

Abstract
Previous studies have shown that acid (H+) retention in patients with chronic kidney disease (CKD) but without metabolic acidosis increases as the estimated glomerular filtration rate (eGFR) decreases over time. The present study examined whether changes in urine excretion of the pH-sensitive metabolite citrate predicted changes in H+ retention over time in similar patients with CKD that were followed for 10 yr. We randomized 120 CKD2 nondiabetic, hypertension-associated nephropathy patients with plasma total CO2 of >24 mM to receive 0.5 meq·kg body wt−1·day−1 NaHCO3 (; n = 40), 0.5 meq·kg body wt−1·day−1 NaCl (NaCl; n = 40), or usual care (UC; n = 40). We assessed eGFR (CKD-EPI) and H+ retention by comparing the observed with expected plasma total CO2 increase 2 h after an oral NaHCO3 bolus (0.5 meq/kg body wt). Although 10 yr versus baseline eGFR was lower for each group, 10-yr eGFR was higher (P < 0.01) in (59.6 ± 4.8 ml·min−1·1.73 m−2) than NaCl and UC (52.1 ± 5.9 and 52.3 ± 4.1 ml·min−1·1.73 m−2, respectively) groups. Less eGFR preservation was associated with higher 10-yr versus baseline H+ retention in the NaCl group (26.5 ± 13.1 vs. 18.2 ± 15.3 mmol, P < 0.01) and UC group (24.8 ± 11.3 vs. 17.7 ± 10.9 mmol, P < 0.01) and with lower 10-yr versus baseline 8-h urine citrate excretion (UcitrateV) for the NaCl group (162 ± 47 vs. 196 ± 52 mg, respectively, P < 0.01) and UC group (153 ± 41 vs. 186 ± 42 mg, respectively, P < 0.01). Conversely, better eGFR preservation in the group was associated with no differences in 10-yr versus baseline H+ retention (14.2 ±13.5 vs. 16.1 ± 15.1 mmol, P = 1.00) or UcitrateV (212 ± 45 vs. 203 ± 49 mg, respectively, P = 0.74). An overall generalized linear model for repeated measures showed that UcitrateV predicted H+ retention (P < 0.01). Less eGFR preservation in patients with CKD2 without metabolic acidosis was associated with increased H+ retention that was predicted by decreased UcitrateV.
Keywords: acidosis, acid retention, bicarbonate, chronic kidney disease, glomerular filtration rate
INTRODUCTION
The kidneys are responsible primarily for excreting “fixed” acid (as opposed to “volatile” acid derived from CO2 excreted by the lungs) from acid (H+)-producing diets typical of developed societies (38). Individuals without known kidney disease given dietary acid H+ excrete it and eventually achieve steady-state urine H+ excretion sufficient to avoid progressive metabolic acidosis (27, 32) but cumulatively excrete less H+ than ingested, consistent with H+ retention (28, 29). Indeed, patients with reduced estimated glomerular filtration rate (eGFR) who eat the high-H+ diets typical of developed societies can have H+ retention (19, 20, 41, 47) with the magnitude inversely related to eGFR (19), and this is not necessarily reflected by plasma acid-base parameters characteristic of metabolic acidosis. High-H+ diets increase indexes of kidney injury in patients with reduced eGFR but no metabolic acidosis (16) and are associated with increased CKD incidence (4).
Overall, chronic kidney disease (CKD) incidence in the United States has not increased in recent analyses, yet the incidence of prevalent CKD progressing to more advanced stages (40), including to end-stage kidney disease (ESKD) (34), has nevertheless increased, as has its associated death rate (7). The higher ESKD incidence in at-risk United States population groups is due more to faster progression of prevalent CKD to ESKD than to greater CKD incidence (22), emphasizing the importance of identifying modifiable factors for CKD progression to ESKD (34). Because NaHCO3 slows the eGFR decline in patients with reduced eGFR who are eating high-H+ diets but have no metabolic acidosis (33), the underlying H+ retention in patients with reduced eGFR might contribute to the subsequent eGFR decline that such patients often experience (3, 33). Epidemiological studies showing that high-H+ diets increase serum anion gap (6) and are associated with an increased risk for CKD progression to ESKD (5, 50) support this hypothesis. Therefore, dietary H+ reduction that reduces underlying H+ retention might limit the progression of prevalent CKD to ESKD.
Dietary H+ reduction treatment of underlying H+ retention might include Na+-based alkali and/or base-producing diets along with their associated challenges. Na+ accompanying alkali salts can be problematic for the Na+-sensitive comorbidities that often accompany CKD (8), requiring additional or increased doses of natriuretic agents (12). Base-producing diets reduce the underlying H+ retention (20), but only a minority of patients can sustain adherence (15). The challenges described for these two available interventions support identification of underlying H+ retention in patients with CKD without metabolic acidosis before these largely asymptomatic patients are subjected to these treatments.
Current methods for identifying H+ retention in patients with CKD are invasive and cumbersome, and so they are not useful in clinical settings. Recent studies have supported that reduced urine citrate excretion (UcitrateV) can identify patients with CKD with reduced eGFR without metabolic acidosis, yet they have H+ retention (20). The present study tested the hypothesis that changes in UcitrateV identify changes in H+ retention as eGFR changes over time (19). If this hypothesis tests true, UcitrateV might be a clinically useful biomarker for tracking the risk for nephropathy progression in patients with CKD, thereby revealing their candidacy for potentially kidney-protective dietary H+ reduction therapy.
MATERIALS AND METHODS
This longitudinal study examined the effects of oral NaHCO3 (group; 0.5 meq·kg body wt−1·day−1) or oral NaCl (NaCl group; 0.5 meq·kg body wt−1·day−1) compared with usual care (UC group), as recommended by guidelines at study initiation (9, 30), on the cystatin C-calculated (CKD-EPI) eGFR (23) over 10 yr as the primary analysis. Secondary analyses included the longitudinal effect(s) of these interventions on H+ retention (see below) and the association of changes in H+ retention with changes in eGFR. Post hoc, we measured 8-h UcitrateV and assessed the association of changes in UcitrateV with changes in H+ retention. Interim analysis at 5 yr showed better eGFR preservation in the group (33), and we advised all patients of this outcome and gave them the opportunity to receive oral NaHCO3 as described. Thirty-four patients elected to receive NaHCO3 from years 6−10 of the protocol in substitution for the regimen to which they had been randomized in blinded fashion (UC, NaCl, or NaHCO3), of whom 14 patients were in the UC group, 9 patients were in the NaCl group, and 11 patients were in the group. Each unblinded patient received NaHCO3 from years 6−10 but remained in their originally assigned group in intention-to-treat fashion as per the protocol. Nevertheless, the analysis reported censored those 34 participants who received NaHCO3 in an unblinded fashion.
Figure 1 shows that we identified, through clinic screening (33), 491 macroalbuminuric, hypertensive, nondiabetic CKD2 [eGFR: 60–89 ml·min−1·1.73 m−2 by the Modification of Diet in Renal Disease Study formula (25)] patients without clinical evidence of glomerulonephritis or evidence of a systemic disease associated with glomerulonephritis, from whom 120 triplicates were matched for age (±2 yr), sex, ethnicity (black vs. Hispanic or white), eGFR (±4 ml·min−1·1.73 m−2), and urine albumin (±40 mg/g creatinine). We measured plasma cystatin C and subsequently calculated eGFR with this parameter (23), and eGFR is so reported because earlier reports have supported that cystatin C compared with creatinine estimated eGFR more accurately at the comparatively higher eGFR levels of our recruited patients (39). We chose macroalbuminuric patients because they are at higher risk of eGFR progression despite recommended kidney-protective interventions than those with lower levels of albuminuria (42). This selection enhanced our ability to examine the effect of changes of H+ retention on the course of eGFR. Other inclusion criteria were 1) nonmalignant hypertension; 2) at least two primary care physician visits in the preceding year, showing compliance with clinic visits; and 3) age ≥ 18 yr and ability to give consent. Exclusion criteria were 1) primary kidney disease or findings consistent thereof, such as at least 3 red blood cells/high-powered field or urine cellular casts; 2) history of diabetes or fasting blood glucose ≥ 110 mg/dl; 3) history of malignancies, chronic infections, pregnancy, or clinical evidence of cardiovascular disease; 4) peripheral edema or diagnoses associated with edema such as heart/liver failure or nephrotic syndrome; and 5) inability to tolerate angiotensin-converting enzyme inhibition because extant guidelines at the start of the study recommended this therapy for patients with macroalbuminuria (9). None of the participants had a kidney biopsy to exclude other CKD causes. Secondary causes of hypertension such as renal artery stenosis and hyperaldosteronism were excluded clinically. We did not do kidney Doppler or plasma aldosterone-to-renin ratio studies. We recruited most participants from those referred to our academic nephrology clinic for management of hypertension and/or evaluation of reduced kidney function. Participant systolic blood pressure (SBP) was reduced pharmacologically toward a goal of <130 mmHg, as recommended by extant guidelines for those with albuminuria (9). We measured 8-h urine net acid excretion (NAE) from urine titratable acidity (TA), ammonium (NH4+), and HCO3 ([NH4+] + [TA] – []) (47).
Fig. 1.
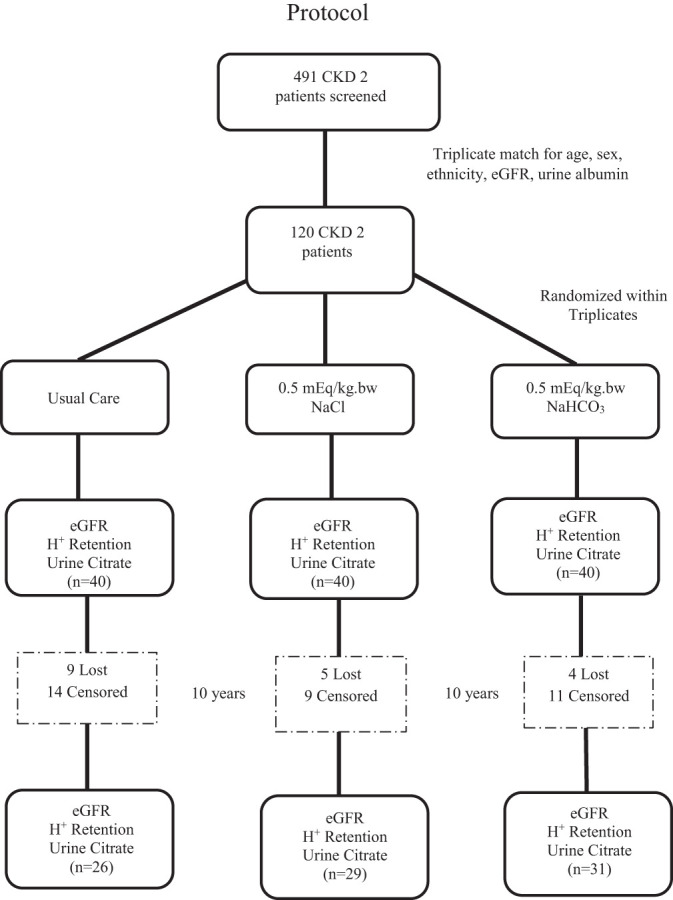
Outline of protocol to longitudinally assess the association between changes in acid (H+) retention and changes in estimated glomerular filtration rate (eGFR) among patients with chronic kidney disease stage 2 (CKD2; eGFR: 60–89 ml·min−1·1.73 m−2) undergoing usual care or treatment with oral NaCl or oral NaHCO3 [meq·kg body wt (bw)−1·day−1].
We assessed H+ retention as “unaccounted ” at baseline and at 10 yr by comparing the difference between the expected and observed change in plasma total CO2 in response to retained (dose minus excretion) 2 h after an oral bolus of 0.5 meq·kg−1·body wt−1 NaHCO3 with the following equation: H+ retention = [(retained /0.5 × body weight) – observed increase in plasma ] × 0.5 body weightt, assuming 50% body weight space distribution for (2).
We assessed steady-state H+ retention by measuring 2-h NAE and venous plasma total CO2 in the three CKD groups after an oral 0.5 meq/kg lean body wt bolus of NaHCO3. We told participants to take nothing by mouth after midnight, except for medications before reporting, after which they voided to empty, had venous blood drawn for pH, Pco2, and total CO2, and had venous access established. They received the NaHCO3 bolus at 8 AM and had urine collected for NAE and venous blood drawn for acid-base parameters at 10 AM. They received 8 oz of chilled distilled H2O, but no other intake, every 2 h to promote urine output. Our assessment of H+ retention assumed that higher H+ retention would manifest by greater H+ titration of the administered , as reflected by a smaller observed compared with expected plasma total CO2 increase (assuming 50% space of distribution) (2) and/or by less urine excretion. This method also assumes similar buffering capacity among CKD stages, an assumption supported by no differences in levels of the major contributors to whole blood buffer base capacity reported in previous studies (47). Because measured parameters did not permit comparison of intracellular buffering capacity, we assumed that this parameter was similar among CKD groups, as done in previous studies (19, 20, 47).
Because patients were recruited before 2006, our study is not registered with clinicaltrials.gov. Our local Institutional Review Board approved the study protocols.
Analytic methods.
We measured plasma and urine creatinine and urine albumin using the Sigma Diagnostics Creatinine Kit (procedure no. 555, Sigma Diagnostics) (36). We measured plasma cystatin C with a particle-enhanced immunonephelometric assay (N Latex cystatin C, Dade Behring, Somerville, NJ) and a nephelometer (BNII, Dade Bering) (14). The IRMA SL Series 2000 blood analysis system (Diametrics, Edison, NJ) measured venous blood pH and Pco2, and [] was calculated from these two parameters. Urine and plasma total CO2 were measured using ultrafluoremetry (43). Urine TA was measured by correction to the ambient blood pH by NaOH addition and NH4+ by the formalin titrametric (to ambient blood pH) method (10). We measured potential renal acid load (PRAL) using a 3-day dairy assessment of the type and amount of foods ingested and scoring them as to their H+ or base content as previously described (37) and as previously performed (19, 20, 47). This calculation does not include an estimate of organic acid excretion that combines with PRAL to estimate urine NAE (20); further details regarding this measurement have been reported in an earlier study from our laboratory (20).
Statistical methods.
Characteristics of the sample are described using descriptive statistics. We used frequencies and percentages to describe categorical variables and used means and SDs to describe continuous variables. Group comparisons were made using one-way ANOVA (or a Kruskal-Wallis test when continuous data were not distributed approximately normally). We used a generalized linear model for repeated-measures data to evaluate change in H+ retention, as measured by unaccounted , from baseline to 10 yr. Tukey-Kramer adjustments were applied for post hoc comparisons between groups and between time points. We used similar models to evaluate changes from baseline for cystatin C-calculated eGFR and plasma total CO2. We used simple linear regression to assess the effect of the net change in H+ retention on the net change in eGFR between baseline and 10 yr, with a statistical significance level set to 0.05. Followed by the intent-to-treat analyses, we carried out sensitivity analyses after excluding the 34 participants who were unblinded and/or changed their randomization status from years 6−10. Similar to the intent-to-treat analyses, the sensitivity analyses used generalized linear models for repeated measures along with Tukey-Kramer adjustments for post hoc comparisons between groups and between time points.
RESULTS
Figure 1 shows the protocol for the 120 patients with CKD2 randomized to the UC (n = 40), NaCl (0.5 meq·kg body wt−1·day−1, n = 40), and (0.5 meq·kg body wt−1·day−1, n = 40) groups and followed with the indicated measurements at baseline and at 10 yr. Table 1 shows that there were no difference in sex, age, body weight (used to calculate H+ retention), or body mass index among groups. Table 2 shows there were no differences in SBP among groups at baseline and 10 yr; all three groups had lower than baseline SBP at 10 yr. Table 2 also shows there were no difference in baseline 8-h urine Na+ excretion (UNaV) and urine K+ excretion (UKV). Ten-year values for UNaV and UKV were higher than baseline in the NaCl and groups but not in the UC group, showing that the two intervention groups ingested the provided NaCl and NaHCO3. Ten-year UKV was higher than the respective baseline in the group but not in the NaCl or UC group.
Table 1.
General participant characteristics
| Usual Care Group | NaCl Group | Group | P Value | |
|---|---|---|---|---|
| Men/women, % | 47.5/52.5 | 47.5/52.5 | 47.5/52.5 | 1.0 |
| Ethnicity | ||||
| Black, % | 62.5 | 62.5 | 62.5 | |
| Hispanic, % | 25.0 | 22.5 | 17.5 | 0.97 |
| White, % | 12.5 | 15.0 | 20.0 | |
| Age, yr | 51.3 (8.5) | 51.5 (8.3) | 51.2 (8.2) | 0.98 |
| Body weight, kg | 84.1 (4.5) | 83.8 (5.2) | 83.2 (5.4) | 0.75 |
| Body mass index | 28.8 (2.3) | 28.1 (2.2) | 28.6 (2.2) | 0.81 |
Values are means (SD) where appropriate; n = 40 participants/group. Patients with chronic kidney disease stage 2 were divided into the following three groups: usual care or treatment with oral NaCl or NaHCO3 ().
Table 2.
Baseline and followup values for systolic blood pressure and 8-h urine excretion of Na+ and K+
| Usual Care Group | NaCl Group | Group | P Value | |
|---|---|---|---|---|
| Systolic blood pressure, mmHg | ||||
| Year 0 | 153.7 (13.3) | 156.1 (15.7) | 155.3 (13.7) | 0.75* |
| Year 10 | 134.1 (8.9) | 133.0 (7.6) | 135.6 (7.1) | 0.25* |
| P value† | <0.01 | <0.01 | <0.01 | |
| Urine Na+ excretion, mmol/8 h | ||||
| Year 0 | 78.4 (5.8) | 77.3 (6.0) | 75.9 (6.1) | 0.82* |
| Year 10 | 75.8 (6.0) | 87.6 (5.9) | 84.3 (6.2) | <0.01* |
| P value† | 0.25 | <0.01 | <0.01 | |
| Urine K+ excretion, mmol/8 h | ||||
| Year 0 | 36.8 (5.1) | 34.6 (5.3) | 35.5 (5.0) | 0.85* |
| Year 10 | 35.3 (5.2) | 36.0 (5.2) | 37.8 (5.1) | 0.23* |
| P value† | 0.63 | 0.19 | 0.039 |
Values are means (SD); n = 40 participants/group. Patients with chronic kidney disease stage 2 were divided into the following three groups: usual care or treatment with oral NaCl or NaHCO3 (). P values at the end of the rows compare values in that row.
Kruskal-Wallis test for comparing three groups. All other P values were from one-way ANOVA.
P values below year 0 and year 10 values compare values for those years.
Table 3 shows there were no difference in baseline plasma levels of cystatin C or creatinine but higher than baseline values at 10 yr for each parameter in all three groups. Nevertheless, both plasma levels of cystatin C and creatinine were lower in the group than in the remaining groups at 10 yr (P < 0.01). Figure 2 shows that cystatin C-calculated eGFR was not different among groups at baseline but was lower at 10 yr (P < 0.01) than at baseline for the UC (52.3 ± 4.1 vs. 73.9 ± 6.4 ml·min−1·1.73 m−2), NaCl (52.1 ± 5.9 vs. 73.7 ± 6.6 ml·min−1·1.73 m−2), and (59.6 ± 4.8 vs. 72.9 ± 6.3 ml·min−1·73 m−2) groups. Ten-year eGFR was higher in the group than in the NaCl and UC groups (P < 0.01) but was not different between the NaCl and UC groups (P = 0.99).
Table 3.
Baseline and followup values for plasma cystatin C and creatinine
| Usual Care Group | NaCl Group | Group | P Value | |
|---|---|---|---|---|
| Plasma cystatin C, mg/dl | ||||
| Year 0 | 1.04 (0.09) | 1.04 (0.09) | 1.05 (0.10) | 0.90* |
| Year 10 | 1.33 (0.11) | 1.34 (0.11) | 1.24 (0.10) | <0.02* |
| P value† | <0.01 | <0.01 | <0.01 | |
| Plasma creatinine, mg/dl | ||||
| Year 0 | 0.95 (0.08) | 0.96 (0.08) | 0.97 (0.07) | 0.95* |
| Year 10 | 1.23 (0.08) | 1.24 (0.08) | 1.14 (0.07) | <0.01* |
| P value† | <0.01 | <0.01 | <0.01 |
Values are means (SD); n = 40 participants/group. Patients with chronic kidney disease stage 2 were divided into the following three groups: usual care or treatment with oral NaCl or NaHCO3 (). P values at the end of the rows compare values in that row.
Kruskal-Wallis test for comparing three groups. All other P values were from one-way ANOVA.
P values below year 0 and year 10 values compare values for those years.
Fig. 2.

Dot plots showing cystatin C-calculated estimated glomerular filtration rate (eGFR) for patients with chronic kidney disease stage 2 (CKD2) undergoing usual care (UC group) or additionally treated with oral NaCl (NaCl group; 0.5 meq·kg body wt−1·day−1) or NaHCO3 ( group; 0.5 meq·kg body wt−1·day−1) at baseline (light gray circles) and at 10 yr (dark gray circles). Black circles show mean ± SD values for the respective groups. *P < 0.05, year 10 vs. the respective baseline value within that group; +P < 0.05 vs. the respective UC group.
Table 4 shows that there were no differences among groups in PRAL at any time point. Table 5 shows 8-h urine NAE and its components at the three time points. Baseline 8-h NAE, 8-h excretion, 8-h TA, and 8-h urine excretion were not different among groups. Five- and ten-year values for 8-h NAE, 8-h NH4+ excretion, and 8-h TA were not different among groups. In contrast, as shown in Table 5, there were higher values of 8-h urine excretion for patients at 5 and 10 yr. Compared with baseline, 5- and 10-yr values for 8-h NH4+ excretion were lower and those for 8-h TA were higher in patients but not in NaCl or UC patients (Table 5). Five- and ten-year 8-h urine excretion were higher than baseline in patients but not in NaCl or UC patients. There was also higher 8-h urine excretion in years 5 and 10 compared with baseline in patients but not in NaCl or UC patients (Table 5). Although baseline plasma total CO2 was not different among groups at baseline, 5-yr plasma total CO2 was lower than baseline in the NaCl and UC groups and higher than baseline in the group (Table 6). Ten-year plasma total CO2 was lower still than the 5-yr value in NaCl and UC groups but was not different in the group compared with plasma total CO2 at 5 yr, and 10-yr plasma total CO2 for the group was not different from its respective baseline.
Table 4.
Changes in potential renal acid load during followup
| Usual Care Group | NaCl Group | Group | P Value | |
|---|---|---|---|---|
| Year 0 | 65.7 (17.3) | 64.2 (11.7) | 63.4 (12.0) | 0.78* |
| Year 5 | 64.1 (15.3) | 62.9 (11.3) | 66.4 (8.3) | 0.67* |
| Year 10 | 61.8 (11.8) | 63.4 (12.3) | 64.8 (8.3) | 0.59* |
| Difference: year 5 vs. year 0 | −1.6 (13.4) | −1.3 (16.7) | 3.0 (14.1) | 0.67 |
| P value† | 0.79 | 0.64 | 0.66 | |
| Difference: year 10 vs. year 0 | −3.9 (13.2) | −0.8 (15.7) | 1.4 (13.0) | 0.43 |
| P value† | 0.26 | 0.81 | 0.85 | |
| Difference: year 10 vs. year 5 | −2.3 (12.5) | 0.5 (15.8) | −1.6 (14.1) | 0.74 |
| P value† | 0.45 | 0.83 | 0.75 |
Values are means (SD) of potential renal acid load (in mmol/day); n = 40 participants/group. Patients with chronic kidney disease stage 2 were divided into the following three groups: usual care or treatment with oral NaCl or NaHCO3 (). Mean differences were determined as follows: mean difference = average (SD) of the year 10 value minus baseline, year 5 value minus baseline, and year 10 value minus year 5 value, respectively, for each patient.
Kruskal-Wallis test for comparing three groups. All other P values are from one-way ANOVA.
Signed-rank test for assessing the within-group difference between baseline and year 5, baseline and year 10, and year 5 and year 10.
Table 5.
Changes in 8-h urine NAE and its components during followup
| 8-h Net Acid Excretion |
8-h Urine Excretion† |
8-h Urine Titratable Acid Execretion |
8-h Urine Excretion |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Usual caregroup | NaCl group | group | P value | Usual caregroup | NaCl group | grop | P value | Usual caregroup | NaCl group | grop | P value | Usual caregroup | NaCl group | group | P value | |
| Year 0 | 24.8 (5.1) | 24.5 (4.3) | 25.3 (4.2) | 0.37* | 14.1 (3.1) | 14.0 (2.9) | 14.6 (2.9) | 0.92* | 10.9 (2.5) | 10.5 (2.3) | 10.8 (2.2) | 0.94* | 0.2 (0.1) | 0.1 (0.1) | 0.1 (0.1) | 0.77* |
| Year 5 | 23.9 (5.3) | 24.1 (4.2) | 25.4 (3.5) | 0.29* | 14.3 (3.3) | 14.5 (2.8) | 13.5 (2.5) | 0.07* | 9.7 (2.3) | 9.6 (2.4) | 12.4 (2.5) | 0.06* | 0.1 (0.2) | 0.1 (0.2) | 0.6 (0.2) | 0.02* |
| Year 10 | 23.7 (5.4) | 24.0 (4.0) | 25.4 (3.2) | 0.17* | 14.7 (3.2) | 14.9 (2.9) | 13.3 (2.4) | 0.06* | 9.2 (2.4) | 9.2 (2.4) | 12.5 (2.7) | 0.06* | 0.1 (0.2) | 0.1 (0.1) | 0.4 (0.2) | 0.04* |
| Difference: year 5 vs. year 0 | −0.9 (13.4) | −0.4 (16.7) | 0.1 (14.1) | 0.06 | 0.2 (2.6) | 0.5 (3.0) | −1.1 (3.2) | −1.2 (2.9) | −0.9 (3.2) | 1.6 (4.2) | 0.03 | −0.1 (1.2) | 0.0 (1.1) | 0.5 (0.3) | 0.02 | |
| P value† | 0.34 | 0.51 | 0.73 | 0.64 | 0.61 | 0.03 | 0.26 | 0.35 | 0.05 | 0.34 | 0.89 | <0.01 | ||||
| Difference: year 10 vs. year 0 | −1.1 (12.5) | −0.5 (15.8) | 0.1 (14.1) | 0.08 | 0.6 (3.7) | 0.9 (2.9) | −1.3 (3.3) | 0.02 | −1.7 (3.1) | −1.3 (2.9) | 1.7 (4.0) | 0.03 | −0.1 (1.1) | 0.0 (1.2) | 0.3 (0.2) | 0.03 |
| P value† | 0.54 | 0.71 | 0.73 | 0.42 | 0.5 | 0.02 | 0.19 | 0.23 | 0.04 | 0.45 | 0.92 | <0.01 | ||||
| Difference: year 10 vs. year 5 | −0.2 (13.2) | −0.1 (15.7) | 0.0 (13.0) | 0.41 | 0.4 (3.5) | 0.4 (2.5) | −0.2 (2.5) | 0.21 | −0.5 (2.9) | −0.4 (2.2) | 0.1 (2.6) | 0.08 | 0.0 (1.2) | 0.0 (1.0) | −0.2 (0.3) | 0.18 |
| P value† | 0.61 | 0.72 | 0.79 | 0.33 | 0.71 | 0.59 | 0.39 | 0.47 | 0.66 | 0.85 | 0.87 | 0.18 | ||||
Values are means (SD); n = 40 participants/group. Patients with chronic kidney disease stage 2 were divided into the following three groups: usual care or treatment with oral NaCl or NaHCO3 (). Mean differences were calculated as follows: mean difference = average (SD) of the year 10 value minus baseline, year 5 value minus baseline, and year 10 value minus year 5 value for each patient.
Kruskal-Wallis test for comparing three groups. All other P values were from one-way ANOVA.
Signed-rank test for assessing the within-group difference between baseline and year 5, baseline and year 10, and year 5 and year 10.
Table 6.
Changes in plasma total CO2 during followup
| Usual Care Group | NaCl Group | Group | P Value | |
|---|---|---|---|---|
| Year 0 | 26.1 (0.8) | 25.8 (0.5) | 25.9 (0.6) | 0.42* |
| Year 5 | 26.0 (0.7) | 25.7 (0.5) | 26.2 (0.7) | <0.01 |
| Year 10 | 25.2 (0.4) | 25.1 (0.5) | 26.1 (0.7) | <0.01 |
| Difference: year 5 vs. year 0 | −0.1 (0.2) | −0.1 (0.1) | 0.3 (0.4) | <0.01 |
| P value† | <0.01 | <0.01 | <0.01 | |
| Difference: year 10 vs. year 0 | −0.9 (0.7) | −0.7 (0.3) | 0.2 (0.4) | <0.01 |
| P value† | <0.01 | <0.01 | <0.01 | |
| Difference: year 10 vs. year 5 | −0.8 (0.7) | −0.6 (0.2) | −0.1 (0.2) | <0.01 |
| P value† | <0.01 | <0.01 | 0.20 |
Values are means (SD) of plasma total CO2 (in mM); n = 40 participants/group. Patients with chronic kidney disease stage 2 were divided into the following three groups: usual care or treatment with oral NaCl or NaHCO3 (). Mean differences were calculated as follows = average (SD) of the year 10 value minus baseline, year 5 value minus baseline, and year 10 value minus year 5 value for each patient.
Kruskal-Wallis test for comparing three groups. All other P values were from one-way ANOVA.
Signed-rank test for assessing the within-group difference between baseline and year 5, baseline and year 10, and year 5 and year 10.
Table 7 shows venous plasma electrolytes and acid-base parameters, with the reported calculated from measured pH and Pco2. There were no differences in plasma values for Na+, K+, or Cl− among groups at baseline or at 10 yr, and 10 yr compared with baseline values for plasma Na+ or Cl− was not different. Plasma K+ was lower at 10 yr compared with baseline in patients but was not different in NaCl or UC patients. Baseline plasma , pH, Pco2, and plasma anion gap were not different among groups. Ten-year , pH, and Pco2 were all lower than baseline for NaCl and UC patients but were not different for patients. Ten-year compared with baseline plasma anion gap was greater in NaCl and UC patients but was not different for patients.
Table 7.
Baseline and followup plasma electrolytes and acid-base parameters
| Na+, meq/l | K+, meq/l | Cl−, meq/l | , meq/l | pH | Pco2, mmHg | Anion Gap, meq/l | |
|---|---|---|---|---|---|---|---|
| Usual care group | |||||||
| Year 0 | 139.3 (1.3) | 4.1 (0.3) | 103.1 (1.4) | 24.9 (0.6) | 7.405 (0.019) | 40.8 (0.2) | 11.3 (0.1) |
| Year 10 | 139.7 (1.4) | 4.2 (0.3) | 103.6 (1.4) | 23.9 (0.6) | 7.393 (0.020) | 40.3 (0.1) | 12.2 (0.1) |
| P value* | 0.32 | 0.79 | 0.21 | <0.01 | <0.01 | 0.04 | <0.01 |
| NaCl group | |||||||
| Year 0 | 139.1 (1.3) | 4.1 (0.2) | 103.3 (1.4) | 24.6 (0.5) | 7.403 (0.022) | 40.5 (0.1) | 11.2 (0.1) |
| Year 10 | 139.7 (1.3) | 4.1 (0.2) | 103.9 (1.4) | 23.7 (0.5) | 7.391 (0.021) | 40.1 (0.1) | 12.1 (0.1) |
| P value* | 0.25 | 0.96 | 0.22 | <0.01 | <0.01 | 0.03 | <0.01 |
| group | |||||||
| Year 0 | 139.2 (1.2) | 4.1 (0.2) | 103.2 (1.4) | 24.7 (0.6) | 7.404 (0.020) | 40.6 (0.1) | 11.3 (0.1) |
| Year 10 | 139.5 (1.2) | 4.0 (0.2) | 103.5 (1.4) | 24.8 (0.7) | 7.405 (0.020) | 40.7 (0.1) | 11.2 (0.1) |
| P value* | 0.95 | 0.047 | 0.78 | 0.62 | <0.01 | 0.46 | 0.54 |
Values are means (SD); n = 40 participants/group. Patients with chronic kidney disease stage 2 were divided into the following three groups: usual care or treatment with oral NaCl or NaHCO3 ().
P values were from a signed-rank test for assessing the within-group difference between baseline and year 10.
Figure 3A shows that baseline H+ retention was not different among groups, and the 10-year compared with baseline value was not different for the group (14.2 ± 13.5 vs. 16.1 ± 15.1 mmol, respectively, P = 1.00). In contrast, 10-yr compared with baseline H+ retention was higher in the NaCl group (26.5 ± 13.1 vs. 18.2 ± 15.3 mmol, respectively, P < 0.01) and UC group (24.8 ± 11.3 vs. 17.7 ± 10.9 mmol, P < 0.01). Overall, there was no association between H+ retention and eGFR across time for the three treatment groups (P = 0.13).
Fig. 3.

Dot plots showing acid (H+) retention in patients with chronic kidney disease stage 2 (CKD2) undergoing usual care (UC group) or treatment with oral NaCl (NaCl group; 0.5 meq·kg body wt−1·day−1) or NaHCO3 ( group; 0.5 meq·kg body wt−1·day−1) at baseline (light gray circles) and at 10 yr (dark gray circles). Black circles show mean ± SD values for the respective groups. *P < 0.05, year 10 vs. the respective baseline value within that group; +P < 0.05 vs. the respective UC group.
Figure 3B also shows that baseline UcitrateV was not different among groups and that the 10-yr compared with baseline value, respectively, was not different for the group (212 ± 45 vs. 203 ± 49 mg, respectively, P = 0.74). In contrast, 10-yr compared with baseline UcitrateV was lower in the NaCl group (162 ± 47 vs. 196 ± 52 mg, respectively, P < 0.01) and UC group (153 ± 41 vs.186 ± 42 mg, respectively, P < 0.01). Although UcitrateV at 10 yr was not different between NaCl and UC group (P = 1.00), 10-yr UcitrateV in the group was higher than both NaCl (P < 0.01) and UC (P < 0.01) groups. A generalized linear model for repeated measures, adjusted for time, showed that UcitrateV predicted H+ retention overall when the data for all three groups were combined (P < 0.01; visual depiction shown in Fig. 4). When this model was applied to the individual groups, UcitrateV did not predict H+ retention for the UC group (P = 0.06) but did so for the NaCl (P < 0.01) and (P < 0.01) groups.
Fig. 4.

Generalized linear model for repeated measures, adjusted for time, for H+ retention versus 8-h urine citrate excretion (UcitrateV) at baseline (left) and at 10 yr (right) combining the data for usual care (UC), NaCl (0.5 meq·kg body wt−1·day−1), and NaHCO3 (; 0.5 meq·kg body wt−1·day−1) groups.
DISCUSSION
This longitudinal analysis shows that better eGFR preservation with chronic oral NaHCO3 than oral NaCl or UC in patients with CKD2 with reduced eGFR and no metabolic acidosis was associated with no further increase in H+ retention, whereas H+ retention increased with NaCl and UC. In addition, unchanged H+ retention in the group was associated with no change in UcitrateV, but UcitrateV decreased with increased H+ retention in the NaCl and UC groups. Furthermore, decreased UcitrateV predicted increased H+ retention over time. Although increased H+ retention in the NaCl and UC groups was associated with significant decreases in plasma total CO2 and plasma anion gap, the quantitative decreases were small, and 10-yr plasma total CO2 and anion gap remained within normal ranges of clinical laboratories. The data support that unlike plasma total CO2 or plasma anion gap, the change in UcitrateV (decrease) is quantitatively sufficient for clinicians to identify increasing H+ retention as eGFR declines in patients with CKD and reduced eGFR but no metabolic acidosis (19), thereby designating them as candidates for dietary H+ reduction to mitigate against further eGFR decline.
The data showing that measurable increases in H+ retention in patients with CKD with reduced eGFR but no metabolic acidosis was associated with only minimal decreases in plasma total CO2 and minimal increases in anion gap challenge clinicians as to how to identify patients with CKD who are potential candidates for dietary H+ reduction therapy. Although high-H+ diets more likely reduce plasma total CO2 in patients with CKD with decreased rather than normal eGFR (1), marked changes in dietary H+ yield only modest changes in plasma acid-base parameters within normal ranges for clinical laboratories (26). This robust protection of plasma against acid-base challenges shows that clinicians cannot rely upon changes in plasma total CO2 to assess underlying H+ retention or to identify if patients are eating a high-H+ diet. The method used to identify H+ retention in the present study and previous studies (19, 20, 47) is invasive and cumbersome, and so is without clinical utility. Recent studies have supported that reduced urine excretion of the pH-sensitive metabolite citrate might be a clinically useful, noninvasive way by which to identify underlying H+ retention in patients with CKD without metabolic acidosis (20). The present study supports that decreases in UcitrateV predict increases in H+ retention in patients with CKD without metabolic acidosis followed over time and might aid clinical decisions as to whether to institute or increase dietary H+ reduction therapy for kidney protection.
There is growing evidence that an acid milieu contributes to CKD progression and that its correction with dietary H+ reduction is kidney protective. Treatment of CKD-related metabolic acidosis in patients with plasma total CO2 < 22 mM, as recommended by current guidelines (24), slows the decline rate of creatinine clearance (11) and eGFR (35) in individuals with very low GFR. Correction of metabolic acidosis that is less severe than that for which current guidelines recommend treatment (i.e., plasma total CO2 = 22–24 mM) and also slows eGFR decline (18). In addition, alkali treatment of individuals with reduced eGFR without metabolic acidosis but eating the high-H+ diets of developed societies (38) slows eGFR decline (33). Furthermore, an epidemiological study has shown that high-H+ diets are associated with increased CKD incidence in a general population (4). Together, these studies support that the full spectrum of “H+ stress” that ranges from high dietary H+ with initially normal plasma total CO2 in individuals with normal eGFR to those with very low eGFR and metabolic acidosis by plasma acid-base parameters is harmful to kidneys and that its amelioration with dietary H+ reduction is kidney protective. Kidney-protective interventions such as dietary H+ reduction will likely provide greater benefit to the large cadre of patients with CKD with with better preserved eGFR than in those with less preserved eGFR, like the initial patients with CKD2 of the present study, to reduce the increasing incidence of progression of prevalent CKD to its more advanced stages (40).
Modest dietary H+ reduction in patients with CKD with reduced eGFR but no metabolic acidosis incompletely corrected underlying H+ retention (47) and slowed but did not stop eGFR decline (33). A cross-sectional study has shown that lower eGFR was associated with higher H+ retention and showed longitudinally that as eGFR decreased, H+ retention increased and was associated with a faster rate of eGFR decline (19). The present study shows that this same modest dietary H+ reduction regimen prevented a further increase in H+ retention as eGFR declined but did not decrease it. Together, these studies support examining whether more aggressive dietary H+ reduction in patients with CKD with initially lower eGFR, and thus possibly greater initial H+ retention, more completely reduces H+ retention and thereby provides greater kidney protection in patients at increased risk for CKD progression. Dietary H+ reduction accomplished by adding base-producing food components decreased H+ retention in patients with CKD without metabolic acidosis (20) and improved CKD-related metabolic acidosis (17, 18). These data support a combination of a low-H+ diet composed of base-producing food components and Na+-based alkali as one strategy by which to enhance dietary H+ reduction. Adding base-producing food components might spare the amount of Na+-based alkali needed to achieve the acid-base goal given the untoward effects of Na+-based alkali (8, 12, 31).
Animal and some patient studies have provide insights as to the mechanisms by which H+ retention might promote CKD progression. Animals with reduced GFR without metabolic acidosis eating a H+-producing diet have H+ retention by microdialysis (45, 46, 48, 49) and high kidney levels of endothelin, angiotensin II, and aldosterone (46, 48, 49). These agents increase nephron acidification in the setting of overall reduced nephron mass (46, 48) but also mediate kidney injury and nephropathy progression (46, 49, 50). Some patients with CKD and reduced eGFR but no metabolic acidosis who eat the high-H+ diets of developed societies have H+ retention associated with high urine levels of endothelin and aldosterone that decrease in response to dietary H+ reduction (47). These data support that increased kidney levels of these three agents mediate the short-term benefit of increased per nephron acidification to maintain overall kidney H+ excretion in a setting of reduced nephron mass and high dietary H+. On the other hand, these agents appear to mediate long-term progressive nephropathy when dietary H+ intake is high.
Present methods for identifying H+ retention in patients with CKD (19, 20, 47) are cumbersome and invasive and thus are not clinically practical. A recent study has shown that decreased urine excretion of the pH-sensitive metabolite citrate that is commonly and easily measured in clinical laboratories noninvasively identifies H+ retention in patients with CKD with reduced eGFR but no metabolic acidosis (20). The present longitudinal analysis shows that reductions in UcitrateV noninvasively identify increases in H+ retention that occur in response to decreasing eGFR over time in patients with CKD but no metabolic acidosis. These data support leveraging the pathophysiological insight that H+ retention reduces UcitrateV to identify patients with CKD without metabolic acidosis who during followup develop worsening H+ retention that is not manifested by noticeable reductions in plasma total CO2 or increases in plasma anion gap and might benefit from dietary H+ reduction for kidney protection. Furthermore, decreasing UcitrateV in patients receiving dietary H+ reduction for kidney protection might indicate a need for more aggressive dietary H+ reduction.
Emerging data support an earlier stage in the spectrum of H+ accumulation that is insufficient to manifest with changes in plasma acid-base parameters characteristic of metabolic acidosis yet is sufficient to increase urine levels of substances associated with progressive eGFR decline like endothelin and aldosterone (47) and increase urine biomarkers of kidney injury (16, 33). Retaining citrate through proximal tubule reabsorption rather than its urine excretion helps in the setting of H+ accumulation because its metabolism consumes H+ and yields (21). Small-scale studies have shown that dietary H+ reduction with chronic oral NaHCO3 reduces urine excretion of endothelin, aldosterone, and biomarkers of kidney injury (16, 33). Nevertheless, current guidelines (24) recommend alkali therapy only for patients with CKD at the metabolic acidosis stage of the H+ stress spectrum, so patients at this earlier stage of H+ retention would not be candidates for such therapy. Larger-scale studies will better determine the utility of dietary H+ reduction for kidney protection in patients with CKD with H+ retention but without metabolic acidosis. Using UcitrateV rather than the present invasive method to identify and track H+ retention in response to interventions would greatly facilitate conduct of such studies.
A spectrum of H+ stress, which includes H+ retention without metabolic acidosis, questions where the accumulated H+ resides before it attains levels that manifest with changes in plasma acid-base parameters characteristic of metabolic acidosis. A dietary H+ increment sufficient to increase urine H+ excretion but not to decrease plasma total CO2 in animals with intact nephron mass increased H+ addition to microdialysate perfused against the kidney cortical interstitium (44), consistent with increased interstitial fluid H+ content. The H+ entering the microdialysate might also have derived from plasma buffers and/or intracellular stores. Other studies showing that this dietary protocol increased H+ content in skeletal muscle have suggested that H+ retention is a systemic phenomenon (45). These data support the interstitial fluid compartment as at least one locus of retained H+ in these settings. Other investigators have reported a direct relationship between extracellular fluid volume and interstitial fluid volume and pressure and that each of the latter were higher patients with reduced GFR (13), suggesting that the interstitial fluid compartment reflects systemic status of other kidney-regulated phenomenon. Together, these data reveal that interstitial fluid can reflect systemic Na+ and H+ status before their respective manifestation in plasma, including pathological excess of each observed with decreased GFR.
Study limitations include our indirect method of assessing H+ retention. In addition, the study population was limited to nondiabetic patients with CKD and contained few white patients, so future studies with larger, more diverse sample sizes will determine whether these findings apply to those with CKD to other etiologies and apply equally across ethnic groups.
In summary, this study shows that better eGFR preservation in NaHCO3-treated patients with CKD2 without metabolic acidosis was associated with no increase in their baseline level of H+ retention, but H+ retention increased in patients that received eqimolar NaCl or placebo that had less eGFR preservation. In addition, decreasing UcitrateV identified those patients with increasing H+ retention during followup. These data support using longitudinal reductions in UcitrateV to identify increasing H+ retention in patients with CKD despite not having metabolic acidosis but who might nevertheless benefit from dietary H+ reduction or who require more aggressive dietary H+ reduction.
GRANTS
This work was supported by funds from the Larry and Jane Woirhaye Memorial Endowment in Renal Research the Texas Tech University Health Sciences Center, by the Statistics Department of Texas A&M University, and by the Academic Operations Division of Baylor Scott and White Health. This work was also supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R21-DK-11344.
DISCLOSURES
D. E. Wesson has a portion of his salary paid through his employer to serve as a consultant for Tricida (San Francisco, CA). None of the other authors have conflicts to disclose.
AUTHOR CONTRIBUTIONS
N.E.M. and D.E.W. conceived and designed research; N.G. and J.S. performed experiments; J.S., L.N.S., and A.M. analyzed data; N.G. and D.E.W. interpreted results of experiments; L.N.S. prepared figures; D.E.W. drafted manuscript; N.G., N.E.M., and D.E.W. edited and revised manuscript; N.G., A.M., N.E.M., and D.E.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the nursing and clerical staff of the Internal Medicine Clinic of the Department of Internal Medicine at Texas Tech University Health Sciences Center for their assistance and the Inside Out Community Outreach Program of Lubbock, TX, for making this study possible.
REFERENCES
- 1.Adeva MM, Souto G. Diet-induced metabolic acidosis. Clin Nutr 30: 416–421, 2011. doi: 10.1016/j.clnu.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 2.Adrogué HJ, Brensilver J, Cohen JJ, Madias NE. Influence of steady-state alterations in acid-base equilibrium on the fate of administered bicarbonate in the dog. J Clin Invest 71: 867–883, 1983. doi: 10.1172/JCI110841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Appel LJ, Wright JT Jr, Greene T, Kusek JW, Lewis JB, Wang X, Lipkowitz MS, Norris KC, Bakris GL, Rahman M, Contreras G, Rostand SG, Kopple JD, Gabbai FB, Schulman GI, Gassman JJ, Charleston J, Agodoa LY; African American Study of Kidney Disease and Hypertension Collaborative Research Group . Long-term effects of renin-angiotensin system-blocking therapy and a low blood pressure goal on progression of hypertensive chronic kidney disease in African Americans. Arch Intern Med 168: 832–839, 2008. doi: 10.1001/archinte.168.8.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banerjee T, Crews DC, Wesson DE, Tilea A, Saran R, Rios Burrows N, Williams DE, Powe NR; Centers for Disease Control and Prevention Chronic Kidney Disease Surveillance Team . Dietary acid load and chronic kidney disease among adults in the United States. BMC Nephrol 15: 137–149, 2014. doi: 10.1186/1471-2369-15-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banerjee T, Crews DC, Wesson DE, Tilea AM, Saran R, Ríos-Burrows N, Williams DE, Powe NR; Centers for Disease Control and Prevention Chronic Kidney Disease Surveillance Team . High dietary acid load predicts ESRD among US adults with CKD. J Am Soc Nephrol 26: 1693–1700, 2015. doi: 10.1681/ASN.2014040332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banerjee T, Crews DC, Wesson DE, McCulloch CE, Johansen KL, Saydah S, Rios Burrows N, Saran R, Gillespie B, Bragg-Gresham J, Powe NR. Elevated serum anion gap in adults with moderate chronic kidney disease increases risk for progression to end-stage renal disease. Am J Physiol Renal Physiol 316: F1244–F1253, 2019. doi: 10.1152/ajprenal.00496.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowe B, Xie Y, Li T, Mokdad AH, Xian H, Yan Y, Maddukuri G, Al-Aly Z. Changes in the US burden of chronic kidney disease from 2002 to 2016. An analysis of the global burden of disease study. JAMA Netw Open 1: e184412, 2018. doi: 10.1001/jamanetworkopen.2018.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bushinsky DA. Tolerance to sodium in patients with CKD-induced metabolic acidosis: Does the accompanying anion matter? Am J Kidney Dis 73: 858.–, 2018. doi: 10.1053/j.ajkd.2018.09.004. [DOI] [PubMed] [Google Scholar]
- 9.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr, Jones DW, Materson BJ, Oparil S, Wright JT Jr, Roccella EJ; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee . The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 289: 2560–2572, 2003. doi: 10.1001/jama.289.19.2560. [DOI] [PubMed] [Google Scholar]
- 10.Cunarro JA, Weiner MW. A comparison of methods for measuring urinary ammonium. Kidney Int 5: 303–305, 1974. doi: 10.1038/ki.1974.41. [DOI] [PubMed] [Google Scholar]
- 11.de Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol 20: 2075–2084, 2009. doi: 10.1681/ASN.2008111205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dubey AK, Sahoo J, Vairappan B, Haridasan S, Parameswaran S, Priyamvada PS. Correction of metabolic acidosis improves muscle mass and renal function in chronic kidney disease stages 3 and 4: a randomized controlled trial. Nephrol Dial Transplant. In press. doi: 10.1093/ndt/gfy214. [DOI] [PubMed] [Google Scholar]
- 13.Ebah LM, Wiig H, Dawidowska I, O’Toole C, Summers A, Nikam M, Jayanti A, Coupes B, Brenchley P, Mitra S. Subcutaneous interstitial pressure and volume characteristics in renal impairment associated with edema. Kidney Int 84: 980–988, 2013. doi: 10.1038/ki.2013.208. [DOI] [PubMed] [Google Scholar]
- 14.Erlandsen EJ, Randers E, Kristensen JH. Evaluation of the Dade Behring N Latex Cystatin C assay on the Dade Behring Nephelometer II System. Scand J Clin Lab Invest 59: 1–8, 1999. doi: 10.1080/00365519950185940. [DOI] [PubMed] [Google Scholar]
- 15.Garneata L, Stancu A, Dragomir D, Stefan G, Mircescu G. Ketoanalogue-supplemented vegetarian very low-protein diet and CKD progression. J Am Soc Nephrol 27: 2164–2176, 2016. doi: 10.1681/ASN.2015040369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goraya N, Simoni J, Jo C-H, Wesson DE. Dietary acid reduction with fruits and vegetables or sodium bicarbonate reduces kidney injury in subjects with moderately reduced GFR due to hypertensive nephropathy. Kidney Int 81: 86–93, 2012. doi: 10.1038/ki.2011.313. [DOI] [PubMed] [Google Scholar]
- 17.Goraya N, Simoni J, Jo CH, Wesson DE. A comparison of treating metabolic acidosis in CKD stage 4 hypertensive kidney disease with fruits and vegetables or sodium bicarbonate. Clin J Am Soc Nephrol 8: 371–381, 2013. doi: 10.2215/CJN.02430312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goraya N, Simoni J, Jo CH, Wesson DE. Treatment of metabolic acidosis in individuals with stage 3 CKD with fruits and vegetables or oral NaHCO3 reduces urine angiotensinogen and preserves GFR. Kidney Int 86: 1031–1038, 2014. doi: 10.1038/ki.2014.83. [DOI] [PubMed] [Google Scholar]
- 19.Goraya N, Simoni J, Sager LN, Pruszynski J, Wesson DE. Acid retention in chronic kidney disease is inversely related to GFR. Am J Physiol Renal Physiol 314: F985–F991, 2018. doi: 10.1152/ajprenal.00463.2017. [DOI] [PubMed] [Google Scholar]
- 20.Goraya N, Simoni J, Pruszynski J, Madias NE, Wesson DE. Urine citrate excretion as a marker of acid retention in patients with chronic kidney disease without overt metabolic acidosis. Kidney Int 95: 1190–1196, 2019. doi: 10.1016/j.kint.2018.11.033. [DOI] [PubMed] [Google Scholar]
- 21.Hamm LL, Simon EE. Roles and mechanisms of urinary buffer excretion. Am J Physiol 253: F595–F605, 1987. doi: 10.1152/ajprenal.1987.253.4.F595. [DOI] [PubMed] [Google Scholar]
- 22.Hsu CY, Lin F, Vittinghoff E, Shlipak MG. Racial differences in the progression from chronic renal insufficiency to end-stage renal disease in the United States. J Am Soc Nephrol 14: 2902–2907, 2003. doi: 10.1097/01.ASN.0000091586.46532.B4. [DOI] [PubMed] [Google Scholar]
- 23.Inker LA, Schmid CH, Tighiouart H, Eckfeldt JH, Feldman HI, Greene T, Kusek JW, Manzi J, Van Lente F, Zhang YL, Coresh J, Levey AS; CKD-EPI Investigators . Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med 367: 20–29, 2012. doi: 10.1056/NEJMoa1114248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kidney International Supplements . Chapter 3: management of progression and complications of CKD. Kidney Int Suppl 3: 73–90, 2013. doi: 10.1038/kisup.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klahr S, Levey AS, Beck GJ, Caggiula AW, Hunsicker L, Kusek JW, Striker G; Modification of Diet in Renal Disease Study Group . The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. N Engl J Med 330: 877–884, 1994. doi: 10.1056/NEJM199403313301301. [DOI] [PubMed] [Google Scholar]
- 26.Kurtz I, Maher T, Hulter HN, Schambelan M, Sebastian A. Effect of diet on plasma acid-base composition in normal humans. Kidney Int 24: 670–680, 1983. doi: 10.1038/ki.1983.210. [DOI] [PubMed] [Google Scholar]
- 27.Lemann J Jr, Lennon EJ, Goodman AD, Litzow JR, Relman AS. The net balance of acid in subjects given large loads of acid or alkali. J Clin Invest 44: 507–517, 1965. doi: 10.1172/JCI105164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemann J Jr, Litzow JR, Lennon EJ. The effects of chronic acid loads in normal man: further evidence for the participation of bone mineral in the defense against chronic metabolic acidosis. J Clin Invest 45: 1608–1614, 1966. doi: 10.1172/JCI105467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lemann J Jr, Bushinsky DA, Hamm LL. Bone buffering of acid and base in humans. Am J Physiol Renal Physiol 285: F811–F832, 2003. doi: 10.1152/ajprenal.00115.2003. [DOI] [PubMed] [Google Scholar]
- 30.Levey AS, Coresh J, Balk E, Kausz AT, Levin A, Steffes MW, Hogg RJ, Perrone RD, Lau J, Eknoyan G; National Kidney Foundation . National Kidney Foundation practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Ann Intern Med 139: 137–147, 2003. doi: 10.7326/0003-4819-139-2-200307150-00013. [DOI] [PubMed] [Google Scholar]
- 31.Loniewski I, Wesson DE. Bicarbonate therapy for prevention of chronic kidney disease progression. Kidney Int 85: 529–535, 2014. doi: 10.1038/ki.2013.401. [DOI] [PubMed] [Google Scholar]
- 32.MacClean AJ, Hayslett JP. Adaptive change in ammonia excretion in renal insufficiency. Kidney Int 17: 595–606, 1980. doi: 10.1038/ki.1980.70. [DOI] [PubMed] [Google Scholar]
- 33.Mahajan A, Simoni J, Sheather SJ, Broglio KR, Rajab MH, Wesson DE. Daily oral sodium bicarbonate preserves glomerular filtration rate by slowing its decline in early hypertensive nephropathy. Kidney Int 78: 303–309, 2010. doi: 10.1038/ki.2010.129. [DOI] [PubMed] [Google Scholar]
- 34.McCullough KP, Morgenstern H, Saran R, Herman WH, Robinson BM. Projecting ESRD incidence and prevalence in the United States through 2030. J Am Soc Nephrol 30: 127–135, 2019. doi: 10.1681/ASN.2018050531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phisitkul S, Khanna A, Simoni J, Broglio K, Sheather S, Rajab MH, Wesson DE. Amelioration of metabolic acidosis in patients with low GFR reduced kidney endothelin production and kidney injury, and better preserved GFR. Kidney Int 77: 617–623, 2010. doi: 10.1038/ki.2009.519. [DOI] [PubMed] [Google Scholar]
- 36.Regalado M, Yang S, Wesson DE. Cigarette smoking is associated with augmented progression of renal insufficiency in severe essential hypertension. Am J Kidney Dis 35: 687–694, 2000. doi: 10.1016/S0272-6386(00)70017-5. [DOI] [PubMed] [Google Scholar]
- 37.Remer T, Manz F. Potential renal acid load of foods and its influence on urine pH. J Am Diet Assoc 95: 791–797, 1995. doi: 10.1016/S0002-8223(95)00219-7. [DOI] [PubMed] [Google Scholar]
- 38.Remer T. Influence of nutrition on acid-base balance−metabolic aspects. Eur J Nutr 40: 214–220, 2001. doi: 10.1007/s394-001-8348-1. [DOI] [PubMed] [Google Scholar]
- 39.Stevens LA, Coresh J, Schmid CH, Feldman HI, Froissart M, Kusek J, Rossert J, Van Lente F, Bruce RD III, Zhang YL, Greene T, Levey AS. Estimating GFR using serum cystatin C alone and in combination with serum creatinine: a pooled analysis of 3,418 individuals with CKD. Am J Kidney Dis 51: 395–406, 2008. doi: 10.1053/j.ajkd.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.United States Renal Data System . USRDS 2017 Annual Data Report. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 2018. [Google Scholar]
- 41.Vallet M, Metzger M, Haymann J-P, Flamant M, Gauci C, Thervet E, Boffa J-J, Vrtovsnik F, Froissart M, Stengel B, Houillier P; NephroTest Cohort Study group . Urinary ammonia and long-term outcomes in chronic kidney disease. Kidney Int 88: 137–145, 2015. doi: 10.1038/ki.2015.52. [DOI] [PubMed] [Google Scholar]
- 42.Warmoth L, Regalado MM, Wesson DE, Simoni J, Harrist RB. Cigarette smoking enhances increased urine albumin excretion as a risk factor for glomerular filtration rate decline in primary hypertension. Am J Med Sci 330: 111–119, 2005. doi: 10.1097/00000441-200509000-00003. [DOI] [PubMed] [Google Scholar]
- 43.Wesson DE. Dietary HCO3 reduces distal tubule acidification by increasing cellular HCO3 secretion. Am J Physiol 271: F132–F142, 1996. doi: 10.1152/ajprenal.1996.271.1.F132. [DOI] [PubMed] [Google Scholar]
- 44.Wesson DE. Dietary acid increases blood and renal cortical acid content in rats. Am J Physiol 274: F97–F103, 1998. doi: 10.1152/ajprenal.1998.274.1.F97. [DOI] [PubMed] [Google Scholar]
- 45.Wesson DE, Simoni J. Increased tissue acid mediates a progressive decline in the glomerular filtration rate of animals with reduced nephron mass. Kidney Int 75: 929–935, 2009. doi: 10.1038/ki.2009.6. [DOI] [PubMed] [Google Scholar]
- 46.Wesson DE, Simoni J. Acid retention during kidney failure induces endothelin and aldosterone production which lead to progressive GFR decline, a situation ameliorated by alkali diet. Kidney Int 78: 1128–1135, 2010. doi: 10.1038/ki.2010.348. [DOI] [PubMed] [Google Scholar]
- 47.Wesson DE, Simoni J, Broglio K, Sheather S. Acid retention accompanies reduced GFR in humans and increases plasma levels of endothelin and aldosterone. Am J Physiol Renal Physiol 300: F830–F837, 2011. doi: 10.1152/ajprenal.00587.2010. [DOI] [PubMed] [Google Scholar]
- 48.Wesson DE, Jo C-H, Simoni J. Angiotensin II receptors mediate increased distal nephron acidification caused by acid retention. Kidney Int 82: 1184–1194, 2012. doi: 10.1038/ki.2012.267. [DOI] [PubMed] [Google Scholar]
- 49.Wesson DE, Jo CH, Simoni J. Angiotensin II-mediated GFR decline in subtotal nephrectomy is due to acid retention associated with reduced GFR. Nephrol Dial Transplant 30: 762–770, 2015. doi: 10.1093/ndt/gfu388. [DOI] [PubMed] [Google Scholar]
- 50.Wesson DE, Pruszynski J, Cai W, Simoni J. Acid retention with reduced glomerular filtration rate increases urine biomarkers of kidney and bone injury. Kidney Int 91: 914–927, 2017. doi: 10.1016/j.kint.2016.10.023. [DOI] [PubMed] [Google Scholar]