Ko J., Mcnamara M., Marcela Diaz-Montero C., Gastman B. Talimogene Laherparepvec combined with anti-PD-1 based immunotherapy for unresectable stage III-IV melanoma: a case series. J. Immunother. Cancer. 2018;6:36. doi: 10.1186/s40425-018-0337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andtbacka R.H., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 11.Heo J., Reid T., Ruo L., Breitbach C.J., Rose S., Bloomston M., Cho M., Lim H.Y., Chung H.C., Kim C.W. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013;19:329–336. doi: 10.1038/nm.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyers D.E., Wang A.A., Thirukkumaran C.M., Morris D.G. Current Immunotherapeutic Strategies to Enhance Oncolytic Virotherapy. Front. Oncol. 2017;7:114. doi: 10.3389/fonc.2017.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirahmadi N., Babaei M.H., Vali A.M., Dadashzadeh S. Effect of liposome size on peritoneal retention and organ distribution after intraperitoneal injection in mice. Int. J. Pharm. 2010;383:7–13. doi: 10.1016/j.ijpharm.2009.08.034. [DOI] [PubMed] [Google Scholar]
- 14.Evgin L., Acuna S.A., Tanese de Souza C., Marguerie M., Lemay C.G., Ilkow C.S., Findlay C.S., Falls T., Parato K.A., Hanwell D. Complement inhibition prevents oncolytic vaccinia virus neutralization in immune humans and cynomolgus macaques. Mol. Ther. 2015;23:1066–1076. doi: 10.1038/mt.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aboody K.S., Najbauer J., Metz M.Z., D’Apuzzo M., Gutova M., Annala A.J., Synold T.W., Couture L.A., Blanchard S., Moats R.A. Neural stem cell-mediated enzyme/prodrug therapy for glioma: preclinical studies. Sci. Transl. Med. 2013;5:184ra59. doi: 10.1126/scitranslmed.3005365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aboody K.S., Brown A., Rainov N.G., Bower K.A., Liu S., Yang W., Small J.E., Herrlinger U., Ourednik V., Black P.M. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc. Natl. Acad. Sci. USA. 2000;97:12846–12851. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Portnow J., Synold T.W., Badie B., Tirughana R., Lacey S.F., D’Apuzzo M., Metz M.Z., Najbauer J., Bedell V., Vo T. Neural Stem Cell-Based Anticancer Gene Therapy: A First-in-Human Study in Recurrent High-Grade Glioma Patients. Clin. Cancer Res. 2017;23:2951–2960. doi: 10.1158/1078-0432.CCR-16-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahmed A.U., Thaci B., Tobias A.L., Auffinger B., Zhang L., Cheng Y., Kim C.K., Yunis C., Han Y., Alexiades N.G. A preclinical evaluation of neural stem cell-based cell carrier for targeted antiglioma oncolytic virotherapy. J. Natl. Cancer Inst. 2013;105:968–977. doi: 10.1093/jnci/djt141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mooney R., Majid A.A., Batalla-Covello J., Machado D., Liu X., Gonzaga J., Tirughana R., Hammad M., Lesniak M.S., Curiel D.T., Aboody K.S. Enhanced Delivery of Oncolytic Adenovirus by Neural Stem Cells for Treatment of Metastatic Ovarian Cancer. Mol. Ther. Oncolytics. 2018;12:79–92. doi: 10.1016/j.omto.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahmed A.U., Thaci B., Alexiades N.G., Han Y., Qian S., Liu F., Balyasnikova I.V., Ulasov I.Y., Aboody K.S., Lesniak M.S. Neural stem cell-based cell carriers enhance therapeutic efficacy of an oncolytic adenovirus in an orthotopic mouse model of human glioblastoma. Mol. Ther. 2011;19:1714–1726. doi: 10.1038/mt.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morshed R.A., Gutova M., Juliano J., Barish M.E., Hawkins-Daarud A., Oganesyan D., Vazgen K., Yang T., Annala A., Ahmed A.U. Analysis of glioblastoma tumor coverage by oncolytic virus-loaded neural stem cells using MRI-based tracking and histological reconstruction. Cancer Gene Ther. 2015;22:55–61. doi: 10.1038/cgt.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thaci B., Ahmed A.U., Ulasov I.V., Tobias A.L., Han Y., Aboody K.S., Lesniak M.S. Pharmacokinetic study of neural stem cell-based cell carrier for oncolytic virotherapy: targeted delivery of the therapeutic payload in an orthotopic brain tumor model. Cancer Gene Ther. 2012;19:431–442. doi: 10.1038/cgt.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Leary M.P., Choi A.H., Kim S.I., Chaurasiya S., Lu J., Park A.K., Woo Y., Warner S.G., Fong Y., Chen N.G. Novel oncolytic chimeric orthopoxvirus causes regression of pancreatic cancer xenografts and exhibits abscopal effect at a single low dose. J. Transl. Med. 2018;16:110. doi: 10.1186/s12967-018-1483-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Leary M.P., Warner S.G., Kim S.I., Chaurasiya S., Lu J., Choi A.H., Park A.K., Woo Y., Fong Y., Chen N.G. A Novel Oncolytic Chimeric Orthopoxvirus Encoding Luciferase Enables Real-Time View of Colorectal Cancer Cell Infection. Mol. Ther. Oncolytics. 2018;9:13–21. doi: 10.1016/j.omto.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chaurasiya S., Chen N.G., Lu J., Martin N., Shen Y., Kim S.-I., Warner S.G., Woo Y., Fong Y. A chimeric poxvirus with J2R (thymidine kinase) deletion shows safety and anti-tumor activity in lung cancer models. Cancer Gene Ther. 2020;27:125–135. doi: 10.1038/s41417-019-0114-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hengstschläger M., Knöfler M., Müllner E.W., Ogris E., Wintersberger E., Wawra E. Different regulation of thymidine kinase during the cell cycle of normal versus DNA tumor virus-transformed cells. J. Biol. Chem. 1994;269:13836–13842. [PubMed] [Google Scholar]
- 27.Lopez Perez R., Münz F., Vidoni D., Rühle A., Trinh T., Sisombath S., Zou B., Wuchter P., Debus J., Grosu A.L. Mesenchymal stem cells preserve their stem cell traits after exposure to antimetabolite chemotherapy. Stem Cell Res. (Amst.) 2019;40:101536. doi: 10.1016/j.scr.2019.101536. [DOI] [PubMed] [Google Scholar]
- 28.Weagel E.G., Burrup W., Kovtun R., Velazquez E.J., Felsted A.M., Townsend M.H., Ence Z.E., Suh E., Piccolo S.R., Weber K.S. Membrane expression of thymidine kinase 1 and potential clinical relevance in lung, breast, and colorectal malignancies. Cancer Cell Int. 2018;18:135. doi: 10.1186/s12935-018-0633-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bowen N.J., Walker L.D., Matyunina L.V., Logani S., Totten K.A., Benigno B.B., McDonald J.F. Gene expression profiling supports the hypothesis that human ovarian surface epithelia are multipotent and capable of serving as ovarian cancer initiating cells. BMC Med. Genomics. 2009;2:71. doi: 10.1186/1755-8794-2-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tothill R.W., Tinker A.V., George J., Brown R., Fox S.B., Lade S., Johnson D.S., Trivett M.K., Etemadmoghadam D., Locandro B. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008;14:5198–5208. doi: 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- 31.Cao P., Mooney R., Tirughana R., Abidi W., Aramburo S., Flores L., Gilchrist M., Nwokafor U., Haber T., Tiet P. Intraperitoneal Administration of Neural Stem Cell-Nanoparticle Conjugates Targets Chemotherapy to Ovarian Tumors. Bioconjug. Chem. 2017;28:1767–1776. doi: 10.1021/acs.bioconjchem.7b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galanis E. Therapeutic potential of oncolytic measles virus: promises and challenges. Clin. Pharmacol. Ther. 2010;88:620–625. doi: 10.1038/clpt.2010.211. [DOI] [PubMed] [Google Scholar]
- 33.Zheng M., Huang J., Tong A., Yang H. Oncolytic Viruses for Cancer Therapy: Barriers and Recent Advances. Mol. Ther. Oncolytics. 2019;15:234–247. doi: 10.1016/j.omto.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng K.W., Dogan A., Vrana J., Liu C., Ong H.T., Kumar S., Dispenzieri A., Dietz A.B., Russell S.J. Tumor-associated macrophages infiltrate plasmacytomas and can serve as cell carriers for oncolytic measles virotherapy of disseminated myeloma. Am. J. Hematol. 2009;84:401–407. doi: 10.1002/ajh.21444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mader E.K., Butler G., Dowdy S.C., Mariani A., Knutson K.L., Federspiel M.J., Russell S.J., Galanis E., Dietz A.B., Peng K.W. Optimizing patient derived mesenchymal stem cells as virus carriers for a phase I clinical trial in ovarian cancer. J. Transl. Med. 2013;11:20. doi: 10.1186/1479-5876-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ong H.T., Hasegawa K., Dietz A.B., Russell S.J., Peng K.W. Evaluation of T cells as carriers for systemic measles virotherapy in the presence of antiviral antibodies. Gene Ther. 2007;14:324–333. doi: 10.1038/sj.gt.3302880. [DOI] [PubMed] [Google Scholar]
- 37.Matsuda T., Karube H., Aruga A. A Comparative Safety Profile Assessment of Oncolytic Virus Therapy Based on Clinical Trials. Ther. Innov. Regul. Sci. 2018;52:430–437. doi: 10.1177/2168479017738979. [DOI] [PubMed] [Google Scholar]
- 38.Mader E.K., Maeyama Y., Lin Y., Butler G.W., Russell H.M., Galanis E., Russell S.J., Dietz A.D., Peng K.-W. Mesenchymal stem cell carriers protect oncolytic measles viruses from antibody neutralization in an orthotopic ovarian cancer therapy model. Clin. Cancer Res. 2009;15:7246–7255. doi: 10.1158/1078-0432.CCR-09-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahmed A.U., Tyler M.A., Thaci B., Alexiades N.G., Han Y., Ulasov I.V., Lesniak M.S. A comparative study of neural and mesenchymal stem cell-based carriers for oncolytic adenovirus in a model of malignant glioma. Mol. Pharm. 2011;8:1559–1572. doi: 10.1021/mp200161f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shmeleva E.V., Smith G.L., Ferguson B.J. Enhanced Efficacy of Vaccination With Vaccinia Virus in Old vs. Young Mice. Front. Immunol. 2019;10:1780. doi: 10.3389/fimmu.2019.01780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grossardt C., Engeland C.E., Bossow S., Halama N., Zaoui K., Leber M.F., Springfeld C., Jaeger D., von Kalle C., Ungerechts G. Granulocyte-macrophage colony-stimulating factor-armed oncolytic measles virus is an effective therapeutic cancer vaccine. Hum. Gene Ther. 2013;24:644–654. doi: 10.1089/hum.2012.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peters D., Freund J., Ochs R.L. Genome-wide transcriptional analysis of carboplatin response in chemosensitive and chemoresistant ovarian cancer cells. Mol. Cancer Ther. 2005;4:1605–1616. doi: 10.1158/1535-7163.MCT-04-0311. [DOI] [PubMed] [Google Scholar]
- 44.Kendall S.E., Najbauer J., Johnston H.F., Metz M.Z., Li S., Bowers M., Garcia E., Kim S.U., Barish M.E., Aboody K.S., Glackin C.A. Neural stem cell targeting of glioma is dependent on phosphoinositide 3-kinase signaling. Stem Cells. 2008;26:1575–1586. doi: 10.1634/stemcells.2007-0887. [DOI] [PubMed] [Google Scholar]
- 45.Najbauer J., Huszthy P.C., Barish M.E., Garcia E., Metz M.Z., Myers S.M., Gutova M., Frank R.T., Miletic H., Kendall S.E. Cellular host responses to gliomas. PLoS ONE. 2012;7:e35150. doi: 10.1371/journal.pone.0035150. [DOI] [PMC free article] [PubMed] [Google Scholar]