

Abstract
Oxidative stress and inflammation have long been recognized to contribute to Parkinson's disease (PD), a common movement disorder characterized by the selective loss of midbrain dopaminergic neurons (mDAn) of the substantia nigra pars compacta (SNpc). The causes and mechanisms still remain elusive, but a complex interplay between several genes and a number of interconnected environmental factors, are chiefly involved in mDAn demise, as they intersect the key cellular functions affected in PD, such as the inflammatory response, mitochondrial, lysosomal, proteosomal and autophagic functions. Nuclear factor erythroid 2 -like 2 (NFE2L2/Nrf2), the master regulator of cellular defense against oxidative stress and inflammation, and Wingless (Wnt)/β-catenin signaling cascade, a vital pathway for mDAn neurogenesis and neuroprotection, emerge as critical intertwinned actors in mDAn physiopathology, as a decline of an Nrf2/Wnt/β-catenin prosurvival axis with age underlying PD mutations and a variety of noxious environmental exposures drive PD neurodegeneration. Unexpectedly, astrocytes, the so-called “star-shaped” cells, harbouring an arsenal of “beneficial” and “harmful” molecules represent the turning point in the physiopathological and therapeutical scenario of PD. Fascinatingly, “astrocyte's fil rouge” brings back to Nrf2/Wnt resilience, as boosting the Nrf2/Wnt resilience program rejuvenates astrocytes, in turn (i) mitigating nigrostriatal degeneration of aged mice, (ii) reactivating neural stem progenitor cell proliferation and neuron differentiation in the brain and (iii) promoting a beneficial immunomodulation via bidirectional communication with mDAns. Then, through resilience of Nrf2/Wnt/β-catenin anti-ageing, prosurvival and proregenerative molecular programs, it seems possible to boost the inherent endogenous self-repair mechanisms. Here, the cellular and molecular aspects as well as the therapeutical options for rejuvenating glia-neuron dialogue will be discussed together with major glial-derived mechanisms and therapies that will be fundamental to the identification of novel diagnostic tools and treatments for neurodegenerative diseases (NDs), to fight ageing and nigrostriatal DAergic degeneration and promote functional recovery.
Keywords: Glia-neuron crosstalk, Parkinson's disease, Gene-environment interactions, Nrf2 signaling, Wnt signaling, Ageing, Oxidative/inflammatory stress, Astrocyte therapies
Graphical abstract
Astrocyte’s fil rouge” targeting Nrf2/Wnt resilience cascades rejuvenates glia-neuron crosstalk for neurorepair and regeneration in Parkinson’s disease.
Highlights
-
•
Nrf2/Wnt signalosome dysfunction mediates the effect of gene × environment interactions in PD.
-
•
Nrf2/Wnt resilience boosts anti-oxidant, anti-inflammatory and prosurvival astrocyte's properties.
-
•
Astrocytes focused therapies to fight ageing and PD and promote functional recovery.
1. Introduction
Oxidative stress and inflammation have long been recognized to contribute to Parkinson's disease (PD), the most prevalent age-dependent movement disorder and the second most common neurodegenerative disease (ND) [[1], [2], [3], [4], [5], [6], [7], [8]]. A first characteristic hallmark of PD is the selective and progressive loss of midbrain dopaminergic neurons (mDAn) of the substantia nigra pars compacta (SNpc), and their terminals in the striatum, responsible for the gradual impairment of motor function leading to the classical motor features of PD (i.e., bradykinesia, rest tremor, rigidity and postural instability) [[7], [8], [9]]. The second pathologic feature is the presence of cytoplasmic inclusions, called Lewy bodies (i.e. eosinophilic intracellular inclusions composed of amyloid-like fibers and α-synuclein), and distrophic neurites, called Lewy neurites, in the SN and other areas of the brain [9,10] (Fig. 1). Along with SNpc-mDAns, other neural populations of the central (CNS) and peripheral nervous systems (PNS) are affected in PD, including DAn of the enteric nervous system (ENS) [11,12]. Accordingly, a number of non-motor symptoms including, autonomic, sleep, cognitive, and mental health disorders, often precede and/or accompany PD onset and progression [13].
Fig. 1.

Nigrostriatal dopaminergic pathway and neuropathological hallmarks of Parkinson's disease (PD). A. In the left hand-side, a sagittal schematic view of nigrostriatal dopaminergic (DAergic) neurons originating (in red) in the subtantia nigra pars compacta (SNpc) of the mesesencephalon, and projecting to the corpus striatum (CPu), which includes the caudate and putamen nuclei. The major neuropathological hallmarks of PD are boxed on the right-hand side. B. Schematic drawing of coronal brain sections at the level of the striatum and SNpc showing the trajectory (in red) of the nigrostriatal DAergic pathway. C-D: Confocal laser scanning microcoscopic images of Cpu and SNpc in coronal brain sections stained with the dopamine marker tyrosine hydroxylase (TH, in green) in intact, saline-treated mice (C) and after exposure to the PD neurotoxin, MPTP (D). Note the severe loss of TH+ fibers in Cpu and of TH+ cell bodies in SNpc, occurring in MPTP-induced PD (D). Scale bars, panel C (Striatum: 25 μm; SNpc: 100 μm), panel D (Striatum: 25 μm; SNpc: 100 μm).
Unfortunately, by the time clinical manifestations appear, about 70% of the dopaminergic (DAergic) fibers in the caudate-putamen (CPu) and almost 50% of the mDAns in SNpc are already lost. Although slow in most cases, progression of the disease is irreversible and current therapies, such as L-3,4-dihydroxyphenylalanine (l-DOPA), are mainly directed towards replacing dopamine (DA) levels in the brain, and as such, provide only symptomatic relief [[14], [15], [16]]. These drugs do not modify the progressive neurodegenerative cell loss associated with PD that, in many cases, results in debilitating side-effects [[14], [15], [16]]. Thus, different lines of research are being pursued to develop novel therapeutic regimens for PD, including pharmacological, cellular and molecular therapies, aimed at protecting the dysfunctional mDAns and/or enhancing their intrinsic regenerative potential [[14], [15], [16]].
Yet, the causes and mechanisms of mDAn degeneration are not completely understood, but current evidence indicates that PD is a multifactorial disease, where a complex interplay between several genes and many environmental factors, especially ageing, oxidative stress and inflammation, contribute to mDAn demise [[14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26], [27]].
Notably, PD is the fastest growing neurological disorder in the world, and as Dorsey and collaborators recently highlighted “demography and by-products of industrialization have now created a Parkinson pandemic …“, with the number of individuals affected expected to growth exponentially from 6.9 million in 2015 to 14.2 million in 2040 [28]. Actually, with the emergence of the Coronavirus Disease-2019 (COVID-19) pandemia, continuing to spread around the world, these numbers are inevitably destined to increase, causing a most severe health care, social and economical burden. Especially, COVID-19 pandemia [29] intersect most critical environmental risk factors for PD and other NDs, namely, ageing, male gender and exacerbated inflammatory response (the so-called “cytokine storm”) [29,30], representing conditions recognized to drive and/or worsen Parkinson's symptoms, as a result of an harmfull impact of peripheral inflammatory cytokines and their crosstalk with brain macrophage/microglia and astrocytes, the key conspirators to mDAn death (reviewed in next sections).
Indeed, after the first compelling demonstration of the importance of glial reaction in PD by Mc Geer and coworkers [1], and during the last three decades, an increasing body of evidence, including work from our laboratory, underscored the pivotal role of astrocytes and microglia in the parkinsonian brain, as critical sources of oxidative and inflammatory mediators, documented in epidemiological, post-mortem, and animal studies [[31], [32], [33], [34], [35], [36], [37], [38], [39], [40], [41], [42], [43], [44], [45], [46]]. While the primum movens in the establishment of the neurodegenerative process is yet to be defined, oxidative stress exacerbation with the complicity of astrocyte and microglia activation in the presence of a PD-specific, genetic and environmental background, appear to trigger a complex series of events causing more reactive oxygen (ROS) and reactive nitrogen species (RNS) generation, in turn amplifying the production of a panel pro-inflammatory cytokines and chemokines, forming a vicious cycle finally leading to the progressive mDAn degeneration, as summarized in next sections. Within the long list of conspirators, both the innate and adaptive immune systems, with the contribution of both cellular and humoral peripheral immune responses appear to play unsuspected roles, as revealed by a long series of clinical research and disease models [[46], [47], [48], [49], [50]].
Increasing evidence implicates a dysfunction of nuclear factor erythroid 2 -like 2 (NFE2L2/Nrf2), a member of the cap ‘n’ collar subfamily of transcription factors, as an important contributor to neurodegeneration [[51], [52], [53]]. Nrf2 is the master regulator of cellular defense that controls the redox state and cell homeostasis coordinating the transcription of more than 200 cytoprotective genes. All these genes contain a common promoter enhancer called the antioxidant response element (ARE) and are transactivated by Nrf2. Nrf2-ARE axis can have an impact on numerous cell functions, ranging from cell differentiation and development to proliferation and inflammation, thus influencing neurodegenerative disease, cardiovascular disease, and cancer [51–53]. Importantly Nrf2 is central to mitochondrial function as it contributes to the overall mitochondrial homeostasis, impacting on mitochondrial membrane potential and respiration, oxidative phosphorylation and the synthesis of ATP, mitochondrial biogenesis and mitochondrial integrity [[54], [55], [56], [57]] (Fig. 2A). Notably, Nrf2 and mithochondrial dysfunction are pivotal for PD, as mDAns are particularly vulnerable to oxidative stress [5,6,8,57,58].
Fig. 2.

The Nrf2-ARE and Wnt/β-catenin/GSK-3β intertwined signaling cascades. A. In normal conditions, Nrf2 is inactive (Nrf2-Off”) and resides in the cytoplasm bound to Keap1. In response to oxidative stress and inflammation, the modification of Keap1 cysteine residues leads to inhibition of Nrf2 ubiquitylation and stabilization of Nrf2, allowing Nrf2 to accumulate in the cytosol and then to translocate into the nucleus where it binds to a small Maf protein and activates transcription of genes containing antioxidant response elements (AREs) in their regulatory regions (Nrf2-On”) [[76], [77], [78]]. In addition to its interaction with Nrf2, Keap1 also binds Cullin 3 (Cul3), which forms a core E3 ubiquitin ligase complex through an association with Ring-box1 protein (Rbx1, also called Roc1) [[76], [77], [78]]. Besides Keap1-mediated regulation, two other E3 ubiquitin ligases have been found to regulate the protein level of Nrf2. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by glycogen synthase kinase 3β (GSK-3β) activity phosphorylating a group of Ser residues in the Neh6 domain of Nrf2 [81, 82, see text]. B. In Wnt/β-catenin pathway, Wnt signal activation is tightly controlled by a dynamic signaling complex, constituted by class Frizzled (Fzd) of the G-protein-coupled receptor (GPCRs) superfamily, the LDL receptor-related protein (LRP) 5/6 coreceptors and Dishevelled (Dvl) and Axin adapters [75]. In the absence of a Wnt ligand, (Wnt-off) the signaling cascade is inhibited. Cytoplasmic β-catenin is phosphorylated and degraded via proteasome mediated destruction, which is controlled by the “destruction complex”, consisting of GSK3β, casein kinase 1α (CK1α), the scaffold protein AXIN, and the tumor suppressor adenomatous polyposis coli (APC) [75]. As a result, the translocation into nucleus is inhibited. Interruption of Wnt/β-catenin signaling also occurs in the presence of the Dkk’ and secreted FZD-related proteins (sFRPs) families of Wnt-antagonists, or Wnt inhibitory protein, WIF. Conversely, Wnt ligand binding to Fzd receptors at the surface of target cells (Wnt-on) triggers a chain of events aimed at disrupting the degradation complex via Dvl phosphorylation [75]. Then β-catenin is separated from the destruction complex, resulting in its accumulation and stabilization in the cytoplasm. Subsequently, β-catenin is imported into the nucleus where it can interact with the TCF/LEF family of transcription factors and recruit transcriptional co-activators, p300 and/or CBP (CREB-binding protein), as well as other components to transcribe a panel of downstream target genes. Conditions that can direct to Nrf2/Wnt-On (Nrf2-activators, GSK-3-antagonists, Wnt1-agonists.) or to Nrf2/Wnt-/Off (PD gene mutations, ageing, inflammation, environmental toxins.) are indicated. Because GSK-3β crosstalk with both Nrf-ARE and canonical Wnt-signaling, inhibition of GSK-3β activity by molecular compounds and various enzymes represents a potential means to activate the anti-oxidant, anti-inflammatory, prosurvial, neuroprotective and neurogenic downstream Nrf2/Wnt gene cascades (for details, see the text).
In this context, and within the ventral midbrain (VM), Nrf2-ARE axis restricted to astrocytes is sufficient to protect against neurotoxin-induced mDAn toxicity [59], whereas Nrf2-deficiency and alpha-Synuclein (α-Syn) expression [60], cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson's disease. Research from our laboratory put forward the hypothesis of a close collaboration between the Nrf2-ARE axis, glial inflammatory pathways and wingless-type MMTV integration site1 (Wnt1)/β-catenin signaling network [[61], [62], [63], [64], [65], [66], [67]]. Notably, the Wnt/β-catenin pathway is a vital cascade promoting mDAn neurogenesis [[68], [69], [70]], mDAn survival and immunomodulation, via bidirectional glia-neuron crosstalk [[61], [62], [63], [64], [65], [66],[71], [72], [73], [74]]. Especially, Nrf2 is implicated in the homeostatic regulation of neural stem progenitor cells (NSCs), both in health and physiopathological disease states, including ageing, inflammation and PD degeneration, via an intense crosstalk with the Wnt signaling pathway, recently reviewed by Marchetti and coworkers [73].
The hallmark of Wnt/β-catenin signaling after binding the Wnt's receptors, Frizzleds (Fzds), is the cytoplasmic accumulation of β-catenin and its nuclear translocation, finally activating the transcription of Wnt target genes involved in DAergic neurogenesis and neuroprotection [71]. β-catenin is tightly regulated via phosphorylation by the ‘destruction complex', consisting of glycogen synthase kinase 3β (GSK-3β), casein kinase 1α (CK1α), the scaffold protein Axin-1, and the tumour suppressor adenomatous polyposis coli (APC) [74] (Fig. 2B). In the absence of a Wnt ligand, the signaling cascade is inhibited as a result of GSK-3β-dependent β-catenin phosphorylation and degradation via proteasome-mediated destruction, which is controlled by the destruction complex (Fig. 2B). As a result, translocation into the nucleus is inhibited. Fascinatingly, in PD, the two major homeostatic systems, i.e.; the Nrf2-anti-oxidant/immuno-protective axis, and the Wnt/β-catenin pro-survival and pro-neurogenic signaling cascade appear intertwined, thus providing a robust self-defense resilience program to fight ageing and nigrostriatal DAergic degeneration, as summarized in the next sections.
Against this background, efforts are being pursued to “rejuvenate” neuron-glial dialogue during ageing, inflammation and basal ganglia injury which forms the focus of this work. After a summary of the principal actors in Nfr2/Wnt signaling dialogue, an overview of the impact of PD mutations intersecting critical glial and mDAn functions interacting with key environmental factors and impacting on oxidative stress, mitochondrial dysfunction, inflammation and Wnt signaling (i.e.,” the key interactors”), is presented. Next the dual harmful/beneficial role of astrocytes and microglia, their mediators and signaling mechanisms will be discussed in light of Nrf2/Wnt crosstalk, together with the therapeutical potential to switch the harmful glial phenotype by pharmacological and cellular therapies centered on glia as a means to promote neuroprotection and incite neurorestoration in the injured PD brain. Fascinatingly, “astrocyte's fil rouge” brings back to Nrf2/Wnt resilience, as a potential way to boost anti-oxidant, anti-ageing, self-protective and pro-regenerative programs for NDs.
2. The Nrf2-ARE/Wnt/β-catenin/GSK-3β intertwined signaling cascades: potential convergence check points for mDAn salvage in PD
Owing to their critical role in the safeguard of tissue and cell homeostasis against a panel of noxious stimuli, both Nrf2 and β-catenin transcriptional activity must be kept under a strict control within the cell cytoplasmic compartment, as a prolonged Nrf2 and/or Wnt signaling activation may lead to various detrimental effects. Accordingly, under basal conditions, both Nrf2 and β-catenin are subjected to ubiquitination and proteasomal degradation (Fig. 2A–B).
Regarding Nrf2, as a member of the basic leucine zipper (bZIP) family of transcription factors, its transcription is negatively regulated through binding to Kelch-like erythroid cell-derived protein with CNC association protein 1 (Keap1), a ubiquitin E3 ligase complex, which mediates Nrf2 degradation by the proteasome [[76], [77], [78]] (Fig. 2A). In normal conditions, Nrf2 is inactive and resides in the cytoplasm bound to Keap1. In response to oxidative stress and inflammation, the modification of Keap1 cysteine residues leads to inhibition of Nrf2 ubiquitylation and stabilization of Nrf2, allowing Nrf2 to accumulate in the cytosol and then to translocate into the nucleus where it binds to a small Maf protein and activates transcription of genes containing antioxidant response elements (AREs) in their regulatory regions [[76], [77], [78]] (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4628872/figure/F1/?report = objectionalFig. 2A). In addition to its interaction with Nrf2, Keap1 also binds Cullin 3 (Cul3), which forms a core E3 ubiquitin ligase complex through an association with Ring-box1 protein (Rbx1, also called Roc1) [[76], [77], [78]]. Besides Keap1-mediated regulation, two other E3 ubiquitin ligases have been found to regulate the protein level of Nrf2.
Notably, Rada and coworkers [79], firstly showed the ability of glycogen synthase kinase-3 (GSK3), a serine/threonine kinase, to phosphorylate a group of Ser residues in the Neh6 domain of mouse Nrf2, that overlap with an SCF/β-TrCP destruction motif (DSGIS, residues 334 to 338), then promoting its degradation in a Keap1-independent manner. Studies of Chowdhry and coworkers [80] further showed that Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity [81, 82, for review]. Another Keap1-independent component is Hrd1, an integral endoplasmic reticulum (ER) membrane E3 ligase, that negatively regulates Nrf2 [83]. Hrd1 is involved in endoplasmic reticulum (ER) stress degrading unfolded proteins that accumulates in the ER due to loss of function of Parkin, a causative factor in familial PD [83], and next section). Notably, mRNA and protein levels of Hrd1 are upregulated in response to ER stress, and Hrd1 ER stress-induced cell death [[83], [84], [85]]. Importantly, recent data implicate a close interactions between p62 (an autophagy adaptor protein) and Keap1, with dysregulation of autophagy promoting a prolonged Nrf2 activation in a p62-dependent fashion [86,87]. Especially, this interaction allows p62 to sequester Keap1 into the autophagosomes, which impairs the ubiquitylation of Nrf2, leading to activation of the Nrf2 signaling pathway [86,87].
Of specific mention, GSK3 is a multifunctional critical regulator of a panel of signaling pathways. GSK3 refers to two isoforms, GSK3α and GSK3β, that are primarily regulated by inhibitory phosphorylation on Ser21- GSK3α and Ser9GSK3β [88,89]. In Wnt canonical pathway, GSK-3β holds a pivotal position, where it phosphorylates β-catenin in concert with APC, Fzd, and axin, causing β-catenin degradation via the ubiquitin-proteasome pathway [71], leading to a “Wnt-Off “conditon (Fig. 2B). On the other hand, activation of canonical Wnt signaling, leads to GSK-3β down-regulation, and un-phosphorylated/activated β-catenin accumulates in the cytoplasm [75]. Subsequently, β-catenin is imported into the nucleus where it can interact with the TCF/LEF family of transcription factors and recruit transcriptional co-activators, p300 and/or CREB-binding protein (CBP) [75], as well as other components to transcribe a panel of downstream Wnt-target genes involved in cell proliferation, differentiation and survival, thereby promoting a “Wnt-On” condition [71] (Fig. 2B).
Converging evidence implicates GSK-3β as a key signaling molecule involved in the maintenance and function of adult mDA neurons [[88], [89], [90], [91], [92], [93], [94], [95]]. Additionally, GSK-3β activation plays a central role in regulating the neuroinflammatory and astroglial response to PD neurodegeneration [[62], [63], [64], [65],[88], [89], [90], [91], [92], [93], [94]]. Notably, GSK-3β plays a role in the phosphorylation of Tau (MAPT), triggering cytoskeleton destabilization, Tau aggregation and neuronal dysfunction/death [[92], [93], [94], [95]]. Because over-expression of GSK-3β promotes astroglial activation, astrocyte and microglia migration and increased expression of proinflammatory mediators, all these processes may impair neuron-glial and glial-NSC interactions leading to exacerbation of neuronal vulnerability/loss [[93], [94], [95]]. Therefore GSK-3β dysregulation in both neurons and glia represents a crucial vulnerability factor and a potential target for mitigating the progression of pathology of PD and other important NDs. For example, the studies of Duka and coworkers [90], using several PD experimental models, showed that α-Syn contributes to GSK3-β-catalyzed Tau phosphorylation and neuron death [90]; and Petit-Paitel et al. [91] studied the involvement of cytosolic and mitochondrial GSK3-β in mitochondrial neuronal dysfunction and cell death upon treatment with PD neurotoxins. Moreover, Creddle and associates [92] investigating both rodent and post-mortem human PD brains, clearly indicated GSK-3β dysregulation contributing to parkinson's-like pathophysiology and associated a region-specific phosphorylation and accumulation of Tau and α-syn, as a result of GSK-3 overactivation, causing neuronal death [92].
Given that the enzyme GSK-3β inhibits both Nrf-ARE and canonical Wnt-signaling, the inhibition of GSK-3β activity by molecular compounds and various enzymes represents a potential means to activate the anti-oxidant, anti-inflammatory, prosurvial and neurogenic downstream gene cascade (Fig. 2A–B). Accordingly, p-GSK-3β (tyr216) can phosphorylate Nrf2 to promote Nrf2 degradation by a kelch-like ECH-associated protein 1 (Keap1)-independent pathway [95]. Conversely, Wnt1 and Wnt1-like agonists such as Wnt-3a can stabilize Nrf2 by preventing its GSK-3-dependent phosphorylation and subsequent SCF/b-TrCP-dependent ubiquitination and proteasomal degradation [96].
In support of a critical functional role of a balanced GSK-3β activity in preclinical models of PD, its pharmacological inhibition, in vivo, by a chronic treatment with a specific GSK-3β antagonist, efficiently mitigated nigrostriatal dysfunctions, both at the SNpc and striatal levels, increased β-catanin gene expression and protein levels within the SNpc, and reverted the MPTP-induced motor dysfunction observed in ageing mice [61,62].
Then, GSK-3β appears as a pivotal kinase contributing to limit both Nrf2 and β-catenin transcriptional activity, whereas Wnt activation efficiently up-regulates the Nrf2-ARE and Wnt/β-catenin neuroprotective axis.
Additionally, GSK3β communicates with both the ERK/MAPK and the phosphatidylinositol 3-kinase/Akt (PI3K/AKT), also known as protein kinase B pathways, which have fundamental roles in mDAn death/survival [61,62,[97], [98], [99], [100]]. Notably, PI3K promotes the phosphorylation and activation of Akt. By making use of phosphorylating-dependent mechanisms, Akt can inhibit apoptosis induced by several stimuli in a multitude of cell types including mDAns [[97], [98], [99], [100]]. Of importance, the PI3K/Akt pathway mediates the effect of various neurotrophic and pro-neurogenic growth factors [96]. Indeed, trophic factors improve neuronal survival largely through PI3K/Akt signaling pathway, and after p-AKT activation, it can inhibit GSK-3β activation. Hence, PI-3K/Akt-mediated GSK-3β inhibition is in turn associated with the activation of cell adaptive and survival pathways in different types of cells, by contrast, GSK-3β activation by phosphorylation of the tyrosine 216 residue (p-Tyr216) located in the kinase domain, is implicated in oxidative stress induced neuronal cell death, including DAergic neuron death, and stem neuroprogenitor cell homeostasis (SNCs), being involved in NSC survival, proliferation and differentiation [73].
Hence, Wnt/β-catenin signaling activation by antagonizing active GSK-3β, can mediate neuroprotection and translate into improved neurological function during ageing, oxidant stress and inflammation and brain injury via Nrf2/PI3–K/Akt-Wnt/Fzd-1/β-catenin cooperation, and as part of a feedback loop regulating cellular homeostasis [61–67, 71–74,92-99, and section 5).
Last, but not least, epigenetic modifications are increasingly emerging as critical regulators of Nrf2-and Wnt-dependent signaling. The complex regulation of Nrf2-and Wnt/β-catenin signaling via epigenetic factors is out of the scope of this work, and these fields were recently reviewed [101,102] Actually, several epigenetic mechanisms including DNA methylation, covalent modification of histones in a promoter, or acetylation have been associated with Nrf2 epigenetic regulation [101]. Additionally, an increasing number of micro-RNA (miRNA) have been reported to both up-or-down regulate Nrf2 function [101 and Refs herein]. However, currently, it is not clearly established if Nrf2 decreased activity in several models results from disruption of epigenetic regulation, albeit evidences have been provided showing that changes in the levels or activity of principal Nrf2 negative regulators including Keap1, GSK-3β, and Hrd1, may impact on Nrf2 activity, thus contributing to the loss of Nrf2 function during ageing and/or inflammation [101].
Also, emerging evidence implicates several miRNAs in controlling Wnt/β-catenin signaling [102]. In a key finding, Anderegg and colleagues uncovered a regulatory circuit between LIM homeobox transcription factor 1-beta (LMX1B) and miR-135a2 that modulates Wnt1/Wnt signaling which in turn determines the size of the midbrain DAergic progenitor pool [103]. On the basis of bioinformatics and luciferase assay data, the authors suggested that miR-135a2 modulates LMX1B and many genes in the Wnt signaling pathway, with both miRNAs and Wnt-signaling pathways forming a network that is likely to play a significant role in adult neurogenesis and adult neuronal mDAns maintenance [103].
Altogether, molecular mechanisms of Nrf2-and Wnt/β-catenin/GSK-3β signaling regulation highlight an intense crosstalk. Importantly in PD, the significance of this circuitry is suggested, by both in vivo and in vitro model systems indicating an Nrf2/PI3–K/Akt-Wnt/Fzd-1/β-catenin cooperation in the regulation of mDAn homeostasis, immunomodulation, and neurogenesis, reviewed in next sections.
Nonetheless, the complexity of the Nrf2/Wnt signaling cascades clearly anticipates that the final outcome of activation is context-dependent, with different and sometimes opposing genetic programs depending on tissue/cellular specificity, the availability of receptor/co-receptors and signaling partners, pathological conditions, and the age of the host. Intuitively, due to the vital action of these signaling pathway in development, systems maintenance, redox homeostatic balance and immune regulation, their dysregulation may culminate in a broad range of diseases, including neurodegeneration and cancer [104,105].
3. Gene-environment interactions converge in the modulation of oxidative stress and inflammation: focus on Nrf2/Wnt/β-catenin interconnected pathways in PD
As recalled in the introduction, both genetic mutations and exposure to environmental risk factors are linked to Parkinson's disease, with approximately 10% of PD cases that can be directly attributed to genetic factors, associated with mutations in genes including α-synuclein (SNCA), E3 ubiquitin-protein ligase parkin (PRKN), ubiquitin C-terminal hydrolase L1 (UCHL1), PTEN-induced putative kinase (PINK1), deglycase gene DJ-1 (PARK7), leucine-rich-repeat kinase 2 (LRRK2), vacuolar protein sorting 35 homolog gene (VPS35), and β-glucocerebrosidase 1 (GBA1), linked to autosomal dominant late-onset [26]. In contrast, the etiology of the vast majority (up to 90%) of so called “idiopathic” cases, is multifactorial, likely arising from a combination of polygenic inheritance and environmental exposures (Fig. 3). Accumulating evidence indicates the expression of mutated genes, including SNCA, PRKN, PINK1, DJ-1, and LRRK2 in astrocytes and/or microglial cells and their implication in glial biology [22,106–112]. Importantly, the pathways regulated by these genes intersect the key cellular functions affected in Parkinson's disease, namely, the inflammatory response, endoplassmic reticulum (ER) stress, mitochondrial, lysosomal, proteosomal, autophagic and Wnt signaling functions [[106], [107], [108], [109], [110], [111], [112], [113], [114], [115], [116], [117], [118], [119], [120], [121], [122]]. Supporting evidence also come from genome-wide (GWA) and genome wide methylation data analysis, further suggesting that immune, mitochondrial and Wnt signaling pathways are associated not only with PD risk but also with PD progression [[123], [124], [125], [126], [127]] Strikingly, VPS35 gene located at 16q13-q21 chromosomal position and the two pathways, the Wnt signaling pathway, and retromer-mediated DMT1 missorting are proposed for the basis of VPS35 related PD [117,118].
Fig. 3.

NRf2/Wnt/β-catenin interconnected pathways and gene-environment interactions in Parkinson's disease (PD). Scheme of the reciprocal gene-environment interactions impacting on nuclear factor erythroid 2 -like 2 (NFE2L2/Nrf2) and Wnt/β-catenin signaling cascades in PD. The expression of SNCA, PRKN, PINK1, DJ-1, and LRRK2 in astrocytes and microglial cells affect the inflammatory response, endoplassmic reticulum (ER) stress, mitochondrial, lysosomal, ubiquitin-proteasome system (UPS), autophagic and Wnt signaling functions. Genetic mutations powerfully interact with a panel of environmental factors, including ageing, neurotoxic exposures (i.e., rotenone, paraquat, MPTP, drugs of abuse), the hormonal background (the stress and reproductive hormones) and life style. Central to the dopaminotoxic cascades, is the dysfunction of Nf2/Wnt signaling axis, critically involved in providing anti-oxidant and anti-inflammatory self-defenses, and promoting the survival and protection of the vulnerable midbrain dopamine neurons (mDAns) via bidirectional astrocyte-neuron crosstalk. In light of the intrinsic vulnerability of mDAns as a result of DA oxidative metabolism associated to the specific microglial environment within the SNpc, a combination of genetic and environmental factors, leading to astrocyte and microglia overactivation, and consequent generation of a panel of cytotoxic mediators, further exacerbates inflammation and oxidative stress. PD mutations via their impact in astrocyte and microglia cells biology, their inter-relations with mitochondrial Nrf2 and Wnt/β-catenin/GSK-β signaling predispose the brain to reach a critical threshold of inflammation and mitochondrial dysfunction, in turn acting as a driving force to exacerbate the progression of inflammation-mediated neurodegeneration of PD.
Here, we summarize the glial-specific functional consequences of the genetic mutations linked to PD and highlight immune, mitochondrial and Wnt/β-catenin interconnected pathways (Fig. 3).
3.1. Impact of genetic mutations on glial immune and mitochondrial functions via Nrf2/Wnt signaling cascades
Both earlier and more recent studies reported the harmful/beneficial consequences of gain (GOF) or loss-of-function (LOF) mutations, as well as their interactions with the ageing process, oxidative stress and inflammation. Alpha synuclein (α-Syn) is a central player in the pathogenesis of sporadic and familial PD [41,43,[45], [46], [47]]. The aggregation of α-Syn and oxidative stress are associated and enhance each other's toxicity [54]. Hence, dysfunctional α-Syn coupled to a proinflammatory, called “M1” microglial phenotype, can potentiate each other and promote the progression of mDAn death [32–35, 41, 43, 45–47). Additionally, high levels of exogenous α-Syn can initiate a Toll like receptor 4 (TLR4) signaling cascade in astrocytes [[128], [129], [130]]. Notably, impairment of the autophagy-lysosome pathway is implicated with the changes in α-synuclein and mitochondrial dysfunction observed in Parkinson's disease (PD). Damaged mitochondria accumulate PINK1, which then recruits parkin, resulting in ubiquitination of mitochondrial proteins. Accordingly, recent evidence linking α-synuclein and mitochondrial dysfunction to inflammation and PD neurodegeneration [132], supports the notion of a critical dysfunction of the astroglial cell compartment, preceding and/or contributing to PD neurodegeneration, with the important contribution of a failure of the Nrf2, which is intimately linked to mitochondrial biogenesis and the autophagy-lysosome pathway [131, 132 and next sections). Also, the genetic evidence suggests that α-Syn can synergistically interact with Wnt/β-catenin pathway components, such as GSK-3β, and together with microtubule-associated protein (MAPT) Tau, may drive neurodegeneration [[90], [91], [92]].
Reportedly, on the one hand, activation/overexpression of GSK-3β present in PD [[92], [93], [94],121], has a role in the phosphorylation of Tau, triggering cytoskeleton destabilization, Tau aggregation and neuronal dysfunction or death. Given, that GSK-3 activation in astrocytes and microglial cells promotes the expression of a panel of harmful proinflammatory markers, this may establish a feedforward cycle of inflammation-dependent neuronal death (Fig. 3).
Recently, in the study of Duffy and coworkers [45], the authors investigted the temporal relationship of neuroinflammation in a model of synucleinopathy following intrastriatal injection of pre-formed alpha-synuclein fibrils (α-Syn PFFS). Importantly, by systematically investigating the temporal profile of Lewy body-like phosphorylated α-Syn inclusion load, reactive microglial morphology, MHC-II antigen presentation, and degeneration in the SN, it was shown that reactive microglia and increased microglial MHC-II expression in association with peak load of α-Syn PFFS in SNpc, months prior to degeneration, thereby supporting the concept that neuroinflammation may precede and then contribute to nigrostriatal degeneration [45]. Castro-Sanchez and coworkers [131] recently analyzed whether overexpression of wild-type α-Syn (α-SynWT) or mutated α-Syn (α-SYNA53T) contributed to the neuronal dopaminergic loss and inflammation process, and studied the role of the chemokine fractalkine (CX3CL1) and its receptor (CX3CR1). Using either in vivo murine models overexpressing human α-SynWT or α-SynA53T in wild type (Cx3cr1+/+) or deficient (Cx3cr1−/−) mice for CX3CR1, coupled to unilateral intracerebral injection of adeno-associated viral vectors, the authors identified microglia CX3CR1 as a critical factor in the modulation of microglial dynamics in response to α-SYNWT or α-SYNA53T, indicating that CX3CR1 plays an essential role in neuroinflammation induced by α-SYNA53T [131]. Of note, not only too much α-Syn, but also too little may dramatically impact on inflammation and nigral degeneration, as silencing α-Syn in mature mDAns can promote a rapid neuroinflammation and subsequent DAergic toxicity [132].
Regarding parkin, the earlier studies of Frank-Cannon et al. [133], showed that PRKN gene deficiency increases the vulnerability of mDAn to various risk factors including inflammation-dependent degeneration. In PRKN deficient mice, Solano and co. [134−136] reported also an increased astrocyte vulnerability to death when challenged by various oxidative stress insults, including H2O2-induced stress, which resulted in abrogation of astrocyte'ability to exert neuroprotective functions. Additionally, in the presence of microglia, rotenone-induced dopamine cell loss of PRKN-KO midbrain neuronal cultures was sharply increased [[134], [135], [136]]. Conversely, over-expression of PRKN protected from excitotoxicity induced by the exitotoxin, kainic acid, thereby demonstrating a critical role for PRKN in the response of glial cells to noxious stimuli [[134], [135], [136]]. Supporting evidence on the role of PRKN in astroglial functionality come from recent studies [137], showing that parkin may regulate astrocyte ER stress and inflammation to control neuronal homeostasis, via modulation of NOD2, nucleotide-oligomerization domain receptor 2 (NOD2), a cytosolic receptor integrating ER stress and inflammation [137]. Notably, PRKN can also regulate Wnt signaling [138]. Hence, in conditon of excessive Wnt signaling, PRKN protect mDAn against β-catenin-induced cell death [138].
PINK1 encodes a highly conserved, 581-amino acid, putative serine-threonine protein kinase that modulates mitochondrial network homeostasis and quality control [139]. Both PRKN and PINK1 orchestrate a protective mitophagic response that ensures the safe disposal of damaged mitochondria. PINK1 phosphorylates ubiquitin (Ub) at the conserved residue S65, in addition to modifying the E3 ubiquitin ligase, PRKN [140]. Given the pivotal role of Nrf2 in mitochondrial function, PINK1 and Nrf2 signaling pathways are believed to cooperate to control mitochondrial homeostasis [141]. Glial PINK1 is critical for the long-term survival of mDAn, as primary astrocytes derived from PINK1–KO mice have increased pro-inflammatory cytokines and higher nitric oxide production upon stimulation of the innate immune response with lipopolysaccharide (LPS) plus interferon-γ [142]. Furthermore, reduced expression of the anti-inflammatory cytokine interleukin-10 (IL-10) from primary microglia derived from PINK1–KO mice was detected as compared to WT [142]. This suggests that PINK1 deficiency alters oxidative stress and inflammatory gene expression in both astrocytes and microglia, either directly or indirectly via cytokine signaling from other cells. Other studies have shown that PINK1 deficiency impairs both the formation of GFAP+- astrocytes during development and the proliferation of astrocytes upon stimulation with epidermal growth factor (EGF) or fetal bovine serum [143]. Recently, Barodia et al. [108] found that PINK1-dependent ubiquitin phosphorylation is predominantly in astrocytes as compared to neuronal and other non-neuronal cell types, supporting the contribution astrocyte dysfunction to PD pathogenesis.
Owing to the relation of mitochondrial function to Wnt signaling [74,[144], [145], [146], [147], [148]], Pink-1 is also linked to Wnt, as Wnt2 overexpression protects against PINK1 mutant-induced mitochondrial dysfunction and oxidative stress [149]. Hence, in PINK-1B9 transgenic flies, which is a PD model, Xia and co [149] recently reported that overexpression of Wnt2 reduced the abnormality rate of PD transgenic Drosophila and improved their flight ability, while other intervention groups had no significant effect. Wnt2 normalized mitochondrial morphology, and increased the mRNA expression levels of NADH-ubiquinone oxidoreductase chain 1 (ND1), ND42, ND75, succinate dehydrogenase complex subunits B, Cytochrome b and Cyclooxygenase 1, which are associated with Wnt2 overexpression [149]. Moreover, overexpression of Wnt2 in PD transgenic Drosophila resulted in the downregulation of ROS and malondialdehyde production, increased manganese superoxide dismutase (MnSOD), as well as the expression levels of PPARG coactivator 1α (PGC-1α) and forkhead box sub-group O (FOXO), suggesting that Wnt2 overexpression may be related to the PGC-1α/FOXO/MnSOD signaling pathway in PINK1 mutant transgenic Drosophila [149].
Interestingly, TOMM40, a mitochondrial translocase that resides between the putative transmembrane domain and the mitochondrial targeting sequence, is required for PINK1-induced localization in the mitochondria, and its phosphorylation of critical serines in ubiquitin results in PRKN recruitment, which then leads to mitophagy [150]. Therefore, dysfunction of PINK1 causes defects in its localization as well as impaired mitophagy. DJ-1 has a recognized role for the maintenance of astrocytic mitochondrial functions and the regulation of oxidative stress and inflammatory pathways [110, 111, 151,152]. Hence, DJ-1 deficiency impairs astrocyte ability to protect DAergic neurons against rotenone [153] and 6-OHDA [154], and selectively enhances mitochondrial Complex I inhibitor-induced neurotoxicity [155]. Opposedly, astrocytic over-expression of DJ-1, in vitro, prevented oxidative stress and mitochondrial dysfunction in primary neurons [156]. Further studies of De Miranda and co [157] showed that astrocyte-specific DJ-1 overexpression, protected against rotenone-induced neurotoxicity in a rat model of Parkinson's disease, thus providing the first direct evidence of a cell non-autonomous protective function of astrocyte DJ-1 in vivo [157].
Another important connection is the one between LRRK2-G2019S (LRRK2-GS), a pathogenic mutation in the PD-associated gene LRRK2, biochemically linked to the intertwined pathways regulating inflammation, mitochondrial function, and autophagy/lysosomal function [[158], [159], [160]]. Here LRRK2-GS and the activation of M1 proinflammatory phenotype [109], act in synergy to amplify dopaminergic neurotoxicity. By contrast, when LRRK2 is inhibited, this in turn reduces the production of microglial harmful mediators and reverses dopaminergic neurotoxicity [161,162].
Notably, a reciprocal LRRK2-Wnt signaling dialogue do occurs, as (i) LRRK2 interacts with proteins of Wnt signalosome [115]; (ii) LRRK2 is recruited to membranes following Wnt stimulation, where (iii) it binds to the Wnt co-receptor LRP6 in cellular models [115]. Of specific interest, pathogenic LRRK2 mutations disrupted Wnt signaling, implicating binding to LRP6-mediated Wnt signaling caused by reduced binding to LRP6 as a potential factor underlying neurodegeneration observed in PD [116]. On the other hand, the protective LRRK2 R1398H variant enhanced GTPase and Wnt signaling activity [119], underlying the complexity of LRRK2/Wnt signaling cross-talk in PD [113]. Likewise, pivotal PD mutations were demonstrated to negatively affect Wnt/β-catenin signaling and to inhibit human induced pluripotent stem cells (iPSCs)’ ability to differentiate into DAergic neurons [120], whereas pharmacological Wnt activation restored their dopaminergic developmental potential [120], thus supporting a robust link between PD mutations and downregulated Wnt/β-catenin signaling.
The ER, a subcellular site of protein folding and maturation, and the main intracellular Ca2+ store of the cell, is another critical link between the dysfunction astrocyte-neuron interactions and increased neuronal vulnerability. Hence, recent studies of Lee et al. [163], indicated that in astrocytes, LRRK2-GS impairs ER Ca2+ homeostasis, which determines cell survival, and, as a result, could contribute to the development of the disease [163]. Also perturbations of lysosome function can result in dysfunctional astroglial biology, as expression of LRRK2-G2019S in astrocytes produced enlarged lysosomes and diminished the lysosomal capacity of these cells, whereas selective LRRK2 kinase inhibitor can correct defects in lysosome function associated with LRRK2 mutations, highlighting the therapeutic potential of LRRK2 kinase inhibitors in the treatment of PD [163].
Together, PD mutations via their impact in astrocyte and microglia cells biology, their inter-relations with mitochondrial/Nrf2 and Wnt/β-catenin/GSK-β signaling may well predispose the brain to reach a critical threshold of inflammation and mitochondrial dysfunction, in turn acting as a driving force to exacerbate the progression of inflammation-mediated neurodegeneration (Fig. 3).
3.1.1. Environmental risk factors cooperate to exacerbate glial dysfunction in PD: an Nrf2/Wnt “liaison” in PD?
3.1.1.1. Ageing and the Nrf2/Wnt-immune link in PD
Ageing, interacting with a myriad of environmental noxious factors, represents a most crucial event, linking increased inflammation and oxidative stress to mitochondrial deficits and dysregulation of lysosomal, proteosomal and autophagic functions, robustely contributing to the chronic mDAn deterioration in the PD brain [[164], [165], [166], [167], [168], [169], [170]]. Notably, ageing is characterized by a loss of homeostatic mechanisms, as underscored by Viña and co-workers, when a disbalance in these mechanisms leads to the development of “frailty”, i.e., an increased vulnerability to a panel of noxious events [171, 172 and Refs herein). Importantly, “the free radical theory of frailty (revised by the authors) “proposes that oxidative damage is associated with frailty, but not with chronological age itself [[172], [173], [174]]. Notably, “frailty”, considered one of the major geriatric syndromes, robustely impact on anti-oxidant self-defense and inflammatory homeostasis [175]. Here we focus on a Nrf2/Wnt signaling failure and its link to inflammation, with consequences for neuron–glia crosstalk, mDA neuron plasticity and repair.
In fact, with advancing age, the nigrostriatal DAergic system progressively declines and the “adaptive” or compensatory capacity of mDAns gradually fails, thus rendering mDAn more “frail” or vulnerable/susceptible to both endogenous and exogenous noxious stimuli, likely contributing to the slow nigrostriatal degeneration of PD, with the late appearance of clinical signs [[176], [177], [178], [179], [180], [181], [182], [183], [184], [185], [186], [187], [188], [189]]. An increasing body of earlier and more recent evidence suggests a prominent role of astrocytes and microglia as main players in mediating the harmful effects of ageing interacting with a specific genetic background and different environmental factors.
Notably, oxidative stress and low-grade inflammation are the hallmarks of ageing, and both processes are even further up-regulated upon injury, neurotoxin exposure, male gender and PD genetic mutations. With age, microglial cells become “primed”, i.e. capable to produce exacerbated levels of a set of pro-inflammatory mediators when challenged with immune or neurotoxic stimuli [169,[181], [182], [183], [184], [185], [186], [187], [188], [189]]. This microglial cell shift to the harmful, M1 phenotype promotes the release of an array of factors that are detrimental for the vulnerable mDAns. Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-ĸB, a protein complex that controls cytokine production and cell survival), is a key actor and the first signal for inflammasome induction [189], together with major pro-inflammatory cytokines, such as tumor necrosis factor α (TNF-α), interleukin 1β (IL-1β) and IL-6 [169]. This inflammatory microenvironment associates with enhanced generation ROS and RNS, that in turn amplifies microglial activation, which results in increased mDAn vulnerability, and/or neuronal death [93,187,188].
The Wnt/β-catenin signaling pathway is of utmost importance owing to its ability to promote tissue repair and regeneration of stem cell activity in diverse organs, and in light of its crucial role in age-related pathogenesis and therapy of disease [71,74, [188], [189], [190], [191], [192]], as the harmful proinflammatory milieu critically intersects the age-dependent decline of Wnts and their inefficient signaling mechanisms in a wide number of tissues and cells [65,72,73], resulting in a further “frailty” factor, thus increasing the vulnerasbility to a panel of noxious challenges, finally leading to a failure to orchestrate a self-protective and reparative program. Hence, with the ageing process, endogenous astrocyte-derived Wnts decline in the hippocampus [[188], [189], [190], [191]], striatum and mesencephalon (64–66), whereas the expression of endogenous antagonists of Wnt/β-catenin signaling, including Dickkopf1 (Dkk1) and GSK-3β, is up-regulated thereby contributing to the reduced neuronal survival and neurorepair capacity, and to the marked impairment of neurogenesis, linking the decline of Wnts to the failure of midbrain dopaminergic neurons to compensate or to adapt to injury [73,74].
As reviewed in this work, two critical risk factors, ageing and MPTP exposure, promoted a long-lasting decrease of Wnt/β-catenin signaling components accompanied by upregulation of active GSK-3β, likely contributing to a chronic proinflammatory status, underlying increased DAergic neuron vulnerability.
Furthermore, age-dependent environmental stressors appear to lead to epigenetic DNA modifications, which cause aberrant gene expression. Specifically, loss of DNA methylation in the promoter of Keap1 gene decreases Nrf2-dependent antioxidant protection and results in a redox imbalance altered towards oxidation [101].
3.1.2. Failure of anti-oxidant self-defenses is directed by Nrf2/Wnt defeat in PD
In fact, in addition to the decline of Wnt/β-catenin pathway, a major self-adaptive system, the Nrf2-ARE-axis decline with age and neurodegeneration. In fact, the response of antioxidants to oxidative stress is a primary defense mechanism to combat noxious effects of oxidative insults and Nrf2 is the master regulator of the oxidative stress response due to its ability to induce the transcription of antioxidant, anti-inflammatory and phase 2 proteins, such as heme oxygenase (HO1), NAD(P)H quinone oxidoreductase (NQO1), superoxide dismutases (SOD1, SOD2), glutathione S-transferase (GST), glutathione peroxidase (GPx), glutathione reductase (GR) and catalase (CAT), which together are capable to regulate the cellular redox state by decreasing ROS levels [59,60,[193], [194], [195]]. Notably, oxidative stress can up-regulate the rate-limiting enzyme in GSH production (i.e. glutamate cysteine ligase) and also increases the expression and membrane targeting of multidrug-resistance associated protein-1 (MRP1) export pump, thus facilitating the efflux of GSH from astrocytes, which promotes a robust protective response to the changing redox milieu [195]. Importantly, the accelerated ageing phenotype in Sod1−/− mice is correlated to increased cellular senescence associated with high levels of circulating proinflammatory cytokines, IL-1β and IL-6, as compared to Wt mice, suggesting that the accelerated ageing phenotype shown by the Sod1−/− mice could result from increased inflammation arising from an accelerated accumulation of senescent cells, thereby supporting increased inflammation and increased pathology as key features of ageing [196]. Of significance, Sod1−/− mice are more frail than controls, whereas protection against oxidative damage by overexpression of antioxidant enzymes, delays the onset of frailty, resulting in beneficial outcomes [172].
Reportedly, in PD patients, the genetic associations show that a functional haplotype in the human NFE2L2 gene promoter, which confers a certain increase in transcriptional activity, is associated with decreased risk and delayed onset of PD [197]. Significantly, the Nrf2 signature in PD patient brain, represented by expression of NQO1, and HO-1 is up-regulated, suggesting a likely effort to activate the self-defense Nrf2-ARE axis to combat oxidative stress exacerbation [198 and section 6].
Within this scenario, a key feature of astrocyte neuroprotective properties, is the activation of the Nrf2-ARE anti-oxidant self-defense program, but this response is sharply impaired with age. Indeed, DA oxidative metabolism represents a vulnerability factor for PD pathogenesis [8], whereby astrocytes play a critical antioxidant self-protective role. In fact, while oxidative stress can up-regulate the expression of astrocytic NF-E2-related factor 2 (Nrf2), which translocates to the nucleus and binds to anti-oxidant responsive elements, ARE, this response declines with age. Together, these functions are critical for mDAn, which are known to be particularly vulnerable to oxidative damage [8,[199], [200], [201]]. Then, in addition to the Wnt/β-catenin signaling failure, also the Nrf2-antioxidant axis fails, leading to accumulation of ROS/RNS, and oxidative stress, which is either causally linked or associated with numerous health problems including diabetes, cardiovascular disease, neurodegenerative conditions (Alzheimer's, Parkinson's, and Huntington), thus targeting Nrf2 has been suggested as a promising therapeutical avenue in neurodegeneration [[51], [52], [53]]. As recalled in section 2, Nrf2-deficiency is implicated in regulating proteasome activity via Nrf2-induced regulation of autophagosome cargo protein p62 [86,87], the defeat of the Nrf2/ARE axis also importantly contributes to the overall dysregulation of proteosomal and autophagy functions observed in PD [197].
Of note, ageing-induced decline of astrocytic Nrf2-ARE axis promotes an up-regulated expression of major microglial proinflammatory cytokines, such as TNF-α, IL1β, IL-6 and Nos2 both at striatal [64] and SNpc [61,72,74], levels, in face of a down-regulation of the anti-inflammatory IL-4 and IL-10, thus exacerbating oxidative stress and inflammation with harmful consequences for DAergic neuronal survival (reviewed in next sections).
3.1.3. Environmental exposures, life style and the Nrf2/Wnt-immune link in PD
Following the first and most highly compelling evidence revealing a profound parkinsonian syndrome after intravenous use of street preparations of meperidine analogues contaminated with MPTP, recently reviewed by Langston [24], an increasing number of environmental neurotoxins has been shown to affect astrocyte and microglial functions, exacerbating oxidative stress and the production of proinflammatory mediators, via down-regulation of Nrf2/Wnt signaling axis, thus contributing to DAergic degeneration [[17], [18], [19], [20], [21], [22], [23], [24],40,196,[199], [200], [201], [202], [203]]. Furthermore, life style, especially physical activity, but also dietary factors, alcohol or drug of abuse consumption, in both pre-natal or post-natal life, are recognized to influence idiopathic PD, as an harmful “exposome” markedly impacts in Nrf2/Wnt signaling network (Fig. 2).
Various toxicants such as herbicides and pesticides, related to rural living/occupation in agriculture, implicated as risk factors in PD, as well as certain drugs can affect glial cell function and many of these compounds recapitulate PD pathology in animal models [[204], [205], [206]]. Remarkably, environmental PD toxins and pesticides downregulate the Nrf2/Wnt/β-catenin signaling cascade in rodent, non human primate and human PD, corroborating dysfunctional Wnt/β-catenin signaling in PD physiopathology [71].
Recently, the herbicide paraquat (PQ) was found to induce astrocytic senescence and pro-inflammatory senescence-associated secretory phenotype (SASP), capable of damaging neighboring cells [207]. By contrast, senescent cell depletion can protect against PQ-induced neuropathology [207]. These data suggest that exposure to certain environmental toxins promotes accumulation of senescent cells in the ageing brain, which can contribute to dopaminergic neurodegeneration [207].
Also the hormonal background plays an additional modulatory role in PD physiopathology. Hence, early-life stress is a risk factor for later life development of PD. Due to the physiological vital role of the hypothalamic-hypophyseal-adrenocortical axis (HPA) and glucocorticoids (GCs) in restraining inflammation and oxidative stress, glucocorticoid receptor (GR) deficient astrocytes and GR-deficient microglia of transgenic mice bearing from early embryonic life a GR-antisense RNA [208], fail to protect mDAn when exposed to an immunologic or neurotoxic challenge, due to the blockade of the GR-mediated down-modulation of oxidative stress and inflammation, resulting in exacerbated ROS and RNS [209]. Here the altered crosstalk between GR and oxidative and inflammatory signaling pathways (i.e. GR-bound GCs-iNOS/NO crosstalk) promotes increased mDAn vulnerability and mDAn death [[209], [210], [211]]. One of the strongest identified risk factors for development of PD is male gender. The prevalence of PD in males is higher in most populations studied, and data on disease incidence suggest that men have at least a two-fold greater risk of PD at all ages [see 212–214 and Refs herein]. Interestingly, looking at gene expression profiling in SN of female and male post mortem PD brain, Cantuti-Castelvetri [212] documented that genes upregulated in females relative to males are mainly involved in signal transduction and neuronal maturation, protein kinase activity, and Wnt signaling pathway [212]. Importantly, sex steroids, particularly estrogens, are important in protecting midbrain astrocytes and dopaminergic neurons against oxidative and inflammatory insult, whereas their abrogation in estrogen-deprived mice, exacerbates the vulnerability of dopaminergic neurons, via a dysfunctional astrocyte-microglia-neuron crosstalk [[213], [214], [215]]. Of note, gender differences are also present in the sensitivity to most environmental PD neurotoxin including rotenone and MPTP. Hence, females displayed significant loss of DAergic neurons in the substantia nigra and less inflammation when exposed to MPTP [[213], [214], [215], [216]] or rotenone [217]. Also at higher doses of the neurotoxin, females did show less inflammation, and less accumulation of α-synuclein and transferrin, possibly as a result of preserved autophagy [217].
Thus, genetic factors interacting with early life events such as exposure to hormones, endotoxins or neurotoxicants, modifying astroglial functions, may finally influence disease predisposition and/or severity [17,[218], [219], [220]]. As a consequence, an altered dialogue between the neuroendocrine and the immune systems via the HPA and reproductive axes, during development, may irreversibly shape glial cells and «program » long-term effects in the mechanisms regulating immune responsiveness to inflammation and oxidative stress [17]. In this connection, and most interestingly, circadian rhythms and clock genes significantly impact on oxidative stress, thus playing roles on critical homeostaic mechanisms, regulating lifespan, neurodegeneration, and cancer, possibly via major adaptive pathways, including the Nrf2-ARE and Wnt/β-catenin pathway, to foster cell survival during injury or block tumor cell growth [100].
Last, but not least, life style, especially physical activity, but also dietary factors, alcohol or drug consumption, besides others, are well known to influence idiopathic PD and also impact in Nrf2/Wnt signaling. Regarding physical activity, the hypothesis that exercising promotes health and longevity is well recognized by earlier and more recent studies as reviewed by Vina and coworkers [[171], [172], [173], [174]]. Hence, the effects of exercise in health, have been reported for several physiopathological conditions including ageing, osteoporosis, diabetes, depression, atherosclerosis and PD [[173], [174], [175],[221], [222], [223], [224], [225], [226], [227], [228]]. Given its role in reducing ageing-associated “frailty”, exercise has been proposed to be considered as an important supplement to other treatments for improving healthy ageing [173,174], having also beneficial effects in modifying the harmful neuroimmune responses [175]. Currently, exercise is increasingly being considered to be a complementary strategy to PD medications [see 224]. Notably, physical activity appears particularly important to combat mitochondrial alterations and oxidative stress contributing to PD progression, and also to reduce the risk of PD, having positive impacts on both motor and nonmotor symptoms of PD [229,230]. Importantly, emerging data increasingly link Nrf2-mediated redox adaptations to beneficial effects of exercise, with this response being significantly impaired with age [[223], [231], [232]]. Especially, physical activity during/following exercise affect the Wnt signaling path of the locomotor system [233].
Notably, exercise activates Nrf2 antioxidant system to protect the nigrostriatal DAergic neurons from MPP+ toxicity [225]. Hence, while the neurotoxin MPP + induced early decreases in total glutathione level and Nrf2/γGCLC (catalytic subunit of γGCL) expression, treadmill exercise for 4 weeks induced upregulation of Nrf2 and γGCLC expression, and also prevented the MPP+-induced nigrostriatal DAergic degeneration. Accordingly, the protective effect of exercise was blocked by the knockdown of Nrf2 using a lentivirus-carried shNrf2 delivery system, supporting an essential role of Nrf2 in the exercise-mediated DAergic protection against the MPP+-induced toxicity [225]. Using the 6-hydroxydopamine (6-OHDA) rat model of PD, Chuang and co [225] evaluated the effect of treadmill training and observed improved performance of gait parameters and also reduced methamphetamine-induced rotation. Importantly, this training improved DAergic neuron viability associated to the recovery of mitochondrial function, mitigating oxidative stress in PD rats. The mechanism has been suggested to be associated with the facilitation of mitochondrial turnover, including facilitation of mitochondrial fusion, fission, and clearance accompanying increased quantities of mitochondria [[225], [226]]. Finally, exercise, enriched environment and dietary factors, modulate brain maintenance and plasticity including neurogenesis, synaptogenesis, enhanced metabolism and angiogenesis, at least in part via a beneficial modulation of oxidative stress, inflammation and Wnt signaling with consequent effect on neuronal survival, behavioral and cognitive functions [65,73,223,[226], [227], [228], [229], [230], [231], [232], [233], [234], [235], [236]].
Therefore, an ever-growing panel of harmful and beneficial environmental factors can modulate the response of the nigrostriatal DAergic system to basal ganglia injury, via a major impact on the Nrf2-mediated response to oxidative stress, and Wnt/inflammatory cascades.
All together, gene-environment interactions may drive a vicious cycle of oxidative stress and inflammation. Notably, such feedforward cycle of chronic glia activation and persistent damage of dopaminergic neurons are likely to play a decisive role for the severity of nigrostriatal DAergic lesion and the overall detrimental effects upon SNpc neurons, including their capacity for neurorescue/neurorepair. Within this frame, astrocytes can either cooperate with microglia to exacerbate M1 phenotype and the consequent neurotoxicity, or in the contrary, they can downregulate microglia activation, to support the imperilled/dysfunctional DAergic neurons and activate intrinsic cues for neuropair/neurorestoration. Yet, the factors determining whether astrocytes will assume a beneficial or harmful phenotype are actively investigated, as reviewed in next sections. The good news is the plasticity of this intersystem crosstalk and the possibility to revert/rejuvenate the dysfunctional neuron-glia communication network to promote neuron survival and functional rescue in PD-based models (reviewed in next sections).
4. Glia-neuron crosstalk links neuroinflammation to dopaminergic neuroprotection/repair in PD: old and novel actors
4.1. The glial world of harmful and beneficial mediators
Astroglial cells have been increasingly recognized as important regulators of brain function and disease via bidirectional interactions with neurons [[237], [238], [239], [240], [241], [242], [243], [244]]. Notably, neurons and glial cells communicating with each other by an array of molecules (e.g., neurotransmitters, neuromodulators, neuropeptides, neuroimmune regulatory molecules) can enhance or inhibit neuronal vulnerabilty against various noxious stimuli, which poses the “To be or not to be inflammed” dilemma [17]. Astrocytes and microglia can protect neurons by scavenging radicals and glutamate, by harboring receptors for endogenous antiinflammatory molecules, by providing energy support, trophic factors, and ‘protective’ cytokines, by stimulating neurorepair also by activating neurogenesis by expressing neurogenic factor. Especially, “resting” microglia thanks to their ramified protrusions continuously scan the neuronal microenvironment representing the most dynamic surveillants of brain parenchyma in vivo [[245], [246], [247], [248]].
As summarized in previous sections, according to the physiopathological condition, the genetic background, together with a panel of environmental factors, astrocytes and microglia loose their neuroprotective functions and turn into an “harmful” proinflammatory phenotype to PD injury. Accordingly, a wide number of preclinical researches demonstrates that anti-inflammatory treatment may be effective to ease PD symptoms [17,39,169,[183], [184], [185], [186], [187],249,250].
However, whether neuroinflammation and oxidative stress can be considered as contributors to, or the consequence of neurodegeneration, still remains to be defined. Notably, astrocytes and microglial cells are pivotal in modulating the stem cell niche that promote neurogenesis, including the survival and identity of neural stem/progenitor cell (NSC)-derived mDAn, thereby regulating adult NSC plasticity in neurogenic niches in the PD brain, but these functions are sharply downregulated with the ageing process and PD degeneration with harmful consequences for mDAn rescue/repair [73]. Recent investigations have provided substantial evidence that a proinflammatory microglial cytokine cocktail containing TNF-α, IL-1α and C1qa reprograms a subset of astrocytes to change their expression profile and phenotype, thus becoming neurotoxic (designated as A1-astrocytes). Knockout or antibody blockage of the three cytokines abolish formation of A1-astrocytes, therefore, this pathway is of high therapeutic interest in neurodegenerative diseases [251].
Hence, accumulating evidence clearly indicates the ability of astrocytes and microglial cells to exert critical neuroprotective and neuroreparative functions. Then, astrocytes harbor a powerful arsenal of neurotrophic and neuroprotective antioxidative molecules and neurotrophic factors, and express receptors for neurotransmitters, cytokines, chemokines, and hormones in cooperation with those produced by microglia [[252], [253], [254], [255], [256], [257]].
Activated astrocytes can support neuron survival and recovery of their synaptic input following moderate neuronal damage [256,257]. Astrocyte inflammatory signaling through STAT3 plays a crucial role in these repair mechanisms, and is a hallmark of the protective astrocyte phenotye [[255], [256], [257], [258]]. Especially, the relationship between reactive astrocytes and microglia is bidirectional, with astrocyte activating microglia acting onto the astrocytes to modulate the extent of the inflammatory response, and microglia, in turn, activating both neuroprotective or detrimental pathways for the neighboring neurons, according to the glial genotype and a plethora of environmental factors. Notably, a prolonged dysfunction of astrocytes and microglia activation have been shown to accelerate the degeneration of SNpc dopaminergic neurons, blocking the compensatory mechanisms of neuronal repair during early dysfunction induced by 6-OHDA lesion in rats [259]). Reportedly, the M2 polarized microglia associates with the production of anti-inflammatory cytokines (e.g., IL-4 and IL-10), neurotrophic factors (e.g., BDNF and IGF-1), and extracellular matrix proteins (e.g., fibronectin) [260].
In the last decade, several lines of evidence pointed to Wingless-type MMTV integration site (Wnt)/β-catenin the Wnt/β-catenin signaling, a chief player in dopaminergic neurodevelopment [[67], [68], [69], [70], [71],261], as an emerging pathway involved in bidirectional astrocyte-neuron crosstalk contributing to dopaminergic neuron survival. Astrocytes are known to release various region specific signaling molecules, such as sonic hedgehog (Shh) and Wnts, which may interact with each others to dictate the neurogenic behavior in the adult CNS [[262], [263], [264], [265], [266]]. Importantly, astrocytes have pivotal roles for defining the stem cell niche. Hence, E13.5 VM astrocytes, but not cortex (Cx) astrocytes, express Wnt1 and Wnt5a and different DA-specific transcription factors such as Pax-2, En-1, and Otx-2 and increase the differentiation of VM embryonic precursors into tyrosine-hydroxylase positive (TH+) neurons, in vitro, suggesting that VM astrocytes constitute part of the neurogenic niche that play a key role in VM-DA neurogenesis [73, for review].
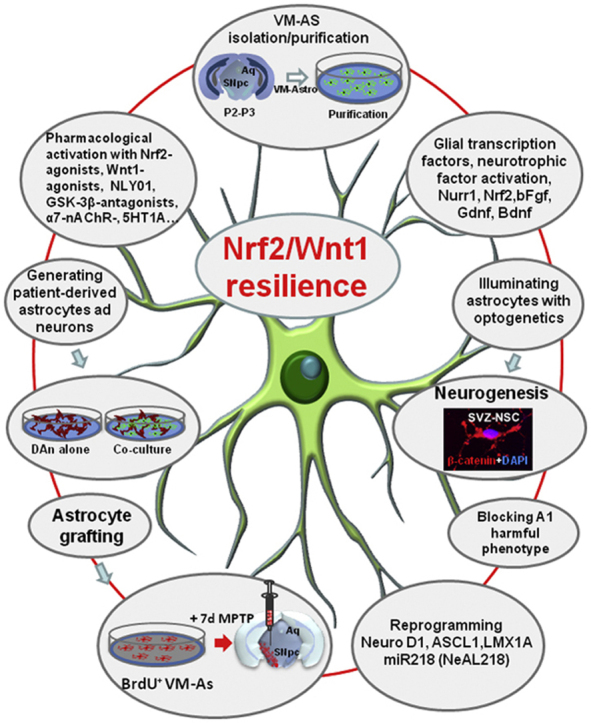
5. Astrocyte-derived Wnt signaling and the NRf2/ARE axis orchestrate resilience of DAergic neurons in PD
5.1. Wnt1 is a bidirectional signal for neuron survival and immunomodulation in basal ganglia injured mice
In the MPTP-based mouse model of basal ganglia injury, a wide gene expression analysis of 92 mRNA species involved in inflammation, immunity, stemness, self-renewal, dopaminergic neurodevelopment, and dopaminergic metabolism, indicated a major upregulation of certain pro-inflammatory chemokines, i.e., CCl3, CXCl10 and CxCl11, and a specific member of the Wnt signaling cascade, i.e., Wnt1, during neurotoxin (i.e, MPTP)-induced nigrostriatal degeneration and self-recovery, suggesting Wnt signaling as an intrinsic response to dopaminergic neuron injury [61]. In situ hybridization histochemistry demonstrated colocalization of Wnt1 with reactive GFAP+ astrocytes within the MPTP-injured midbrain associating to the rescue of the imperilled/dysfunctional nigrostriatal neurons [61]. Additionally, increased Wnt1 mRNA transcription was detected in astrocytes derived ex vivo from MPTP-injured midbrain, and chemokine-activated astrocytes expressed Wnt1, in vitro (Fig. 4).
Fig. 4.

Wnt1 is a novel actor in astrocyte-neuron crosstalk. A: scheme of ventral midbrain astrocytes (VM-AA) isolation, purification and direct (co-culture) or indirect (AS-conditioned medium, ACM) culture paradigms with purified primary mesencephalic dopaminergic neurons (mDAn), in the absence or the presence of the PD neurotoxin, MPP+. Survival, dopamine (DA) uptake and caspase 3 assays are used to monitor mDAn death and functionality. B:In situ hybridization histochemistry coupled to confocal laser scanning microscopy and dual immunofluorescent staining with the astrocyte cytoskeleton marker, glial fibrillary acidic protein (GFAP, in red) and Wnt1 mRNA (in green) showing the expression of Wnt1 in primary GFAP+ cells (orange-yellow). C: Astrocyte-neuron crosstalk with primary mDAns. Confocal image of TH+ neurons (in green) in co-culture with VM-AS (in red), showing TH+ neurons with long and branched TH+ neuronal processes, interacting with GFAP+ star-shaped astrocytes. Fig. 4. Scale bars, panel B: 25 μm, Box: 10 μm, panel C: 50 μm.
Of specific interest, another chief actor of Wnt/β-catenin signaling, GSK-3β, was over-expressed both preceding and during the active phase of DAergic degeneration together with the up-regulation of the active protein, pGSK-3-Tyr216 [61,62]. Such an over expression and increased protein levels corresponded to the peak of DAergic degeneration in SNpc, and correlated to DAergic striatal degeneration, striatal DA uptake levels and motor deficit as revealed by behavioral analyses [61,62]. On the other and significantly, the time-dependent histochemical and functional recovery of nigrostriatal DAergic neurons associated with a time-dependent up-regulation of principal Wnt/β-catenin signaling components, in face of a marked downregulation of active GSK3-β, both at a gene and protein expression levels within the SNpc [61]. The previous evidence that Wnt signaling may be reinduced in the adult CNS after injury [267], coupled to earlier findings showing Wnt's expression in astrocytes both during development and in adult brain [268,269], then suggested a potential glial compensatory mechanism implicated in dopaminergic neuroprotection and/or neurorescue [61]. We then hypothesized that such a mechanism might contribute to the recognized capacity of nigrostriatal neurons to mount a compensatory/self-adaptive response upon injury [164, 177, 178,[270], [271], [272], [273]]. In fact, astrocyte and microglia activation, including the expression of proinflammatory cytokines and neurotrophic factors during dopaminergic nigrostriatal recovery upon injury were previously underlined [[274], [275], [276], [277]]. Strikingly, a persistent increase of GFAP+ astrocytes in VM, and a robust GFAP+astrocyte-TH neuron crosstalk characterized the nigrostriatal DAergic recovery phase, as evidenced by biochemical, molecular and motor behavior data [61]. Especially, the remarkable TH+ fiber sprouting and GFAP-to-TH neurons cell-to cell contacts, accompanied nigrostriatal neurorepair, which persisted long after MPTP insult (Fig. 5).
Fig. 5.

Dopaminergic neurorepair upon MPTP injury is directed by glial fibrillary acid protein (GFAP+) astrocytes. A reconstruction of representative confocal images of midbrain coronal sections at the SNpc level, stained with tyrosine hydroxylase positive neurons (TH+, in green) and GFAP (in red) from MPTP-treated mice, 65 days post injury is shown. Note the robust TH neurorepair, as revealed by fluorescence immunohistochemistry. Within the rescued SNpc, bright TH+ neurons extending long processes can be observed running together with bright reactive GFAP+ astrocytes, coursing intermingled with TH+ neurons (boxed magnification) and seemingly guiding the dopaminergic neurorestorative process. VTA: ventral tegmental area. Scale bar:100 μm.
Intuitively, the astroglial cell compartment appeared a critical actor for mDAn resilience, as many adaptive changes occurring at this level serve to increase the defense against oxidative stress, to reduce inflammation, to improve mitochondrial performance, to increase neurotrophic support, and to activate adult neurogenesis [240, [274], [275], [276], [277], [278], [279], and Refs in previous sections]. Activation of endogenous compensatory mechanisms is recognized to mask the of PD before the appearance of the first clinical symptoms [274], which raises the possibility that some individuals with PD suffer from a reduction of these neuroprotective mechanisms and that treatments that boost these mechanisms may provide therapeutic benefit [272]. The striking increase of astrocyte's Wnt1 and microglial-derived chemokines, CCl3, CXCl10 and CxCl11, further linked reactive astrocytes and Wnt/β-catenin signaling to nigrostriatal injury/and repair, and suggested astroglial Wnt1 as a novel compensatory rescue signal for mesencephalic DAergic neurons [reviewed in 43]. In support of a vital role of Wnt/β-catenin pathway, pharmacological activation of Wnt signaling by in vivo treatment with a specific GSK-3β antagonist efficiently mitigated nigrostriatal dysfunctions, both at the SNpc and striatal levels, and reverted the MPTP-induced motor dysfunction observed in ageing mice [61,62].
Corroborating a pro-survival role for Wnt/β-catenin signaling, in vivo, antagonism of Wnt1 signal transduction by blocking Fzd1 receptor with Dkk1 injection in the intact young adult SNpc, sharply reduced dopaminergic neuron survival [62], indicating astrocyte-neuron crosstalk via Wnt signaling as a potential astrocytic neuroprotective mechanism in the adult midbrain [61,62]. Several lines of evidence then supported these in vivo findings, as Wnt1 was reported to exert robust neuroprotective effects in “in vitro” PD cellular models (i.e., primary mesencphalic DAergic neurons, expressing the dopamine transporter, DAT, and exposed to different oxidative stressors and specific PD neurotoxins, such as MPP+ and 6-OHDA) [62], whereas knocking down of either β-catenin or Fzd-1 receptor [62] resulted in the abrogation of neuroprotection. These findings underscored that Fzd receptors and β-catenin are ‘physiological check-points’ for DAergic neuron survival, and corroborated the possibility that astroglial-derived Wnt1 might provide a compensatory mechanism to limit the degenerative process and/or activate the spontaneous SNpc self-repair program, as observed “in vivo“ [61,62].
So far, a wide panel of conditions affecting midbrain dopaminergic neurons in rodent models of basal ganglia injury have shown to strongly impair canonical Wnt/β-catenin signaling, while an increasing number of pharmacological, immunomodulatory agents, and cell therapies affording neuroprotection have been recognized to activate the canonical Wnt/β-catenin signaling pathway, promoting DAergic neurorescue and immunomodulation, and counteracting the impairment of neurogenesis in PD injured brain [summarized in section 6.1.2].
Of special interest, Wnt signaling also contributes to the modulation of inflammation in the midbrain via bidirectional glia-neuron crosstalk, thus providing neuroprotection also via immunomodulation [65]. Hence, atrocytes and macrophage/microglial cells in the brain, and immune cells in the periphery express Wnts and harbour a panel of Wnt's receptors thereby modulating in an autocrine/paracrine fashion immune responses both at central and peripheral levels [65,[280], [281], [282], [283], [284], [285], [286], [287], [288]]. In turn, Wnt receptors are present in glial cells and Wnt ligands can exert both anti- and pro-inflammatory effects. Then, when microglia is activated in vivo by MPTP exposure and acquires the M1 phenotype, expression levels of cytokines (TNF-α and IL-1β) and chemokines, together with the concurrent generation of ROS and RNS, are rapidly and robustly upregulated as a result of NF-κB induction (Fig. 6). MPTP also induces upregulation of the pro-inflammatory GSK-3β that further exacerbates microglial reaction [65,74,93,94,289]. Reportedly, the NF-κB and the Wnt/β-catenin pathway interact to differentially regulate inflammation, with GSK-3β playing a central role in between (Fig. 6). While GSK-3β is a negative regulator of β-catenin, it positively regulates NF-κB by targeting IkB, the major inhibitor of NF-κB, to proteasomal degradation [65,93]. On the other hand, β-catenin itself can form a complex with the p50 subunit of NF-κB, thereby preventing NF-κB transcriptional activity. The complexity of Wnt's immunomodulation is further underscored by the proinflammatory role of the non-canonical Wnt5a, which constitutes one part of a self-perpetrating cycle, via autocrine Wnt5A/CamKII activation and paracrine stimulation of T-helper1 (Th-1)- cytokines, inducible nitric oxide synthase (iNOS) and cicloxygenases (COX2) [[280], [281], [282], [283], [284], [285], [286], [287], [288]].
Fig. 6.

Nrf2/Wnt/immune crosstalk in oxidative stress and inflammation in PD. Schematic illustration of astrocyte-microglia crosstalk. Upon activation by neurotoxins, endotoxins, brain injury and ageing, macrophage/microglia produce a panel of pro-inflammatory cytokines (TNF-α and IL-1β) and chemokines (CCL3, CXCl10 and CXCL11). Up-regulation of microglial PHOX-derived ROS, iNOS-derived NO, and GSK-3β, a known regulator of NF-kB-dependent gene transcription, further exacerbates microglia reaction. Wnt5a constitutes one part of a self-perpetrating cycle, via autocrine Wnt5A/CamKII activation and paracrine stimulation of Th-1- cytokines, iNOS and COX2 [280–2282]. To restrain microglia exacerbation, up-regulation of astrocyte- Nrf2/HO-1 and Wnt1/β-catenin, mitigate the inflammatory milieu and favor a down-regulation of cytokines expression. NF-κB and the Wnt/β-catenin pathway also interact to differentially regulate inflammation, with GSK-3β playing a central role in between. While GSK-3β is a negative regulator of β-catenin, it positively regulates NF-κB by targeting IkB, the major inhibitor of NF-κB, to proteasomal degradation. On the other hand, β-catenin itself can form a complex with the p50 subunit of NF-κB, thereby preventing NF-κB transcriptional activity. Besides, HO-1 indirect modulation, Nrf2-NF-κB interplay contributes to the regulation of immune response under oxidative stress and inflammation aimed at counterbalancing the exacerbated inflammation. Then, astrocyte upregulation of Nrf2/HO-1 and Wnt1/β-catenin during oxidative stress and inflammation represent a critical regulatory level, whereby astrocytes can mitigate M1 exacerbated phenotype and the heighthened levels of proinflammatory cytokines.
5.2. Nrf2/HO-1/NF-κB/Wnt crosstalk restrains astrocyte-microglia exacerbated proinflammatory phenotype in PD
Within the Nrf2-ARE axis, HO-1 is a key mediator of cellular adaptive (i.e. antioxidant and anti-inflammatory) responses [59,193,194]. This protein is induced by hypoxia, cytokines, and oxidative stress, amongst other factors. HO-1 is itself an antioxidant protein that protects cells from oxidative damage by downregulating ROS levels. In turn, HO-1 plays a down-regulatory role in NF-κB nuclear translocation, thereby downmodulation NF-κB-dependent proinflammatory cytokine expression [290]. Therefore, astrocyte upregulation of HO-1 during oxidative stress and inflammation represents a critical regulatory level, whereby astrocytes can mitigate M1 exacerbated phenotype and the heighthened levels of proinflammatory cytokines [6].
Besides, HO-1 indirect modulation, Nrf2-NF-κB interplay is recognized to contribute to regulation of immune response under oxidative stress and inflammation aimed at counterbalancing the exacerbated inflammation. Of specific mention, Nrf2 and NF-κB are central transcriptional activators and Keap1 and IκB-α are regulatory proteins which induce proteasomal degradation of these transcriptional factors under stress conditions [291]. Interestingly, during inflammation, both Nrf2 and NF-κB are coordinated effectors of the Rho family, GTP-binding Protein RAC1, a mediator in the execution of the inflammatory innate program, including NADPH oxidase-dependent production of ROS [291]. Hence, adding a further level of control, RAC1 can induce the anti-inflammatory Nrf2/HO-1 pathway. Additionally, NF-κB activity is induced by active RAC1, and in turn, Nrf2 can modulate this effect, thus uncovering a new mechanism of regulation of inflammatory events trough a RAC1/NRF2/HO-1 axis [291].
Of note, aged microglia over-expressing ROS, RNS and a panel of proinflammatory cytokines, when challenged with inflammatory and/or neurotoxic challenges, also exhibits up-regulated GSK-3β levels in face of β-catenin downmodulation [72] (Fig. 6). Here, astrocyte-microglia dialogue is likely to play an important restraining role, via both Nrf2-ARE axis activation and via Wnt immunomodulation. Accordingly, activation of Wnt signaling with either GSK-3β antagonists, or astrocyte-derived Wnt1 results in a significant reversal of oxidative stress and inflammation, both in vivo and in vitro [72]. Additionally, given the high sensitivity of microglial cells to Nrf2 activation, astrocyte Nrf2-ARE signaling further provide a down-regulatory mechanism to shut down inflammation (Fig. 6).
Together, a close interrelationship between Nrf2-ARE, Wnt signaling and glial pathways are at play and collaborate to monitor oxidative stress and inflammation, to maintain and protect the vulnerable DAergic cell population, via bidirectional astrocyte-neuron and astrocyte microglia reciprocal crosstalk [Fig. 6].
5.3. Resilience of Nrf2-HO1/Wnt/β-catenin neuroimmune axis shape DAergic neuron plasticity in PD
With age, the described failure of the astrocytic Nrf2-antioxidant axis response upon inflammation and oxidative stress dramatically impact in VM astrocyte-microglia-neuron interactions [[64], [65], [74], [101], [292]]. At the SNpc level, ageing-induced decline of astrocytic Nrf2 gene expression promotes an up-regulation of major microglial proinflammatory gene transcripts, such as TNF-α, IL1β, IL-6 and Nos2 both at striatal [64,187,188] and SNpc [61,65,72,74] levels, exacerbating oxidative stress and inflammation. Concurrently, Wnt/β-catenin genes are sharply downregulated in face of an overexpression of endogenous Nrf2-and Wnt-antagonist genes, including, the expression of GSK-3β, several Dickkopf (Dkk) and the Fzd-related members (sFRPs), whose proteins are able to bind to Wnts directly, thus inhibiting Wnt signaling transduction and β-catenin nuclear translocation and transcriptional activity [[72], [73], [74]] (Fig. 7). Particularly, ageing and MPTP exposure downregulated the antioxidant gene, HO1, together with SOD1 in the aged MPTP-injured VM as compared to younger and saline-treated counterparts, underscoring that with age, failure to activate an anti-oxidant self-defense response to the MPTP challenge contributed to mDAn death [74]. Further evidence for such a failure with age and MPTP exposure was the observation that the nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, Nox2, a predominant oxidase family member expressed in astrocytes [201], both the mRNA and protein level, were robustely up-regulated by an almost 3-fold over non MPTP-treated aged (saline-injected) mice, supporting failure of Nrf2-HO1 and Wnt/β-catenin axis with age and basal ganglia injury as pivotal factors for mDAn degeneration [72,74] (Fig. 7).
Fig. 7.

Astrocyte-microglia interactions and Nf2/Wnt1/β-catenin resilience in mDAn neuroprotection. Major environmental factors including ageing, inflammation, neurotoxin exposure (MPTP/MPP+, 6-OHDA, pesticides), in synergy with genetic mutations results in dysfunctional astrocyte-microglial crosstalk associated to the exacerbated production of proinflammatory mediators. The glial switch to the A1/M1 harmful astrocyte and microglial phenotype is the result of the inhibition of Nrf2/Wnt/β-catenin signaling (“Nrf2/Wnt off”). In these conditions, reactive astrocytes no longer mount an efficient resilience program for the vulnerable mDAns. Hence, the crucial anti-oxidant and anti-inflammatory Nrf2/HO-1 and dopaminotrophic factors, namely Wnt1, are sharply inhibited. As a result, active GSK-3β is up-regulated in mDAns, leading to β-catenin degradation. Then, in the absence of an efficient Nrf2-ARE axis at play, the “frailty” of mDAns increases in turn leading to mDAn degeneration. By contrast, astrocyte upregulation of Nrf2/HO-1 and Wnt1/β-catenin during oxidative stress and inflammation represent a critical resilience program for DAns. Then, increased astrocyte-derived Wnt1 (and Wnt1-like agonists, such as Wnt1, Rspo or Norrin) activates Fzd-1 receptors (“Wnt on”), leading to the blockade of GSK-3β-induced phosphorylation (P) and proteosomal degradation of the neuronal pool of β-catenin. Stabilized β-catenin can translocate into the nucleus and associate with a family of transcription factors and regulate the expression of Wnt target genes involved in DA neuron survival/plasticity, neuroprotection and repair. β-catenin may also function as a pivotal defense molecule against oxidative stress, and can act as a coactivator for several nuclear receptors involved in the maintenance/protection of DA neurons. The hypothetical contribution of various endogenous Wnt agonists (Responding, Rspo, Norrin) or antagonists (Dkkopf, Dkk1, Wif, frizzled-related proteins, SFRp) are also indicated. Resilience of Nrf2/Wnt/β-catenin program can be activated by several treatments, including GSK-3β antagonists, Wnt1-like agonists, nitric-oxide–(NO)–anti-inflammatory non-steroidal drugs (NSAID). Different conditions/treatments can inhibit Nrf2/Wnt beneficial signaling cascades, including gene mutations, ageing, inflammation, endogenous Wnt-antagonist expression, leading mDAn degeneration (see the text for details).
Accordingly, when VM astrocytes from aged-MPTP-treated mice were isolated “ex vivo”, their metabolic activity showed a significant reduction vs saline-treated counterparts, in line with the upregulation of both ROS and RNS produced by VM astrocytes derived from aged and MPTP treated mice [74]. By contrast, the specific iNOS-NO inhibitor, L-Nil, efficiently counteracted the sharp increase in oxidative and nitrosative stress of aged astrocytes, with beneficial effect on mitochondrial reductase activity [74].
Given the decline of astrocyte-derived Wnts with age, we asked whether Wnt/β-catenin signaling activation might affect the exacerbated redox status of aged astrocytes. Remarkably, we found a significant counteraction of both ROS and RNS up-regulation when Wnt/β-catenin signaling was activated by treament with a specific GSK-3β-antagonist, in turn resulting in increased astrocyte metabolic activity [74]. Thus, the exacerbated oxidative/nitrosative status of astrocytes from aged MPTP-injured SNpc can be reversed by activation of Wnt signaling. In turn, this resulted in increased of Nrf2-antioxidant gene expression, driving a switch in the proinflammatory and oxidative SN microenvironment towards and A2 and M2 anti-inflammatory, neuroprotective phenotype [74] (Fig. 7).
All together, these findings argue in favor of reciprocal astrocytes/microglial/neuron interactions, and implicate resilience of the Nrf2/HO1/Wnt/β-catenin axis as a critical mediator in promoting neuroprotection.
Regarding neurogenesis in PD, the role of Wnt/Nrf2 signaling pathways in subventricular (SVZ), hippocampal subgranular (SGZ), and midbrain peri-aqueductal NSC niches of the adult and ageing brain has been recently reviewed and the contribution of the exacerbated oxidative and inflammatory status within the ageing niches expanded to the decline of astrocyte Wnt-dependent regulation, ultimately leading to NSC neurogenic impairment and loss of NSC plasticity [73 for an extensive review].
As highlighted in this article, Wnts and the components of Wnt/β-catenin signaling are not only widely expressed in the adult niches, but most importantly they respond to MPTP injury and are required to trigger neurorepair programs in MPTP-induced PD thanks to the “Wnt/immune crosstalk dialogue” with glial cells in strict collaboration with Nrf2/HO1 axis, as a means to boost neurogenesis promoting mDAn neurorestoration [[63], [64], [65], [66]]. Particularly, ageing-dependent mitochondrial dysfunction in synergy with neurotoxin exposure negatively impact on the astrocytic Nrf2-driven HO-1 response within the SVZ niche [[63], [64], [65], [66],73].
6. The therapeutic impact: a glial avenue for nigrostriatal resilience in PD?
From the presented findings, astrocytes, the star-shaped cells, extensively interacting with neurons and microglia, are at the forefront of neurorescue, neurorepair and regeneration therapies. In particular, midbrain astrocytes are uniquely positioned to drive neuroprotective and regenerative programs in PD. Therefore, targeting not only diseased neurons, but specifically manipulating astroglial cells may provide a new approach for drug development for the treatment of neurological diseases, as PD [[293], [294], [295], [296], [297], [298], [299]] (Fig. 8). As herein reviewed, an increasing number of studies indicates the possibility to convert the hostile astrocyte's and microglial midbrain environment to rescue the injured mDAns harnessing Nrf2/Wnt signaling, by either molecular or pharmacological correction of glial cell harmful phenotype, to improve neuronal survival and promote DAergic neurogenesis. Astrocytes may be targeted with existing and novel-anti-oxidant, anti-inflammatory, and anti-apoptotic drugs, serving as “chaperones” to promote optimal recovery, and may also be replaced by exogenous cell transplantation or by increasing the production of endogenous astrocyte precursors, as herein discussed.
Fig. 8.

Astrocyte's “fil rouge” targeting Nrf2/Wnt resilience cascades. Manipulating astrocytes provides a new approach for drug development in neurological diseases. Schematic representation of a panel of manipulations of astrocytes for DAn neurorepair and regeneration. The potential exists to revert some A1 age-dependent changes, including pharmacological correction of glial dysfunction harnessing astrocyte-derived Nrf2/Wnts and neurotrophic factors, or blocking A1 harmful phenotype with glucagon-like peptide-1 receptor agonist, NLY01; activating glial Nurr1, or activating astrocyte neurotransmitter receptors; antagonizing GSK-3β in either neurons and glial cells by GSK-3β-antagonists, as well as physical activity and exercising. Novel frontiers regard the use optogenetics to illuminate astrocytes, promoting their neuroprotective and proneurogenic functions. Additionally, genetic manipulation of astrocytes and co-grafting techniques to improve the injured microenvironment, activate dopaminergic neurogenesis and incite neurorepair are being studied. Derivation of astrocyte differentiated from NSCs or hiPSC sources; astrocyte reprogramming into neurons, represent some of these very challenging new research areas. Additionally, generating patient-specific astrocytes capable of recapitulate a patient's genetic background and disease phenotype and using co-culture techniques with PD-specific neurons, may help screening new molecules for drug discovery and therapeutical applications to treat neurological diseases.
6.1. Harnessing the Nrf2/Wnt signaling axis in PD
6.1.1. Astrocyte-specific overexpression of Nrf2 and Nrf2 activators to fight PD
Following the earlier studies of Vargas and co [300] introducing the therapeutic potential for astrocyte Nrf2 activation to mitigate neurodegeneration in experimental models of familial amyotrophic lateral sclerosis (ALS), several reports focused on the ability of astrocyte-Nrf2 to counteract PD neurodegeneration in various experimental models of the disease. Using transgenic mice over-expressing Nrf2 selectively in astrocytes, the authors showed the ability of Nrf2-astrocytes to reverse the toxicity of astrocytes expressing ALS-linked mutant hSOD1 when cocultured with motor neurons. This effect resulted from increased glutathione secretion from astrocytes. The authors suggested that Nrf2 activation in astrocytes is a viable therapeutic target to prevent chronic neurodegeneration [300]. In the alpha-synuclein mutant (A53T) mouse model [200], increased Nrf2 selectively overexpressing Nrf2 in astrocytes (GFAP-Nrf2) delayed the onset and extended the life span of the hSYN(A53T) mice, which correlated with increased motor neuron survival, reduced oxidative stress and gliosis [200]. Furthermore, Nrf2 in astrocytes delayed chaperone-mediated autophagy and macroautophagy dysfunction observed in the hSYN(A53T) mice, indicating that Nrf2 in astrocytes provides neuroprotection against hSYN(A53T)-mediated toxicity by promoting the degradation of hSYN(A53T) through the autophagy-lysosome pathway in vivo [200]. Interestingly, the study of Skibinski and co-workers [132], further expands the biological role of Nrf2 in neuroprotection, by showing its ability to mitigate LRRK2- and α-Syn-induced neurodegeneration by potently promoting neuronal protein homeostasis [132]. Here, Nrf2 reduced PD-associated protein toxicity and activated distinct mechanisms to handle different misfolded proteins [132]. Nrf2 decreased steady-state levels of α-Syn in part by increasing α-Syn degradation. In contrast, Nrf2 sequestered misfolded diffuse LRRK2 into more insoluble and homogeneous inclusion bodies [132].
Together, both astrocyte-dependent and neuron-specific Nrf2 mechanisms are at play in improving neuronal survival against PD toxicity caused by both mutant LRRK2 and α-synuclein.
In the study of Sigfridsson et al. [301], astrocyte-specific overexpression of Nrf2 can protect against optic tract damage and behavioural alterations in a mouse model of cerebral hypoperfusion. Here, transgenic mice overexpressing Nrf2 (GFAP-Nrf2) and wild type littermates were subjected to bilateral carotid artery stenosis or sham surgery. The results of the study showed that while pro-inflammatory gene expression was significantly upregulated in the optic tract of hypoperfused Wt mice, in Tg mice, overexpression of Nrf2 in astrocytes repressed inflammation, supporting the potential use of Nrf2-activators in the amelioration of cerebrovascular-related inflammation and white matter degeneration [301].
A vast panel of Nrf2 activators are being studied thanks to their ability to modulate all critical mDAn processes and to maintain mitochondrial homeostasis in neurons [[51], [52], [53], [302], [303], [304]]. Hence, Alarcón-Aguilar et al. [305] determined if cortical astrocytes derived from old rats are able to respond to tertbuthyl-hydroquinene (tBHQ) pretreatment to stimulate the Nrf2-antioxidant response pathway thus inducing an antioxidant strategy against MPP+ toxicity. This study showed an age-dependency in astrocyte susceptibility to MPP+ toxicity, and further reported that a pretreatment with tBHQ efficiently transactivated Nrf2, increasing antioxidant enzymes and developing cellular protection [305]. Synthetic triterpenoids (TPs) are robust activators of Nrf2 activity. Using a novel Neh2-luciferase reporter, Kaidery et al. [306], showed that TP upregulated a number of cytoprotective genes, including those involved in glutathione biosynthesis, in vitro. When structurally modified to penetrate the brain, TP-induced messenger RNA and protein levels for a battery of Nrf2-dependent cytoprotective genes [306]. This effect associated to reduced MPTP-induced oxidative stress and inflammation, and ameliorated dopaminergic neurotoxicity in mice [306]. The neuroprotective effect of these TPs against MPTP neurotoxicity was dependent on Nrf2, since treatment with TP in Nrf2 knockout mice failed to block against MPTP neurotoxicity and induce Nrf2-dependent cytoprotective genes [306].
Dimethyl fumarate (DMF), a drug already in use for the treatment of multiple sclerosis, was shown to increase nuclear levels of active Nrf2, with subsequent upregulation of antioxidant target genes and to reduce the 6-OHDA-induced neuronal death in mice, in vivo, and in the human DAergic cell line SH-SY5Y, used as an in vitro model of the disease [307]. These effects associated with a reduction of ROS induced d by 6-OHDA treatment. By contrast, treatment with Nrf2 siRNA failed to block 6-OHDA neurotoxicity in SH-SY5Y cells [307]. By alkylating cysteine residues in Keap1 DMFO disrupts the interaction between Keap1 and Nfr2, in turn resulting in Nrf2 translocation to the nucleus and activation of mitochondrial biogenesis in multiple sclerosis [307] and MPTP-induced model of PD. Recently, pharmacological targeting of Nrf2 was tested in rodent after stereotaxic injection of recombinant adeno-associated viral vector expressing human α-synuclein, showing α-synucleinopathy, mDAn death, oxidative stress, and neuroinflammation [198]. Here, DMF, protected mDAns against α-Syn toxicity and decreased astrocytosis and microgliosis, whereas the protective effect was not observed in Nrf2-knockout mice [198]. Importantly, in vitro studies indicated that DMF-induced neuroprotection associated with altered regulation of several autophagy markers with a “shift in microglial dynamics toward a less pro-inflammatory and a more wound-healing phenotype” [198]. Supporting the critical role of an efficient Nrf2-axis, in postmortem samples of PD patients, the cytoprotective proteins associated with Nrf2 expression, NQO1 and p62, were partly sequestered in Lewy bodies, thus supporting impaired neuroprotective capacity of the NRF2 signature in PD [198].
The molecular mechanism of DMF action was further studied in mouse hippocampus, and shown to involve KEAP1 but also PI3K/AKT/GSK-3-dependent pathways [95]. Reportedly, DMF modulates TAU phosphorylation, neuronal impairment measured by calbindin-D28K and BDNF expression, as well as the associated inflammatory processes involved in astrogliosis, microgliosis and pro-inflammatory cytokines production, thereby highlighting neuroprotective effects of DMF via crosstalk of the KEAP1/NRF2/GSK3 signaling [95,[308], [309]].
[5-(3,4-Difluorophenyl)-3-(6-methylpyridin-3-yl)-1,2,4-oxadiazole], is a novel Nrf2 activator targeting the brain, that has been recently shown to protect against MPTP-induced subacute Parkinson's disease in mice by inhibiting the NLRP3 inflammasome, in vivo, and to protect PC12 cells against oxidative stress [310]. Here, DDO-7263 improved the behavioral abnormalities induced by MPTP in mice, significantly attenuated MPTP-induced Daergic neuron death in SN and striatum associating with a downmodulation of inflammatory markers [310]. In a cellular system (PC12 cells) widely used as an in vitro cell culture model for PD, DDO-7263 was able to promote neuroprotection against H2O2-induced oxidative damage via the activation of Nrf2-ARE signaling pathway and the inhibition of NLRP3 inflammasome activation, suggesting a therapeutic potential for this novel Nrf2 activator [310].
Tauroursodeoxycholic acid (TUDCA) has been shown [311] to prevent MPP+- and α-Syn-induced oxidative stress, through Nrf2 activation, in vitro, in human DAergic cell (SH-SY5Y cells) model and in vivo, in the MPTP mouse model of PD, TUDCA treatment increased the expression of Nrf2, Nrf2 stabilizer DJ-1, and Nrf2 downstream target antioxidant enzymes HO-1 and GPx, suggesting that TUDCA may be a promising agent to limit ROS-mediated damage, in different models of PD [311].
Tert-butylhydroquinone (tBHQ) enhanced angiogenesis and astrocyte activation through Nrf2 pathway in mice with permanent distal middle cerebral artery occlusion (dMCAO) [312]. tBHQ significantly reduced the infarct volume, enhanced post-stroke angiogenesis and astrocytic endfeet covered ratio in the peri-infarct area [312]. The Nrf2/HO-1/VEGF pathway was activated by tBHQ in the angiogenesis process, but not in Nrf2−/− mice, where the Nrf2 deficiency blocked the effects of tBHQ on angiogenesis process and neurological recovery indicating a beneficial effect of activating Nrf2 pathway after cerebral ischemia [312].
Matrix metalloproteinase (MMP)-8 inhibitor, M8I, can control neuroinflammation in lipoteichoic acid (LTA)-stimulated rat primary astrocytes [312]. Here, treatment with LTA, a major cell wall component of Gram-positive bacteria, led to astrocyte activation and induced the expression of inflammatory molecules such as iNOS, COX-2, and pro-inflammatory cytokines, as well as MMP-1, MMP-3, MMP-8, MMP-9, and MMP-13 in rat primary astrocytes [312]. M8I inhibited LTA-induced NF-κB, MAP kinase, and Akt activities, while it increased the anti-inflammatory PPAR-γ activities. Moreover, M8I showed antioxidant effects by suppressing ROS production in LTA- or H2O2-stimulated astrocytes. M8I increased the expression of phase II antioxidant enzymes such as hemeoxygenase-1, NQO1, catalase, and MnSOD by modulating the Nrf2/ARE signaling pathway [312]. Reportedly, several Nrf2 activators regulate mitochondrial biogenesis in different tissues and model systems, as recently reviewed by Gureev and coll [304,313].
Piperine analogues activating the Nrf2/keap1 pathway have been recently identified and optimized by Wang et al. [314], evaluated as neuroprotectant against hydrogen peroxide (H2O2) induced damage in the neuron-like PC12 cells. Among these analogues, 3b exhibited the most potent protection afforded by phase II antioxidant enzymes, such as HO-1 and NQO1 activation [314]. In in vivo study, oral administration attenuated PD-associated behavioral deficits in MPTP-induced mouse model of PD and protected TH+ dopaminergic neurons. These results provided evidence that compound “3b” might be a promising candidate for Parkinson's disease treatment [314].
6.1.2. Harnessing astrocyte-derived Wnt and Wnt/β-catenin activators in PD experimental models “in vivo” and “in vitro”
Notably, astrocyte-derived Wnts can rejuvenate the microenvironment thus promoting neuroprotection, immunomodulation, and activates DAergic neurogenesis. During ageing and inflammatory conditions, endogenous astrocytes reduce Wnt1 expression loosing their neuroprotective functions against MPP+-induced DAergic neuron death. Hence, co-cultures with aged-astrocytes induced an inhibition of TH+ neurons and DA uptake levels compared with co-culture with young-astrocytes [61,62,72,74], which promoted instead a significant increase in TH+ neuron survival and functionality, as a result of Wnt1/β-catenin signaling activation [66,72,74]. Young-astrocytes pre-treated with TNF- α + IL-1β or conditioned media from aged-microglia significantly decreased both TH+ neuron survival and DA uptake levels, whereas aged-astrocytes pre-treated with CCL3 + CXCL11, wich induced an increased expression of Wnt1, efficiently counteracted the MPP+-induced reduction of TH neuron survival and DA uptake [72]. In stark contrast, pre-treatment of neurons with the Wnt antagonist, Dkk1, inhibited these effects, thereby supporting that astrocyte-derived Wnt1/β-catenin signaling contributed to TH neuroprotection [72].
Studies in human NSCs implicated Wnt activation in neurotrophin-induced NSC growth, as Yang et al. [315] showed the ability of BDNF to promote their growth via GSK-3β-mediated crosstalk with the Wnt/β-catenin signaling pathway, and Li et al. [316] involved the contribution of the PI3K/Akt/GSK-3β/β-catenin pathway in BDNF-induced neuron and NSC growth. Corroborating these studies, cultured rat midbrain astrocytes abundantly expressing Wnts, spondin-2 (SPO-2), a secreted protein of the R-spondin family, which activates Wnt/β-catenin signaling, by preventing clearance of the Frizzled–LRP-Wnt receptor complex promoted DAergic neurogenesis from embryonic (E10) VM precursors [317]. Additionally, a number of anti-inflammatory cytokines and multiple antioxidant genes was upregulated in the cultured astrocytes [317]. Of specific importance, the forced Nurr1+Foxa2 expression in VM-astrocytes further promoted the astrocyte-mediated dopaminotrophic actions [317]. Hence, VM-astrocytes transduced with Nurr1+Foxa2-expressing lentiviruses a greater neuroprotective ability, as TH+ neurons were more resistant to the toxic insult induced by H2O2 treatment than differentiated TH+DA neurons cocultured with the control-astrocytes [317]. Consistently, significantly greater intracellular glutathione levels were manifested in the Nurr1+Foxa2-VM-astrocytes compared with the control VM-astrocytes, in accord with the upregulated secretory ROS scavenging factors Sod3 and Gpx3, ROS levels in mDAns [317]. These findings collectively indicated that the enhanced ROS scavenging capacity in VM-astrocytes by Nurr1+Foxa2 also contributed to VM-astrocyte–mediated neuroprotective actions [317].
In accord with the contribution of Wnt signaling promoting neuroprotection, an ever growing repertoire of DAergic neuroprotective drugs in different rodent models of PD are increasingly being discovered to act via the activation of astrocyte-derived Wnt signaling in mDAns, promoting neuroprotection/neurorescue, mitigating inflammation, and/or activating neurogenesis. Different studies focused on the neuroprotective capacity of Wnt1-agonists and pharmacological inhibitors of GSK-3β.
Wei et al. [318] supported the ability of exogenous Wnt1 to protect SH-SY5Y neurons against 6-OHDA-induced dopaminergic toxicity via the activation of Wnt/β-catenin pathway, and Zhang and co [319] corroborated the protective role of enhancing β-catenin activity to afford neuroprotection of PC12 cells against rotenone toxicity. Here, GSK-3β inhibitors LiCl and SB216763 leading to β-catenin stabilization afforded neuroprotection via the induction of the mDAergic transcription factor, orphan nuclear receptor, Nurr1, crucially involved in the survival and maintenance of mDAergic neurons [319]. Amongst others GSK-3β inhibitors, bromoinduru-30-oxime-(6-BIO) was shown to protect hippocampal neurons from the apoptotic effects of amyloid-β (Aβ) oligomers via a direct activation of Wnt/β-catenin pathway [320]. Interestingly enough, different classes of pharmacological agents including statins (simvastin) [321], opioids [322], nicotinic receptor modulators [323], were reported to protect neuronal cells, including mDAns, against apoptosis, in either in vivo or in vitro models of PD, via the activation of Wnt/β-catenin signaling pathway, thus supporting the critical role of this signaling system for the protection of mDAergic neurons against cytotoxicity.
Other studies indicated the potential of Wnt1-like agonist, such as Wnt1 inducible signaling pathway protein 1 (WISP1), a downstream target in the Wnt1 pathway, to block neurodegeneration [324]. WISP1, also known as CCN4, is a member of the six secreted extracellular matrix associated CCN family of proteins that mediate a wide panel of critical functions including the ability to prevent apoptosis, control caspase activation, and oversee autophagy [324]. The neuroprotective mechanism of WISP1 was shown to involve pivotal pathways controlling neuronal death/survival, such as phosphoinositide 3 kinase/Akt1, apoptotic mitochondrial signaling and included Bad, Bax, Bim, and Bcl-xL [324]. Thus, targeting downstream pathways of Wnt1, such as WISP1, may represent po tential avenues for neurorepair upon CNS injury. Additionally, antagonizing Wnt signaling inhibitors, such as sFRP3, can improve age‐related cellular changes in BubR1 progeroid ageing mouse [325].
Wnts and Wnt-agonists may offer a therapeutical potential, albeit there are risks and concerns for direct modulation of Wnt/βeta-catenin signaling, regarding both the safety and selectivity [105]. The field of small molecules as potential tools to selectively activate or inhibit Wnt/β-catenin signaling is increasingly recognized [[326], [327], [328], [329]], with a number of both established and novel modulators, including Wnt3a-like agonists, siRNAs and inhibitors targeting GSK-3β, Axin-LRP5/6 or transcription factor complexes (recently reviewed in 73]. Additionally, manipulation of Wnt/βeta-catenin signaling has become an attractive strategy to ameliorate in vitro differentiation protocols for increasing the fraction of midbrain DAergic neurons [see 260 and Refs herein].
6.1.3. Herbal derivatives targeting Nrf2/Wnt signaling cascades against PD
Herbal derivatives, (primarily from the Traditional Chinese Medicine), endowed with pharmacological properties (including anti-cancer, anti-bacterial, and anti-oxidant activities) are being studied for their neuroprotective potential, at least in part via a robust effect on Nrf2/Wnt signaling cascades with beneficial effects on neuron survival, proliferation, immunomodulation and neurogenesis [[330], [331], [332], [333], [334], [335], [336], [337], [338], [339], [340], [341], [342]]. Flavonoids, such as Curcumin ameliorates DAergic neuronal oxidative damage via activation of the Akt/Nrf2/Nfkb pathway and Wnt/β-catenin crosstalk [[333], [334], [335]]. In particular, in a 6-OHDA rodent model of PD, Curcumin enhanced viability, survival and attenuated apoptosis of primary cells by activating the Wnt/β-catenin signaling pathway [335]. Higher Wnt3a and β-catenin mRNA and protein expressions, enhanced SOD and glutathione peroxidase (GSH-Px) contents, and elevated mitochondrial membrane potential were observed, by contrast, antagonizing Wnt signaling with Dkk1 efficiently reversed curcumin-induced neuroprotection [335]. Resveratrol, is well recognized to alleviate oxidative stress and inflammation; and in SAMP8 mice, treatment with resveratrol reverted ageing and neurodegenerative conditions mitigating mitochondrial dysfunction and immune overactivity, and activated Wnt signaling [336]. Rhodiola extracts and salidroside exhibit anti-oxidant properties activating the Wnt/β-Catenin signaling pathway in rats with Parkinson's Disease [337]. Naringenin (NAR), displays anti-oxidant, cardioprotective, anti-inflammatory and neuroprotective activities, and may also confer neuroprotection in primary rat midbrain neuron-glia co-cultures, via the activation of astroglial Nrf2 [338], thus targeting astroglial Nrf2 to support dopaminergic neurons. Bruceine D, one of the active components of Brucea javanica, which is widely used to treat cancer in China, was recently shown [339] to activate Nrf2 to restrain Parkinson's disease in mice through suppressing oxidative stress and inflammatory response. Ginsenoside Rg1 is a major bioactive ingredient in Panax ginseng that has low toxicity and has been shown to have neuroprotective effects also through the Wnt/β-catenin signaling pathway in both in vivo and in vitro models of Parkinson's disease [340].
6.1.4. Blocking A1 astrocyte exacerbated phenotype in PD
Specifically targeting glial activation states with NSAIDs and other anti-inflammatory drugs as a therapeutic option to mitigate DAergic degeneration of PD has long been studied in an ever-increasing number pre-clinical/clinical models [33,[37], [38], [39],93,166,[184], [185], [186], [187]] “polarizing” the interest in targeting inflammation-dependent neurodegeneration, recently reviewed in light of the dual harmful/beneficial switch of glial phenotypes [260], and often intersecting the modulation of Wnt signaling patway [65,67,[72], [73], [74],343]. A vast panel of therapeutic opportunities has been disclosed, disease-modifying and symptomatic therapies under development for PD, including anti-inflammatory drugs recently discussed by Elkouzi and co. [344 and Refs herein]. Recent studies target the A1 proinflammatory status, with novel compounds, exploiting astrocyte immunomodulatory potential of neurotransmitters receptors and transcription factors, as summarized.
As recalled in section 4, LPS-activated microglia secreting proinflammatory cytokines, including IL-1β and TNF- α, can switch astrocyte beneficial status into a highly cytotoxic phenotype, resulting in the inhibition of astrocyte's property to promote neuronal survival, outgrowth, and synaptogenesis [251]. Conversely, blocking of A1 astrocyte conversion by microglia exert neuroprotective functions in models of PD [72,74]. For example, a novel glucagon-like peptide-1 receptor agonist, NLY01 [345] proved its neuroprotective effects in two mouse models of PD, in a glia-dependent manner [346]. NLY01 prevented microglia from releasing inflammatory mediators known to convert astrocytes into a neurotoxic A1 reactive subtype [346]. Also, both ex vivo and in vitro studies indicated the possibility to rejuvenate aged microglia cells by treatment conditioned media from young astrocytes, that efficiently reverse the up-regulated levels of TNF-α, IL1-β and RNS of microglial cells acutely isolated from the aged midbrain, whereas Wnt/β-catenin antagonism abolished the Wnt-induced cytokine suppression, supporting Wnt intermediacy (72,74].
6.1.5. Activating astrocyte neurotransmitter receptors in PD
Activation of astrocytic neurotransmitter receptors has been recently gained attention in light of a number of findings showing a robust immuno-modulatory role of astrocyte neurotrasmitter receptors [[347], [348], [349], [350], [351], [352], [353]]. Hence, the α7 nicotinic acetylcholine receptors (α7-nAChRs) expressed in glial cells [348] has been reported to represent a potential link between inflammation and neurodegeneration in PD, with a potential intermediacy of the Nrf2/Wnt/β-catenin signaling activation [[348], [349], [350]]. Also, nicotine can exert a protective effect on H2O2-induced astrocyte apoptosis and glial cell-derived neurotrophic factor (GDNF) downregulation, while this effect was abolished by an α7-nAChR-selective antagonist [349]. The underlying mechanisms might involve alleviation of mitochondrial membrane potential loss, stabilization of the Bax/Bcl-2 balance, and inhibition of cleaved caspase-9 activity and Nrf2/Wnt activation, through α7-nAChR activation [349]. Recently, the same authors showed the ability of nicotine to increase expression levels of Wnt/β-catenin signaling proteins in the PD mouse model or in the SH-SY5Y cells treated by 1-methyl-4-phenylpyridinium, and these effects were also reversed by α7-siRNA treatment in vivo or in vitro, which suggested a contribution of Wnt/β-catenin signaling in endogenous α7-nAChR neuroprotective mechanisms [323,349].
Serotonin receptors on astrocytes have been also proposed as potential therapeutic targets in PD [354,355]. Stimulation of astrocyte serotonin 1A (5-HT1A) receptors promotes astrocyte proliferation and upregulation of the antioxidant molecules metallothionein (MT)-1,2, which protect dopaminergic neurons against oxidative stress [[351], [352], [353]]. Rotigotine, an anti-parkinsonian drug that can bind to dopamine and 5-HT1A receptors, was reported to increase the number of astrocytes and MT-1,2 expression in cultured astrocytes. Pretreatment with conditioned media from rotigotine-treated astrocytes significantly inhibited 6-OHDA-induced dopaminergic toxicity, and these effects were blocked by co-administration with a 5-HT1A antagonist [353]. Thus, astrocytes targeting 5-HT1A receptors may contribute to neuroprotection, through upregulation of MT expression in astrocytes [[351], [352], [353]].
6.1.6. Activating astrocyte transcription factors
Activation of glial transcription factors, such as Nurr1 (originally known as a transcription factor specific for developing and adult mDAn), has been shown to protect neighboring mDAns by reducing synthesis and release of astroglial proinflammatory cytokines [354,355]. Two antimalarial drugs, amodiaquine (AQ) and chloroquine stimulated the transcriptional function of Nurr1 and enhanced the Nurr1-dependent transcriptional activation of DA-specific genes. Interestingly, they further enhanced transrepression of neurotoxic proinflammatory gene expression in microglia [354,355]. Of specific interest, pharmacological stimulation of Nurr1 causes both neuroprotection and anti-inflammatory effects in the 6-OHDA lesion model of PD [354]. In this study a novel Nurr1 agonist, SA00025, was tested for both its efficiency to induce the transcription of dopaminergic target genes in vivo and prevent dopaminergic neuron degeneration in an inflammation exacerbated 6-OHDA-lesion model of PD [354]. Here, the neuroprotective effects of SA00025 in this mDAn degeneration model were associated with changes in microglial morphology indicative of a resting state and a decrease in microglial specific IBA-1 staining intensity in the SNpc and reduced astrocyte IL-6 levels, underscoring the potential of small molecules targeting neuronal and glial Nurr1 as neuroprotective strategy for PD [354].
6.1.7. Illuminating astrocytes
Light stimulation of astrocytes with optogenetics has now being applied to reveal the function of astrocytes in physiology and pathology. This novel approach modulates the bidirectional interactions between astrocytes and neurons in both synaptic and neuronal networks [356,357]. This strategy allows specific cell stimulation by external illumination which may remotely manipulate intracellular pathways in single cells, using channelrhodopsin-2 (ChR2) activation to allow cationic currents to depolarize genetically targeted cells [356, 357, and Refs herein]. Optogenetic activation of VM astrocytes was used by Yang et al. [358], showing that it can enhance the DAergic differentiation of stem cells and promote brain repair in PD models in vivo and in vitro with basic fibroblast growth factor (bFGF) being identified as a prominent mediator. As recently reviewed by Xie et al. [357], recent findings highlight the possibility to use optogenetics to control the release of gliotransmitters and regulate astrocytic membrane channels. Thus, the capability of modulating the bidirectional interactions between astrocytes and neurons in both synaptic and neuronal networks via optogenetics represents a novel way to manipulate astrocytes that might represent a feasible and be an effective way to investigate the potential therapeutic strategy for PD and other NDs [356,357]. Mederos and co [356] used a new approach based on selective expression of melanopsin, a G-protein-coupled photopigment, in astrocytes to trigger Ca2+ signaling. Using the genetically encoded Ca2+ indicator GCaMP6f and two-photon imageing, the authors showed that melanopsin, a G-protein-coupled photopigment expressed by a small subset of mammalian retinal ganglion cells, is both competent to stimulate robust IP3-dependent Ca2+ signals in astrocyte fine processes, and to evoke an ATP/Adenosine-dependent transient boost of hippocampal excitatory synaptic transmission [356]. In vivo, melanopsin-astrocyte activation enhances episodic-like memory, suggesting melanopsin as an optical tool that could recapitulate the wide range of regulatory actions of astrocytes on neuronal networks in behaving animals [356].
Interestingly, Zhao et al. [359] reported that optical depolarization of young neuroblast, i.e., DCX-expressing cells, induced cognitive recovery and maturation of newborn neurons after traumatic brain injury via Wnt/βeta-catenin signaling pathway activation. Moreover, Wnt signaling also plays a key role in controlling neuron activity-regulated neurotrophic factor (i.e. Bdnf) expression [360], supporting Wnt signaling as a potential actor in neural stimulation, and likely underlying beneficial effects on neurogenesis and cognitive functions [reviewed in 73].
6.1.8. Astrocyte genetic manipulation for GF delivery, co-transplantation and reprogramming therapies for PD
The possibility of modifying the pathologic brain environment by utilizing the neurotrophic properties of astrocytes is being actively pursued using different approaches, including derivation of astrocyte differentiated from NSCs or hiPSC sources, or conversion of fibroblasts directly into astrocytes, astrocyte co-transplantaion and astrocyte reprogramming [[293], [294], [295], [296], [297], [298], [299]]. Genetic manipulation of astrocytes to deliver at the location of active neuropathology potential neuroprotective molecules was studied by different authors Gene therapy approaches for PD can deliver neurotrophic factors such as GDNF or neurturin via neuronal transgene expression. Drinkut and co [361] expressed GDNF exclusively in astrocytes and evaluated the efficacy of this approach in the mouse MPTP- and rat 6-OHDA models of PD. Here, astrocytic GDNF expression showed a localized but efficient alternative to current gene therapeutic strategies for the treatment of PD, suggesting astrocyte neurotrophic factor expression as novel venues for neurotrophic factor-based gene therapy targeting severe diseases of the brain [361].
In the study of de Pin et al. [362], the conditional BDNF delivery from astrocytes was shown to rescue memory deficits, spine density, and synaptic properties in the 5xFAD mouse model of AD. Brulet et al. [363] used the transcription factor NEUROD1, previously shown to convert reactive glial cells to neurons in the cortex, to determine whether astrocyte-to-neuron transdifferentiation can occur under physiological conditions. Here, using adeno-associated virus 9 (AAV9), which crosses the blood-brain barrier without injury, to deliver NEUROD1 to astrocytes through an intravascular route, the athors found that a small, but significant number of non-reactive astrocytes converted to neurons in the striatum, but not the cortex, and suggested that a single transcription factor can induce astrocyte-to-neuron conversion under physiological conditions, potentially facilitating future clinical approaches long after the acute injury phase [363].
Transplantation of astrocytes generated in vitro by directed differentiation of glial precursor (GPA) cells, producing multiple agents including BDNF, GDNF, neurturin and IGF1, into the 6-OHDA-lesioned rat striatum was addressed by Proshel et al. [364]. Hence, a rescue of parvalbumin+ GABAergic interneurons, the expression of the synaptic modulatory proteins thrombospondin-1 and 2, and the increased expression of the synaptic protein synaptophysin was observed in 6-OHDA-lesioned striatum of GPA-transplanted rats, suggesting multiple benefits of GPA transplants without requiring prior genetic manipulation [364].
Co-grafting astrocytes derived from the midbrain remarkably enhance NSC-based cell therapeutic outcomes along with robust DA neuron engraftment in PD rodents [295]. To improve the therapeutic outcomes of NSC transplantation, Song and coworkers [295], exploited the neurotrophic actions of astrocytes coupled to Nurr1/Foxa2 functions in this cell type, using the 6-OHDA rat model of PD. Hence, Nurr1+Foxa2 engineering in astrocytes further improved astrocytic function to protect midbrain neurons against toxins, in vitro [295]. The authors identified potential neurotrophic cytokines, Wnt signaling molecules, extracellular matrix (ECM) proteins, and antiinflammatory and antioxidant factors that may mediate the actions of co-transplantation of VM astrocytes with embryonic VM precursors in the rat striatum [295]. Hence, several growth/neurotrophic factors (Gdnf, Nt3, Shh, Wnt1, Wnt3, Wnt5), trophic ECM proteins (Col6a2, Fn1, Thbs-1), and antioxidant proteins (Gpx3 and Sod3) was upregulated in the striatum transplanted with VM-astrocytes compared with the striatum grafted with control cortex astrocytes/NPCs (or cortex astrocytes) at 1 month after transplantation [295]. In addition, the expression of a panel of anti-inflammatory markers (Ifnb, Ccl17, Il-1r2, Il-1rn, Ym1, and Il-10) was also greater in brains grafted with VM-Ast, while the expression of pro-inflammatory genes did not increase, and some genes (iNOS, Il-1β, Cxcl11) were downregulated [295].
Adult NSC transplantation into the aged MPTP-injured SN showed [72] that a large fraction of transplanted NSCs acquired an astrocytic phenotype both at the SN level and at the midbrain peri-aqueductal regions, where a robust migration of NSCs and NSC-derived astrocytes to the Wnt-sensitive midbrain dopaminergic niche accompanied a time dependent dopaminergic neurorescue [72]. Remarkably, in NSC-grafted mice, the NSC-derived astrocytes and the endogous astrocytes expressed Wnt1, mediating dopaminergic neurorescue and microglia down-modulation. Here, the expression levels of various pro-inflammatory genes involved in inflammation-dependent DA neurotoxicity were up-regulated 4-8-fold in SNpc tissues from MPTP/PBS mice, vs. saline/PBS mice [72]. In contrast, MPTP/NSC mice showed a significant down-regulation of all these inflammatory mRNA species, including Tnf and Tnfrsf1a, Il1, Nos2, Nfkb and the phagocyte oxidase Cybb (gp91phox), vs. MPTP/PBS controls [72]. Additionally, looking at the astrocytic Nrf2-Hmox axis, it was found NSC grafts promoted a significant up-regulation of both genes vs. MPTP/PBS mice. This finding suggested an ability of NSC grafts to normalize the unbalance of pro-inflammatory and oxidative stress markers in the aged MPTP-lesioned SN milieu [72]. The overall gene and protein expression profiling data suggested that NSC grafts induce major changes in several oxidative/inflammatory and Nrf2/Wnt/β-catenin-dependent genes within the SN of aged MPTP mice [72].
Grafting primary VM astrocytes was also approached [74] to address the ability of transplanted VM astrocytes, by themselves, to promote DAergic neurorescue, in vivo. Using primary mouse post-natal (P2-3) VM astrocytes as a graft source for unilateral transplantation above the subtantia nigra (SN) of middle-aged MPTP mice after the onset of motor symptoms, Serapide and co [74] addressed their potential to ameliorate the aged and MPTP-injured microenvironment, thus mitigating nigrostriatal toxicity. Here, grafting VM astrocytes was shown to rejuvenate the SN microenvironment via a downmodulation of microglial pro-inflammatory status and the resilience of astrocyte Nrf2/Wnt-signaling axis [74]. Importantly VM-As-grafts promoted an enriched expression of canonical Wnt signature genes in the middle-aged MPTP-injured VM, which included Wnt1, β-catenin, and Fzd1 receptor, thus triggering a “Wnt-on” state, likely contributing to DAergic neurorescue [74]. Accordingly, a number of endogenous Wnt signaling antagonists, such as GSK-3β, Dkk1 and sFrp1 were downregulated by 1 wpt in the grafted VM-As SN tissues and As-cultures, when compared to their MPTP/PBS-As counterparts exhibiting a significant up-regulation of endogenous Wnt antagonists, supporting the “Wnt off” state of the aged MPTP-injured midbrain [74].
Activating Nrf2/Wnt/β-catenin signaling, as observed in VM-As-grafted mice and VM-As-derived cultures, or MPTP/PBS-As-derived cultures treated with the GSK-3β-antagonist, powerfully reverted oxidative/nitrosative stress markers promoting DAn survival and growth. Hence, Wnt/β-catenin activation in VM astrocyte–grafted mice was suggested to promote a beneficial effect, switching the microglial M1 phenotype to a likely more quiescent anti-inflammatory state, whereas a lack of astrocyte-derived Wnt-microglial dialogue as observed in aged MPTP-injured SNpc, likely contributed to the loss of major Nrf2-antioxidant genes, in turn responsible for astrocyte failure to protect and rescue/repair the injured DAn of middle-aged MPTP mice [74]. Together, these findings argue in favor of reciprocal astrocyte/microglial/neuron interactions, supporting resilience Nrf2/HO1/Wnt/β-catenin axis as a critical mediator in promoting neuroprotection in astrocyte-grafted mice [74].
Reprogramming has emerged as a powerful approach for cell replacement therapy that would avoid the use of cell transplantation. Earlier and more recent studies, provided strong evidence that a persistent expression of neurogenic fate determinants, driven by silencing-resistant retroviral vectors, can instruct astroglia from the postnatal cortex, in vitro, to mature into fully functional, synapse-forming neurons [369−374]. Additionally, both rodent and human fibroblasts have been reprogrammed into induced dopaminergic neurons capable of promoting some functional recovery after transplantation to animal models of basal ganglia injury [[365], [366], [367], [368], [369], [370]].
In the work of Rivetti di Val Cervo et al. [294], three transcription factors, NEUROD1, ASCL1 and LMX1A, and the microRNA miR218 (designated NeAL218), were used to reprogram human astrocytes in vitro, and mouse astrocytes in vivo, into induced dopamine neurons [294]. The reprogramming efficiency was also improved by treatment in vitro with small molecules activating Wnt signaling, besides other pathways. Especially, in a mouse model of Parkinson's disease, NeAL218 alone reprogrammed adult striatal astrocytes into induced dopamine neurons that are excitable, being also capable to correct some aspects of motor behavior in vivo [294]. With the optimization of this approach, the authors suggest a novel therapeutical potential that may enable clinical therapies for Parkinson's disease by delivery of genes rather than cells [294]. Importantly, the possibility to generate patient-specific astrocytes capable of recapitulating a patient's genetic background and disease phenotype now permits to investigate the role of astrocyte dysfunction in human disease when addressing neuron crosstalk in coculture paradigms with patient derived neurons [371, 372]. Accordingly, PD patient-derived cells and tissue show increased oxidative stress, impaired mitochondrial function and downregulation of Wnt/β-catenin signaling, at least in part resulting from astrocyte dysfunction. Hence, healthy astrocytes can protect and attenuate mitochondrial dysfunctions in human iPSC-derived dopaminergic neurons from PD patients [371, 372], thus corroborating the critical role of astrocyte-neuron crosstalk, oxidative stress and Wnt signaling in human PD physiopathology. Then, using both patient-derived neurons and astrocytes represent a challenging approach for drug-screening and discovery.
Together, and increasing number of treatments in pre-clinical PD models, are being reported to up-regulate Nrf2/Wnt pathways, exerting anti-oxidant/anti-inflammatory actions, associated with neuroprotective activities, supporting astrocyte-Nrf2/Wnt resilience as therapeutic target in neuroinflammatory-dependent neurodegeneration.
7. Conclusive remarks and future directions to “cure” PD
Oxidative stress and inflammation-driven neurotoxicity have long been suggested to play a central role in the progression of various NDs, including PD, where activation of innate immune responses engender a dangerous crosstalk with harmful consequences for the vulnerable mDAns innervating the striatum, and responsible for the inesorable degeneration of the nigrostriatal dopaminergic pathway responsible for the neuropathological hallmarks leading to the classical motor features of PD. PD treatment options are conventionally focused on dopamine replacement and provision of symptomatic relief, but do not modify the progressive neurodegenerative cell loss associated with PD that, in many cases, results in debilitating side-effects.
Herein, in summarizing the compelling evidence underscoring glia and its mediators as vital actors in PD, we highlight Nrf2, the master regulator of cellular defense against oxidative stress and inflammation, and Wnt/β-catenin signaling cascade, a vital pathway for mDAn neurogenesis and neuroprotection, emerging as critical intertwinned actors in mDAn resilience. Hence, molecular mechanisms of Nrf2/Wnt/β-catenin/GSK-3β signaling regulation highlight an intense crosstalk. Importantly in PD, the significance of this circuitry is suggested in different PD model systems indicating an intricate Nrf2/PI3–K/Akt-Wnt/β-catenin cooperation in the regulation of mDAn homeostasis, immunomodulation, and neurogenesis. Hence a decline of an Nrf2/Wnt prosurvival axis with age underlies PD mutations and a variety of noxious environmental exposures driving PD neurodegeneration. Notably major PD-associated genes, including SNCA, PRKN, LRRK2, PINK1, DJ-1, and VPS35 are linked to the Nrf2/Wnt signaling axis, and contribute to abnormal immune responses, via a dysfunction of mitochondrial, lysosomal, proteosomal and autophagic pathways. Such a pathological interplay finally triggers a self-perpetuating cycle of oxidative stress, inflammation and neuronal death. Importantly enough, besides the vast array of harmful environmental factors, life style, especially exercising, may promote a panel of self-protective anti-ageing mechanisms with beneficial effect in PD, at least in part via activation of Nrf2/Wnt resilience.
In fact, the Nrf2/Wnt signaling system crosstalks with major pathways regulating astrocyte-microglia and glia-neuron interactions, in response to ageing, inflammation and PD neurodegeneration. Remarkably on the one hand, Nrf2/HO-1 and NF-ĸB affect each other to coordinate anti-oxidative and inflammatory responses determining the fate of innate response. On the other hand, M1 pro-inflammatory microglia status, can be mitigated by astrocyte-microglia crosstalk via Wnt/GSK-3 and Nr2/HO-1 interplay, aimed at counterbalancing the hostile inflammatory microenvironment.
Resilience of Nrf2/Wnt signaling axis crosstalk can restrict inflammation and oxidative stress, prevents neuronal loss, rescuing the dysfunctional or imperilled neurons and contributing to dopaminergic plasticity in the adult brain. Rejuvenating the dysfunctional astrocyte-neuron communication network in PD-based models by specifically manipulating astrocytes, now provides a new approach for drug development for the treatment of neurological diseases, as PD. A remarkable potential exists to revert some of the described age-dependent changes, including molecular, cellular and pharmacological correction of glial dysfunction. Harnessing astrocyte-derived Nrf2/Wnts and neurotrophic factors, or blocking A1 harmful phenotype with glucagon-like peptide-1 receptor agonist, NLY01; activating glial Nurr1, or activating, astrocyte neurotransmitter receptors; as well as by antagonizing GSK-3β in either neurons and glial cells. Fascinatingly, “astrocyte's fil rouge” brings back to Nrf2/Wnt resilience, as a potential and robust mean to boost anti-oxidant, anti-ageing, self-protective and pro-regenerative programs for NDs, and in particular PD (Fig. 8).
Novel frontiers regard the use optogenetics to illuminate astrocytes, thus promoting their neuroprotective and proneurogenic functions. Additionally, genetic manipulation of astrocytes and co-grafting techniques to improve the injured microenvironment, activate dopaminergic neurogenesis and incite neurorepair are being studied. Hence, derivation of astrocyte differentiated from NSCs or hiPSC sources, or conversion of fibroblasts directly into astrocytes, astrocyte reprogramming into neurons, represent some of these very challenging new research areas. Coupled to the possibility to generate patient-specific astrocytes capable of recapitulate a patient's genetic background and disease phenotype on the one hand, and using co-culture techniques with PD-specific neurons, it is also possible to screen new molecules for drug discovery and therapeutical applications to treat neurological diseases. Al together efforts in the underpinning of astrocyte-neuron crosstalk at a molecular and cellular levels will be fundamental to the identification of novel diagnostic tools and treatments for NDs to promote functional recovery (Fig. 8).
Conflict of interest
No conflict of interest to declare.
Acknowledgements
I wish to aknowledge the contribution of past and present members of the Marchetti laboratory; S. Pluchino and members of its laboratory.
In particular I wish to aknowledge, Dr C Tirolo for the art work and all original images produced in this Review, Dr F LEpiscopo, for the careful revision of the Reference section. I am endebted with all of my collaborators, including Maria F Serapide, Carmela Giachino, Nunzio Testa and Salvatore Caniglia for their unvaluable work documented in the Reference section.
This research program has received support from different funding agencies, the Italian Ministry of Health (Ricerca Corrente 2009–2020), the Italian Ministry of Education, University and Research (MIUR), the University of Catania (Chance); OASI Research Institute-IRCCS, Troina, Italy OASI (IRCCS), Troina (EN) Italy and BIOMETEC, at the University of Catania, Medical School, Catania, Italy.
Abbreviation
- 6-OHDA
6-hydroxydopamine
- α-syn
α-synuclein
- ALS
amiotrophic lateral sclerosis
- APC
adenomatous polyposis coli
- AQ
amodiaquine
- Aq-PVR
aqueduct-periventricular region
- AR
AR-AO14418
- bFGF
basic fibroblast growth factor
- BDNF
brain-derived neurotrophic factor
- BrdU
bromodeoxyuridine
- CHIR
CHIR99021
- CK1α
casein kinase 1α
- CPu
corpus striatum
- CRD
cysteine-rich domain
- CSF
cerebrospinal fluid
- DA
dopamine
- DAn
dopaminergic neurons
- DAT
dopamine transporter
- DDO-7263
[5-(3,4-Difluorophenyl)-3-(6-methylpyridin-3-yl)-1,2,4-oxadiazole]
- EGF
epidermal growth factor
- DCX
doublecortin
- Dkk
Dickkopf
- DG
dentate gyrus
- Dvl
Dishevelled
- Fzd
Frizzled
- GBA1
β-glucocerebrosidase 1
- GFAP
glial fibrillary acidic protein
- GDNF
glial-derived neurotrophic factor
- GF
growth factor
- GSH
Glutathione
- GSK-3β
glycogen synthase kinase 3β
- Hmox1
heme oxygenase 1
- IBA1
ionized calcium-binding adapter molecule 1
- icv
intracerebroventricular
- iNOS
inducible nitric oxide synthase
- iPSC
induced pluripotent stem cell
- Keap1
Kelch-like ECH-associated protein 1
- l-DOPA
levodopa
- LEF
lymphoid enhancer binding factor
- LGR
leucine-rich repeat-containing G-protein coupled receptors
- LRP
low-density lipoprotein receptor-related protein
- LRRK2
leucine-rich repeat kinase 2
- MAP2a
microtubule-associated protein 2a
- MMF
Monomethylfumarate
- mDAn
midbrain dopaminergic neurons
- mNSC
mid brain neural stem progenitor cells
- MPP+
1-methyl-4-phenylpyridinium
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- mtDNA
mitochondrial DNA
- ND
neurodegenerative disorder
- NLY0
glucagon-like peptide-1 receptor agonist
- Nrf2
nuclear factor erythroid-2-related factor 2
- NSAID
non-steroidal anti-inflammatory drug
- NSC
neural stem cell
- Nurr1
nuclear receptor-related factor 1
- PCP
planar cell polarity
- PD
Parkinson's disease
- PHOX
phagocyte oxidase
- PI3K
phosphoinositide 3-kinase
- PINK1
PTEN-induced putative kinase
- PP2A
protein phosphatase-2A
- PTX
paclitaxel
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- Rspo
R-spondin
- SAMP8
senescence associated mouse prone 8
- sFRP
secreted Fzd-related protein
- SGZ
subgranular zone
- Shh
sonig hedgehog
- SN
substantia nigra
- SNpc
substantia nigra pars compacta
- SPO-2
spondin-2
- Cpu
caudate putament
- SVZ
subventricular zone
- TCF
T cell factor
- TH
tyrosine hydroxylase
- TNFα
tumor necrosis factor α
- VM
ventral midbrain
- VTA
ventral tegmental area
- WIF
Wnt inhibitory factor
- WIP1
wild-type p53-induced phosphatase 1
- Wnt1
wingless-type mouse mammary tumor virus integration site1
References
- 1.McGeer P.L., Itagaki S., Boyes B.E. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 2.Schapira A.H., Cooper J.M., Dexter D. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 3.Glinka Y., Tipton K.F., Youdim M.B. Mechanism of inhibition of mitochondrial respiratory complex I by 6-hydroxydopamine and its prevention by desferrioxamine. Eur. J. Pharmacol. 1998;351:121–129. doi: 10.1016/s0014-2999(98)00279-9. [DOI] [PubMed] [Google Scholar]
- 4.Langston J.W., Forno L.S., Tetrud J. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen M., Wong Y.C., Ysselstein D. Synaptic, mitochondrial, and lysosomal dysfunction in Parkinson's disease. Trends Neurosci. 2019;42:140–149. doi: 10.1016/j.tins.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Subramaniam S.R., Chesselet M.F. Mitochondrial dysfunction and oxidative stress in Parkinson's disease. Prog. Neurobiol. 2013;106–107:17–32. doi: 10.1016/j.pneurobio.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Del Tredici K., Braak H. Lewy pathology and neurodegeneration in premotor Parkinson's disease. Mov. Disord. 2012;27:597–607. doi: 10.1002/mds.24921. [DOI] [PubMed] [Google Scholar]
- 8.Blesa J., Trigo-Damas I., Quiroga-Varela A. Oxidative stress and Parkinson's disease. Front. Neuroanat. 2015;9:91. doi: 10.3389/fnana.2015.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Killinger B.A., Kordower J.H.J. Spreading of alpha-synuclein - relevant or epiphenomenon? Neurochem. Sep. 2019;150(5):605–611. doi: 10.1111/jnc.14779. Epub 2019 Jul 9.PMID: 31152606 Review. [DOI] [PubMed] [Google Scholar]
- 10.Chu Y., Muller S., Tavares A. Intrastriatal alpha-synuclein fibrils in monkeys: spreading, imageing and neuropathological changes. 2019;142(11):3565–3579. doi: 10.1093/brain/awz296. 2019 Nov 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braak H., Ghebremedhin E., Rüb U. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- 12.Garrido-Gil P., Rodriguez-Perez A.I., Dominguez-Meijide A. Bidirectional neural interaction between central dopaminergic and gut lesions in Parkinson's disease models. Mol. Neurobiol. 2018;55:7297–7316. doi: 10.1007/s12035-018-0937-8. [DOI] [PubMed] [Google Scholar]
- 13.Reichmann H., Brandt M.D., Klingelhoefer L. The nonmotor features of Parkinson's disease: pathophysiology and management advances. Curr. Opin. Neurol. 2016;29(4):467–473. doi: 10.1097/WCO.0000000000000348. https://doi:10.1097/WCO.0000000000000348 [DOI] [PubMed] [Google Scholar]
- 14.Obeso J.A., Stamelou M., Goetz C.G. Past, present, and future of Parkinson's disease: a special essay on the 200th Anniversary of the Shaking Palsy. Mov. Disord. 2017;32:1264–1310. doi: 10.1002/mds.27115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schapira A.H., Olanow C.W., Greenamyre J.T. Slowing of neurodegeneration in Parkinson's disease and Huntington's disease: future therapeutic perspectives. Lancet. 2014;384:545–555. doi: 10.1016/S0140-6736(14).61010-2. [DOI] [PubMed] [Google Scholar]
- 16.Jankovic J. Pathogenesis-targeted therapeutic strategies in Parkinson's disease. Mov. Disord. 2019;34:866–875. doi: 10.1002/mds.27534. [DOI] [PubMed] [Google Scholar]
- 17.Marchetti B., Abbracchio M.P. To be or not to be (inflammed). Is that the question in anti-inflammatory drug therapy of neurodegenerative diseases? Trends in Pharmacol. Sci. 2005;26:517–525. doi: 10.1016/j.tips.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 18.Betarbet R., Canet-Aviles R.M., Sherer T.B. Intersecting pathways to neurodegeneration in Parkinson's disease: effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system. Neurobiol. Dis. 2006;22:404–420. doi: 10.1016/j.nbd.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 19.Edwards T.M., Myers J.P. Environmental exposures and gene regulation in disease etiology. Environmental Health Perspectives. Environ. Health Perspect. 2007;115:1264–1270. doi: 10.1289/ehp.9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cannon J.R., Greenamyre J.T. Gene-environment interactions in Parkinson's disease: specific evidence in humans and mammalian models. Neurobiol. Dis. 2013;57:38–46. doi: 10.1016/j.nbd.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirsch E.C., Jenner P., Przedborski S. Pathogenesis of Parkinson's disease. Mov. Disord. 2013;28:24–30. doi: 10.1002/mds.25032. [DOI] [PubMed] [Google Scholar]
- 22.Dzamko N., Geczy C.L., Halliday G.M. Inflammation is genetically implicated in Parkinson's disease. Neuroscience. 2015;302:89–102. doi: 10.1016/j.neuroscience.2014.10.028. [DOI] [PubMed] [Google Scholar]
- 23.Liu H.F., Ho P.W., Leung G.C. Combined LRRK2 mutation, ageing and chronic low dose oral rotenone as a model of Parkinson's disease. Sci. Rep. 2017;18:40887. doi: 10.1038/srep40887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langston J.W. The MPTP story. J. Parkinsons Dis. 2017;7:S11–S19. doi: 10.3233/JPD-179006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collier T.J., Sortwell C.N., Mercado N.M. Cell therapy for Parkinson's disease: why it doesn't work every time. Mov. Disord. 2019;34:1120–1127. doi: 10.1002/mds.27742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blauwendraat C., Heilbron K., Vallerga C.L. Parkinson's disease age at onset genome-wide association study: defining heritability, genetic loci, and α-synuclein mechanisms. Mov. Disord. 2019;34:866–875. doi: 10.1002/mds.27659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fernando M.B., Ahfeldt T., Brennand K.J. Modeling the complex genetic architectures of brain disease. Nat. Genet. 2020;52:363–369. doi: 10.1038/s41588-020-0596-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dorsey E.R., Sherer T., Okun M.S. The emerging evidence of the Parkinson pandemic. J. Parkinsons Dis. 2018;8:S3–S8. doi: 10.3233/JPD-181474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang C., Wang Y., Li X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao M. Cytokine storm and immunomodulatory therapy in COVID-19: role of chloroquine and anti-IL-6 monoclonal antibodies. Int. J. Antimicrob. Agents. 2020 Jun;55(6):105982. doi: 10.1016/j.ijantimicag.2020.105982. Epub 2020 Apr 16.PMID: 32305588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langston J.W., Ballard P.A., Jr. Parkinson's disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N. Engl. J. Med. 1983;309:310. doi: 10.1056/nejm198308043090511. [DOI] [PubMed] [Google Scholar]
- 32.Gao H.M., Liu B., Zhang W. Critical role of microglia NADPH-oxidase-derived free radicals in the in vitro MPTP model of Parkinson's disease. Faseb. J. 2003;17:1954–1966. doi: 10.1096/fj.03-0109fje. [DOI] [PubMed] [Google Scholar]
- 33.Whitton P.S. Inflammation as a causative factor in the aetiology of Parkinson's disease. Br. J. Pharmacol. 2007;150:963–976. doi: 10.1038/sj.bjp.0707167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu X., Zhang D., Pang H. Macrophage antigen complex-1 mediates reactive microgliosis and progressive dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. J. Immunol. 2008;181:7194–7204. doi: 10.4049/jimmunol.181.10.7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanaan N.M., Kordower J.H., Collier T.J. Age-related changes in dopamine transporters and accumulation of 3-nitrotyrosine in rhesus monkey midbrain dopamine neurons: relevance in selective neuronal vulnerability to degeneration. Eur. J. Neurosci. 2008;27:3205–3215. doi: 10.1111/j.1460-9568.2008.06307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGeer P.L., McGeer E.G. Glial reactions in Parkinson's disease. Mov. Disord. 2008;23:474–483. doi: 10.1002/mds.21751. [DOI] [PubMed] [Google Scholar]
- 37.Hirsch E.C., Hunot S. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–397. doi: 10.1016/S1474-4422(09).70062-6. [DOI] [PubMed] [Google Scholar]
- 38.Przedborski S. Inflammation and Parkinson's disease pathogenesis. Mov. Disord. 2010;25:S55–S57. doi: 10.1002/mds.22638. [DOI] [PubMed] [Google Scholar]
- 39.Whitton P.S. Neuroinflammation and the prospects for anti-inflammatory treatment of Parkinson's disease. Curr. Opin. Invest. Drugs. 2010;11:788–794. PMID: 20571974. [PubMed] [Google Scholar]
- 40.Gao H.M., Hong J.S. Gene-environment interactions: key to unraveling the mystery of Parkinson's disease. Prog. Neurobiol. 2011;94:1–19. doi: 10.1016/j.pneurobio.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao H.M., Zhang F., Zhou H. Neuroinflammation and α-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson's disease. Environ. Health Perspect. 2011;119:807–814. doi: 10.1289/ehp.1003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marchetti B., L'Episcopo F., Morale M.C. Uncovering novel actors in astrocyte-neuron crosstalk in Parkinson's disease: the Wnt/β-catenin signaling cascade as the common final pathway for neuroprotection and self-repair. Eur. J. Neurosci. 2013;37:1550–1563. doi: 10.1111/ejn.12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barcia C., Guillemin G.J., Curtin J.F. Editorial: glial cells: managers of neuro-immunity. Front. Cell. Neurosci. 2016;10:60. doi: 10.3389/fncel.2016.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGeer P.L., Rogers J., McGeer E.G. Inflammation, antiinflammatory agents, and alzheimer's disease: the last 22 years. J Alzheimers Dis. 2016;54:853–857. doi: 10.3233/JAD-160488. [DOI] [PubMed] [Google Scholar]
- 45.Duffy M.F., Collier T.J., Patterson J.R. Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J. Neuroinflammation. 2018;15:129. doi: 10.1186/s12974-018-1171-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tansey M.G., Romero-Ramos M. Immune system responses in Parkinson's disease: early and dynamic. Eur. J. Neurosci. 2019;49:364–383. doi: 10.1111/ejn.14290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanchez-Guajardo V., Barnum C.J., Tansey M.G., Romero-Ramos M. Neuroimmunological processes in Parkinson's disease and their relation to α-synuclein: microglia as the referee between neuronal processes and peripheral immunity. ASN Neuro. 2013;5:113–139. doi: 10.1042/AN20120066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kannarkat G.T., Boss J.M., Tansey M.G. The role of innate and adaptive immunity in Parkinson's disease. J. Parkinsons Dis. 2013;3:493–514. doi: 10.3233/JPD-130250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harms A.S., Thome A.D., Yan Z. Peripheral monocyte entry is required for alpha-Synuclein induced inflammation and Neurodegeneration in a model of Parkinson disease. Exp. Neurol. 2018;300:179–187. doi: 10.1016/j.expneurol.2017.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Campos-Acuña J., Elgueta D., Pacheco R. T-Cell-Driven inflammation as a mediator of the gut-brain Axis involved in Parkinson's disease. Front. Immunol. 2019;10:239. doi: 10.3389/fimmu.2019.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abdalkader M., Lampinen R., Kanninen K.M. Targeting Nrf2 to suppress ferroptosis and mitochondrial dysfunction in neurodegeneration. Front. Neurosci. 2018;10:466. doi: 10.3389/fnins.2018.00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson D.A., Johnson J.A. Nrf2-a therapeutic target for the treatment of neurodegenerative diseases. Free Radic. Biol. Med. 2015;88:253–267. doi: 10.1016/j.freeradbiomed.2015.07.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cuadrado A., Rojo A.I., Wells G. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019;18:295–317. doi: 10.1038/s41573-018-0008-x. [DOI] [PubMed] [Google Scholar]
- 54.Holmstrom K.M., Kostov R.V., Dinkova-Kostova A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicology. 2016;1:80–91. doi: 10.1016/j.cotox.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dinkova-Kostova A.T., Abramov A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015;88:179–188. doi: 10.1016/j.freeradbiomed.2015.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ryoo I.G., Kwak M.K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 2018;359:24–33. doi: 10.1016/j.taap.2018.09.014. [DOI] [PubMed] [Google Scholar]
- 57.Kaidery A.M., Ahuja M., Thomas B. Crosstalk between Nrf2 signaling and mitochondrial function in Parkinson's disease. Mol. Cell. Neurosci. 2019;101:103413. doi: 10.1016/j.mcn.2019.103413. Dec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Surmeier D.J. Determinants of dopaminergic neuron loss in Parkinson's disease. FEBS J. 2018;285(19):3657–3668. doi: 10.1111/febs.14607. https://doi:10.1111/febs.14607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen P.C., Vargas M.R., Pani A.K. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson's disease: critical role for the astrocyte. Proc. Natl. Acad. Sci. U.S.A. 2009;106:2933–2938. doi: 10.1073/pnas.0813361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lastres-Becker I., Ulusoy A., Innamorato N.G. α-Synuclein expression and Nrf2-deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson's disease. Hum. Mol. Genet. 2012;21:3173–3192. doi: 10.1093/hmg/dds143. [DOI] [PubMed] [Google Scholar]
- 61.L'Episcopo F., Tirolo C., Testa N. Reactive astrocytes and Wnt/β-catenin signaling link nigrostriatal injury to repair in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Neurobiol. Dis. 2011;41:508–527. doi: 10.1016/j.nbd.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.L'Episcopo F., Serapide M.F., Tirolo C. A Wnt1 regulated Frizzled-1/β-catenin signaling pathway as a candidate regulatory circuit controlling mesencephalic dopaminergic neuron-astrocyte crosstalk: therapeutical relevance for neuron survival and neuroprotection. Mol. Neurodegener. 2011;13:6–49. doi: 10.1186/1750-1326-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.L'Episcopo F., Tirolo C., Testa N. Plasticity of subventricular zone neuroprogenitors in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) mouse model of Parkinson's disease involves crosstalk between inflammatory and Wnt/β-catenin signaling pathways: functional consequences for neuroprotection and repair. J. Neurosci. 2012;32:2062–2085. doi: 10.1523/JNEUROSCI.5259-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.L'Episcopo F., Tirolo C., Testa N. Ageing-induced Nrf2-ARE pathway disruption in the subventricular zone (SVZ) drives neurogenic impairment in parkinsonian mice via PI3K-Wnt/β-catenin dysregulation. J. Neurosci. 2013;33:1462–1485. doi: 10.1523/JNEUROSCI.3206-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marchetti B., Pluchino S. Wnt your brain be inflamed? Yes, it Wnt! Trends Mol. Med. 2013;19:144–156. doi: 10.1016/j.molmed.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.L'Episcopo F., Tirolo C., Testa N. Wnt/β-catenin signaling is required to rescue midbrain dopaminergic progenitors and promote neurorepair in ageing mouse model of Parkinson's disease. Stem Cell. 2014;32:2147–2163. doi: 10.1002/stem.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harvey K., Marchetti B. Regulating Wnt signaling: a strategy to prevent neurodegeneration and induce regeneration. J. Mol. Cell Biol. 2014;6:1–2. doi: 10.1093/jmcb/mju002. [DOI] [PubMed] [Google Scholar]
- 68.Arenas E. Wnt signaling in midbrain dopaminergic neuron development and regenerative medicine for Parkinson's disease. J. Mol. Cell Biol. 2014;6:42–53. doi: 10.1093/jmcb/mju001. [DOI] [PubMed] [Google Scholar]
- 69.Wurst W., Prakash N. Wnt-1 regulated genetic networks in midbrain dopaminergic neuron development. J. Mol. Cell Biol. 2014;6:34–41. doi: 10.1093/jmcb/mjt046. [DOI] [PubMed] [Google Scholar]
- 70.Joksimovic M., Awatramani R. Wnt/β-catenin signaling in midbrain dopaminergic neuron specification and neurogenesis. J. Mol. Cell Biol. 2014;6:27–33. doi: 10.1093/jmcb/mjt043. [DOI] [PubMed] [Google Scholar]
- 71.Marchetti B. 2018. Wnt/β-Catenin signaling pathway governs a full program for dopaminergic neuron survival, neurorescue and regeneration in the MPTP mouse model of Parkinson's disease. Int. J. On mol. Sci., 19. Pii: E3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.L'Episcopo F., Tirolo C., Peruzzotti-Jametti L. Neural stem cell grafts promote astroglia-driven neurorestoration in the aged parkinsonian brain via wnt/β-catenin signaling. Stem Cell. 2018;36(8):1179–1197. doi: 10.1002/stem.2827. [DOI] [PubMed] [Google Scholar]
- 73.Marchetti B., Tirolo C., L'Episcopo F. Parkinson's Disease, Ageing and Adult Neurogenesis: wnt/β-catenin signaling as the key to unlock the mystery of endogenous brain repair. Ageing Cell. 2020;19 doi: 10.1111/acel.13101. e13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Serapide M.F., L'Episcopo F., Tirolo C. Boosting anti-oxidant self-defenses by grafting astrocytes rejuvenates the aged microenvironment and mitigates nigrostriatal toxicity in parkinsonian brain via an Nrf2-driven Wnt/β-catenin pro-survival axis. Frontiers Ageing Neuroscience. 2020;12:24. doi: 10.3389/fnagi.2020.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Janda C.Y., Waghray D., Levin A.M., Thomas C., Garcia K.C. Structural basis of wnt recognition by frizzled. Science. 2012;337:59–64. doi: 10.1126/science.1222879. https://doi:10.1126/science.1222879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cullinan S.B., Gordan J.D., Jin J. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1ligase. Mol. Cell Biol. 2004;24:8477–8486. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kaspar J.W., Niture S.K., Jaiswal A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009;47:1304. doi: 10.1016/j.freeradbiomed.2009.07.035. 1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Itoh K., Wakabayashi N., Katoh Y. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rada P., Rojo A.I., Chowdhry S. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell Biol. 2011;31:1121–1133. doi: 10.1128/MCB.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chowdhry S., Zhang Y., McMahon M. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. 2013;32:3765–3781. doi: 10.1038/onc.2012.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cuadrado A. Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/beta-TrCP. Free Radic. Biol. Med. 2015;2015(88):147–157. doi: 10.1016/j.freeradbiomed.2015.04.029. [DOI] [PubMed] [Google Scholar]
- 82.Hayes J.D., Chowdhry S., Dinkova-Kostova A.T., Sutherland C. Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of β-TrCP and GSK-3. Biochem Soc Trans. 2015 Aug. 2015;43(4):611–620. doi: 10.1042/BST20150011. Epub 2015 Aug 3.PMID: 26551701 Review. [DOI] [PubMed] [Google Scholar]
- 83.Wu T., Zhao F., Gao B. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014;28:708–722. doi: 10.1101/gad.238246.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kaneko M., Nomura Y. ER signaling in unfolded protein response. Life Sci. 2003;74:199–205. doi: 10.1016/j.lfs.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 85.Omura T., Kaneko M., Okuma Y. Endoplasmic reticulum stress and Parkinson's disease: the role of HRD1 in averting apoptosis in neurodegenerative disease. Oxid Med Cell Longev. 2013;2013:239854. doi: 10.1155/2013/239854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jiang T., Harder B., Rojo de la Vega M. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015;88:199–204. doi: 10.1016/j.freeradbiomed.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pajares M., Jimenez-Moreno N., Garcia-Yague A.J. Transcription factor NFE2L2/Nrf2 is a regulator of macroautophagy genes. Autophagy. 2016;12:1902–1916. doi: 10.1080/15548627.2016.1208889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jope R.S., Cheng Y., Lowell J. Stressed and inflamed, can GSK3 be blamed? Trends Biochem. Sci. 2017;42:180–192. doi: 10.1016/j.tibs.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beurel E., Grieco S.F., Joper R.S. Glycogen synthase kinase-3 (GSK-3): regulation, actions, and disease. Pharmacol. Ther. 2015;148:114–131. doi: 10.1016/j.pharmthera.2014.11.016. https://doi:10.1016/j.pharmthera.2014.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Duka T., Duka V., Joyce J.N. Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson's disease models. Faseb. J. 2009;23:2820–2830. doi: 10.1096/fj.08-120410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Petit-Paitel A1, Brau F., Cazareth J. Involvment of cytosolic and mitochondrial GSK-3beta in mitochondrial dysfunction and neuronal cell death of MPTP/MPP-treated neurons. PloS One. 2009;4 doi: 10.1371/journal.pone.0005491. e5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Credle J.J., George J.L., Wills J. GSK-3β dysregulation contributes to Parkinson's-like pathophysiology with associated region-specific phosphorylation and accumulation of tau and α-synuclein. Cell Death Differ. 2015;22:838–851. doi: 10.1038/cdd.2014.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.L'Episcopo F., Tirolo C., Caniglia S. Targeting Wnt signaling at the neuroimmune interface for dopaminergic neuroprotection/repair in Parkinson's disease. J. Mol. Cell Biol. 2014;6:13–26. doi: 10.1093/jmcb/mjt053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.L'Episcopo F., Drouin-Ouellet J., Tirolo C., Pulvirenti A., Giugno R., Testa N., Caniglia S., Serapide M.F., Cisbani G., Barker R.A., Cicchetti F., Marchetti B. GSK-3β-induced Tau pathology drives hippocampal neuronal cell death in Huntington's disease: involvement of astrocyte-neuron interactions. Cell Death Dis. 2016;7:e2206. doi: 10.1038/cddis.2016.104. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cuadrado A., Kügler S., Lastres-Becker I. Pharmacological targeting of GSK-3 and NRF2 provides neuroprotection in a preclinical model of tauopathy. Redox Biol. 2018 Apr;14:522–534. doi: 10.1016/j.redox.2017.10.010. Epub 2017 Nov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rada P., Rojo A.I., Offergeld A. WNT-3A regulates an Axin1/NRF2 complex that regulates antioxidant metabolism in hepatocytes. Antioxidants Redox Signal. 2015;22:555–571. doi: 10.1089/ars.2014.6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ojeda L., Gao J., Hooten K.G. 2011. Critical role of PI3K/Akt/GSK3β in motoneuron specification from human neural stem cells in response to FGF2 and EGF. PLoS One, 6:e23414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nair V.D., Olanow C.W. Differential modulation of Akt/Glycogen synthase kinase-3beta pathway regulates apoptotic and cytoprotective signaling responses. J. Biol. Chem. 2008;283:15469–15478. doi: 10.1074/jbc.M707238200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chong Z.Z., Shang Y.C., Hou J. Wnt1 neuroprotection translates into improved neurological function during oxidant stress and cerebral ischemia through AKT1 and mitochondrial apoptotic pathways. Oxid. Med. Cell Longev. 2010;3:153–165. doi: 10.4161/oxim.3.2.11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maiese K. Moving to the rhythm with clock (circadian) genes, autophagy, mTOR, and SIRT1 in degenerative disease and cancer. Curr. Neurovascular Res. 2017;14:299–304. doi: 10.2174/1567202614666170718092010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Silva-Palacios A., Ostolga-Chavarría M., Zazueta C., Königsberg M. Nrf2: molecular and epigenetic regulation during ageing. Ageing Res Rev.Nov. 2018;47:31–40. doi: 10.1016/j.arr.2018.06.003. Epub 2018 Jun 18. [DOI] [PubMed] [Google Scholar]
- 102.Song J.L., Nigam P., Tektas S.S., Selva E. microRNA regulation of Wnt signaling pathways in development and disease. Cell. Signal. 2015;27(7):1380–1391. doi: 10.1016/j.cellsig.2015.03.018. https://doi:10.1016/j.cellsig.2015.03.018 Jul. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Anderegg A., Awatramani R. Making a mes: a transcription factor-microRNA pair governs the size of the midbrain and the dopaminergic progenitor pool. Neurogenesis (Austin) 2015;2(1):e998101. doi: 10.1080/23262133.2014.998101. https://doi:10.1080/23262133.2014.998101 Mar 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sajadimajd S., Khazaei M. Oxidative stress and cancer: the role of Nrf2. Curr. Cancer Drug Targets. 2018;18(6):538–557. doi: 10.2174/1568009617666171002144228. 2018. [DOI] [PubMed] [Google Scholar]
- 105.Nusse R., Clevers H. Wnt/β-Catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169(6):985–999. doi: 10.1016/j.cell.2017.05.016. https://doi:10.1016/j.cell.2017.05.016 [DOI] [PubMed] [Google Scholar]
- 106.Schmidt S. Genetic mouse models for Parkinson's disease display severe pathology in glial cell mitochondria. Hum. Mol. Genet. 2011;20:1197–1211. doi: 10.1093/hmg/ddq564. [DOI] [PubMed] [Google Scholar]
- 107.Booth H.D.E., Hirst W.D., Wade-Martins R. The role of astrocyte dysfunction in Parkinson's disease pathogenesis. Trends Neurosci. 2017;40:358–370. doi: 10.1016/j.tins.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Barodia S.K., McMeekin L.J., Creed R.B. PINK1 phosphorylates ubiquitin predominantly in astrocytes. NPJ Parkinsons Dis. 2019;5:29. doi: 10.1038/s41531-019-0101-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gillardon F., Schmid R., Draheim H. Parkinson's disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience. 2012;208:41–48. doi: 10.1016/j.neuroscience.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 110.Kim J.H., Choi D.J., Jeong H.K. DJ-1 facilitates the interaction between STAT1 and its phosphatase, SHP-1, in brain microglia and astrocytes: a novel anti-inflammatory function of DJ-1. Neurobiol. Dis. 2013;60:1–10. doi: 10.1016/j.nbd.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 111.Ashley A.K. DJ-1 mutation decreases astroglial release of inflammatory mediators. Neurotoxicology. 2016;52:198–203. doi: 10.1016/j.neuro.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 112.Choi I., Choi D.J., Yang H. PINK1 expression increases during brain development and stem cell differentiation, and affects the development of GFAP-positive astrocytes. Mol. Brain. 2016;9:5. doi: 10.1186/s13041-016-0186-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Berwick D.C., Harvey K. The importance of Wnt signaling for neurodegeneration in Parkinson's disease. Biochem. Soc. Trans. 2012;40:1123–1128. doi: 10.1042/BST20120122. [DOI] [PubMed] [Google Scholar]
- 114.Galli S., Lopes D.M., Ammari R. Deficient Wnt signaling triggers striatal synaptic degeneration and impaired motor behaviour in adult mice. Nat. Commun. 2014;5 doi: 10.1038/ncomms5992. 4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Berwick D.C., Harvey K. The regulation and deregulation of Wnt signaling by PARK genes in health and disease. J. Mol. Cell Biol. 2014;6:3–12. doi: 10.1093/jmcb/mjt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Berwick D.C., Javaheri B., Wetzel A. Pathogenic LRRK2 variants are gain-of-function mutations that enhance LRRK2-mediated repression of β-catenin signaling. Mol. Neurodegener. 2017;12:9. doi: 10.1186/s13024-017-0153-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Williams E.T., Chen X., Moore D.J. VPS35, the retromer complex and Parkinson's disease. J. Parkinsons Dis. 2017;7:219–233. doi: 10.3233/JPD-161020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Belenkaya T.Y., Wu Y., Tang X. The retromer complex influences Wnt secretion by recycling wntless from endosomes to the trans-Golgi network. Dev. Cell. 2008;14:120–131. doi: 10.1016/j.devcel.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 119.Nixon-Abell J., Berwick D.C., Grannó S. Protective LRRK2 R1398H variant enhances GTPase and wnt signaling activity. Front. Mol. Neurosci. 2016;9:18. doi: 10.3389/fnmol.2016.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Awad O., Panicker L.M., Deranieh R.M. Altered differentiation potential of Gaucher's disease iPSC neuronal progenitors due to Wnt/β-catenin downregulation. Stem Cell Rep. 2017;9:1853–1867. doi: 10.1016/j.stemcr.2017.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kwok J.B., Hallupp M., Loy C.T. GSK3β polymorphism alter transcription and splicing in Parkinson's disease. Ann. Neurol. 2005;58:829–839. doi: 10.1002/ana.20691. [DOI] [PubMed] [Google Scholar]
- 122.Lee J.H., Han J.H., Kim H. Parkinson's disease-associated LRRK2-G2019S mutant acts through regulation of SERCA activity to control ER stress in astrocytes. Acta Neuropathol Commun. 2019;7:68. doi: 10.1186/s40478-019-0716-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hamza T.H., Zabetian C.P., Tenesa A. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat. Genet. 2010;42:781–785. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Latourelle J.C., Dimitriu A., Hadzi T.C. Evaluation of Parkinson disease risk variants as expression-QTLs. PloS One. 2012;7:e46199. doi: 10.1371/journal.pone.0046199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang L., Deng J., Pan Q. Targeted methylation sequencing reveals dysregulated Wnt signaling in Parkinson disease. J. Genet. Genom. 2016;43:587–592. doi: 10.1016/j.jgg.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 126.Chuang Y.H., Paul K.C., Bronstein J.M. Parkinson's disease is associated with DNA methylation levels in human blood and saliva. Genome Med. 2017;9:76. doi: 10.1186/s13073-017-0466-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chuang Y.H., Lu A.T., Paul K.C. Longitudinal epigenome-wide methylation study of cognitive decline and motor progression in Parkinson's disease. J. Parkinsons Dis. 2019;9:389–400. doi: 10.3233/JPD-181549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Harms A.S., Cao S., Rowse A.L. MHCII is required for α-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J. Neurosci. 2013;33:9592–9600. doi: 10.1523/JNEUROSCI.5610-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fellner L., Irschick R., Schanda K. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia. 2013;61:349–360. doi: 10.1002/glia.22437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rannikko E.H., Weber S.S., Kahle P.J. Exogenous alpha-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015;16:57. doi: 10.1186/s12868-015-0192-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rocha E.M., De Miranda B., Sanders L.H. Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson's disease. Neurobiol. Dis. 2018;109(Pt B):249–257. doi: 10.1016/j.nbd.2017.04.004. Epub 2017 Apr 8. [DOI] [PubMed] [Google Scholar]
- 132.Skibinski G., Hwang V., Ando D.M. Nrf2 mitigates LRRK2- and α-synuclein-induced neurodegeneration by modulating proteostasis. Proc. Natl. Acad. Sci. U. S. A. 2017;114(5):1165–1170. doi: 10.1073/pnas.1522872114. 2017 Jan 31. Epub 2016 Dec 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Frank-Cannon T.C., Tran T., Ruhn K.A. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J. Neurosci. 2008;28:10825–10834. doi: 10.1523/JNEUROSCI.3001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Solano R.M., Menéndez J., Casarejos M.J. Midbrain neuronal cultures from parkin mutant mice are resistant to nitric oxide-induced toxicity. Neuropharmacology. 2006;51:327–340. doi: 10.1016/j.neuropharm.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 135.Solano R.M., Casarejos M.J., Menéndez-Cuervo J. Glial dysfunction in parkin null mice: effects of ageing. J. Neurosci. 2008;28:598–611. doi: 10.1523/JNEUROSCI.4609-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Casarejos M.J., Menéndez J., Solano R.M. Susceptibility to rotenone is increased in neurons from parkin null mice and is reduced by minocycline. J. Neurochem. 2006;97:934–946. doi: 10.1111/j.1471-4159.2006.03777.x. [DOI] [PubMed] [Google Scholar]
- 137.Singh K., Han K., Tilve S. Parkin targets NOD2 to regulate astrocyte endoplasmic reticulum stress and inflammation. Glia. 2018;66:2427–2437. doi: 10.1002/glia.23482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rawal N., Corti O., Sacchetti P. Parkin protects dopaminergic neurons from excessive Wnt/beta-cateninsignaling. BBRC (Biochem. Biophys. Res. Commun.) 2009;388:473–478. doi: 10.1016/j.bbrc.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 139.Petit A., Kawarai T., Paitel E. Wild-type PINK1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by Parkinson disease-related mutations. J. Biol. Chem. 2005;280:34025–34032. doi: 10.1074/jbc.M505143200. [DOI] [PubMed] [Google Scholar]
- 140.Fiesel F.C., Ando M., Hudec R. (Patho-)physiological relevance of PINK1-dependent ubiquitin phosphorylation. EMBO Rep. 2015;16:1114–1130. doi: 10.15252/embr.201540514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Murata H., Takamatsu H., Liu S. 2015. NRF2 regulates PINK1 expression under oxidative stress conditions. PLoS One, 10:e0142438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sun L., Shen R., Agnihotri S.K. Lack of PINK1 alters glia innate immune responses and enhances inflammation-induced, nitric oxide-mediated neuron death. Sci. Rep. 2018;8:383. doi: 10.1038/s41598-017-18786-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Choi I., Kim J., Jeong H.K. PINK1 deficiency attenuates astrocyte proliferation through mitochondrial dysfunction, reduced AKT and increased p38 MAPK activation, and downregulation of EGFR. Glia. 2013;61:800–812. doi: 10.1002/glia.22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Palomera-Avalos V., Griñán-Ferré C., Puigoriol-Ilamola D. Resveratrol protects SAMP8 brain under metabolic stress: focus on mitochondrial function and wnt pathway. Mol. Neurobiol. 2017;54:1661–1676. doi: 10.1007/s12035-016-9770-0. [DOI] [PubMed] [Google Scholar]
- 145.Zhang Q., Wu X., Chen P. The mitochondrial unfolded protein response is mediated cell-non-autonomously by retromer-dependent wnt signaling. Cell. 2018;174:870–883. doi: 10.1016/j.cell.2018.06.029. e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bernkopf D.B., Jalal K., Brückner M. Pgam5 released from damaged mitochondria induces mitochondrial biogenesis via Wnt signaling. J. Cell Biol. 2018;217:1383–1394. doi: 10.1083/jcb.201708191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rasmussen M.L., Ortolano N.A., Romero-Morales A.I. Wnt signaling and its impact on mitochondrial and cell cycle dynamics in pluripotent stem cells. Genes. 2018 doi: 10.3390/genes9020109. 9: pii: E 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Singh S., Mishra A., Mohanbhai S.J. Axin-2 knockdown promote mitochondrial biogenesis and dopaminergic neurogenesis by regulating Wnt/β-catenin signaling in rat model of Parkinson's disease. Free Radical Biol. Med. 2018;129:73–87. doi: 10.1016/j.freeradbiomed.2018.08.033. [DOI] [PubMed] [Google Scholar]
- 149.Xia S.R., Wen X.Y., Fan X.L. Wnt2 overexpression protects against PINK1 mutant-induced mitochondrial dysfunction and oxidative stress. Mol. Med. Rep. 2020;21:2633–2641. doi: 10.3892/mmr.2020.11066. [DOI] [PubMed] [Google Scholar]
- 150.Bertolin G., Ferrando-Miguel R., Jacoupy M. The TOMM machinery is a molecular switch in PINK1 and PARK2/PARKIN-dependent mitochondrial clearance. Autophagy. 2013;9:1801–1817. doi: 10.4161/auto.25884. [DOI] [PubMed] [Google Scholar]
- 151.Waak J., Weber S.S., Waldenmaier A. Regulation of astrocyte inflammatory responses by the Parkinson's disease-associated gene DJ-1. Faseb. J. 2009;23:2. doi: 10.1096/fj.08-125153. [DOI] [PubMed] [Google Scholar]
- 152.Kim K.S., Kim J.S., Park J.Y. DJ-1 associates with lipid rafts by palmitoylation and regulates lipid rafts-dependent endocytosis in astrocytes. Hum. Mol. Genet. 2013;22:4805–4817. doi: 10.1093/hmg/ddt332. [DOI] [PubMed] [Google Scholar]
- 153.Mullett S.J., Hinkle D.A. DJ-1 knock-down in astrocytes impairs astrocyte-mediated neuroprotection against rotenone. Neurobiol. Dis. 2009;33:28–36. doi: 10.1016/j.nbd.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lev N., Barhum Y., Ben-Zur T. Knocking out DJ-1 attenuates astrocytes neuroprotection against 6-hydroxydopamine toxicity. J. Mol. Neurosci. 2013;50:542–550. doi: 10.1007/s12031-013-9984-9. [DOI] [PubMed] [Google Scholar]
- 155.Mullett S.J., Hinkle D.A. DJ-1 deficiency in astrocytes selectively enhances mitochondrial Complex I inhibitor-induced neurotoxicity. J. Neurochem. 2011;117:375–387. doi: 10.1111/j.1471-4159.2011.07175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mullett S.J., Di Maio R., Greenamyre J.T. DJ-1 expression modulates astrocyte-mediated protection against neuronal oxidative stress. J. Mol. Neurosci. 2013;49(3):507–511. doi: 10.1007/s12031-012-9904-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.De Miranda B.R., Rocha E.M., Bai Q. Astrocyte-specific DJ-1 overexpression protects against rotenone-induced neurotoxicity in a rat model of Parkinson's disease. Neurobiol. Dis. 2018;115:101–114. doi: 10.1016/j.nbd.2018.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Dzamko N., Gysbers A., Perera G. Toll-like receptor 2 is increased in neurons in Parkinson's disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol. 2017;133:303–319. doi: 10.1007/s00401-016-1648-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lee H., James W.S., Cowley S.A. LRRK2 in peripheral and central nervous system innate immunity: its link to Parkinson's disease. Biochem. Soc. Trans. 2017;45:131–139. doi: 10.1042/BST20160262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kim B., Yang M.S., Choi D. 2012. Impaired inflammatory responses in murine Lrrk2–knockdown brain microglia. PLoS One, 7:e34693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Moehle M.S., Webber P.J., Tse T. LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci. 2012;32:1602–1611. doi: 10.1523/JNEUROSCI.5601-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Henry A.G., Aghamohammadzadeh S., Samaroo H. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum. Mol. Genet. 2015;24:6013–6028. doi: 10.1093/hmg/ddv314. [DOI] [PubMed] [Google Scholar]
- 163.Lee J.H., Han J.H., Kim H. Parkinson's disease-associated LRRK2-G2019S mutant acts through regulation of SERCA activity to control ER stress in astrocytes. Acta Neuropathol Commun. 2019;7(1):68. doi: 10.1186/s40478-019-0716-4. 2019 May 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Collier T.J., Lipton J., Daley B.F. Ageing-related changes in the nigrostriatal dopamine system and the response to MPTP in nonhuman primates: diminished compensatory mechanisms as a prelude to parkinsonism. Neurobiol. Dis. 2007;26:56–65. doi: 10.1016/j.nbd.2006.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Boger H.A., Granholm A.C., McGinty J.F. A dual-hit animal model for age-related parkinsonism. Prog. Neurobiol. 2010;90:217–229. doi: 10.1016/j.pneurobio.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.L'Episcopo F., Tirolo C., Testa N. Switching the microglial harmful phenotype promotes lifelong restoration of subtantia nigra dopaminergic neurons from inflammatory neurodegeneration in aged mice. Rejuvenation Res. 2011;14:411–424. doi: 10.1089/rej.2010.1134. [DOI] [PubMed] [Google Scholar]
- 167.Rodriguez M., Rodriguez-Sabate C., Morales I. Parkinson's disease as a result of ageing. Ageing Cell. 2015;14:293–308. doi: 10.1111/acel.12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Giguère N., Burke Nanni S., Trudeau L.E. On cell loss and selective vulnerability of neuronal populations in Parkinson's disease. Front. Neurol. 2018;19:455. doi: 10.3389/fneur.2018.00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Niraula A., Sheridan J.F., Godbout J.P. Microglia priming with ageing and stress. Neuropsychopharmacology. 2017;42:318–333. doi: 10.1038/npp.2016.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Collier T.J., Kanaan N.M., Kordower J.H. Ageing and Parkinson's disease: different sides of the same coin? Mov. Disord. 2017;32:983–990. doi: 10.1002/mds.27037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Viña J., Borras C., Gomez-Cabrera M.C. A free radical theory of frailty. Free Radic. Biol. Med. 2018;124:358–363. doi: 10.1016/j.freeradbiomed.2018.06.028. [DOI] [PubMed] [Google Scholar]
- 172.Vina J., Borras C., Abdelaziz K.M. The free radical theory of ageing revisited: the cell signaling disruption theory of ageing. Antioxidants Redox Signal. 2013;19:779–787. doi: 10.1089/ars.2012.5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Vina J., Borras C., Sanchis-Gomar F. Pharmacological properties of physical exercise in the elderly. Curr. Pharmaceut. Des. 2013;20:3019–3029. doi: 10.2174/13816128113196660704. [DOI] [PubMed] [Google Scholar]
- 174.Vina J., Sanchis-Gomar F., Martinez-Bello V. Exercise acts as a drug; the pharmacological benefits of exercise. Br. J. Pharmacol. 2012;167:1–12. doi: 10.1111/j.1476-5381.2012.01970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rietman M.L., Spijkerman A.M.W., Wong A. Antioxidants linked with physical, cognitive and psychological frailty: analysis of candidate biomarkers and markers derived from the MARK-AGE study. Mech. Ageing Dev. 2019;177:135–143. doi: 10.1016/j.mad.2018.04.007. Epub 2018 Apr 30. [DOI] [PubMed] [Google Scholar]
- 176.Hornykiewicz O. Parkinson's disease and the adaptive capacity of the nigrostriatal dopamine system: possible neurochemical mechanisms. Adv. Neurol. 1993;60:140–147. PMID: 8420131. [PubMed] [Google Scholar]
- 177.Bezard E., Gross C.E. Compensatory mechanisms in experimental and human parkinsonism: towards a dynamic approach. Prog. Neurobiol. 1998;55:93–116. doi: 10.1016/s0301-0082(98)00006-9. [DOI] [PubMed] [Google Scholar]
- 178.Hindle J.V. Ageing, neurodegeneration and Parkinson's disease. Age Ageing. 2010;39:156–161. doi: 10.1093/ageing/afp223. [DOI] [PubMed] [Google Scholar]
- 179.de la Fuente-Fernández R., Schulzer M., Kuramoto L. Age-specific progression of nigrostriatal dysfunction in Parkinson's disease. Ann. Neurol. 2011;69:803–810. doi: 10.1002/ana.22284. [DOI] [PubMed] [Google Scholar]
- 180.Boche D., Perry V.H., Nicoll J.A. Review: activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013;39(1):3–18. doi: 10.1111/nan.12011. 2013 Feb. [DOI] [PubMed] [Google Scholar]
- 181.Streit W.J. Microglial activation and neuroinflammation in Alzheimer's disease: a critical examination of recent history. Front. Ageing Neurosci. 2010;3:2–22. doi: 10.3389/fnagi.2010.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Njie E.G., Boelen E., Stassen F.R. Ex vivo cultures of microglia from young and agent rodent brain reveal age-related changes in microglial function. Neurobiol. Ageing. 2012;33:195e1–19512. doi: 10.1016/j.neurobiolageing.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Perry V.H., Teeling J. Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 2013;35:601–612. doi: 10.1007/s00281-013-0382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tansey M.G., McCoy M.K., Frank-Cannon T.C. Neuroinflammatory mechanisms in Parkinson's disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 2007;208:1–25. doi: 10.1016/j.expneurol.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tansey M.G., Goldberg M.S. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.L'Episcopo F., Tirolo C., Testa N. Glia as a turning point in the therapeutic strategy of Parkinson's disease. CNS Neurol. Disord. Drug Targets. 2010;9:349–372. doi: 10.2174/187152710791292639. [DOI] [PubMed] [Google Scholar]
- 187.L'Episcopo F., Tirolo C., Caniglia S. Combining nitric oxide release with anti-inflammatory activity preserves nigrostriatal dopaminergic innervation and prevents motor impairment in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J. Neuroinflammation. 2010;7:83. doi: 10.1186/1742-2094-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Okamoto M., Inoue K., Iwamura H.M. Reduction in paracrine Wnt3 factors during ageing causes impaired adult neurogenesis. Faseb. J. 2011;25:3570–3582. doi: 10.1096/fj.11-184697. [DOI] [PubMed] [Google Scholar]
- 189.Orellana A.M., Vasconcelos A.R., Leite Age-related neuroinflammation and changes in AKT-GSK-3β and WNT/β-CATENIN signaling in rat hippocampus. Age. 2015;7:1094–1108. doi: 10.18632/aging.100853. https://doi:10.18632/ageing.100853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Seib D.R.M., Corsini N.S., Ellwanger K. Loss of Dickkopf-1 restores neurogenesis in old age and counteracts cognitive decline. Cell Stem Cell. 2013;12:204–214. doi: 10.1016/j.stem.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 191.Bayod S., Felice P., Andrés P. Downregulation of canonical Wnt signaling in hippocampus of SAMP8 mice. Neurobiol. Ageing. 2015;36:720–729. doi: 10.1016/j.neurobiolageing.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 192.Garcìa-Velasquez L., Arias C. The emerging role of Wnt signaling dysregulation in the understanding and modification of age-associated diseases. Ageing Res. Rev. 2017;37:135–145. doi: 10.1016/j.arr.2017.06.001. https://doi:10.1016/j.arr.2017.06.001 [DOI] [PubMed] [Google Scholar]
- 193.Surh Y.J., Kundu J.K., Li M.H. Role of Nrf2-mediated heme oxygenase-1 upregulation in adaptive survival response to nitrosative stress. Arch Pharm. Res. (Seoul) 2009;32(8):1163–1176. doi: 10.1007/s12272-009-1807-8. [DOI] [PubMed] [Google Scholar]
- 194.Bitar M.S., Al-Mulla F. A defect in Nrf2 signaling constitutes a mechanism for cellular stress hypersensitivity in a genetic rat model of type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2012;301:E1119–E1129. doi: 10.1152/ajpendo.00047.2011. [DOI] [PubMed] [Google Scholar]
- 195.Gennuso F., Fernetti C., Tirolo C. Bilirubin protects astrocytes from its own toxicity inducing up-regulation and translocation of multidrug resistance-associated protein 1 (Mrp 1) Proc. Natl. Acad. Sci. U.S.A. 2004;101:2470–2475. doi: 10.1073/pnas.0308452100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zhang Y., Unnikrishnan A., DeepaS A new role for oxidative stress in ageing: the accelerated ageing phenotype in Sod1-/- mice is correlated to increased cellular senescence. Redox Biol. 2017;11:30–37. doi: 10.1016/j.redox.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.von Otter M., Landgren S., Nilsson S. Association of Nrf2-encoding NFE2L2 haplotypes with Parkinson's disease. BMC Med. Genet. 2010;11(36):2010. doi: 10.1186/1471-2350-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lastres-Becker I., García-Yagüe A.J., Scannevin R.H. Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson's disease. Antioxidants Redox Signal. 2016;25:61–77. doi: 10.1089/ars.2015.6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Innamorato N.G., Jazwa A., Rojo A.I. Different susceptibility to the Parkinson's toxin MPTP in mice lacking the redox master regulator Nrf2 or its target gene heme oxygenase-1. PloS One. 2010;5:e11838. doi: 10.1371/journal.pone.0011838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Gan L., Vargas M.R., Johnson D.A. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. Neuroscience. 2012;32 doi: 10.1523/JNEUROSCI.3049-12.2012. 17775–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Belarbi K., Cuvelier E., Destée A. NADPH oxidases in Parkinson's disease: a systematic review. Mol. Neurodegener. 2017;12:84. doi: 10.1186/s13024-017-0225-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Warner T.T., Schapira A.H.V. Genetic and environmental factors in the cause of Parkinson's disease. Ann. Neurol. 2003;53:S16–S25. doi: 10.1002/ana.10487. [DOI] [PubMed] [Google Scholar]
- 203.Peng J., Peng L., Stevenson F.F. Iron and paraquat as synergistic environmental risk factors in sporadic Parkinson's disease accelerate age-related neurodegeneration. J. Neurosci. 2007;27:6914–6922. doi: 10.1523/JNEUROSCI.1569-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Shaerzadeh F., Streit W.J., Heysieattalab S. Methamphetamine neurotoxicity, microglia, and neuroinflammation. J. Neuroinflammation. 2018;15:341. doi: 10.1186/s12974-018-1385-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Costello S., Cockburn M., Bronstein J. Parkinson's disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am. J. Epidemiol. 2009;169:919–926. doi: 10.1093/aje/kwp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Paul K.C., Sinsheimer J.S., Cockburn M. Organophosphate pesticides and PON1 L55M in Parkinson's disease progression. Environ. Int. 2017;107:75–81. doi: 10.1016/j.envint.2017.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chinta S.J., Woods G., Demaria M. Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson's disease. Cell Rep. 2018;22:930–940. doi: 10.1016/j.celrep.2017.12.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Morale M.C., Batticane N., Gallo F. Disruption of hypothalamic-pituitary-adrenocortical system in transgenic mice expressing type II glucocorticoid receptor antisense ribonucleic acid permanently impairs T cell function: effects on T cell trafficking and T cell responsiveness during postnatal development. Endocrinology. 1995;136:3949–3960. doi: 10.1210/endo.136.9.7649104. [DOI] [PubMed] [Google Scholar]
- 209.Morale M.C., Serra P., Delogu M.R. Glucocorticoid receptor deficiency increases vulnerability of the nigrostriatal dopaminergic system: critical role of glial nitric oxide. Faseb. J. 2004;18:164–166. doi: 10.1096/fj.03-0501fje. [DOI] [PubMed] [Google Scholar]
- 210.Marchetti B., Serra P.A., L'Episcopo F. Hormones are key actors in gene x environment interactions programming the vulnerability to Parkinson's disease: glia as a common final pathway. Ann. NY. Acad. Sci. 2005;1057:296–318. doi: 10.1196/annals.1356.023. [DOI] [PubMed] [Google Scholar]
- 211.Marchetti B., Kettenmann H., Streit W.J. Glia-neuron crosstalk in neuroinflammation, neurodegeneration and neuroprotection. Brain res. Rev. Special Issue. 2005;482:129–489. doi: 10.1016/j.brainresrev.2004.12.002. [DOI] [Google Scholar]
- 212.Cantuti-Castelvetri I., Keller-McGandy C., Bouzou B. Effects of gender on nigral gene expression and Parkinson disease Neurobiol Dis. 2007 Jun. 2007;26(3):606–614. doi: 10.1016/j.nbd.2007.02.009. Epub 2007 Mar 3.PMID: 17412603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Morale M.C., Serra P.A., L'Episcopo F. Estrogen, neuroinflammation and neuroprotection in Parkinson's disease: glia dictates resistance versus vulnerability to neurodegeneration. Neuroscience. 2006;138:869–878. doi: 10.1016/j.neuroscience.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 214.Morale M.C., L'Episcopo F., Tirolo C. Loss of aromatase Cytochrome P450 function as a risk factor for Parkinson's disease? Brain res. Rev. 2008;57:431–443. doi: 10.1016/j.brainresrev.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 215.Marchetti B. Estrogen, neuroinflammation and neuroprotection in Parkinson's disease: role of neuron-gla crosstalk. In: Di Giovanni G., Esposito E., editors. The Basal Ganglia Pathophysiology : Recent Advances.”, Chapter 10. 2007. pp. 253–284. Transworld Research Network-ISBN:81-7895-268-8. [Google Scholar]
- 216.Marchetti B., L'Episcopo F., Tirolo C. Vulnerability to Parkinson's disease: towards an unifying theory of disease etiology. In: Nriagu Jerome O., editor. Encyclopedia of Environmental Health. Elsevier Inc; 2011. pp. 690–704. [DOI] [Google Scholar]
- 217.De Miranda B.R., Fazzari M., Rocha E.M. Sex differences in rotenone sensitivity reflect the male-to-female ratio in human Parkinson's disease incidence. Toxicol. Sci. 2019;170:133–143. doi: 10.1093/toxsci/kfz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Marchetti B., Morale M.C., Testa N. Stress, the immune system and vulnerability to degenerative disorders of the central nervous system in transgenic mice expressing glucocorticoid receptor antisense RNA. Brain Res. Rev. 2001;37:259–272. doi: 10.1016/s0165-0173(01)00130-8. [DOI] [PubMed] [Google Scholar]
- 219.Marchetti B., Morale M.C., Brouwer J. Exposure to a dysfunctional glucocorticoid receptor from early embryonic life programs the resistance to experimental autoimmune encephalomyelitis via nitric oxide-induced immunosuppression. J. Immunol. 2002;168:5848–5859. doi: 10.4049/jimmunol.168.11.5848. [DOI] [PubMed] [Google Scholar]
- 220.Marchetti B. Cross-talk signals in the CNS: role of neurotrophic and hormonal factors, adhesion molecules and intercellular signaling agents in luteinizing hormone-releasing hormone (LHRH) neuron-astroglia interactive network. Trends in Biosci. 1997;2:1–32. doi: 10.2741/a177. [DOI] [PubMed] [Google Scholar]
- 221.Tajiri N., Yasuhara T., Shingo T. Exercise exerts neuroprotective effects on Parkinson's disease model of rats. Brain Res. 2010;1310:200–207. doi: 10.1016/j.brainres.2009.10.075. [DOI] [PubMed] [Google Scholar]
- 222.Yoon M.C., Shin M.S., Kim T.S. Treadmill exercise suppresses nigrostriatal dopaminergic neuronal loss in 6-hydroxydopamine-induced Parkinson's rats. Neurosci. Lett. 2007;423:12–17. doi: 10.1016/j.neulet.2007.06.031. [DOI] [PubMed] [Google Scholar]
- 223.Anastasía A., Torre L., de Erausquin G.A. Enriched environment protects the nigrostriatal dopaminergic system and induces astroglial reaction in the 6-OHDA rat model of Parkinson's disease. Journal of neurochem. 2009;109:755–765. doi: 10.1111/j.1471-4159.2009.06001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Xu X., Fu F., Le W. Exercise and Parkinson's disease. Int. Rev. Neurobiol. 2019;147:45–74. doi: 10.1016/bs.irn.2019.06.003. [DOI] [PubMed] [Google Scholar]
- 225.Tsou Y.H., Shih C.T., Ching C.H. Treadmill exercise activates Nrf2 antioxidant system to protect the nigrostriatal dopaminergic neurons from MPP+ toxicity. Exp. Neurol. 2015;263:50–62. doi: 10.1016/j.expneurol.2014.09.021. [DOI] [PubMed] [Google Scholar]
- 226.Chuang C.S., Chang J.C., Cheng F.C. Modulation of mitochondrial dynamics by treadmill training to improve gait and mitochondrial deficiency in a rat model of Parkinson's disease. Life Sci. 2017;191:236–244. doi: 10.1016/j.lfs.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 227.Chen Y.H., Kuo T.T., Kao J.H. Exercise ameliorates motor deficits and improves dopaminergic functions in the rat hemi-Parkinson's model. Sci. Rep. 2018;8:3973. doi: 10.1038/s41598-018-22462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Palasz E., Niewiadomski W., Gasiorowska A. Exercise-induced neuroprotection and recovery of motor function in animal models of Parkinson's disease. Front. Neurol. 2019;10:1143. doi: 10.3389/fneur.2019.01143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Tomlinson C.L., Patel S., Meek C. Physiotherapy intervention in Parkinson's disease: systematic review and meta-analysis. BMJ. 2012;345:e5004. doi: 10.1136/bmj.e5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Ridgel A.L., Peacock C.A., Fickes E.J. Active-assisted cycling improves tremor and bradykinesia in Parkinson's disease. Arch. Phys. Med. Rehabil. 2012;93:2049–2054. doi: 10.1016/j.apmr.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 231.Done A.J., Gage M.J., Nieto T. Exercise-induced Nrf2-signaling is impaired in ageing. Free Radic. Biol. Med. 2016;96:130–138. doi: 10.1016/j.freeradbiomed.2016.04.024. [DOI] [PubMed] [Google Scholar]
- 232.Done A.J., Traustadóttir T. Nrf2 mediates redox adaptations to exercise. Redox Biology. 2016;10:191–199. doi: 10.1016/j.redox.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Leem Y.H., Kato M., Chang H.J. Regular exercise and creatine supplementation prevent chronic mild stress-induced decrease in hippocampal neurogenesis via Wnt/GSK3β/β-catenin pathway. Exerc Nutrition Biochem. 2018;22:1–6. doi: 10.20463/jenb.2018.0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Jang Y., Koo J.H., Kwon I. Neuroprotective effects of endurance exercise against neuroinflammation in MPTP-induced Parkinson's disease mice. Brain Res. 2017;1655:186–193. doi: 10.1016/j.brainres.2016.10.029. [DOI] [PubMed] [Google Scholar]
- 235.Wadhwa M., Prabhakar A., Ray K. Inhibiting the microglia activation improves the spatial memory and adult neurogenesis in rat hippocampus during 48 h of sleep deprivation. J. Neuroinflammation. 2017;14:222. doi: 10.1186/s12974-017-0998-z. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 236.Lin T.W., Tsai S.F., Kuo Y.M. Physical exercise enhances neuroplasticity and delays alzheimer's disease. Brain Plast. 2018;4:95–110. doi: 10.3233/BPL-180073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Gallo F., Morale M.C., Avola R. Cross-talk between luteinizing hormone-releasing hormone (LHRH) neurons and astroglial cells: developing glia release factors that accelerate neuronal differentiation and stimulate LHRH release from GT(1-1) neuronal cell line and LHRH neurons induce astroglia proliferation. Endocrine. 1995;3:863–874. doi: 10.1007/BF02738891. [DOI] [PubMed] [Google Scholar]
- 238.Magistretti P.J. Role of glutamate in neuron-glia metabolic coupling. Am. J. Clin. Nutr. 2009;90:875S–880S. doi: 10.3945/ajcn.2009.27462CC. [DOI] [PubMed] [Google Scholar]
- 239.Bélanger M., Magistretti P.J. The role of astroglia in neuroprotection. Dialogue Clin. Neuroscience. 2009;11:281–295. doi: 10.31887/DCNS.2009.11.3/mbelanger. PMCID: PMC3181926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Sofroniew M., Vinters H.B. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Bélanger M., Allaman I., Magistretti P.J. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metabol. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 242.Magistretti P.J., Allaman I. A cellular perspective on brain energy metabolism and functional imageing. Neuron. 2015;86:883–901. doi: 10.1016/j.neuron.2015.03.035. [DOI] [PubMed] [Google Scholar]
- 243.Mamczur P., Borsuk B., Paszko J. Astrocyte-neuron crosstalk regulates the expression and subcellular localization of carbohydrate metabolism enzymes. Glia. 2015;63:328–340. doi: 10.1002/glia.22753. [DOI] [PubMed] [Google Scholar]
- 244.Magistretti P.J., Allaman I. Lactate in the brain: from metabolic end-product to signaling molecule. Nat. Rev. Neurosci. 2017;19:235–249. doi: 10.1038/nrn.2018.19. [DOI] [PubMed] [Google Scholar]
- 245.Ghosh A., Streit W.J., Minghetti L. Microglia in development and disease. Clin. Dev. Immunol. 2013:736459. doi: 10.1155/2013/736459. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Davalos D., Grutzendler J., Yang G. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 247.Streit W.J. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40:133–139. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- 248.Nimmerjahn A., Kirchhoff F., Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 249.Schiess M. Non-steroidal anti-inflammatory drugs protect against Parkinson neurodegeneration: can an NSAID a day keep Parkinson disease away? Arch. Neurol. 2003;60:1043–1044. doi: 10.1001/archneur.60.8.1043. [DOI] [PubMed] [Google Scholar]
- 250.Chen H., Jacobs E.J., Schwarzschild M.A. Non-steroidal anti-inflammatory drug use and the risk for Parkinson's disease. Ann. Neurol. 2005;58:963–967. doi: 10.1002/ana.20682. [DOI] [PubMed] [Google Scholar]
- 251.Liddelow S.A., Guttenplan K.A., Clarke L.E. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Gallo F., Morale M.C., Spina-Purrello V. Basic fibroblast growth factor (bFGF) acts on both neurons and glia to mediate the neurotrophic effects of astrocytes on LHRH neurons in culture. Synapse. 2000;36:233–253. doi: 10.1002/(SICI).1098-2396(20000615).36:4<233::AID-SYN1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 253.Jha M.K., Kim J.H., Song G.J. Functional dissection of astrocyte-secreted proteins: implications in brain health and diseases. Prog. Neurobiol. 2017 doi: 10.1016/j.pneurobio.2017.12.003. pii: S0301–0082(17)30124-7. [DOI] [PubMed] [Google Scholar]
- 254.Ayadi A.E., Zigmond M.J., Smith A.D. IGF-1 protects dopamine neurons against oxidative stress: association with changes in phosphokinases. Exp. Brain Res. 2016;234:1863–1873. doi: 10.1007/s00221-016-4572-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Burda J.E., Sofroniew M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229–248. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Tyzack G.E., Sitnikov S., Barson D. Astrocyte response to motor neuron injury promotes structural synaptic plasticity via STAT3-regulated TSP-1 expression. Nat. Commun. 2014;5:4294. doi: 10.1038/ncomms5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Faulkner J.R., Herrmann J.E., Woo M.J. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J. Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Anderson M.A., Burda J.E., Ren Y., Ao Y., O'Shea T.M., Kawaguchi R. Astrocyte scar formation aids central nervous system axon regeneration. Nature. 2016;532:195–200. doi: 10.1038/nature17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Kuter K., Olech L., Glowacka U. Prolonged dysfunction of astrocytes and activation of microglia accelerate degeneration of dopaminergic neurons in the rat substantia nigra and block compensation of early motor dysfunction induced by 6-OHDA. Mol. Neurobiol. 2018;55:3049–3066. doi: 10.1007/s12035-017-0529-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Subramaniam S.R., Federoff H.J. Targeting microglial activation states as a therapeutic avenue in Parkinson's disease. Front Ageing Neurosci. 2017;9:176. doi: 10.3389/fnagi.2017.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Brodski C., Blaess S., Partanen J. 2019. Crosstalk of intercellular signaling pathways in the generation of midbrain dopaminergic neurons in vivo and from stem cells. J. Dev. Biol., 7. Pii: E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Alvarez-Builla A., Garcia-Verdugo J.M., Tramontin A.D. A unified hypothesis on the lineage of neural stem cells. Nat. Rev. Neurosci. 2001;2:2287–2293. doi: 10.1038/35067582. [DOI] [PubMed] [Google Scholar]
- 263.Lie D.C., Colamarino S.A., Song H.G. Wnt signaling regulates adult hippocampal neurogenesis. Nature. 2005;473:1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- 264.Barkho B.Z., Song H., Aimone J.B. Identification of astrocyte-expressed factors that modulate neural stem/progenitor cell differentiation. Stem Cell. Dev. 2006;15:407–421. doi: 10.1089/scd.2006.15.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Cahoy J.D., Emery B., Kaushal A. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Jiao J., Chen D.F. Induction of neurogenesis in nonconventional neurogenic regions of the adult central nervous system by niche astrocyte-produced signals. Stem Cells. 2008 May;26(5) doi: 10.1634/stemcells.2007-0513. 1221-30. Epub 2008 Mar 6.PMID: 18323412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Osakada F., Ooto S., Akagi T. Wnt signaling promotes regeneration in the retina of adult mammals. J. Neurosci. 2007;27:4210–4219. doi: 10.1523/JNEUROSCI.4193-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Castelo-Branco G., Rawal N., Arenas E. GSK-3beta inhibition/beta-catenin stabilization in ventral midbrain precursors increases differentiation into dopamine neurons. J. Cell Sci. 2004;117(Pt 24):5731–5737. doi: 10.1242/jcs.01505. [DOI] [PubMed] [Google Scholar]
- 269.Castelo-Branco G., Sousa K.M., Bryja V. Ventral midbrain glia express region-specific transcription factors and regulate dopaminergic neurogenesis through Wnt-5a secretion. Mol. Cell. Neurosci. 2006;31:251–262. doi: 10.1016/j.mcn.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 270.Inestrosa N.C., Arenas E. Emerging role of Wnts in the adult nervous system. Nat. Rev. Neurosci. 2010;11:77–86. doi: 10.1038/nrn2755. [DOI] [PubMed] [Google Scholar]
- 271.Batchelor P.E., Liberatore G.T., Wong J.Y. Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J. Neurosci. 1999;19:1708–1716. doi: 10.1523/JNEUROSCI.19-05-01708.1999. PMCID: PMC6782182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Zigmond M.J., Cameron J.L., Leak R.K. Triggering endogenous neuroprotective processes through exercise in models of dopamine deficiency. Park. Relat. Disord. 2009;3(15 Suppl):S42–S45. doi: 10.1016/S1353-8020(09)70778-3. [DOI] [PubMed] [Google Scholar]
- 273.Arkadir D., Bergman H., Fahn S. Redundant dopaminergic activity may enable compensatory axonal sprouting in Parkinson disease. Neurology. 2014;82:1093–1098. doi: 10.1212/WNL.0000000000000243. [DOI] [PubMed] [Google Scholar]
- 274.Blesa J., Trigo-Damas I., Dileone M. Compensatory mechanisms in Parkinson's disease: circuits adaptations and role in disease modification. Exp. Neurol. 2017;298(Pt B):148–161. doi: 10.1016/j.expneurol.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 275.Ho A., Blum M. Induction of interleukin-1 associated with compensatory dopaminergic sprouting in the denervated striatum of young mice: model of ageing and neurodegenerative disease. J. Neurosci. 1998;18:5614–5629. doi: 10.1523/JNEUROSCI.18-15-05614.1998. PMID:9671653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Parish C.L., Finkelstein D.I., Tripanichkul W. The role of interleukin-1, interleukin-6, and glia in inducing growth of neuronal terminal arbors in mice. J. Neurosci. 2002;22:8034–8041. doi: 10.1523/JNEUROSCI.22-18-08034.2002. PMCID: PMC6758077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Stanic D., Finkelstein D.I., Bourke D.W. Time-course of striatal re-innervation following lesions of dopaminergic SNpc neurons of the rat. Eur. J. Neurosci. 2003;18:1175–1188. doi: 10.1046/j.1460-9568.2003.02800.x. [DOI] [PubMed] [Google Scholar]
- 278.Horner P.J., Palmer T.D. New roles for astrocytes: the night life of an “astrocyte”. La vida loca! Trends Neurosci. 2003;26:597–603. doi: 10.1016/j.tins.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 279.Sandhu J.K., Gardaneh M., Iwasiow R. Astrocyte-secreted GDNF and glutathione antioxidant system protect neurons against 6OHDA cytotoxicity. Neurobiol. Dis. 2009;33:405–414. doi: 10.1016/j.nbd.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 280.Pereira C.P., Bachli E.B., Schoedon G. The wnt pathway: a macrophage effector molecule that triggers inflammation. Curr. Atherosclerosis Rep. 2009;11:236–242. doi: 10.1007/s11883-009-0036-4. [DOI] [PubMed] [Google Scholar]
- 281.Neumann J., Schaale K., Farhat K. Frizzled1 is a marker of inflammatory macrophages, and its ligand Wnt3a is involved in reprogramming Mycobacterium tuberculosis-infected macrophages. Faseb. J. 2010;24:4599–4612. doi: 10.1096/fj.10-160994. [DOI] [PubMed] [Google Scholar]
- 282.Schaale K., Neumann J., Schneider D. Wnt signaling in macrophages: augmenting and inhibiting mycobacteria-induced inflammatory responses. Eur. J. Cell Biol. 2011;90:553–559. doi: 10.1016/j.ejcb.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 283.Chae W.J., Ehrlich A.K., Chan P.Y. The wnt antagonist dickkopf-1 promotes pathological type 2 cell-mediated inflammation. Immunity. 2016;44:246–258. doi: 10.1016/j.immuni.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Chong Z.Z., Maiese K. Cellular demise and inflammatory microglial activation during beta-amyloid toxicity are governed by Wnt1 and canonical signaling pathways. Cell. Signal. 2007;19:1150–1162. doi: 10.1016/j.cellsig.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Kilander M.B., Halleskog C., Schulte G. Recombinant WNTs differentially activate β-catenin-dependent and -independent signaling in mouse microglia-like cells. Acta Physiol. 2011;203:363–372. doi: 10.1111/j.1748-1716.2011.02324.x. [DOI] [PubMed] [Google Scholar]
- 286.Halleskog C., Mulder J., Dahlström J. WNT signaling in activated microglia is proinflammatory. Glia. 2011;59:119–131. doi: 10.1002/glia.21081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Halleskog C., Dijksterhuis J.P., Kilander M.B. Heterotrimeric G protein-dependent WNT-5A signaling to ERK1/2 mediates distinct aspects of microglia proinflammatory transformation. J. Neuroinflammation. 2012;9:111. doi: 10.1186/1742-2094-9-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Halleskog C., Schulte G. WNT-3A and WNT-5A counteract lipopolysaccharide-induced pro-inflammatory changes in mouse primary microglia. Neurochem. 2013;125:803–808. doi: 10.1111/jnc.12250. [DOI] [PubMed] [Google Scholar]
- 289.Jope R.S., Yuskaitis C.J., Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem. Res. 2007;32:577–595. doi: 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Yang Y.C., Lii C.K., Wei Y.L., Li C.C., Lu C.Y., Liu K.L., Chen H.W. Docosahexaenoic acid inhibition of inflammation is partially via cross-talk between Nrf2/heme oxygenase 1 and IKK/NF-κB pathways. J. Nutr. Biochem. 2013;24(1):204–212. doi: 10.1016/j.jnutbio.2012.05.003. 2013 Jan. [DOI] [PubMed] [Google Scholar]
- 291.Cuadrado A., Martín-Moldes Z., Ye J., Isabel Lastres-Becker I. transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family. GTP-binding Protein RAC1 During Inflammation. 2014;289(22):15244–15258. doi: 10.1074/jbc.M113.540633. 014 May 30. Epub 2014 Apr 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Chinta S.J., Lieu C.A., Demaria M. Environmental stress, ageing and glial cell senescence: a novel mechanistic link to Parkinson's disease? The Journal of International Medicine. 2013;273:429–436. doi: 10.1111/joim.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Guo Z., Zhang L., Wu Z. In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer's disease model. Cell Stem Cell. 2014;14:188–202. doi: 10.1016/j.stem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Rivetti di Val Cervo P., Romanov Ra, Rivetti di Val Cervo P., Romanov Ra., Spigolon G., Spigolon G. Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson's disease model. Nat. Biotechnol. 2017;35:444–452. doi: 10.1038/nbt.3835. [DOI] [PubMed] [Google Scholar]
- 295.Song J.J., Oh S.M., Kwon O.C. Cografting astrocytes improves cell therapeutic outcomes in a Parkinson's disease. J. Clin. Invest. 2018;128:463–482. doi: 10.1172/JCI93924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Gorshkov K., Aguisanda F., Thorne N. Astrocytes as targets for drug discovery. Drug Discov. Today. 2018;23:673–680. doi: 10.1016/j.drudis.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Scott K.M., Williams-Gray C.H. Targeting aged astrocytes may Be a new therapeutic strategy in Parkinson's disease. Mov. Disord. 2018;33:758–759. doi: 10.1002/mds.27387. [DOI] [PubMed] [Google Scholar]
- 298.Antic S.D., Baker B.J., Canepari M. Editorial: new insights on neuron and astrocyte function from cutting-edge optical techniques. Front. Cell. Neurosci. 2019;13:463. doi: 10.3389/fncel.2019.00463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Janowska J., Gargas J., Ziemka-Nalecz M. Directed glial differentiation and transdifferentiation for neural tissue regeneration. Exp. Neurol. 2019;319:112813. doi: 10.1016/j.expneurol.2018.08.010. [DOI] [PubMed] [Google Scholar]
- 300.Vargas M.R., Johnson D.A., Sirkis D.W. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. Version 2. J Neurosci. 2008;28(50):13574–13581. doi: 10.1523/JNEUROSCI.4099-08.2008. 2008 Dec 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Sigfridsson M., Marangoni M., Johnson J.A. Astrocyte-specific overexpression of Nrf2 protects against optic tract damage and behavioural alterations in a mouse model of cerebral hypoperfusion. 2018;8(1):12552. doi: 10.1038/s41598-018-30675-4. Aug 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Jazwa A., Cuadrado A. Targeting heme oxygenase-1 for neuroprotection and neuroinflammation in neurodegenerative diseases. Curr. Drug Targets. 2010;11:1517–1531. doi: 10.2174/1389450111009011517. [DOI] [PubMed] [Google Scholar]
- 303.Jazwa A., Rojo A.I., Innamorato N.G. Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxidants Redox Signal. 2011;14:2347–2360. doi: 10.1089/ars.2010.3731. 2011. [DOI] [PubMed] [Google Scholar]
- 304.Gureev A.P., Popov V.N. Nrf2/ARE pathway as a therapeutic target for the treatment of Parkinson diseases. Neurochem. Res. 2019;44:2273–2279. doi: 10.1007/s11064-018-02711-2. [DOI] [PubMed] [Google Scholar]
- 305.Alarcón-Aguilar A., Luna-López A., Ventura-Gallegos J.L. Primary cultured astrocytes from old rats are capable to activate the Nrf2 response against MPP+ toxicity after tBHQ pretreatment. Aug. 2014;35(8):1901–1912. doi: 10.1016/j.neurobiolageing.2014.01.143. Epub 2014 Feb 7. [DOI] [PubMed] [Google Scholar]
- 306.Kaidery N.A., Banerjee R., Smirnova N.A. Targeting nrf2-mediated gene transcription by extremely potent synthetic triterpenoids attenuate dopaminergic neurotoxicity in the MPTP mouse model of Parkinson's disease. 2013;18(2):139–157. doi: 10.1089/ars.2011.4491. Jan 10. Epub 2012 Aug 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Jing X., Shi H., Zhang C. Dimethyl fumarate attenuates 6-OHDA-induced neurotoxicity in SH-SY5Y cells and in animal model of Parkinson's disease by enhancing Nrf2 activity. Neuroscience. 2015;131–140:2015. doi: 10.1016/j.neuroscience.2014.11.047. [DOI] [PubMed] [Google Scholar]
- 308.Hayashi G., Jasoliya M., Sahdeo S. Dimethyl fumarate mediates Nrf2-dependent mitochondrial biogenesis in mice and humans. Hum. Mol. Genet. 2017;26(15):2864–2873. doi: 10.1093/hmg/ddx167. 2017 Aug 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Hayes J.D., Chowdhry S., Dinkova-Kostova A.T. Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of β-TrCP and GSK-3. Biochem. Soc. Trans. 2016;43(4):611–620. doi: 10.1042/BST20150011. 2015 Aug. Epub 2015 Aug 3.PMID: 26551701 Review. [DOI] [PubMed] [Google Scholar]
- 310.Xu L.L., WuY-F, Yan F. 5-(3,4-Difluorophenyl)-3-(6-methylpyridin-3-yl)-1,2,4-oxadiazole (DDO-7263), a novel Nrf2 activator targeting brain tissue, protects against MPTP-induced subacute Parkinson's disease in mice by inhibiting the NLRP3 inflammasome and protects PC12 cells against oxidative stress. Apr. 2019;134:288–303. doi: 10.1016/j.freeradbiomed.2019.01.003. Epub 2019 Jan 5. [DOI] [PubMed] [Google Scholar]
- 311.Moreira S., Fonseca I., Nunes M.J. Nrf2 activation by tauroursodeoxycholic acid in experimental models of Parkinson's disease. Exp. Neurol. 2017;295:77–87. doi: 10.1016/j.expneurol.2017.05.009. 2017 Sep. Epub 2017 May 25. [DOI] [PubMed] [Google Scholar]
- 312.Lee E.J., Park J.S., Lee Y.Y. Anti-inflammatory and anti-oxidant mechanisms of an MMP-8 inhibitor in lipoteichoic acid-stimulated rat primary astrocytes: involvement of NF-κB, Nrf2, and PPAR-γ signaling pathways. J. Neuroinflammation. 2018;15(1):326. doi: 10.1186/s12974-018-1363-6. Nov 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Gureev A.P., Shaforostova E.A., Popov V.N. Regulation of mitochondrial biogenesis as a way for active longevity: interaction between the Nrf2 and PGC-1α signaling pathways. Front. Genet. 2019;14(10):435. doi: 10.3389/fgene.2019.00435. 2019 May. eCollection 2019.PMID: 31139208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Wang L., Cai X., Shi M. Identification and optimization of piperine analogues as neuroprotective agents for the treatment of Parkinson's disease via the activation of nrf2/keap1 pathway. May. 2020;5(199):112385. doi: 10.1016/j.ejmech.2020.112385. Online ahead of print) [DOI] [PubMed] [Google Scholar]
- 315.Yang J.W., Ma W., Luo T. BDNF promotes human neural stem cell growth via GSK-3β-mediated crosstalk with the wnt/β-catenin signaling pathway. Growth Factors. 2016;34(1–2):19–32. doi: 10.3109/08977194.2016.1157791. https://doi:10.3109/08977194.2016.1157791 [DOI] [PubMed] [Google Scholar]
- 316.Li X.T., Liang Z., Wang T.T. Brain-derived neurotrophic factor promotes growth of neurons and neural stem cells possibly by triggering the phosphoinositide 3-kinase/AKT/glycogen synthase kinase-3β/β-catenin pathway. CNS Neurol. Disord. - Drug Targets. 2017;16(7):828–836. doi: 10.2174/1871527316666170518170422. https://doi:10.2174/1871527316666170518170422 [DOI] [PubMed] [Google Scholar]
- 317.Song J.J., Oh S.M., Kwon O.C. Cografting astrocytes improves cell therapeutic outcomes in a Parkinson's disease. J. Clin. Invest. 2018;128(1):463–482. doi: 10.1172/JCI93924. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Wei L., Sun C., Lei M. Activation of Wnt/β-catenin pathway by exogenous Wnt1 protects SH-SIY cells against 6-hydroxydopamine toxicity. J. Mol. Neurosci. 2013;49:105–115. doi: 10.1007/s12031-012-9900-8. 2013. [DOI] [PubMed] [Google Scholar]
- 319.Zhang L., Cen L., Qu S. Enhancing beta-catenin activity via GSK3beta inhibition protects PC12 cells against rotenone toxicity through Nurr1 induction. PloS One. 2016;11(4):e0152931. doi: 10.1371/journal.pone.0152931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Silva-Alvarez C., Arrazola M.S., Godoy J.A. Canonical Wnt signaling protects hippocampal neurons from Aβ oligomers: role of non-canonicalWnt-5a/Ca(2+) in mitochondrial dynamics. Front. Cell Neurosci. 2013;7:97. doi: 10.3389/fncel.2013.00097. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Gao K., Shen Z., Yuan Y. Simvastatin inhibits neural cell apoptosis and promotes locomotor recovery via activation of Wnt/β-catenin signaling pathway after spinal cord injury. J. Neurochem. 2016;138(1):139–149. doi: 10.1111/jnc.13382. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Wang J., Gu J., Wu H. Pentazocine protects SN4741 cells against MP+-Induced cell damage via up-regulation of the canonical wnt/β-catenin signaling pathway. Front Ageing Neurosci. 2017;2017(9):196. doi: 10.3389/fnagi.2017.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Liu Y., Hao S., Yang B. Wnt/β-catenin signaling plays an essential role in α7 nicotinic receptor-mediated neuroprotection of dopamnergic neurons in a mouse model of Parkinson's disease. Biochem. Pharmacol. 2017;140:115–123. doi: 10.1016/j.bcp.2017.05.017. 2017. [DOI] [PubMed] [Google Scholar]
- 324.Maiese K. Novel applications of trophic factors, Wnt and WISP for neuronal repair and regeneration in metabolic disease. Neural Regeneration Research. 2015;10:518–528. doi: 10.4103/1673-5374.155427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Cho C.H., Yoo K.Y., Oliveros A. sFRP3 inhibition improves age-related cellular changes in BubR1 progeroid mice. Ageing Cell. 2019;18:e12899. doi: 10.1111/acel.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Wei Z.Z., Zhang J.Y., Taylor T.M. Neuroprotective and regenerative roles of intranasal Wnt-3a administration after focal ischemic stroke in mice. Cereb Blood Flow Metab. 2018;38(3):404–421. doi: 10.1177/0271678X17702669. 2018 Mar. Epub 2017 Apr 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Chen D.Z., Yang B.J., He X.L. Design, synthesis and structure-activity relationship optimization of phenanthridine derivatives as new Wnt/β-catenin signaling pathway agonists. Bioorg. Chem. 2019;84:285–294. doi: 10.1016/j.bioorg.2018.11.020. https://doi:10.1016/j.bioorg.2018.11.020 [DOI] [PubMed] [Google Scholar]
- 328.Banerjee A., Jothimani G., Prasad S.V. Targeting wnt signaling through small molecules in governing stem cell fate and diseases. 2019. https://www.ingentaconnect.com/content/ben/emiddt;jsessionid=3jua2cpouit9p.x-ic-live-01https://doi:10.2174/1871530319666190118103907 HYPERLINK. \o "link to all issues of this title" Endocrine, Metabolic & Immune Disorders Drug Targets, 19(3), 233-246. [DOI] [PubMed]
- 329.Janda C., Dang L.T., You C. Surrogate Wnt agonists that phenocopy canonical Wnt/β-catenin signaling. Nature. 2017;545(7653):234–237. doi: 10.1038/nature22306. https://doi:10.1038/nature22306 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Pallauf K., Duckstein N., Hasler M. Flavonoids as putative inducers of the transcription factors Nrf2, FoxO, and PPARγ. Oxid Med Cell Longev. 2017 doi: 10.1155/2017/4397340. 2017:4397340. Epub 2017 Jul 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Liu D., Chen L., Zhao H. Small molecules from natural products targeting the Wnt/β-catenin pathway as a therapeutic strategy. Biomed. Pharmacother. 2019;117:108990. doi: 10.1016/j.biopha.2019.108990. https://doi:10.1016/j.biopha.2019.108990 Jun 18. [DOI] [PubMed] [Google Scholar]
- 332.Ghosh A.K., Rao V.R., Wisniewski V.J. Differential activation of glioprotective intracellular signaling pathways in primary optic nerve head astrocytes after treatment with different classes of antioxidants. Antioxidants. 2020;9(4) doi: 10.3390/antiox9040324. 2020 Apr 16. pii: E324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Daverey A., Agrawal S.K. Curcumin protects against white matter injury through NF-κB and Nrf2 cross talk. J. Neurotrauma. 2020;37(10):1255–1265. doi: 10.1089/neu.2019.6749. 2020 May 15. Epub 2020 Feb 5. [DOI] [PubMed] [Google Scholar]
- 334.Cui Q., Li X., Zhu H. Curcumin ameliorates dopaminergic neuronal oxidative damage via activation of the Akt/Nrf2 pathway. Mol. Med. Rep. 2016;13(2):1381–1388. doi: 10.3892/mmr.2015.4657. 2016 Feb. Epub 2015 Dec 8.PMID: 26648392. [DOI] [PubMed] [Google Scholar]
- 335.Yl Ju B., Zhang Y.Z. Protective effect of curcumin against oxidative stress-induced injury in rats with Parkinson's disease through the wnt/β-catenin signaling pathway. Cell. Physiol. Biochem. 2017;43:2226–2241. doi: 10.1159/000484302. [DOI] [PubMed] [Google Scholar]
- 336.Palomera-Avalos V., Griñán-Ferré C., Puigoriol-Ilamola D. Resveratrol protects SAMP8 brain under metabolic stress: focus on mitochondrial function and wnt pathway. Mol. Neurobiol. 2017;54(3):1661–1676. doi: 10.1007/s12035-016-9770-0. Apr. Epub 2016 Feb 12.PMID: 26873850. [DOI] [PubMed] [Google Scholar]
- 337.Wu D.M., Han X.R., Wen X. Salidroside protection against oxidative stress injury through the wnt/β-catenin signaling pathway in rats with Parkinson's disease. Cell. Physiol. Biochem. 2018;46:1793–1806. doi: 10.1159/000489365. [DOI] [PubMed] [Google Scholar]
- 338.Wang G.Q., Zhang B., He X.M. Naringenin targets on astroglial Nrf2 to support dopaminergic neurons. Pharmacol. Res. 2019;139:452–459. doi: 10.1016/j.phrs.2018.11.043. [DOI] [PubMed] [Google Scholar]
- 339.Yang Y., Kong F., Ding Q. Bruceine D elevates Nrf2 activation to restrain Parkinson's disease in mice through suppressing oxidative stress and inflammatory response. Biochem. Biophys. Res. Commun. 2020;526(4):1013–1020. doi: 10.1016/j.bbrc.2020.03.097. 2020 Jun 11. [DOI] [PubMed] [Google Scholar]
- 340.Zhou T., Zu G., Zhang X. Neuroprotective effects of ginsenoside Rg1 through the Wnt/β-catenin signaling pathway in both in vivo and in vitro models of Parkinson's disease. Neuropharmacology. 2016;2016(101):480–489. doi: 10.1016/j.neuropharm.2015.10.024. [DOI] [PubMed] [Google Scholar]
- 341.Gao K., Wang Y.S., Yuan Y.J. Neuroprotective effect of rapamycin on spinal cord injury via activation of the Wnt/β-catenin signaling pathway. Neural. Regen. Res. 2015:951–957. doi: 10.4103/1673-5374.158360. 2015;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Zhang B.Q., Zheng G.Y., Han Y. Ilexonin A promotes neuronal proliferation and regeneration via activation of the canonical wnt signaling pathway after cerebral ischemia reperfusion in rats. Evid. base Compl. Alternative Med. 2016;2016:9753189. doi: 10.1155/2016/9753189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Singh S., Mishra A., Shukla S. ALCAR exerts neuroprotective and pro-neurogenic effects by inhibition of glial activation and oxidative stress via activation of the wnt/β-catenin signaling in parkinsonian rats. Mol. Neurobiol. 2016;2016(53):4286–4301. doi: 10.1007/s12035-015-9361-5.Wang. [DOI] [PubMed] [Google Scholar]
- 344.lkouzi A., Vedam-Mai V., Eisinger Rs. Emerging therapies in Parkinson disease - repurposed drugs and new approaches. Nat Rev Neurol. Apr. 2019;15(4):204–223. doi: 10.1038/s41582-019-0155-7.P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Yun S.P., Kam T.I., Panicker N. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson's disease. Nat. Med. 2018;24:931–938. doi: 10.1038/s41591-018-0051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Athauda D., Foltynie T. The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson's disease: mechanisms of action. Drug Discov. Today. 2016;21:802–818. doi: 10.1016/j.drudis.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 347.Zhang X., Lao K., Qiu Z. Potential Astrocytic Receptors and Transporters in the Pathogenesis of Alzheimer's Disease. J. Alzheimers Dis. 2019;67(4):1109–1122. doi: 10.3233/JAD-181084. PMID: 30741675. [DOI] [PubMed] [Google Scholar]
- 348.Yang Y.H., Li D.L., Bi X.Y. Acetylcholine inhibits LPS-induced MMP-9 production and cell migration via the α7 nAChR-JAK2/STAT3 pathway in RAW264.7 cells. Cell physiol biochem. 2015. 2015;36(5):2025–2038. doi: 10.1159/000430170. [DOI] [PubMed] [Google Scholar]
- 349.Liu Y., Zeng X., Hui Y. Activation of α7 nicotinic acetylcholine receptors protects astrocytes against oxidative stress-induced apoptosis: implications for Parkinson's disease. Neuropharmacology. 2015 Apr. 2015;91:87–96. doi: 10.1016/j.neuropharm.2014.11.028. [DOI] [PubMed] [Google Scholar]
- 350.Patel H., McIntire J., Ryan S. Anti-inflammatory effects of astroglial α7 nicotinic acetylcholine receptors are mediated by inhibition of the NF-κB pathway and activation of the Nrf2 pathway. J Neuroinflammation. 2017;14(1):192. doi: 10.1186/s12974-017-0967-6. 2017 Sep. 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Miyazaki I., Asanuma M., Murakami S. Targeting 5-HT(1A) receptors in astrocytes to protect dopaminergic neurons in Parkinsonian models. Neurobiol. Dis. 2013;59:244–256. doi: 10.1016/j.nbd.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 352.Miyazaki I., Asanuma M. Serotonin 1A receptors on astrocytes as a potential target for the treatment of Parkinson's disease. Curr. Med. Chem. 2016;23:686–700. doi: 10.2174/0929867323666160122115057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Isooka N., Miyazaki I., Kikuoka R. Dopaminergic neuroprotective effects of rotigotine via 5-HT1A receptors: possibly involvement of metallothionein expression in astrocytes. Neurochem. Int. 2020;132:104608. doi: 10.1016/j.neuint.2019.104608. [DOI] [PubMed] [Google Scholar]
- 354.Smith G.A., Rocha E.M., Rooney T., Barneoud P., McLean J.R., Beagan J.…Isacson O. A Nurr1 agonist causes neuroprotection in a Parkinson's disease lesion model primed with the toll-like receptor 3 dsRNA inflammatory stimulant poly(I: C) PloS One. 2015;27(3):10. doi: 10.1371/journal.pone.0121072. https://doi:10.1371/journal.pone.0121072 e0121072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Kim C.H., Han B.S., Moon J. Nuclear receptor Nurr1 agonists enhance its dual functions and improve behavioral deficits in an animal model of Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 2015;112:8756–8761. doi: 10.1073/pnas.1509742112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Mederos S., Hernández-Vivanco A., Ramírez-Franco J. Melanopsin for precise optogenetic activation of astrocyte-neuron networks. Glia. 2019;67:915–934. doi: 10.1002/glia.23580. [DOI] [PubMed] [Google Scholar]
- 357.Xie Z., Yang Q., Song D. Optogenetic manipulation of astrocytes from synapses to neuronal networks: a potential therapeutic strategy for neurodegenerative diseases. Glia. 2020;68(2):215–226. doi: 10.1002/glia.23693. 2020 Feb. Epub 2019 Aug 10. [DOI] [PubMed] [Google Scholar]
- 358.Yang F., Liu Y., Tu J. Activated astrocytes enhance the dopaminergic differentiation of stem cells and promote brain repair through bFGF. Nat. Commun. 2014;5:5627. doi: 10.1038/ncomms6627. https://doi:10.1038/ncomms6627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Zhao M.L., Chen S.J., Li X.H. Optical depolarization of DCX-expressing cells promoted cognitive recovery and maturation of newborn neurons via the wnt/β-catenin pathway. J. Alzheim. Dis. 2018;63(1):303–318. doi: 10.3233/JAD-180002. https://doi:10.3233/JAD-180002 [DOI] [PubMed] [Google Scholar]
- 360.Zhang W., Shi Y., Peng Y. Neuron activity-induced Wnt signaling up-regulates expression of brain-derived neurotrophic factor in the pain neural circuit. J. Biol. Chem. 2018;293(40):15641–15651. doi: 10.1074/jbc.RA118.002840. https://doi:10.1074/jbc.RA118.002840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Drinkut A., Tereshchenko Y., Schulz J.B. Efficient gene therapy for Parkinson's disease using astrocytes as hosts for localized neurotrophic factor delivery. Mol Ther. 2012 Mar. 2011;20(3):534–543. doi: 10.1038/mt.2011.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.de Pins B., Cifuentes-Díaz C., Farah A.T. Conditional BDNF delivery from astrocytes rescues memory deficits, spine density, and synaptic properties in the 5xFAD mouse model of alzheimer disease. J. Neurosci. 2019;39(13):2441–2458. doi: 10.1523/JNEUROSCI. Mar 27. 2121-18.2019. Epub 2019 Jan 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Brulet R., Matsuda T., Zhang L. NEUROD1 instructs neuronal conversion in non-reactive astrocytes. Stem Cell Reports. 2017;8:1506–1515. doi: 10.1016/j.stemcr.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Proschel C., Stripay J.L., Shih C.H. Delayed transplantation of precursor cell-derived astrocytes provides multiple benefits in a rat model of Parkinsons. EMBO Mol. Med. 2014;6:504–518. doi: 10.1002/emmm.201302878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Heinrich C., Gascón S., Masserdotti G. Generation of subtype-specific neurons from postnatal astroglia of the mouse cerebral cortex. Nat. Protoc. 2011;6:214–228. doi: 10.1038/nprot.2010.188. [DOI] [PubMed] [Google Scholar]
- 366.Caiazzo M., Dell'Anno M.T., Dvoretskova E. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature. 2011;476:224–227. doi: 10.1038/nature10284. [DOI] [PubMed] [Google Scholar]
- 367.Kim J., Su S.C., Wang H. Functional integration of dopaminergic neurons directly converted from mouse fibroblasts. Cell Stem Cell. 2011;9:413–419. doi: 10.1016/j.stem.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Liu X., Li F., Stubblefield E.A. Direct reprogramming of human fibroblasts into dopaminergic neuron-like cells. Cell Res. 2012;22:321–332. doi: 10.1038/cr.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Pfisterer U., Kirkeby A., Torper O. Direct conversion of human fibroblasts to dopaminergic neurons. Proc. Natl. Acad. Sci. U.S.A. 2011;108:10343–10348. doi: 10.1073/pnas.1105135108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Addis R.C., Hsu F.C., Wright R.L. Efficient conversion of astrocytes to functional midbrain dopaminergic neurons using a single polycistronic vector. PloS One. 2011;6:e28719. doi: 10.1371/journal.pone.0028719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Ishii M.N., Yamamoto K., Shoji M. Human induced pluripotent stem cell (hiPSC).-derived neurons respond to convulsant drugs when co-cultured with hiPSC-derived astrocytes. Toxicology. 2017;389:130–138. doi: 10.1016/j.tox.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 372.Du F., Yu Q., Chen A. Astrocytes attenuate mitochondrial dysfunctions in human dopaminergic neurons derived from iPSC. Stem Cell Reports. 2018;10:366–374. doi: 10.1016/j.stemcr.2017.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]