

Abstract
Although CCCTC binding factor (CTCF) has been demonstrated to play a variety of often contradictory roles in tumorigenesis, little is known about its function in the tumorigenesis of ocular melanoma. Here, we generated two artificial CTCF peptides (Decoy-CTCFs) combining the zinc finger domain of wild-type CTCF and artificial marker region. This Decoy-CTCF retained the DNA binding region but lost the functional regions of wild-type CTCF. Transferring artificial CTCF into ocular melanoma cells suppressed proliferation and migration in the tumor cells, while no effect was observed in normal cells. Intriguingly, we first showed that decoy-CTCF inhibited tumorigenesis by preventing the histone acetyltransferase EP300 from binding to the promoter of SELL. Thus SELL was a novel oncogene in the tumorigenesis of ocular melanoma. These studies provide efficient decoy CTCF-based therapeutic concept in malignant ocular melanoma and reveal the potential mechanism underlying decoy-based tumor therapy.
Keywords: decoy CTCF, ocular melanoma, therapeutic efficacy
Graphical Abstract
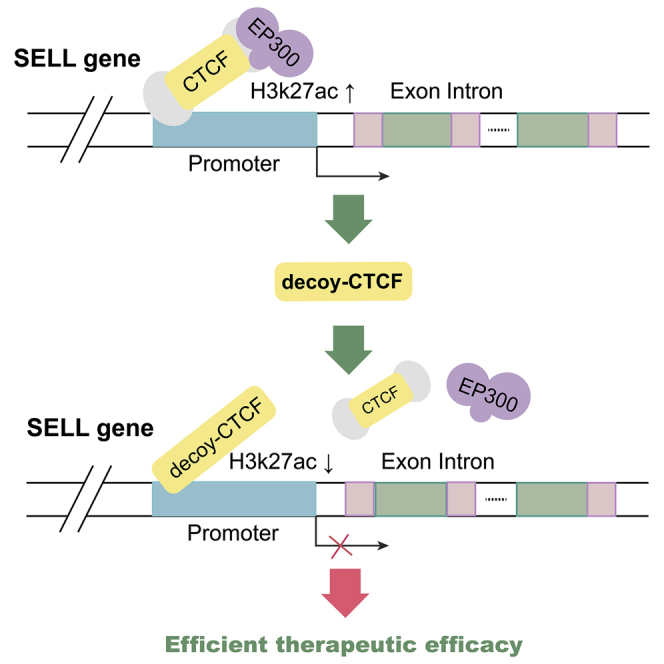
An artificial CTCF peptide (Decoy-CTCFs) prevents the histone acetyltransferase EP300 to interact the promoter of SELL for inactivating SELL expression and triggering efficient therapeutic efficacy in ocular tumors. These studies delineate a novel decoy CTCF-guided mechanism in tumor therapy and provide a promising therapeutic concept for ocular melanoma.
Introduction
CCCTC-binding factor (CTCF) is a special protein that binds to tens of thousands of sites throughout the genome. Initially, it was characterized as a transcription factor capable of activating or repressing gene expression.1,2 Later, CTCF was shown to act as an insulator, indicating that it can block the interaction between promoters and enhancers.3 CTCF was found to be an important architectural protein bridging the genome. In addition to its function as a chromatin barrier, this protein can interact with several other architectural proteins to form topologically associating domains (TADs).4,5 At a more local scale, CTCF can regulate various aspects of gene expression by facilitating or constraining interactions between genes and their regulatory elements.6, 7, 8
Many studies have demonstrated that CTCF is involved in gene transcription, DNA repair, cell mitosis, and other functions.9 The dysfunction of CTCF may cause several diseases and even lead to tumorigenesis. Schroeder et al.10 found that the expression of CTCF is linked to poor prognosis in prostate cancer. The progression of neuroblastoma was found to be stimulated by PARP1-mediated CTCF activation.11 In contrast, LINC00346 was shown to interact with CTCF to disrupt the CTCF-mediated repression of c-Myc and lead to the tumorigenesis of pancreatic cancer.12 The loss of CTCF was demonstrated to play a B-ALL cell line-specific role in maintaining MYC expression.13 These studies indicated that CTCF may play various often contradictory roles in different tumors.
However, the effect of CTCF in ocular melanoma is still being investigated. Ocular melanoma is the most common malignant ocular tumor in adults.14 It grows fast and has a strong propensity toward fatal metastasis.15 Over the past few decades, researchers have studied the mechanism of ocular melanoma, and several gene mutations have been found to be related to its tumorigenesis.16,17 However, these genetic changes are difficult to address. With the development of research techniques, an increasing number of epigenetic changes, such as DNA methylation and changes in chromosome structure, have been found to play an important role in the genesis and development of ocular melanoma.18,19 These findings suggest that the architectural protein CTCF may play an important role in the tumorigenesis of ocular melanoma, which is likely to be reversed by medical treatments.
In this study, we constructed an artificial CTCF peptide (Decoy-CTCF) combining the zinc-finger domain of wild-type CTCF and the CpG methyltransferase Sss1. This Decoy-CTCF retained the DNA binding region but lost the functional regions of wild-type CTCF so that it could disrupt the function of wild-type CTCF but retain its DNA binding capacity. We demonstrated that this Decoy-CTCF can significantly inhibit both the proliferation and migration of ocular melanoma in vitro and in vivo. We also found the novel decoy CTCF-mediated mechanism by inhibiting SELL expression in tumorigenesis.
Results
The Decoy-CTCF Represses Tumor Proliferation and Migration In Vitro
By exploring The Cancer Genome Atlas (TCGA) database, we found that high CTCF levels positively correlated with a poor prognosis in all kinds of tumor patients (Figure 1A). Since CTCF plays an important role in bridging genome topology, simple knockout of CTCF may cause several unknown changes. We generated a Decoy-CTCF (dsCTCF) vector (Figure 1B, middle panel). This dsCTCF has the zinc-finger domain of wild-type CTCF and thus retains the ability to bind target DNA sequences. The deletion of the N-terminal and C-terminal domains abolished the protein recruiting function of dsCTCF. In addition, the Sss1 domain of dsCTCF allows it to methylate CpG islands of some DNA sequences near the region where it binds. Thus, dsCTCF has a similar mass as wild-type CTCF, and its structure may help to prevent the binding of wild-type CTCF due to its DNA methylation sensitivity.20 Meanwhile, we constructed a marked Decoy-CTCF (deCTCF), which has a zinc-finger domain and an EGFP domain (Figure 1B, bottom panel). Then, we used the pCDH-CMV plasmid to construct Decoy-CTCF lentivirus and transfected it into ocular tumor cells and normal cells. After screening for 3 weeks by puromycin, we detected the expression of dsCTCF or deCTCF by qPCR, fluorescence microscopy, and western blot analysis. The results showed that all the transfected cell lines stably expressed deCTCF (Figures 1C and 1D; Figure S1A) or dsCTCF (Figures 1E and 1F; Figures S1B and 1C). We next tested whether the Decoy-CTCF could inhibit tumor proliferation. An in vitro cell proliferation assay was carried out by a cell counting kit and plate clone formation assays. The results showed that the proliferation of the dsCTCF- or deCTCF-transfected ocular melanoma cells was significantly reduced (Figures 1G and 1H; Figure S2A–2C), while no effect was found in the normal cells (Figure 1G). Then, we explored the effect of the Decoy-CTCF on tumor migration. Transwell migration assays and scratch tests showed that dsCTCF or deCTCF could significantly reduce the migration of ocular melanoma (Figures 1I and 1J; Figure S3). These data showed that the Decoy-CTCF could significantly repress tumor proliferation and migration in vitro. However, there were no differences between the dsCTCF- and deCTCF-expressing tumor cells.
Figure 1.

Decoy-CTCF Repress Tumor Proliferation and Migration In Vitro
(A) The TCGA database of overall tumor demonstrated prolonged survival time in patients with low CTCF expression. (B) Schematic diagram of decoy-CTCF. Top panel: the wild-type CTCF with zinc-finger (ZF) domain, N-terminal (NT), and C-terminal (CT) domain. Middle panel: dsCTCF with ZF domain and Sss1 domain. Bottom panel: deCTCF with ZF domain and EGFP domain. (C) Fluorescence microscope showed the deCTCF expressed in both tumor and normal transfected cells. (D and E) qPCR showed the deCTCF (D) and dsCTCF (E) expressed in both tumor and normal transfected cells. (F) Western blot verified that the dsCTCF expressed in both tumor and normal transfected cells. (G) CCK8 assay demonstrated that dsCTCF could significantly reduce the proliferation of transfected ocular melanoma but have no effect on normal cells. (H) Plate clone formation assay verified that dsCTCF or deCTCF could significantly reduce the proliferation of transfected ocular melanoma. (I) Transwell migration assay showed that dsCTCF or deCTCF could significantly reduce the migration ability of ocular melanoma. (J) Scratch test suggested that dsCTCF or deCTCF could significantly reduce the migration ability.
The Decoy-CTCF Represses Tumorigenesis In Vivo
To further investigate the effect of the Decoy-CTCF in tumorigenesis, we carried out a soft agar assay. We observed that fewer and smaller colonies were formed in the Decoy-CTCF-expressing ocular melanoma cells than in the control cells either under a microscope or in whole well testing (Figure 2A; Figure S4). In contrast, the normal cells did not form colonies regardless of Decoy-CTCF expression (data not shown). To examine this phenomenon in vivo, we established a subcutaneous xenograft model in nude mice using the dsCTCF-expressing ocular melanoma cells. We then evaluated the size of the resultant tumors two times a week for 28 days. As expected, the tumor volume of the dsCTCF-expressing groups was significantly reduced compared with that of the empty vector group (Figure 2B). The mice were sacrificed on the 28th day, and the tumor tissues were collected. By determining the tumor weight, we observed that the Decoy-CTCF-expressing group underwent a conspicuous reduction in tumor weight (Figure 2C). Hematoxylin and eosin staining of the tumor tissues showed more necrotic regions in the Decoy-CTCF-expressing group (Figure 2D). Taken together, these results demonstrated that the Decoy-CTCF can repress the tumorigenesis of ocular melanoma cells in vivo.
Figure 2.

Decoy-CTCFs Repress Tumorigenesis In Vitro and In Vivo
(A) Soft agar assay showed fewer and smaller colonies in dsCTCF or deCTCF expressed ocular melanoma cells. (B and C) Subcutaneous xenograft model demonstrated that the tumor volume (B) and weight (C) of dsCTCF expressed groups was significantly reduced. (D) Hematoxylin and eosin stain of tumor tissues showed more necrosis regions in dsCTCF expressed group.
Decoy-CTCF Suppressed Tumor Proliferation and Migration by Inhibiting SELL
To explore the mechanism of decoy CTCF-derived tumor inhibition, we carried out RNA sequencing (RNA-seq) and CTCF chromatin immunoprecipitation (ChIP)-seq assays (Figure S5A). We observed a significant enrichment of the CTCF signal at the promoter of selectin L (SELL) in ocular melanoma cells, while the expressed peak and CTCF signal at the SELL locus disappeared in the Decoy-CTCF transfected cells. To verify this bioinformatics analysis, we examined the expression of SELL. The results showed that SELL was highly expressed in the ocular melanoma cells at both the RNA and protein levels (Figures 3A and 3B; Figure S5B). Next, we explored the role of SELL in ocular melanoma. Real-time PCR and western blot assays demonstrated that SELL expression was significantly decreased after short hairpin RNA (shRNA) interference (Figures 3C and 3D; Figure S5C). The plate clone formation assay showed that the proliferation of the SELL-interfered group was significantly reduced (Figure 3E; Figure S6A). The Transwell migration assay showed that SELL interference could significantly reduce the migration of ocular melanoma (Figure 3F; Figure S6B). Similarly, we observed fewer and smaller colonies in the SELL-interfered groups, indicating that SELL is sufficient for tumorigenesis of ocular melanoma (Figure 3G; Figures S6C and S6D). We then verified this in vitro result by subcutaneous xenograft models. The tumor volume and weight in the SELL-silenced group were significantly reduced (Figures 3H and 3I). Furthermore, by utilizing TCGA database, we investigated the overall survival probability in the SELL high expression group and the low expression group in patients with uveal melanoma. The results showed that a high SELL level positively correlated with a poor prognosis (Figure 3J). These data showed that SELL plays an oncogenic role in the tumorigenesis of ocular tumors.
Figure 3.

Decoy-CTCFs Suppressed Tumor Proliferation and Migration by Inhibiting SELL
(A and B) qPCR (A) and western blot (B) showed that SELL was highly expressed in ocular melanoma cells. (C and D) qPCR (C) and western blot (D) showed the expression of SELL was significantly decreased in ocular tumor cells after shRNA interference. (E) Plate clone formation assay verified that the proliferation of SELL-interfered group was significantly reduced. (F) Transwell migration assay showed that SELL interference could significantly reduce the migration ability of ocular melanoma. (G) Soft agar assay investigated fewer and smaller colonies in SELL-interfered groups. (H and I) Subcutaneous xenograft model demonstrated that the tumor volume (H) and weight (I) of SELL-interfered group was significantly reduced. (J) The TCGA database of uveal melanoma demonstrated prolonged survival time in patients with low SELL expression.
Decoy CTCF Repeals the Histone Acetyltransferase EP300 at the SELL Promoter
Next, we explored how decoy CTCF can control the expression of SELL. Through a STRING protein interaction (https://string-db.org/cgi/input.pl) network, we determined that CTCF could bind to several transcription factors, such as EZH2 and EP300 (Figure S7). Since H3K27me3 mediated by EZH2 is related to gene repression, we focused on EP300, which could lead to gene activation by H3K27ac modification. To validate our hypothesis, we first verified the binding of CTCF at the SELL promoter region. Two kinds of CTCF protein antibodies were used; one can recognize the N terminus of CTCF and the other can recognize the zinc finger region. Thus, we could distinguish wild-type CTCF and Decoy-CTCF (Figure 4A). As expected, CTCF could interact with the promoter of SELL in the ocular melanoma cells. In addition, Decoy-CTCF transfection prevented the interaction with wild-type CTCF (Figures 4B and 4C). Then, we verified whether CTCF recruited EP300. The results showed that EP300 was recruited to the promoter of SELL and led to H3K27ac modification at this region (Figures 4D and 4E). The transfection of the Decoy-CTCF abolished this recruitment and reduced the level of H3K27ac modification (Figures 4D and 4E). Finally, coimmunoprecipitation (coIP) assays were carried out to examine the interaction between CTCF and EP300. As expected, the wild-type CTCF could be pulled down by the EP300 protein, and EP300 could also be pulled down by the CTCF protein, while the Decoy-CTCF was not found to interact with Ep300 (Figure 4G). These data indicate that decoy CTCF could repeal EP300 off the promoter of SELL and inhibited SELL expression by H3K27ac modification, thereby highlight the regulatory role of histone acetylation in maintaining SELL activation.
Figure 4.

Decoy CTCF Repeals the Histone Acetyltransferase EP300 at the SELL Promoter
(A) Schematic diagram of decoy-CTCF ChIP assay. Two kinds of CTCF protein antibody were used; one can recognize the N-terminal of CTCF (blue) and the other one can recognize the zinc-finger region (red). And the testing site of ChIP assay was shown on the bottom. (B and C) Semi-quantitative (B) and quantitative (C) ChIP assay demonstrated that CTCF could combine to the promoter of SELL in ocular melanoma cells. And decoy-CTCF transfection could reject this combination of wild-type CTCF. (D and E) Semi-quantitative (D) and quantitative (E) ChIP assay showed that EP300 was recruited to the promoter of SELL and lead to the H3K27ac modification at this region. Decoy-CTCF could abolish this recruitment and let down the H3K27ac modification level. (F) Coimmunoprecipitation (coIP) assays suggested that the wild-type CTCF could be pulled out by baiting the EP300 protein (panels 1–3), and EP300 could also be pulled out by baiting the CTCF protein (panels 4–6). Decoy-CTCF could not be detected to interact with Ep300 (panels 7–8).
Discussion
The genesis and development of malignant tumors are due to abnormalities in gene expression derived from both genetic and epigenetic lesions.21,22 In recent decades, researchers have concentrated on studying genetic lesions and have identified many gene targets that induce the development and metastasis of tumors.23, 24, 25 Recently, increasing epigenetic lesions have been found to play an important role in tumorigenesis.26, 27, 28, 29 Abnormal chromosome conformation was especially recognized as a key factor in the tumorigenesis of several tumors.30, 31, 32
As a special transcription factor, CTCF has three structural domains: the N-terminal, C-terminal, and zinc-finger domains. CTCF functions as an architectural protein that contributes to the establishment of genome topology. At a more local scale, CTCF can mediate both short- and long-range interactions between promoters and enhancers/repressors by colocalizing with cohesin.33 In this study, we generated a novel Decoy-CTCF lacking the functional N-terminal and C-terminal domains that maintained the DNA binding domain zinc finger.20 Owing to this unique structure, the Decoy-CTCF could competitively bind to CTCF binding sites in the genome sequence and may help to reverse the effects of the abnormal wild-type CTCF by partly reducing its function. In addition, since CTCF was proven to be a DNA methylation-sensitive protein, a Sss1 domain was added to the Decoy-CTCF to construct a competitiveness-enhanced subtype (dsCTCF). An EGFP domain was added to the Decoy-CTCF to construct a marked subtype (deCTCF).
Notably, this Decoy-CTCF could significantly repress tumor proliferation and migration in ocular melanoma. This protein could also repress tumorigenesis in vivo. However, little difference was found between the two subtypes dsCTCF and deCTCF. We suppose the reason for this phenomenon is that the Decoy-CTCF has the same DNA binding domain as the wild-type CTCF, indicating that the Sss1 domain in dsCTCF may not help to enhance the blockade of wild-type CTCF. This structure may help dsCTCF maintain a similar molecular size as wild-type CTCF. This assumption was confirmed by genome-wide methylation microarrays comparing the wild-type and dsCTCF-transfected ocular melanoma cells (OCM1, OCM1a). The results showed that dsCTCF only led to a slight change in the methylation levels near its binding sites (Figure S8A), indicating that the repression of tumorigenesis caused by the Decoy-CTCF might not be caused directly by DNA methylation (Figure S8B).
Notably, selectin L (SELL) encodes a cell surface adhesion molecule that has been demonstrated to be related to cell adhesion and tethering or rolling.34,35 Many studies have reported that increased selectin ligand expression on tumor cells correlates with enhanced metastasis and poor prognosis for cancer patients.36 However, the mechanism for this pathology is still unclear. In this study, we revealed for the first time that SELL acts as an oncogene in the tumorigenesis of ocular melanoma. We also demonstrated for the first time that the CTCF-mediated H3K27ac contributes to high SELL expression in ocular melanoma. CTCF can bind to the promoter of SELL and recruit the histone acetyltransferase EP300, which increases H3K27ac at the SELL promoter and leads to the high expression of SELL.
It is important and a challenge to commercially produce dCTCF protein by using in vitro synthetic peptide systems for use in the pharmaceutical industry, and another advantage is that the synthetic peptide could direct inject into tumor by eye micromanipulation for avoiding risk of intravenous or intra-arterial injection. Although our artificial CTCF peptide obtains an efficient therapeutic efficacy in vitro and in vivo, it might take a long time to translate this pre-clinic study into real clinic.
In summary, our results provide a novel therapeutic strategy in which the artificial Decoy-CTCF could significantly repress tumorigenesis both in vitro and in vivo. This Decoy-CTCF-mediated tumor suppression is achieved by competitively inhibiting wild-type CTCF recruitment of the histone acetyltransferase EP300 to activate the oncogene SELL. Most importantly, since the increased selectin ligand expression on tumor cells has been demonstrated to correlate with poor prognosis for cancer patients, this new treatment for ocular melanoma may show promise for curing and studying the pathogenesis of other tumors.
Materials and Methods
Cell Culture
The human normal cell line HDF (human fibroblast cell line) and PIG1 (human melanocyte cell line) were cultured in DMEM (GIBCO) supplemented with 10% certified heat-inactivated fetal bovine serum (FBS, GIBCO), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37°C in a humidified 5% CO2 atmosphere. The ocular melanoma cell line OCM1 and OCM1A were also cultured in DMEM (GIBCO) as described above.
Lentivirus Package
The 293T cells were cultured in Dulbecco’s modified Eagle’s medium (GIBCO, USA) supplemented with 10% (vol/vol) FBS, maintained at 37°C at a concentration of 5,000,000 cells and transfected using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) with 3 μg of the Decoy-CTCF plasmid and 3 μg of pMD2.D and 6.0 μg of PsPax. After incubation overnight with 293T cells, the medium was replaced with 10 mL of fresh medium. The virus-containing supernatants were collected at 48 h and 72 h after transfection and then mixed and filtered through a 0.45-μm cellulose acetate filter (Sartorius). The viral supernatants were concentrated with Amicon Ultra-15 centrifugal filter units (Millipore) at 4°C and at 4,000 rpm for 30 min. Selection was performed by incubating with 4 μg/mL puromycin for 2 weeks.
RNA Isolation and cDNA Synthesis
Total RNA was extracted from the cultured cells using TRIzol reagent (Sigma-Aldrich). The purified RNA was quantified using a Nanodrop 2000 UV-Vis Spectrophotometer (Thermo Scientific). The same amounts of total RNA were reverse transcribed into cDNA using the PrimeScript RT Reagent Kit (TaKaRa Biotechnology).
Quantitative Real-Time RT-PCR
We validated the expression levels of highly methylated genes in OCM1, OCM1a, OCM1-decoy, OCM1a-decoy, and HDF by quantitative real-time RT-PCR. The expression levels were validated by real-time PCR using an ABI Prism 7500 system (Applied Biosystems) for 40 cycles (95°C for 15 s and 60°C for 30 s) after an initial 10 min incubation at 95°C, using Power SYBR Green PCR Master Mix (Applied Biosystems). All of the genes were normalized to the control gene glyceraldehyde phosphate dehydrogenase (GAPDH).
Cell Proliferation Assay
The cell proliferation assay (cell counting kit-8 [CCK8] assay) was performed as previously described.37 In short, cells were seeded at 5,000 cells per well in flat-bottomed 96-well plates. At the end of the incubation time, 10 μL of CCK8 solution (Dojindo, Japan) was added to each well. The samples were incubated for 4 h, and then the absorbance was measured at 450 nm in a microplate reader (Varioskan Flash; Thermo, USA) for 3 consecutive days.
Transwell Assay
The migratory ability of the cells was evaluated using a 24-well Transwell system (Corning, USA) equipped with 8-μm pore size polycarbonate filters according to the manufacturer’s instructions. The upper compartment contained 10,000 cells suspended in appropriate medium supplemented with 5% FBS, and the lower compartment contained 10% FBS. After the appropriate incubation time at 37°C, the lower compartment was fixed with 100% methanol and stained with 0.1% crystal violet before photographing. The crystal violet was washed from the migrated cells using 100 μL of 33% acetic acid. The absorbance of the washed liquid was determined with a microplate reader at 490 nm.
Scratch Assays
Cells were seeded at 200,000 cells per well in 6-well plates. For scratch wound assays, freshly confluent monolayers of cells were scraped off manually using a 200 μL sterile pipette tip. The cell culture medium was then replaced with fresh medium without FBS. The wound was monitored every 24 h under a phase contrast microscope at 200× magnification. The experiments were repeated three times.
Soft Agar Tumor Formation Assay
A soft agar colony formation assay was performed in 6-well plates. One milliliter of the bottom layer comprising 0.6% agar in complete medium was spread in each 6-well plate. A total of 20,000 cells were suspended in 1.0 mL of complete medium containing 0.3% agar and seeded into each well. The cultures were fed every 3 to 4 days with 300 μL of complete medium for 3–4 weeks. For quantification, the colonies grown in soft agar were stained with 0.005% crystal violet. The size of the colonies was determined using Adobe Photoshop.
Genome-wide DNA Methylation Array
Total DNA was prepared from OCM1, OCM1-decoy, OCM1a, and OCM1a-decoy cells using a QIAamp DNA Mini Kit (QIAGEN). Genome-wide DNA methylation was analyzed using the Infinium HD Methylation 450K assay (Oe Biotech). The methylation levels were compared between the tumor and tumor-decoy groups. Then, genes with positive DiffScores were analyzed to determine which were the same.
Animal Experiments
All procedures were approved by the Animal Research Committee of the Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine. Six nude mice were subcutaneously injected to transplant tumors in each group. The nude mice were able to function normally after this procedure. The transplanted tumors were measured twice a week for length and width.
Author Contributions
In this report, X.W., H.W., P.C., and J.F. designed and performed the experiments and drafted the manuscript; X.Z. and T.D. collected and analyzed the data; R.J. and S.G. discussed the manuscript; H.Z. and X.F. revised and approved the manuscript. All the authors approved this manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Key Research and Development Plan (2017YFE0196300); the National Natural Science Foundation of China (grants 31870748 and 81802739); the ShuGuang Project of Shanghai Municipal Education Commission and Shanghai Education Development Foundation (17SG19); the Outstanding Yong Medical Scholar of Shanghai Municipal Commission of Health and Family Planning (2017YQ067); the Shanghai Science and Technology Development Funds (19QA1405100); the Outstanding Yong Scholar Grant of Tongji University (PA2019000239); and the Startup Funding of Frontier Science Research Center for Stem Cells & Shanghai East Hospital of Tongji University.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.07.004.
Contributor Information
He Zhang, Email: zhanghe@tongji.edu.cn.
Xianqun Fan, Email: fanxq@sjtu.edu.cn.
Supplemental Information
References
- 1.Baniahmad A., Steiner C., Köhne A.C., Renkawitz R. Modular structure of a chicken lysozyme silencer: involvement of an unusual thyroid hormone receptor binding site. Cell. 1990;61:505–514. doi: 10.1016/0092-8674(90)90532-j. [DOI] [PubMed] [Google Scholar]
- 2.Lobanenkov V.V., Nicolas R.H., Adler V.V., Paterson H., Klenova E.M., Polotskaja A.V., Goodwin G.H. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene. 1990;5:1743–1753. [PubMed] [Google Scholar]
- 3.Bell A.C., West A.G., Felsenfeld G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell. 1999;98:387–396. doi: 10.1016/s0092-8674(00)81967-4. [DOI] [PubMed] [Google Scholar]
- 4.Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flavahan W.A., Drier Y., Johnstone S.E., Hemming M.L., Tarjan D.R., Hegazi E., Shareef S.J., Javed N.M., Raut C.P., Eschle B.K. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature. 2019;575:229–233. doi: 10.1038/s41586-019-1668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bossen C., Mansson R., Murre C. Chromatin topology and the regulation of antigen receptor assembly. Annu. Rev. Immunol. 2012;30:337–356. doi: 10.1146/annurev-immunol-020711-075003. [DOI] [PubMed] [Google Scholar]
- 7.Zhang H., Jiao W., Sun L., Fan J., Chen M., Wang H., Xu X., Shen A., Li T., Niu B. Intrachromosomal looping is required for activation of endogenous pluripotency genes during reprogramming. Cell Stem Cell. 2013;13:30–35. doi: 10.1016/j.stem.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 8.Merkenschlager M., Odom D.T. CTCF and cohesin: linking gene regulatory elements with their targets. Cell. 2013;152:1285–1297. doi: 10.1016/j.cell.2013.02.029. [DOI] [PubMed] [Google Scholar]
- 9.Ong C.T., Corces V.G. CTCF: an architectural protein bridging genome topology and function. Nat. Rev. Genet. 2014;15:234–246. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoflmayer D., Steinhoff A., Hube-Magg C., Kluth M., Simon R., Burandt E. Expression of CCCTC-binding factor (CTCF) is linked to poor prognosis in prostate cancer. Mol. Oncol. 2019;14:129–138. doi: 10.1002/1878-0261.12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao X., Li D., Huang D., Song H., Mei H., Fang E., Wang X., Yang F., Zheng L., Huang K., Tong Q. Risk-Associated Long Noncoding RNA FOXD3-AS1 Inhibits Neuroblastoma Progression by Repressing PARP1-Mediated Activation of CTCF. Mol. Ther. 2018;26:755–773. doi: 10.1016/j.ymthe.2017.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Peng W.X., He R.Z., Zhang Z., Yang L., Mo Y.Y. LINC00346 promotes pancreatic cancer progression through the CTCF-mediated Myc transcription. Oncogene. 2019;38:6770–6780. doi: 10.1038/s41388-019-0918-z. [DOI] [PubMed] [Google Scholar]
- 13.Hyle J., Zhang Y., Wright S., Xu B., Shao Y., Easton J., Tian L., Feng R., Xu P., Li C. Acute depletion of CTCF directly affects MYC regulation through loss of enhancer-promoter looping. Nucleic Acids Res. 2019;47:6699–6713. doi: 10.1093/nar/gkz462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mariani P., Servois V., Piperno-Neumann S. Therapeutic options in metastatic uveal melanoma. Dev. Ophthalmol. 2012;49:166–181. doi: 10.1159/000328333. [DOI] [PubMed] [Google Scholar]
- 15.Singh A.D., Turell M.E., Topham A.K. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology. 2011;118:1881–1885. doi: 10.1016/j.ophtha.2011.01.040. [DOI] [PubMed] [Google Scholar]
- 16.Hou C., Xiao L., Ren X., Tang F., Guo B., Zeng W., Liang C., Yan N. Mutations of GNAQ, GNA11, SF3B1, EIF1AX, PLCB4 and CYSLTR in Uveal Melanoma in Chinese Patients. Ophthalmic Res. 2019;63:358–368. doi: 10.1159/000502888. [DOI] [PubMed] [Google Scholar]
- 17.Smit K.N., Jager M.J., de Klein A., Kiliҫ E. Uveal melanoma: Towards a molecular understanding. Prog. Retin. Eye Res. 2020;75:100800. doi: 10.1016/j.preteyeres.2019.100800. [DOI] [PubMed] [Google Scholar]
- 18.Fan J., Xing Y., Wen X., Jia R., Ni H., He J., Ding X., Pan H., Qian G., Ge S. Long non-coding RNA ROR decoys gene-specific histone methylation to promote tumorigenesis. Genome Biol. 2015;16:139. doi: 10.1186/s13059-015-0705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan J., Xu Y., Wen X., Ge S., Jia R., Zhang H., Fan X. A Cohesin-Mediated Intrachromosomal Loop Drives Oncogenic ROR lncRNA to Accelerate Tumorigenesis. Mol. Ther. 2019;27:2182–2194. doi: 10.1016/j.ymthe.2019.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H., Niu B., Hu J.F., Ge S., Wang H., Li T., Ling J., Steelman B.N., Qian G., Hoffman A.R. Interruption of intrachromosomal looping by CCCTC binding factor decoy proteins abrogates genomic imprinting of human insulin-like growth factor II. J. Cell Biol. 2011;193:475–487. doi: 10.1083/jcb.201101021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez L.A., Naguibneva I., Lehrmann H., Vervisch A., Tchénio T., Lozano G., Harel-Bellan A. Synthetic small inhibiting RNAs: efficient tools to inactivate oncogenic mutations and restore p53 pathways. Proc. Natl. Acad. Sci. USA. 2002;99:14849–14854. doi: 10.1073/pnas.222406899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reed J.C. Apoptosis-based therapies. Nat. Rev. Drug Discov. 2002;1:111–121. doi: 10.1038/nrd726. [DOI] [PubMed] [Google Scholar]
- 23.Eldholm V., Haugen A., Zienolddiny S. CTCF mediates the TERT enhancer-promoter interactions in lung cancer cells: identification of a novel enhancer region involved in the regulation of TERT gene. Int. J. Cancer. 2014;134:2305–2313. doi: 10.1002/ijc.28570. [DOI] [PubMed] [Google Scholar]
- 24.Toki H., Inoue M., Minowa O., Motegi H., Saiki Y., Wakana S., Masuya H., Gondo Y., Shiroishi T., Yao R., Noda T. Novel retinoblastoma mutation abrogating the interaction to E2F2/3, but not E2F1, led to selective suppression of thyroid tumors. Cancer Sci. 2014;105:1360–1368. doi: 10.1111/cas.12495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodrigues M., Mobuchon L., Houy A., Fiévet A., Gardrat S., Barnhill R.L., Popova T., Servois V., Rampanou A., Mouton A. Outlier response to anti-PD1 in uveal melanoma reveals germline MBD4 mutations in hypermutated tumors. Nat. Commun. 2018;9:1866. doi: 10.1038/s41467-018-04322-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J., Benavente C.A., McEvoy J., Flores-Otero J., Ding L., Chen X., Ulyanov A., Wu G., Wilson M., Wang J. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012;481:329–334. doi: 10.1038/nature10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kagey M.H., Newman J.J., Bilodeau S., Zhan Y., Orlando D.A., van Berkum N.L., Ebmeier C.C., Goossens J., Rahl P.B., Levine S.S. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suvà M.L., Riggi N., Bernstein B.E. Epigenetic reprogramming in cancer. Science. 2013;339:1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanber D., Berulava T., Ammerpohl O., Mitter D., Richter J., Siebert R., Horsthemke B., Lohmann D., Buiting K. The human retinoblastoma gene is imprinted. PLoS Genet. 2009;5:e1000790. doi: 10.1371/journal.pgen.1000790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kandoth C., Schultz N., Cherniack A.D., Akbani R., Liu Y., Shen H., Robertson A.G., Pashtan I., Shen R., Benz C.C., Cancer Genome Atlas Research Network Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denholtz M., Bonora G., Chronis C., Splinter E., de Laat W., Ernst J., Pellegrini M., Plath K. Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell. 2013;13:602–616. doi: 10.1016/j.stem.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wen X., Lu L., He Z., Fan X. Orchestrating epigenetic roles targeting ocular tumors. OncoTargets Ther. 2016;9:1001–1009. doi: 10.2147/OTT.S93995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Splinter E., Heath H., Kooren J., Palstra R.J., Klous P., Grosveld F., Galjart N., de Laat W. CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev. 2006;20:2349–2354. doi: 10.1101/gad.399506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dwir O., Kansas G.S., Alon R. An activated L-selectin mutant with conserved equilibrium binding properties but enhanced ligand recognition under shear flow. J. Biol. Chem. 2000;275:18682–18691. doi: 10.1074/jbc.M001103200. [DOI] [PubMed] [Google Scholar]
- 35.Mehta-D’souza P., Klopocki A.G., Oganesyan V., Terzyan S., Mather T., Li Z., Panicker S.R., Zhu C., McEver R.P. Glycan Bound to the Selectin Low Affinity State Engages Glu-88 to Stabilize the High Affinity State under Force. J. Biol. Chem. 2017;292:2510–2518. doi: 10.1074/jbc.M116.767186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borsig L. Selectins in cancer immunity. Glycobiology. 2018;28:648–655. doi: 10.1093/glycob/cwx105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y., Xu Z., Jiang J., Xu C., Kang J., Xiao L., Wu M., Xiong J., Guo X., Liu H. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev. Cell. 2013;25:69–80. doi: 10.1016/j.devcel.2013.03.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.