

Abstract
Aryl hydrocarbon receptor (AHR) is a transcription factor that binds to DNA as a heterodimer with the AHR nuclear translocator (ARNT) after interaction with ligands, such as polycyclic and halogenated aromatic hydrocarbons and other xenobiotics. The endogenous ligands and functions of AHR have been the subject of many investigations. In the present study, the potential role of AHR signaling in the development of left ventricular hypertrophy and cardiac fibrosis by angiotensin II (Ang II) infusion was investigated in mice lacking the AHR gene (Ahr−/−). We also assessed the hypothesis that fenofibrate, a peroxisome proliferator-activated receptor-α (PPARα) activator, reduces cardiac fibrosis through the c-Jun signaling. Male Ahr−/− and age-matched wild-type mice (n = 8 per group) were infused with Ang II at 100 ng/kg/min daily for 2 weeks. Treatment with Ang II increased systolic blood pressure to comparable levels in Ahr−/− and wild-type mice. However, Ahr−/− mice developed severe cardiac fibrosis after Ang II infusion compared with wild-type mice. Ang II infusion also significantly increased the expression of endothelin in the left ventricles of Ahr−/− mice, but not in wild-type mice, and significantly increased the c-Jun signaling in Ahr−/− mice. Ang II infusion also significantly enhanced the expression of hypoxia-inducible factor-1α (HIF-1α) and the downstream target vascular endothelial growth factor (VEGF) in the left ventricles of Ahr−/− mice. These results suggested pathogenic roles for the AHR signaling pathway in the development of cardiac fibrosis. Treatment with fenofibrate reduced cardiac fibrosis and abrogated the effects of Ang II on the expression of endothelin, HIF-1α, and VEGF. The inhibitory effect of fenofibrate on cardiac fibrosis was mediated by suppression of VEGF expression through modulation of c-Jun/HIF-1α signaling.
Keywords: AHR, Cardiac hypertrophy, Fibrosis, Angiotensin II, PPARα, HIF-1α, Vascular endothelial growth factor
Introduction
The aryl hydrocarbon receptor (AHR) is a DNA-binding protein and a member of the helix–loop–helix/Per–ARNT–SIM (bHLH/PAS) family (Nebert 2017). It is known as a multifunctional protein that mediates xenobiotic metabolism and environmental responses. AHR is expressed in a wide variety of tissues at various levels from fetus through adult, suggesting that it has diverse and tissue-specific physiological functions (Puga et al. 2009). Activation of the cytoplasmic AHR by various natural and synthetic ligands is followed by translocation into the nucleus and dimerization with the AHR nuclear translocator (ARNT). Various environmental factors, such as halogenated aromatic hydrocarbons (HAH) and polycyclic aromatic hydrocarbons (PAH) found in tobacco smoke and pollutants, can bind and activate the AHR. The bestknown environmental ligand of the AHR is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (Kolluri et al. 2017). The nuclear AHR/ARNT complex transactivates the expression of various target genes, such as cytochrome P450 family 1 subfamily A member 1 (CYP1A1) through binding to xenobiotic response elements (XREs) in the promoter regions of the AHR targets (Sherr 2011).
The exact physiological roles of the AHR are not fully understood partly due to the wide variety of the target genes and involved tissues. However, a number of studies have suggested the involvement of AHR in cardiovascular development and homeostasis. For example, the activation of AHR by TCDD or related chemicals was reported to disrupt cardiovascular development in animals (Carreira et al. 2015). Furthermore, mice lacking the AHR exhibited vascular abnormalities in the liver, kidney, and peripheral vessels (Ichihara 2011; Lahvis et al. 2000), as well as cardiac hypertrophy (Fernandez-Salguero et al. 1997; Lund et al. 2003). Notwithstanding these studies, the exact role of the AHR in normal cardiovascular development and/or homeostasis remains unclear although the phenotype of Ahr-null mice suggests that the AHR may play a significant role in the heart and vasculature.
The peroxisome proliferator-activated receptor (PPAR) family and their target genes appear to contribute to the regulation of lipid and glucose homeostasis, and are considered regulators of inflammation and atherosclerosis (Ogata et al. 2002). Notably, PPARα is the major cardiac PPAR in adults and Pparα-null mice develop severe cardiac hypertrophy in response to chronic pressure overload (Smeets et al. 2008). Previous studies reported that the activation of PPARα reduced cardiac hypertrophy, decreased cardiac fibrosis, attenuated cardiac dysfunction, and improved survival by the inhibition of profibrotic, proinflammatory, and prohypertrophic genes (Barger et al. 2000; Ichihara et al. 2006). Fenofibrate, a specific PPARα activator, was reported to reduce endothelin 1 (ET-1 )-induced cardiac hypertrophy through downregulation of activating protein-1 (AP-1) binding and inhibition of p38 mitogen-activated protein kinase (MAPK) signaling (Balakumar et al. 2011; Duhaney et al. 2007).
The present study revealed a novel role for the AHR and PPARα signaling pathways in the cardiovascular system. Specifically, we defined the link between AHR and the PPARα signaling in the development of left ventricular (LV) hypertrophy and cardiac fibrosis in angiotensin II (Ang-induced hypertrophic wild-type mice and Ahr-null mice (Ahr−/−).
Materials and methods
Animal experiments
Ahr-null mice were obtained from National Cancer Institute, MD, USA (Fernandez-Salguero et al. 1995) in 2003. Then, Ahr-null mice had been maintained by the Animal Resource Facility at Nagoya University Graduate School of Medicine. To induce cardiac hypertrophy, osmotic minipump system (ALZET Osmotic Pumps; DURECT, Cupertino, CA, USA) was implanted dorsally in male Ahr−/− and age-matched wild-type mice (n = 8, per group). The mice were treated with Ang II (Sigma-Aldrich, St. Louis, MO, USA), which was dissolved in saline and infused subcutaneously at a rate of 50 or 100 ng/kg/min every day for 2 weeks (Peng et al. 2006), in a preliminary study. Then, male Ahr−/− and age-matched wild-type mice infused Ang II at 100 ng/kg/min were also treated simultaneously with fenofibrate (30 mg/kg/day; Kaken Pharmaceutical, Tokyo, Japan) for 2 weeks (n = 8, per group).
The animals were housed in temperature- and light-controlled environment at 25 °C with a 12-h light–dark cycle. The experimental protocol was approved by the animal care committee of Nagoya University Graduate School of Medicine and all animal procedures were conducted in accordance with the guidelines for the care and use of laboratory animals approved by the university.
Measurements of systolic blood pressure
Systolic blood pressure was measured in conscious mice by the tail-cuff method (BP-98A, MCP-1; Softron, Tokyo, Japan) after 2 weeks of Ang II treatment using the method described previously (Huang et al. 2017). The reported blood pressure and heart rate values represent the mean of four or five determinations taken at the same period.
Tissue sample collection and histological analysis
After blood pressure measurement, mice were killed by deep anesthesia. The heart was quickly removed via thoracotomy and weighed. The left ventricle was separated from the atria and right ventricle, weighed, and cut into 2-mm-thick slices. The apex and base of the left ventricles were then immediately frozen in liquid nitrogen for quantitative real-time polymerase chain reaction (RT-PCR) analysis and enzymelinked immunosorbent assay (ELISA). Midventricular slices were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS). The heart tissues were embedded in paraffin, sectioned (thickness, 4 μm), and stained with hematoxylin-eosin solution to measure the cross-sectional areas of myocardium. The heart tissues were also stained with Sirius red solution to evaluate the extent of fibrosis. The cross-sectional areas of myocardium and the area of the fibrosis in the interstitial and perivascular regions were calculated with the cellSens image system (Olympus, Tokyo, Japan).
Expression of fetal-type cardiac genes
Total RNA was subsequently extracted from the apex of LV tissues using ReliaPrep RNA tissue miniprep system (Promega, Madison, WI, USA) using the protocol provided by the manufacturer. The concentration of total RNA was quantified by spectrophotometry (ND-1000; NanoDrop Technologies, Wilmington, DE, USA). RNA was reverse transcribed to single-strand cDNA using SuperScript III First-Strand Synthesis System for RT-PCR (Life Technologies, Carlsbad, CA, USA). cDNA was subjected to quantitative RT-PCR analysis with FastStart Universal Probe Master Mix (Roche, Basel, Switzerland) and primers for atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), collagen I (COL1A1) and collagen III (COL3A1) using an ABI 7000 Real-Time PCR system (Life Technologies), as described previously (Yokoyama et al. 2018). The gene expression level was normalized to that of β-actin (ACTB) in the same cDNA.
To confirm the changes of AHR expression after Ang II treatment in wild-type mice, cDNA was also subjected to quantitative PCR analysis with primers for AHR and the gene expression level was normalized to that of β-actin in the same cDNA.
Measurement of endothelin-1 and c-Jun levels
The expression levels of endothelin-1 (EDN1) and c-Jun were measured by quantitative RT-PCR analysis using primers specific for the mRNAs encoding these two molecules. The Actb mRNA was used as an internal standard. Endothelin-1 levels were determined in protein extracts prepared from homogenates of LV tissues using ET-1 ELISA kit (R&D Systems, Minneapolis, MN, USA), according to the guidelines supplied by the manufacturer. Nuclear extracts of LV tissue were prepared according to the instructions provided with the nuclear extraction kit (Active Motif, Carlsbad, CA, USA). c-Jun levels were determined in nuclear extracts from LV tissues using the TransAM AP-1 c-Jun assay kit (Active Motif) and were used to quantify the binding of c-Jun to the AP-1 site, according to the protocol supplied by the manufacturer.
Western blot analysis
Nuclear extracts from LV tissues were subjected to immunoblot analysis with a rabbit polyclonal antibody to HIF-1α (1:200 dilutions; Santa Cruz Biotechnology, Dallas, TX, USA) as well as with a mouse monoclonal antibody to lamin B (1:500 dilution; Chemicon, Santa Monica, CA, USA). Immune complexes were detected with enhanced chemiluminescence (ECL) reagents (GE Healthcare Bio-Science, Piscataway, NJ, USA). Band intensity was quantified using Quantity One Image software (Bio-Rad, Hercules, CA, USA). Protein extracts prepared from homogenates of LV tissues were also subjected to immunoblot analysis with goat polyclonal antibody to vascular endothelial growth factor (VEGF) (dilution of 1:200; Santa Cruz Biotechnology) as well as with a mouse monoclonal antibody to β-actin (1:500 dilution; Chemicon).
Immunohistochemical analysis
Capillary vessels were stained with a rabbit polyclonal antibody to CD 31 (1:100 dilutions; Abcam, Cambridge, UK) to estimate the capillary density. Sections were photographed with BX50 Olympus microscope and then CD31-positive cells were counted in number with the cellSens image system. The capillary density was averaged and expressed as the number of capillaries per unit area.
Statistical analysis
All parameters were expressed as mean ± standard error of the mean (SEM). Statistical analyses were performed using oneway analysis of variance (ANOVA) followed by Dunnett’s post hoc analysis. P values less than 0.05 denoted the presence of statistical significance. All statistical analyses were performed using the JMP 8.0 software (SAS Institute, Cary, NC, USA).
Results
Effects of Ang II on body, heart, and LV weights and blood pressure
Administration of Ang II at 50 or 100 ng/kg/min daily for 2 weeks did not change body (Fig. 1a) and heart weights (Fig. 1c) in both wild-type and Ahr−/− mice. Systolic blood pressure was significantly higher in mice treated with high-dose Ang II than the control mice of both strains, but there was no significant difference in systolic blood pressure of mice treated with low-dose Ang II compared with the control mice (Fig. 1b). Left ventricle of mice treated with high-dose Ang II was significantly heavier than that of the control mice of the two strains (Fig. 1d). LV weight of Ahr−/− mice treated with high-dose Ang II was also significantly higher than that of wild-type mice treated with the same dose of Ang II. There were no significant differences in all parameters measured in the control wild-type and Ahr−/− mice. When heart and LV weights were adjusted for body weight, the same results were observed (Supplementary Fig. 1a, b).
Fig. 1.
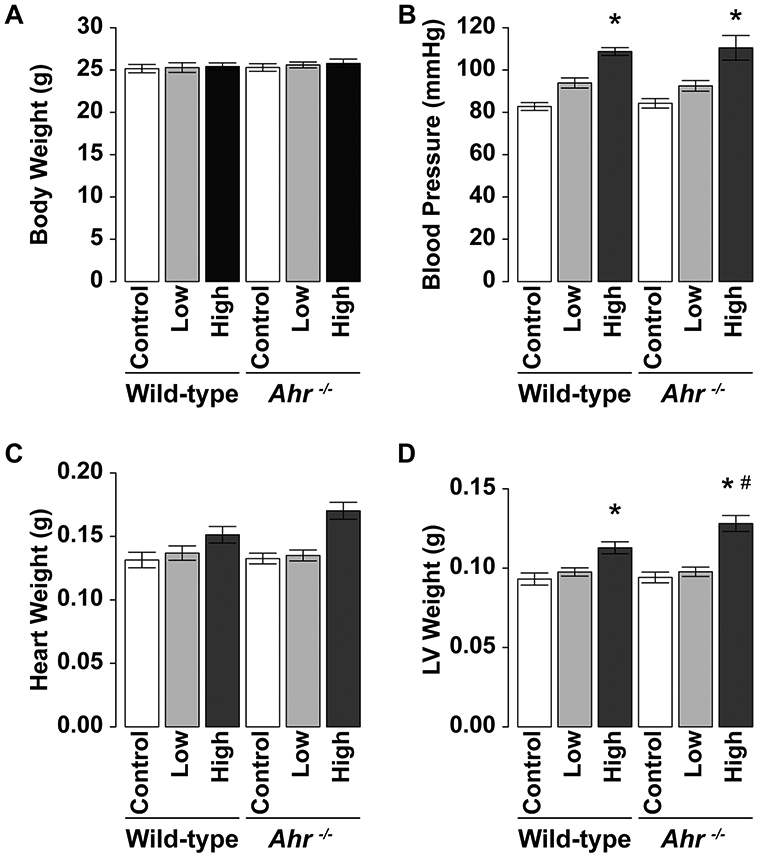
Effects of administration of Ang II on a body weight, b blood pressure, c heart weight, and d LV weight in wild-type and Ahr−/− mice treated with low- or high-dose of Ang II. Data are mean ± SEM of eight mice per group. *P < 0.05 vs. wild-type control mice; #P < 0.05 vs. wild-type Ang II mice
Next, Ang II was infused at a rate of 100 ng/kg/min daily for 2 weeks to test the hypothesis that fenofibrate, a PPARα activator, can reduce cardiac fibrosis. Administration of Ang II at a rate of 100 ng/kg/min had no significant effect on body weight (Fig. 2a) but was associated with a significant increase in systolic blood pressure. Interestingly, co-administration of Ang II and fenofibrate had no effect on blood pressure compared with both wild-type and Ahr−/− mice (Fig. 2b). Treatment with Ang II was associated with a significant increase in heart weight and this effect was abrogated by fenofibrate in both strains (Fig. 2c). Furthermore, administration of Ang II was associated with a significant increase in LV weight compared with the control mice of both strains (Fig. 2d), and the increase was significantly larger in Ahr−/− mice than wild-type mice. Fenofibrate significantly abrogated the increase in LV weight in Ahr−/− mice (Fig. 2d). When adjusted for body weight, LV weight/body weight of the mice treated with Ang II was significantly higher than that of the control mice of the two strains and the increase was significantly larger in Ahr−/− mice than wild-type mice. Fenofibrate abrogated the increase in LV weight/body weight in Ahr−/− mice, but there was no significant difference statistically (Supplementary Fig. 1c, d) because body weight of the mice treated with fenofibrate was slightly smaller than that in others.
Fig. 2.
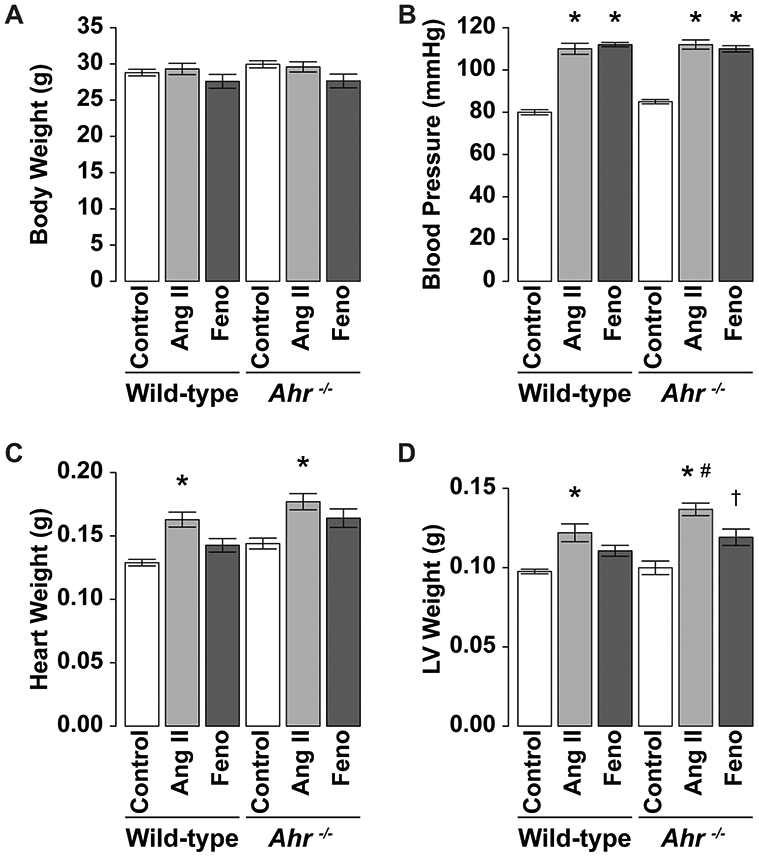
Effects of administration of Ang II and fenofibrate on a body weight, b blood pressure, c heart weight, and d LV weight in wild-type and Ahr−/− mice treated with Ang II (at 100 ng/ kg/min) and fenofibrate. Data are mean ± SEM of eight mice per group. *P < 0.05 vs. wild-type control mice; #P < 0.05 vs. wild-type Ang II mice; †P < 0.05 vs. Ahr−/− Ang II mice
Effects of Ang II and fenofibrate on cardiac hypertrophy and fibrosis
Anp and Bnp mRNA expression levels were significantly higher in the left ventricles of mice treated with Ang II than the control mice in both wild-type and Ahr−/− mice (Fig. 3a, b). Furthermore, Anp mRNA level was significantly greater in Ang II-treated Ahr−/− mice than Ang II-treated wild-type mice (Fig. 3a). Fenofibrate abrogated the effects of Ang II on both proteins. Furthermore, a similar pattern was noted in Col1a1 and Col3a1 mRNA levels; Ang II upregulated their levels, with a preferential effect on Ahr−/− mice compared with the wild-type, and treatment with fenofibrate abrogated these effects (Fig. 3c, d).
Fig. 3.
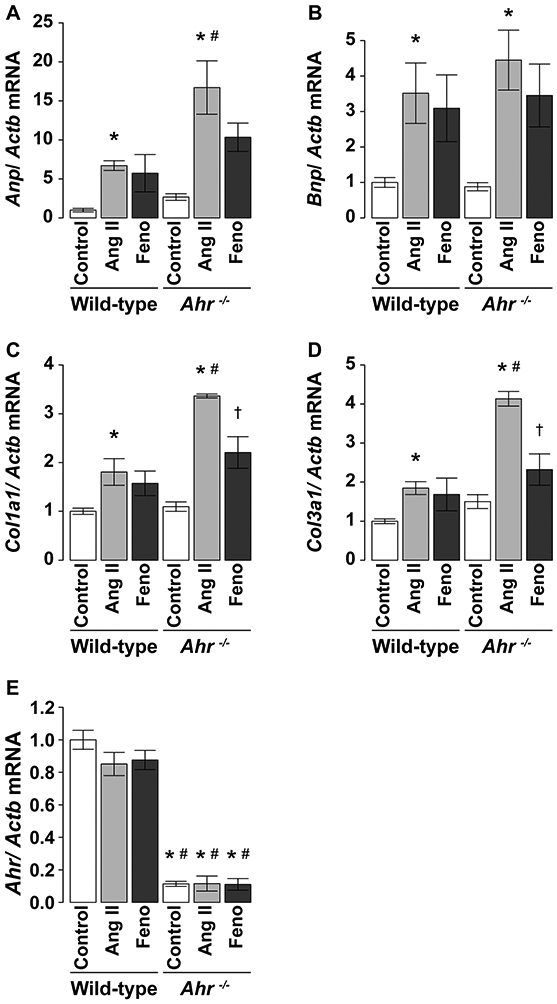
Expression levels of a Anp, b Bnp, c Colla1, d Col3a1, and e Ahr mRNAs in the left ventricle of wild-type and Ahr−/− mice treated with Ang II (at 100 ng/kg/min) and fenofibrate. The expression levels were determined by quantitative RT-PCR analysis and expressed relative to the level of Actb mRNA. Data are mean ± SEM of eight mice per group. *P < 0.05 vs. wild-type control mice; #P < 0.05 vs. wild-type Ang II mice; †P < 0.05 vs. Ahr−/− Ang II mice
Light microscopic analysis demonstrated that the cross-sectional area of cardiomyocytes (Fig. 4a, b), as well as interstitial (Fig. 4c, d) and perivascular fibrosis (Fig. 4e, f), was significantly greater in the left ventricles of mice treated with Ang II than the control mice in both genotypes of wild type and Ahr−/−. Among them, the increase in the extent of interstitial fibrosis was significantly larger in Ahr−/− mice than wild-type mice (Fig. 4d). The treatment with fenofibrate reduced the increase in the cross-sectional area of cardiomyocytes and the extent of interstitial fibrosis (Fig. 4b, d).
Fig. 4.
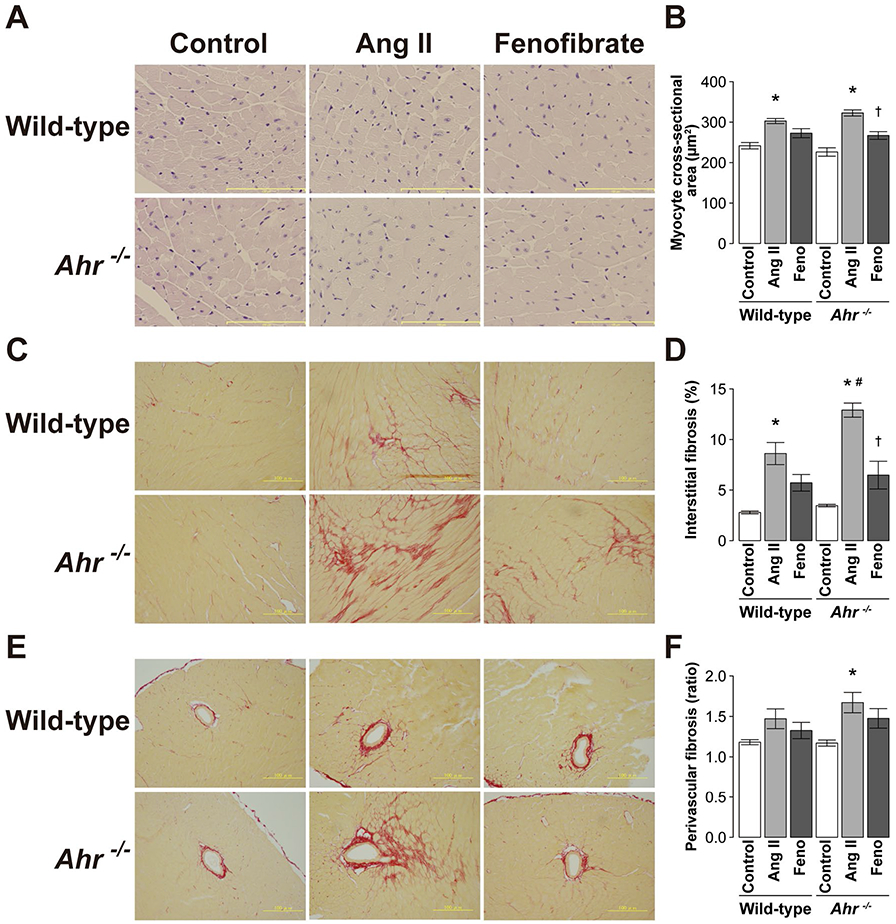
Light micrographs of a myocytes in hematoxylin–eosinstained sections and c interstitial and e perivascular fibrosis in Sirius red-stained sections of the LV wall in representative wild-type and Ahr−/− mice treated with Ang II and fenofibrate. Scale bars, 100 μm. Quantitative analysis of b myocyte cross-sectional area, c interstitial fibrosis, and f perivascular fibrosis and in the left ventricle of wild-type and Ahr−/− mice treated with Ang II and fenofibrate. Data are mean ± SEM of six mice per group. *P < 0.05 vs. wild-type control mice; #P < 0.05 vs. wild-type Ang II mice; †P < 0.05 vs. Ahr−/− Ang II mice
Effects of Ang II and fenofibrate on endothelin-1 and c-Jun levels
There were no significant differences in Ahr mRNA expression levels after Ang II treatment in the left ventricles of wild-type mice (Fig. 3e). Administration of Ang II was associated with a significant upregulation of Edn1 and c-Jun mRNA expression levels in the left ventricles of Ahr−/− mice but not in the control mice (Fig. 5a, b). These increases were significantly inhibited by fenofibrate in Ahr−/− mice. Furthermore, endothelin-1 levels in protein extracts from LV tissues were higher in Ang II-treated than the control Ahr−/− mice. The increases tended to be prevented by treatment with fenofibrate (Fig. 5c). The c-Jun levels in nuclear extracts from LV tissues were greater in mice treated with Ang II than the control in both genotypes of wild-type and Ahr−/−. Treatment with fenofibrate tended to reduce the increase but the effect was not significant (Fig. 5d).
Fig. 5.
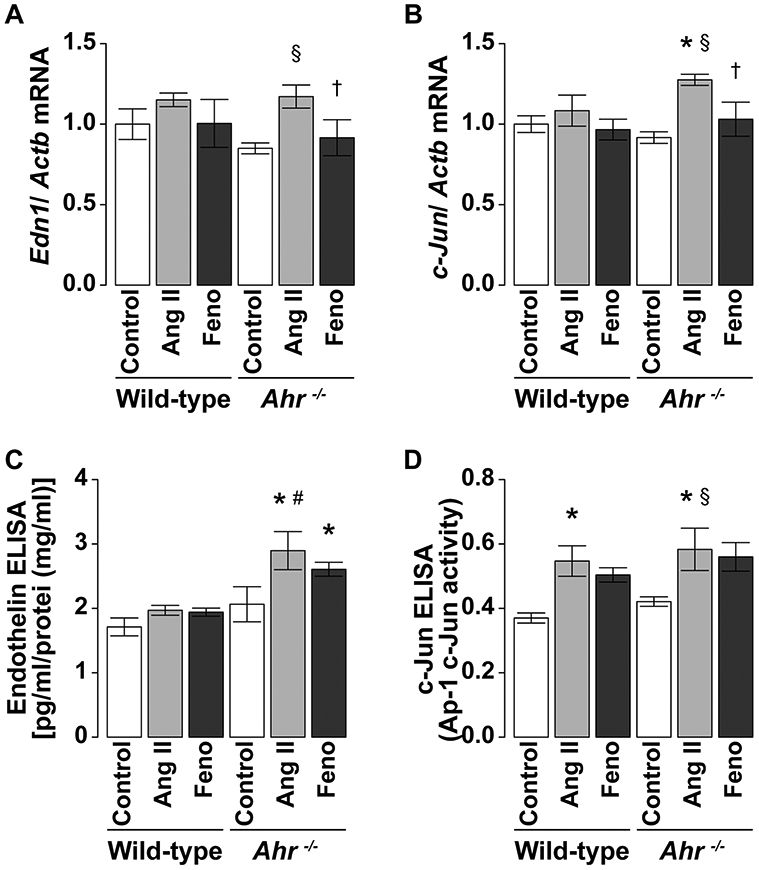
Expression levels of a endothelin-1 and b c-Jun in the left ventricle of wild-type and Ahr−/− mice treated with Ang II (100 ng/kg/min) and fenofibrate. The expression levels were determined by quantitative RT-PCR analysis and expressed relative to the level of Actb mRNA. The activities of c endothelin-1 and d c-Jun in the left ventricle were also determined by ELISA. Data are mean ± SEM of eight mice per group. *P < 0.05 vs. wild-type control mice; #P < 0.05 vs. wild-type Ang II mice; §P < 0.05 vs. Ahr−/− control mice; †P < 0.05 vs. Ahr−/− Ang II mice
Effects of Ang II and fenofibrate on HIF-1α and VEGF expression levels
Immunohistochemical analysis indicated the upregulation of HIF-1α expression in LV nuclear extracts of mice treated with Ang II compared with the control mice in both genotypes of wild type and Ahr−/− (Fig. 6a). A higher expression level was noted in Ahr−/− mice treated with Ang II. The expression of VEGF was also greater in mice treated with Ang II than the control mice in both strains and was significantly greater in Ang II-treated Ahr−/− mice than their wild-type counterparts. Treatment with fenofibrate abrogated the effect of Ang II on VEGF expression in Ahr−/− mice.
Fig. 6.
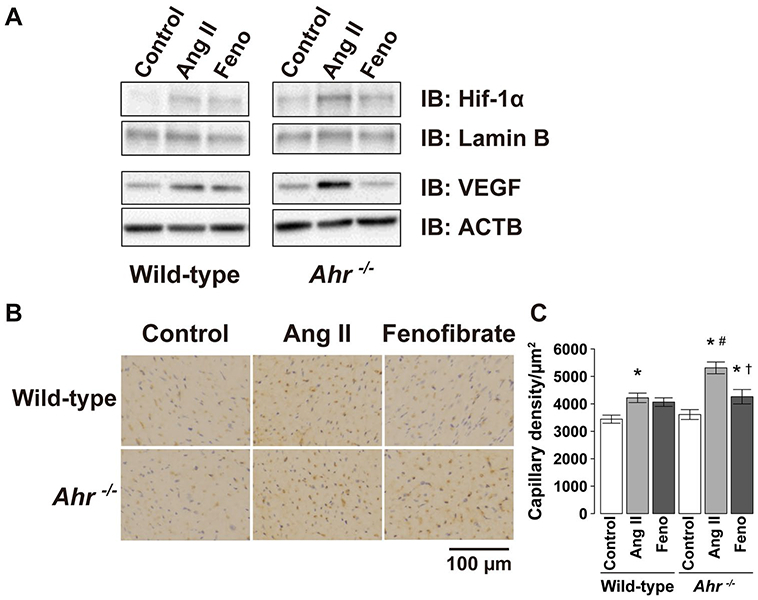
Representative blots of a immunoblot analysis of the amounts of HIF-1α in nuclear extracts from LV tissues and VEGF in protein extracts from homogenates of LV tissues of wild-type and Ahr−/− mice treated with Ang II and fenofibrate. b Capillary detected by immunohistochemical staining with anti-CD31 antibody in the LV wall. Scale bars, 100 μm. c Quantitative analysis of the capillary density in the left ventricle of wild-type and Ahr−/− mice treated with Ang II and fenofibrate. Data are mean ± SEM of six mice per group. *P < 0.05 vs. wild-type control mice; #P < 0.05 vs. wild-type Ang II mice; †P < 0.05 vs. Ahr−/− Ang II mice
Immunohistochemical analysis revealed that capillary density was significantly greater in the left ventricles of mice treated with Ang II than the control mice in both genotypes of wild type and Ahr−/− (Fig. 6b, c). The increase in the capillary density was significantly larger in Ahr−/− mice than wild-type mice. The treatment with fenofibrate reduced the increase in the capillary density (Fig. 6c).
Discussion
We showed in the present study the development of severe cardiac fibrosis after Ang II infusion in Ahr−/− mice compared with wild-type mice. Ang II infusion significantly enhanced the expression of endothelin in the left ventricles of Ahr−/− mice, but not in wild-type mice. Furthermore, the left ventricles of Ahr−/− mice showed a significant increase in the transcriptional activity of c-Jun, as well as the upregulation of HIF-1α and VEGF expression after Ang II infusion. Fenofibrate abrogated the effects of Ang II on HIF-1α and VEGF expression, resulting in the reduction of capillary density. Our results suggest that ablation of AHR was associated with enhanced Ang II-induced cardiac fibrosis and this effect was likely mediated through the associated enhancement of c-Jun transcriptional activity.
AHR was reported to be involved in the pathogenesis of abnormal cardiovascular structure and function (Carreira et al. 2015; Ichihara 2011). We reported previously that ablation of the AHR in a surgical animal model of ischemia was associated with the enhancement of ischemia-induced angiogenesis (Ichihara et al. 2007). As demonstrated the lack of effect of AHR ablation on systolic blood pressure in the same study (Ichihara et al. 2007), the present study also demonstrated no significant effect of AHR ablation on blood pressure. On the other hand, another study (Lund et al. 2008) reported the finding of systemic hypertension in Ahr-null mice at modest altitude. Interestingly, another study showed that maintaining a colony of Ahr-null mice for multiple generations at modest altitude was associated with adaption and development of hypotension in Ahr-null mice (Zhang et al. 2010). While mice used by Zhang et al. were obtained from the same laboratory (National Cancer Institute) as ours, they were maintained in a different laboratory since 2002. Therefore, the phenotype might be somewhat different compared with the mice used in the present study, possibly due to diet or influences of the gut microbiota.
Endothelin is involved in mechanical stress-induced cardiomyocyte hypertrophy and is an important modulator of gene expression in Ang II-based cardiac hypertrophy (Yamazaki et al. 1996). Our study demonstrated significant enhancement of ET-1 expression in the left ventricles of Ahr−/− mice after Ang II infusion. This finding is in agreement with that of a previous study which showed increased plasma ET-1 level in Ahr−/− mice (Lund et al. 2003). There is ample evidence to suggest the involvement of AHR in gross enlargement of the heart and fibrosis of the myocardium (Maayah et al. 2014; Sauzeau et al. 2011). In the present study, basic endothelin levels were not higher in the control Ahr−/− mice than in wild-type mice, however, it is likely that endothelin plays an essential role in cardiac fibrosis induced by Ang II in Ahr−/− mice. A previous study also showed increased plasma Ang II level in Ahr−/− mice (Lund et al. 2003). It has been shown that ET-1 enhances the conversion of Ang I to Ang II (Houde et al. 2013). Conversely, the treatment of Ang II was not associated with the upregulation of Ahr mRNA expression levels in the present study, suggesting that interaction between AHR and Ang II signaling have yet to be elucidated. Further studies are needed to understand the mechanisms of interaction between them.
Angiogenesis is promoted during cardiac remodeling associated with hypertension to compensate for the increased distances between capillaries and cardiomyocytes (Sano et al. 2007; Xu et al. 2005). HIF-1α and activation of VEGF are crucial for the transition from cardiac remodeling, including hypertrophy and fibrosis, to heart failure. VEGF induces the proliferation and movement of endothelial cells, remodeling of the extracellular matrix and formation of capillary tubules. A previous study showed that VEGF-mediated signals are essential in the pathogenesis of Ang II infusion-induced inflammation and remodeling (Zhao et al. 2004). Cardiac angiogenesis induced by overload plays an important role in cardiac adaptation to maladaptive hypertrophy (Li et al. 2018) and blockage of vascular growth attenuated the development of cardiac hypertrophy. As one of the mechanisms, superfluous delivery of nutrients and oxygen, transported in response to an increase in capillary mass, might promote the hypertrophic growth of cardiomyocytes (Oka et al. 2014). The present study showed a significant Ang II-induced upregulation of HIF-1α and VEGF expression in the left ventricles of Ahr−/− mice, suggesting the involvement of HIF-1α in Ang II-induced expression of VEGF, which promotes the development of cardiac fibrosis. Our previous study showed that the DNA-binding activity of the HIF-1α–ARNT complex and the amounts of ARNT and HIF-1α associated with the VEGF gene promoter were markedly upregulated in the ischemic hindlimb of Ahr−/− mice (Ichihara et al. 2007). Although the role of AHR signaling in the regulation of HIF-1α expression remains to be elucidated, an increased abundance and activity of the HIF-1α–ARNT heterodimer might promote the cardiac fibrosis after treatment with Ang II in Ahr−/− mice. Besides, ET-1 is known to activate JNK signaling and induce the expression of c-Jun in cardiomyocytes (Recchia et al. 2009). Furthermore, previous studies suggested that both c-Jun and HIF-1α functionally cooperate in transcription activation, by protecting HIF-1α against degradation by c-Jun (Yu et al. 2009). The significant Ang II-induced upregulation of c-Jun and HIF-1α expression might be due to the enhancement of ET-1 expression in the left ventricles of Ahr−/− mice after Ang II infusion.
The present study showed that Ang II greatly enhanced the development of cardiac fibrosis. Treatment with fenofibrate prevented the increase in Col1a1 and Col3a1 mRNA levels in the left ventricles although fenofibrate did not suppress the increase in blood pressure induced by Ang II infusion. Fenofibrate was reported previously to inhibit cardiac hypertrophy through a negative regulation of the link between nuclear factor of activated T-cells c4 (NFATc4) and p65 subunit of nuclear factor-kappa B (p65-NFκB) (Zou et al. 2013). Fenofibrate also inhibited the development of compensated hypertensive LV hypertrophy, attenuated the LV relaxation abnormality and systolic dysfunction, and improved the survival rate in the rat model through the inhibition of the inflammatory response and of activation of redox-regulated transcription factors in the left ventricle (Ichihara et al. 2006). These results suggest that the effects of fenofibrate were not due to its antihypertensive properties but rather due to antihypertrophic action and the consequent beneficial effects on ventricular structure. Fenofibrate was reported to attenuate ET-1-induced cardiomyocyte hypertrophy through several pathways (Duhaney et al 2007). Inhibition of ET-1-induced cardiac hypertrophy by fenofibrate is considered to be mediated through the negative regulation of AP-1-binding activity (Irukayama-Tomobe et al. 2004). In the present study, fenofibrate reduced cardiac fibrosis and suppressed the increase in the expression of endothelin, c-Jun, HIF-1α, and VEGF genes, which are consistent with the above previous findings. However, the present study has not revealed whether PPARα signaling directly interacts with AHR signaling.
In conclusion, the present study demonstrated that ablation of the AHR was associated with extensive fibrosis after Ang II infusion. Our results suggest that these effects are mediated, at least in part, by overexpression of endothelin and that transcriptional activity of c-Jun, as well as the expression of HIF-1α and VEGF in the heart enhanced the development of cardiac fibrosis in Ahr-null mice. Moreover, we also demonstrated that fenofibrate reduced cardiac fibrosis, probably by under-expression of VEGF through the suppression of c-Jun/HIF-1α activity.
Supplementary Material
Acknowledgements
The authors thank Ms Yuka Sakamaki for the secretarial support and Ms Yumiko Tateno for the experimental support. This work was supported in part by a grant from the JSPS KAKENHI Grant Number JP26293149.
Footnotes
Conflict of interest The authors declare no conflict of interest.
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s00204-019-02446-1) contains supplementary material, which is available to authorized users.
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Balakumar P, Rohilla A, Mahadevan N (2011) Pleiotropic actions of fenofibrate on the heart. Pharmacol Res 63:8–12 [DOI] [PubMed] [Google Scholar]
- Barger PM, Brandt JM, Leone TC, Weinheimer CJ, Kelly DP (2000) Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J Clin Invest 105:1723–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira VS, Fan Y, Kurita H, Wang Q, Ko CI, Naticchioni M, Jiang M, Koch S, Zhang X, Biesiada J, Medvedovic M, Xia Y, Rubinstein J, Puga A (2015) Disruption of Ah receptor signaling during mouse development leads to abnormal cardiac structure and function in the adult. PLoS One 10:e0142440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhaney TA, Cui L, Rude MK, Lebrasseur NK, Ngoy S, De Silva DS, Siwik DA, Liao R, Sam F (2007) Peroxisome proliferator-activated receptor alpha-independent actions of fenofibrate exac-erbates left ventricular dilation and fibrosis in chronic pressure overload. Hypertension 49:1084–1094 [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kim S, Nebert DW, Rudikoff S, Ward JM, Gonzalez FJ (1995) Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 268:722–726 [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero PM, Ward JM, Sundberg JP, Gonzalez FJ (1997) Lesions of aryl-hydrocarbon receptor-deficient mice. Vet Pathol 34:605–614 [DOI] [PubMed] [Google Scholar]
- Houde M, Jamain MD, Labonté J, Desbiens L, Pejler G, Gurish M, Takai S, D’Orléans-Juste P (2013) Pivotal role of mouse mast cell protease 4 in the conversion and pressor properties of Bigendothelin-1. J Pharmacol Exp Ther 346:31–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Ichihara S, Yamada Y, Banu S, Ichihara G (2017) Effect of 4-week inhalation exposure to 1-bromopropane on blood pressure in rats. J Appl Toxicol 37:331–338 [DOI] [PubMed] [Google Scholar]
- Ichihara S, Obata K, Yamada Y, Nagata K, Noda A, Ichihara G, Yamada A, Kato T, Izawa H, Murohara T, Yokota M (2006) Attenuation of cardiac dysfunction by a PPAR-alpha agonist is associated with down-regulation of redox-regulated transcription factors. J Mol Cell Cardiol 41:318–329 [DOI] [PubMed] [Google Scholar]
- Ichihara S, Yamada Y, Ichihara G, Nakajima T, Kondo T, Gonzalez FJ, Murohara T (2007) A role for the aryl hydrocarbon receptor in regulation of ischemia-induced angiogenesis. Arterioscler Thromb Vasc Biol 27:1297–1304 [DOI] [PubMed] [Google Scholar]
- Ichihara S (2011) Role of AHR in the development of the liver and blood vessels In: Pohjanvirta J (ed) The AH receptor in biology and toxicology. Wiley, Oxford, pp 413–422 [Google Scholar]
- Irukayama-Tomobe Y, Miyauchi T, Sakai S, Kasuya Y, Ogata T, Takanashi M, Iemitsu M, Sudo T, Goto K, Yamaguchi I (2004) Endothelin-1-induced cardiac hypertrophy is inhibited by activation of peroxisome proliferator-activated receptor-alpha partly via blockade of c-Jun NH2-terminal kinase pathway. Circulation 109:904–910 [DOI] [PubMed] [Google Scholar]
- Kolluri SK, Jin UH, Safe S (2017) Role of the aryl hydrocarbon receptor in carcinogenesis and potential as an anti-cancer drug target. Arch Toxicol 91:2497–2513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahvis GP, Lindell SL, Thomas RS, McCuskey RS, Murphy C, Glover E, Bradfield CA (2000) Portosystemic shunting and persistent fetal vascular structures in aryl hydrocarbon receptor-deficient mice. Proc Natl Acad Sci USA 97:10442–10447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zeng J, Wu L, Tao L, Liao Z, Chu M, Li L (2018) Loss of P53 regresses cardiac remodeling induced by pressure overload partially through inhibiting HIF1α signaling in mice. Biochem Biophys Res Commun 501:394–399 [DOI] [PubMed] [Google Scholar]
- Lund AK, Goens MB, Kanagy NL, Walker MK (2003) Cardiac hypertrophy in aryl hydrocarbon receptor null mice is correlated with elevated angiotensin II, endothelin-1, and mean arterial blood pressure. Toxicol Appl Pharmacol 193:177–187 [DOI] [PubMed] [Google Scholar]
- Lund AK, Agbor LN, Zhang N, Baker A, Zhao H, Fink GD, Kanagy NL, Walker MK (2008) Loss of the aryl hydrocarbon receptor induces hypoxemia, endothelin-1, and systemic hypertension at modest altitude. Hypertension 51:803–809 [DOI] [PubMed] [Google Scholar]
- Maayah ZH, Ansari MA, El Gendy MA, Al-Arifi MN, Korashy HM (2014) Development of cardiac hypertrophy by sunitinib in vivo and in vitro rat cardiomyocytes is influenced by the aryl hydrocarbon receptor signaling pathway. Arch Toxicol 88:725–738 [DOI] [PubMed] [Google Scholar]
- Nebert DW (2017) Aryl hydrocarbon receptor (AHR): "pioneer member" of the basic-helix/loop/helix per-Arnt-sim (bHLH/PAS) family of "sensors" of foreign and endogenous signals. Prog Lipid Res 67:38–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata T, Miyauchi T, Sakai S, Irukayama-Tomobe Y, Goto K, Yamaguchi I (2002) Stimulation of peroxisome-proliferator-activated receptor alpha (PPAR alpha) attenuates cardiac fibrosis and endothelin-1 production in pressure-overloaded rat hearts. Clin Sci (Lond) 103:284s–288s [DOI] [PubMed] [Google Scholar]
- Oka T, Akazawa H, Naito AT, Komuro I (2014) Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ Res 114:565–571 [DOI] [PubMed] [Google Scholar]
- Peng X, Kraus MS, Wei H, Shen TL, Pariaut R, Alcaraz A, Ji G, Cheng L, Yang Q, Kotlikoff MI, Chen J, Chien K, Gu H, Guan JL (2006) Inactivation of focal adhesion kinase in cardiomyocytes promotes eccentric cardiac hypertrophy and fibrosis in mice. J Clin Invest 116:217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puga A, Ma C, Marlowe JL (2009) The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem Pharmacol 77:713–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recchia AG, Filice E, Pellegrino D, Dobrina A, Cerra MC, Maggiolini M (2009) Endothelin-1 induces connective tissue growth factor expression in cardiomyocytes. J Mol Cell Cardiol 46:352–359 [DOI] [PubMed] [Google Scholar]
- Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I (2007) p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 446:444–448 [DOI] [PubMed] [Google Scholar]
- Sauzeau V, Carvajal-González JM, Riolobos AS, Sevilla MA, Men-acho-Márquez M, Román AC, Abad A, Montero MJ, Fernández-Salguero P, Bustelo XR (2011) Transcriptional factor aryl hydrocarbon receptor (Ahr) controls cardiovascular and respiratory functions by regulating the expression of the Vav3 protooncogene. J Biol Chem 286:2896–2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr DH (2011) Another important biological function for the aryl hydrocarbon receptor. Arterioscler Thromb Vasc Biol 31:1247–1248 [DOI] [PubMed] [Google Scholar]
- Smeets PJ, Teunissen BE, Willemsen PH, van Nieuwenhoven FA, Brouns AE, Janssen BJ, Cleutjens JP, Staels B, van der Vusse GJ, van Bilsen M (2008) Cardiac hypertrophy is enhanced in PPAR alpha−/− mice in response to chronic pressure overload. Cardiovasc Res 78:79–89 [DOI] [PubMed] [Google Scholar]
- Xu J, Nagata K, Obata K, Ichihara S, Izawa H, Noda A, Nagasaka T, Iwase M, Naoe T, Murohara T, Yokota M (2005) Nicorandil promotes myocardial capillary and arteriolar growth in the failing heart of Dahl salt-sensitive hypertensive rats. Hypertension 46:719–724 [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Komuro I, Kudoh S, Zou Y, Shiojima I, Hiroi Y, Mizuno T, Maemura K, Kurihara H, Aikawa R, Takano H, Yazaki Y (1996) Endothelin-1 is involved in mechanical stress-induced cardiomyocyte hypertrophy. J Biol Chem 271:3221–3228 [DOI] [PubMed] [Google Scholar]
- Yokoyama Y, Mise N, Suzuki Y, Tada-Oikawa S, Izuoka K, Zhang L, Zong C, Takai A, Yamada Y, Ichihara S (2018) MicroRNAs as potential mediators for cigarette smoking induced atherosclerosis. Int J Mol Sci 19(4):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Miao ZH, Jiang Y, Li MH, Yang N, Li T, Ding J (2009) c-Jun protects hypoxia-inducible factor-1alpha from degradation via its oxygen-dependent degradation domain in a nontranscriptional manner. Cancer Res 69:7704–7712 [DOI] [PubMed] [Google Scholar]
- Zhang N, Agbor LN, Scott JA, Zalobowski T, Elased KM, Trujillo A, Duke MS, Wolf V, Walsh MT, Born JL, Felton LA, Wang J, Wang W, Kanagy NL, Walker MK (2010) An activated renin-angiotensin system maintains normal blood pressure in aryl hydrocarbon receptor heterozygous mice but not in null mice. Biochem Pharmacol 80:197–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Egashira K, Hiasa K, Ishibashi M, Inoue S, Ohtani K, Tan C, Shibuya M, Takeshita A, Sunagawa K (2004) Essential role of vascular endothelial growth factor and Flt-1 signals in neointimal formation after periadventitial injury. Arterioscler Thromb Vasc Biol 24:2284–2289 [DOI] [PubMed] [Google Scholar]
- Zou J, Le K, Xu S, Chen J, Liu Z, Chao X, Geng B, Luo J, Zeng S, Ye J, Liu P (2013) Fenofibrate ameliorates cardiac hypertrophy by activation of peroxisome proliferator-activated receptor-α partly via preventing p65-NFκB binding to NFATc4. Mol Cell Endocrinol 370:103–112 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.