

Abstract
Although a persistent inflammatory state has long been associated with aging and negative health outcomes, the underlying mechanisms remain unclear. Mitochondrial dysfunction has been proposed as a cause of inflammaging, but evidence of an association in humans is lacking. In this study, we analyzed the cross-sectional association between inflammatory biomarkers and mitochondrial oxidative capacity in skeletal muscle, assessed as post-exercise phosphocreatine recovery time constant by phosphorus magnetic resonance spectroscopy, in a population of 669 adults (mean age 67 years) from the Baltimore Longitudinal Study of Aging. We observed that participants with lower mitochondrial oxidative capacity exhibited hallmarks of inflammation, specifically markedly higher levels of interleukin-6 and C-reactive protein, as well as increased erythrocyte sedimentation rate when compared with participants with better oxidative capacity, independent of age and sex. We speculate that this association reflects the observation that products of damaged mitochondria, such as mitochondrial DNA, activate multiple pathways that lead to inflammation. Furthermore, excess production of oxidative species (ROS) by dysfunctional mitochondria could trigger inflammation either directly via NF-κB or through oxidative damage to proteins, lipids, and nucleic acids. Longitudinal studies are necessary to ascertain whether and through which mechanisms mitochondrial dysfunction activate inflammation or whether both these phenomena derive from a common root.
Keywords: Mitochondria, Inflammaging, Oxidative stress, Phosphorus magnetic resonance spectroscopy
Introduction
A chronic, low-grade inflammatory state, accompanied by high blood levels of pro-inflammatory biomarkers, is often observed with aging and has been associated with a higher risk of sarcopenia, cardiovascular and neurodegenerative diseases, anemia, multimorbidity, disability, frailty, and premature death (Fabbri et al. 2014; Ferrucci and Fabbri 2018; Franceschi et al. 2000; Franceschi and Campisi, 2014; Wang et al. 2017). Despite extensive research and several hypotheses aimed toward explaining the origin of “inflammaging,” causal mechanisms remain unclear. An emerging hypothesis focuses on mitochondrial health and the interrelationship of age-related deterioration in mitochondrial function and generation of reactive oxygen species (ROS) (Conley et al. 2000; Porter et al. 2015; Sun et al. 2016). ROS can upregulate inflammation directly, by triggering the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and indirectly, by inducing macromolecular damage. Such damage can trigger a cascade of events that ultimately result in the production of type I interferons (α and β) and other proinflammatory cytokines. If the dysfunctional mitochondria are not removed, the pro-inflammatory state becomes chronic (Green et al. 2011).
The connection between poor mitochondrial health and chronic inflammation is further supported by evidence that diseases characterized by a strong mitochondrial dysfunction, such as type 2 diabetes, Alzheimer’s disease, and Parkinson’s disease, are associated with systemic inflammation (Beal 2003; De Felice and Ferreira 2014). Whether age-associated mild decline in mitochondrial function explains in part elevated blood levels of pro-inflammatory markers observed in many older persons is unknown.
In this study, we examined whether poorer or sub-optimal mitochondrial oxidative capacity assessed in vivo by phosphorus magnetic resonance spectroscopy (31P-MRS) in relatively healthy persons was associated with higher blood levels of interleukin (IL)-6 and C-reactive protein (CRP) and with increased erythrocyte sedimentation rate (ERS), among the most widely used biomarkers of inflammaging.
Methods
Participants
The study population consisted of 669 men and women aged 22 to 97 years from the Baltimore Longitudinal Study of Aging (BLSA), seen between April 2013 and December 2019. The BLSA is a prospective open cohort study of community-dwelling volunteers. Since 1958, the BLSA has been continuously enrolling participants, primarily from the Baltimore, MD, and Washington, DC, area. BLSA participants are free of major chronic conditions and functional impairments at enrollment and are followed for life regardless of changes in health and functional status, as described in detail elsewhere (Kuo et al. 2020; Stone and Norris 1966). Assessments were performed at the Clinical Research Unit of the Intramural Research Program of the National Institute on Aging during an on-unit clinic visit over 2.5 to 3.5 days. Certified nurse practitioners and certified technicians conducted all assessments according to standardized procedures. The study protocol was approved by the Institutional Review Board of the National Institute of Environmental Health Sciences (NIEHS, NC). All participants consented to participate in the study, after receiving detailed descriptions at every visit.
Demographic and health characteristics were ascertained either through self-report questionnaires or using standard criteria and algorithms (Guralnik et al. 1995). Physical activity was determined through a standardized questionnaire that enquired about different types of activities performed during a typical week. A measure considering all the high intensity activities, including brisk walking, was adopted in our models (Brach et al. 2004).
Body weight was measured in kilograms using a calibrated scale to the nearest 0.1 kg. Body height was measured in centimeters by a stadiometer to the nearest 0.1 cm (Lohman et al. 1988). Body mass index was calculated by dividing body weight by the square of height in meters.
IL-6, CRP, and ESR
IL-6 (pg/mL) and CRP (μg/mL) serum concentrations were quantified using enzyme-linked immunosorbent assays (ELISA, R&D Systems, Minneapolis, MN, and Alpha Diagnostic International, San Antonio, TX). ESR (mm/h) is defined as the rate at which erythrocytes (red blood cells) settle at the bottom of a test tube of anticoagulated whole blood in one hour. Higher ESR occurs when acute phase reactant proteins are high, indicating a pro-inflammatory condition. In this study, ESR was measured using a standard BD vacutainer tube (BD, Franklin Lakes, NJ). IL-6, CRP, and ESR values of 0 (non-detectable values) were excluded, and all measures were logarithmically transformed in the analyses to minimize the influence of skewed distribution.
Phosphorus magnetic resonance spectroscopy
Using a 3T MR scanner (Achieva, Philips Healthcare, Andover, MA), in vivo 31P-MRS measurements of the concentrations of the phosphorus-containing metabolites phosphocreatine (PCr), inorganic phosphate (Pi), and ATP were obtained from the vastus lateralis muscle of the left thigh, following a standardized protocol described previously (Choi et al. 2016; Coen et al. 2012). Spectra of phosphorous-containing metabolites were acquired before, during, and after a ballistic knee extension exercise performed for an average duration of 30 s. A series of pulse-acquire 31P spectra were obtained before, during, and after the exercise with a repetition time of 1.5 s, using a 10-cm 31P-tuned surface coil (PulseTeq, Surrey, UK) fastened above the left thigh. Signals were averaged over four successive acquisitions for signal-to-noise ratio enhancement, so that the data consisted of 75 spectra obtained with a temporal resolution of 6 s. The duration of exercise was optimized by consistently requiring a depletion in PCr of 33 to 67% relative to initial baseline values, in order to standardize the measure of oxidative function across different subjects and to provide sufficient dynamic range to fit the PCr recovery curve. If intramuscular acidosis, defined as intracellular pH lower than 6.8, was detected, the test was repeated at a lower intensity (Paganini et al. 1997). The pH was determined according to the chemical shift of Pi relative to PCr (Taylor et al. 1986). Spectra were processed with jMRUI software (MRUI Consortium, version 5.2), and metabolite concentrations were calculated by nonlinear least squares fitting implemented through AMARES (Naressi et al. 2001; Vanhamme et al. 1999).
Post-exercise PCr recovery rates were calculated by fitting time-dependent changes in PCr peak area to the monoexponential recovery function:
where PCr(0) is the end-of-exercise PCr signal area (i.e., the PCr signal area at the beginning of the recovery period); ΔPCr is the decrease in signal area from its pre-exercise baseline value, averaged from the multiple baseline scans, to PCr(0) resulting from in-magnet exercise; and τPCr is the PCr exponential recovery time constant, measured in seconds (Choi et al. 2016). This time constant is inversely proportional to the maximum in vivo oxidative capacity of skeletal muscle, with longer τPCr reflecting slower recovery and therefore lower oxidative capacity (Prompers et al. 2014). Since the energy demands during post-exercise PCr resynthesis are minimal, 1/τPCr reflects the maximum mitochondrial ATP production rate (Arnold et al. 1984; Choi et al. 2016; Conley et al. 2000).
Statistical analysis
Variables were described as mean values and standard deviations or proportions as appropriate. The time constant τPCr was expressed as an ordinal variable, through assignment to quintiles. Participants in the first quintile of τPCr (best mitochondrial function, τPCr below 39.7 s) were considered the reference group and compared with those in the higher quintiles using dummy variables in a linear regression analysis adjusted for sex (coded as 1 = male, 0 = female) and age. Body mass index (BMI) and physical activity (min/week) were initially considered in the model but later removed as they did not contribute significantly to model fit (P = 0.22 and P = 0.24, respectively). The analyses were performed using RStudio version 1.2.1335. P < 0.05 was considered statistically significant.
Results
Table 1 reports the demographic and health characteristics of the 669 study participants. Most subjects were Caucasian (67%), approximately half women (55%), and the majority had completed college (82%). The prevalence of chronic diseases was relatively low. The mean (standard deviation) for τPCr was 49.8 (11.7). Table 2 presents study population characteristics by quintile of mitochondrial oxidative capacity (τPCr). Age, BMI, physical activity, IL-6, CRP, and ESR differed across quintiles of τPCr, with participants in the highest quintile of τPCr (poorer oxidative capacity) being older, having higher BMI, reporting less physical activity, and having higher IL-6 and CRP blood levels and higher ESR.
Table 1.
Demographic and health characteristics of 669 adults from the Baltimore Longitudinal Study of Aging
| Characteristic | Mean (SD), median (IQR), or percent | |
|---|---|---|
| Age (years)a | 66.9 (15.2) | |
| Race (%) | White | 67.1 |
| Black | 23.5 | |
| Other | 9.4 | |
| Sex (% female) | 55.3 | |
| Education, < 16 years (%) | 17.1 | |
| Smoking status (%) | Never | 68 |
| Former | 30.5 | |
| Current | 1.5 | |
| BMI (kg/m2)a | 26.77 (4.49) | |
| Physical activity (min/week)a | 107.9 (165.3) | |
| τPCr (s)a | 49.80 (11.72) | |
| Interleukin 6 (pg/mL)b | 3.7 (2.9–4.98) | |
| CRP (μg/mL)b | 1.09 (0.57–2.31) | |
| ESR (mm/h)b | 8 (4–15) | |
| Hypertension (%) | 33.6 | |
| Myocardial infarction (%) | 2.1 | |
| Cerebrovascular accident (%) | 3.9 | |
| Peripheral artery disease (%) | 6.7 | |
| Diabetes mellitus (%) | 13.9 | |
| Cancer (%) | 26.1 | |
aMean (SD)
bMedian (IQR)
Table 2.
Characteristics of 669 study participants by quintile of mitochondrial oxidative capacity (τPCr, seconds)
| Characteristic | Quintile 1 | Quintile 2 | Quintile 3 | Quintile 4 | Quintile 5 | P value* |
|---|---|---|---|---|---|---|
| < 39.7 | 39.7–46.1 | 46.1–52.1 | 52.1–59.5 | > 59.5 | ||
| (n = 134) | (n = 134) | (n = 134) | (n = 134) | (n = 133) | ||
| Age (years)a | 56.9 (16) | 64.8 (14.7) | 66 (15.1) | 72.6 (12.6) | 74.4 (10.3) | < 0.0001 |
| Female sex (%) | 55.2 | 50 | 55.2 | 55.2 | 60.9 | 0.52 |
| BMI (kg/m2)a | 26 (4.1) | 26.6 (4.5) | 26.9 (4.5) | 27.2 (4.5) | 27.1 (4.7) | 0.025 |
| Physical activity (min/week)a | 165.9 (221.4) | 131.1 (160) | 90.2 (147.8) | 87.8 (142.9) | 67.4 (124.8) | < 0.0001 |
| IL-6 (pg/mL)b | 3.24 (2.6–4.15) | 3.45 (2.9–4.5) | 3.7 (2.9–4.68) | 3.8 (3.1–5) | 4.1 (3.2–5.5) | 0.02 |
| CRP (μg/mL)b | 0.8 (0.44–1.76) | 1.08 (0.48–1.96) | 1.06 (0.64–2.55) | 1.17 (0.61–2.54) | 1.76 (0.73–3.87) | < 0.0001 |
| ESR (mm/h)b | 6 (2–10) | 6 (3–12) | 7 (4–12) | 8.5 (5–18) | 14 (6–24) | < 0.0001 |
aMean (SD)
bMedian (IQR) or percent values
*ANOVA for continuous variables; Pearson chi-square test for categorical variables
Table 3 reports results of the linear regression model assessing the relationship between τPCr and log(IL-6) within quintiles of τPCr, using dummy variables and adjusting for age and sex. Participants in the first quintile of τPCr, those with the best oxidative capacity, served as the reference group. Participants with the lowest oxidative capacity (upper quintile of τPCr, with τPCr > 59.5 s) had significantly higher log(IL-6) levels than the reference group. None of the other quintiles differed from the reference quintile. Table 4 shows a similar linear regression treating log(CRP) as the outcome of interest. Participants in the third, fourth, and fifth quintile of τPCr had significantly and progressively higher log(CRP) than the reference group, independent of covariates. Finally, Table 5 reports the results of the regression with log(ESR) as outcome: subjects in the fourth and fifth quintiles of τPCr had larger log(ESR) than those in the first quintile, independent of age and sex (P = 0.06 and P = 0.0002 respectively). Figure 1 represents the relationships between τPCr and log(IL-6), log(CRP), and log(ESR).
Table 3.
Linear regression model with dummy variables for quintiles of τPCr assessing the relationship between log(IL-6) and τPCr, adjusting for covariates
| Variable | Beta coefficient | Standard error | P value |
|---|---|---|---|
| Quintile 2 | 0.03 | 0.04 | 0.47 |
| Quintile 3 | 0.04 | 0.04 | 0.38 |
| Quintile 4 | 0.07 | 0.04 | 0.12 |
| Quintile 5 | 0.10 | 0.04 | 0.02 |
| Age | 0.004 | 0.0009 | 0.0002 |
| Sex | 0.05 | 0.03 | 0.05 |
Table 4.
Linear regression model with dummy variables for quintiles of τPCr assessing the relationship between log(CRP) and τPCr, adjusting for covariates
| Variable | Beta coefficient | Standard error | P value |
|---|---|---|---|
| Quintile 2 | 0.08 | 0.06 | 0.18 |
| Quintile 3 | 0.13 | 0.06 | 0.03 |
| Quintile 4 | 0.15 | 0.06 | 0.02 |
| Quintile 5 | 0.24 | 0.06 | 0.0002 |
| Age | 0.003 | 0.001 | 0.05 |
| Sex | − 0.07 | 0.03 | 0.08 |
Table 5.
Linear regression model with dummy variables for quintiles of τPCr assessing the relationship between log(ESR) and τPCr, adjusting for covariates
| Variable | Beta coefficient | Standard error | P value |
|---|---|---|---|
| Quintile 2 | 0.01 | 0.05 | 0.87 |
| Quintile 3 | 0.02 | 0.05 | 0.67 |
| Quintile 4 | 0.09 | 0.05 | 0.06 |
| Quintile 5 | 0.19 | 0.05 | 0.0002 |
| Age | 0.006 | 0.001 | < 0.0001 |
| Sex | − 0.26 | 0.03 | < 0.0001 |
Fig. 1.
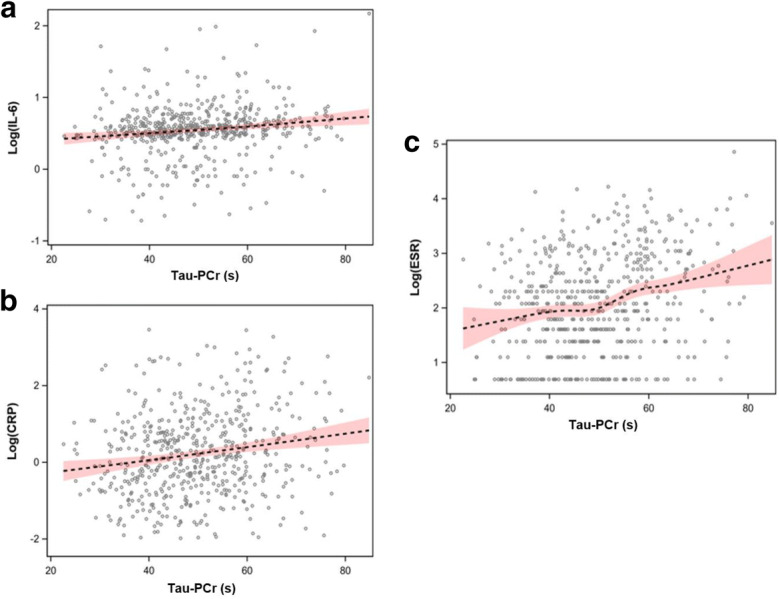
Scatterplots of the relationship between τPCr and log(IL-6) (a), log(CRP) (b), and log(ESR) (c) with loess, or locally weighted, regression lines
Discussion
The present study found that a greater proportion of persons with very low in vivo skeletal muscle mitochondrial oxidative capacity had higher levels of IL-6 and CRP as compared with subjects with better oxidative capacity.
For decades, IL-6 has been considered the “cytokine for gerontologists,” as high IL-6 levels have been consistently associated with higher prevalence and incidence of multiple chronic diseases and many adverse health outcomes (Ershler 1993; Maggio et al. 2006). For example, elevated IL-6 levels are often found in older patients with severe multimorbidity, disability, and frailty, with additional evidence that high levels of IL-6 predict the development of these outcomes, independent of other established risk factors (Cohen et al. 1997; Harris et al. 1999). IL-6 is widely recognized as a hallmark of inflammaging (Franceschi and Campisi 2014).
CRP is an acute-phase protein produced by hepatocytes in response to IL-6. Its main function is to activate the complement system after binding to dying cells or bacteria. Similar to CRP, ESR is a non-specific and widely used marker of inflammation. ESR is determined by the balance between factors that enhance sedimentation of red blood cells, such as fibrinogen, and factors resisting sedimentation. During an inflammatory process, fibrinogen increases, causing a shift of the balance toward sedimentation.
Several potential mechanisms have been proposed to explain the potential role of mitochondria in the genesis of inflammaging. In healthy mitochondria, the process of oxidative phosphorylation produces small amounts of ROS, which currently are considered important signaling molecules and are easily removed by multiple scavenging systems. However, if mitochondria become dysfunctional, production of ROS exceeds scavenging capacity and thus induces macromolecular damage. Mechanistic work, primarily conducted using model organisms, has shown that through oxidative stress, mitochondrial dysfunction triggers inflammation through multiple pathways (López-Armada et al. 2013). ROS directly activate NF-κB and other redox-sensitive transcription factors and induce production of cytokines, chemokines, eicosanoids, adhesion molecules, and inducible nitric oxide synthase (Chung et al. 2009; López-Armada et al. 2013). Further, free radicals can damage lipids, proteins, and nucleic acids, causing major structural and functional alterations of membranes, enzymes, signaling molecules, genes, and RNAs. Mitochondrial macromolecules are particularly susceptible to oxidative damage, due to proximity and reduced protective barriers (Sastre et al. 2000). Cardiolipin, a phospholipid specific to mitochondrial membranes and essential to preservation of the mitochondrial cristae and correct functioning of the electron transport chain (ETC) complexes, is a main target of free radical attack. Peroxidation of cardiolipin disrupts energy production, regulation of apoptosis, and other mitochondrial functions and has been implicated in the pathogenesis of sarcopenia and frailty (Ferrucci and Zampino 2020; Semba et al. 2019). Through impairment of the ETC, oxidized cardiolipin could induce further generation of ROS, mainly produced by complexes I and III (Murphy 2009).
When mitochondria sustain damage, mitochondrial components are released into the cytoplasm and extracellular space. These components include mtDNA, formyl peptides, and lipids, which can act as damage-associated molecular pattern (DAMP) agents, through a connection that can be traced back to the bacterial origins of the mitochondrion. Since mtDNA is not methylated, it is recognized by the immune system through members of the toll-like receptor family, activating an inflammatory response via stimulation of the NF-κB pathway (Sun et al. 2016). Formyl peptides can trigger the immune response by interacting with the formyl peptide receptor-1 (Sun et al. 2016).
Furthermore, mtDNA and increased levels of ROS activate the NLR family pyrin domain containing 3 (NLRP3) inflammasome, the major immune sensor for stress signals, which induces a pro-inflammatory response mainly through activation of caspase-1 and subsequent secretion of the cytokines IL-1β and IL-18 (Tschopp 2011). Notably, inflammasomes are implicated in the pathogenesis of age-related diseases such as metabolic syndrome, atherosclerosis, and Alzheimer’s disease (Salminen et al. 2012). mtDNA can also activate cyclic GMP-AMP synthase (cGAS), a cytosolic sensor for genomes of viruses and bacteria. cGAS catalyzes the formation of a messenger that activates the adaptor protein stimulator of interferon genes (STING) with consequent stimulation of the transcription factor interferon regulatory factor 3 (IRF3) and expression of innate immune response genes, including type I interferon (IFN) cytokines (Dhanwani et al. 2018; White et al. 2014). Among its functions, type I IFN enhances production of IL-6 (Zimmermann et al. 2016). An increase in plasma mtDNA concentration with age has been observed in past studies and has been associated with increased concentrations of IL-6 and other pro-inflammatory cytokines (Pinti et al. 2014).
In this study, we demonstrated that poor mitochondrial oxidative capacity is associated with elevated serum levels of IL-6 and CRP and with faster ESR. Because of the cross-sectional, observational nature of this analysis, our data cannot establish whether inflammation is a function of a health condition that also causes mitochondrial damage, or conversely that dysfunctional mitochondria directly cause inflammation through the mechanisms reported above and illustrated in Fig. 2. Nevertheless, our study shows that relatively healthy individuals, mainly those with poor mitochondrial oxidative capacity measured in vivo, show signs of systemic inflammation status. Of note, several subjects with poor oxidative capacity did not show elevated inflammatory markers (Fig. 1), suggesting that mitochondrial energetic dysfunction is not synonymous with systemic inflammation and may reflect non-disease factors such as low physical activity or exercise tolerance (Adelnia et al. 2019). Future studies should test the hypothesis that improving mitochondrial health through exercise and/or pharmacological interventions may reduce inflammation and its consequences on health. Another limitation of this study is that the mitochondrial oxidative capacity was measured exclusively in skeletal muscle, while systemic inflammation was assessed via blood samples.
Fig. 2.

Mechanisms through with damaged mitochondria may trigger inflammation. ROS can directly activate NF-kB or oxidize phospholipids (such as cardiolipin), proteins (such as ETC complexes), or mtDNA. Mutated mtDNA can translate into altered ETC protein complexes. Oxidized cardiolipins and altered ETC complexes generate more ROS. mtDNA can be released and activate the cGAS/STING pathway with induction of IFN production, activate the TLR of the neutrophils, or stimulate the NLRP3 inflammasome. ROS reactive oxygen species, mtDNA mitochondrial DNA, NFκB nuclear factor κB, ETC electron transport chain, cGAS cyclic GMP-AMP synthase, STING stimulator of interferon genes, IFN interferon, CRP C-reactive protein, NLRP3 NLR family pyrin domain containing 3, IL interleukin, TNFα tumor necrosis factor α, TLR toll-like receptor
Strengths of the study include a large and well-characterized cohort with rigorously standardized collection of in vivo 31P-MRS measurements, as well as accurate measurements of serum concentrations of IL-6 and CRP, and of ESR. The results of this study may not be generalizable to other populations, as the ethnic composition, exceptional health status, and high educational attainment of this cohort differ from the general population. Moreover, it is worth noting that since this population is healthy, the signals from mitochondrial dysfunction and inflammation observed in this study may be weaker than those that would be detected in the general population. Further longitudinal research is needed to gain insight into the temporal relationship between the occurrence of mitochondrial dysfunction and chronic inflammation. These studies should include older persons who are frail or pre-frail as these are most likely to show substantial impairment of mitochondrial oxidative capacity and oxidative stress.
Funding information
This work was supported by the Intramural Research Program of the National Institute on Aging.
Compliance with ethical standards
The study protocol was approved by the Institutional Review Board of the National Institute of Environmental Health Sciences (NIEHS, NC). All participants consented to participate in the study, after receiving detailed descriptions at every visit.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Adelnia F, Urbanek J, Osawa Y, Shardell M, Brennan NA, Fishbein KW, Spencer RG, Simonsick EM, Schrack JA, Ferrucci L. Moderate-to-vigorous physical activity is associated with higher muscle oxidative capacity in older adults. J Am Geriatr Soc. 2019;67:1695–1699. doi: 10.1111/jgs.15991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold D, Matthews P, Radda G. Metabolic recovery after exercise and the assessment of mitochondrial function in vivo in human skeletal muscle by means of 31P NMR. Magn Reson Med. 1984;1:307–315. doi: 10.1002/mrm.1910010303. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:120–131. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- Brach JS, Simonsick EM, Kritchevsky S, Yaffe K, Newman AB, Health A, Group BCSR The association between physical function and lifestyle activity and exercise in the health, aging and body composition study. J Am Geriatr Soc. 2004;52:502–509. doi: 10.1111/j.1532-5415.2004.52154.x. [DOI] [PubMed] [Google Scholar]
- Choi S, et al. 31P magnetic resonance spectroscopy assessment of muscle bioenergetics as a predictor of gait speed in the Baltimore Longitudinal Study of Aging. J Gerontol A Biol Sci Med Sci. 2016;71:1638–1645. doi: 10.1093/gerona/glw059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HY, et al. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res Rev. 2009;8:18–30. doi: 10.1016/j.arr.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coen PM, et al. Skeletal muscle mitochondrial energetics are associated with maximal aerobic capacity and walking speed in older adults. J Gerontol A Biol Sci Med Sci. 2012;68:447–455. doi: 10.1093/gerona/gls196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen HJ, Pieper CF, Harris T, Rao KMK, Currie MS. The association of plasma IL-6 levels with functional disability in community-dwelling elderly. J Gerontol A Biol Sci Med Sci. 1997;52:M201–M208. doi: 10.1093/gerona/52A.4.M201. [DOI] [PubMed] [Google Scholar]
- Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. J Physiol. 2000;526:203–210. doi: 10.1111/j.1469-7793.2000.t01-1-00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Ferreira ST. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes. 2014;63:2262–2272. doi: 10.2337/db13-1954. [DOI] [PubMed] [Google Scholar]
- Dhanwani R, Takahashi M, Sharma S. Cytosolic sensing of immuno-stimulatory DNA, the enemy within. Curr Opin Immunol. 2018;50:82–87. doi: 10.1016/j.coi.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ershler WB. Interleukin-6: a cytokine for gerontolgists. J Am Geriatr Soc. 1993;41:176–181. doi: 10.1111/j.1532-5415.1993.tb02054.x. [DOI] [PubMed] [Google Scholar]
- Fabbri E, et al. Aging and the burden of multimorbidity: associations with inflammatory and anabolic hormonal biomarkers. J Gerontol A Biol Sci Med Sci. 2014;70:63–70. doi: 10.1093/gerona/glu127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15:505–522. doi: 10.1038/s41569-018-0064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrucci L, Zampino M. A mitochondrial root to accelerated ageing and frailty. Nat Rev Endocrinol. 2020;16:133–4. [DOI] [PMC free article] [PubMed]
- Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging: an evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69:S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy–inflammation–cell death axis in organismal aging. Science. 2011;333:1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guralnik JM, Fried LP, Simonsick EM, Lafferty ME, Kasper JD. The Women’s Health and Aging Study: health and social characteristics of older women with disability: DIANE Publishing; 1995.
- Harris TB, et al. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am J Med. 1999;106:506–512. doi: 10.1016/S0002-9343(99)00066-2. [DOI] [PubMed] [Google Scholar]
- Kuo PL, et al. A roadmap to build a phenotypic metric of ageing: insights from the Baltimore Longitudinal Study of Aging. J Intern Med. 2020;287:373–94. [DOI] [PMC free article] [PubMed]
- Lohman TG, Roche AF, Martorell R. Anthropometric standardization reference manual. Champaign, IL: Human kinetics books; 1988. [Google Scholar]
- López-Armada MJ, Riveiro-Naveira RR, Vaamonde-García C, Valcárcel-Ares MN. Mitochondrial dysfunction and the inflammatory response. Mitochondrion. 2013;13:106–118. doi: 10.1016/j.mito.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Maggio M, Guralnik JM, Longo DL, Ferrucci L. Interleukin-6 in aging and chronic disease: a magnificent pathway. J Gerontol A Biol Sci Med Sci. 2006;61:575–584. doi: 10.1093/gerona/61.6.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naressi A, Couturier C, Castang I, De Beer R, Graveron-Demilly D. Java-based graphical user interface for MRUI, a software package for quantitation of in vivo/medical magnetic resonance spectroscopy signals. Comput Biol Med. 2001;31:269–286. doi: 10.1016/S0010-4825(01)00006-3. [DOI] [PubMed] [Google Scholar]
- Paganini A, Foley J, Meyer R. Linear dependence of muscle phosphocreatine kinetics on oxidative capacity. Am J Physiol. 1997;272:C501–C510. doi: 10.1152/ajpcell.1997.272.2.C501. [DOI] [PubMed] [Google Scholar]
- Pinti M, et al. Circulating mitochondrial DNA increases with age and is a familiar trait: implications for “inflamm-aging”. Eur J Immunol. 2014;44:1552–1562. doi: 10.1002/eji.201343921. [DOI] [PubMed] [Google Scholar]
- Porter C, et al. Mitochondrial respiratory capacity and coupling control decline with age in human skeletal muscle. Am J Physiol Endocrinol Metab. 2015;309:E224–E232. doi: 10.1152/ajpendo.00125.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prompers JJ, Wessels B, Kemp GJ, Nicolay K. MITOCHONDRIA: investigation of in vivo muscle mitochondrial function by 31P magnetic resonance spectroscopy. Int J Biochem Cell Biol. 2014;50:67–72. doi: 10.1016/j.biocel.2014.02.014. [DOI] [PubMed] [Google Scholar]
- Salminen A, Ojala J, Kaarniranta K, Kauppinen A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: impact on the aging process and age-related diseases. Cell Mol Life Sci. 2012;69:2999–3013. doi: 10.1007/s00018-012-0962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre J, Pallardó FV, de la Asunción García J, Viña J. Mitochondria, oxidative stress and aging. Free Radic Res. 2000;32:189–198. doi: 10.1080/10715760000300201. [DOI] [PubMed] [Google Scholar]
- Semba RD, Moaddel R, Zhang P, Ramsden CE, Ferrucci L. Tetra-linoleoyl cardiolipin depletion plays a major role in the pathogenesis of sarcopenia. Med Hypotheses. 2019;127:142–149. doi: 10.1016/j.mehy.2019.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JL, Norris AH. Activities and attitudes of participants in the Baltimore Longitudinal Study. J Gerontol. 1966;21:575–580. doi: 10.1093/geronj/21.4.575. [DOI] [PubMed] [Google Scholar]
- Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell. 2016;61:654–666. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor D, Styles P, Matthews P, Arnold D, Gadian D, Bore P, Radda G. Energetics of human muscle: exercise-induced ATP depletion. Magn Reson Med. 1986;3:44–54. doi: 10.1002/mrm.1910030107. [DOI] [PubMed] [Google Scholar]
- Tschopp J. Mitochondria: sovereign of inflammation? Eur J Immunol. 2011;41:1196–1202. doi: 10.1002/eji.201141436. [DOI] [PubMed] [Google Scholar]
- Vanhamme L, Van Huffel S, Van Hecke P, van Ormondt D. Time-domain quantification of series of biomedical magnetic resonance spectroscopy signals. J Magn Reson. 1999;140:120–130. doi: 10.1006/jmre.1999.1835. [DOI] [PubMed] [Google Scholar]
- Wang J, Leung K-S, Chow SK-H, Cheung W-H. Inflammation and age-associated skeletal muscle deterioration (sarcopaenia) J Orthop Transl. 2017;10:94–101. doi: 10.1016/j.jot.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159:1549–1562. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, et al. IFNα enhances the production of IL-6 by human neutrophils activated via TLR8. Sci Rep. 2016;6:19674. doi: 10.1038/srep19674. [DOI] [PMC free article] [PubMed] [Google Scholar]