

Abstract
Background
Previous studies have identified and validated a risk-associated Active transcriptome phenotype commonly expressed in the cancer-adjacent and histologically normal epithelium, stroma, and adipose containing peritumor microenvironment of clinically established invasive breast cancers, conferring a 2.5- to 3-fold later risk of dying from recurrent breast cancer. Expression of this Active transcriptome phenotype has not yet been evaluated in normal breast tissue samples unassociated with any benign or malignant lesions; however, it has been associated with increased peritumor adipocyte composition.
Methods
Detailed histologic and transcriptomic (RNAseq) analyses were performed on normal breast biopsy samples from 151 healthy, parous, non-obese (mean BMI = 29.60 ± 7.92) women, ages 27–66 who donated core breast biopsy samples to the Komen Tissue Bank, and whose average breast cancer risk estimate (Gail score) at the time of biopsy (1.27 ± 1.34) would not qualify them for endocrine prevention therapy.
Results
Full genome RNA sequencing (RNAseq) identified 52% (78/151) of these normal breast samples as expressing the Active breast phenotype. While Active signature genes were found to be most variably expressed in mammary adipocytes, donors with the Active phenotype had no difference in BMI but significantly higher Gail scores (1.46 vs. 1.18; p = 0.007). Active breast samples possessed 1.6-fold more (~ 80%) adipocyte nuclei, larger cross-sectional adipocyte areas (p < 0.01), and 0.5-fold fewer stromal and epithelial cell nuclei (p < 1e−6). Infrequent low-level expression of cancer gene hotspot mutations was detected but not enriched in the Active breast samples. Active samples were enriched in gene sets associated with adipogenesis and fat metabolism (FDR q ≤ 10%), higher signature scores for cAMP-dependent lipolysis known to drive breast cancer progression, white adipose tissue browning (Wilcoxon p < 0.01), and genes associated with adipocyte activation (leptin, adiponectin) and remodeling (CAV1, BNIP3), adipokine growth factors (IGF-1, FGF2), and pro-inflammatory fat signaling (IKBKG, CCL13).
Conclusions
The risk-associated Active transcriptome phenotype first identified in cancer-adjacent breast tissues also occurs commonly in healthy women without breast disease who do not qualify for breast cancer chemoprevention, and independently of breast expressed cancer-associated mutations. The risk-associated Active phenotype appears driven by a pro-tumorigenic adipocyte microenvironment that can predate breast cancer development.
Keywords: Risk-associated normal breast tissue, Active transcriptome, Activated adipocytes
Background
Previous studies of normal-appearing cancer-adjacent breast tissue have identified at least two different transcriptional phenotypes [1, 2], including an Active transcriptome phenotype associated with near 3-fold excess risk of future death from breast cancer [3, 4]. The histologically normal but molecularly altered nature of cancer-adjacent tissue has long been recognized and was originally referred to as “field cancerization.” More recently characterized by gene expression studies, this peritumor tissue microenvironment was thought to be unlike that found in normal healthy tissues and able to promote the growth and invasiveness of diverse tumor types due to its enrichment in wound healing, hypoxia, epithelial-mesenchymal transition, and pro-inflammatory gene signals [5]. While histologically similar to other peritumor microenvironments, cancer-adjacent normal breast samples were also thought to be transcriptionally distinct from reduction mammoplasty breast samples [1]. A microarray study of 79 non-malignant breast biopsies prompted by mammography, interrogating over 9000 variably expressed genes, identified two normal breast tissue subtypes including one that shared gene expression features with stromal, stem and mesenchymal cells [2]. Curiously, the biopsied females possessing these different transcriptome phenotypes did not significantly differ by age, BMI (body mass index), or mammographic density at the time of breast biopsy. Investigators therefore concluded that additional studies were needed to further characterize the cellular and molecular biology underlying the two different transcriptome phenotypes and to clarify their potential links to future breast cancer development [2].
Focusing on observed transcriptional differences among histologically normal cancer-adjacent breast tissues, Roman-Perez et al. first described a multi-gene (> 3700) signature capable of distinguishing normal breast samples expressing either an Active or Inactive transcriptome phenotype, the former characterized by increased expression of genes linked to cell motility, inflammation, fibrosis, and chemotaxis as well as decreased expression of cell adhesion, differentiation, and cell-cell contact genes [3]. While these two normal breast transcriptome phenotypes were not associated with any specific hormonal or intrinsic subtypes with respect to their adjacent breast tumors, patients bearing the Active peritumor microenvironment were 2.5-fold more likely to relapse and die of estrogen receptor (ER)-positive breast cancer over the next decade [3]. Using this same multi-gene classifying signature to interrogate cancer-adjacent normal breast samples acquired by The Cancer Genome Atlas (TCGA) program, Troester et al. identified 40% of samples with the Active transcriptome phenotype and showed in multivariate analysis that ER-positive patients with this peritumor microenvironment were 3-fold more likely to die of breast cancer within the next 10 years [4]. This last TCGA study made the surprising additional observation that histologically normal samples expressing the Active phenotype were composed of significantly more fat (mean 85% vs. 70%, p = 8.8e−05), fewer stromal (mean 8% vs. 19%, p = 0.00013), and epithelial (mean 7% vs. 9%, p = 0.027) cells, suggesting that the excess breast cancer mortality risk associated with the Active peritumor phenotype might be due to its adipocyte composition [4].
To explore the possibility that the breast cancer risk-associated Active transcriptome phenotype might actually be a pre-existent condition within an otherwise healthy-appearing woman’s breast tissue, and not simply induced by a nearby breast neoplasm, we studied the transcriptional phenotypes of normal breast biopsies donated by healthy parous women with no history of breast disease or specific mammographic abnormalities. Given the TCGA association of this Active phenotype with increased mammary fat content [4], we also examined the possibility that altered normal breast adipocyte populations contribute to this risk-associated Active transcriptome phenotype.
Methods
Donor population, breast biopsies, and histologic analyses
Paraffin-archived normal breast tissue samples were obtained from 151 parous, non-Hispanic white women (ages 27–66), without recent mammographic abnormality or history of breast cancer, who provided upper outer quadrant core biopsies for research purposes collected and archived by the Susan G. Komen Tissue Bank (KTB; Indiana University Simon Cancer Center, Indianapolis IN 46202). Donors supplied written informed consent and were recruited under a protocol approved by the Indiana University Institutional Review Board, and completed questionnaires with reproductive histories sufficient to calculate Gail 5-year risk scores (for all except 19 who were < age 35). Core biopsies were processed by standardized KTB operating protocols (KTB SOP) [6]; however, during our transcriptome analysis of these samples, we learned from KTB that the first 96 biopsy samples had been obtained and processed by 10% formalin fixation and paraffin embedding (FFPE), after which a change in the KTB SOP protocol occurred such that the next 55 sequentially obtained biopsies were processed by a formalin- and crosslinking-free PAXgene tissue preservation system (PreAnalytiX-Qiagen/BD, Switzerland) prior to paraffin embedding (PFPE). Given the two different tissue processing protocols, transcriptome analyses and donor characteristics were first performed and compared in a batch-specific manner (FFPE, F = 96 samples; PFPE, P = 55 samples); subsequently, an effort to batch correct and integrate all the gene expression data was also undertaken (see below). KTB provided digitized hematoxylin and eosin (H&E)-stained sections for every sample, from which cell composition was scored (% nuclei and area for fat, stromal and epithelial content, with additional comment about extent of leukocyte infiltration) by a dedicated breast pathologist blinded to all donor and batch details. Fat, stromal, and epithelial areas were quantitated using Aperio Image Scope software (version 12.3.2.8013, Leica Biosystems, Buffalo Grove, IL). This histologic analysis confirmed that no sample contained preneoplastic or neoplastic cells. Furthermore, terminal duct lobular unit (TDLU) counts were independently and blindly determined on each H&E slide as recently reported [7]. For adipocyte cross-sectional area assessment, four representative images from each H&E slide were analyzed at × 50 magnification (measuring ≥ 50 adipocytes per image) using Fiji imaging software with the open-source Adiposoft v1.13 plugin, as previously described [8].
RNA sequencing for gene expression and gene signature scoring
Contiguous thick paraffin sections (10 μm each) sufficient to extract ~ 100 ng of total breast RNA per sample were sent to NantOmics, LLC (Culver City, CA 90232) for full transcriptome ribo-deplete RNA sequencing (RNAseq), performed on the Illumina NovaSeq platform. Reads were aligned using Bowtie2 v2.2.6 and RSEM v1.2.25 to RefSeq build 73 on hg19, generating both untransformed and log2-scaled TPM values for all expressed genes, publically deposited at https://xenabrowser.net/datapages/?cohort=Normal%20Breast%20(Benz%202020). Normalized, median centered, log2-scaled TPM values from the 96 F and 55 P batch samples were independently interrogated by unsupervised hierarchical clustering using 1318 variably expressed genes (IQR > 0.8) mapped onto our dataset from the > 3500 previously validated Active/Inactive classifying genes (see Supplement Table 1) to assign samples within each batch as having either the Active or Inactive transcriptome phenotype. This Active vs. Inactive phenotype classification was associated with donor risk factors, breast tissue composition, expression of other candidate genes, and gene signatures (Supplement Table 4). Other gene expression signatures scored included cAMP lipolysis, adipocyte browning, SASP, AST, IGF1, IGF1R, IFN, TGFβ, and CSR activities; all signatures, their gene components, and methods of score calculation are provided in Supplement Table 1. In addition, a numeric Active/Inactive signature score was calculated for correlation with other batch-specific numeric sample characteristics as a sign-corrected average using the following mathematical formula where Ss = signature score for that sample, Tg = TPM of the gene, D = set of downregulated signature genes, and U = set of upregulated signature gene values as listed in Supplement Table 1:
Transcriptome expression of cancer gene hotspot mutations
Given the lack of available KTB germline sequence data (e.g., peripheral blood DNAseq), transcript-level BAM files were analyzed for potential somatic variants using a proprietary loci-based variant caller, Locus, against two curated lists of human cancer mutation hotspots: (i) the Memorial Sloan Kettering (MSK-IMPACT) list of actionable cancer targets [9] and (ii) the curated set of ~ 1760 cancer gene hotspot mutations recently found expressed in the bulk RNAseq data of ~ 6700 normal human tissue samples from ~ 500 different individuals donating to the Genotype-Tissue Expression (GTEx) project [10]. The lists of all cancer gene hotspot mutations/variants detected in the KTB RNAseq data present in both hotspot databases are provided in Supplement Table 2, and the heuristics used to graphically score these hotspot mutations in relation to other KTB sample characteristic included mutation likelihood score ≥ 5, variant allele frequency (AF) ≥ 0.02 and ≤ 0.40, variant allele read depth (AD) ≥ 2, and predicted non-silent amino acid (AA) change.
Batch-specific phenotype assignment and batch-corrected transcriptome values associated with mammary adipocytes and fat metabolism
Table 1 shows the breast biopsy donor characteristics by sample batch, indicating that F and P batches were well balanced for donor age at biopsy, age at menarche, age at first birth, parity, BMI, family history, and Gail 5 year risk scores. After batch-specific classification of each sample’s transcriptome as either Active or Inactive, F and P batches demonstrated partially overlapping gene sets differentially expressed between their Active and Inactive phenotypic subsets (FDR p < 0.05): 7679 genes differed between the F batch phenotypes (Wilcoxon p = 1e−14), and 6614 genes differed between the P batch phenotypes (Wilcoxon p = 6.5e−08). However, in order to perform maximally powered gene set enrichment analysis (GSEA) and TumorMap analyses on the combined (F + P = 151) collection of Active (F = 47/96, P = 31/55) and Inactive (F = 49/96, P = 24/55) sample transcriptomes, we mapped all P transcriptome data into the F transcriptome RNAseq space using a quantile normalization procedure to minimize batch-specific gene expression differences [11]. This batch-correction approach used the F sample’s expression quantiles as the target distribution and the P sample’s expression quantiles as the source distribution, and this quantile normalization was performed separately for each HUGO gene, excluding zero expression genes from both source and target datasets (attaching them after the mapping was complete) so that both source and target quantiles were computed using only expressed gene values. GSEA and TumorMap analyses were then performed on the combined batch-integrated gene expression dataset across 151 samples after each sample was phenotyped within its batch as either Active or Inactive.
Table 1.
Donor characteristics by breast sample batch
| Characteristic | F batch | P batch | p valuea |
|---|---|---|---|
| N = 96 | N = 55 | ||
| Age at biopsy (years) | .73 | ||
| Mean | 44.75 | 45.36 | |
| Age at menarche (years) | .73 | ||
| Mean | 12.57 | 12.49 | |
| Parity (live births) | .21 | ||
| Mean | 2.18 | 2.00 | |
| Age at first birth (years) | .04 | ||
| Mean | 27.56 | 25.82 | |
| BMI (kg/m2) | .31 | ||
| Mean | 30.32 | 28.87 | |
| Family historyb | .76 | ||
| Percent positive | 25.0% | 23.62% | |
| Gail 5 year risk scores | .51 | ||
| Mean | 1.34% | 1.17% |
ap value is the chi-squared p value for difference in means between samples
bAt least one first-degree relative with breast cancer
GSEA (https://www.gsea-msigdb.org/gsea/index.jsp; v2.2.4) was used to identify gene sets upregulated within the Active samples. A compendium of 18,408 gene sets was assessed for enrichment, including gene sets and pathways collected from a variety of sources (GO, HumanCyc, IOB, MSigdb, NCI, NetPath, Panther, Reactome, WikiPathways, and KEGG). The batch-corrected expression matrix from the KTB samples was used to compare the Active with the Inactive phenotypes, and 1000 phenotype permutations were used. Adipose-associated gene sets were identified by filtering gene set names with “fat,” “adip,” and “lip” and then removing names with terms such as “sulfate” and “sulfation,” resulting in 505 (2.7%) adipose-associated gene sets from the full set of 18,408. Gene sets enriched at FDR ≤ 10% were identified (Supplement Table 3), supporting further hypothesis-driven comparison of the Active transcriptome phenotype for mammary adipocyte characteristics and fat-specific metabolism pathways. Those expression signatures interrogated included cAMP-dependent adipocyte lipolysis [8, 12], white adipose tissue browning [13], senescence-associated secretory phenotype (SASP), and autophagy-to-senescence transition (AST) as found associated with aging mammary tissue [14], IGF1R and IGF1 ligand activation signatures [15, 16], and immune/inflammatory modules (IFN, TGFB, CSR) previously associated with malignant breast tissue [17, 18]. Single gene transcripts interrogated include those associated with varying adipocyte functions (FGF2, IGF1, CAP1, BNIP3, CAV1, LEP, LEPR, ADIPOQ, IKBKG, CCL13, SERPIN1) as well as general mesenchymal (CD38, CD68, IL6, SERPINE1, WIF1, IGF1R) and epithelial (KIT, MYB, TRPS1) cell states.
TumorMap visualization of Active and Inactive normal breast transcriptomes and attribute mapping
As recently described and frequently used to compare transcriptome similarities and differences between tumor types and/or their molecular subtypes, the TumorMap is an interactive website tool enabling visualization of multi-dimensional patient sample data in a two-dimensional layout [19]. Here we used TumorMap to compare both batch-specific and batch-integrated transcriptome layouts for our 151 KTB normal breast samples, related these to 1096 TCGA-determined breast cancer transcriptomes across their five different molecular subtypes [20], and overlayed the map with KTB normal breast attributes. A sample-by-sample similarity matrix was first computed from the sample-by-gene mRNA expression matrix. The TumorMap’s force-directed layout engine then used the similarity matrix to position all samples in a two-dimensional space; samples that are close to each other in the multi-dimensional RNAseq space are also close to each other in the two-dimensional TumorMap layout. Locality patterns in the distribution of various sample attributes are revealed by coloring the samples in the layout based on their attribute scores; this form of spatial correlation analysis helps find pattern associations between attributes (e.g., co-occurring or mutually exclusive pairs) not easily found by direct sample correlations.
Statistics
RNAseq transcriptome data, derived as described earlier, were analyzed using Bioconductor R (www.bioconductor.org) software programs, and normalized RSEM TPM values were used to compare batch-specific single gene expression levels and multi-gene signature scores, the latter calculated as described above. Histologic and gene expression co-variates subjected to statistical association for all batch (F, P) and Active/Inactive classified KTB samples are summarized in Supplement Table 4. Gail 5-year risk scores were calculated using the Breast Cancer Risk Assessment Tool (https://bcrisktool.cancer.gov). All tabulated data (Supplement Table 4) were compared for median and mean value differences between sample batches and according to their Active/Inactive classifications by chi-square and T test. All graphical plots show single, median or mean (± SD) values as described; grouped measures (e.g., box-whisker plots) were statistically compared by Wilcoxon rank-sum test and correlations evaluated by Pearson’s linear regression (Rp) and/or Spearman (Rs) analyses. Significant differences were determined as *p < 0.05 or **p < 0.01.
Results
Unsupervised clustering of the 96 F and 55 P batch healthy breast RNAseq transcriptomes using their 1318 most variably expressed genes from the previously validated Active/Inactive multi-gene classifying signature (Supplement Tables 1; 3, 4) yielded the two normal breast heat maps shown in Fig. 1a; these two heat maps identified 47/96 F batch samples and 31/55 P batch breast samples for a total of 52% of all KTB samples as having the Active transcriptome phenotype. Batch-specific and combined cohort (F + P) box-whisker plots show that the Active samples possess 1.6-fold more (~ 80%, Wilcoxon p = 3.9e−11) adipocyte nuclei and 0.5-fold fewer stromal (Wilcoxon p = 4.3e−07) and epithelial (Wilcoxon p = 1.2e−10) cell nuclei (Fig. 1b). KTB donors with the Active phenotype had no difference in BMI but significantly higher Gail scores (1.46 vs. 1.18; p = 0.007).
Fig. 1.

Classification of normal breast transcriptomes into Active and Inactive phenotypes and increased adipocyte association with Active samples. a Unsupervised clustering heat map (red = increased gene expression, blue = decreased gene expression) showing batch-specific assignment of Active (orange covariate bar) and Inactive (blue co-variate bar) phenotype samples. Horizontal dendrogram = samples, vertical dendrogram = 1318 classifying genes (gold bars = genes upregulated for Active, gray bars = genes upregulated for Inactive phenotype). b Box plots of normal breast cell composition (% adipocyte, stromal, epithelial nuclei) across all 151 samples relative to their transcriptome phenotype assignments (expression cluster Active or Inactive). c Left panel: two-dimensional TumorMap layouts [19] of all KTB normal breast full transcriptomes after batch-integration and projected relative to 1096 TCGA breast cancer full transcriptomes. Batch-integrated normal breast transcriptome samples colored according to batch (blue = F, yellow = P), and TCGA breast cancer transcriptomes colored according to their PAM50 subtype [20]. Right panel: normal breast full transcriptome TumorMaps presented in isolation before and after batch-correction, showing good sample set integration of both F and P batch samples and preserved spatial separation of Active and Inactive phenotypes, and overlay of the integrated set with color scores (red = high) of their individual cell compositions (% nuclei)
After using a quantile normalization batch-correction procedure to integrate the two sets of normal breast transcriptome values, we used a two-dimensional visualization tool, TumorMap [19], to compare both the batch-specific and batch-integrated normal breast transcriptional landscapes to that of 1096 TCGA breast cancer samples spatially separated into their five different intrinsic subtypes (Fig. 1c): LumA, LumB, Her2, Normal-like, and Basal [20]. By generating isolated TumorMaps of the KTB normal breast transcriptomes we could superimpose individual attributes onto each sample including batch identity (F, P), Active/Inactive phenotype assignment, percent cell type composition (adipocyte, stromal, epithelial nuclei), and various single or multi-gene expression scores. From these attribute maps we see that the integrated normal breast transcriptional landscape still preserves the spatial distinction between Active and Inactive phenotype samples; as well, they serve to visually illustrate enrichment of the Active samples with higher adipocyte content and Inactive samples with higher stromal and epithelial cell content (Fig. 1c). Likewise, microscopically determined TDLU scores or mRNA expression of epithelial-specific genes like KIT and TRPS1 are seen to map preferentially over those normal breast samples possessing the highest epithelial content (Supplement Figure 1). In contrast, while macrophage and CD68 gene module signatures indicate that these immune attributes associate best with adipocyte content, a CD8 T cell immune signature preferentially associates with higher epithelial content samples (Supplement Figure 2). The numeric correlation coefficients and p values supporting these attribute maps, as well as all other study covariates for each sample, are summarized in Supplement Table 4.
Transcript-level BAM files within each batch were also analyzed independently and in depth to identify the expression of > 5140 low-frequency gene loci variants across a curated (MSK-IMPACT) list of actionable cancer targets [9], recording the variant nucleotide mutation, a calculated likelihood score for the rarely expressed variant, total read depths at both the reference and variant allele sites, and the resulting variant allele frequency (Supplement Table 2). These variant calls were then filtered against an independent listing of ~ 1760 cancer gene hotspot mutations identified by RNAseq analysis as being expressed in ~ 6700 normal tissue samples donated to the GTEx project, including 180 different normal breast samples [10]. This filtered call list of 567 hotspot mutations, subjected to the heuristics described in the “Methods” section, thus represents a conservative identification of low-frequency cancer gene mutation events expressed in our combined cohort of KTB normal breast samples: 464 in F batch and 103 in P batch samples (Supplement Table 2). Despite the significantly different number of mutation calls in F and P batch samples, the cancer gene hotspot mutation burden (counts/sample) was not significantly different between Active and Inactive normal breast samples (Fig. 2a), but there appeared to be a weakly positive correlation between this mutation burden and sample adipocyte content (Spearman r = + 0.21, p = 0.01), and insignificant negative correlation between mutation burden and sample epithelial content (Spearman r = − 0.14, p = 0.11). As previously reported in the GTEx set of human breast tissue samples [10], TP53 hotspot mutations occurred most frequently; EGFR and PIK3CA mutations were also detected, and, of these two epithelium-enriched mutation events, only the infrequent PIK3CA hotspot mutations showed any statistical association with the Active phenotype (Fig. 2b, p = 0.03).
Fig. 2.

Normal breast sample scatterplots and TumorMaps of their transcriptome-expressed cancer gene hotspot mutations. As described in the “Methods” section (and detailed in Supplement Table 2), frequencies of the filtered list of detected 567 cancer gene hotspot mutations (variant counts/sample) are plotted: a according to sample batch (F, P) and prior transcriptome assignment as either Active or Inactive, and relative to each sample’s adipocyte or epithelial content (% nuclei) and b onto batch-integrated transcriptome TumorMaps with each sample overlaid by its relative hotspot variant frequency score (red = high) and mapped according to the cancer gene identity (total, TP53, EGFR, or PIK3CA variants)
GSEA performed on the batch-integrated (F + P) transcriptome values identified 186 gene sets upregulated in the Active set compared to the Inactive set, at FDR ≤ 10% (Supplemental Table 3). Of these, 21 (11.2%) were adipose-associated pathways, including “TRANSCRIPTIONAL REGULATION OF WHITE ADIPOCYTE DIFFERENTIATION” (from Reactome) and “HALLMARK_ADIPOGENESIS” (from MSigDB) which showed the second and third lowest FDR. Of the total 18,408 gene sets in the compendium, 505 are adipose-associated (2.7%), suggesting that adipose-associated gene sets are well represented among those gene sets significantly upregulated in the Active relative to Inactive samples.
To orthogonally investigate this GSEA observed association of Active samples with differentially expressed fat pathways, the top 200 upregulated genes used to classify the Active transcriptome phenotype were sent to Human Cell Atlas investigators to interrogate two reduction mammoplasty samples freshly disaggregated into discrete epithelial, fibroblast, adipocyte, endothelial, and immune/inflammatory cell fractions and subjected to 10× single-cell RNAseq (scRNAseq) analysis, as previously described [21]. These investigators reported that only the mammary adipocyte fractions from the reduction mammoplasty samples showed above baseline expression with varying degrees of overexpression of the 200 most upregulated Active signature genes, signifying that these Active signature genes primarily report on altered mammary adipocyte expression (personal communication, Kai Kessenbrock, Ph.D., kai.kessenbrock@uci.edu). Given both the GSEA and scRNAseq findings, as well as the significantly increased number of adipocytes enumerated in the Active phenotype samples, further microscopic and gene-specific analyses of the KTB samples were undertaken to compare potential differences in adipocyte morphology and fat metabolizing pathways between the Active and Inactive samples.
Mean adipocyte cross-sectional areas were determined by automated analysis of each sample’s H&E slide, as described in the “Methods” section and previously reported [8], with adipocyte area delineation for a representative KTB sample slide illustrated in Fig. 3a. The calculated mean adipocyte area (μm2) for each breast sample correlated with that donor’s BMI (r = 0.48, p < 0.0001), and normal breast samples classified as having the Active transcriptome phenotype showed significantly larger adipocytes (p < 0.01) than those with the Inactive phenotype (Fig. 3a). Multi-gene signatures corresponding to previously reported breast cancer-associated adipocyte changes, including cAMP-dependent lipolysis [8, 12] and white adipose tissue browning [13], were also significantly elevated (Wilcoxon p ≤ 0.002) in the Active samples relative to the Inactive samples within both F and P batches (Fig. 3b, c). Likewise, expression of single genes associated with adipocyte activation (leptin, leptin receptor, adiponectin), pro-inflammatory fat signaling (IKBKG, CCL13), fat remodeling (CAV1, BNIP3), and adipokine growth factors (IGF-1, FGF2) were all significantly elevated in the Active samples relative to the Inactive samples within both batches (Fig. 4 for F batch results, Supplement Figure 3 for P batch results). Summary representations of all microscopic features and gene expression continuous values, and their correlations with one another across all samples within each batch, are shown in Fig. 5. All numeric correlation coefficients and p values supporting these (Fig. 5) co-variate associations are provided in Supplement Table 4.
Fig. 3.

Normal breast adipocyte size and gene expression signatures in relation to Active and Inactive transcriptome phenotypes. a Representative H&E image (sample K102670) showing automated adipocyte delineation for cross-sectional area determination on each sample, and distribution plots of mean adipocyte areas for all 151 samples according to their donor’s BMI (r = 0.48, p < 0.0001) and the sample’s transcriptome phenotype as either Active or Inactive (**p < 0.01). b Batch-specific (F, P) box plot distributions of a previously reported breast cancer-associated cAMP-dependent lipolysis signature [8, 12] according to sample transcriptome phenotype assignment (expression cluster) as either Active or Inactive. c Batch-specific (F, P) box plot distributions of a previously reported breast cancer-associated white adipocyte browning signature [13] shown according to sample transcriptome phenotype assignment (expression cluster) as either Active or Inactive
Fig. 4.

Normal breast expression of genes representing fat activation, remodeling, pro-inflammatory signaling, and growth factor expression. F batch-specific box plot distributions of single genes reflecting adipocyte activation (leptin, leptin receptor, adiponectin), adipocyte pro-inflammatory signaling (IKBKG, CCL13), remodeling (CAV1, BNIP3), and adipokine growth factor expression (IGF1 score, FGF2) shown according to sample transcriptome phenotype assignment, Active or Inactive. Comparable P batch-specific box plot distributions are shown in Supplement Figure 3
Fig. 5.
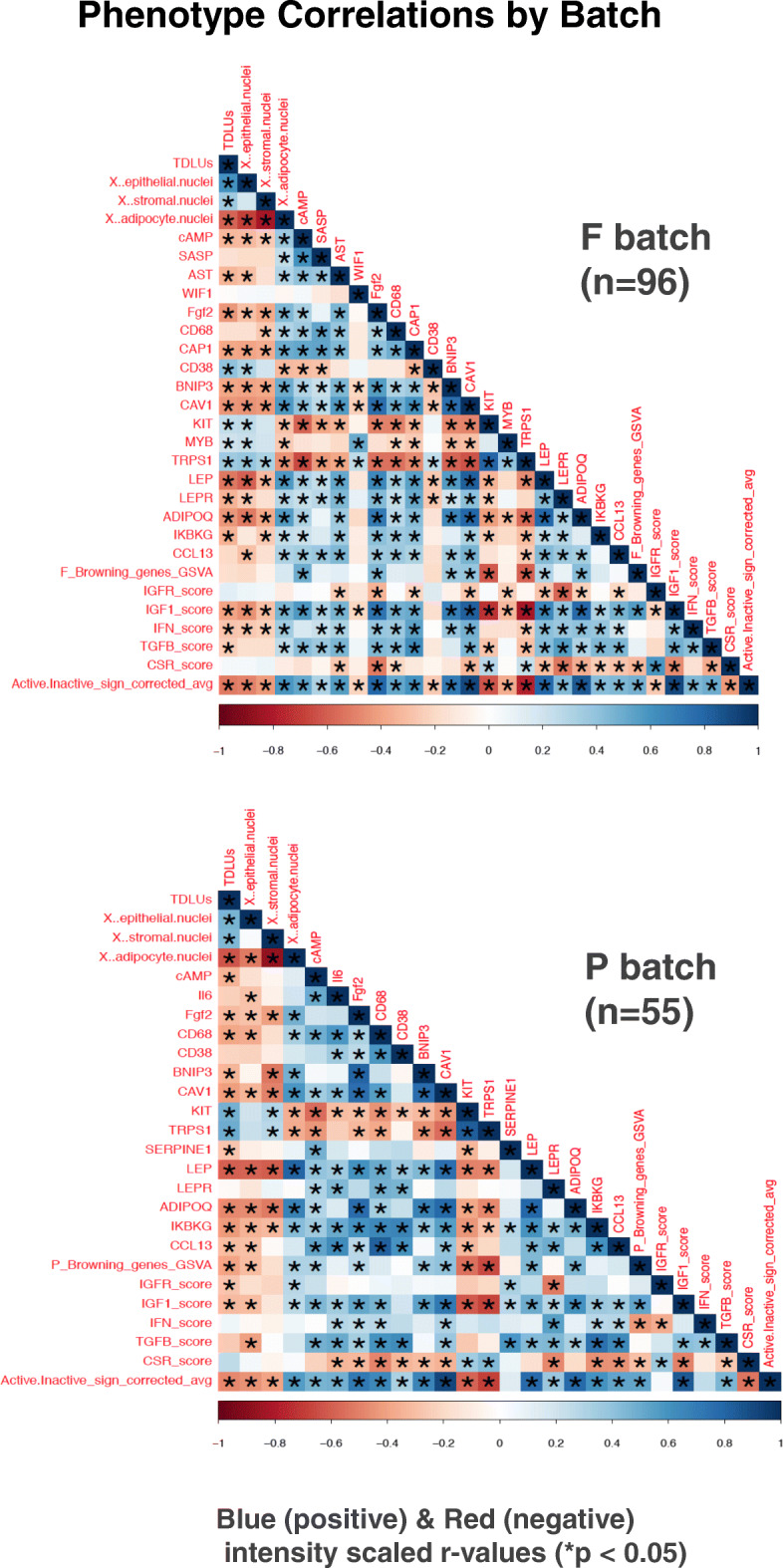
Batch-specific phenotype correlations summarizing all normal breast sample microscopic features and their measured gene expression relationships. Matrices show Pearson correlation heatmap relationships between all histologic, single gene and gene signature expression values, determined separately on the F (n = 96) and P (n = 55) batch normal breast samples, with correlations shown as blue (positive) or red (negative) intensity scaled r values (− 1 to + 1) and asterisks signifying p < 0.05 significance
Discussion
In this KTB cohort of predominantly non-obese Caucasian women without any current or past history of breast disease (characterized in Tables 1 and 2), more than half were found to possess the same Active mammary transcriptome phenotype previously shown to characterize the peritumor microenvironment of ~ 40% of newly diagnosed breast cancers associated with a near 3-fold excess risk of later-life breast cancer mortality [3, 4]. While > 80% of our donor cohort possessed Gail scores less than 1.65 and thus not recommended for breast cancer chemoprevention, those with Active transcriptomes possessed significantly higher Gail scores relative to those without, supporting their greater future risk for developing breast cancer (Table 2; Supplement Table 4). For this KTB cohort, however, at least another decade of follow-up will be necessary to prospectively test this predicted outcome. In the interim, the finding of a risk-associated normal breast Active transcriptome phenotype evident before any histologic evidence of breast neoplasia offers a unique opportunity to discern the composition and molecular nature of a potentially pro-tumorigenic breast microenvironment.
Table 2.
Donor characteristics by breast sample phenotype
| Characteristic | Active | Inactive | p valuea |
|---|---|---|---|
| N = 78 | N = 73 | ||
| Age at biopsy (years) | .01 | ||
| Mean | 47.13 | 42.25 | |
| Age at menarche (years) | .06 | ||
| Mean | 12.78 | 12.32 | |
| Parity (live births) | .13 | ||
| Mean | 2.01 | 2.22 | |
| Age at first birth (years) | .95 | ||
| Mean | 26.89 | 26.92 | |
| BMI (kg/m2) | .75 | ||
| Mean | 29.80 | 29.41 | |
| Family historyb | .54 | ||
| Percent positive | 25.6% | 23.3% | |
| Gail 5 year risk scores | .01 | ||
| Mean | 1.46% | 1.18% |
ap value is the chi-squared p value for difference in means between samples
bAt least one first-degree relative with breast cancer
The recent TCGA study confirming a 3-fold excess 10-year survival risk associated with the cancer-adjacent Active phenotype revealed that this multi-gene expression signature is closely mirrored by a microRNA (miR) signature composed of > 300 different miRs, including some associated with breast cancer and several (e.g., miR-200 members) known to directly regulate epithelial-to-mesenchymal transition (EMT), angiogenesis, and metastasis [4]. As well, that TCGA study demonstrated that both Active miR and mRNA classifying signatures correlated strongly with increased adipocyte content [4], in keeping with the current study’s microscopic and transcriptomic GSEA findings as well as the independent scRNAseq findings by Human Cell Atlas investigators that our Active gene classifying signature reports selectively on the variable expression of adipocytes as isolated from reduction mammoplasty samples (personal communication, K. Kessenbrock).
While the transcriptomes of normal breast samples from cancer-adjacent and reduction mammoplasty samples have previously been shown to be distinguishable [1], this study of KTB donated biopsy samples from women without any breast disease illustrates the potential introduction of technical batch artifacts when samples are differentially processed and fixed prior to RNA extraction and RNAseq assessment, although such artifacts may be largely corrected using advanced batch-integration algorithms as employed here. Fortunately, we observed excellent F-to-P batch concordance between patient and sample characteristics (Tables 1 and 2), and for almost all gene-gene and gene-attribute correlations (Figs. 3, 4, and 5). Using the batch-integrated transcriptome values and a new two-dimensional visualization tool, TumorMap [19], we could spatially integrate and map all the samples into a common transcriptional landscape while still preserving their spatial differentiation between Active and Inactive phenotypes (Fig. 1c). This visualization tool also illustrated the marked distinction between expression profiles of fat and stromal cell predominant normal breast samples relative to the transcriptional landscape of > 1000 different epithelium-enriched (> 60%) TCGA breast cancers representing all five intrinsic/PAM50 subtypes (Fig. 1c). Overlying various KTB sample attributes onto the integrated normal breast transcriptome landscape enabled sample-by-sample visual maps of how the Active normal breast samples are more enriched with adipocytes while the Inactive samples are more enriched in stromal and epithelial cells (Fig. 1c). Confirmed by histological scoring (Fig. 1b), the Active KTB normal samples possessed 1.6-fold more (80% vs. 50% mean nuclei, p = 3.9e−11) adipocytes and 0.5-fold fewer stromal and epithelial cells (p < 1e−6).
Prior studies have shown that normal female mammary composition can vary from < 10 to > 70% adipocytes, and, despite the acknowledged absolute dependence of normal mammary gland development on a mature mammary fat pad, there remains poor understanding behind the marked variations observed in mammary gland white adipose tissue (WAT) content across premenopausal females as well as its weak and inconsistent positive association with female age and obesity [22–24]. Notable here, neither age nor BMI scores were significantly different between KTB donors with Active vs. Inactive normal breast phenotypes (Table 2, Supplement Table 4). Unexpectedly, our 151 samples from parous and predominantly peri- or post-menopausal women showed no consistent evidence for an aging effect on their composite breast tissue as assessed by gene signatures specific for DNA damage response, cellular senescence, senescence-associated secretory profile, or autophagy-to-senescence transition, although our KTB samples possessed a minority (< 50%) of stromal cells whereas earlier studies describing these aging signatures specifically interrogated breast stroma excluding adipocytes [14].
Microscopic evaluation of our KTB tissue sections detected only rare leukocyte infiltrates and no crown-like structures within these adipocyte-rich normal breast samples (Supplement Table 4), yet the RNA from multiple adjacent thick sections revealed immune/inflammatory cell signatures [17, 18] suggesting (i) more CD68+ macrophages mapping onto the TGFβ+, adipocyte-rich Active samples and (ii) more CD8+ T cells mapping onto CSR+, KIT+, and TRPS1+ epithelial- and stromal-rich Inactive samples (Fig. 5c, Supplement Figures 1 and 2, Supplement Table 4). In addition to their quantitative excess of adipocytes, Active normal breast sample signatures showed greater TGFβ expression and more M2 tumor-promoting (CD68+) macrophages associating with their hypertrophied mature adipocytes (Fig. 3a), a previously described feature of post-weaning early-stage mammary involution [24]. The observed upregulation of NOD signaling intermediates, IKBKG and CCL13 (Fig. 4), is also consistent with a pro-inflammatory adipocyte population within these Active breast samples [25]. WAT hypertrophy in association with pro-inflammatory adipocyte signaling characterizes the obesity-driven microenvironment thought to commonly promote breast, endometrial, prostate, and gastrointestinal cancers [26–29]. While adipose tissue-induced inflammation in the absence of cancer is most typically a feature of visceral WAT, mammary gland WAT inflammation has been proposed as a breast cancer risk factor [30]. Although others believe that the adipokines hormonally secreted by hypertrophic visceral WAT are the transforming mediators between truncal obesity and epithelial malignancies like breast cancer [31, 32], it is relevant to note that imaging studies now indicate that > 90% of all breast cancers, unlike benign breast lesions, arise at the fat-gland interface [33].
By their hypertrophic feature alone, Active normal breast adipocytes appear akin to visceral WAT but distinct from what have been called cancer-associated adipocytes, which are typically smaller, more spindly, and show gap junction connections to adjacent malignant breast epithelium—the latter enabling lipolytic depletion of triglyceride stores to fuel tumor mitochondria by supplying free fatty acids and metabolites for malignant cell growth and progression [8, 27]. On the other hand, the hypertrophied adipocytes spanning the small islands of normal breast epithelium in our KTB Active samples exhibit a dysregulated transcriptional pattern quite typical of cancer-associated adipocytes [8, 13, 27–29], including increased expression of leptin (and its receptor) and adiponectin (Fig. 4), lipoprotein digesting and adipocyte remodeling genes like CAV1 and BNIP3 (Fig. 4), expression signatures defining increased cAMP-dependent lipolysis (Fig. 3b) and adipocyte “browning” (Fig. 3c), and multi-fold overexpression of the potent breast tumor-promoting adipokine growth factors, IGF-1 and FGF2 (Fig. 4). Leptin, produced mainly by adipocytes, has been proposed as a mediator between obesity, inflammation, and breast cancer development [34]. IGF-1, produced by adipocytes and essential for normal mammary gland growth and development, has also been implicated as a mechanistic link between obesity and breast cancer development [35–37]. In addition to the chemo-attractant and macrophage-recruiting properties of other adipokines, adiponectin has been shown to cross-talk with the IGF-1 axis and thereby potentiate its growth-promoting effect on breast cancer cells [38]. Perhaps the most potent of adipokines capable of initiating and promoting malignant transformation of breast epithelium is FGF2, essential for normal mammary gland development and stem cell function [39] yet a proven driver of various cancers and a target for new anti-cancer therapeutics [40], experimentally capable of inducing both skin and mammary epithelial cancers [31, 32].
The above-described properties of pro-tumorigenic adipokine growth factors may either mutationally initiate breast tumorigenesis or simply promote the growth of an already initiated but microscopically occult population of pre-neoplastic breast epithelial cells, much like the well-described properties of cancer-associated fibroblasts [41]. A recent deep RNAseq study of normal GTEx organs and tissues has revealed that, despite their healthy histologic appearance, a significant proportion of the 180 different normal breast samples in that collection actually expressed from 1 to 30 cancer-driving gene mutations per sample, the sample’s overall mutational burden dependent in part on the donor’s age but with many of the expressed mutations occurring within proven cancer gene hotspots [10]. Our KTB normal breast RNAseq study supports this surprising albeit infrequent finding of expressed cancer gene hotspot mutations (Fig. 2, Supplement Table 2), and while we did not observe any difference in overall mutation burden or frequency between the Active and Inactive normal breast samples, further studies are needed to confirm our preliminary observations that normal breast mutation burden may correlate with adipocyte content and that Active samples may indeed possess more PIK3CA hotspot mutations.
Conclusions
In summary, our findings of a risk-associated Active transcriptome linked to an activated adipocyte population, along with infrequent but detectable cancer gene hotspot mutations, all expressed in histologically normal-appearing adult breast tissue, suggest that a pro-tumorigenic adipocyte microenvironment can not only pre-exist the development of breast neoplasia but also provide fertile soil for a newly seeded, mutated and replication-competent mammary epithelial cell, whose growth is further promoted toward the development of a clinical breast cancer. Moving forward, breakthrough advances in personalized breast cancer risk assessment and prevention strategies would seem to depend on being able to screen mature women for both their normal breast tissue mutational burden as well as the presence of a pro-tumorigenic microenvironment that includes activated mammary adipocytes.
Supplementary information
Additional file 1: Supplement Figure 1. Isolated TumorMap of the batch-integrated normal breast transcriptomes overlain with color-scaled intensity scores (red = high) for their various epithelial attributes including % epithelial nuclei, TDLU scores, KIT and TRPS1 gene expression levels. All numeric sample scores can be found in Supplement Table 4. Supplement Figure 2. Isolated TumorMap of the batch-integrated normal breast transcriptomes overlain with color-scaled intensity scores (red = high) for their various tissue compositions (% stromal, adipocyte, epithelial nuclei) and transcriptome gene expression modules representing specific immune cell signatures (macrophage, CD68, CD8 Tcell). Modules are defined in Supplement Table 1 and their numeric signature scores are listed in Supplement Table 4. Supplement Figure 3. Normal breast expression of genes representing adipocyte activation, remodeling, and pro-inflammatory signaling for all P batch samples according to their sample transcriptome phenotype assignment as either Active or Inactive (as described in Fig. 4 legend).
Additional file 2: Supplement Table 1. Lists of genes included within each gene expression signature analyzed to determine Active/Inactive, cAMP lipolysis, adipocyte browning, SASP, AST, IGF1, IGF1R, IFN, TGFβ, and CSR activity scores. Also indicated is their reference sources and method of score calculation.
Additional file 3: Supplement Table 2. RNAseq cancer gene hotspot mutations detected in 151 KTB samples. Sheet 1 identifies the > 5100 unthresholded cancer mutation hotspot calls from the KTB RNAseq analysis occurring in the combined set of MSK-IMPACT curated cancer hotspot clinical targets [10] and the experimentally determined RNAseq identified set of expressed cancer gene hotspot mutations within > 6700 normal GTEx human tissue samples as recently described [11]. Sheet 2 (“normal_breast_rna.getz_list.092”) lists only those > 1760 cancer gene hotspot mutations from sheet 1 meeting the threshold mutation likelihood score > 5, while sheets 3 and 4 list the thresholded hotspot mutations according to F and P sample batches with each sample phenotyped as either Active or Inactive. Sheet 5 is a sample key mapping all KTB identification numbers, barcodes, and UUID numbers pertaining to the RNAseq results and cancer gene hotspot mutation calls. Of note, Fig. 2 panels (“Breast sample scatterplots and TumorMaps of RNA expressed cancer hotspot mutations”) include plots derived from the curated and thresholded hotspot mutations as listed in sheet 2.
Additional file 4: Supplement Table 3. Rank ordered GSEA (www.gsea-msigdb.org/gsea) analysis showing 186 gene sets upregulated (from total set of 18,408 gene sets) in the batch-integrated Active vs. Inactive transcriptome samples, at FDR ≤ 10%. Also shown are their individual nominal p-values, FDR q-values, their gene set size, and the 21 (11.2%) that are specifically involved in adipose-associated pathways.
Additional file 5: Supplement Table 4. Master spreadsheet listing showing sample and donor covariates for each of the 151 KTB barcodes including batch assignment (F or P), Active/Inactive phenotype assignment and score, donor features (including age, BMI, 5 year Gail risk scores), percentages of adipocyte/stromal/epithelial cell nuclei, TDLU counts, mean adipocyte areas, immune modules, and all gene signatures and single genes values used to calculate the Pearson correlations shown in Fig. 5.
Acknowledgements
We appreciate the contributions of cytotechnologist, Renata Cora (Mt. Kisco, NY), who performed all TDLU counts, as well as Kai Kessenbrock (UC Irvine) and his Human Cell Atlas colleagues who evaluated our Active gene signature in their single nuclei RNAseq analysis of reduction mammoplasty samples. Since samples from the Susan G. Komen Tissue Bank at the IU Simon Cancer Center were used in this study, we thank Indiana University contributors who collected the samples and data used in this study as well as all donors and their families whose help and participation made this work possible. We thank Zero Breast Cancer (San Rafael, CA) for their administrative assistance, and we also thank Jingchun Zhu and colleagues at the Genomics Institute, UC Santa Cruz, for uploading and hosting all the annotated transcriptomic data supporting this study on their public, interactive and web-based functional genomics platform, UCSC Xena (https://xena.ucsc.edu).
Abbreviations
- AA
Amino acid
- AD
Allele read depth
- AF
Allele frequency
- AST
Autophagy-to-senescence transition
- BMI
Body mass index
- ER
Estrogen receptor
- FDR
False discovery rate
- F batch
FFPE batch
- FFPE
Formalin fixed, paraffin embedded
- FGF2
Fibroblast growth factor-2
- GSEA
Gene set enrichment analysis
- GTEx
Genotype-Tissue Expression
- H&E
Hematoxylin and eosin
- IGF-1
Insulin-like growth factor-1
- KTB
Komen Tissue Bank
- mIR
MicroRNA
- P batch
PFPE batch
- PFPE
PAXgene fixed, paraffin embedded
- RNAseq
Full transcriptome RNA sequencing
- SASP
Senescence-associated secretory phenotype
- TCGA
The Cancer Genome Atlas
- TDLU
Terminal duct lobular unit
- TPM
Transcripts per million
- WAT
White adipose tissue
Authors’ contributions
CCB, CY, and MP conceived of and designed the study. JEH, MP, SCB, GK, and RC acquired data. TK, CY, CKW, JZS, YN, CV, SCB, JS, and CCB analyzed/interpreted data. CCB drafted the manuscript. All authors revised the manuscript and read and approved the final manuscript.
Funding
NIH/NCI U24-CA210990 (CCB, CY, CW, JS); Elizabeth MA Stevens memorial funding (CCB); NIH/NCI F99CA212488 (RC); and NIH/NIA training grant AG052374 (TK).
Availability of data and materials
The primary transcriptome (RNAseq) data obtained on all tissue samples have been deposited on the public UCSC Xena functional genomics platform: https://xenabrowser.net/datapages/?cohort=Normal%20Breast%20(Benz%202020).
All other primary data analyzed and presented in this study are located in the Supplementary tables attached to this manuscript.
Ethics approval and consent to participate
Coded tissue samples, histology sections, and reproductive histories were provided for this study by the Susan G. Komen Tissue Bank (KTB; Indiana University Simon Cancer Center, Indianapolis IN 46202) who, under a research protocol approved by the Indiana University Institutional Review Board, recruited and enrolled all donors under informed and written consent, collected, processed, and archived all the coded and de-identified samples including questionnaire data used for sample annotation.
Consent for publication
Not applicable.
Competing interests
SCB, CV, JZS, and YN are/were employees of ImmunityBio (Santa Cruz). SCB, CV, and JZS also have ownership interests (including stocks and patents) in NantCell, NantOmics, and NantHealth. All other co-authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13058-020-01322-6.
References
- 1.Troester MA, Lee MH, Carter M, Fan C, Cowan DV, Roman-Perez E, Pirone JR, Perou CM, Jerry DJ, Schneider SS. Activation of host wound responses in breast cancer microenvironmnet. Clin Cancer Res. 2009;15:7020–7028. doi: 10.1158/1078-0432.CCR-09-1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haakensen VD, Lingjaerde OC, Luders T, Riis M, Prat A, Troester MA, Holmen MM, Frantzen JO, Romundstad L, Navjord D, Bukholm IK, Johannesen TB, Perou CM, Ursin G, Kristensen VN, Borresen-Dale A-L, Helland A. Gene expression profiles of breast biopsies from healthy women identify a group with claudin-low features. BMC Med Genomics. 2011;4:77. doi: 10.1186/1755-8794-4-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roman-Perez E, Casbas-Hernandez P, Pirone JR, Rein J, Carey LA, Lubet RA, Mani SA, Amos KD, Troester MA. Gene expression in extratumoral microenvironment predicts clinical outcome in breast cancer patients. Breast Cancer Res. 2012;14:R51. doi: 10.1186/bcr3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Troester MA, Hoadley KA, D’Arcy M, Cherniack AD, Stewart C, Koboldt DC, Robertson AG, Mahurkar S, Shen H, Wilkerson MD, Sandhu R, Johnson NB, Allison KH, Beck AH, Yau C, Bowen J, Sheth M, Hwang ES, Perou CM, Laird PW, Ding L, Benz CC. DNA defects, epigenetics, and gene expression in cancer-adjacent breast: a study from The Cancer Genome Atlas. npj-Breast Cancer. 2016;2:16007. doi: 10.1038/npjbcancer.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aran D, Camarda R, Odegaard J, Paik H, Oskotsky B, Krings G, Goga A, Sirota M, Butte AJ. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat Commun. 2017;8:1077. doi: 10.1038/s41467-017-01027-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The Komen Tissue Bank at the IU Simon Cancer Center. “Standard Operating Procedures” . https://komentissuebank.iu.edu/researchers/sop/ (accessed 25 Jan 2019).
- 7.Powell MJ, Dufault SM, Henry JE, Allison AC, Cora R, Benz CC. Pregnancy hypertension and a commonly inherited IGF1R variant (rs2016347) reduce breast cancer risk by enhancing mammary gland involution. J Oncol. 2019;2019:6018432. doi: 10.1155/2019/6018432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Camarda R, Williams J, Malkov S, Zimmerman LJ, Manning S, Aran D, Beardsley A, Van de Mark D, Chen Y, Berdan CA, Louie SM, Mahieu C, Winkler J, Willey E, Gagnon JD, Shinoda K, Ansel KM, Werb Z, Nomura DK, Kajimura S, Butte AJ, Sanders ME, Liebler DC, Rugo H, Krings G, Shepherd JA, Goga A. Tumor cell-adipocyte gap junctions activate lipolysis and are essential for breast tumorigenesis. bioRxiv. 2018; 10.1101/277939.
- 9.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, Brannon AR, O’Reilly C, Sadowska J, Casanova J, Yannes A, Hechtman JF, Yao J, Song W, Ross DS, Oultache A, Dogan S, Borsu L, Hameed M, Nafa K, Arcila ME, Ladanyi M, Berger MF. Memorial Sloan Kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT) J Mol Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yizhak K, Aguet F, Kim J, Hess JM, Kubler K, Grimsby J, Frazer R, Zhang H, Haradhvala NJ, Rosebrock D, Livitz D, Li X, Arich-Landkof E, Shoresh N, Stewart C, Segre AV, Branton PA, Polak P, Ardlie KG, Getz G. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. 2019;364:eaaw0726. doi: 10.1126/science.aaw0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shinoda K, Luijten IHN, Hasegawa Y, Hong H, Sonne SB, Kim M, Xue R, Chondronikola M, Cypess AM, Tseng Y-H, Nedergaard J, Sidossis LS, Kajimura S. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nat Med. 2015;21:389–394. doi: 10.1038/nm.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang F, Gao S, Chen F, Fu Z, Yin H, Lu X, Yu J, Lu C. Mammary fat of breast cancer: gene expression profiling and functional characterization. PLoS One. 2014;9:e109742. doi: 10.1371/journal.pone.0109742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brouwers B, Fumagailli D, Brohee S, Hatse S, Govaere O, Floris G, van den Eynde K, Bareche Y, Schoffski P, Smeets A, Neven P, Lambrechts D, Sotirious C, Wildiers H. The footprint of the ageing stroma in older patients with breast cancer. Breast Cancer Res. 2017;19:78. doi: 10.1186/s13058-017-0871-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Creighton CJ, Casa A, Lazard Z, Huang S, Tsimelzon A, Hilsenbeck SG, Osborne CK, Lee AV. Insulin-like growth factor-1 activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol. 2008;26:4078–4085. doi: 10.1200/JCO.2007.13.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mu L, Tuck D, Katsaros D, Lu L, Schulz V, Perincheri S, Menato G, Scarampi L, Harris L, Yu H. Favorable outcome associated with an IGF-1 ligand signature in breast cancer. Breast Cancer Res Treat. 2012;133:321–331. doi: 10.1007/s10549-012-1952-5. [DOI] [PubMed] [Google Scholar]
- 17.Amara D, Wolf DM, van’t Veer L, Esserman L, Campbell M, Yau C. Co-expression modules identified from published immune signatures reveal five distinct immune subtypes in breast cancer. Breast Cancer Res Treat. 2017;161:41–50. doi: 10.1007/s10549-016-4041-3. [DOI] [PubMed] [Google Scholar]
- 18.Danaher P, Warren S, Dennis L, D’Amico L, White A, Disis ML, Geller MA, Odunsi K, Beechem J, Fling SP. Gene expression markers of tumor infiltrating leukocytes. J Immunother Cancer. 2017;5:18. doi: 10.1186/s40425-017-0215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newton Y, Novak AM, Swatloski T, McColl DC, Chopra S, Graim K, Weinstein AS, Baertsch R, Salama SR, Ellrott K, Chopra M, Goldstein TC, Haussler D, Morozova O, Stuart JM. TumorMap: exploring the molecular similarities of cancer samples in an interactive portal. Cancer Res. 2017;77:e111–e114. doi: 10.1158/0008-5472.CAN-17-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, Taylor AM, Cherniak AD, Thorsson V, Akbani R, Bowlby R, Wong CK, Wiznerowicz M, Sanchez-Vega F, Robertson AG, Schneider BG, Lawrence MS, Noushmehr H, Malta TM, Network CGAR, Stuart JM, Benz CC, Laird PW. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173:291–304. doi: 10.1016/j.cell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyen QH, Pervolarakis N, Blake K, Ma D, Davis RT, James N, Phung AT, Willey E, Kumar R, Jabart E, Driver I, Rock J, Goga A, Khan SA, Lawson DA, Werb Z, Kessenbrock K. Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Nat Commun. 2018;9:2028. doi: 10.1038/s41467-018-04334-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hovey RC, Aimo L. Diverse and active roles for adipocytes during mammary gland growth and function. J Mammary Gland Biol Neoplasia. 2010;15:279–290. doi: 10.1007/s10911-010-9187-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun X, Sandhu R, Figueroa JD, Gierach GL, Sherman ME, Troester MA. Benign breast tissue composition in breast cancer patients: associations with risk factors, clinical variables, and gene expression. Cancer Epidemiol Biomark Prev. 2014;23:2810–2818. doi: 10.1158/1055-9965.EPI-14-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zwick RK, Rudolph MC, Shook BA, Holtrup B, Roth E, Lei V, Van Keymeulen A, Seewaldt V, Kwei S, Wysolmerski J, Rodeheffer MS, Horsley V. Adipocyte hypertrophy and lipid dynamics underlie mammary gland remodeling after lactation. Nat Commun. 2018;9:3592. doi: 10.1038/s41467-018-05911-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin Z, Deng T, Peterson LE, Yu R, Lin J, Hamilton DJ, Reardon PR, Sherman V, Winnier GE, Zhan M, Lyon CJ, Wong STC, Hsueh WA. Transciptome analysis of human adipocytes implicates the NOD-like receptor pathway in obesity-induced adipose inflammation. Mol Cell Endocrinol. 2014;394:80–87. doi: 10.1016/j.mce.2014.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Himbert C, Delphan M, Scherer D, Bowers LW, Hursting S, Ulrich CM. Signals from the adipose microenvironment and the obesity-cancer link- a systematic review. Cancer Prev Res. 2017;10:494–506. doi: 10.1158/1940-6207.CAPR-16-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoy AJ, Balaban S, Saunders DN. Adipocyte-tumor cell metabolic crosstalk in breast cancer. Trends Mol Med. 2017;23:381–392. doi: 10.1016/j.molmed.2017.02.009. [DOI] [PubMed] [Google Scholar]
- 28.Dirat B, Bochet L, Dabek DD, Dauvillier S, Majed B, Wang YY, Meulle A, Salles B, Gonidec SL, Garrido I, Escourrou G, Valet P, Muller C. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011;71:2455–2465. doi: 10.1158/0008-5472.CAN-10-3323. [DOI] [PubMed] [Google Scholar]
- 29.Park J, Morley TS, Kim M, Clegg DJ, Scherer PE. Obesity and cancer- mechanisms underlying tumour progression and recurrence. Nat Rev Endocrinol. 2014;10:455–465. doi: 10.1038/nrendo.2014.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carter JM, Hoskin TL, Pena MA, Brahmbhatt R, Winham SJ, Frost MH, Stallings-Mann M, Radisky DC, Knutson KL, Visscher DW, Degnim AC. Macrophagic “crown-like structures” are associated with an increased risk of breast cancer in benign breast disease. Cancer Prev Res. 2018;11:113–119. doi: 10.1158/1940-6207.CAPR-17-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chakraborty D, Benham V, Bullard B, Kearney T, Hsia HC, Gibbon D, Demireva EY, Lunt SY, Bernard JJ. Fibroblast growth factor receptor is a mechanistic link between visceral adiposity and cancer. Oncogene. 2017;36:6668–6679. doi: 10.1038/onc.2017.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benham V, Chakraborty D, Bullard B, Bernard JJ. A role of FGF2 in visceral adiposity-associated mammary epithelial transformation. Adipocyte. 2018;7:113–120. doi: 10.1080/21623945.2018.1445889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim WH, Li M, Han W, Ryu HS, Moon WK. The spatial relationship of malignant and benign breast lesions with respect to the fat-gland interface on magnetic resonance imaging. Sci Rep. 2016;6:39085. doi: 10.1038/srep39085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niu J, Jiang L, Guo W, Shao L, Liu Y, Wang L. The association between leptin level and breast cancer: a meta analysis. PLoS One. 2013;8:e67349. doi: 10.1371/journal.pone.0067349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garten A, Schuster S, Kiess W. The insulin-like growth factors in adipogenesis and obesity. Endocrinol Metab Clin N Am. 2012;41:283–295. doi: 10.1016/j.ecl.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 36.Kleinberg DL, Ruan W. IGF-1, GH, and sex steroid effects in normal mammary gland development. J Mammary Gland Biol Neoplasia. 2008;13:353–360. doi: 10.1007/s10911-008-9103-7. [DOI] [PubMed] [Google Scholar]
- 37.Christopoulos PF, Msaouel P, Koutsilieris M. The role of the insulin-like growth factor-1 system in breast cancer. Mol Cancer. 2015;14:43. doi: 10.1186/s12943-015-0291-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lauro L, Naimo GD, Ricchio E, Panno ML, Ando S. Cross-talk between adiponectin and IGF-1R in breast cancer. Front Oncol. 2015;5:157. doi: 10.3389/fonc.2015.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pond AC, Bin X, Batts T, Roarty K, Hilsenbeck S, Rosen JM. Fibroblast growth factor receptor signaling is essential for normal mammary gland development and stem cell function. Stem Cells. 2013;31:178–189. doi: 10.1002/stem.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akl MR, Nagpal P, Ayoub NM, Tai B, Prabhu SA, Capac CM, Gliksman M, Goy A, Suh KS. Molecular and clinical significance of fibroblast growth factor 2 (FGF2/bFGF) in malignancies of solid and hematological cancers for personalized therapies. Oncotarget. 2016; 7:44735-62. [DOI] [PMC free article] [PubMed]
- 41.Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 2016; 30:1002-19. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplement Figure 1. Isolated TumorMap of the batch-integrated normal breast transcriptomes overlain with color-scaled intensity scores (red = high) for their various epithelial attributes including % epithelial nuclei, TDLU scores, KIT and TRPS1 gene expression levels. All numeric sample scores can be found in Supplement Table 4. Supplement Figure 2. Isolated TumorMap of the batch-integrated normal breast transcriptomes overlain with color-scaled intensity scores (red = high) for their various tissue compositions (% stromal, adipocyte, epithelial nuclei) and transcriptome gene expression modules representing specific immune cell signatures (macrophage, CD68, CD8 Tcell). Modules are defined in Supplement Table 1 and their numeric signature scores are listed in Supplement Table 4. Supplement Figure 3. Normal breast expression of genes representing adipocyte activation, remodeling, and pro-inflammatory signaling for all P batch samples according to their sample transcriptome phenotype assignment as either Active or Inactive (as described in Fig. 4 legend).
Additional file 2: Supplement Table 1. Lists of genes included within each gene expression signature analyzed to determine Active/Inactive, cAMP lipolysis, adipocyte browning, SASP, AST, IGF1, IGF1R, IFN, TGFβ, and CSR activity scores. Also indicated is their reference sources and method of score calculation.
Additional file 3: Supplement Table 2. RNAseq cancer gene hotspot mutations detected in 151 KTB samples. Sheet 1 identifies the > 5100 unthresholded cancer mutation hotspot calls from the KTB RNAseq analysis occurring in the combined set of MSK-IMPACT curated cancer hotspot clinical targets [10] and the experimentally determined RNAseq identified set of expressed cancer gene hotspot mutations within > 6700 normal GTEx human tissue samples as recently described [11]. Sheet 2 (“normal_breast_rna.getz_list.092”) lists only those > 1760 cancer gene hotspot mutations from sheet 1 meeting the threshold mutation likelihood score > 5, while sheets 3 and 4 list the thresholded hotspot mutations according to F and P sample batches with each sample phenotyped as either Active or Inactive. Sheet 5 is a sample key mapping all KTB identification numbers, barcodes, and UUID numbers pertaining to the RNAseq results and cancer gene hotspot mutation calls. Of note, Fig. 2 panels (“Breast sample scatterplots and TumorMaps of RNA expressed cancer hotspot mutations”) include plots derived from the curated and thresholded hotspot mutations as listed in sheet 2.
Additional file 4: Supplement Table 3. Rank ordered GSEA (www.gsea-msigdb.org/gsea) analysis showing 186 gene sets upregulated (from total set of 18,408 gene sets) in the batch-integrated Active vs. Inactive transcriptome samples, at FDR ≤ 10%. Also shown are their individual nominal p-values, FDR q-values, their gene set size, and the 21 (11.2%) that are specifically involved in adipose-associated pathways.
Additional file 5: Supplement Table 4. Master spreadsheet listing showing sample and donor covariates for each of the 151 KTB barcodes including batch assignment (F or P), Active/Inactive phenotype assignment and score, donor features (including age, BMI, 5 year Gail risk scores), percentages of adipocyte/stromal/epithelial cell nuclei, TDLU counts, mean adipocyte areas, immune modules, and all gene signatures and single genes values used to calculate the Pearson correlations shown in Fig. 5.
Data Availability Statement
The primary transcriptome (RNAseq) data obtained on all tissue samples have been deposited on the public UCSC Xena functional genomics platform: https://xenabrowser.net/datapages/?cohort=Normal%20Breast%20(Benz%202020).
All other primary data analyzed and presented in this study are located in the Supplementary tables attached to this manuscript.