

Coronaviruses (CoVs) include a wide range of important human and animal pathogens. Examples of human CoVs include severe acute respiratory syndrome coronavirus (SARS-CoV-1), Middle East respiratory syndrome coronavirus (MERS-CoV), and the most recently emerged SARS-CoV-2. Examples of pig CoVs include porcine epidemic diarrhea virus (PEDV), porcine deltacoronavirus (PDCoV), and swine enteric alphacoronavirus (SeACoV). There are no vaccines or antiviral drugs for most of these viruses. All known CoVs encode a bifunctional nsp14 protein which possesses ExoN and guanine-N-7 methyltransferase (G-N-7 MTase) activities, responsible for replication fidelity and RNA cap G-N-7 methylation, respectively. Here, we biochemically characterized G-N-7 MTase of PEDV nsp14 and found that G-N-7 MTase-deficient PEDV was defective in replication and induced greater responses of type I and III interferons. These findings highlight that CoV G-N-7 MTase may be a novel target for rational design of live attenuated vaccines and antiviral drugs.
KEYWORDS: Porcine epidemic diarrhea virus, RNA methylation, coronavirus
ABSTRACT
The 5′ cap methylation of viral RNA plays important roles in RNA stability, efficient translation, and immune evasion. Thus, RNA cap methylation is an attractive target for antiviral discovery and development of new live attenuated vaccines. For coronaviruses, RNA cap structure is first methylated at the guanine-N-7 (G-N-7) position by nonstructural protein 14 (nsp14), which facilitates and precedes the subsequent ribose 2′-O methylation by the nsp16-nsp10 complex. Using porcine epidemic diarrhea virus (PEDV), an Alphacoronavirus, as a model, we showed that G-N-7 methyltransferase (G-N-7 MTase) of PEDV nsp14 methylated RNA substrates in a sequence-unspecific manner. PEDV nsp14 can efficiently methylate RNA substrates with various lengths in both neutral and alkaline pH environments and can methylate cap analogs (GpppA and GpppG) and single-nucleotide GTP but not ATP, CTP, or UTP. Mutations to the S-adenosyl-l-methionine (SAM) binding motif in the nsp14 abolished the G-N-7 MTase activity and were lethal to PEDV. However, recombinant rPEDV-D350A with a single mutation (D350A) in nsp14, which retained 29.0% of G-N-7 MTase activity, was viable. Recombinant rPEDV-D350A formed a significantly smaller plaque and had significant defects in viral protein synthesis and viral replication in Vero CCL-81 cells and intestinal porcine epithelial cells (IPEC-DQ). Notably, rPEDV-D350A induced significantly higher expression of both type I and III interferons in IPEC-DQ cells than the parental rPEDV. Collectively, our results demonstrate that G-N-7 MTase activity of PEDV modulates viral replication, gene expression, and innate immune responses.
IMPORTANCE Coronaviruses (CoVs) include a wide range of important human and animal pathogens. Examples of human CoVs include severe acute respiratory syndrome coronavirus (SARS-CoV-1), Middle East respiratory syndrome coronavirus (MERS-CoV), and the most recently emerged SARS-CoV-2. Examples of pig CoVs include porcine epidemic diarrhea virus (PEDV), porcine deltacoronavirus (PDCoV), and swine enteric alphacoronavirus (SeACoV). There are no vaccines or antiviral drugs for most of these viruses. All known CoVs encode a bifunctional nsp14 protein which possesses ExoN and guanine-N-7 methyltransferase (G-N-7 MTase) activities, responsible for replication fidelity and RNA cap G-N-7 methylation, respectively. Here, we biochemically characterized G-N-7 MTase of PEDV nsp14 and found that G-N-7 MTase-deficient PEDV was defective in replication and induced greater responses of type I and III interferons. These findings highlight that CoV G-N-7 MTase may be a novel target for rational design of live attenuated vaccines and antiviral drugs.
INTRODUCTION
The 5′ cap structure of eukaryotic mRNAs is essential for mRNA stability and efficient translation initiation (1, 2). The 5′ cap of cellular mRNAs is formed through a series of enzymatic steps: first, the 5′-triphosphate end (pppN-) of the nascent transcript is processed into a diphosphate by an RNA triphosphatase (RTPase); second, a GMP moiety from GTP is transferred to the diphosphate mRNA by an RNA guanylyltransferase (GTase) to generate the cap core structure (GpppN-); lastly, the guanosine cap is methylated at the guanine-N-7 position by S-adenosyl-l-methionine (SAM)-dependent cap guanine-N-7 methyltransferase (G-N-7 MTase) to generate a cap 0 structure (m7GpppN-) in the presence of the methyl group donor SAM. In higher eukaryotes and some viruses, the cap 0 structure is further methylated at the 2′-OH position of the ribose by cap ribose-2′-O-MTase to form the cap 1 structure (m7GpppNm-) and type 2 cap (m7GpppNmNm-) (3, 4). These mRNA capping and methylation reactions are conserved among all eukaryotes.
Many viruses have evolved their own mRNA cap-forming machinery and utilize different mechanisms. It has been known for many years that the genomic and subgenomic RNAs of coronaviruses (CoVs) carry a 5′ cap structure (5–7). However, the mechanism and the enzymes involved in CoV RNA cap formation remained unclear until the outbreak of the severe acute respiratory syndrome coronavirus (SARS-CoV-1) in 2003 and the subsequent biochemical characterization of SARS-CoV cap-forming machinery (8–15). Our current understanding of RNA cap formation of CoVs is summarized as follows. In response to a cis element in viral genome RNA, the virally encoded RNA-dependent RNA polymerase (RdRP) synthesizes nascent genome and subgenome RNA, which is hydrolyzed by nsp13, an RTPase, to yield ppN-RNA (16). Subsequently, the ppN-RNA is capped by an unknown CoV capping enzyme to form GpppN-RNA. The cap structure can then be further methylated at G-N-7 position by CoV nsp14, a bifunctional protein, which includes an N-terminal exoribonuclease activity domain (ExoN) (17) and a C-terminal G-N-7 MTase domain (8). Finally, CoV nsp16 together with its stimulatory factor nsp10 forms a complex acting as the 2′-O-MTase to raise cap 0 to cap 1 structure (9, 11, 12). Recently, crystal structures of several cap-forming enzymes in SARS-CoV-1, including nsp14, nsp16, and the nsp16-nsp10 complex, have been determined, and their catalytic sites involved in the enzymatic activities have been mapped to the amino acid level (11–13, 15).
The family Coronaviridae includes many important human and animal pathogens, which can be classified into Coronavirinae and Torovirinae subfamilies. The Coronavirinae subfamily can be further subdivided into four genera, Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus. The genus Alphacoronavirus includes several economically important pig CoVs such as porcine epidemic diarrhea virus (PEDV), transmissible gastroenteritis virus (TGEV), and swine enteric alphacoronavirus (SeACoV) (18, 19). The genus Betacoronavirus includes many important human pathogens such as SARS-CoV-1, Middle East respiratory syndrome coronavirus (MERS-CoV), and the 2019 newly emerged SARS-CoV-2 (20, 21). An example of a Gammacoronavirus is avian infectious bronchitis virus (IBV). The genus Deltacoronavirus includes porcine deltacoronavirus (PDCoV) and avian deltacoronavirus (22). Despite major efforts, most of these CoVs do not have effective vaccines or antiviral drugs. PEDV is the causative agent of porcine epidemic diarrhea (PED), an acute and highly contagious enteric disease characterized by vomiting, diarrhea, and dehydration, leading to high mortality in newborn piglets. PEDV was discovered in the United Kingdom in 1971 and subsequently reported in many swine-producing countries of Europe and Asia (23, 24). Since 2010, massive PED outbreaks have been reported in China, with high mortality rates in suckling piglets (25, 26). Quickly, the new emerging PED outbreaks spread worldwide, and cases were reported in Vietnam, Thailand, South Korea (27), Japan (28), the United States (29), Mexico, and Canada (28). Globally, PEDV causes huge economic losses to the pork industry every year.
RNA cap methylation is an attractive target for antiviral discovery and development of new live attenuated vaccines. Although RNA cap methylation in SARS-CoV-1, a member of genus Betacoronavirus, is relatively well studied, whether the mechanism involved in cap formation is conserved in other CoV members of this genus remains largely unknown. In addition, the roles of RNA cap methylation in CoV replication, gene expression, and pathogenesis remain poorly understood. In this study, we characterized RNA cap G-N-7 MTase activity of PEDV nsp14 and determined its roles in viral replication, gene expression, and innate immune response. We found that PEDV nsp14 methylated the viral RNA at the G-N-7 position in a sequence-independent manner. Interestingly, PEDV nsp14 can utilize single or dinucleotide cap analogs as the substrates. Subsequently, we established an infectious cDNA clone of PEDV and recovered a recombinant PEDV carrying a D350A mutation in nsp14 (rPEDV-D350A). The rPEDV-D350A is deficient in RNA cap G-N-7 methylation, shows defects in replication and gene expression in cell culture, and induces significantly higher type I and type III interferon responses than the parental rPEDV. Our results demonstrate that the G-N-7 MTase activity of nsp14 plays an important role in PEDV replication, gene expression, and innate immune response.
RESULTS
PEDV nsp14 methylates RNA substrate at the G-N-7 position in a wide range of pHs.
To begin to examine the G-N-7 MTase activity of PEDV nsp14, we first generated highly purified 6×His-tagged recombinant nsp14 protein from an Escherichia coli expression system (Fig. 1A). A 43-nucleotide (nt) triphosphated RNA (5′-pppA-RNA43) representing the 5′-terminal 43 nucleotides of the PEDV genome was synthesized by T7 RNA polymerase, capped by the ScriptCap m7G capping system in the presence of [α-32P]GTP (the GMP donor) with or without SAM (the methyl donor), which yielded an α-32P-labeled G-N-7 unmethylated RNA (Gp*ppA-RNA43) or methylated RNA (m7Gp*ppA-RNA43) (* indicates [α-32P] label). The unmethylated GpppA-RNA43 was used as the RNA substrate for the G-N-7 methylation assay and also served as a negative control. The G-N-7 methylated m7Gp*ppA-RNA43 was used as a positive control. For the G-N-7 methylation assay, 50 ng of Gp*ppA-RNA43 was incubated with 0.5 μg of nsp14 in the presence of the methyl donor SAM for 3 h. RNA was purified and subjected to nuclease P1 digestion followed by thin-layer chromatography (TLC) on polyethyleneimine (PEI) cellulose F sheets. Nuclease P1 cleaves the bond between the 3′-hydroxyl and 5′-phosphoryl group of adjacent nucleosides. Cleavage of RNA by nuclease P1 should yield GpppA or m7GpppA, representing G-N-7 unmethylated and methylated cap structures, respectively. Nuclease P1 digestion of RNA substrate Gp*ppA-RNA43 yielded a product which comigrated with a GpppA cap analog (NEB), confirming that it was an unmethylated cap (Fig. 1B, lane 1). Nuclease P1 digestion of m7Gp*ppA-RNA43 (positive control) yielded a product which comigrated with the m7GpppA cap analog (NEB), confirming that it was a fully G-N-7-methylated cap (Fig. 1B, lane 2). PEDV nsp14 efficiently methylated the RNA substrate at the G-N-7 position under various pH conditions, demonstrating that nsp14 is an RNA G-N-7 MTase (Fig. 1B, lanes 3 to 9). However, PEDV nsp14 was more active in neutral and basic environments (Fig. 1B, lanes 5 to 9) than in an acidic environment (Fig. 1B, lanes 3 and 4). After 3 h of incubation, Gp*ppA-RNA43 was completely converted into m7Gp*ppA-RNA43 under neutral and basic conditions, whereas a small portion of RNA substrate remained unmethylated under acidic conditions.
FIG 1.

Biochemical analyses of G-N-7 MTase activity of PEDV nsp14 in vitro. (A) SDS-PAGE analysis of purified PEDV nsp14 protein. The 6×His-tagged PEDV nsp14 was expressed and purified from E. coli. (B) Analysis of G-N-7 MTase activity of PEDV nsp14 in different pHs. One microgram of purified PEDV nsp14 was incubated with 2 × 103 cpm of Gp*ppA-RNA43 substrate in methylation buffer from pH 4.0 to 10.0 (lanes 3 to 9) at 37°C for 4 h. The products were digested with nuclease P1 at 50°C for 30 min and separated on a TLC plate. The migration of the markers m7Gp*ppA and Gp*ppA (lanes 1 and 2) is indicated. Spots were dried and visualized with a phosphorimager.
PEDV nsp14 G-N-7 MTase does not require a minimal length of the RNA substrate.
Many viral MTases require a minimal length of the RNA substrate for methylation. The minimal length of the RNA substrate for vesicular stomatitis virus (VSV) G-N-7 and 2′-O MTases was shown to be 5 nt (30). The flavivirus G-N-7 and 2′-O MTases required the RNA substrates to be longer than 20 nucleotides (31). Since PEDV nsp14 efficiently methylated 43-nt RNA, we first examined whether PEDV nsp14 can methylate a longer RNA substrate. To do this, a 296-nt RNA corresponding to the 5′ end of the PEDV genome was synthesized and tested for G-N-7 methylation. PEDV nsp14 efficiently methylated 296-nt PEDV RNA similarly to 43-nt RNA (Fig. 2A). To dissect the minimal length requirement for PEDV nsp14, we synthesized 30-, 20-, and 10-nt RNA corresponding to the 5′ end of the PEDV genome. As shown in Fig. 2B, PEDV nsp14 efficiently methylated 30-, 20-, and 10-nt RNA. We next examined the G-N-7 MTase activity using RNA substrates ranging from 9 nt to 3 nt. Again, all of these short RNA substrates were efficiently methylated by PEDV nsp14 (Fig. 2C). Finally, we further narrowed down the RNA substrate to 2 nt. In the methylation assay, PEDV nsp14 was incubated with dinucleotide cap analogs (GpppA, GpppG, m7GpppA, and m7GpppG) in the presence of 3H-SAM. The cap analogs were purified by DEAE-Sephadex columns, and the amount of 3H-SAM incorporation was quantitated by a scintillation counter. PEDV nsp14 efficiently methylated both GpppA and GpppG cap analogs (Fig. 2D). As controls, no 3H-SAM was incorporated into G-N-7-methylated cap analogs (m7GpppA and m7GpppG). Using a similar assay, we determined whether PEDV nsp14 can methylate single nucleotides. In this assay, vaccinia virus G-N-7 MTase was used as a positive control, whereas bovine serum albumin (BSA) was used as a negative control. As shown in Fig. 2E, PEDV nsp14 can methylate GTP, although the efficiency was lower than that of vaccinia virus G-N-7 MTase. However, PEDV nsp14 cannot methylate ATP, CTP, or UTP.
FIG 2.

Length requirement of RNA substrate for G-N-7 MTase activity of PEDV nsp14. Comparison of G-N-7 MTase activity of PEDV nsp14 using RNA substrates with lengths of 296 nt and 43 nt (A), 10 nt to 30 nt (B), and 3 nt to 9 nt (C). One microgram of PEDV nsp14 protein was incubated with 2 × 103 cpm of Gp*ppA-RNA with different lengths in methylation buffer (pH 7.0) at 37°C for 4 h. The products were digested with nuclease P1 at 50°C for 30 min and separated on a TLC plate. (D) Methylation of dinucleotide cap analogs by PEDV nsp14. One microgram of PEDV nsp14 protein was incubated with dinucleotide cap analogs at 37°C for 4 h. The products were purified in small DEAE-Sephadex columns and quantitated by a scintillation counter. Data are the averages from three independent experiments ± standard deviations. (E) Methylation of NTPs by PEDV nsp14. One microgram of PEDV nsp14 protein was incubated with NTPs at 37°C for 4 h. The products were purified in small DEAE-Sephadex columns and quantitated by a scintillation counter. Data are the averages from three independent experiments ± standard deviations. Vaccinia virus cap G-N-7 MTase was used as a positive control, and BSA was used as a negative control.
Longer RNA substrate facilitates PEDV G-N-7 methylation.
We also compared the kinetics of the N-7 MTase activity of PEDV nsp14 using 43-nt (Fig. 3A) and 9-nt (Fig. 3B) RNA substrates. Briefly, PEDV nsp14 was incubated with Gp*ppA-RNA43 or Gp*ppA-RNA9 in the presence of SAM, and reaction was stopped every 30 min and analyzed in a TLC plate. After a 30-min incubation, approximately 12% of the 43-nt substrate cap was G-N-7 methylated, whereas methylation products from the 9-nt RNA substrate were only 3%. After each 30-min interval, the percentage of G-N-7 methylation using the 43-nt substrate cap was higher than when using the 9-nt substrate cap. After 4 h, 90% of the 43-nt RNA substrate and 73% of the 9-nt RNA substrate were methylated. These results suggest that PEDV nsp14 prefers longer RNA substrates with the same 5′ terminal sequence.
FIG 3.

Comparison of G-N-7 MTase activity of PEDV nsp14 using 43-nt and 9-nt RNA substrates. (A) Dynamics of G-N-7 MTase using a 43-nt RNA substrate. (B) Dynamics of G-N-7 MTase using a 9-nt RNA substrate. One microgram of PEDV nsp14 protein was incubated with 2 × 103 cpm of 43-nt or 9-nt Gp*ppA-RNA at 37°C for the indicated time points, and the reaction was stopped by addition of 1% sodium dodecyl sulfate and 0.5 mM EDTA. The products were digested with nuclease P1 and separated by TLC. The migration of the markers m7Gp*ppA and Gp*ppA is indicated. The density of spots was quantified by ImageJ. The percentage of each methylated cap species was calculated as follows: density of methylated cap m7Gp*ppA/density of methylated cap m7Gp*ppA and unmethylated cap Gp*ppA.
PEDV nsp14 methylates RNA substrate in a sequence-unspecific manner.
Many viral MTases methylate RNA in a sequence-specific manner (30, 32). We next determined whether PEDV nsp14 is a sequence-specific G-N-7 MTase. To do this, RNA substrates with 43 nucleotides of 5′ terminal genomic RNA from TGEV, SARS-CoV, PDCoV, and IBV were synthesized to represent viruses in Alphacoronavirus, Betacoronavirus, Deltacoronavirus, and Gammacoronavirus genera, respectively (Fig. 4A). The homology of these RNA sequences with PEDV ranges from 35.5% to 46.5%. All these RNA substrates share the same first nucleotide (adenine). The second nucleotide of the RNA substrate from TGEV is an A, for SARS-CoV is a T, and for the other three viruses is a C. As shown in Fig. 4B, PEDV nsp14 had a similar methylation efficiency using all these RNA substrates from the Coronavirinae subfamily (Fig. 4B, lanes 4 to 8). Finally, we synthesized two random RNA substrates, a 43-nt poly(A) RNA [Gppp(A)43] and a 43-nt poly(G) RNA [Gppp(G)43]. As shown in Fig. 4C, nsp14 can methylate both Gppp(A)43 and Gppp(G)43, although the efficiency seems slightly less than for the authentic PEDV RNA substrate (Fig. 4C). These results suggest that PEDV nsp14 methylates RNA in a sequence-unspecific manner.
FIG 4.

PEDV nsp14 methylates RNA substrate in a sequence-unspecific manner. (A) Sequences of RNA substrate used for G-N-7 MTase assay. RNA was synthesized by T7 RNA polymerase and capped by vaccinia virus capping enzyme as described in Materials and Methods. Methylation of RNA sequences from different coronaviruses (B) or random poly(A) and poly(G) sequences (C) by PEDV nsp14. 5′-Terminal 43-nt RNA substrates from PEDV, TGEV, SARS-CoV-1, PDCoV, and IBV or 43-nt poly(A) (Gp*ppA43) and poly(G) (Gp*ppG43) sequences were synthesized in vitro. One microgram of PEDV nsp14 protein was incubated with 2 × 103 cpm of 43-nt RNA substrates from different coronaviruses (B) or a random sequence (C) at 37°C for 4 h. The products were digested with nuclease P1 and separated by TLC. The migration of the markers m7Gp*ppA and Gp*ppA is indicated.
Mutations in the SAM binding site in PEDV nsp14 abolished G-N-7 MTase activity.
Our ultimate goal is to generate recombinant PEDVs that are defective in G-N-7 MTase. Since knockout of G-N-7 methylation in viral RNA may be lethal to virus, our mutagenesis strategy is to select some amino acids in PEDV nsp14 that are completely or partially defective in G-N-7 methylation based on the studies in SARS-CoV nsp14. Sequence alignment of nsp14 proteins of representative members of Alphacoronavirus (PEDV and TGEV), Betacoronavirus (SARS-CoV-1 and SARS-CoV-2), Deltacoronavirus (PDCoV), and Gammacoronavirus (IBV) genera showed that the ExoN and G-N-7 MTase domains are highly conserved and located at the N and C termini of nsp14, respectively (see Fig. S1A in the supplemental material). SAM-dependent G-N-7 MTases typically present a D/ExGxGxG motif (where x means any amino acid), the hallmark of the SAM binding site (33). Sequence alignment in the core G-N-7 MTase domain of nsp14 protein in all CoVs clearly identified a DxGxPxG/A motif corresponding to the D/ExGxGxG motif in G-N-7 MTase (Fig. 5A and Fig. S1A). Structural homology analysis found that nsp14 of PEDV is similar to nsp 14 of SARS-CoV-1 (PDB identifier [ID] 5C8S) (Fig. S1B and C). In SARS-CoV-1 nsp14, individual alanine mutations to D331 and G333 within the SAM binding site (DIGNPKA) completely abolished G-N-7 methylation, whereas alanine mutations to P335 and K336 resulted in nsp14 mutants that retained 61% and 50% of G-N-7 methylation, respectively (8, 34). The equivalent amino acids in the SAM binding site (DIGNPKG) of PEDV nsp14 are D330, G332, P334, and K335. Thus, we first generated two single PEDV nsp14 mutants, D330A and G332A, designed to knock out the G-N-7 MTase completely. We also generated a double mutant, P334A-K335A, designed to further reduce the G-N-7 MTase. We also generated a fourth mutant, D350A, designed to retain partial MTase activity. In SARS-CoV-1 nsp14, the equivalent amino acid residue D351A retained 47% of G-N-7 MTase activity (15). Recombinant PEDV nsp14s carrying these mutations were expressed in E. coli and purified (Fig. 5B). Each nsp14 mutant was incubated with a 296-nt RNA substrate in the presence of 3H-SAM, and G-N-7 MTase activity was quantitatively measured by a scintillation counter. As shown in Fig. 5C, nsp14 mutants D330A and G332A only retained 3% to 5% of G-N-7 MTase activity, whereas the double mutant (P334A-K335A) and D350A retained approximately 15% and 29% of MTase activity, respectively.
FIG 5.

Mutations to the SAM binding site abolish G-N-7 MTase. (A) Sequence alignment of CoV nsp14 proteins. The G-N-7 MTase regions of PEDV nsp14 (gi: 557844763) and its homologs from Alphacoronavirus genus TGEV (gi: 110746821), Betacoronavirus genus SARS-CoV-1 (gi: 40795428) and SARS-CoV-2 (gi: 1806553187), Deltacoronavirus genus PDCoV (gi: 668361756), and Gammacoronavirus genus IBV (gi: 9626535) were chosen for alignment by ClustalW2. Identical residues are shown in boxes with a solid red background, and conserved residues are highlighted in red. Amino acid residues in the SAM binding residue chosen for mutagenesis are marked with blue triangles. (B) SDS-PAGE analysis of PEDV nsp14 mutants. 6×Hig-tagged PEDV nsp14 mutants were expressed and purified from E. coli. Protein marker is indicated on the left. (C) Analysis of G-N-7 MTase activity of nsp14 mutants. One microgram of PEDV nsp14 protein was incubated with a 296-nt RNA substrate at 37°C for 4 h in the presence of 3H-SAM. The incorporation of 3H-SAM into the RNA substrate was measured by a scintillation counter. Data are the averages from three independent experiments ± standard deviations.
Construction of PEDV infectious cDNA clone and recovery of recombinant PEDV carrying mutations in nsp14.
We have developed a plasmid-based reverse genetics system for the PEDV USA/Colorado/2013 strain (Fig. 6A). The full-length cDNA clone of PEDV was assembled into a pSMART-BAC vector in a single step using seven overlapping fragments (designated from A to G) by using a yeast recombination system (Fig. 6A). To introduce mutations in the nsp14 in the pSMART-BAC-PEDV, we used two-step selection/counterselection recombineering technique to replace the wild-type nsp14 with the nsp14 mutations described in Materials and Methods. To recover recombinant PEDV, 4 μg of pSMART-BAC-PEDV (wild-type or mutation) and 2 μg of pCAGGS-N were transfected into Vero CCL-81 cells using Lipofectamine 3000 reagent. For recovery of the parental rPEDV, syncytia were typically observed at 48 to 72 h after transfection. At 96 h posttransfection, cell culture supernatants were collected to inoculate fresh Vero CCL-81 cells. When extensive syncytia were observed, supernatants were collected for further passage. Recovery was considered unsuccessful if no syncytia were observed at the 4th passage. A mutation was considered to be lethal to virus if recovery was not successful after eight independent transfection experiments. The recovery of the parental rPEDV was successful for all eight trials, indicating that the plasmid-based reverse genetics system was highly efficient. After three trials, we recovered two mutants, rPEDV-D350A and rPEDV-D330A. However, we failed to recover rPEDV-G332A and rPEDV-P334A-K335A after eight trials. Subsequently, plaque purification was performed for rPEDV-D350A and rPEDV-D330A. After 48 h of incubation, rPEDV-D350A formed significantly smaller plaques than parental rPEDV (Fig. 7A). However, plaques for rPEDV-D330A were invisible after 48 h of incubation. A new plaque assay was performed for rPEDV-D330A. After incubation for 5 days, small plaques were observed for rPEDV-D330A. Six plaques were picked from each recombinant virus and were grown in a T25 flask of Vero CCL-81 cells. The recombinant virus was further passaged 2 to 3 times in Vero CCL-81 cells. At passage 3, total RNA was extracted from cell culture supernatants, and the nsp14 gene was amplified by reverse transcription-PCR (RT-PCR) and sequenced. All plaques picked from rPEDV-D350A retained the desired D350A mutation (Fig. 7B). Thus, we sequenced the full-length genome of rPEDV-D350A. No additional mutations were found elsewhere in the genome. Unfortunately, all plaques picked from rPEDV-D330A did not contain the D330A mutation. Subsequently, we sequenced the virus stocked from the first and second passages, and the nsp14 genes from all of these stocks were wild type. Since rPEDV-D330A was unstable, we decided not to pursue this virus further. To further test the genetic stability of rPEDV-D350A, we continuously passaged the rPEDV-D350A in Vero CCL-81 cells for 13 passages. At passages 8 and 13, the virus stocks were sequenced. Results showed that rPEDV-D350A retained the D350A mutation. These results suggest that rPEDV-D350A is genetically stable in Vero CCL-81 cells for at least 13 passages.
FIG 6.

Strategy to construct the infectious PEDV cDNA clone and mutants. (A) Assembly of the full-length genomic cDNA of PEDV in the pSMART-BAC vector. The pSMART-BAC vector was modified as described in Materials and Methods. The full-length genomic cDNA of PEDV was amplified by RT-PCR using seven overlapping fragments designated A to G, and was assembled by using the GeneArt High-order genetic assembly system. The diagram of PEDV genes encoding ORF1a, ORF1b, spike (S), ORF3, envelope (E), membrane (M), and nucleocapsid (N) is shown. Regulatory elements, including the cytomegalovirus (CMV) promoter, hepatitis delta virus (HDV) ribozyme sequence, and bovine growth hormone (BGH) polyadenylation and terminator, are indicated in the plasmid. (B) Strategy to introduce mutations to PEDV genome. Diagrams of the two-step selection recombineering are shown. The first step is to place the rpsl+-Kana cassette at the locus of nsp14 via positive kanamycin selection. The second step is to replace the rpsl+-Kana cassette in the BAC intermediate construct with an nsp14 mutant cassette containing the same homology arms via negative selection of streptomycin. A and B indicate homology arms; nsp14 and nsp14-mutation indicate wild-type nsp14 and nsp14 mutations, respectively.
FIG 7.

G-N-7 MTase-defective rPEDV is defective in replication in Vero CCL-81 cells. (A) rPEDV-D350A forms smaller plaques. Plaque assay was performed in Vero-CCL-81 cells. After 50 h of incubation, plaques were fixed in 4% formaldehyde and stained with crystal violet. The plaques were scanned, and the diameters of plaques were measured by Image J Software. Plaque size is the average from 20 plaques ± standard deviations. (B) Sequence analysis of rPEDV-D350A. Virus stock at passage 5 was sequenced. (C) Growth kinetics of recombinant PEDVs. Confluent Vero CCL-81 cells were infected with the indicated viruses at an MOI of 1.0 or 0.1. Cell culture supernatants were harvested at 8, 13, and 18 h postinoculation, and viral titers were determined by a plaque assay. Data are the averages from three independent experiments ± standard deviations. (D) Detection of PEDV nucleocapsid (N) protein by Western blotting. Confluent Vero CCL-81 cells were infected with the indicated viruses at an MOI of 1.0 or 0.1. Cell lysates were collected at 8, 13, and 18 h postinoculation; PEDV N protein and β-actin were detected by Western blotting. (E and F) RNA synthesis of recombinant PEDVs. Confluent Vero CCL-81 cells were infected with the indicated viruses at an MOI of 0.1 or 1.0. Total RNA was extracted from cells at 8, 13, and 18 h postinoculation, and viral genomic (E) and subgenomic (F) RNA and GAPDH mRNA were quantified by real-time RT-PCR. Viral genomic and subgenomic RNA copies were normalized to GAPDH mRNA copies. Data are the averages from three independent experiments ± standard deviations.
Recombinant rPEDV-D350A was defective in replication in cell culture.
Next, we characterized replication of rPEDV-D350A in Vero CCL-81 cells. We first monitored the kinetics of release of infectious virus by a single-step growth assay. Briefly, Vero CCL-81 cells were infected with each of the designated recombinants at a multiplicity of infection (MOI) of 1.0 or 0.1, and viral titer was determined at time points from 0 to 18 h postinoculation. At an MOI of 1.0, rPEDV-D350A replicated slower than rPEDV, although there was no statistical difference (P > 0.05) (Fig. 7C). At an MOI of 0.1, rPEDV-D350A had a significant defect in replication compared to that of rPEDV (P < 0.05) (Fig. 7C). Next, total cell lysates were harvested in Vero CCL-81 cells infected by each virus at an MOI of 1.0 or 0.1. As shown in Fig. 7D, rPEDV-D350A synthesized less viral N protein than rPEDV at both MOIs, although the difference was more obvious at an MOI of 0.1. We also measured viral genome and subgenome RNA synthesis in Vero CCL-81 cells. rPEDV-D350A synthesized less genome RNA than rPEDV (Fig. 7E). In addition, rPEDV-D350A synthesized less subgenomic RNA than rPEDV, although there was no significant difference between these two viruses (P > 0.05) (Fig. 7F). In addition, rPEDV-D350A developed similar kinetics of syncytial formation in virus-infected cells (Fig. 8). We also compared the replication of rPEDV-D350A and rPEDV in porcine intestinal epithelial cells (IPEC-DQ), the primary target cells for PEDV in pigs. Similarly to that in Vero CCL-81 cells, rPEDV-D350A had significantly less N protein synthesis at MOIs of 1.0 and 0.1 than rPEDV (Fig. 9A). A single step growth assay showed that rPEDV-D350A had a significant defect in replication in IPEC-DQ cells (Fig. 9B). In addition, rPEDV-D350A synthesized less genome (Fig. 9C) and subgenome (Fig. 9D) RNA than rPEDV in virus-infected cells. Therefore, these results demonstrated that rPEDV-D350A was significantly defective in replication in cell culture.
FIG 8.
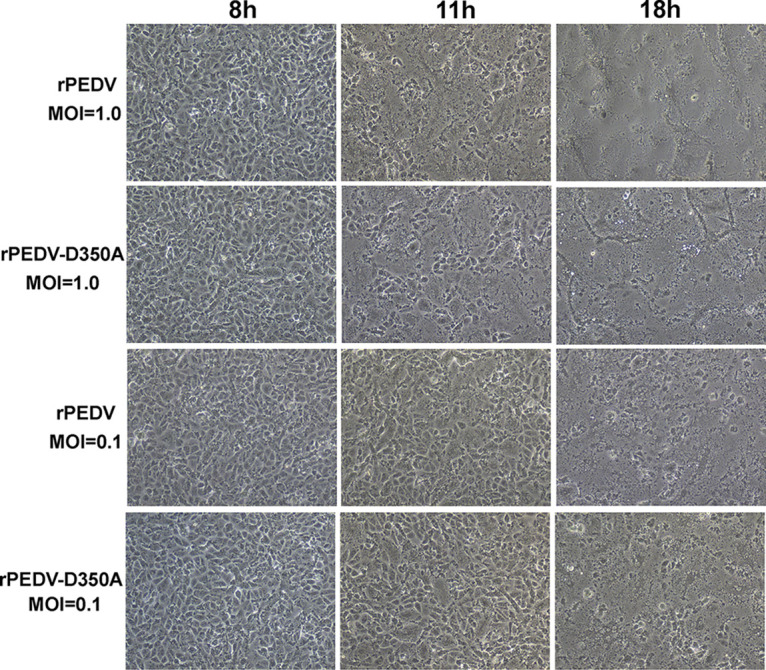
Progression of PEDV-induced cytopathic effects in Vero CCL-81 cells. Confluent Vero CCL-81 cells were infected with rPEDV and rPEDV-D350A at an MOI of 1.0 or 0.1. Syncytial formation was imaged at the indicated time points.
FIG 9.

G-N-7 MTase-defective rPEDV is defective in replication in IPEC-DQ cells. (A) Detection of PEDV N protein by Western blotting. Confluent IPEC-DQ cells were infected with the indicated viruses at an MOI of 1.0 or 0.1. Cell lysates were collected at 8, 13, and 18 h postinoculation; PEDV N protein and β-actin were detected by Western blotting. (B) Growth kinetics of recombinant PEDVs. Confluent IPEC-DQ cells were infected with the indicated viruses at an MOI of 1.0 or 0.1. Cell culture supernatants were harvested at 8, 13, and 18 h postinoculation, and viral titers were determined by a plaque assay. Data are the averages from three independent experiments ± standard deviations. (C and D) RNA synthesis of recombinant PEDVs. Confluent IPEC cells were infected with the indicated viruses at an MOI of 0.1 or 1.0. Total RNA was extracted from cells at 8, 13, and 18 h postinoculation, and viral genomic RNA (C), subgenomic RNA (D), and GAPDH mRNA were quantified by real-time RT-PCR. Viral genomic and subgenomic RNA copies were normalized to GAPDH mRNA copies. Data are the averages from three independent experiments ± standard deviations.
Recombinant rPEDV-D350A induces significantly higher type I and III interferon responses in IPEC-DQ cells.
RNA cap methylation has been shown to be important for innate immune evasion (35, 36). Notably, it was shown that a recombinant mouse hepatitis virus (rMHV) carrying a mutation (G332A) in the SAM binding site in nsp14 induced a higher type I interferon response than the parental rMHV (37). Thus, we determined whether rPEDV-D350A had a similar effect. PEDV targets intestinal epithelial cells (IECs) and causes enteric infection. It is known that IECs respond poorly to type I interferons (IFNs), as these cells express low levels of type I IFN receptors (38, 39). However, recent studies showed that IECs produce abundant type III IFNs which were capable of restricting PEDV replication (38, 39). In humans, type III IFNs include IFN-λ1 (interleukin 29 [IL-29]), IFN-λ2 (IL-28A), IFN-λ3 (IL-28B), and IFN-λ4. In pigs, IFN-λ1, IFN-λ3, and IFN-λ4 have been characterized, whereas it is unclear whether IECs express IFN-λ2 (40). Thus, we compared the type I and III IFN responses of rPEDV and rPEDV-D350A in IPEC-DQ cells. Briefly, IPEC-DQ cells were infected by rPEDV-D350A or rPEDV at an MOI of 1.0 or 0.1, total RNA was extracted from cell lysates at 11 and 18 h postinoculation, and swine type I (IFN-α and IFN-β gene) and type III (IFN-λ1 and IFN-λ3) responses were determined by real-time RT-PCR and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). At an MOI of 1.0, rPEDV-D350A induced significantly higher expression of IFN-α (Fig. 10A), IFN-β (Fig. 10B), IFN-λ1 (Fig. 10C), and IFN-λ3 (Fig. 10D) at 18 h postinoculation than rPEDV (P < 0.05). Interestingly, IFN-λ3 had the most dramatic increase (approximately 5 times). At an MOI of 0.1, rPEDV-D350A induced higher expression of IFN-α (Fig. 10E) and IFN-β (Fig. 10F) than rPEDV, but there was no significant difference between these two viruses (P > 0.05). However, IFN-λ1 (Fig. 10G) and IFN-λ3 (Fig. 10H) expression in rPEDV-D350A was significantly higher than that in rPEDV (P < 0.05). Again, IFN-λ3 expression in rPEDV-D350A increased 13 times at 18 h postinoculation compared to that in rPEDV. Collectively, these results demonstrated that G-N-7 MTase-deficient PEDV induced significantly higher expression of type I and III IFNs.
FIG 10.

G-N-7 MTase-defective rPEDV induces higher type I and type III interferon responses in IPEC-DQ cells. Confluent IPEC-DQ cells were infected with rPEDV or rPEDV-D350A at an MOI of 1.0 or 0.1. Total RNA was extracted from cells at 11 and 18 h postinoculation. Swine IFN-α, IFN-β, IFN-λ1, IFN-λ3, and GAPDH mRNA were quantified by real-time RT-PCR. Swine IFN-α, IFN-β, IFN-λ1, and IFN-λ3 mRNA copies were normalized to GAPDH mRNA copies. Data are the averages from three independent experiments ± standard deviations. (A) IFN-α at MOI of 1.0; (B) IFN-β at MOI of 1.0; (C) IFN-λ1 at MOI of 1.0; (D) IFN-λ3 at MOI of 1.0; (E) IFN-α at MOI of 0.1; (F) IFN-β at MOI of 0.1; (G) IFN-λ1 at MOI of 0.1; and (H) IFN-λ3 at MOI of 0.1.
Virion RNA of rPEDV-D350A induces significantly higher type I and III interferon responses in IPEC-DQ cells.
Finally, we determined whether virion RNA of rPEDV-D350A induces higher IFN expression than rPEDV. Briefly, RNA was extracted from rPEDV-D350A or rPEDV virions purified by sucrose gradient ultracentrifugation. Equal amounts of each virion RNA were transfected into IPEC-DQ cells, and swine type I and type III responses were determined by real-time RT-PCR. Unlike virus infection, transfection of viral RNA in cells results in little viral protein synthesis, particularly at early time points, thereby reducing the effects of viral proteins on IFN production. As a positive control, poly(I·C) induced robust expression of type I and III IFNs (Fig. 11). Virion RNA of rPEDV-D350A induced higher expression of IFN-α, IFN-β, and IFN-λ3 than virion RNA of rPEDV (Fig. 11). These results demonstrate that G-N-7-deficient PEDV RNA directly induces higher type I and III responses, suggesting that virion RNA lacking G-N-7 and 2′-O methylation is recognized by host cells as nonself RNA, thereby inducing higher IFN responses.
FIG 11.

G-N-7 MTase-defective PEDV virion RNA induces higher type I and type III interferon responses in IPEC-DQ cells. Confluent IPEC cells were transfected with 1010 RNA copies of virion RNA of rPEDV or rPEDV-D350A or 5 μg of poly(I·C). Total RNA was extracted from cells at 11 and 18 h postinoculation. Swine IFN-α (A), IFN-β (B), IFN-λ1 (C), and IFN-λ3 (D) were quantified by real-time RT-PCR and normalized by GAPDH mRNA copies. Data are the averages from three independent experiments ± standard deviations.
DISCUSSION
CoVs contain the largest known RNA genomes (27 to 32 kb) and share many common strategies in replication and gene expression. One unique characteristic is that all CoVs encode a bifunctional nsp14 protein, which contain a metal ion-dependent 3′-to-5′ exoribonuclease activity (ExoN) at the N terminus (17) and a SAM-dependent G-N-7 MTase activity at the C terminus (8, 15). The ExoN is a proofreading enzyme essential for replication fidelity, whereas G-N-7 MTase activity is critical for mRNA stability, translation, and immune evasion. Studies in SARS-CoV-1 nsp14 showed that the ExoN and G-N-7 MTase domains were functionally distinct and physically independent (8, 15). The unique combination of ExoN and G-N-7 MTase activity in nsp14 provides an attractive target for antiviral drug development against CoVs. Currently, most of our knowledge on CoV nsp14 came from the studies of SARS-CoV-1, a Betacoronavirus (8, 13, 15, 41). Given the high diversity and wide host range of CoVs, there is a critical need to understand the function of nsp14s of CoVs in other genera, allowing us to identify the similarities and differences among different CoV genera.
Eukaryotic cellular G-N-7 MTase typically acts in a sequence-independent manner and can methylate polyribonucleotides and dinucleotide GpppG (42). Many RNA viruses replicate in cytoplasm and do not have access to the nucleus. Unlike host G-N-7 MTases, many virally encoded G-N-7 MTases specifically methylate virus-specific mRNA and require a minimal length for RNA substrates. For example, flavivirus NS5 protein possesses both G-N-7 and 2′-O-MTases and methylates RNA in a sequence-dependent manner, with the minimal length requirement of 20 nucleotides (31). Similarly, the conserved region VI (CR-VI) of large (L) polymerase protein possesses both 2′-O and G-N-7 methylase activities that act specifically on RNA substrates that contain the conserved gene start sequences of VSV mRNA at the 5′ terminus (30). Both VSV G-N-7 and 2′-O MTase activities have a maximum activity at pH 7.0 at 30°C, and the G-N-7 MTase activity was significantly inhibited at alkaline pH (30). In addition, VSV G-N-7 and 2′-O MTases efficiently methylated the 10-, 51-, and 110-nt VSV-specific RNAs (30). However, the 5-nt RNA was not methylated by the VSV L protein at either the G-N-7 or the ribose 2′-O position (30). This result suggests that the minimal length requirement for VSV mRNA cap methylation is between 5 and 10 nt. In contrast, several viral G-N-7 MTases are not sequence specific and do not require a minimal length. Among them, the vaccinia virus D1-D12 complex is the best-characterized viral G-N-7 MTase, which can efficiently methylate single nucleotide, dinucleotide, and polynucleotide RNA (43, 44). Alphavirus nsP1 acts as both an RNA capping enzyme and a G-N-7 MTase. The nsP1 is able to methylate GTP and dGTP, as well as some 5′–5′ dinucleotides containing guanosine (45). Methylation of the capping guanosine precedes its transfer to the 5′ end of mRNA through an nsP1-m7GMP covalent intermediate (45). In addition, alphavirus Semliki Forest virus (SFV) can utilize GTP and GpppG but not GpppA as the substrates (46).
Among CoVs, SARS-CoV-1 nsp14 is the best-characterized G-N-7 MTase. It was shown that SARS-CoV-1 nsp14 can methylate GTP and dGTP as well as cap analogs GpppG, GpppA, and m7GpppG but not ATP, CTP, UTP, dATP, dCTP, dUTP, or cap analog m7GpppA (41). Similarly, MHV nsp14 and TGEV nsp14 can also methylate GTP and dGTP as efficiently as SARS-CoV-1 nsp14 (41). We now added PEDV nsp14 to this list. PEDV nsp14 can efficiently methylate GTP and cap analogs GpppA and GpppG but not ATP, CTP, or UTP. In addition, CoV G-N-7 MTase can recognize different lengths of RNA substrates. It is unclear how these substrate recognition characteristics are relevant to CoV replication and pathogenesis. It is possible that CoV nsp14 efficiently methylates different lengths of transcripts in order to enhance the stability of the nascent transcripts for efficient elongation and synthesis of full-length RNA. Another interesting observation of CoV G-N-7 MTase is that it is highly active in a wide range of pHs, being more active with alkaline pH than acidic pH. Many CoVs can replicate efficiently in multiple organs. For example, although the major target organ for SARS-CoV-1 is the lung, it can also infect the intestine, an alkaline environment. Clinically, many SARS patients experienced both pneumonia and acute gastroenteritis (47). Similar clinical manifestations were reported for the newly emerged SARS-CoV-2 (48). Perhaps, CoVs evolve their own G-N-7 MTases to be functional in a wide range of pHs for efficient methylation of their RNA in order to replicate in different organs.
It appears that ExoN and G-N-7 MTase activities of CoV nsp14 are important for viral replication but play distinct roles in regulating innate immunity. Deletion or mutation of the conserved ExoN DEDD motif in nsp14 is lethal or attenuates the viral replication (17, 49) and results in reduced fidelity of replication in MHV (50) and SARS-CoV-1 (51). Working with TGEV, a porcine Alphacoronavirus, it was shown that a recombinant TGEV (rTGEV-ZF-C) carrying a point mutation in zinc finger 1 (ZF-C) of ExoN triggered a reduced antiviral response in cell culture, including interferon beta (IFN-β), tumor necrosis factor (TNF), and interferon-stimulated genes, compared to that of the parental virus (52). Thus, ExoN positively regulates innate immunity in TGEV. In contrast, CoV G-N-7 MTase activity suppresses the innate immune response of MHV, a murine Betacoronavirus. Case et al. generated two rMHVs (D330A and G332A) carrying mutations in the SAM binding site (DxG motif) (37). Recombinant rMHV carrying a G332A substitution in nsp14 displayed delayed replication kinetics, decreased peak titers, and decreased translation efficiency. Interestingly, rMHV-G332A induced significantly higher expression of type I interferon, similarly to the nsp16 D130A virus, which was only defective in 2′-O methylation (35). In addition, replication of the G332A virus was diminished following treatment of cells with IFN-β. Since CoV RNA cap G-N-7 methylation is required for 2′-O methylation, the rMHV-G332A mutant was likely defective in both G-N-7 and 2′-O methylation, which resulted in a phenotype similar to that of a viral mutant (nsp16 D130A) only defective in 2′-O methylation. In contrast, the rMHV-D330A mutant did not affect viral replication or inhibition by IFN-β. However, Case and his colleagues did not biochemically determine whether D330A and G332A were indeed defective in G-N-7 methylation in MHV (37). One possible explanation for their results is that the G332A mutant in MHV nsp14 lacked G-N-7 MTase, whereas the D330A mutant retained G-N-7 MTase. It should be noted that both mutations (D330A and G332A) in nsp14 of SARS-CoV-1, another Betacoronavirus, abolished G-N-7 MTase activity (15).
Unlike that in MHV, the conserved SAM binding site residues (DxG) in nsp14 are required for PEDV viability. We showed that individual mutations (D330A and G332A) abolished the G-N-7 MTase. We were unable to recover these viral mutants after multiple attempts. However, D350A, which retained 29% of G-N-7 MTase activity, was viable. Compared to the parental rPEDV, rPEDV-D350A was significantly defective in cell culture: formed smaller plaques and had reduced viral replication, protein synthesis, and RNA synthesis. The attenuation characteristics of rPEDV-D350A were similar to those of rMHV-G332A but not rMHV-D330A. This suggests that G-N-7 MTase is important for PEDV protein translation and replication.
Notably, rPEDV-D350A was capable of inducing significantly higher expression of type I and III IFNs than the parental rPEDV. In addition, virion RNA of rPEDV-D350A induced higher type I and III IFN expression than that of rPEDV. The increase in type III IFN was more dramatic than that of the type I IFN response. Thus, it is likely that PEDV RNA lacking G-N-7 and 2′-O methylation is recognized by host cells as nonself RNA, thereby inducing higher IFN expression. Recently, it was shown that recombinant PEDV carrying mutations in the KDKE motif in nsp16, the signature motif of 2′-O MTase, triggered stronger type I and type III IFN production in IPEC-DQ cells (53). Presumably, these PEDV mutants were defective in 2′-O MTase, although they did not experimentally demonstrate the 2′-O MTase activity in their study (53). Since it is known that G-N-7 methylation precedes the 2′-O methylation in CoV, rPEDV-D350A is likely defective in both G-N-7 and 2′-O methylation. Our results suggest that viral RNA G-N-7 and/or 2′-O methylations regulate type I and III interferons in PEDV. The type III IFN responses, mediated by IFN lambda (IFN-λ), exert antiviral activity mainly in epithelial cells at the mucosal surfaces, such as in gut or respiratory epithelial cells. It has been shown that many enteric viruses, including PEDV, have evolved to evade the type III IFN responses during infection and that type III IFNs play a vital role in maintaining the antiviral state of the mucosal epithelial surface in the gut (38–40). In addition, it has been shown that the swine type III IFNs have greater antiviral activities against PEDV than IFN-α (54). We also found that the increase of IFN-λ3 induced by rPEDV-D350A was more dramatic than that of IFN-λ1. Interestingly, it was shown that IFN-λ3 restricts PEDV infection more efficiently than IFN-λ1 (39). Nevertheless, the finding that G-N-7 MTase-deficient PEDV induces higher type I and III IFNs may have important applications in vaccine and antiviral development for PEDV.
One important application of this study is to develop a new live attenuated vaccine candidate for PEDV. PEDV causes significant economic losses in the swine industry worldwide. Currently, there is no effective vaccine or antiviral drug for this virus. Since the first outbreak of PEDV CV777 strain in the United Kingdom in 1971, an inactivated vaccine has been developed but was not sufficient to protect piglets from diarrhea (55). Also, a live attenuated PEDV CV777 strain has been developed by a traditional attenuation approach: passing the virus repeatedly in Vero cells. However, the CV777-based live attenuated vaccine cannot provide sufficient protection against the current prevalent PEDV strains (55, 56). We hypothesized that recombinant PEDV lacking RNA cap methylation is a novel approach for rational design of new live attenuated vaccines for PEDV and other CoVs. In fact, we and others have proved this concept in several RNA viruses, including paramyxoviruses (57), pneumoviruses (58, 59), rhabdoviruses (60), flaviviruses (61), and coronaviruses (53, 62). In this study, we showed that rPEDV-D350A, which only retained 29% of G-N-7 MTase activity, was significantly defective in replication and gene expression in both Vero cells and pig intestinal cells compared to the parental rPEDV. Another advantage is that rPEDV-D350A induced significantly higher type I and III IFN responses, which may induce stronger adaptive immunity. A future direction is to test the attenuation and immunogenicity of rPEDV-D350A in piglets.
In summary, we have biochemically characterized G-N-7 MTase of PEDV, a member of the Alphacoronavirus genus. We found that PEDV G-N-7 MTase methylates RNA substrates in a sequence-unspecific manner and that G-N-7 MTase-deficient PEDV is defective in replication and induces higher type I and III IFN responses. These findings highlight that inhibition of CoV G-N-7 MTase may be a novel approach for rational design of live attenuated vaccine candidates and antiviral drugs.
MATERIALS AND METHODS
Cells and viruses.
Vero CCL-81 cells (ATCC CCL-81) were grown in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Corning) supplemented with 10% fetal bovine serum (FBS). The intestinal porcine epithelial cells (IPEC-DQ), a subline of IPEC-J2 cells, were generally provided by Qiuhong Wang (The Ohio State University) and maintained in RPMI 1640 supplemented with 10% FBS (53). The Colorado strain of PEDV (USA/Colorado/2013; GenBank accession number KF272920) was obtained from APHIS-NVSL. PEDV was grown in Vero CCL-81 cells in DMEM supplemented with 0.018% (wt/vol) tryptose phosphate broth (TPB) (Sigma), 0.02% yeast extract (Sigma), 5 μg/ml trypsin 1:250 (Sigma, catalog number T0646), 10 UI/ml penicillin-streptomycin, 0.05 mg/ml gentamicin, and 0.05 mg/ml kanamycin. At 48 h postinfection, cell culture supernatant was harvested, and virus titer was determined by plaque assay in Vero CCL-81 cells.
Chemical and cap analogs.
Nucleoside triphosphates (NTPs) and cap analogs (GpppG, GpppA, m7GpppG, and m7GpppA) were purchased from NEB. α-32P-GTP (3,000 Ci/mmol), and S-[methyl-3H]-adenosyl-l-methionine (0.55 mCi/ml) were purchased from PerkinElmer.
Cloning, expression, and purification of recombinant PEDV nsp14.
The PEDV nsp14 flanked by NdeI and BamHI was amplified by RT-PCR with an N-terminal 6×His tag from the PEDV isolate USA/Colorado/2013 and cloned into the pET-21b vector. The cloned PCR products and plasmids (pET-21b-nsp14) were sequenced to ensure accuracy. E. coli Rosetta (DE3) transformed with expression plasmids was grown at 37°C until the absorbance value reached 0.6 to 0.8 at 600 nm, and the cells were chilled on ice for 5 min. The cells were grown for an additional 20 h at 10°C after inducing nsp14 expression by adding 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG), and the final products were harvested by centrifugation. Bacterial pellets were resuspended in 30 ml lysis buffer (50 mM NaH2PO4 [pH 8.0], 300 mM NaCl, 10 mM imidazole, 1% Triton X-100, 5 mM β-mercaptoethanol, 0.2 mg/ml lysozyme, and 10% glycerol). After sonication of the bacterial cells, soluble recombinant proteins were loaded to a prebalanced TALON resin-loaded column (Clontech) and washed with 200× bed volume washing buffer (50 mM NaH2PO4 [pH 8.0], 300 mM NaCl, 5 mM β-mercaptoethanol, 10% glycerol) containing 20 mM or 40 mM imidazole. The proteins were eluted from the columns with an elution buffer (50 mM NaH2PO4 [pH 8.0], 300 mM NaCl, 250 mM imidazole, 5 mM β-mercaptoethanol, 10% glycerol). The purified proteins were loaded onto a PD-10 desalting column (Amersham Bioscience), which was washed with the desalting buffer (50 mM Tris-HCl [pH 8.0], 50 mM NaCl, 0.1% Triton X-100, 2 mM dithiothreitol [DTT], 10% glycerol). The proteins were analyzed by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and quantified by Bradford protein assay (Sigma Chemical Co., St. Louis, MO). All the purified proteins were stored at −80°C until use.
Site-directed mutagenesis.
The nsp14 mutants (D330A, G332A, P334A-K335A, and D350A) of PEDV in pET-21b-nsp14 were generated by site-directed mutagenesis using QuikChange methodology (Stratagene, La Jolla, CA). All constructs were sequenced to confirm the presence of the introduced mutations.
Synthesis of 5′-triphosphorylated oligo-RNAs.
An oligonucleotide (5′-TAATACGACTCACTATT/A) corresponding to the T7 promoter sequence (−17 to −1) was annealed to each template oligonucleotide at the promoter region. Template oligonucleotide, 5′-AGAAAAAGAGCTAACTATCCGTAGATAGAAAATCTTTTTAACTAATAGTGAGTCGTATTA-3′ (promoter region underlined), was used to synthesize 43-nt A start 5′ triphosphate (pppA-) RNA, which indicates the first 43-nt of PEDV 5′ genome RNA. Oligo 5′-AGAAAAAGAGCTAACTATCCGTAGATAGAAAATCTTTTTAACCTATAGTGAGTCGTATTA-3′ was the template to synthesize 43-nt G start 5′ triphosphate (pppG-) RNA. Using the same strategy, 30-, 20-, 10-, 9-, 8-, 7-, 6-, 5-, 4-, 3-, and 2-nt A starting 5′ triphosphate RNA templates were also synthesized and purified. A 296-nt RNA containing the 5′ untranscribed region (UTR) of the PEDV genome was also generated and purified using the same method. Lastly, 43-nt RNA templates of the 5′ termini of IBV TGEV (gi: 110746821), SARS (gi: 40795428), PDCoV (gi: 668361756), and IBV (gi: 9626535) and poly(A)43 and poly(G)43 were synthesized in vitro. To prepare substrates for the RNA capping reaction, the transcription reactions were carried out with T7-Scribe Standard RNA IVT kit (CellScript LLC) using regular NTPs. The transcripts were extracted with phenol-chloroform-isoamyl alcohol (25:24:1) and precipitated with ethanol three times in the presence of glycogen (Sigma). The resulting RNA pellet was washed with 80% ethanol, dried, and dissolved in 40 μl of RNase-free H2O.
Preparation of capped RNA substrates.
In vitro-synthesized RNAs were capped using a ScriptCap m7G capping system according to the manufacturer’s protocol (CellScript) to generate 32P-labeled cap structures (m7Gp*ppA- or m7Gp*ppG- and Gp*ppA- or Gp*ppG-) (* indicates 32P label). The methyl donor SAM was absent for preparing the Gp*ppA/Gp*ppG-RNA, and inorganic pyrophosphatase (1 U; NEB) was added to enhance the yield of 32P-labeled Gp*ppA/Gp*ppG-RNA, because the capping reaction is reversible in the absence of the methyl donor SAM. After 2 h at 37°C, the reaction mixtures were treated with 10 U of calf intestinal alkaline phosphatase (NEB) for 1 h, followed by purification with phenol-chloroform-isoamyl alcohol (25:24:1) three times and precipitation with ethanol in the presence of glycogen (Sigma). The resulting RNA pellets were washed with 80% ethanol, dried, and dissolved in various amounts RNase-free water to make 2 kcpm/μl RNA substrate stocks.
Biochemical assays for G-N-7 MTase activity.
One microgram of purified recombinant nsp14 protein and 2 × 103 cpm of 32P-labeled RNA substrates were added to an 8.5-μl reaction mixture (50 mM Tris-HCl [pH 7.0], 6 mM KCl, 2 mM DTT, 1.25 mM MgCl2, 10 U RNase inhibitor, 0.2 mM SAM) and incubated at 37°C for 3 h. Reactions were stopped by the addition of 1% sodium dodecyl sulfate (SDS) and 0.5 mM EDTA. Then, RNA substrates were extracted with phenol-chloroform and precipitated with ethanol. RNA cap structures were liberated with 5 μg of nuclease P1 (Sigma) in a buffer containing 10 mM Tris-HCl (pH 7.5) and 1 mM ZnCl2 at 50°C for 30 min, spotted onto polyethyleneimine cellulose F plates (Merck) for thin-layer chromatography (TLC), and developed in 0.4 M ammonium sulfate for 1 h. The extent of the 32P-labeled cap was determined by scanning the chromatogram with a phosphorimager. The MTase activity assay with 3H-labeling was carried out in a 30-μl reaction mixture containing 1 μg of purified recombinant nsp14, 14.9 pM of 3H-SAM (67.3 Ci/mmol, 0.5 μCi/μl), and 2 mM NTPs or cap analogs (m7GpppA/m7GpppG/GpppA/GpppG) in methylation buffer (50 mM Tris-HCl [pH 7.0], 2 mM DTT, 1.25 mM MgCl2, and 10 U RNase inhibitor) at 37°C for 3 h. The 3H-labeled NTPs or cap analogs were purified in small DEAE-Sephadex columns and quantitated by a scintillation counter (PerkinElmer).
Prediction 3D structure model of PEDV nsp14.
The crystal structure of SARS-CoV nsp14 (PDB ID 5C8S) was chosen as the template to generate a three-dimensional (3D) model of PEDV nsp14. Protein structure predictions were carried out via MODELLER (63) and using a publicly available service for fold recognition: mGenTHREADER (64). Structural figures were drawn with Chimera (65). Structural based sequence alignments were displayed with ESPRIPT (66).
Assembly of the full-length genomic cDNA of PEDV.
The full-length genomic cDNA of PEDV Colorado strain was assembled into a bacterial artificial chromosome (BAC) backbone vector pSMART-BAC-BamHI (CopyRight v2.0 BAC Cloning kits; Lucigen). The pSMART-BAC vector was modified to insert a yeast replication origin from the plasmid pYES1L (Thermo Fisher Scientific), a cytomegalovirus (CMV) promoter from pCI vector (Promega), a hepatitis delta virus ribozyme (HDVRz) sequence, and a bovine growth hormone (BGH) polyadenylation and terminator. The full-length cDNA clone of PEDV was constructed using seven overlapping fragments (designated from A to G) by using the GeneArt High-order genetic assembly system according to the protocol provided by Thermo Fisher Scientific. Briefly, 100 ng of vector was mixed with 200 ng of each PEDV DNA fragment in polyethylene glycol (PEG)-lithium acetate (LiAc) solution; the ligation products were transformed into MaV 203 competent yeast cells by electroporation and plated on CSM-Trp agar plates. After incubation for 2 days at 30°C, an individual colony was picked for yeast colony PCR analysis. For initial screening, the connection regions between fragments were amplified by PCR and sequenced. The positive plasmid was then transformed into TOP10B competent cells, and plasmid DNA was verified by restriction enzyme digestion, PCR analysis, and sequencing to confirm that no additional mutations were introduced during the assembly. The final plasmid was designated pSMART-BAC-PEDV. Primers used in this study are listed in Table 1.
TABLE 1.
Primers used in this study
| Primer | Nucleotide sequence (5′→3′) |
|---|---|
| Cloning | |
| 1-4004-F | GCGCAGCGGCGGCCGCGCTGATACCGCCGCCTTGACATTGATTATTGACTAGTT |
| 1-4004-R | TCAGGAGCTACATTAGGACCATCATCCTCAAGCTCAGAG |
| 4005-7993-F | TGAGGATGATGGTCCTAATGTAGCTCCTGAAACAAATGT |
| 4005-7993-R | ACACACTAACAAAAGCACTATGTAAGCTTGCACCAAAAT |
| 7994-12384-F | CAAGCTTACATAGTGCTTTTGTTAGTGTGTTGTCGAATA |
| 7994-12384-R | GGTTACCTCCTTAACGTGTACCACCTTGCCATCATTGTC |
| 12385-16986-F | GGCAAGGTGGTACACGTTAAGGAGGTAACCGCACAGAAT |
| 12385-16986-R | ACTCAACACGAGCGCGCTGTGGTATGATGCGTGAACATT |
| 16987-21901-F | GCATCATACCACAGCGCGCTCGTGTTGAGTGTTATGATG |
| 16987-21901-R | ATAAGAAATACCATCCTCACCAGCACTAGTAACATTAAG |
| 21902-26980-F | ACTAGTGCTGGTGAGGATGGTATTTCTTATCAACCCTGT |
| 21902-26980-R | ACTGACAGAAGCCATAAAGTTTCTGTTTAGACTAAATGA |
| 26981-28980-F | CTAAACAGAAACTTTATGGCTTCTGTCAGTTTTCAGGAT |
| 26981-28980-R | GCTCACTGACTTTAATTAACTGCGGCGAGGGGCGCGCCACCGCATCCCCAGCAT |
| Mutagenesis | |
| D350A-F | AAATGGTTTTGCTTTGCCAAGAATCCTACTAATTC |
| D350A-R | GAATTAGTAGGATTCTTGGCAAAGCAAAACCATTT |
| G332A-F | GCCATATATGATATTGCCAATCCTAAGGGCATTAG |
| G332A-R | CTAATGCCCTTAGGATTGGCAATATCATATATGGC |
| P334A-K335A-F | GATATTGGCAATGCTGCGGGCATTAGATGTGCC |
| P334A-K335A-R | GGCACATCTAATGCCCGCAGCATTGCCAGCATC |
| D330A-F | GAAAGCCATATATGCTATTGGCAATCCTAAGGGC |
| D330A-R | GCCCTTAGGATTGCCAATAGCATATATGGCTTTC |
| RT-qPCR | |
| IFN-λ1F | GGTGCTGGCGACTGTGATG |
| IFN-λ1R | GATTGGAACTGGCCCATGTG |
| IFN-λ3F | ACTTGGCCCAGTTCAAGTCT |
| IFN-λ3R | CATCCTTGGCCCTCTTGA |
| IFN-λ4F | GCTATGGGACTGTGGGTCTT |
| IFN-λ4R | AGGGAGCGGTAGTGAGAGAG |
| IFN-αF | ACCTTTGCTTTACTGGTGGCC |
| IFN-αR | ATCTGTGCCAGGAGCATCAAG |
| IFN-βF | TAGGCGACACTGTTCGTGTTG |
| IFN-βR | CCAAGCAAGTTGTAGCTCATGG |
| GAPDH-F | CAATGACCCCTTCATTGACC |
| GAPDH-R | GACAAGCTTCCCGTTCTCAG |
| Subgenome-F | AATCCAGGGCCACTTCGAA |
| Subgenome-R | TTCGCCCTTGGGAATTCTC |
| Genome-F | TGAAGCCGTCTCATACTATTCTG |
| Genome-R | AATCCCTCAACAGTGTCAGC |
Engineering nsp14 mutations in PEDV full-length genomic cDNA.
Site-directed mutagenesis was used to engineer nsp14 mutations in PEDV full-length genome cDNA plasmid using a two-step selection/counterselection recombineering method as described previously (67, 68). The first step of the BAC modification was to place the 30S ribosomal subunit protein S12 (rpsl+)-kanamycin (Kana) cassette at the locus of interest via positive kanamycin selection. SW102 bacteria harboring the pSMART-BAC-PEDV construct were incubated at 42°C for 15 min to induce the recombinases. For recombineering, 100 ng of the expression cassette was added to electrocompetent bacteria. Drug-resistant recombinants were first selected by using kanamycin resistance and then analyzed with PCR to verify that the insertion was in the proper location. The second step was to replace the rpsl+-Kana cassette in the BAC intermediate construct with a genome cassette carrying the desired mutant nucleotide via negative selection with streptomycin. The induction of recombination and electroporation were performed as for the first step. Following electroporation, diluted SW102 bacteria were plated on LB agar plates supplemented with streptomycin to select for bacteria harboring BACs that lost the rpsl+-Kana cassette. Using this strategy, D330A, G332A, P334A-K335A, and D350A nsp14 mutants were engineered in pSMART-BAC-PEDV. All plasmids were sequenced to confirm that no additional mutations were introduced.
Recovery of recombinant PEDV.
Confluent Vero CCL-81 cells in 6-well plates were transfected with 4 μg of pSMART-BAC-PEDV or pSMART-BAC-PEDV-mutation and 2 μg of a plasmid encoding the PEDV N gene (pCAGGS-N) using Lipofectamine 3000 reagent according to the manufacturer’s instructions. At 24 h posttransfection, cell culture medium was replaced with 2 ml of DMEM supplemented with 0.018% (wt/vol) tryptose phosphate broth (TPB), 0.02% yeast extract, and 5 μg/ml trypsin 1:250 and incubated at 37°C for 4 days. Cells were harvested and subjected to freeze-thaw three times and mixed with cell culture supernatant. After centrifugation at 2,000 × g for 5 min, supernatants were collected for further passage in fresh Vero CCL-81 cells. Successful recovery of rPEDV was initially characterized by syncytium formation and further confirmed by plaque assay and sequencing.
Purification of PEDV and virion RNA extraction.
rPEDV-D350A and rPEDV were purified by sucrose gradient ultracentrifugation. Briefly, 10 T150 flasks of Vero CCL-81 cells were infected with rPEDV-D350A or rPEDV. At 48 h, cell culture supernatants were harvested, and cell debris was removed by centrifugation (3,000 × g for 20 min). Virus was then concentrated by ultracentrifugation in a Ty50.2 rotor (Beckman Coulter, Fullerton, CA) at 30,000 × g for 2 h at 4°C. The pellet was resuspended in 500 μl of NTE buffer (100 mM NaCl, 10 mM Tris, 1 mM EDTA, pH 7.4) and further purified through a 10% (wt/vol) sucrose NTE cushion by centrifugation for 2 h at 50,000 × g at 4°C in an SW50.1 rotor (Beckman). The final virus-containing pellet was resuspended in 500 μl of NTE buffer overnight. Virion RNA was extracted from purified virions with TRIzol reagent, and genome RNA copies were quantified by real-time RT-PCR.
Single-cycle growth curves.
Confluent Vero CCL-81 or IPEC-DQ cells were infected with PEDV at a multiplicity of infection (MOI) of 1.0 or 0.1. After 1 h of absorption, the inoculum was removed, the cells were washed twice with DMEM, fresh DMEM (supplemented with 5 μg/ml trypsin 1:250) was added, and the infected cells were incubated at 37°C. Cell culture fluids were collected at the designated intervals, and virus titers were determined by plaque assay in Vero CCL-81 cells.
Plaque assays.
Confluent Vero CCL-81 cells in 6-well plates were infected with serial dilutions of rPEDV or mutant in DMEM containing tryptose phosphate broth (0.3% [vol/vol]) and trypsin (5 μg/ml). After absorption for 1 h at 37°C, cells were washed three times with DMEM and overlaid with 2 ml of DMEM containing low-melting agarose (1% [wt/vol]), tryptose phosphate broth (0.3% [vol/vol]), and trypsin (5 μg/ml). After incubation at 37°C for 2 to 5 days, cells were fixed with 4% paraformaldehyde for 2 h. The overlays were removed, and the plaques were visualized after staining with crystal violet. The diameter of plaques for each virus were measured using Image J software.
Quantification of type I and III IFNs by real-time RT-PCR.
For PEDV infection, confluent IPEC-DQ cells were infected with each virus at an MOI of 0.1 or 1.0 at 37°C in DMEM for 1 h. Inocula were removed, cells were washed with PBS, and fresh medium was added. For virion RNA transfection, confluent IPEC-DQ cells were transfected with 1010 RNA copies of each virion RNA. At 11 and 18 h postinfection or transfection, cell culture supernatants were aspirated and cell lysates were harvested by adding TRIzol reagent. Total RNA was purified using the phenol-chloroform method. cDNA was generated by reverse transcription (RT)-PCR using 1 μg of total RNA according to the protocol with random primers. The cDNA was subjected to quantitative PCR using SYBR green PCR master mix according to the manufacturer’s instructions (Life Technologies, CA) with the ABI 7500 real-time PCR system. The primer sequences are described in Table 1; the GAPDH gene was used as an internal control. The threshold cycle (CT) values for target genes and the differences in their CT values were determined. Relative transcription levels of target genes are presented as fold changes relative to the respective control by using the 2−ΔΔCT method.
Western blotting.
Confluent Vero CCL-81 or IPEC-DQ cells were infected with PEDV. At the designated time postinfection, cells were lysed in lysis buffer containing 5% β-mercaptoethanol, 0.01% NP-40, and 2% SDS. Equal amounts of samples were analyzed by 12% SDS-PAGE and transferred to a Hybond enhanced chemiluminescence nitrocellulose membrane (Amersham) in a Mini Trans-Blot electrophoretic transfer cell (Bio-Rad). The blot was probed with mouse monoclonal antibody (MAb) against PEDV-N protein (clone S-1-5; Medgene Labs) at a dilution of 1:5,000 and secondary anti-mouse antibody conjugated to horseradish peroxidase (HRP) (Abcam) at dilution of 1:10,000. The blot was developed with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) and exposed to Kodak BioMax MR film.
Statistical analysis.
Quantitative analysis was performed by either densitometric scanning of autoradiographs or by using a phosphorimager (Typhoon; GE Healthcare, Piscataway, NJ) and ImageQuant TL software (GE Healthcare, Piscataway, NJ) or Image J software (NIH, Bethesda, MD). Statistical analysis was performed by one-way multiple comparisons using SPSS (version 8.0) statistical analysis software (SPSS Inc., Chicago, IL). A P value of <0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank Qiuhong Wang for providing IPEC-DQ cells for this study and members of the J. Li laboratory for critical readings of the manuscript.
J. Gu was supported by Key Research & Development Program of China (2016YFD0500102). Work in J. Li’s lab was supported by start-up funds from the Department of Veterinary Biosciences at The Ohio State University.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Muthukrishnan S, Morgan M, Banerjee AK, Shatkin AJ. 1976. Influence of 5′-terminal m7G and 2′–O-methylated residues on messenger ribonucleic acid binding to ribosomes. Biochemistry 15:5761–5768. doi: 10.1021/bi00671a012. [DOI] [PubMed] [Google Scholar]
- 2.Furuichi Y, LaFiandra A, Shatkin AJ. 1977. 5′-Terminal structure and mRNA stability. Nature 266:235–239. doi: 10.1038/266235a0. [DOI] [PubMed] [Google Scholar]
- 3.Furuichi Y, Shatkin AJ. 2000. Viral and cellular mRNA capping: past and prospects. Adv Virus Res 55:135–184. doi: 10.1016/s0065-3527(00)55003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cougot N, van Dijk E, Babajko S, Séraphin B. 2004. ‘Cap-tabolism’. Trends Biochem Sci 29:436–444. doi: 10.1016/j.tibs.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Lai MM, Stohlman SA. 1981. Comparative analysis of RNA genomes of mouse hepatitis viruses. J Virol 38:661–670. doi: 10.1128/JVI.38.2.661-670.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lai MM, Patton CD, Stohlman SA. 1982. Further characterization of mRNA’s of mouse hepatitis virus: presence of common 5′-end nucleotides. J Virol 41:557–565. doi: 10.1128/JVI.41.2.557-565.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Vliet AL, Smits SL, Rottier PJ, de Groot RJ. 2002. Discontinuous and non-discontinuous subgenomic RNA transcription in a nidovirus. EMBO J 21:6571–6580. doi: 10.1093/emboj/cdf635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Cai H, Pan J, Xiang N, Tien P, Ahola T, Guo DY. 2009. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc Natl Acad Sci U S A 106:3484–3489. doi: 10.1073/pnas.0808790106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouvet M, Debarnot C, Imbert I, Selisko B, Snijder EJ, Canard B, Decroly E. 2010. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog 6:e1000863. doi: 10.1371/journal.ppat.1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lugari A, Betzi S, Decroly E, Bonnaud E, Hermant A, Guillemot JC, Debarnot C, Borg JP, Bouvet M, Canard B, Morelli X, Lecine P. 2010. Molecular mapping of the RNA cap 2′-O-methyltransferase activation interface between severe acute respiratory syndrome coronavirus nsp10 and nsp16. J Biol Chem 285:33230–33241. doi: 10.1074/jbc.M110.120014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Decroly E, Debarnot C, Ferron F, Bouvet M, Coutard B, Imbert I, Gluais L, Papageorgiou N, Sharff A, Bricogne G, Ortiz-Lombardia M, Lescar J, Canard B. 2011. Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2′-O-methyltransferase nsp10/nsp16 complex. PLoS Pathog 7:e1002059. doi: 10.1371/journal.ppat.1002059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y, Su CY, Ke M, Jin X, Xu LR, Zhang Z, Wu AD, Sun Y, Yang ZN, Tien P, Ahola T, Liang Y, Liu XQ, Guo DY. 2011. Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2′-O-methylation by nsp16/nsp10 protein complex. PLoS Pathog 7:e1002294. doi: 10.1371/journal.ppat.1002294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma Y, Wu L, Shaw N, Gao Y, Wang J, Sun Y, Lou Z, Yan L, Zhang R, Rao Z. 2015. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc Natl Acad Sci U S A 112:9436–9441. doi: 10.1073/pnas.1508686112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Sun Y, Wu AD, Xu S, Pan RG, Zeng C, Jin X, Ge XY, Shi ZL, Ahola T, Chen Y, Guo DY. 2015. Coronavirus nsp10/nsp16 methyltransferase can be targeted by nsp10-derived peptide in vitro and in vivo to reduce replication and pathogenesis. J Virol 89:8416–8427. doi: 10.1128/JVI.00948-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Tao J, Sun Y, Wu A, Su C, Gao G, Cai H, Qiu S, Wu Y, Ahola T, Guo D. 2013. Structure-function analysis of severe acute respiratory syndrome coronavirus RNA cap guanine-N7-methyltransferase. J Virol 87:6296–6305. doi: 10.1128/JVI.00061-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ivanov KA, Ziebuhr J. 2004. Human coronavirus 229E nonstructural protein 13: characterization of duplex-unwinding, nucleoside triphosphatase, and RNA 5′-triphosphatase activities. J Virol 78:7833–7838. doi: 10.1128/JVI.78.14.7833-7838.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minskaia E, Hertzig T, Gorbalenya AE, Campanacci V, Cambillau C, Canard B, Ziebuhr J. 2006. Discovery of an RNA virus 3′->5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc Natl Acad Sci U S A 103:5108–5113. doi: 10.1073/pnas.0508200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan Y, Tian X, Qin P, Wang B, Zhao P, Yang YL, Wang L, Wang D, Song Y, Zhang X, Huang YW. 2017. Discovery of a novel swine enteric alphacoronavirus (SeACoV) in southern China. Vet Microbiol 211:15–21. doi: 10.1016/j.vetmic.2017.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma YM, Zhang Y, Liang XY, Oglesbee M, Krakowka S, Niehaus A, Wang GP, Jia AQ, Song HH, Li JR. 2016. Two-way antigenic cross-reactivity between porcine epidemic diarrhea virus and porcine deltacoronavirus. Vet Microbiol 186:90–96. doi: 10.1016/j.vetmic.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W, China Novel Coronavirus Investigating and Research Team. 2020. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Y, Zhang Y, Liang X, Lou F, Oglesbee M, Krakowka S, Li J. 2015. Origin, evolution, and virulence of porcine deltacoronaviruses in the United States. mBio 6:e00064-15. doi: 10.1128/mBio.00064-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pensaert MB, de Bouck P. 1978. A new coronavirus-like particle associated with diarrhea in swine. Arch Virol 58:243–247. doi: 10.1007/BF01317606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coussement W, Ducatelle R, Debouck P, Hoorens J. 1982. Pathology of experimental CV777 coronavirus enteritis in piglets. I. Histological and histochemical study. Vet Pathol 19:46–56. doi: 10.1177/030098588201900108. [DOI] [PubMed] [Google Scholar]
- 25.Sun RQ, Cai RJ, Chen YQ, Liang PS, Chen DK, Song CX. 2012. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg Infect Dis 18:161–163. doi: 10.3201/eid1801.111259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian PF, Jin YL, Xing G, Qv LL, Huang YW, Zhou JY. 2014. Evidence of recombinant strains of porcine epidemic diarrhea virus, United States, 2013. Emerg Infect Dis 20:1735–1738. doi: 10.3201/eid2010.140338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwon HJ, Ryu YB, Kim YM, Song N, Kim CY, Rho MC, Jeong JH, Cho KO, Lee WS, Park SJ. 2013. In vitro antiviral activity of phlorotannins isolated from Ecklonia cava against porcine epidemic diarrhea coronavirus infection and hemagglutination. Bioorg Med Chem 21:4706–4713. doi: 10.1016/j.bmc.2013.04.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alonso C, Goede DP, Morrison RB, Davies PR, Rovira A, Marthaler DG, Torremorell M. 2014. Evidence of infectivity of airborne porcine epidemic diarrhea virus and detection of airborne viral RNA at long distances from infected herds. Vet Res 45:73. doi: 10.1186/s13567-014-0073-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stevenson GW, Hoang H, Schwartz KJ, Burrough ER, Sun D, Madson D, Cooper VL, Pillatzki A, Gauger P, Schmitt BJ, Koster LG, Killian ML, Yoon KJ. 2013. Emergence of Porcine epidemic diarrhea virus in the United States: clinical signs, lesions, and viral genomic sequences. J Vet Diagn Invest 25:649–654. doi: 10.1177/1040638713501675. [DOI] [PubMed] [Google Scholar]
- 30.Rahmeh AA, Li J, Kranzusch PJ, Whelan SP. 2009. Ribose 2′-O methylation of the vesicular stomatitis virus mRNA cap precedes and facilitates subsequent guanine-N-7 methylation by the large polymerase protein. J Virol 83:11043–11050. doi: 10.1128/JVI.01426-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dong H, Ray D, Ren S, Zhang B, Puig-Basagoiti F, Takagi Y, Ho CK, Li H, Shi PY. 2007. Distinct RNA elements confer specificity to flavivirus RNA cap methylation events. J Virol 81:4412–4421. doi: 10.1128/JVI.02455-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong HP, Ren SP, Zhang B, Zhou YS, Puig-Basagoiti F, Li HM, Shi PY. 2008. West Nile virus methyltransferase catalyzes two methylations of the viral RNA cap through a substrate-repositioning mechanism. J Virol 82:4295–4307. doi: 10.1128/JVI.02202-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin JL, McMillan FM. 2002. SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr Opin Struct Biol 12:783–793. doi: 10.1016/s0959-440x(02)00391-3. [DOI] [PubMed] [Google Scholar]
- 34.Stadler J, Zoels S, Fux R, Hanke D, Pohlmann A, Blome S, Weissenbock H, Weissenbacher-Lang C, Ritzmann M, Ladinig A. 2015. Emergence of porcine epidemic diarrhea virus in southern Germany. BMC Vet Res 11:142. doi: 10.1186/s12917-015-0454-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin TY, Schneller S, Zust R, Dong H, Thiel V, Sen GC, Fensterl V, Klimstra WB, Pierson TC, Buller RM, Gale M Jr, Shi PY, Diamond MS. 2010. 2′-O Methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zust R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, Szretter KJ, Baker SC, Barchet W, Diamond MS, Siddell SG, Ludewig B, Thiel V. 2011. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol 12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Case JB, Ashbrook AW, Dermody TS, Denison MR. 2016. Mutagenesis of S-adenosyl-l-methionine-binding residues in coronavirus nsp14 N7-methyltransferase demonstrates differing requirements for genome translation and resistance to innate immunity. J Virol 90:7248–7256. doi: 10.1128/JVI.00542-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng X, van Geelen A, Buckley AC, O’Brien A, Pillatzki A, Lager KM, Faaberg KS, Baker SC. 2019. Coronavirus endoribonuclease activity in porcine epidemic diarrhea virus suppresses type I and type III interferon responses. J Virol 93:e02000-18. doi: 10.1128/JVI.02000-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Q, Ke H, Blikslager A, Fujita T, Yoo D. 2018. Type III interferon restriction by porcine epidemic diarrhea virus and the role of viral protein nsp1 in IRF1 signaling. J Virol 92:e01677-17. doi: 10.1128/JVI.01677-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sang Y, Rowland RR, Blecha F. 2010. Molecular characterization and antiviral analyses of porcine type III interferons. J Interferon Cytokine Res 30:801–807. doi: 10.1089/jir.2010.0016. [DOI] [PubMed] [Google Scholar]
- 41.Jin X, Chen Y, Sun Y, Zeng C, Wang Y, Tao J, Wu A, Yu X, Zhang Z, Tian J, Guo D. 2013. Characterization of the guanine-N7 methyltransferase activity of coronavirus nsp14 on nucleotide GTP. Virus Res 176:45–52. doi: 10.1016/j.virusres.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ensinger MJ, Moss B. 1976. Modification of the 5′ terminus of mRNA by an RNA (guanine-7-)-methyltransferase from HeLa cells. J Biol Chem 251:5283–5291. [PubMed] [Google Scholar]
- 43.Mao X, Shuman S. 1996. Vaccinia virus mRNA (guanine-7-)methyltransferase: mutational effects on cap methylation and AdoHcy-dependent photo-cross-linking of the cap to the methyl acceptor site. Biochemistry 35:6900–6910. doi: 10.1021/bi960221a. [DOI] [PubMed] [Google Scholar]
- 44.Saha N, Shuman S. 2001. Effects of alanine cluster mutations in the D12 subunit of vaccinia virus mRNA (guanine-N7) methyltransferase. Virology 287:40–48. doi: 10.1006/viro.2001.1006. [DOI] [PubMed] [Google Scholar]
- 45.Ahola T, Kaariainen L. 1995. Reaction in alphavirus mRNA capping: formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc Natl Acad Sci U S A 92:507–511. doi: 10.1073/pnas.92.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ahola T, Ahlquist P. 1999. Putative RNA capping activities encoded by brome mosaic virus: methylation and covalent binding of guanylate by replicase protein 1a. J Virol 73:10061–10069. doi: 10.1128/JVI.73.12.10061-10069.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang JT, Sheng WH, Fang CT, Chen YC, Wang JL, Yu CJ, Chang SC, Yang PC. 2004. Clinical manifestations, laboratory findings, and treatment outcomes of SARS patients. Emerg Infect Dis 10:818–824. doi: 10.3201/eid1005.030640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang W, Du RH, Li B, Zheng XS, Yang XL, Hu B, Wang YY, Xiao GF, Yan B, Shi ZL, Zhou P. 2020. Molecular and serological investigation of 2019-nCoV infected patients: implication of multiple shedding routes. Emerg Microbes Infect 9:386–389. doi: 10.1080/22221751.2020.1729071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Almazan F, Dediego ML, Galan C, Escors D, Alvarez E, Ortego J, Sola I, Zuniga S, Alonso S, Moreno JL, Nogales A, Capiscol C, Enjuanes L. 2006. Construction of a severe acute respiratory syndrome coronavirus infectious cDNA clone and a replicon to study coronavirus RNA synthesis. J Virol 80:10900–10906. doi: 10.1128/JVI.00385-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eckerle LD, Lu X, Sperry SM, Choi L, Denison MR. 2007. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J Virol 81:12135–12144. doi: 10.1128/JVI.01296-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eckerle LD, Becker MM, Halpin RA, Li K, Venter E, Lu X, Scherbakova S, Graham RL, Baric RS, Stockwell TB, Spiro DJ, Denison MR. 2010. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog 6:e1000896. doi: 10.1371/journal.ppat.1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becares M, Pascual-Iglesias A, Nogales A, Sola I, Enjuanes L, Zuñiga S. 2016. Mutagenesis of coronavirus nsp14 reveals its potential role in modulation of the innate immune response. J Virol 90:5399–5414. doi: 10.1128/JVI.03259-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hou YX, Ke HZ, Kim J, Yoo D, Su YF, Boley P, Chepngeno J, Vlasova AN, Saif LJ, Wang QH. 2019. Engineering a live attenuated porcine epidemic diarrhea virus vaccine candidate via inactivation of the viral 2′-O-methyltransferase and the endocytosis signal of the spike protein. J Virol 93:e00406-19. doi: 10.1128/JVI.00406-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li L, Fu F, Xue M, Chen W, Liu J, Shi H, Chen J, Bu Z, Feng L, Liu P. 2017. IFN-lambda preferably inhibits PEDV infection of porcine intestinal epithelial cells compared with IFN-alpha. Antiviral Res 140:76–82. doi: 10.1016/j.antiviral.2017.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang D, Fang L, Xiao S. 2016. Porcine epidemic diarrhea in China. Virus Res 226:7–13. doi: 10.1016/j.virusres.2016.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu J, Chai X, Cheng Y, Xing G, Liao A, Du L, Wang Y, Lei J, Gu J, Zhou J. 2018. Molecular characteristics of the spike gene of porcine epidemic diarrhoea virus strains in Eastern China in 2016. Virus Res 247:47–54. doi: 10.1016/j.virusres.2018.01.013. [DOI] [PubMed] [Google Scholar]
- 57.Wang Y, Liu R, Lu M, Yang Y, Zhou D, Hao X, Zhou D, Wang B, Li J, Huang YW, Zhao Z. 2018. Enhancement of safety and immunogenicity of the Chinese Hu191 measles virus vaccine by alteration of the S-adenosylmethionine (SAM) binding site in the large polymerase protein. Virology 518:210–220. doi: 10.1016/j.virol.2018.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Wei Y, Zhang X, Cai H, Niewiesk S, Li J. 2014. Rational design of human metapneumovirus live attenuated vaccine candidates by inhibiting viral mRNA cap methyltransferase. J Virol 88:11411–11429. doi: 10.1128/JVI.00876-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun J, Wei Y, Rauf A, Zhang Y, Ma Y, Zhang X, Shilo K, Yu Q, Saif YM, Lu X, Yu L, Li J. 2014. Methyltransferase-defective avian metapneumovirus vaccines provide complete protection against challenge with the homologous Colorado strain and the heterologous Minnesota strain. J Virol 88:12348–12363. doi: 10.1128/JVI.01095-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma Y, Wei Y, Zhang X, Zhang Y, Cai H, Zhu Y, Shilo K, Oglesbee M, Krakowka S, Whelan SP, Li J. 2014. mRNA cap methylation influences pathogenesis of vesicular stomatitis virus in vivo. J Virol 88:2913–2926. doi: 10.1128/JVI.03420-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li SH, Dong H, Li XF, Xie X, Zhao H, Deng YQ, Wang XY, Ye Q, Zhu SY, Wang HJ, Zhang B, Leng QB, Zuest R, Qin ED, Qin CF, Shi PY. 2013. Rational design of a flavivirus vaccine by abolishing viral RNA 2′-O methylation. J Virol 87:5812–5819. doi: 10.1128/JVI.02806-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Menachery VD, Yount BL Jr, Josset L, Gralinski LE, Scobey T, Agnihothram S, Katze MG, Baric RS. 2014. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′-O-methyltransferase activity. J Virol 88:4251–4264. doi: 10.1128/JVI.03571-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. 2000. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct 29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 64.McGuffin LJ, Bryson K, Jones DT. 2000. The PSIPRED protein structure prediction server. Bioinformatics 16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 65.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 66.Gouet P, Courcelle E, Stuart DI, Metoz F. 1999. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 67.Fehr AR. 2020. Bacterial artificial chromosome-based lambda Red recombination with the I-SceI homing endonuclease for genetic alteration of MERS-CoV. Methods Mol Biol 2099:53–68. doi: 10.1007/978-1-0716-0211-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Narayanan K, Chen QW. 2011. Bacterial artificial chromosome mutagenesis using recombineering. J Biomed Biotechnol 2011:971296. doi: 10.1155/2011/971296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.