

Abstract
Nonalcoholic fatty liver disease (NAFLD) is supposed to manifest its metabolic phenotype in the liver, but it is common to have lean individuals diagnosed with NAFLD, known as lean NAFLD. We conducted a two‐stage analysis to identify NAFLD‐associated loci in Japanese patients. In stage I, 275 metabolically healthy normal‐weight patients with NAFLD were compared with 1,411 non‐NAFLD controls adjusted for age, sex, and alcohol consumption by a genome‐wide association study (GWAS). In stage II, human leukocyte antigen (HLA) in chromosome 6 (chr6) (P = 6.73E‐08), microRNA (MIR) MIR548F3 in chr7 (P = 4.25E‐07), myosin light chain 2 (MYL2) in chr12 (P = 4.39E‐07), and glycoprotein precursor (GPC)6 in chr13 (P = 5.43E‐07), as suggested by the GWAS, were assessed by single nucleotide polymorphism (SNP) association analysis of whole NAFLD against non‐NAFLD in 9,726 members of the general population. A minor allele of the secondary lead SNP in chr6, rs2076529, was significantly associated (odds ratio [OR], 1.19; 95% confidence interval [CI], 1.11‐1.28; P = 2.10E‐06) and the lead SNP in chr7 was weakly associated (OR 1.15; 95% CI, 1.04‐1.27; P = 6.19E‐03) with increased NAFLD risk. Imputation‐based typing of HLA showed a significant difference in the distribution of HLA‐B, HLA‐DR‐beta chain 1 (DRB1), and HLA‐DQ‐beta chain 1 (DQB1) alleles in lean NAFLD GWAS. Next‐generation sequence‐based typing of HLA in 5,649 members of the general population replicated the significant difference of HLA‐B allele distribution and the significant increase of the HLA‐B*54:01 allele in whole NAFLD. Fecal metagenomic analysis of 3,420 members of the general population showed significant dissimilarity in beta‐diversity analysis of rs2076529 and HLA‐B*54:01 allele carriers from noncarriers. Veillonellaceae was increased but Verrucomicrobia was decreased in rs2076529 minor allele and HLA‐B*54:01 allele carriers as in NAFLD. Conclusion: HLA was identified as a novel locus associated with NAFLD susceptibility, which might be affected by the alteration of gut microbiota.

Abbreviations
- ALDH2
aldehyde dehydrogenase 2 family member
- BMI
body mass index
- BTLN2
butyrophilin‐like 2
- chr
chromosome
- CI
confidence interval
- DPB1
DP‐beta chain 1
- DQB1
DQ‐beta chain 1
- DRB1
DR‐beta chain 1
- GWAS
genome‐wide association study
- HbA1c
hemoglobin A1c
- HLA
human leukocyte antigen
- LD
linkage disequilibrium
- LEfSe
linear discriminant analysis effect size
- MHNW
metabolically healthy normal weight
- MYL2
myosin light chain 2
- NAFLD
nonalcoholic fatty liver disease
- OR
odds ratio
- OTU
operational taxonomic unit
- PC
principal component
- PCR
polymerase chain reaction
- PICRUSt
phylogenetic investigation of communities by reconstruction of unobserved states
- PNPLA3
patatin‐like phospholipase domain containing 3
- QC
quality control
- SNP
single nucleotide polymorphism
- TMM
Tohoku Medical Megabank Project
Nonalcoholic fatty liver disease (NAFLD) is defined as liver steatosis without alcoholic liver injury and other liver disease from apparent pathogenic factors, e.g., pharmacological. The global prevalence of NAFLD was estimated at 25.2% by a recent systematic review.( 1 ) The number of patients with NAFLD is increasing annually due to the gradual increment in the prevalence of metabolic syndrome.( 2 )
NAFLD is a multifactorial disorder influenced by genetic and environmental factors. The heritability of NAFLD has been estimated to range from 20% to 70% by population‐based and familial‐aggregation studies.( 3 ) In the last decade, several genome‐wide association studies (GWASs) have been reported and have identified patatin‐like phospholipase domain containing 3 (PNPLA3), the neurocan (NCAN)‐cartilage intermediate layer protein 2 (CILP2) region, protein phosphatase 1 regulatory subunit 3B (PPP1R3B), glucokinase regulator (GCKR), and lyso‐phospholipase‐like 1 (LYPLAL1) genes associated with NAFLD.( 4 , 5 ) Identification of these loci has promoted the elucidation of molecular pathways involved in NAFLD development. PNPLA3 encodes a protein with acyltransferase and hydrolase activities that is involved in triglyceride mobilization from hepatic lipid storage.( 6 ) Trans‐membrane 6 superfamily member 2 (TM6SF2) has been identified as a functional gene in the NCAN‐CILP2 region, which is involved in NAFLD through efficient lipid transfer into very low‐density lipoprotein.( 7 ) Variants of GCKR and LYPLAL1 are reported to be involved in NAFLD through alterations of hepatic glucose metabolism.( 8 , 9 ) However, epidemiological and experimental studies have proposed further mechanisms, i.e., inflammation, oxidative stress, and gut microbiota.( 10 ) It has been speculated that the molecules and their variants involved in these pathways are still obscure and there are potential missing heritable factors of NAFLD.
NAFLD is considered to be a metabolic phenotype of the liver because metabolic syndrome and related comorbidities, such as type 2 diabetes, dyslipidemia, and hypertension, are strongly associated with NAFLD.( 11 ) However, it is common for lean individuals to be diagnosed with NAFLD, a condition known as lean NAFLD.( 12 , 13 ) The pathogenesis of lean NAFLD is not well known. The prevalence of lean NAFLD ranges widely from 3% to 30%,( 14 ) and it has been reported that the prevalence in Asia is higher than in Western countries.( 15 ) This variation suggests that genetic factors play an important role in the pathogenesis of lean NAFLD.
Some studies have categorized lean NAFLD due to differentiated profiles.( 14 , 16 ) The condition can be separated into two categories: a metabolically obese normal‐weight (MONW) type and a metabolically healthy normal‐weight (MHNW) type. The MONW type has increased waist circumference and visceral adipose accumulation, which is the principal variance associated with NAFLD, probably through metabolic pathophysiology and insulin resistance.( 17 ) We have thus speculated that MHNW‐lean NAFLD may be caused by genetic burden instead of insulin resistance. The aim of the present study was to clarify the genetic background of lean NAFLD followed by testing whether the identified genetic factors were also associated with whole NAFLD. The replicated locus in whole NAFLD, human leukocyte antigen (HLA), was further assessed for its correlation with gut microbiota to elucidate potential pathways.
Participants and Methods
Study Design
We conducted a two‐stage analysis to identify NAFLD‐associated loci in Japanese patients. In stage I, 48 of the leanest male and 48 of the leanest female nonalcohol‐abuse (<30 g/day) MHNW NAFLD cases were chosen from three cross‐sectional studies referring to each of the following obesity measures. These parameters were the best predictors of NAFLD in the data of each population: visceral fat accumulation level for population 1 (n = 2,229), body mass index (BMI) for population 2 (n = 4,077), and waist circumstance for population 3 (n = 3,420) (Supporting Table S1). Stage I GWAS analyzed the leanest NAFLD panel as a case and the general population not diagnosed with NAFLD from the Tohoku Medical Megabank Project (TMM)( 18 ) as a control. Populations 1 and 3 were collected from the Kanto area, population 2 was collected from across Japan except Okinawa, and the TMM sample was collected mainly from Miyagi Prefecture. In stage II, populations 1 and 3, a total of 5,649 samples, were genotyped for the candidate loci that emerged from the stage I analysis. In addition, population 2 was genotyped to confirm the genetic association. Diagnosis of NAFLD was made by ultrasonography for patients who were negative for hepatitis virus and drug‐induced hepatitis and did not abuse alcohol according to a questionnaire for alcohol consumption. Part (17.1%, n = 795) of populations 1 and 3 underwent abdominal computed tomography (CT) scans, and the hepatosplenic (L/S) ratio of CT values showed that sensitivity and specificity of ultrasonographic diagnoses were 85.1% and 83.4%, respectively, when an L/S ratio <1.12 was used as a threshold for the diagnosis of steatosis. We used the following categories: the leanest NAFLD as the case of GWAS, nonobese NAFLD (BMI <25 kg/m2), obese NAFLD (BMI ≥25 kg/m2), and whole NAFLD, which included all categories of NAFLD. The Jichi Medical University Human Genomes/genetic Analysis Research Committee approved the study, and informed consent was obtained from all participants.
GWAS
The leanest NAFLD cases initially numbered 288. Whole‐genome single nucleotide polymorphism (SNP) genotyping was performed using an SNP array optimized for ethnic Japanese (Japonica array version 1; Toshiba Corporation).( 19 ) Genotype calling was conducted using Affymetrix Genotyping Console Software (version 4.2.0.26; Thermo Fisher Scientific). After genotyping, samples were subjected to quality control (QC). The criteria were dish QC >0.82 and sample call ratio >0.97. Next, the following subjects were excluded: (1) five cases showing kinship by the relatedness measure PI‐HAT and (2) eight cases with insufficient call rates (less than 0.95). These QC procedures resulted in reducing the leanest NAFLD case number to 275. The genotyping results were compared with the genotype data from 1,411 non‐NAFLD controls enrolled in the TMM and adjusted for age, sex, and alcohol consumption. A logistic regression model was applied for a genome‐wide scan using sex as a covariate. Population stratification was adjusted for 10 principal components (PCs) calculated using a tool for genome‐wide complex trait analysis.( 20 )
SNP Genotyping
We conducted individual SNP typing in populations 1, 2, and 3 by using TaqMan probes: rs2076529 (C___2488469_1_), rs2189883 (C___2620047_10), rs2301610 (C___2538069_1), rs66781047 (C__97772748_10), rs58542926 (C__89463510_10), and rs738409 (C______7241_10) (LifeTechnologies, Carlsbad, CA). SNP association studies analyzed whole NAFLD as a case and non‐NAFLD as a control in participants without alcohol abuse.
HLA Typing
Imputation‐based HLA typing was performed for a GWAS panel by using the Japonica array, as described.( 21 ) Next‐generation sequencing‐based HLA typing was conducted by two‐step polymerase chain reaction (PCR) for genome panels of populations 1 and 3. In the first step, exons 2 and 3 of HLA‐A, ‐B, ‐C, ‐DR‐beta chain 1 (DRB1), ‐DQ‐beta chain 1 (DQB1), and ‐DP‐beta chain 1 (DPB1) were amplified in a single tube using Platinum Multiplex PCR Master Mix (LifeTechnologies) and specific primer pairs tailed by adaptor sequences (Supporting Table S2). PCR products were provided for the second step tagmentation PCR using the Nextera system (Illumina, San Diego, CA). PCR products were purified and normalized using Agencourt AMPure XP beads (Beckman Coulter, Brea, CA). The mixed library was sequenced using the MiSeq system (Illumina). The paired‐end fastq files were provided for HLA allele calling, using HLA‐HD version 1.1.02 software.( 22 )
16s Metagenomic Analysis
Bacterial genomic DNA was extracted from bacteria suspended in fecal occult blood test tubes (Eiken Chemical Co., Ltd., Tokyo, Japan). We transferred 200 μL fecal suspension into a test tube containing 300 µL bacterial DNA extraction buffer (5 mL 1 M Tris‐HCl [pH 9.0], 4 mL 0.5 M ethylene diamine tetraacetic acid, 5 mL 10% sodium dodecyl sulfate, 16 mL H2O), 500 μL TE saturated phenol, and a microspoon of glass beads.( 23 ) After 10 minutes of vigorous shaking, the tubes were centrifuged, and bacterial DNA from the aqueous phase was purified by ethanol precipitation. Sequencing of the v3‐4 regions of 16S ribosomal RNA was performed following the 16S Metagenomic Sequencing Library Preparation Guide (Illumina). The paired‐end fastq files were merged using MacQIIME version 1.9.1. From the merged fastq files, operational taxonomic units (OTUs) were selected, and a Biological Observation Matrix (BIOM)‐formatted OTU table and phylogenetic tree were made using the application QIIME version 1.9.1( 24 ) installed in a Linux workstation. OTU table and phylogenetic tree files were analyzed further for alpha and beta diversity according to the sample profile. The OTU table was further analyzed using the linear discriminant analysis effect size (LEfSe) application( 25 ) in the Galaxy web server to identify biologically relevant differences in the sample profile. Further, we analyzed the OTU table using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt)( 26 ) and Kyoto Encyclopedia of Genes and Genomes pathways to predict the functional enrichment of microbiota. Predicted categories and the statistical inferences between genotypic groups through PICRUSt analysis were visualized using the software Statistical Analysis of Metagenomic Profiles (STAMP) version 2.1.3( 27 ) with default settings.
Statistical Analysis
Distributions of HLA alleles were calculated by Fisher’s exact test. SNP association studies for whole NAFLD were evaluated by logistic regression models. Statistical analyses were performed using R version 3.1.2. For all analyses, statistical significance was taken at P < 0.05.
Results
Characteristics of the Leanest NAFLD Case for GWAS
Anthropometric and clinical data profiles of the non‐NAFLD panel, leanest NAFLD panel, and the parental three population panels are shown in Supporting Table S1 and Supporting Fig. S1. The leanest NAFLD panel showed significantly lower BMI, waist circumference, and hemoglobin A1c (HbA1c), but higher low‐density lipoprotein (LDL) cholesterol levels compared to the general population. When compared against the non‐NAFLD panel, the leanest NAFLD panel showed equivalent HbA1c, waist, and lower BMI (P < 0.01) but lower high‐density lipoprotein cholesterol (P < 0.01), higher LDL (P < 0.01), and higher triglycerides (P < 0.01), indicating that the leanest NAFLD panel was of normal weight and euglycemic but slightly impaired at lipid metabolism.
Suggestive Loci Identified Through GWAS for the Leanest NAFLD
GWAS of the leanest NAFLD without adjustment by population stratification showed that four loci were associated at the suggestive level (P < 1.0E‐06) (Fig. 1A). The most significant association was observed in chromosome 6 (chr6), rs2076530 (P = 6.73E‐08), which is localized in the butyrophilin‐like 2 (BTLN2) gene mapped between HLA‐B and HLA‐DRB1 (Supporting Fig. S2A). The second locus was detected in chr7, rs2189883 (P = 4.25E‐07), which is localized in a core sequence of uncharacterized microRNA (MIR), MIR548F3 (Supporting Fig. S2B). The third locus was in chr12, rs2071629 (P = 4.39E‐07), which is localized in the myosin light chain 2 (MYL2) gene and is related to a functional SNP of aldehyde dehydrogenase 2 family member (ALDH2), rs671, in a large linkage disequilibrium (LD) block (Supporting Fig. S2C). The fourth association was detected in chr13, rs59980018 (P = 5.43E‐07), which is localized in intron 6 of the glypican 6 (GPC6) gene (Supporting Fig. S2D). In contrast to these loci, two loci robustly associated with NAFLD in previous reports, PNPLA3 and GATA zinc finger domain containing 2A (GATAD2A), showed P values under the threshold of suggestive significance (Supporting Fig. S2E,F).
Fig. 1.
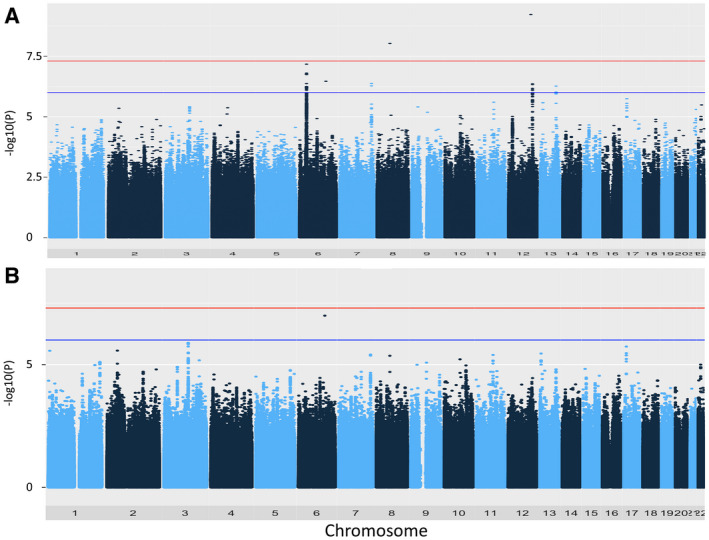
Manhattan plot of genome‐wide association results. (A) Plot without adjustment by population stratification; (B) with adjustment by population stratification using 10 PCs. Applying a regression analysis with sex as a covariate, the 275 leanest NAFLD cases were assessed against 1,411 nonalcohol abuse non‐NAFLD controls. The red line indicates the GWAS significance threshold (P = 5.0E‐08), and the blue line indicates the suggestive level (P = 1.0E‐06). Signals exceeding the significance threshold resulted from mistyping and should be ignored.
After adjusting the population stratification using 10 PCs, all the suggestive association signals disappeared, even with a mild inflation factor (λ = 1.093) (Fig. 1B). However, HLA and ALDH2 are known to be associated with lifestyle‐related disorders, e.g., obesity and dyslipidemia.( 28 , 29 , 30 ) Thus, we further evaluated whether the four loci were also associated with whole NAFLD in the populations of the Kanto area (populations 1 and 3, n = 5,649), in all Japan (population 2, n = 4,077), and in the combined analysis (n = 9,726). Logistic regression analysis using sex and HbA1c as covariates showed a significant association of rs2076529 (the secondary lead SNP in chr6; D′ = 1.00 and r 2 = 0.78 with rs2076530) and a weak association with rs2189883. Minor alleles of rs2076529 (odds ratio [OR], 1.19; 95% confidence interval [CI], 1.11‐1.28; P = 2.10E‐06) and rs2189883 (OR 1.15; 95% CI, 1.04‐1.27; P = 6.19E‐03) increased NAFLD risk in the combined analysis (Table 1). As reported,( 5 ) missense SNPs of TM6SF2 (rs5854292 [OR, 1.30; 95% CI, 1.14‐1.48; P = 6.30E‐5]) and PNPLA3 (rs738409 [OR, 1.34; 95% CI, 1.24‐1.45; P = 1.26E‐14]) were closely associated with whole NAFLD (Table 1). The association of rs2076529 and rs2189883 was weakened when an obesity indicator (BMI) was included as a covariate, while the associations of rs738409 were strengthened (Table 1). We then evaluated the SNPs by analysis of variance using BMI as an objective variable. Four lead SNPs, rs2076529, rs2189883, rs2301610 (secondary lead SNP in chr12), and rs66781047 (secondary lead SNP in chr13), showed weak correlations with BMI in male and female individuals (Supporting Table S3), suggesting that the four loci found in the leanest NAFLD GWAS were confounding variables with obesity.
Table 1.
Statistical Significances of Lead SNPs in Lean NAFLD GWAS and Replication Studies
| SNP | chr | Leanest NAFLD GWAS* | Replication by Model 1 † | Replication by Model 2 ‡ | ||||
|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | HWE P | ||
| rs2076529 | 6 | 1.66 (1.37‐2.00) | 1.73E‐07 | 1.22 (1.11‐1.34) § | 2.84E‐05 § | 1.19 (1.07‐1.33) § | 1.34E‐03 § | 0.167 § |
| 1.17 (1.03‐1.33) || | 1.36E‐02 || | 1.20 (1.04‐1.38) || | 1.29E‐02 || | 0.941 || | ||||
| 1.19 (1.11‐1.28) ¶ , * | 2.10E‐06 ¶ , * | 1.19 (1.09‐1.30) ¶ , * | 5.48E‐05 ¶ , * | 0.271 ¶ , * | ||||
| rs2189883 | 7 | 1.77 (1.42‐2.22) | 4.25E‐07 | 1.17 (1.04‐1.32) § | 1.02E‐02 § | 1.21 (1.05‐1.39) § | 1.03E‐02 § | 0.668 § |
| 1.11 (0.94‐1.32) || | 2.19E‐01 || | 1.10 (0.91‐1.33) || | 3.12E‐01 || | 0.500 || | ||||
| 1.15 (1.04‐1.27) ¶ , * | 6.19E‐03 ¶ , * | 1.16 (1.04‐1.30) ¶ , * | 9.76E‐03 ¶ , * | 0.458 ¶ , * | ||||
| rs2301610 | 12 | 1.70 (1.38‐2.09) | 7.67E‐07 | 1.04 (0.94‐1.15) § | 4.52E‐01 § | 1.10 (0.97‐1.25) § | 1.21E‐01 § | 0.560 § |
| 1.05 (0.91‐1.23) || | 4.90E‐01 || | 1.10 (0.99‐1.21) || | 3.83E‐01 || | 0.285 || | ||||
| 1.02 (0.93‐1.11) ¶ , * | 6.92E‐01 ¶ , * | 1.10 (0.99‐1.21) ¶ , * | 6.35E‐02 ¶ , * | 0.796 ¶ , * | ||||
| rs66781047 | 13 | 1.74 (1.39‐2.17) | 9.59E‐07 | 1.07 (0.95‐1.21) § | 2.73E‐01 § | 1.03 (0.90‐1.18) § | 6.81E‐01 § | 0.835 § |
| 1.17 (1.01‐1.37) || | 4.13E‐02 || | 1.39 (1.17‐1.66) || | 1.71E‐04 || | 0.121 || | ||||
| 1.09 (0.99‐1.20) ¶ , * | 6.15E‐02 ¶ , * | 1.16 (1.04‐1.28) ¶ , * | 7.10E‐03 ¶ , * | 0.409 ¶ , * | ||||
| rs58542926 | 19 | n.s. | n.s. | 1.34 (1.14‐1.58) § | 4.47E‐04 § | 1.50 (1.24‐1.82) § | 4.16E‐05 § | 0.084 § |
| 1.32 (1.06‐1.63) || | 1.13E‐02 || | 1.27 (1.00‐1.61) || | 4.58E‐02 || | 0.117 || | ||||
| 1.30 (1.14‐1.48) ¶ , * | 6.30E‐05 ¶ , * | 1.35 (1.16‐1.57) ¶ , * | 7.48E‐05 ¶ , * | 0.201 ¶ , * | ||||
| rs738409 | 22 | 1.25 | 0.04862 | 1.37 (1.25‐1.51) § | 3.77E‐11 § | 1.55 (1.38‐1.73) § | 1.99E‐14 § | 0.466 § |
| 1.33 (1.17‐1.51) || | 1.33E‐05 || | 1.42 (1.23‐1.64) || | 1.29E‐06 || | 0.581 || | ||||
| 1.34 (1.24‐1.45) ¶ , * | 1.26E‐14 ¶ , * | 1.47 (1.35‐1.61) ¶ , * | 1.65E‐18 ¶ , * | 0.072 ¶ , * | ||||
Statistical values for GWAS were obtained by logistic regression analysis in which sex was used as a covariate.
Statistical values for replication by model 1 were obtained by logistic regression analysis in which whole NAFLD was used as an objective variant and sex and HbA1c were used as covariates.
Covariates in model 2 were sex, HbA1c, and BMI.
Replication analysis in populations 1 and 3.
Replication analysis in population 2.
Replication analysis in the combined three populations.
Abbreviations: HWE, Hardy‐Weinberg equilibrium; n.s., not studied.
Association With the HLA Locus
The lead SNP in chr6 is localized between HLA‐B and HLA‐DRB1. We therefore conducted imputation‐based HLA typing in the Japonica array panel to determine whether HLA loci or alleles were associated with the leanest NAFLD. Significant differences of allele distribution between the leanest NAFLD and controls were observed in HLA‐B (P = 0.028), HLA‐DQB1 (P = 0.0065), and HLA‐DRB1 (P = 0.00050) (Table 2; Supporting Table S4). HLA allele frequencies of HLA‐B*40:01, HLA‐B*54:01, HLA‐C*03:04, HLA‐DRB1*04:04, HLA‐DRB1*04:05, HLA‐DRB1*12:02, and HLA‐DQB1*04:01 were significantly different between the leanest NAFLD and controls.
Table 2.
Association Studies of HLA‐B Alleles
| HLA‐B Allele | Study 1 | Study 2 | ||||||
|---|---|---|---|---|---|---|---|---|
| Control | Leanest NAFLD | OR | P † | Control | Whole NAFLD | OR | P † | |
| B*07:02 | 153 | 32 | 1.08 | 0.6818 | 261 | 210 | 1.12 | 0.2500 |
| B*13:01 | 23 | 7 | 1.58 | 0.3158 | 46 | 42 | 1.26 | 0.2790 |
| B*15:01 | 246 | 45 | 0.95 | 0.8031 | 407 | 332 | 1.14 | 0.0941 |
| B*15:07 | 8 | 1 | 0.65 | 1.0000 | 26 | 30 | 1.59 | 0.1024 |
| B*15:11 | 17 | 3 | 0.92 | 1.0000 | 36 | 41 | 1.58 | 0.0487 |
| B*15:18 | 50 | 9 | 0.93 | 1.0000 | 78 | 53 | 0.94 | 0.7892 |
| B*35:01 | 238 | 49 | 1.07 | 0.6766 | 366 | 281 | 1.07 | 0.4564 |
| B*37:01 | 19 | 5 | 1.36 | 0.5750 | 23 | 22 | 1.32 | 0.3662 |
| B*39:01 | 108 | 20 | 0.96 | 1.0000 | 161 | 121 | 1.04 | 0.7594 |
| B*40:01 | 155 | 29 | 0.97 | 1.0000 | 243 | 166 | 0.94 | 0.5728 |
| B*40:02 | 279 | 32 | 0.59 | 0.0054 | 340 | 236 | 0.96 | 0.6311 |
| B*40:03 | 3 | 1 | 1.73 | 0.5065 | 24 | 8 | 0.46 | 0.0711 |
| B*40:06 | 117 | 24 | 1.06 | 0.8152 | 183 | 120 | 0.90 | 0.4074 |
| B*44:02 | 15 | 1 | 0.35 | 0.4941 | 24 | 21 | 1.21 | 0.5471 |
| B*44:03 | 167 | 39 | 1.21 | 0.2862 | 360 | 246 | 0.94 | 0.4951 |
| B*46:01 | 154 | 20 | 0.67 | 0.1105 | 238 | 155 | 0.90 | 0.3205 |
| B*48:01 | 96 | 10 | 0.54 | 0.0781 | 143 | 108 | 1.04 | 0.7459 |
| B*51:01 | 221 | 48 | 1.13 | 0.4915 | 383 | 250 | 0.89 | 0.1951 |
| B*51:02 | 2 | 2 | 5.19 | 0.1255 | 9 | 10 | 1.54 | 0.3612 |
| B*52:01 | 266 | 64 | 1.25 | 0.1386 | 690 | 520 | 1.05 | 0.4708 |
| B*54:01 | 177 | 53 | 1.55 | 0.0100 | 316 | 281 | 1.25 | 0.0099 |
| B*55:02 | 23 | 2 | 0.45 | 0.4117 | 115 | 65 | 0.78 | 0.1096 |
| B*56:01 | 20 | 1 | 0.26 | 0.2330 | 50 | 25 | 0.69 | 0.1581 |
| B*56:03 | 5 | 0 | 0.00 | 1.0000 | 11 | 2 | 0.25 | 0.0875 |
| B*58:01 | 18 | 3 | 0.86 | 1.0000 | 29 | 21 | 1.00 | 1.0000 |
| B*59:01 | 51 | 13 | 1.32 | 0.3909 | 84 | 61 | 1.00 | 1.0000 |
| B*67:01 | 55 | 5 | 0.47 | 0.1112 | 51 | 27 | 0.73 | 0.2056 |
| Other | 136 | 32 | 0.81 | 0.30 | 415 | 246 | 0.81 | 0.0098 |
| Total | 2,686 | 518 | 5,112 | 3,700 | ||||
| P (multi‐allelic) ¶ , * | 0.0285 | 0.0130 | ||||||
P values were obtained by Fisher's exact tests for the multi‐allelic contingency table of allele numbers observed in study 1 of lean NAFLD versus TMM control and in study 2 of whole NAFLD versus control population.
P values of individual alleles were obtained by Fisher's exact tests for a 2 × 2 contingency table of the number of the left column allele and the number of all remaining alleles.
To evaluate further the association of the HLA locus with whole NAFLD in the general population, we sequenced amplicons of exons 2 and 3 in HLA‐A, ‐B, ‐C, ‐DRB1, ‐DQB1, and ‐DPB1 genes of 5,649 members of a cross‐sectional study population using next‐generation sequencing. Allele frequencies of HLA‐A, ‐B, ‐C, ‐DQB1, and ‐DPB1 were equivalent to Japanese statistics,( 31 , 32 ) but those of HLA‐DRB1 were different, suggesting a miscalling. We thus excluded HLA‐DRB1 from the analysis. The distribution of alleles of HLA‐B was significantly different between non‐NAFLD and whole NAFLD (P = 0.013) but that of HLA‐DQB1 was not (P = 0.433) (Table 2; Supporting Table S4). In individual alleles of the HLA‐B locus, B*54:01 was significantly frequent in whole NAFLD (OR, 1.25; P = 0.0099), similar to its frequency in the leanest NAFLD (Table 2).
Metagenomic Analysis of NAFLD and Related Loci
The HLA locus has been genetically shown to have a robust association with several lifestyle‐related diseases, although the mechanisms have not been resolved. Considering the important immunologic role of HLA and recent suggestions that the microbiome is involved in NAFLD pathogenesis, we speculated that HLA locus variations affect NAFLD through gut microbiota.
As a DNA source for massive gut microbiota analysis in population 3, we chose fecal occult blood tests, which contain a solution stabilizing human hemoglobin. Our experiment using 26 paired samples of freshly extracted fecal DNA and samples extracted after 1 day of suspension in fecal occult blood test tubes showed almost identical beta‐diversity and taxonomic plots (Supporting Fig. S3). This indicated that the 16s metagenomic data obtained from fecal occult blood tests reflected freshly prepared fecal gut microbiota and are applicable to gut microbiota analysis in a large population.
From the total metagenomic analysis population (n = 3,420), nonalcohol abuse and sufficient 16s read coverage (>5,000) samples (n = 2,616) were included in the subsequent statistical analysis. Alpha‐diversity analysis showed that fecal OTUs of NAFLD were significantly decreased compared to the control, as reported (Fig. 2A).( 33 , 34 ) Alpha‐diversity analysis of nonobese NAFLD revealed significantly decreased OTUs of an equivalent level of obese NAFLD (Fig. 2B). Beta diversity also showed significant dissimilarity between nonobese NAFLD and controls (Table 3). LEfSe analysis showed significant enrichment of Epsilonproteobacteria and Campylobacterales and significant decreases of Ruminococcaceae, clostridia, Firmcutes, and Faecalibacterium in nonobese NAFLD (Fig. 2D), similar to that shown in whole NAFLD (Fig. 2C). These data suggest that gut microbiota of nonobese NAFLD were as dysbiotic as in obese NAFLD, despite the BMI average of nonobese NAFLD being just 1.6 and that of obese NAFLD being 6.7 over the control (Supporting Table S5).
Fig. 2.
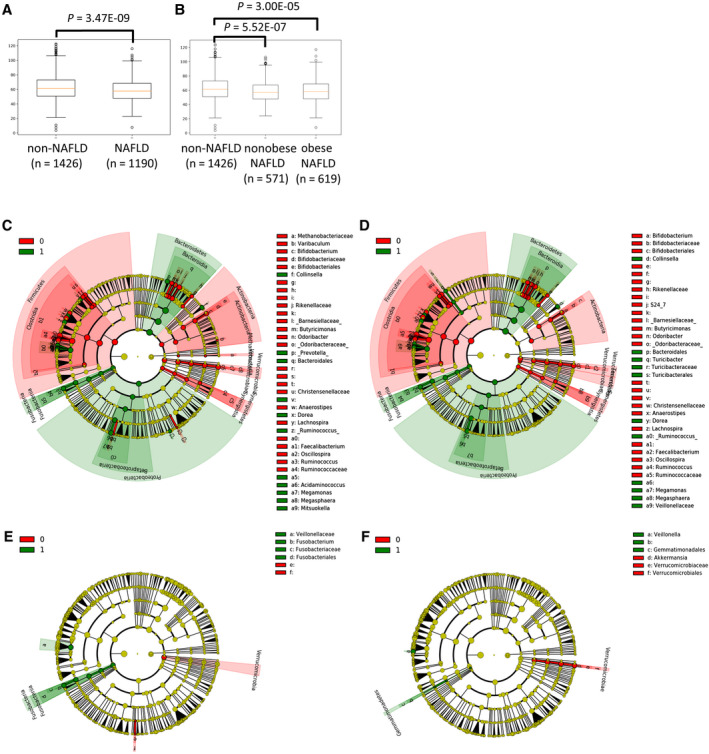
Fecal metagenomic analysis. (A) Bacterial alpha diversity assessed by phylogenetic diversity (PD) whole‐tree index between non‐NAFLD and whole NAFLD. Statistical significance P values were assessed by the Student t test. (B) PD whole‐tree index among non‐NAFLD, nonobese NAFLD, and obese NAFLD. Plots in A and B show interquartile range (box), median (vertical line), and outliers (whiskers). (C) LEfSe cladogram in the comparison between non‐NAFLD (0) and whole NAFLD (1). (D) Comparison between non‐NAFLD (0) and non‐obese NAFLD (1). (E) Comparison between rs2076529 noncarriers (0) and carriers (1). (F) Comparison between HLA‐B*54:01 noncarriers (0) and carriers (1). Red and green colors indicate increased taxa in 0 and 1 groups, respectively.
Table 3.
Comparison of UniFrac Distance Within Control Groups versus Between Control and Case Groups
| Group 1 | Group 2 | Unweighted | Weighted | ||
|---|---|---|---|---|---|
| t Statistic | P Value | t Statistic | P Value | ||
| Non‐NAFLD vs. non‐NAFLD | non‐NAFLD vs. NAFLD | −58.80 | <0.000001 | −47.00 | <0.000001 |
| Non‐NAFLD vs. non‐NAFLD | non‐NAFLD vs. non obese NAFLD | −30.79 | <0.000001 | −28.05 | <0.000001 |
| Non‐NAFLD vs. non‐NAFLD | non‐NAFLD vs. obese NAFLD | −70.70 | <0.000001 | −55.36 | <0.000001 |
| Noncarrier vs. noncarrier | non‐carrier vs. rs2076529 carrier | −7.59 | <0.000001 | 3.85 | 0.0012 |
| Noncarrier vs. noncarrier | non‐carrier vs. HLA‐B*54:01 carrier | 28.98 | <0.000001 | 9.11 | <0.000001 |
Tests of significance were performed using a two‐sided Student two‐sample t test with Bonferroni correction.
Minor allele carriers of rs2076529 showed equivalent alpha diversity with homozygotes of the major allele, but beta diversity was different between them in both unweighted and weighted UniFrac distances (Table 3). Veillonellaceae and Fusobacteria were increased, but Verrucomicrobia was decreased in rs2076529 minor allele carriers, the direction of which was equivalent to that of NAFLD and nonobese NAFLD (Fig. 2E). HLA‐B*54:01 carriers showed equivalent alpha diversity with noncarriers, but beta diversity was different from noncarriers (Table 3). Veillonella and Gemmatimonadales were increased but Verrucomicrobia and Akkermansia were decreased in HLA‐B*54:01 carriers, the direction of which was the same in NAFLD and nonobese NAFLD (Fig. 2F; Supporting Table S6). PICRUSt analysis showed that nonobese NAFLD had significantly increased taxonomic groups functionally enriched in metabolic pathways of carbohydrates, glycans, and amino acids. The rs2076529 carriers had enriched taxonomic groups involved in carbohydrate metabolism (Supporting Fig. S4).
Discussion
In this study, we performed GWAS for lean patients with NAFLD. This appears to be the first report of a genome‐wide search for lean NAFLD‐associated genes. The case group was selected from MHNW individuals to minimize the possibility of detecting obesity‐ or insulin resistance‐associated genes and to discover new genetic factors of NAFLD. As shown in the profiles of the subpopulations, the nonobese NAFLD group showed significantly higher metabolic parameters compared to the non‐NAFLD group while those in the leanest NAFLD group were almost equivalent with non‐NAFLD.
Through the leanest NAFLD GWAS, we detected four novel loci on chr6, chr7, chr12, and chr13 that have not been reported to have a genetic association with NAFLD in previous studies. While adjustment of population stratification concealed the association signals in the leanest NAFLD GWAS, further studies of four lead SNPs in the general population showed that rs2076529 on chr6 was strongly and rs2189883 on chr7 was weakly associated with whole NAFLD. Variations in the HLA locus are known to be correlated with geographic background in European ancestry and influenced by population stratification.( 35 ) Furthermore, a nonsignificant but substantial association signal has been shown in the HLA region by a Japanese NAFLD GWAS.( 36 ) Therefore, we considered that the association signal observed on chr6 in our leanest NAFLD GWAS was not ignorable as a false‐positive signal. However, the loci that were robustly associated with NAFLD in previous reports, PNPLA3 and GATAD2A,( 4 , 5 ) did not show a significant correlation with the leanest NAFLD. We speculated that the statistical model, which excluded obesity indicators from the covariates, and the small sample number of leanest NAFLD resulted in the subsignificant associations.
All four lead SNPs were correlated with BMI. The HLA locus and MYL2 have been shown to have a robust association with obesity in East Asian GWAS.( 28 , 29 ) HLA and breast cancer suppressor protein associated protein (BRAP) (related to MYL2 and ALDH2 in a large LD block; Fig. 2C) loci are also associated with plasma lipid levels.( 30 ) Risk alleles of these lead SNPs increased the risk of the leanest NAFLD group, indicating that HLA and MYL2 loci affect the pathogenesis of obesity and NAFLD simultaneously through undiscovered mechanisms.
The most closely associated locus with lean NAFLD, which was replicated in whole NAFLD, was rs2076529 localized in the BTLN2 gene, which is a member of the immunoglobulin superfamily that has been implicated as a regulator of T‐cell activation.( 37 ) The minor allele of rs2076530 produces a premature truncation of the BTLN2 protein and loses the function of T‐cell activation, which has been reported to impact the susceptibility of sarcoidosis.( 38 ) However, due to the long‐range LD across the HLA region, it is difficult to determine the primary pathogenic gene because other diseases are associated with rs2076530, e.g., type 1 diabetes, rheumatoid arthritis, and systemic lupus erythematosus.( 39 ) Although several studies have suggested that variations beside HLA genes affect antigen expression levels,( 40 ) considering its extremely high allelic variability, HLA is thought to play a principal role in the pathogenesis and progression of NAFLD.
We revealed that the allele distributions of HLA‐B, HLA‐DQB1, and HLA‐DRB1 were significantly different between non‐NAFLD and the leanest NAFLD in the GWAS study. The difference in allele frequency of HLA‐B was replicated in whole NAFLD by our sequence‐based study. In addition, we showed that the B*54:01 allele was associated with NAFLD in both the leanest and whole NAFLD panel. To clarify how it affects the pathogenesis of NAFLD, we evaluated the association between the B*54:01 allele and microbiome. Some reports have shown that particular HLA alleles are associated with NAFLD,( 41 , 42 ) but we have no knowledge about the pathogenic roles of HLA alleles in NAFLD development. One possibility is HLA affects liver injury through the immune system directly, and another is that HLA might regulate NAFLD indirectly through the microbiome. Recently, studies have placed emphasis on the relationship between NAFLD and the microbiome. The gut microbiota have been shown to contribute to NAFLD through various bacterial metabolites, such as pathogen‐associated molecular patterns, lipopolysaccharide, and short‐chain fatty acids.( 43 ) Moreover, the gut microbiota have recently been shown to influence several HLA‐linked diseases. A previous study showed that HLA‐B27 and ‐DRB1 alleles, which are associated with ankylosing spondylitis and rheumatoid arthritis, affect intestinal microbiome dysbiosis and influence susceptibility to these diseases.( 44 )
HLA‐B*54:01, a candidate risk allele of NAFLD, showed significant differences in beta diversity between carriers and noncarriers. Moreover, the phylum Verrucomicrobia was reduced and Gemmatimonadetes was enriched in HLA B*54:01 carriers. The phylum Verrucomicrobia was also decreased in NAFLD. A low abundance of Verrucomicrobia has been reported in patients with NAFLD with previous cirrhosis.( 45 ) The genus Akkermansia in the phylum Verrucomicrobia, which was reduced in HLA B*54:01 carriers, has been shown to degrade mucin and to improve obesity, glucose, and fat metabolism.( 46 , 47 ) PICRUSt analysis suggested enrichment of bacteria is involved in carbohydrate metabolism, which may affect the pathogenesis of NAFLD through energy absorption. These data suggest that HLA‐B*54:01 alleles might have the potential to affect susceptibility to NAFLD through the alteration of microbiome composition, especially through a reduction of Akkermansia.
One limitation of this study was the sequencing failure of HLA‐DRB1, probably due to the high degree of sequence similarity with DRB3/4/5 and copy number variation of pseudogenes. Long‐read sequencing studies spanning the entire HLA locus are needed to clarify the association with NAFLD.( 48 ) The small number of leanest NAFLD cases in the GWAS was another limitation because MHNW NAFLD cases were just 3% in our population. Further, ultrasonographic diagnosis of NAFLD has been shown to be relatively inaccurate.( 49 ) A GWAS using a larger cohort population diagnosed using magnetic resonance imaging is required to validate and discover further lean NAFLD loci.
In summary, we report four candidate loci associated with lean NAFLD in a first reported GWAS attempt. One of the four loci was localized in the HLA locus, and the genetic association was replicated in whole NAFLD. Among HLA loci, the HLA‐B locus was associated with both MHNW NAFLD and overall NAFLD. The HLA‐B*54:01 allele increased the risk of NAFLD, which might influence NAFLD pathogenesis through regulation of the microbiome.
Supporting information
Fig S1
Fig S2
Fig S3‐S4
Table S1‐S6
Supplementary Material
Acknowledgment
We thank Takuma Yamaguchi, Kayo Nagashima, and Yukiko Ohashi for their excellent technical assistance.
Supported by the Japanese Ministry of Education, Culture, Sports, Science, and Technology (MEXT) (Grants‐in‐Aid for Scientific Research No. 17K09864 to S.I.), the MEXT‐supported Program for the Strategic Research Foundation at Private Universities (2013‐2017), the Takeda Science Foundation (Grant Code No. 3‐2313‐004 to S.I.), and the Japan Agency for Medical Research and Development (JP17km0405205, JP17km0105001, and JP17km0105002 to M.N.).
Potential conflict of interest: Nothing to report.
References
- 1. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease‐meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73‐84. [DOI] [PubMed] [Google Scholar]
- 2. Khashab MA, Liangpunsakul S, Chalasani N. Nonalcoholic fatty liver disease as a component of the metabolic syndrome. Curr Gastroenterol Rep 2008;10:73‐80. [DOI] [PubMed] [Google Scholar]
- 3. Sookoian S, Pirola CJ. Genetic predisposition in nonalcoholic fatty liver disease. Clin Mol Hepatol 2017;23:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Speliotes EK, Yerges‐Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, et al.; NASH CRN; GIANT Consortium; MAGIC Investigators; GOLD Consortium . Genome‐wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 2011;7:e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kahali B, Halligan B, Speliotes EK. Insights from genome‐wide association analyses of nonalcoholic fatty liver disease. Semin Liver Dis 2015;35:375‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pingitore P, Pirazzi C, Mancina RM, Motta BM, Indiveri C, Pujia A, et al. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim Biophys Acta 2014;1841:574‐580. [DOI] [PubMed] [Google Scholar]
- 7. Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg‐Hansen A, et al. Exome‐wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beer NL, Tribble ND, McCulloch LJ, Roos C, Johnson PR, Orho‐Melander M, et al. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet 2009;18:4081‐4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sliz E, Sebert S, Würtz P, Kangas AJ, Soininen P, Lehtimäki T, et al. NAFLD risk alleles in PNPLA3, TM6SF2, GCKR and LYPLAL1 show divergent metabolic effects. Hum Mol Genet 2018;27:2214‐2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bedi O, Aggarwal S, Trehanpati N, Ramakrishna G, Krishan P. Molecular and pathological events involved in the pathogenesis of diabetes‐associated nonalcoholic fatty liver disease. J Clin Exp Hepatol 2019;9:607‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ballestri S, Zona S, Targher G, Romagnoli D, Baldelli E, Nascimbeni F, et al. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta‐analysis. J Gastroenterol Hepatol 2016;31:936‐944. [DOI] [PubMed] [Google Scholar]
- 12. Das K, Das K, Mukherjee PS, Ghosh A, Ghosh S, Mridha AR, et al. Nonobese population in a developing country has a high prevalence of nonalcoholic fatty liver and significant liver disease. Hepatology 2010;51:1593‐1602. [DOI] [PubMed] [Google Scholar]
- 13. Younossi ZM, Stepanova M, Negro F, Hallaji S, Younossi Y, Lam B, et al. Nonalcoholic fatty liver disease in lean individuals in the United States. Medicine (Baltimore) 2012;91:319‐327. [DOI] [PubMed] [Google Scholar]
- 14. Kim D, Kim WR. Nonobese fatty liver disease. Clin Gastroenterol Hepatol 2017;15:474‐485. [DOI] [PubMed] [Google Scholar]
- 15. Shi Y, Wang Q, Sun Y, Zhao X, Kong Y, Ou X, et al. The prevalence of lean/nonobese nonalcoholic fatty liver disease: a systematic review and meta‐analysis. J Clin Gastroenterol 2020;54:378‐387. [DOI] [PubMed] [Google Scholar]
- 16. Wang AY, Dhaliwal J, Mouzaki M. Lean non‐alcoholic fatty liver disease. Clin Nutr 2019;38:975‐981. [DOI] [PubMed] [Google Scholar]
- 17. Stefan N, Schick F, Häring HU. Causes, characteristics, and consequences of metabolically unhealthy normal weight in humans. Cell Metab 2017;26:292‐300. [DOI] [PubMed] [Google Scholar]
- 18. Kuriyama S, Yaegashi N, Nagami F, Arai T, Kawaguchi Y, Osumi N, et al. The Tohoku Medical Megabank project: design and mission. J Epidemiol 2016;26:493‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kawai Y, Mimori T, Kojima K, Nariai N, Danjoh I, Saito R, et al. Japonica array: improved genotype imputation by designing a population‐specific SNP array with 1070 Japanese individuals. J Hum Genet 2015;60:581‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: A tool for genome‐wide complex trait analysis. Am J Hum Genet 2011;88:76‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Khor SS, Yang W, Kawashima M, Kamitsuji S, Zheng X, Nishida N, et al. High‐accuracy imputation for HLA class I and II genes based on high‐resolution SNP data of population‐specific references. Pharmacogenomics J 2015;15:530‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kawaguchi S, Higasa K, Shimizu M, Yamada R, Matsuda F. HLA‐HD: An accurate HLA typing algorithm for next‐generation sequencing data. Hum Mutat 2017;38:788‐797. [DOI] [PubMed] [Google Scholar]
- 23. Matsuki T. Procedure of DNA extraction from fecal sample for the analysis of intestinal microflora. J Intest Microbiol 2006;20:259‐262. [Google Scholar]
- 24. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high‐throughput community sequencing data. Nat Methods 2010;7:335‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol 2011;12:R60 http://genomebiology.biomedcentral.com/articles/10.1186/gb‐2011‐12‐6‐r60. Accessed November 29, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013;31:814‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 2014;30:3123‐3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wen W, Zheng W, Okada Y, Takeuchi F, Tabara Y, Hwang JY, et al. Meta‐analysis of genome‐wide association studies in East Asian‐ancestry populations identifies four new loci for body mass index. Hum Mol Genet 2014;23:5492‐5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wen W, Kato N, Hwang JY, Guo X, Tabara Y, Li H, et al. Genome‐wide association studies in East Asians identify new loci for waist‐hip ratio and waist circumference. Sci Rep 2016;6:17958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010;466:707‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakajima F, Nakamura J, Yokota T. Analysis of HLA haplotypes in Japanese, using high resolution allele typing. MHC 2001;8:1‐32. [Google Scholar]
- 32. Ikeda N, Kojima H, Nishikawa M, Hayashi K, Futagami T, Tsujino T, et al. Determination of HLA‐A, ‐C, ‐B, ‐DRB1 allele and haplotype frequency in Japanese population based on family study. Tissue Antigens 2015;85:252‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang W, Wu N, Wang X, Chi Y, Zhang Y, Qiu X, et al. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non‐alcoholic fatty liver disease. Sci Rep 2015;5:8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim HN, Joo E‐J, Cheong HS, Kim Y, Kim HL, Shin H, et al. Gut microbiota and risk of persistent nonalcoholic fatty liver diseases. J Clin Med 2019;8pii:E1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Davies RW, Wells GA, Stewart AF, Erdmann J, Shah SH, Ferguson JF, et al. A genome‐wide association study for coronary artery disease identifies a novel susceptibility locus in the major histocompatibility complex. Circ Cardiovasc Genet 2012;5:217‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kawaguchi T, Shima T, Mizuno M, Mitsumoto Y, Umemura A, Kanbara Y, et al. Risk estimation model for nonalcoholic fatty liver disease in the Japanese using multiple genetic markers. PLoS One 2018;13:e0185490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nguyen T, Liu XK, Zhang Y, Dong C. BTNL2, a butyrophilin‐like molecule that functions to inhibit T cell activation. J Immunol 2006;176:7354‐7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morais A, Lima B, Peixoto MJ, Alves H, Marques A, Delgado L. BTNL2 gene polymorphism associations with susceptibility and phenotype expression in sarcoidosis. Respir Med 2012;106:1771‐1777. [DOI] [PubMed] [Google Scholar]
- 39. Orozco G, Eerligh P, Sánchez E, Zhernakova S, Roep BO, González‐Gay MA, et al. Analysis of a functional BTNL2 polymorphism in type 1 diabetes, rheumatoid arthritis, and systemic lupus erythematosus. Hum Immunol 2005;66:1235‐1241. [DOI] [PubMed] [Google Scholar]
- 40. Nishida N, Sugiyama M, Sawai H, Nishina S, Sakai A, Ohashi J, et al. Key HLA‐DRB1‐DQB1 haplotypes and role of the BTNL2 gene for response to a hepatitis B vaccine. Hepatology 2018;68:848‐858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Doganay L, Katrinli S, Colak Y, Senates E, Zemheri E, Ozturk O, et al. HLA DQB1 alleles are related with nonalcoholic fatty liver disease. Mol Biol Rep 2014;41:7937‐7943. [DOI] [PubMed] [Google Scholar]
- 42. Karrar A, Hariharan S, Fazel Y, Moosvi A, Houry M, Younoszai Z, et al. Analysis of human leukocyte antigen allele polymorphism in patients with non alcoholic fatty liver disease. Medicine (Baltimore) 2019;98:e16704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Safari Z, Gérard P. The links between the gut microbiome and non‐alcoholic fatty liver disease (NAFLD). Cell Mol Life Sci 2019;76:1541‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Asquith M, Sternes PR, Costello ME, Karstens L, Diamond S, Martin TM, et al. HLA alleles associated with risk of ankylosing spondylitis and rheumatoid arthritis influence the gut microbiome. Arthritis Rheumatol 2019;71:1642‐1650. [DOI] [PubMed] [Google Scholar]
- 45. Ponziani FR, Bhoori S, Castelli C, Putignani L, Rivoltini L, Del Chierico F, et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology 2019;69:107‐120. [DOI] [PubMed] [Google Scholar]
- 46. Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, et al. Cross‐talk between Akkermansia muciniphila and intestinal epithelium controls diet‐induced obesity. Proc Natl Acad Sci U S A 2013;110:9066‐9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dao MC, Everard A, Aron‐Wisnewsky J, Sokolovska N, Prifti E, Verger EO, et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut 2016;65:426‐436. [DOI] [PubMed] [Google Scholar]
- 48. Suzuki S, Ranade S, Osaki K, Ito S, Shigenari A, Ohnuki Y, et al. Reference grade characterization of polymorphisms in full‐length HLA class I and II genes with short‐read sequencing on the ION PGM system and long‐reads generated by single molecule, real‐time sequencing on the PacBio platform. Front Immunol 2018;9:2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roldan‐Valadez E, Favila R, Martínez‐López M, Uribe M, Méndez‐Sánchez N. Imaging techniques for assessing hepatic fat content in nonalcoholic fatty liver disease. Ann Hepatol 2008;7:212‐220. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3‐S4
Table S1‐S6
Supplementary Material