

Abstract
Lysine specific demethylase-1 (LSD1) has been shown to be critical in acute myeloid leukemia (AML) pathogenesis and this has led to the development of LSD1 inhibitors (LSD1i) which are currently tested in clinical trials. Nonetheless, preclinical studies reported that AML cells frequently exhibit intrinsic resistance to LSD1 inhibition, and the molecular basis for this phenomenon is largely unknown. We explored the potential involvement of mammalian target of rapamycin (mTOR) in mediating the resistance of leukemic cells to LSD1i. Strikingly, unlike sensitive leukemias, mTOR complex 1 (mTORC1) signaling was robustly triggered in resistant leukemias following LSD1 inhibition. Transcriptomic, chromatin immunoprecipitation and functional studies revealed that insulin receptor substrate 1(IRS1)/extracellular-signal regulated kinases (ERK1/2) signaling critically controls LSD1i induced mTORC1 activation. Notably, inhibiting mTOR unlocked the resistance of AML cell lines and primary patient-derived blasts to LSD1i both in vitro and in vivo. In conclusion, mTOR activation might act as a novel pro-survival mechanism of intrinsic as well as acquired resistance to LSD1i, and combination regimens co-targeting LSD1/mTOR could represent a rational approach in AML therapy.
Introduction
Among the novel epigenetic druggable targets in acute myeloid leukemia (AML) therapy, lysine specific histone demethylase 1 (LSD1) has gained attention based on its preferential overexpression in primary AML compared to normal hematopoietic stem and progenitor cells.1,2 In cooperation with the oncogenic MLL-AF9 fusion protein, LSD1 actively sustains AML maintenance.3 Moreover, LSD1 inhibition reactivates an all-trans-retinoic acid (ATRA)-dependent differentiation pathway in AML.4 LSD1 overexpression has also been associated with poor prognosis in various types of tumors including colon and lung cancers.5,6 Mechanistically, LSD1 is a flavin adenine dinucleotide (FAD)-dependent amine oxidase that specifically removes mono- or dimethylated histone H3K4 and H3K9 resulting in context-specific transcriptional repression and activation respectively.7 Apart from chromatin, LSD1 demethylates and hence regulates a wide array of non-histone targets.8,9 All these activities account for the role of LSD1 in regulating tumor proliferation, metastasis and metabolism.2,10,11 We and others have developed LSD1 inhibitors (LSD1i) with potent and selective biochemical profiles and some of which are currently evaluated in clinical trials.12–15 Nonetheless, the preclinical antileukemic activity of LSD1i as a monotherapy is relatively modest.16,17 In solid tumors, specific DNA methylation signatures correlated with the sensitivity to LSD1i.16 However, the molecular mechanisms underlying the differential responsiveness of AML to LSD1i remain largely unknown.
Mammalian target of rapamycin (mTOR) signaling is frequently hyperactive in AML.18 mTOR exists in two distinct complexes; mTORC1 and mTORC2. mTORC1 primarily acts on substrates (as p70 S6 kinase) which controls glycolysis, protein synthesis and lipogenesis.19 mTORC2 regulates actin rearrangement, metabolism and survival (acting on substrates such as AKT).20 Inactivating mTORC1 significantly prolongs the survival of mice transplanted with MLL-AF9 expressing AML cells.21 We and others have previously demonstrated that mTOR activation acts as a fundamental adaptive response exploited by cancer cells to evade the cytotoxic stimuli triggered by several anticancer drugs including epigenetic therapies.22–24 Therefore, in this study, we investigated the potential implication of mTOR in mediating the sensitivity/resistance of AML cells to LSD1i.
Methods
Cell lines and cell culture
AML cell lines were obtained from either DSMZ or ATCC. KASUMI-1, NB4 and THP-1 cells were cultured in RPMI-1640 media supplemented with 2 mM L-glutamine, 10% FBS and 1% penicillin-streptomycin. SKNO-1 cells were cultured in RPMI-1640 media supplemented with 10% FBS, 2 mM L-glutamine, 10 ng/mL GM-CSF and 1% penicillin-streptomycin. UF1 cells were cultured in RPMI-1640 media supplemented with 20% FBS and 2 mM L-glutamine. OCI-AML3 cells were cultured in α-MEM media supplemented with 20% FBS, 2 mM L-glutamine, and 1% penicillin-streptomycin. Phoenix™-Ampho cells were cultured in DMEM media supplemented with 2 mM L-glutamine, 10% FBS and 1% penicillin-streptomycin. Cells were maintained in a humidified tissue culture incubator at 37°C with 5% CO2.
Primary patient-derived AML blast and cord blood-derived CD34+ cells
A primary human AML sample (referred to as AML-IEO20; t(9;11);NPM WT;FLT3 WT) was obtained from the IEO Biobank according to the procedures approved by the Ethical Committee of the European Institute of Oncology. Mononuclear cells were isolated from the peripheral blood/bone marrow samples by Ficoll density centrifugation. For ex vivo studies, AML-IEO20 cells (passage no. 3, ≥90% human leukemic blasts) were thawed and cultured in RPMI-1640 medium supplemented with 20% FBS, 1% S637 and 2mM L-glutamine. Primary human cord blood-derived CD34+ (non-transduced and hMLL-AF9 transduced) cells were cultured in HPGM™ Hematopoietic Growth Medium supplemented with 10% FBS, 2 mM L-glutamine, 100 ng/mL SCF, 100 ng/mL FLT3 and 100 ng/mL thrombopoietin. Before proceeding with in vitro experiments, cryopreserved cells were allowed to recover for at least three days.
In vivo studies
DDP38003 was dissolved in vehicle (40% PEG-400 in 5% glucose solution). A stock solution of rapamycin (10 mg/mL) was prepared in 100% ethanol and stored at -20°C until use. Immediately before administration, rapamycin was diluted in vehicle composed of 5% PEG-400 and 5% Tween-80. AML-IEO20 cells (0.25×106 cells/mouse) were transplanted via tail vein injection of 8-10 weeks old NOD-SCID-IL2Rcγnull (NSG) mice. One week post-transplantation, mice were randomly assigned into four different groups which were treated for five weeks. The first group served as vehicle treated group. The second group was administered DDP38003 (16.8 mg/kg, by oral gavage). The third group received rapamycin (5 mg/kg, intraperitoneally). The fourth group received DDP38003 and rapamycin. The survival of the mice was analyzed and represented by a Kaplan-Meier survival plot. All animal studies were conducted in compliance with the Italian Legislative Decree No.116 dated January 1992 and European Communities Council Directive No.86/609/EEC concerning the protection of animals used for experimental purposes and other scientific purposes according to the institutional policy regarding the care and use of laboratory animals. Mice were housed according to the guidelines set out in Commission Recommendation 2007/526/EC – June 18, 2007, guidelines of the accommodation and care of animals used for experimental and other scientific purposes. The study was approved by both the Ethical Committee of the European Institute of Oncology and Italian Ministry of Health (Project license number 199/2017).
For additional methods, please refer to the Online Supplementary Materials and Methods.
Results
Heterogeneous AML responses to LSD1i do not correlate with basal LSD1 level
To explore the anti-leukemic activity of inhibiting LSD1, we initially used DDP38003 (previously referred to as Compound 15),13 a potent selective and irreversible LSD1i, against a panel of AML cell lines belonging to different subtypes. As previously reported,12,17 AML cells demonstrated heterogeneous responses to LSD1 inhibition. DDP38003 dramatically diminished the proliferation and viability (cellular ATP level) of KASUMI-1, SKNO-1 and UF1 cells (Figure 1A and Online Supplementary Figure S1A). Indeed, DDP38003 induced apoptotic cell death in sensitive AML cells (Figure 1B and Online Supplementary Figure S1B). Conversely, the viability and proliferation of NB4, OCI-AML3 and THP-1 cells were not significantly affected reflecting their resistance to DDP38003 (Figure 1A-B and Online Supplementary Figure S1A-B). Next, we investigated whether DDP38003 was efficiently inhibiting LSD1 in resistant AML. Indeed, genes reported to be directly repressed by LSD112,25 were upregulated post-DDP38003 treatment in both resistant THP-1 and sensitive KASUMI-1 cells confirming efficient LSD1 inhibition (Online Supplementary Figure S1C). We then inquired whether such differential responsiveness of AML cells correlates with the basal level of LSD1. LSD1 levels in both sensitive and resistant AML were comparable ruling out this hypothesis (Figure 1C). Altogether, our findings indicate that assessing the changes in the transcript levels of direct target genes of LSD1 per se and/or basal LSD1 levels do not explain differential vulnerability/responsiveness of AML cells to LSD1i.
Figure 1.

In contrast to acute myeloid leukemia cells sensitive to LSD1 inhibition, mTOR signaling is robustly activated in resistant AML cells in response to their treatment with DDP38003. (A) Growth curves of the indicated acute myeloid leukemia (AML) cell lines treated with either vehicle or DDP38003 (0.5 μM) for the indicated time points of treatment assessed using trypan blue cell counting. Data were statistically analyzed using two way ANOVA followed by Bonferroni post-hoc test. A: P<0.05 compared to vehicle-treated cells (n=3). (B) Representative flowcytometry dot plots depicting the effect of six days of vehicle or DDP38003 (0.5 μM) treatment on the viability/apoptosis of the indicated AML cells assessed by Annexin V/PI staining (left panel) and their quantitation (right panel). (C) Western blot analysis of LSD1 levels in the indicated AML cell lines. β-actin served as the loading control. (D) Immunoblotting analysis of mTOR signaling pathway in LSD1i-sensitive (KASUMI-1, SKNO-1 and UF1) and LSD1i-resistant (NB4, OCI-AML3 and THP-1) AML cells treated for six days with either vehicle or different concentrations of DDP38003 (0.1 and 0.5 μM). Vinculin served as the loading control. The presented blots are derived from replicate samples run on parallel gels and controlled for even loading. LSD1: lysine specific demethylase-1; LSD1i: LSD1 inhibitors.
Activation of mTORC1 correlates with the resistance of AML cells to LSD1 inhibition
mTOR is constitutively activated in AML blasts26 and mediates chemoresistance.23 We therefore investigated the effect on mTOR signaling as a potential mechanism of resistance of AML cells to LSD1i. Indeed, DDP38003 triggered mTORC1 activation in resistant AML cells as shown by increased phosphorylation of its downstream targets: p70 S6 kinase (p70S6K), ribosomal S6 and 4 eukaryotic-binding protein 1 (4E-BP1) (Figure 1D). Conversely, treatment of sensitive AML with DDP38003 inactivated mTORC1 (Figure 1D). The activity of mTORC2 (evaluated by assessing AKT phosphorylation at S473) was not consistently modulated in response to LSD1i in resistant and sensitive AML cells (Figure 1D). Collectively, these findings suggest that distinct fine-tuning of mTORC1 activity correlates with the differential responsiveness of AML cells to DDP38003.
Mirroring the response to DDP38003, mTORC1 was induced in resistant AML cells and inhibited in sensitive AML cells following their treatment with MC2580 (another selective LSD1i previously referred to as Compound 14e27) (Online Supplementary Figure S1D-G). Finally, to confirm that LSD1 was the key molecular target for the phenotypic/molecular responses and exclude potential off-target effects associated with pharmacological inhibition, two different LSD1-targeting small hairpin RNA (shRNA) were used. Consistently, while LSD1 knockdown sharply affected the proliferation of KASUMI-1 cells resulting in preferential counter-selection of one of the shRNA against wild-type cells, THP-1 and OCI-AML3 cells tolerated LSD1 knockdown (Figure 2A-C). shRNA against LSD1 efficiently reduced LSD1 mRNA and protein levels, and subsequently upregulated the transcription of a direct target gene of LSD1 (CD11b) which was also modulated by DDP38003 (Figure 2D-L). Recapitulating the effect of pharmacological LSD1i, mTORC1 was inhibited in sensitive AML cells and induced in resistant AML cells following LSD1 knockdown (Figure 2J-L). Altogether, our data demonstrate that the sensitivity/resistance of AML cells to LSD1 inhibition is associated with distinctive modulation of mTORC1 activity.
Figure 2.

Genetic LSD1 knockdown recapitulates the effects of pharmacological LSD1 inhibition on mTOR signaling in sensitive and irresponsive acute myeloid leukemia cells. (A-C) Growth curves of KASUMI-1 (A), THP-1 (B) and OCI-AML3 (C) cells transduced with retroviral vectors expressing short hairpin RNA (shRNA) against control (scrambled) or LSD1 (shLSD1 #1 and shLSD1 #2) (n=3). (D-F) Normalized LSD1 and CD11b mRNA levels assessed in transduced KASUMI-1 (D), THP-1 (E), OCI-AML3 (F) cells expressing the indicated shRNA using real-time quantitative PCR (RT-qPCR). Data were statistically analyzed using either Student’s t-test (A, D and G) or one way ANOVA followed by Bonferrroni post hoc test (B, C, E, F, H and I). *: P<0.05 compared to control (scrambled). (J-L) Western blot analysis of lysates obtained from transduced KASUMI-1 (J), THP-1 (K), OCI-AML3 (L) cells expressing the indicated shRNA. The presented blots are derived from replicate samples run on parallel gels and controlled for even loading.
Abrogating mTOR signaling counteracts the resistance of AML cells to LSD1 inhibition
Next, we explored the effect of inactivating mTOR on the response of AML cells resistant to LSD1 inhibition using different strategies. Inhibiting mTOR pharmacologically using either rapamycin (allosteric mTOR inhibitor) or AZD8055 (ATP competitive mTOR kinase inhibitor) sensitized resistant THP-1 cells to pharmacological LSD1i or genetic knockdown of LSD1 (Figure 3A-H). Mimicking nutritional stress using 2-deoxyglucose (2DG, a non-metabolizable glucose analogue)28 also counteracted DDP38003-mediated mTOR activation and rendered THP-1 cells responsive to LSD1i (Online Supplementary Figure S2A-C).
Figure 3.

Inhibition of mTOR signaling reverses the resistance of acute myeloid leukemia cells to LSD1 inhibition. (A) Growth curves of THP-1 cells treated with vehicle, DDP38003 (0.5 μM), rapamycin (10 nM) or DDP38003 and rapamycin for the indicated time points of treatment. Data were statistically analyzed using two way ANOVA followed by Bonferrroni post hoc test, a,b,c: P<0.05 compared to vehicle, DDP38003 or rapamycin alone treated groups respectively (n=3). Note that we have previously demonstrated that LSD1 inhibition affects THP-1 cells in clonogenic but not in liquid culture assays.35 Indeed, co-inhibiting mTOR significantly augmented the anti-clonogenic activity of DDP38003 further promoting myeloid lineage differentiation of THP-1 cells (Online Supplementary Figure S3A-B). (B) Western blot analysis of lysates obtained from THP-1 cells (A) following 72 hours (h) of treatment. β-actin served as the loading control. (C) Proliferation curves of THP-1 cells treated with vehicle, DDP38003 (0.5 μM), AZD8055 (20 nM) or DDP38003 and AZD8055 for the indicated time points of treatment. Data were statistically analyzed using two way ANOVA followed by Bonferrroni post hoc test, a,b,c: P<0.05 compared to vehicle, DDP38003 or AZD8055 alone treated groups respectively (n=3). (D) Western blot analysis of lysates obtained from THP-1 cells (C) following 72 h of treatment. β-actin served as the loading control. (E) Growth curves of transduced THP-1 cells expressing control shRNA or shRNA against LSD1 treated with vehicle or rapamycin for the indicated time points of treatment. Data were statistically analyzed using two way ANOVA followed by Bonferrroni post hoc test, a,b,c:P<0.05 compared to vehicle, shLSD1 or rapamycin alone treated groups respectively (n=3). (F) Western blot analysis of lysates obtained from THP-1 cells (E) following 144 h of treatment. β-actin served as the loading control. (G) Growth curves of transduced THP-1 cells expressing control shRNA or shRNA against LSD1 treated with vehicle or AZD8055 for the indicated time points of treatment. Data were statistically analyzed using two way ANOVA followed by Bonferrroni post hoc test, a,b,c: P<0.05 compared to vehicle, shLSD1 or AZD8055 alone treated groups respectively (n=3). (H) Western blot analysis of lysates obtained from THP-1 cells (G) following 144 h of treatment. β-actin served as the loading control.
A substantial proportion of initially responder cancer patients eventually relapses/progresses. Trying to simulate this scenario, parental KASUMI-1 cells (designated as KASUMI-1/P) were continuously exposed to increasing concentrations of DDP38003 for 12 months until they started to proliferate in the presence of DDP38003. Resistant descendent cells (named KASUMI-1/R) demonstrated a resistance index (RI) of 21 against DDP38003, and were also cross-resistant to MC2580 (RI of ~10) (Online Supplementary Figure S4A-C). As shown in the Online Supplementary Figure S4D, mTOR signaling was activated in KASUMI-1/R cells compared to their parental counter-part, and treatment with DDP38003, despite reducing the extent of mTOR activation, did not reach the low levels observed in parental cells. Besides boosting the responses of KASUMI-1/P cells to LSD1i, inhibiting mTOR dramatically reversed the acquired resistance of KASUMI-1/R cells to LSD1i via triggering apoptosis and this thereby indicates that compensatory mTOR activation protects AML cells against LSD1i-induced apoptotic cell death (Online Supplementary Figure S4E-G). Collectively, our data suggest that targeting mTOR counteracts both intrinsic and acquired resistance of AML cells to LSD1i in vitro.
IRS1 and ERK1/2 signaling are involved in mTOR regulation by LSD1
We attempted to gain insights into the mechanism(s) through which LSD1 regulates mTOR. AMP activated protein kinase (AMPK) is a key negative regulator of mTOR.29 Following LSD1i, AMPK activity was increased in both sensitive and resistant AML, as reflected by increased phosphorylation of AMPK and its downstream target, acetyl CoA carboxylase (ACC) (Online Supplementary Figure S5A-D). The levels of Raptor, a regulator and component of mTORC1, were not consistently modulated in response to LSD1 inhibition (Online Supplementary Figure S5E-G). Taken together, these results suggest that these mechanisms might not be critical for the observed modulatory effects on mTOR. We then monitored the activity of mTORC1 signaling in sensitive KASUMI-1 and resistant THP-1 cells at different time points following LSD1i. Even though six hours (h) of DDP38003 treatment were not enough to elicit any remarkable effects on the proliferation of sensitive KASU-MI-1 cells, mTORC1 was dramatically inactivated (Online Supplementary Figure S6A). Conversely, 24 h post-LSD1i in resistant THP-1 cells, mTORC1 was robustly triggered (Online Supplementary Figure S6B). Such modulatory effects were maintained throughout the subsequent time points of treatment. Accordingly, we decided to perform transcriptomic analysis at the earliest detected and last tested time points in which mTORC1 activity was modulated secondary to LSD1i (i.e. 6 and 72 h in KASUMI-1 cells and 24 and 72 h in THP-1 cells). Consistent with the results of cell viability assays (Figure 1A-B and Online Supplementary Figure S1A-B), ingenuity pathway analysis (IPA) showed a significant modulation of gene sets involved in “cellular growth and proliferation” in sensitive KASUMI-1 but not resistant THP-1 cells post-LSD1i treatment (Online Supplementary Figure S7A-B). Notably, IPA predicted extracellular-signal regulated kinases 1 and 2 (ERK1/2) to be activated in resistant but not in sensitive AML following LSD1i (Online Supplementary Table S1A-4). ERK1/2 is reported to be an upstream activator of mTOR.26 In parallel with mTOR modulation, DDP38003 inhibited ERK1/2 in sensitive AML cells (KASUMI-1) and activated ERK1/2 in resistant AML cells (THP-1 and NB4) cells (Online Supplementary Figure S6A-C). Inhibiting ERK1/2 using several unrelated selective MEK1/2 inhibitors as U0126, pimasertib and trametinib rendered resistant AML cells more vulnerable to LSD1 inhibition (Online Supplementary Figure S6D-H). These findings all suggest that ERK1/2 acts upstream of mTOR dysregulation by LSD1.
To further investigate how LSD1 regulates ERK1/2 and mTOR, we analyzed our RNA-Seq data which revealed that a subset of genes was differentially modulated in resistant versus sensitive AML following LSD1i (Figure 4A). Among those differentially expressed genes, insulin receptor substrate 1 (IRS1) was upregulated in resistant but not responsive AML cells after DDP38003 treatment (Figure 4A-B and Online Supplementary Table S5). IRS1 is an adaptor protein which regulates various pathways including ERK1/2 and mTOR.30 Confirming RNA sequencing (RNA-Seq) data, treatment with pharmacological LSD1i or LSD1 knockdown significantly increased IRS1 mRNA and protein levels in resistant AML cells, unlike sensitive AML cells (Figure 4C-K and Online Supplementary Figure S8A). Cistrome database analyses of previously deposited LSD1 ChIp-seq tracks12 showed that LSD1 was associated to the IRS1 promoter in resistant NB4 cells, while it was not bound in sensitive KASUMI-1 and SKNO-1 cells (Online Supplementary Figure S8B). We confirmed LSD1 binding to the IRS1 promoter of LSD1i resistant AML cells by ChIP-qPCR (Figure 4L-M and Online Supplementary Figure S8C-D). Following the induction of IRS1 by DDP38003 treatment, H3K4me3, H3K9Ac and H3K27Ac histone marks at IRS1 promoter were strongly increased compared to vehicle-treated cells (Figure 4N-P). Modest changes in H3K4me2 were found in THP-1 and KASUMI-1 cells following their treatment with LSD1 inhibitors (DDP38003, GSK690 and RN-1) (Online Supplementary Figure S8E-F).
Figure 4.

Insulin receptor substrate 1 is transcriptionally upregulated in resistant acute myeloid leukemia cells following LSD1 inhibition. (A) Venn diagram illustrating the number of differentially as well as commonly expressed genes in THP-1 and KASUMI-1 cells 72 hours (h) following their treatment with DDP38003 (0.5 μM). (B) RNA sequencing tracks of insulin receptor substrate 1 (IRS1) gene following the indicated time points of treating KASUMI-1 and THP-1 cells with either vehicle or DDP38003 (0.5 μM). (C-D) Normalized IRS1 mRNA levels assessed in THP-1 (C) and OCI-AML3 (D) cells following their treatment with either vehicle (Veh) or DDP38003 (DDP, 0.5 μM) and using real-time quantitative PCR (RT-qPCR). Data were statistically analyzed using either Student’s t-test *: P<0.05. (E-F) Normalized IRS1 mRNA levels assessed in THP-1 (E) and OCI-AML3 (F) cells transduced with the indicated short hairpin RNA (shRNA) (control [scrambled] or LSD1 [shLSD1 #1 and shLSD1 #2]) using real-time quantitative PCR (RT-qPCR). Data were statistically analyzed using one way ANOVA followed by Bonferrroni post hoc test.*: P<0.05 compared to control (scrambled). (H-K) Western blot analysis of lysates obtained from THP-1 (H), OCI-AML3 (I), NB4 (J) and SKNO-1 (K) cells treated with either vehicle or DDP38003 (0.5 μM). Vinculin served as a loading control. (L) Schematic outline of the chromatin immunoprecipitation qPCR (ChIP-qPCR) primers designed to analyze the enrichment of LSD1 or histone marks on IRS1 promoter. (M) LSD1 ChIP-qPCR analyses were performed in THP-1 cells using antibody against LSD1 or IgG as a control. Enrichment values at the indicated sites (A–D) were normalized to input DNA. Values are means ± standard deviation (SD). *: P<0.05. (N-P) ChIP-qPCR analyses to assess H3K4me3 (N), H3K9Ac (O) and H3K27Ac (P) histone marks were performed in THP-1 cells 72 h following their treatment with either vehicle or DDP38003 (0.5 μM). Enrichment values at the indicated sites (A–D) were normalized to input DNA. Values are means ± standard deviation (SD). *: P<0.05.
To delineate the hierarchical relationship of IRS1 with ERK1/2 and mTOR, we investigated the effect of NT157, a selective IRS1/2 inhibitor.31 Co-treatment with NT157 reversed LSD1i-induced ERK/mTOR stimulation and sensitized tolerant AML cells to LSD1i, suggesting that IRS1 acts upstream of ERK/mTOR (Figure 5A-C).
Figure 5.

Inhibiting insulin receptor substrate 1 sensitizes resistant acute myeloid leukemia cells to LSD1 inhibition. (A) Relative cell number of NB4 cells treated with either vehicle (Veh), DDP38003 (DDP, 0.5 μM), NT-157 (1.25 μM) or their combination for 72 hours (h). Data were statistically analyzed using one way ANOVA followed by Bonferrroni post hoc test (n=3). *:P<0.05. (C) Relative cell number of THP-1 cells treated with either vehicle (Veh), DDP38003 (DDP, 0.5 μM), NT-157 (1.25 μM) or their combination for 24 h. Data were statistically analyzed using one way ANOVA followed by Bonferrroni post-hoc test (n=3).*: P<0.05. (B) Western blot analysis of THP-1 cells treated as indicated. Vinculin served as a loading control. (D-E) Relative cell number of NB4 (D) and THP-1 (F) cells treated with either vehicle (Veh), DDP38003 (DDP, 0.5 μM), all-trans-retinoic acid (ATRA – 1 μM) or their combination. Data were statistically analyzed using one way ANOVA followed by Bonferrroni post hoc test (n=3).*: P<0.05. (E) Western blot analysis. Vinculin served as a loading control.
All trans-retinoic acid (ATRA) has been reported to downregulate IRS1.32,33 Indeed, ATRA repressed IRS1 transcription and cooperated with LSD1i as previously reported (Online Supplementary Figure S9A-E and Figure 5D-F).4 Consistently, ATRA counteracted LSD1i-mediated induction of IRS1/ERK/mTOR in resistant AML cells and led to reduced H3K4me2 and, to a greater extent, H3K27Ac accumulation on IRS1 promoter (Figure 5F and Online Supplementary Figure S9F-I). Collectively, our data indicate that differential dysregulation of IRS1/ERK signaling might contribute at least partly to the modulation of mTOR following LSD1i.
Targeting mTOR sensitizes resistant primary human AML blasts to LSD1 inhibition in vitro and in vivo
We then checked the effect of LSD1i on primary human cells. mTOR signaling was not modulated in primary human hematopoietic CD34+ progenitor cells which tolerated DDP38003 (Online Supplementary Figure S10A-C). In contrast, mTOR was inhibited by LSD1i treatment in transduced human MLL-AF9 expressing CD34+ cells where LSD1i adversely affected their proliferation and clonogenicity while promoting myeloid differentiation (Online Supplementary Figure S11A-G).
Eventually, we explored the therapeutic value of co-inhibiting mTOR in primary patient derived AML blasts resistant to LSD1i (referred to as AML-IEO20, expressing the oncofusion protein MLL-AF9). Confirming the results seen in resistant AML cell lines, DDP38003 induced mTORC1 in resistant AML-IEO20 cells (Figure 6A-C). Inhibiting mTOR signaling sensitized AML-IEO20 cells to LSD1i (Figure 6D-F). This was associated with increased G0/G1 arrest and apoptotic cell death (Online Supplementary Figure S12A-B).
Figure 6.

Targeting mTOR sensitizes primary patient-derived MLL-AF9 expressing (AML-IEO20) leukemia blasts to LSD1 inhibition in vitro. (A) Growth curves of AML-IEO20 leukemic cells treated with vehicle or DDP38003 (0.1 or 0.5 μM) for the indicated time points of treatment (n=3). (B) Assessment of CD11b mRNA levels in AML-IEO20 leukemic cells following 72 hours (h) of treatment with vehicle or DDP38003 (0.1 and 0.5 μM) using real-time quantitative PCR (RT-qPCR). Data were statistically analyzed using one way ANOVA followed by Bonferrroni post hoc test.*: P<0.05 compared to vehicle-treated cells. (C) Immunoblotting analysis of mTOR signaling pathway in AML-IEO20 leukemic cells treated for 72 h with either vehicle or different concentrations of DDP38003 (0.1 or 0.5 μM). Vinculin served as the loading control. The presented blots are derived from replicate samples run on parallel gels and controlled for even loading. (D-E) Effect of targeting mTOR signaling using an allosteric mTOR inhibitor (rapamycin – 10 nM) (D) or an ATP competitive mTOR kinase inhibitor (AZD8055 – 10 nM) (E) on the growth kinetics of AML-IEO20 leukemic cells treated with vehicle or DDP38003 (0.5 μM). Data were statistically analyzed using two way ANOVA followed by Bonferrroni post hoc test (n=3). a,b,c: P<0.05 compared to vehicle, DDP38003/shLSD1 or rapamycin/AZD8055 alone treated groups respectively. (F) Western blot analysis of lysates obtained from AML-IEO20 cells treated as indicated for 72 h. Vinculin served as the loading control. The presented blots are derived from replicate samples run on parallel gels and controlled for even loading.
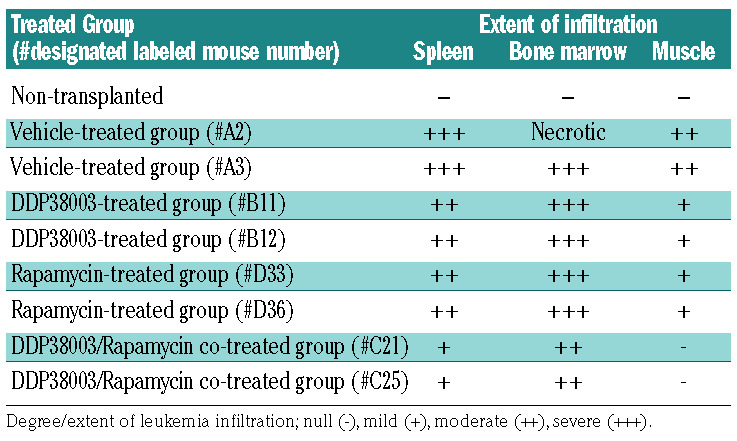
To validate our results in vivo, AML-IEO20 cells were transplanted into NSG mice. As shown in Figure 7A, one week post-transplantation, mice were randomly assigned into four cohorts and treated with: vehicle, DDP38003, rapamycin or their combination. After two weeks of treatment, DDP38003 as a monotherapy failed to lessen the percent of circulating human AML cells, while rapamycin caused a significant decrement (Figure 7C). DDP38003/rapamycin combinatorial regimen significantly reduced the percent of hCD45+ leukemic cells in the peripheral blood as compared to vehicle and DDP38003-treated groups (Figure 7C). Even though DDP38003/rapamycin co-treatment further lessened the percent of human AML cells by almost 60% compared to rapamycin alone, such a decrement was not statistically significant. At this stage of treatment, two mice from each cohort were sacrificed, and spleen and bone marrow tissues were harvested. Rapamycin, but not DDP38003, reduced spleen and bone marrow infiltration by leukemic cells was comparable to the vehicle-treated group (Figure 7D-F). Notably, DDP38003/rapamycin combination elicited an even stronger reduction of leukemic infiltration (Figure 7E-F). May Grunwald/Giemsa-stained cytospin preparations of blood smear, spleen and bone marrow, histopathological and immunohistochemical examinations further confirmed the superior antileukemic the activity of the LSD1i/mTORi combinatorial regimen (Online Supplementary Figure S12C-F and Table 1). After three weeks of treatment, circulating hCD45+ leukemic cells were present in the cohorts treated with vehicle, DDP38003 and rapamycin as monotherapies while they remained dramatically decreased by the combination treatment (Figure 7G). Indeed, the combination treatment significantly prolonged the survival of PDX mice as compared to vehicle (P=0.001), DDP38003 (P=0.0004) and rapamycin (P=0.0024)-treated groups (Figure 7J). Altogether, our results provide the first proof of principle demonstrating preclinical evidence for a therapeutic strategy to restore the efficacy of LSD1i in irresponsive AML patients based on co-inhibiting LSD1/mTOR.
Figure 7.

Targeting mTOR sensitizes primary patient-derived MLL-AF9 expressing (AML-IEO20) leukemia blasts to LSD1 inhibition in vitro and in vivo. (A) Schematic outline of the in vivo studies with the PDX model of AML-IEO20 leukemia. (B-I) Flow cytometric analyses of the percent of human CD45+ leukemic cells in the peripheral blood (PB) obtained from NSG mice transplanted with primary human AML-IEO20 cells and treated for 7 (B), 14 (C), 21 (G) and 28 (H) days with either: vehicle (Veh), DDP38003 (DDP, 16.8 mg/kg, PO), rapamycin (Rapa, 5 mg/kg, IP) or DDP38003 + rapamycin. (D) Effect of different treatments on the spleen index following 15 days of treatment. Data were statistically analyzed using one-way ANOVA followed by Tukey-Kramer post hoc test. a,b,c: P<0.05 compared to non-transplanted and vehicle-treated and DDP38003 alone xenotransplanted groups respectively. Upper right panel: image of spleens harvested from NSG mice 15 days following their treatment with either vehicle or DDP38003 or rapamycin or a combination of DDP38003 and rapamycin (n=2/group). (E-F) Flowcytometric analysis of AML-IEO20 leukemic engraftment depicted as percent of human CD45+ leukemic cells in the spleen (E) and bone marrow (F) after 15 days of treatment initiation (n=2/group). Data were statistically analyzed using one-way ANOVA followed by Tukey-Kramer post hoc test. *: P<0.05. Data represents mean ± standard deviation (SD). Data were statistically analyzed using one-way ANOVA followed by Tukey-Kramer post hoc test. *: P<0.05. (I) Flow cytometric analyses of the percent of human CD45+ leukemic cells in peripheral blood obtained from NSG mice transplanted with primary human AML-IEO20 cells and treated for the indicated time points with either: vehicle, DDP38003, rapamycin or their combination. (J) Kaplan Meier survival curve of mice engrafted with primary AML-IEO20 cells treated with vehicle, DDP38003, rapamycin or DDP38003 + rapamycin. Statistical significance was evaluated using log-rank (Mantel-Cox’s) test. a,b,c: P<0.05 compared to vehicle, DDP38003 or rapamycin-only treated groups respectively. (K) Schematic representation of the proposed mechanism by which modulation of IRS1/ERK/mTOR signaling governs the sensitivity/resistance of acute myeloid leukemia (AML) cells to LSD1 inhibition (LSD1i).
Table 1.
Targeting mammalian target of rapamycin (mTOR) sensitizes primary patient-derived acute myeloid leukemia (AML) blasts (AML-IEO20) to LSD1 inhibition. Degree of leukemic cells infiltration of the spleen, bone marrow and surrounding muscular tissues harvested from NSG mice transplanted with human primary AML-IEO20 cells, sacrificed 15 days after initiation of treatment.
Discussion
AML cells have been reported to elicit heterogeneous responses to LSD1i.12,17 Here, we explored the mechanisms of sensitivity and resistance of AML cells to LSD1 inhibition. Initially, we ruled out the possibility that differential basal LSD1 levels might account for discrepant vulnerability of AML cells to LSD1i, consistent with what was described with T-cell lymphoblastic leukemias.34 Moreover, global transcriptomic changes in the target genes of LSD1 (as CD11b) did not correlate with the discrepant responses of AML cells to LSD1i. Intriguingly, we found that distinctive modulation of mTORC1 activity acts as a key mediator of the susceptibility of AML cells to LSD1i therapy (Figure 7K). We and others have previously demonstrated that mTORC1 contributes to the resistance of diverse types of tumours to targeted anticancer therapies, such as histone deacetylase (HDAC) and tyrosine kinase inhibitors.23,35,36 Likewise, mTORC1 signaling was robustly triggered in AML cells that tolerated LSD1i. In contrast, mTORC1 was inhibited in LSD1i-sensitive AML, as recently described using S2101, another LSD1i, in responsive ovarian carcinoma cells.37 Inhibiting mTOR via direct pharmacological inhibition, or mimicking energetic stress using the non-metabolizable glucose analogue, 2-deoxyglucose, reversed LSD1i-induced mTOR activation and counteracted the resistance of AML cells to LSD1i. Intriguingly, our findings with the glycolytic inhibitor, 2-deoxyglucose, could also be explained by Poulain et al., who demonstrated that heightened mTORC1 activity promotes glycolysis and drives glucose addiction in AML cells.38 Since mTOR acts as a fundamental metabolic checkpoint, LSD1-induced mTOR modulation might contribute to the epigenetic plasticity of cancer cell metabolism.11 The ON/OFF regulatory effects of LSD1i on mTOR could also account for the previously reported regulatory effects of LSD1 on metabolic reprogramming.39,40
Importantly, co-inhibiting LSD1 and mTOR significantly reduced the leukemic burden and prolonged the survival of mice xenotransplanted with primary patient-derived AML (with MLL-AF9 chromosomal translocations) compared to monotherapies. Consistent with the preclinically observed synergy between HDAC inhibitors and mTOR inhibitors, encouraging anticancer activities of vorinostat (HDAC inhibitor) when combined with sirolimus (mTOR inhibitor) have also been reported in patients with refractory Hodgkin lymphoma, perivascular epithelioid tumor, and hepatocellular carcinoma.36,41 The observed synergy between LSD1i and mTOR inhibitors remains to be verified in patient-derived AML blasts exhibiting a diverse genetic background.
In addition, it is worth mentioning that while we have not noticed any potential impact of LSD1 inhibition on the proliferation as well as mTOR signaling of primary human CD34+ cord blood cells following three days of treatment, this does not exclude potential adverse effects on normal hematopoiesis following long-term LSD1 inhibition which was previously reported.42 Hence, this should carefully be considered while designing clinical trials evaluating the efficacy of LSD1i as a monotherapy or in combination regimens.
Acquired resistance is a frequently encountered hurdle in cancer therapy. Despite being formerly responsive, tumor cells have a formidable capability to develop resistance to indefinite spectra of anti-cancer agents when chal lenged for long periods.23 After prolonged exposure of responsive AML (KASUMI-1/P) to DDP38003, secondarily resistant AML cells (KASUMI-1/R) started to grow in the presence of LSD1i. Such KASUMI-1/R cells were cross resistant to another LSD1i. Of note, mTOR activation was observed not only in AML cells intrinsically resistant to LSD1 inhibition, but also as a mechanism of acquired resistance to LSD1 inhibition in primarily sensitive AML cells. Analogously, imatinib triggered mTOR activation in a chronic phase chronic myelogenous leukemia (CML) patient which critically mediated CML survival during the early phase of acquired imatinib resistance before the acquisition of a kinase mutation.22 Although we have not analyzed the eventual genetic alterations in KASUMI-1/R cells, the observation that acquired resistance could be reverted by mTOR inhibition suggests that an adaptive rather than a genetic mechanism is involved in mediating mTOR activation and resistance to LSD1 inhibition. Nonetheless, this shall be systemically investigated in our future studies on a larger subset of secondarily resistant AML.
Delving deeper, we have investigated how LSD1 differentially modulates mTOR in resistant versus sensitive AML cells. Intriguingly, we have observed mTORC1 activation in experimental conditions where AMPK - which in many cases acts as a mTOR inhibitor29 - was activated and thereby excluding its involvement. In line with AMPK stimulation, we found that LSD1i increases the phosphorylation and hence inactivation of the down-stream target of AMPK, ACC which is the rate-limiting enzyme of fatty acid synthesis. In line with our data, LSD1 knockdown has been shown to reduce the triglyceride levels through modulating sterol regulatory element binding protein (SREBP1)-mediated activation of lipogenic gene transcription.43 mTOR also promotes de novo lipogenesis via activating SREBP1 and phosphorylating serine/arginine protein kinases, thereby promoting the splicing of lipogenic pre-mRNA.44 Our data highlighting the modulatory effects of LSD1 on ACC and mTOR enrich the evidences linking LSD1 to the regulation of lipid metabolism.
Upstream regulator IPA predicted ERK1/2 to be activated in resistant but not sensitive AML following LSD1i. Of note, ERK1/2 is known to positively regulate the activity of mTOR signaling via acting both upstream and downstream of mTOR.23,45 Inhibiting ERK1/2 inhibited mTOR and counteracted AML resistance to LSD1i. Moreover, transcriptomic studies showed that IRS1 was upregulated following LSD1i in resistant but not responsive AML cells. We found LSD1 to be associated with the IRS1 promoter only in AML cells resistant to LSD1i, suggesting that AML cells display different modes of regulation of this gene (negative regulation by LSD1 in resistant cells, other mechanisms in sensitive cells). Consistently, LSD1i led to remodeling of the IRS1 promoter in resistant AML cells, with a prominent accumulation of H3K4me3, H3K9Ac and H3K27Ac histone marks. Our findings are consistent with the previously reported studies which demonstrated that the transcriptional consequences of LSD1 inhibition are preceded by the preferential enrichment of H3K9Ac and H3K27Ac marks at LSD1-bound regulatory regions.14,46
Notably, a selective IRS1/2 inhibitor, NT157,31 reversed LSD1i-induced ERK1/2 and mTOR activation and thereby sensitized resistant AML cells to LSD1i. Our data are in accordance with Machado-Neto and colleagues who reported that silencing IRS1 inactivates ERK1/2 and mTOR signaling in K562 CML cells.30 In line with previous studies which reported that ATRA downregulates IRS1,32,33 we have further demonstrated that ATRA reduced H3K4me2 and H3K27Ac accumulation on IRS1 promoter. Indeed, ATRA dramatically abolished LSD1i-mediated IRS1 induction and rendered resistant AML vulnerable to LSD1i. Within this context, we speculate that LSD1i and ATRA cooperate by acting via distinct mechanisms. LSD1i unlocks ATRA-differentiation pathway4 whereas ATRA counteracts LSD1i-mediated upregulation of IRS1. These complementary activities might contribute to the synergistic antileukemic activity of their combination.4 Altogether, our findings imply that LSD1i-mediated modulation of IRS1 and ERK1/2 might contribute –at least partly–to mTOR regulation by LSD1 (Figure 7K).
In conclusion, our data underscore a pro-survival role of mTOR in mediating both intrinsic and acquired resistance of AML cells to LSD1i and provide an objective rationale for considering epigenetic (LSD1i)/metabolic (mTORi) combinatorial regimens for irresponsive AML patients.
Acknowledgments
We would like to thank Dr. Fabio Santoro, Dr. Roberto Ravasio and Prof. Pier Giuseppe Pelicci for their support and constructive discussions. We are also thankful to Dr. Federica Pisati who performed the histopathological and immunohistochemical analyses. AKA has been awarded a fellowship by AIRC (Italian Association for Cancer Research). Work in SM’s lab is supported by AIRC, CNR (Epigen Flahsgip Project), Regione Lombardia (Progetto Neon) and Horizon 2020 (TRAN-SCAN DRAMA) grants. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/105/8/2105
References
- 1.Niebel D, Kirfel J, Janzen V, Holler T, Majores M, Gutgemann I. Lysine-specific demethylase 1 (LSD1) in hematopoietic and lymphoid neoplasms. Blood. 2014;124(1): 151–152. [DOI] [PubMed] [Google Scholar]
- 2.Wada T, Koyama D, Kikuchi J, Honda H, Furukawa Y. Overexpression of the shortest isoform of histone demethylase LSD1 primes hematopoietic stem cells for malignant transformation. Blood. 2015;125(24): 3731–3747. [DOI] [PubMed] [Google Scholar]
- 3.Harris WJ, Huang X, Lynch JT, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21(4):473–487. [DOI] [PubMed] [Google Scholar]
- 4.Schenk T, Chen WC, Göllner S, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18(4):605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lv T, Yuan D, Miao X, et al. Overexpression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS One. 2012;7(4):e35065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jie D, Zhongmin Z, Guoqing L, et al. Positive expression of LSD1 and negative expression of E-cadherin correlate with metastasis and poor prognosis of colon cancer. Dig Dis Sci. 2013;58(6):1581–1589. [DOI] [PubMed] [Google Scholar]
- 7.Setzger E, Wissmann M, Yin N, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437(7057):436–439. [DOI] [PubMed] [Google Scholar]
- 8.Baek SH, Kim K Il. Regulation of HIF-1 alpha stability by lysine methylation. BMB Rep. 2016; 49(5):245–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng J, Xu G, Liu J, et al. Phosphorylation of LSD1 at Ser112 is crucial for its function in induction of EMT and metastasis in breast cancer. Breast Cancer Res Treat. 2016;159 (3):443–456. [DOI] [PubMed] [Google Scholar]
- 10.Hino S, Sakamoto A, Nagaoka K, et al. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat Commun. 2012;3:712–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakamoto A, Hino S, Nagaoka K, et al. Lysine demethylase LSD1 coordinates glycolytic and mitochondrial metabolism in hepatocellular carcinoma cells. Cancer Res. 2015;75(7):1445–1456. [DOI] [PubMed] [Google Scholar]
- 12.McGrath JP, Williamson KE, Balasubramanian S, et al. Pharmacological inhibition of the histone lysine demethylase KDM1A suppresses the growth of multiple acute myeloid leukemia subtypes. Cancer Res. 2016;76(7):1975–1988. [DOI] [PubMed] [Google Scholar]
- 13.Vianello P, Botrugno OA, Cappa A, et al. Discovery of a novel inhibitor of histone lysine-Specific demethylase 1A (KDM1A/LSD1) as orally active antitumor agent. J Med Chem. 2016;59(4):1501–1517. [DOI] [PubMed] [Google Scholar]
- 14.Maes T, Mascaro C, Tirapu I, et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell. 2018;33(3): 495–511. [DOI] [PubMed] [Google Scholar]
- 15.Abdel-Aziz AK, Minucci S. Comparing apples with oranges: Studying LSD1 inhibitors in cellular assays. Pharmacol Res. 2019;146:104345. [DOI] [PubMed] [Google Scholar]
- 16.Mohammad HP, Smitheman KN, Kamat CD, et al. A DNA Hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell. 2015; 28(1):57–69. [DOI] [PubMed] [Google Scholar]
- 17.Ishikawa Y, Gamo K, Yabuki M, et al. A novel LSD1 inhibitor T-3775440 disrupts GFI1B-containing complex leading to trans-differentiation and impaired growth of AML cells. Mol Cancer Ther. 2017;16(2):273–284. [DOI] [PubMed] [Google Scholar]
- 18.Hoshii T, Tadokoro Y, Naka K, et al. mTORC1 is essential for leukemia propagation but not stem cell self-renewal. J Clin Invest. 2012;122(6):2114–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martelli AM, Evangelisti C, Chiarini F, McCubrey JA. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010;1(2): 89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou Z, Chen J, Yang J, Bai X. Targeted inhibition of rictor/mTORC2 in cancer treatment: a new era after rapamycin. Curr Cancer Drug Targets. 2016;16(4):288–304. [DOI] [PubMed] [Google Scholar]
- 21.Gao Y, Gao J, Li M, et al. Rheb1 promotes tumor progression through mTORC1 in MLL-AF9-initiated murine acute myeloid leukemia. J Hematol Oncol. 2016;9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burchert A, Wang Y, Cai D, et al. Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia. 2005;19(10):1774–1782. [DOI] [PubMed] [Google Scholar]
- 23.Elgendy M, Abdel-Aziz AK, Renne SL, et al. Dual modulation of MCL-1 and mTOR determines the response to sunitinib. J Clin Invest. 2017;127(1):153–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malone CF, Emerson C, Ingraham R, et al. mTOR and HDAC inhibitors converge on the TXNIP/thioredoxin pathway to cause catastrophic oxidative stress and regression of RAS-driven tumors. Cancer Discov. 2017;7(12):1450–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang J, Ying H, Mao T, et al. Upregulation of CD11b and CD86 through LSD1 inhibition promotes myeloid differentiation and suppresses cell proliferation in human monocytic leukemia cells. Oncotarget. 2017;8(49): 85085–85101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chow S, Minden MD, Hedley DW. Constitutive phosphorylation of the S6 ribosomal protein via mTOR and ERK signaling in the peripheral blasts of acute leukemia patients. Exp Hematol. 2006;34(9):1183–1191. [DOI] [PubMed] [Google Scholar]
- 27.Binda C, Valente S, Romanenghi M, et al. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J Am Chem Soc. 2010;132(19): 6827–6833. [DOI] [PubMed] [Google Scholar]
- 28.Kovacs K, Decatur C, Toro M, et al. 2-Deoxy-glucose downregulates endothelial AKT and ERK via interference with N-linked glycosylation, induction of endoplasmic reticulum stress, and GSK3b sctivation. Mol Cancer Ther. 2016;15(2): 264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inoki K, Ouyang H, Zhu T, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006; 126(5):955–968. [DOI] [PubMed] [Google Scholar]
- 30.Machado-Neto JA, Favaro P, Lazarini M, Costa FF, Olalla Saad ST, Traina F. Knockdown of insulin receptor substrate 1 reduces proliferation and downregulates Akt/mTOR and MAPK pathways in K562 cells. Biochim Biophys Acta. 2011;1813 (8):1404–1411. [DOI] [PubMed] [Google Scholar]
- 31.Fenerich BA, Machado-Neto JA, Alves AP, et al. The pharmacological IGF1R-IRS1/2 Inhibitor NT157 presents multiple antineoplastic effects in myeloproliferative neoplasms. Blood. 2017;130(Suppl 1):1253. [Google Scholar]
- 32.Del Rincón SV, Guo Q, Morelli C, Shiu HY, Surmacz E, Miller WH. Retinoic acid mediates degradation of IRS-1 by the ubiquitin-proteasome pathway, via a PKC-dependant mechanism. Oncogene. 2004;23(57):9269–9279. [DOI] [PubMed] [Google Scholar]
- 33.Ravikumar S, Perez-Liz G, Del Vale L, Soprano DR, Soprano KJ. Insulin receptor substrate-1 is an important mediator of ovarian cancer cell growth suppression by all-trans retinoic acid. Cancer Res. 2007; 67(19):9266–9275. [DOI] [PubMed] [Google Scholar]
- 34.Goossens S, Peirs S, Loocke WV, et al. Oncogenic ZEB2 activation drives sensitivity towards KDM1A inhibition in T-cell acute lymphoblastic leukemia. Blood. 2017; 129(8):981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abdel-Aziz AK, Abdel-Naim AB, Shouman S, Minucci S, Elgendy M. From resistance to sensitivity: insights and implications of biphasic modulation of autophagy by sunitinib. Front Pharmacol. 2017;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peng X, Zhang D, Li Z, Fu M, Liu H. mTOR inhibition sensitizes human hepatocellular carcinoma cells to resminostat. Biochem Biophys Res Commun. 2016;477(4):556–562. [DOI] [PubMed] [Google Scholar]
- 37.Feng S, Jin Y, Cui MZJ. Lysine-specific demethylase 1 (LSD1) Inhibitor S2101 induces autophagy via the AKT/mTOR pathway in SKOV3 ovarian cancer cells. Med Sci Monit. 2016;22:4742–4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poulain L, Sujobert P, Zylbersztejn F, et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia. 2017;31(11):2326–2335. [DOI] [PubMed] [Google Scholar]
- 39.Duteil D, Metzger E, Willmann D, et al. LSD1 promotes oxidative metabolism of white adipose tissue. Nat Commun. 2014;5:4093:1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hino S, Kohrogi K, Nakao M. Histone demethylase LSD1 controls the phenotypic plasticity of cancer cells. Cancer Sci. 2016;107(9):1187–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park H, Garrido-Laguna I, Naing A, et al. Phase I dose-escalation study of the mTOR inhibitor sirolimus and the HDAC inhibitor vorinostat in patients with advanced malignancy. Oncotarget. 2016;7 (41):67521–67531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kerenyi MA, Shao Z, Hsu YJ, et al. Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. Elife. 2013;2:e00633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abdulla A, Zhang Y, Hsu FN, et al. Regulation of lipogenic gene expression by lysine-specific histone demethylase-1 (LSD1). J Biol Chem. 2014;289(43):29937–29947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee G, Zheng Y, Cho S, et al. Post-transcriptional regulation of de novo post-transcriptional regulation of de novo lipogenesis by mTORC1-S6K1-SRPK2 signaling. Cell. 2017;171(7):1545–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roux PP, Shahbazian D, Vu H, et al. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates Cap-dependent translation. J Biol Chem. 2007;282(19):14056–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maiques-Diaz A, Spencer GJ, Lynch JT, et al. Enhancer activation by pharmacologic displacement of LSD1 from GFI1 induces differentiation in acute myeloid leukemia. Cell Rep. 2018;22(13):3641–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]