

Abstract
Background: Idiopathic Pulmonary Fibrosis (IPF) is a progressive inflammatory disorder driven by a fibrotic cascade of events such as epithelial to mesenchymal transition, extracellular matrix production and collagen formation in the lungs in a sequential manner. IPF incidences were raising rapidly across the world. FDA approved pirfenidone and nintedanib (tyrosine kinase inhibitors) are being used as a first-line treatment drugs for IPF, however, neither the quality of life nor survival rates have been improved because of patient noncompliance due to multiple side effects. Thus, the development of novel therapeutic approaches targeting TGF-β mediated cascade of fibrotic events is urgently needed to improve the survival of the patients suffering from devastating disease.
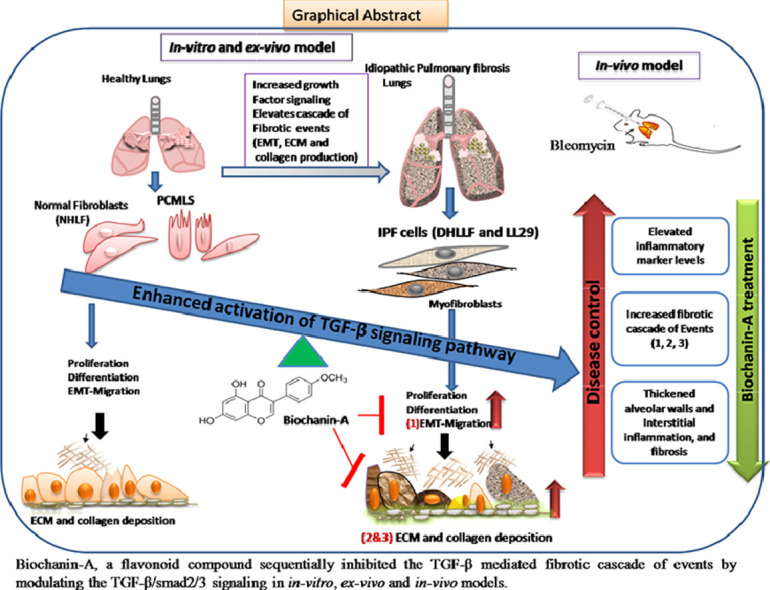
Purpose: The aim of this study was to investigate and validate the anti-fibrotic properties of Biochanin-A (isoflavone) against TGF-β mediated fibrosis in in vitro, ex vivo, in vivo models and to determine the molecular mechanisms that mediate these anti-fibrotic effects.
Methods: The therapeutic activity of BCA was determined in in vitro/ex vivo models. Cells were pre-treated with BCA and incubated in presence or absence of recombinant-TGF-β to stimulate the fibrotic cascade of events. Pulmonary fibrosis was developed by intratracheal administration of bleomycin in rats. BCA treatment was given for 14 days from post bleomycin instillation and then various investigations (collagen content, fibrosis gene/protein expression and histopathological changes) were performed to assess the anti-fibrotic activity of BCA.
Results:In vitro/ex vivo (Primary normal, IPF cell line and primary IPF cells/ Precision cut mouse lung slices) experiments revealed that, BCA treatment significantly (p < 0.001) reduced the expression of TGF-β modulated fibrotic genes/protein expressions (including their functions) which are involved in the cascade of fibrotic events. BCA treatment significantly (p < 0.01) reduced the bleomycin-induced inflammatory cell-infiltration, inflammatory markers expression, collagen deposition and expression of fibrotic markers in lung tissues equivalent or better than pirfenidone treatment. In addition, BCA treatment significantly (p < 0.001) attenuated the TGF-β1/BLM-mediated increase of TGF-β/Smad2/3 phosphorylation and resulted in the reduction of pathological abnormalities in lung tissues determined by histopathology observations.
Conclusion: Collectively, BCA treatment demonstrated the remarkable therapeutic effects on TGF-β/BLM mediated pulmonary fibrosis using IPF cells and rodent models. This current study may offer a novel treatment approach to halt and may be even rescue the devastating lung scarring of IPF.
Keywords: TGF-β, Bleomycin, Inflammation, Myofibroblasts, Extra-cellular matrix, Collagen, BALF
Abbreviations: ALP, Alkaline phosphatase; α-SMA, a-smooth muscle actin; BALF, Bronchoalveolar lavage fluid (BALF); BCA, Biochanin-A; BLM, Bleomycin; COL1A1, Collagen1α1; COL3Al, Collagen3α1; CTCF, Corrected Total Cellular Fluorescence; CTGF, Connective tissue growth factor; DHLF, Diseased (IPF) human lung fibroblasts; E-cad, E-cadherin; ECM, extracellular matrix; EMT, epithelial to mesenchymal transition; FDA, Food and Drug Administration; FN-I, Fibronectin-I; HIF1-α, Hypoxia-Inducible Factor 1-α; hr, hour; IPF, Idiopathic pulmonary fibrosis; LDH, Lactic Acid Dehydrogenase; LOXL-2, Lysyl oxidase-like2; NHLF, Normal human lung fibroblasts; PCMLS, Precision cut mouse lung slices; rTGF-β1, recombinant transforming growth factor-beta 1; RT-qPCR, Reverse transcriptase quantitative PCR; SRB, sulforhodamine-B; TIMP, Tissue inhibitory metalloproteinase
Graphical abstract
Introduction
Idiopathic pulmonary fibrosis (IPF) is a type of pulmonary disorder that results in scarring of the lungs for an unknown reason. It is a form of interstitial lung disease, primarily causing inflammation in lung tissue and space surrounding the air sacs of the lungs, ultimately causing thickened, stiff tissue formation that leads to difficulty in breathing. Apart from its poor prognosis, it was reported that the survival rate is as low as 2–5 years after diagnosis of the IPF (Richeldi et al., 2017). Besides low survival rate, IPF patients face scarcity of effective drugs. Although there are two growth factor inhibitors (pirfenidone and nintedanib) in the clinical practice, their usage is limited because of low efficacy and patient noncompliance (Maher and Strek, 2019). In clinical-trials, pirfenidone and nintedanib did not show any statistically significant difference from placebo in terms of mortality and morbidity. In addition to that, pirfenidone induced the increased incidence of nausea, dyspepsia, vomiting, rashes, and dizziness whereas nintedanib caused severediarrhea, elevated levels of aspartate aminotransferase and alanine aminotransferase (Ikeda et al., 2019). Recent data on COVID-19 suggested that there could be a substantial fibrotic consequences following SARS-CoV-2 infection (George et al., 2020; Spagnolo et al., 2020). Given the scale of the pandemic, the burden of IPF following SARS-CoV-2 infection is likely to be high; hence developing new anti-fibrotic agents may help in such pandemics. Therefore, there is an immediate need for the development of effective treatment against IPF.
One of the initiating dynamic forces behind the cascade of fibrotic events is epithelial to mesenchymal transition (EMT), a process where epithelial cells gain mesenchymal morphology by increasing the expression of mesenchymal markers and reducing the expression of epithelial markers, which are responsible for tight junctions formation (Salton et al., 2019). LOXL-2 is a copper-dependent monoamine oxidase that contributes to the remodelling of the extracellular-matrix (ECM) by cross-linkage of collagen and elastin fibres. ECM/LOXL-2 has emerged as a potential therapeutic target in cancer and fibrosis. EMT, ECM production, and collagen deposition are the three major sequential events, which are actively involved in the progression and maintenance of the IPF (Chen et al., 2019; Tjin et al., 2017).
Biochanin-A (BCA), an organic isoflavone, has been categorized as a special phytoestrogen (Raheja et al., 2018). Despite BCA exhibits poor bioavailability still, BCA exhibited various protective bioactivities such as anti-inflammatory (Kole et al., 2011b; Oh et al., 2016), anti-oxidant, antiallergic, antidiabetic (Harini et al., 2012; Mehrabadi et al., 2018) and anticancer activities against various forms of cancer (Szliszka et al., 2013; Yu et al., 2019). It is well known that EMT and inflammation are the two vital processes in the pathogenesis of IPF. It was reported that, BCA attenuated the EMT process in various cancers (including lung cancer) and showed potent anti-cancer activity by inducing the cell cycle arrest and apoptosis (Wang et al., 2018b). These beneficial effects of BCA prompted us to investigate anti-fibrotic activity against IPF. In the present study, we investigated the anti-fibrotic activity of BCA against TGF-β mediated cellular changes and functional parameters in in vitro/ex vivo systems. Further, we focused on elucidating the therapeutic role and possible mechanism of action of BCA against BLM-induced pulmonary fibrosis in vivo model. Overall, our study highlights the protective role of BCA against IPF in the in vitro, ex vivo and in vivo models and therefore we propose a viable therapeutic approach for this unmet medical need.
Materials and methods
Cell-lines and cell culture
LL29 cells (lung fibroblasts from IPF patient) were purchased from ATCC and cultured in Ham's F-12 K medium with 15% FBS. Primary lung fibroblasts cells, NHLF (Normal human lung fibroblasts) and DHLF (Diseased (IPF) human lung fibroblasts) cells were purchased from LONZA (MD, USA) and cultured in FGF™-2 medium. All the cells were maintained in a humidified atmosphere of 95% air with 5% CO2 at 37 °C. All experiments were performed on cells between passages 2 - 6 and at the confluence of 70–90%. All in vitro experiments were conducted 3 times as a biological repeat and each biological repeat has three technical repeats.
Cell-viability by SRB assay
Cells were seeded in a 96 well plate (8 × 103 cells/well), after 12 hr of attachment, cells were treated with either DMSO (vehicle control) or BCA (TCI, KITA-KU, Tokyo, Japan) at various concentrations (500, 250, 125, 62.5, 31.25, 15.65 and 7.7 µM) and then cells were incubated for 72 h. The purity of BCA was > 98% and the chemical structure of BCA was shown in Fig S1. Further, IC50/cell viability was determined with sulforhodamine-B (Sigma Aldrich, St Louis, MI, USA) assay (Martins et al., 2019). IC50 was calculated by curve-fit method using GraphPad Prism-5.
TGF-β stimulation/hypoxia induction
After reaching 70% of confluence, cells were subjected to serum starvation for 24 h then cells were pre-treated with BCA (10, 20 and 30 µM) for 2 h in serum-free media, thereafter, cells were treated with rTGF-β (R&D Systems, MN, USA) at a concentration of 5 ng/ml for 72 h (Kim et al., 2019; Molina-Molina et al., 2018a). For hypoxia-based experiments, cells were treated with BCA (10, 20 and 30 µM) for 2h and then cultured in presence of 1% oxygen using a tri-gas incubator (Thermo Fisher, Waltham, MA) for 72h.
RNA isolation, cDNA synthesis and RT-qPCR
RNA isolation was carried out using trizol as described earlier (Balaji et al., 2016). Briefly, RNA was isolated using the trizol-chloroform method and total RNA was quantified using nano-drop. cDNA synthesis was performed using 1 μg of RNA with primescript cDNA synthesis kit (Takara bio-India) according to manufacturer's instructions. Primers for EMT (Twist1, Fibronectin-I (FN-I), Vimentin and SNAIL) markers, MET markers (E-cad), ECM markers (TIMP-I, TIMP-III, α-SMA and LOXL-2), collagen markers (CTGF, COL1A1, and COL3A1) and reference markers (β2M, GAPDH and β-actin) were designed using Primer-3 software and sequences were shown in Table S1. RT-qPCR was carried out using SYBR green mix and the relative expression of mRNA was calculated using the comparative Ct (ΔCt) and data was expressed as Mean ±SEM.
Immunofluorescence and western-blot analysis
NHLF, DHLF and LL29 cells were pre-treated with either DMSO or BCA (10 µM, 20 µM or 30 µM) for 2 h and then cells were cultured in presence or absence of TGF-β (5 ng/ml) for another 72 h. After a treatment period, immunofluorescence assay was performed as described in previous study (Saxena et al., 2018). Briefly, cells were fixed with methanol: acetone (24:1) at 4 °C. Then cells were permeabilized with PBS containing Triton-X 100, blocked with 1% BSA and probed with antibodies (E-cad, α-SMA, COL1Al and FN-I (Abcam, USA)). Thereafter, cells were probed with secondary antibody tagged with FITC (1:200 dilution) followed by mounting, thereafter, cells were visualized under confocal microscope FLUOVIEW FV 10i (Olympus, USA) with 60 × magnification or epifluorescence microscope (Olympus, USA) at 20 × magnification. Image processing and quantification were performed using ImageJ software. For Corrected Total Cellular Fluorescence (CTCF) quantification, fluorescence intensity was first normalized to the number of nuclei. Then, change in intensity under various conditions was plotted relative to the control. For western-blot analysis, cells or tissue homogenates were subjected to protein isolation using RIPA lysis buffer, and then protein concentrations were estimated using bicinchoninic acid reagent. Thirty μg of protein per sample was loaded onto SDS-PAGE Bis-Tris 8–10% protein gel for electrophoresis and then transferred onto polyvinylidene difluoride (PVDF) membranes (0.45 μm, Millipore, Billerica, MA, USA). After protein transfer, PVDF membranes were blocked with 5% BSA (Bovine serum albumin (Sigma Aldrich, USA)) for 1 h at room temperature and then blots were incubated with appropriate primary antibodies overnight at 4 °C. β-actin (Cell signalling technology, Massachusetts USA) served as the loading control Secondary antibodies (Jackson Laboratory, USA) and an ECL kit (Pierce, Waltham, MA, USA) were employed to generate chemiluminescent signals. All immunoblot results represent triplicate repeats. Densitometry analysis was performed using ImageJ software.
In vitro-wound-healing assay
An equal number of NHLF, DHLF and LL29 cells were seeded in 6-well plates. On attaining 100% confluence, cells were serum starved for 24 hr in serum-free medium thereafter, subsequently two scratch wounds were made using a sterile P-200 pipette-tip in each well. Then cells were treated with BCA for 2 h, post 2 h incubation, TGF-β (5 ng/ml) was added to all the wells except vehicle control well. Photomicro-graphs were taken at 0 h, 24 h, 48 h and 72 h of post wound generation, wound area was calculated by Image J software and percentage of wound closure was calculated and analyzed by using Graph pad Prism-5.
Experimental animals
Adult female Wistar rats (180–200 g, n = 50) at the age of 8–10 weeks were used to develop bleomycin induced pulmonary fibrosis model. C57BL/6 J mice (6–8 weeks) were used for isolation of precision-cut mouse lung slices and lung derived cells. All animals were kept under constant environmental and nutritional conditions throughout the experimental period. All experimental protocols (in vivo and ex vivo) were approved (IICT/084/2020) in the Institutional Animal Ethics Committee of the CSIR-IICT, Hyderabad, Government of India.
Precision-cut mouse lung slices (PCMLS) and derived cells
Lung tissues were isolated aseptically from C57BL/6 J mice (6–8 weeks old) and transferred into a sterile container and sliced into small pieces of about 10 mm3. PCMLS (~300–600 μm thickness) were prepared manually and cultured in a 6- well plate (5 slices/well) as described previously (Pieretti et al., 2014). After four days of culturing, PCMLS were serum starved for 24 h in serum-free medium and then treated with BCA (20 µM) for 2 h. Further, PCMLS were cultured in the presence or absence of mouse-rTGF-β1 (5 ng/ml) for 72 h. Thereafter, cells were harvested for downstream experiments such as RT-qPCR and western-blot analysis. Cells derived (by trypsinization) from PCMLS were counted and seeded for immunofluorescence assay and wound healing assays.
Induction of pulmonary fibrosis and BALF analysis
Pulmonary fibrosis was induced by a single intratracheal instillation of BLM (5 mg/kg) to the set of animals (n = 40) excluding sham control animals (n = 8) (Akgedik et al., 2012). BLM was dissolved in saline to attain the concentration of 5 mg/ml. Animals were anesthetized with ketamine (80 mg/kg) and xylazine (10 mg/kg). BLM was administered intratracheally (5 mg/kg) to the anesthetized Wistar rats and saline was administered intratracheally in to sham control animals. Post BLM administration, body weight reduction was calculated (on 14th day) and the animals which showed body weight reduction (around 10%) were included in the study. Further, these animals were randomised into four groups (namely, BLM control, BLM ++BCA-5 mg/kg, BLM ++BCA-10 mg/kg and BLM ++pirfenidone-50 mg/kg) apart from sham control (8 rats/group). Rats were treated with 5 mg/kg/day or 10 mg/kg/day of BCA or 50 mg/kg of pirfenidone orally for another 14 days. BCA doses 5 mg/kg/day and 10 mg/kg/day were similar to the doses used by previous studies in literature (Chundi et al., 2016; Ko et al., 2011). BCA was given in a suspension form (gum-acacia 0.25%) and pirfenidone (Kind gift from Sun Pharmaceutical Industries Ltd, India) was given in a solution form (dissolved in sterile water), BLM control group animals were received vehicle suspension (gum-acacia 0.25%) and sham control animals were received saline. The rats were anesthetized 28 days after BLM induction, the bronchoalveolar lavage fluid (BALF) was collected by intratracheal instillation and draining of 1.5 ml saline for two times. The lung tissues were excised for pulmonary coefficient measurement (lung weight/body weight). The left lower lobes were fixed in 10% formalin for the examination of histopathology and the other lung tissue samples were stored at -80 °C for RT-qPCR, western-blot analysis and hydroxylproline assays.
BALF samples were centrifuged at 300 g for 5 min at 4 °C and then cell pellets were re-suspended in 1 ml of PBS to quantify inflammatory cell counts in cell counter (SIEMENS, ADVIA 2120i); supernatant BALF was examined for ALP and LDH parameters using auto-analyzer (Siemens, Germany).
Hydroxyproline assay
Collagen deposition was determined by measuring the total hydroxyproline content in wet lung tissue, which was measured by a hydroxyproline assay kit (Abcam), according to the manufacturer's protocol. Briefly, lungs were homogenized in sterile distilled water and subjected to alkaline hydrolysis using 10 N NaOH and heated at 120 °C for 1 h. Following alkaline hydrolysis, samples were neutralized with 10 N HCL. Then samples were transferred to 96-well plates and kept for evaporation at 65 °C. Further, samples were oxidized by adding an oxidation mixture followed by the addition of the developer and then samples were incubated at 37 °C for 5 min. Further, DMAB was added and incubated at 65 °C for 45 min and absorbance was measured at 560 nm using a microplate reader. The concentration of total hydroxyproline (µg/mg of wet tissue) was calculated.
Histopathology and immunohistochemistry
A portion of the pulmonary lobe was harvested, rinsed with ice-cold phosphate buffer saline, and fixed with 10% neutral-buffered formalin, embedded in paraffin wax, sectioned (6 μm) and stained with H&E and Masson's trichrome. Three serial slices of 3 μm thickness each 50 μm apart of both lung lobes were stained with Masson's Trichrome. The slides were analyzed with a microscope in a random order using an (10 ×) objective. The severity of fibrosis and alveolitis in lung tissues (using H&E and Masson's trichrome staining) was semi-quantitatively assessed by a pathologist in a blinded fashion via Ashcroft scoring system (Ashcroft et al., 1988). The structural alterations of tissue were assessed based on the degree haemorrhage, emphysema, alveolar wall thickening, inflammatory lesions and collagen deposition or fibrosis (Zaghloul et al., 2017).
Immunohistochemistry was performed as described in previous protocol (Liu et al., 2018). In- brief, tissue sections were incubated with primary antibodies (E-cad, FN-I and α-SMA) for overnight. Then sections were washed and stained with secondary anti-rabbit antibody tagged with horseradish peroxidase (HRP) enzyme (Jackson Laboratory, USA) on the following day protein expression was detected using Signal Stain® DAB Chromogen kit (Cell Signaling Technology, USA). The immunohistochemical intensity was semi-quantitatively scored by an experienced pathologist based on the intensity of the primary antibody staining. Highest intensity was graded 'high' (+++), moderate intensity was graded 'medium' (++) and lowest intensity was graded as 'low' (+).
Statistical analysis
Statistical significance was determined using student's t-test, ANOVA and two-way ANOVA. Curve-fit method was used to analyse IC50 value. Results were shown as Mean±SEM. Values of * p < 0.05, ⁎⁎ p < 0.01, *** p < 0.001 were considered significant. NS signifies non-significant.
Results
Fibrotic gene/markers were over-expressed in IPF cells and BCA selectively reduced the expression of fibrotic markers in IPF cells
Fibrosis is a progressive disorder, characterized by the over expression of various fibrotic genes which are involved in differentiation, EMT, ECM production and collagen deposition. In order to estimate the basal gene expression of fibrotic genes in NHLF, DHLF and LL29 cells RT-qPCR assays were employed. Gene expression assays revealed that, majority of the fibrotic genes (TGF-β1, α-SMA, Twist-I, SNAIL, COL3A1 and CTGF) were significantly (p < 0.01) up-regulated in DHLF and LL29 cells compared to NHLF (Fig. 1 A and B). Other fibrotic genes such as TIMP-I, TIMP-III, FN-I, vimentin and COL1A1 were also over-expressed in IPF cells (Fig. 1A and B). In addition to mRNA levels, protein expression of α-SMA, FN-I and COL1A1 (1C-1F) were also significantly (p < 0.001) increased in IPF cells.
Fig. 1.

Fibrotic gene/markers were over-expressed in IPF cells and BCA showed selective inhibition towards IPF cells
NHLF, DHLF and LL29 cells were cultured and subjected to RT-qPCR analysis for the specified transcripts A-B) Gene expression analysis. Parallel dishes were subjected to immunofluorescence for α-SMA, Fibronectin and COL1A1. C-F) Immunofluorescence analysis in NHLF, DHLF and LL29 cells, respectively. Cells (5 × 105) were cultured and treated with BCA or vehicle for 72 h then subjected to RT-qPCR. Gene expression analysis in G) PCLMS, H) NHLF, I) LL29 cells and J) DHLF was performed using specified primers. Results were shown as Mean ± SEM, n = 3. ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ p < 0.001.
To determine the IC50 of BCA, cell viability assay was performed using all three cell types. SRB assay revealed, 50% cell death was observed at a concentration of 73.55 ± 0.3 µM, 79.73 ± 0.22 µM and 103.02 ± 0.3 µM in NHLF, DHLF and LL29 cells respectively (Fig. S1B, S1C and S1D). Based on the results of IC50, non-cytotoxic doses were finalized (10, 20 and 30 µM) and validated (Fig. S1E) in all cell types including cells derived from PCMLS. Similar doses were used in previous studies to demonstrate the anti-inflammatory activity of BCA (Kole et al., 2011a; Ming et al., 2015). To determine the selectivity and specificity of the BCA against IPF cells, gene expression analysis was performed in presence or absence of BCA (20 µM) in normal (PCMLS and NHLF) and IPF lung cells (DHLF and LL29). Surprisingly, results revealed that, majority of the genes (except TIMP-I in LL29) were significantly (p < 0.001) down regulated (~50% gene expression) in IPF cells (Fig. 1G and H) in contrast to non-IPF cells (only a few were down-regulated) (Fig. 1I and J). Hence, this data suggests that, treatment with BCA selectively suppress the expression of fibrotic genes in IPF cells where basal levels of fibrotic genes were highly over expressed.
BCA ameliorated the TGF-β induced expression of EMT genes/markers
In in vitro studies, TGF-β1 plays a major role in the induction of EMT, which is the driving force for myofibroblasts conversion (Chen et al., 2014). Therefore, gene expression analysis was performed to assess the effect of BCA on EMT markers in TGF-β stimulated cells (Fig. 2 ). TGF-β1 treatment resulted in significant increase in the expression of EMT genes (Vim, FN-I, SNAIL and Twist-I) (Fig. 2A-K). Vimentin (in NHLF) and Twist-I (in NHLF and DHLF) levels were not modulated upon treatment with TGF-β1 (Fig. 2A, D and H). On the other-hand BCA treatment significantly (p < 0.001) suppressed the expression of EMT genes in NHLF (Fig. 2B and C), DHLF (Fig. 2E-G) and LL29 cells (Fig. 2I-K). To further confirm the effects of BCA on EMT markers, immunofluorescence assay was carried out for FN-I (Fig. 2L-N) and it revealed that, increased expression of FN-I in TGF-β1 treated cells and significantly (p < 0.001) reduced expression of FN-I in BCA treated NHLF (Fig. 2L and S2A), DHLF (Fig. 2M and S2B) and LL29 cells (Fig. 2N and S2C).
Fig. 2.

BCA treatment attenuated the TGF-β-induced expression of EMT genes/markers: Cells were pre-treated with BCA for 2 h, thereafter rTGF-β added to the cells and cultured for 72 h. Further, cells were subjected to RT-qPCR for the specified transcripts in A-D) NHLF, E-H) DHLF, I-K) LL29. Parallel dishes were subjected to immunofluorescence assay for FN-1 protein. L-N) Immunofluorescence analysis in NHLF, DHLF and LL29 cells, respectively. Results are shown as Mean ± SEM, n = 3. ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ p < 0.001.
BCA abrogated the TGF-β induced expression of ECM markers
ECM plays a distinct role in organizing the tissue architecture and regulation of cell function. This dynamic activity is controlled partly by Matrix-Metallo-Proteinase (MMP) and Tissue Inhibitors of Metalloproteinase (TIMPS). Therefore, we investigated the expression of ECM markers (TIMP-I and TIMP-III) and it is supporting markers (LOXL-2 and α-SMA) levels in cell lines (Fig. 3 ). TGF-β1 stimulation significantly enhanced (Fig. 3A-L) the expression of ECM markers in normal and IPF cells. In-line with EMT marker's expression, BCA treatment also attenuated the expression of α-SMA, TIMP-I, TIMP-III and LOXL-2 in NHLF (Fig. 3A-D), DHLF (Fig. 3E-H) and LL29 (Fig. 3I-L) cells in a dose dependent manner (except in LL29 cells). Additionally, to validate these results, protein expression analysis was performed for α-SMA. Immunofluorescence results revealed that, BCA treatment significantly (p < 0.001) diminished the TGF-β1 mediated α-SMA expression and distribution in NHLF (Fig. 3M, S3A), DHLF (Fig. 3N, S3B) and LL29 (Fig. 3O, S3C) cells. Taken together, BCA treatment significantly (p < 0.01, p < 0.001) reduced the ECM marker expression in TGF-β1 stimulated fibroblast cells.
Fig. 3.

BCA treatment ameliorated the TGF-β-induced expression of ECM markers: Cells were pre-treated with BCA for 2 h, thereafter rTGF-β added to the cells and cultured for 72 h. Further, cells were subjected to RT-qPCR for the specified transcripts in A-D) NHLF, E-H) DHLF, I-L) LL29. Parallel dishes were subjected to immunofluorescence analysis for α-SMA protein. M-O) Immunofluorescence analysis in NHLF, DHLF and LL29 cells, respectively. Results were shown as Mean ± SEM, n = 3. ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ p < 0.001.
BCA attenuated the expression of collagen deposition markers via TGF-β/Smad-3 pathway
TGF-β1/Smad cascade is a key fibrotic axis that drives EMT by increasing the ECM synthesis and collagen deposition. TGF-β1 stimulation phosphorylates Smad2/3 and modulates the target gene expression (Fernandez and Eickelberg, 2012). To investigate the effect of BCA on TGF-β1/Smad signaling pathway, expression levels of Smad2 and Smad7 were measured in TGF-β1 treated cells and results revealed that, Smad2 expression levels (Fig. 4 A, E and I) were increased in TGF-β treated cells and smad7 levels (Fig. 4M, N and O) were significantly reduced, it indicates that, TGF-β/Smad signaling has been activated in the test system. BCA treatment significantly (p < 0.001) attenuated the Smad2 levels and elevated the smad7 mRNA levels in a dose dependent manner in NHLF (Fig. 4A and M), DHLF (Fig. 4E and N) and LL29 (Fig. 4I and O) cells.
Fig. 4.

BCA treatment attenuated the TGF-β/smad3 axis induced expression of collagen depositing markers: Cells were pre-treated with BCA for 2 h, thereafter rTGF-β added to the cells and cultured for 72 h. Further, cells were subjected to RT-qPCR for the specified transcripts in A-D) NHLF, E-H) DHLF, I-L) LL29. M-O) Smad7 mRNA expression in NHLF, DHLF and LL29 cells, respectively. Results were shown as Mean ± SEM, n = 3. ∗ p < 0.05, ∗∗ p < 0.01,∗∗∗ p < 0.001.
Expression of collagen deposition markers were significantly (p < 0.001) elevated in TGF-β treated cells, whereas treatment with BCA significantly reduced the same (CTGF, COL1A1 and COL3A1) in NHLF (Fig. 4B-D), DHLF (Fig. 4F-H) and LL29 cells (Fig. 4J-L). Gene expression results were corroborated with immunofluorescence analysis of COL1A1 in NHLF (Fig. S4A and S4D), DHLF (Fig. S4B and S4E) and LL29 cells (Fig. S4C and S4F). Overall, these results revealed that, BCA treatment attenuated the TGF-β mediated over expression of EMT, ECM and collagen deposition markers in normal and IPF cells.
BCA abrogated the hypoxia induced fibrotic-gene expression in LL29 cells
Hypoxia has been reported to promote TGF-β1 expression in various fibrotic disorders (Hanna et al., 2013). To confirm this gene expression analysis was performed (Using LL29 cells) for fibrotic markers (TGF-β, α-SMA, LOXL-2, COL1A1 and COL3A1) under hypoxic conditions. To confirm the induction of hypoxia, HIF1-α and VEGF mRNA levels were measured. The mRNA expression results revealed that HIF1-α and VEGF levels were significantly (p < 0.01) increased in hypoxia control (Fig. S5A and S5B). Thereafter, cells were treated with BCA and cultured in hypoxia condition for 72 hr. Interestingly, key fibrotic gene TGF-β1 was significantly (p < 0.05) up-regulated along with other fibrotic markers in hypoxia control, however, BCA treatment reduced the expression of fibrotic markers in a dose dependent manner (Fig. S5C-G). Overall, these results further confirmed that, fibrotic cascade activation by TGF-β pathway was significantly reduced by BCA treatment in IPF cells.
BCA attenuated the expression of fibrotic markers in PCMLS
To confirm the in vitro results of BCA, experiment were performed using PCMLS as an ex-vivo model. Similar to in vitro model, gene expression data on PCMLS revealed that, all fibrotic cascade genes were highly up-regulated in TGF-β control. BCA (20 µM) treatment showed significant (p < 0.001) down-regulation of all fibrotic genes (Fig. 5 A-F). In contrast to fibroblast cells, in PCLMS derived cells, we investigated the E-cad gene expression to assess the reversal of EMT, E-cad levels were down-regulated upon TGF-β1 stimulation and BCA treatment rescued the same (Fig. 5G). Further, immunofluorescence results (Fig. 5H-J and S6A-B) revealed that, FN-I, COL1A1 levels were significantly (p < 0.01) attenuated and E-cad levels were elevated upon BCA treatment in ex vivo test system.
Fig. 5.

Role of BCA on TGF-β-induced fibrotic genes/markers expression in PCMLS:
PCMLS were pre-treated with BCA for 2 h, thereafter rTGF-β (mouse) added to the cells and cultured for 72 h. Further, PCMLS were subjected to RT-qPCR for the specified transcripts. A-G) Gene expression analysis. Graphs in panels represent fold change in gene expression normalized to GAPDH. PCMLS derived cells were subjected to immunofluorescence analysis for H) FN-1, I) COL1A1 and J) E-cad. Results were shown as Mean ± SEM, n = 3. ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ p < 0.001.
BCA treatment attenuated the expression and function of fibrotic markers through TGF-β/Smad signaling
To validate the smad2 and smad7 mRNA results obtained in Fig. 4, protein expression analysis for phospho-smad3 was performed upon TGF-β challenge. Western blot analysis revealed that TGF-β treatment increased the expression of phospho-Smad3 in LL29 and PCMLS cells (Fig. 6 A-B and 6E-F). BCA treatment significantly (p < 0.01) reduced the expression of phospho-smad3 in a dose dependent manner. Interestingly, the elevated expression of smad7 (Fig. 4M, N and O), which inhibits p-Smad3, was increased following BCA treatment. Therefore, we speculate that up-regulation of Smad7 is a potential intermediate mechanism for the down-phosphorylation of Smad3 by BCA. Therefore, the anti-fibrotic activity of BCA could be due to modulation of smad7/p-smad3 regulation in IPF cells. As a consequence of phospho-smad3 up-regulation, FN-1 and α-SMA levels were also significantly (p < 0.001) up-regulated upon TGF-β1 stimulation however, BCA treatment attenuated the elevation of FN1- and α-SMA (Fig. 6C-D and 6G-H). On the other hand, E-cad expression (PCMLS) was slightly reduced in TGF-β1 control, surprisingly, BCA treatment rescued the E-cad expression in PCMLS (Fig. 6E and I).
Fig. 6.

BCA treatment attenuated the expression and function of pro-fibrotic markers through TGF-β/smadsignaling cascade: LL29 cells or PCMLS were pre-treated with BCA for 2 h, thereafter rTGF-β added to the cells and cultured for 72 h. Further, cells were subjected to immunoblot analysis A and E) Immunoblot for specified antibodies. The graphs represent densitometric quantification for the specified proteins in B-D) LL29 and F-I)PCMLS. J-M) After attaining 100% confluency of NHLF, PCMLS derived cells, LL29 and DHLF cells, cells were pre-treated with BCA for 2 h and then scratch was made, thereafter cells were cultured for 72h in presence or absence of rTGF-β1. The graphs represent time kinetics of wound confluence percentage, calculated by image J software Results were shown as Mean ± SEM, n = 3. ∗ p < 0.05,∗∗ p < 0.01,∗∗∗ p < 0.001.
To corroborate gene/protein expression data with functional activity of EMT, scratch assay was performed using NHLF, PCMLS derived cells, DHLF and LL29 cells. BCA attenuated the TGF-β induced cell migration (Fig. 6J-M and S7A-D) in a concentration dependent manner. Hence, these results indicated that, BCA treatment not only inhibits the expression of fibrotic cascade of genes (EMT, ECM and Collagen) but also attenuates the functional activities of fibrotic genes.
BCA attenuated BLM-induced pulmonary fibrosis in rats
To further evaluate the anti-fibrotic activity of BCA in in vivo system, we employed widely used BLM induced pulmonary fibrosis model in rats. After 14 days of bleomycin administration by intratracheal route, rats were (orally) treated with BCA-5 mg/kg or BCA-10 mg/kg or pirfenidone-50 mg/kg for a period of 14 days as described in schematic plan (Fig. 7 A). Pro-inflammatory markers such as ALP and LDH levels were significantly increased in BLM control samples, upon treatment with BCA or pirfenidone, ALP (Fig. 7B) and LDH (Fig. 7C) levels were reduced in a dose dependent manner. Further, neutrophil infiltration and WBC count was measured in all the BALF samples of rats, elevated levels of neutrophil and WBC was observed in BLM control group and those levels were reduced significantly (p < 0.01, p < 0.001) in BCA-10 mg/kg treatment group (Fig. 7D and E). Treatment with pirfenidone reduced the BLM elevated WBC count, however, neutrophil levels were unchanged in BALF samples. BCA or pirfenidone treatment markedly ameliorated the BLM induced increase in lung index (Fig. 7F) and collagen levels (Hydroxy proline) in the lung tissues (Fig. 7G). Taken together, these results revealed that BCA treatment could potentially attenuate the BLM-induced infiltration of inflammatory cells and degree of fibrosis in lung tissues compared to pirfenidone treatment.
Fig. 7.

Protective role of BCA on BLM-induced pulmonary fibrosis: A) Schematic representation of BLM Induced pulmonary fibrosis. Post bleomycin (BLM) instillation, rats were orally treated with BCA (5 mg/kg), BCA (10 mg/kg), pirfenidone (50 mg/kg) for a period of two weeks. BLM group and sham control animals received vehicle suspension and saline respectively. After the treatment period, rats were anesthetized and BALF fluid and lung tissue was collected. B) Alkaline phosphatase C) and lactate dehydrogenase (LDH) levels were measured in BALF D) neutrophil count and E) WBC count was estimated in BALF. F) Lung index G) Hydroxyproline content. Data was shown as Mean ± S.E.M (n = 4). * p < 0.05, ** p < 0.01 vs. BLM group.
BCA treatment attenuated the fibrotic marker's expression by modulating the TGF-β/Smad pathway in in-vivo
Elevated expression of TGF-β and fibrotic markers were observed in BLM control samples, on the other-hand, BCA treatments significantly attenuated the BLM induced TGF-β gene expression (Fig. 8 A) and expression of other fibrotic markers (α-SMA, CTGF, TIMP-I, COL1A1, COL3A1 and FN-I) in a dose dependent manner (Fig. 8B-G). Pirfenidone treatment reduced the expression of TGF-β and other fibrotic genes but failed to show a significant reduction of COL1A1 and COL3A1 when compared with BLM control. BCA treatment reversed the BLM induced reduction of E-cad levels in both the treatment groups (Fig. 8H), but E-cad levels remained low in pirfenidone treatment. In addition to gene expression data, protein expression analysis also revealed that, BCA treatment significantly (p < 0.001) reduced the BLM induced phospho-smad3, α-SMA and FN-I expression in lung samples (Fig. 8I-L). These measures correlate with in-vitro investigations of BCA in IPF cells, hence, these results strongly demonstrate the anti-fibrotic activity BCA in in vitro and in vivo models.
Fig. 8.

BCA treatment attenuated the fibrotic marker's expression by modulating the TGF-β/smad pathway in in vivo: After treatment, lung tissues were collected and part of tissues was subjected for RT-qPCR or immunoblot analysis for the specified transcripts or antibodies respectively. A-H) Gene expression analysis I) Representative images of western blot, J-L) Graph represents densitometric quantification of the specified proteins; Results were shown as Mean ± SEM; n = 4. * p < 0.05; ** p < 0.01; *** p < 0.001 vs. the BLM alone group.
BCA treatment mitigated the BLM-induced pathological changes in lungs
Histopathological analysis by H&E (Fibrosis score) and Masson's trichrome staining (Ashcroft score) revealed that increased thickening in alveolar septa and collapse of alveolar spaces excess with proliferation of fibroblasts were observed in BLM group (Fig. 9 A). BCA and pirfenidone treatment ameliorated the BLM induced abnormal changes in the lungs (mild thickened alveolar walls), inflammatory cells infiltration and reduced deposition of collagen fibers compared to BLM control group (Fig. 9A, B, F and G). In addition to histopathology, immunohistochemistry was performed to examine the expression of fibrotic markers in lung sections. BCA treatment reduced the expression of FN-I (Fig. 9C and H), COL1A1 (Fig. 9D and H) and increased the expression of E-cad (Fig. 9E and H) compared to BLM group. These results indicate that, BCA treatment reduced the EMT progression and promoted the reversal of EMT by increasing the E-cad expression (Fig. 9H). Collectively, in-vivo results demonstrated that, BCA could effectively protect the BLM-induced inflammation, fibrotic marker expression and pathological changes in lung tissues.
Fig. 9.

Protective effect of BCA on bleomycin-induced pathological changes
After treatment with BCA (5 mg/kg) or (10 mg/kg) or pirfinidone (50 mg/kg) vehicle, lungs were isolated and subjected for histopathology. A and B) Representative pictures (10 ×) of A) H&E-stained, B) Masson's trichrome-stained sections. The Black arrow points at typical areas of fibrosis, manifesting as group of cells with protruding cytoplasm (myofibroblasts). C, D and E) Immunohistochemistry for FN-I, COL1A1 and E-cad in lung sections. The black arrows at typical areas represent the increased expression of protein. Images were taken under original magnification 20 ×. Graphs in panels represent the score of F) Fibrosis and G) Ashcroft score, score numbers of 0–8, were evaluated by experienced pathologists in a blinded fashion. H) Table represents the semi-quantitative evaluation of protein expression (FN-I, COL1A1 and E-cad) in specified treatment groups. Data was expressed as Mean ± S.E.M. * p < 0.05; ** p < 0.01; *** p < 0.001 vs. the BLM alone group.
Discussion
In humans, IPF is an aging-associated progressive disorder resulting from persistent damage to the alveolar epithelium followed by uncontrolled proliferation and differentiation of fibroblasts. Fibroblasts are converted to myofibroblasts and these myofibroblasts are responsible for the excessive proliferation, EMT, ECM production and lead to overlay the deposition of collagen in affected organs.
Biochanin-A is mainly found in red clover and is known to have various protective effects such as neuroprotective (Tan et al., 2013), anti-inflammatory (Kole et al., 2011b) and antioxidant properties. BCA treatment showed significant anti-inflammatory effects via inhibition of NF-κB and MAPK as well as mitigating the release of inflammatory mediators in various disease models (Sarfraz et al., 2020; Zhang and Chen, 2015). BCA has demonstrated to have anti-cancer activity against different cancers (Raheja et al., 2018),by inhibiting the proliferation of cells and EMT process (Wang et al., 2018a). The main limitation of the BCA is its poor bioavailability and is also reported to undergo entero hepatic recirculation (Moon et al., 2006). We speculate that the increased mean residence time (MRT) due to entero hepatic recirculation might be responsible for therapeutic activity in long dosage schedules. Though anti-fibrotic activity of BCA against CCl4-induced hepatic fibrosis (Breikaa et al., 2013) was reported in earlier, mechanism of action was still unclear, our studydescribed the molecular mechanism of BCA against pulmonary fibrosis.
Since the pathophysiology of IPF is closely related to oxidative stress, inflammation and EMT processes, we speculated that, BCA may have protective effects against IPF because of its potent antioxidant (Sadri et al., 2017) and anti-inflammatory properties. Our study first time demonstrated the anti-fibrotic activity of a BCA using IPF cells and primary human lung cells.
Reports on LL29, NHLF and DHLF cells regarding the expression of IPF markers are scanty. Therefore we investigated the basal expression of fibrotic (EMT, ECM, and collagen) markers with respect to the cascade of events in IPF. We observed that, majority of the markers which are known to over-express in IPF conditions were highly expressed in DHLF and LL29 cells compared to NHLF cells. Surprisingly, BCA treatment (without TGF-β stimulation) showed more specificity towards IPF cells by down-regulating the expression of fibrotic genes compared to normal cells (Fig. 1). It indicates that BCA treatment has more affinity towards IPF cells or fibrotic gene over-expressed cells (exogenous stimulation).
Tanjore et al. 2009 reported that one-third of myofibroblasts were generated from Type-II lung epithelial cells through the process of EMT upon BLM treatment. Hence, targeting the EMT process would be beneficial for ameliorating the IPF. EMT can be activated by a plethora of cues such as hypoxia, TGF-β activation, receptor tyrosine kinases, Notch and Wnt pathways activation (Moustakas and Heldin, 2016). Our results demonstrated that, BCA treatment significantly attenuated the expression of EMT genes and protein markers in both in vitro and ex vivo experiments (Fig. 3).
In addition to an elevation of EMT gene expressions, TGF-β1 activation promotes cellular migration and invasion in various disease conditions (Sun et al., 2017). To corroborate the gene/protein expression data, a functional assay of EMT (Scratch assay) was performed which revealed that BCA treatment attenuated the TGF-β induced migration of cells in all cell types (Fig. 6). Differentiation is mainly regulated by EMT and ECM markers which is characterized by persistence of fibroblast to myofibroblast conversion and its accumulation and in lung tissues (Chanda et al., 2019). It was reported that, there is a close association with excessive inflammation, oxidative stress and abnormal EMT with IPF (Yan et al., 2018). Accumulating evidence suggests that, reactive oxygen species (ROS) as crucial conspirators in EMT engagement (Giannoni et al., 2012). Several studies (Liu et al., 2020; Yang et al., 2019) mainly investigated on the expression of EMT and collagen markers to evaluate the anti-fibrotic activity of test compounds. Since, BCA shows strong anti-oxidant property (Sadri et al., 2017), we investigated the role of BCA against expression of EMT and ECM markers in IPF condition. Gene expression analysis of ECM markers (TIMP-I, TIMP-III and LOXL-2) revealed that BCA treatment significantly (p < 0.01) abrogated the TGF-β mediated expression of ECM markers in a dose-dependent manner (Fig. 3). CTGF plays a key role in pulmonary fibrosis by inducing the COL1A1 expression followed by improving the α-SMA levels in lung tissues through activation of JNK pathway (Lin et al., 2013). Our data showed that BCA treatment significantly reduced the expression of CTGF, COL1A1 and COL3A1 in normal and IPF cells (Fig. 4 and S4). It was reported (Abdalla et al., 2015) that, hypoxia induces fibroblasts proliferation and pulmonary fibrosis in mice lungs. In-refer to confirm the anti-fibrotic activity of BCA against hypoxia challenge, we investigated the expression of fibrotic markers under hypoxic condition. Interestingly, BCA treatment potentially attenuated the hypoxia-challenged fibrotic marker's expression in LL29 cells (Fig. S5).
Though, we used primary cells (NHLF and DHLF) and IPF cell lines to assess the anti-fibrotic activity of BCA, to confirm the anti-fibrotic activity in intact tissues, further experiments were performed using lung slices (PCMLS and PCLMS derived cells) where cells mimic-3D architecture and complex cellularity of the airways and parenchyma. Gene and protein expression analysis results revealed that BCA significantly decreased the TGF-β induced expression of fibrotic cascade markers in ex vivo model (Fig. 5).
Various studies revealed that TGF-β could persuade Smad2/3 phosphorylation to promote fibroblast differentiation (Huang et al., 2019). It was also reported that, smad7 is an inhibitory Smad and plays a protective role in several inflammatory disorders by down regulating the phospho- smad3 (Zhou et al., 2018). These evidences impelled us to focus on the impact of BCA on TGF-β/Smad signaling pathway. These results revealed that BCA treatment significantly increased the Smad7 levels and attenuated the expression of Smad2 mRNA levels (in all cell types) and phospho-smad3 protein in LL29 and PCMLS cells (Fig. 6). The possible mechanism of the therapeutic effects of BCA on pulmonary fibrosis might be attributed to the inhibition of TGF-β/Smad3 signaling (Fig. 2, Fig. 3, Fig. 4, Fig. 5, Fig. 6).
Further, we investigated the effect of BCA on widely used bleomycin induced pulmonary fibrosis in rats (Robbe et al., 2015). In vivo results revealed that, the lung index was significantly increased in BML group and BCA treatment (5 mg/kg or 10 mg/kg) reversed the same significantly. BALF examination showed that inflammatory markers (ALP and LDH) and inflammatory cells (neutrophils count) were significantly reduced in BCA treatment compared to BLM control samples. Comparison of generation of inflammatory markers and infiltration of inflammatory cells between BCA and pirfenidone revealed that the former was superior in terms of inhibition of inflammatory markers (production) and the degree of cell infiltration, suggesting that, BCA represents a strong candidate for ameliorating IPF. These results show that BCA protects the lungs from BLM induced pulmonary inflammation (Fig. 7).
BLM treatment is known to increase the inflammation and growth factor signaling in lung tissues. TGF-β1 induction upon BLM treatment which lead to stimulation of differentiation, proliferation, ECM production and collagen deposition in lung tissues by canonical signaling pathway. In vivo experiments clearly showed that, pirfenidone (50 mg/kg) could suppress the expression of TGF-β, and BCA (5 mg/kg or 10 mg/kg) treatment significantly reduced the expression of TGF-β as well at low dose in a dose dependent manner. Further, our results revealed that BCA treatment modulated the TGFβ/Smad axis and significantly reduced the expression of fibrotic markers (α-SMA, FN-I, CTGF, COL1AL, and COL3A1) in lung tissue samples (Fig. 8). Treatment with pirfenidone did not significantly affect the COL1A1 and COL3A1 expression and failed to recover the synthesis of E-cadherin, this observations is in-line with previous studies(Molina-Molina et al., 2018b). Histopathological evaluation revealed that, BCA treatment significantly reduced the degree of pulmonary fibrosis in BLM-induced rats as represented by H&E and Masson's trichrome staining. Furthermore, BCA treatment significantly reduced the collagen deposition in lung sections by Ashcroft score analysis as evidenced in reduced hydroxyproline content in the lung tissues. In line with previous studies (Gao et al., 2015; Liu et al., 2020)present study also assessed the FN-I and COL1A1 expression in lung tissues, in addition to that we have also assessed the E-cad expression to check the reversal of EMT, BCA treatment dramatically reduced the BLM elevated levels of FN-I and COL1A1 and improved the E-cad levels in lung tissues (Fig. 9). Overall, our study revealed the therapeutic potential of BCA against pulmonary fibrosis by modulating the TGF-β mediated EMT, myofibroblasts differentiation and collagen deposition in IPF cells and bleomycin treated rats.
Conclusion
In conclusion, BCA mitigated the development and progression of pulmonary fibrosis by modulating the TGF-β/Smad3 pathway and ameliorating the fibrotic cascade of events. Further studies are warranted to improve bioavailability of BCA and to determine the in-depth mechanism of action of BCA in comparison to the FDA approved drugs to provide the potential clinical value of the biochanin-A against IPF.
Funding
This work was not funded.
Author contributions
Conceptualization: S.B.A and S.R.K; Methodology: S.B.A, K.G, S.R.K; Validation: S.B.A, K.G., S.R.K; Invitro cell culture: S.B.A, KG. Q-RT-PCR, western-blot analysis, ex-vivo and in-vivo experiments: S.B.A, TSK, K.G. Immunofluorescence analysis: TS, KG. Writing - review & editing: S.B.A,K.G, S.R.; funding acquisition: S.B.A, S.R.K. All data were generated in-house, and no paper mill was used. All authors agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Competing Interest
The authors declare no competing or financial interests.
Acknowledgments
The authors are thankful to the Director, CSIR-IICT, Hyderabad, India, for providing facilities necessary for the completion of this work and for his constant support. We thank Dr. Mahesh Kumar Jerald (Veterinary pathologist) for interpretation of the histopathology results. We thank Sun Pharmaceutical Industries Ltd for providing pirfenidone as a kind gift. We also thank Dr. Ramesh Ummani for availing to use tri-gas cylinder and Dr. Sumana Chakravarty for availing to use confocal microscope. TS thank the Council of Scientific and Industrial Research (CSIR), New Delhi, India, for the award of Junior Research Fellowship (JRF). CSIR-IICT manuscript communication number: IICT/Pubs./2020/0084.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.phymed.2020.153298.
Appendix. Supplementary materials
References
- Abdalla M., Sabbineni H., Prakash R., Ergul A., Fagan S.C., Somanath P.R. The Akt inhibitor, triciribine, ameliorates chronic hypoxia-induced vascular pruning and TGFβ-induced pulmonary fibrosis. Br. J. Pharmacol. 2015;172:4173–4188. doi: 10.1111/bph.13203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akgedik R., Akgedik Ş., Karamanlı H., Uysal S., Bozkurt B., Ozol D., Armutcu F., Yıldırım Z. Effect of Resveratrol on Treatment of Bleomycin-Induced Pulmonary Fibrosis in Rats. Inflammation. 2012;35:1732–1741. doi: 10.1007/s10753-012-9491-0. [DOI] [PubMed] [Google Scholar]
- Ashcroft T., Simpson J.M., Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 1988;41:467–470. doi: 10.1136/jcp.41.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaji S.A., Udupa N., Chamallamudi M.R., Gupta V., Rangarajan A. Role of the Drug Transporter ABCC3 in Breast Cancer Chemoresistance. PLoS ONE. 2016;11 doi: 10.1371/journal.pone.0155013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breikaa, R.M., Algandaby, M.M., El-Demerdash, E., Abdel-Naim, A.B., 2013. Multimechanistic antifibrotic effect of biochanin a in rats: implications of proinflammatory and profibrogenic mediators. PLoS ONE8. [DOI] [PMC free article] [PubMed]
- Chanda D., Otoupalova E., Smith S.R., Volckaert T., De Langhe S.P., Thannickal V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Aspects Med. 2019;65:56–69. doi: 10.1016/j.mam.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Li S., Li W. LOX/LOXL in pulmonary fibrosis: potential therapeutic targets. J. Drug Target. 2019;27:790–796. doi: 10.1080/1061186X.2018.1550649. [DOI] [PubMed] [Google Scholar]
- Chen T., Nie H., Gao X., Yang J., Pu J., Chen Z., Cui X., Wang Y., Wang H., Jia G. Epithelial–mesenchymal transition involved in pulmonary fibrosis induced by multi-walled carbon nanotubes via TGF-beta/Smad signaling pathway. Toxicol. Lett. 2014;226:150–162. doi: 10.1016/j.toxlet.2014.02.004. [DOI] [PubMed] [Google Scholar]
- Chundi V., Challa S.R., Garikapati D.R., Juvva G., Jampani A., Pinnamaneni S.H., Venigalla S. Biochanin-A attenuates neuropathic pain in diabetic rats. J. Ayurveda Integr. Med. 2016;7:231–237. doi: 10.1016/j.jaim.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez I.E., Eickelberg O. The Impact of TGF-βon Lung Fibrosis. Proc. Am. Thorac. Soc. 2012;9:111–116. doi: 10.1513/pats.201203-023AW. [DOI] [PubMed] [Google Scholar]
- Gao L., Tang H., He H., Liu J., Mao J., Ji H., Lin H., Wu T. Glycyrrhizic acid alleviates bleomycin-induced pulmonary fibrosis in rats. Front. Pharmacol. 2015;6 doi: 10.3389/fphar.2015.00215. 215-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George, P.M., Wells, A.U., Jenkins, R.G., Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. The Lancet Respir. Med.2020. [DOI] [PMC free article] [PubMed]
- Giannoni E., Parri M., Chiarugi P. EMT and oxidative stress: a bidirectional interplay affecting tumor malignancy. Antioxid. Redox Signal. 2012;16:1248–1263. doi: 10.1089/ars.2011.4280. [DOI] [PubMed] [Google Scholar]
- Hanna C., Hubchak S.C., Liang X., Rozen-Zvi B., Schumacker P.T., Hayashida T., Schnaper H.W. Hypoxia-inducible factor-2α and TGF-β signaling interact to promote normoxic glomerular fibrogenesis. Am. J. Physiol. Renal Physiol. 2013;305:F1323–F1331. doi: 10.1152/ajprenal.00155.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harini R., Ezhumalai M., Pugalendi K.V. Antihyperglycemic effect of biochanin A, a soy isoflavone, on streptozotocin-diabetic rats. Eur. J. Pharmacol. 2012;676:89–94. doi: 10.1016/j.ejphar.2011.11.051. [DOI] [PubMed] [Google Scholar]
- Huang S., Chen B., Su Y., Alex L., Humeres C., Shinde A.V., Conway S.J., Frangogiannis N.G. Distinct roles of myofibroblast-specific Smad2 and Smad3 signaling in repair and remodeling of the infarcted heart. J. Mol. Cell. Cardiol. 2019;132:84–97. doi: 10.1016/j.yjmcc.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S., Sekine A., Baba T., Katano T., Tabata E., Shintani R., Sadoyama S., Yamakawa H., Oda T., Okuda R., Kitamura H., Iwasawa T., Takemura T., Ogura T. Negative impact of anorexia and weight loss during prior pirfenidone administration on subsequent nintedanib treatment in patients with idiopathic pulmonary fibrosis. BMC Pulm. Med. 2019;19:78. doi: 10.1186/s12890-019-0841-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.S., Yoon Y.M., Meang M.K., Park Y.E., Lee J.Y., Lee T.H., Lee J.E., Kim I.H., Youn B.S. Reversion of in vivo fibrogenesis by novel chromone scaffolds. EBioMedicine. 2019;39:484–496. doi: 10.1016/j.ebiom.2018.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko W.-C., Lin L.-H., Shen H.-Y., Lai C.-Y., Chen C.-M., Shih C.-H. Biochanin A, a Phytoestrogenic Isoflavone with Selective Inhibition of Phosphodiesterase 4, Suppresses Ovalbumin-Induced Airway Hyperresponsiveness. Evid. Based Complement. Alternat. Med. 2011;2011 doi: 10.1155/2011/635058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole L., Giri B., Manna S.K., Pal B., Ghosh S. Biochanin-A, an isoflavon, showed anti-proliferative and anti-inflammatory activities through the inhibition of iNOS expression, p38-MAPK and ATF-2 phosphorylation and blocking NFκB nuclear translocation. Eur. J. Pharmacol. 2011;653:8–15. doi: 10.1016/j.ejphar.2010.11.026. [DOI] [PubMed] [Google Scholar]
- Kole L., Giri B., Manna S.K., Pal B., Ghosh S. Biochanin-A, an isoflavon, showed anti-proliferative and anti-inflammatory activities through the inhibition of iNOS expression, p38-MAPK and ATF-2 phosphorylation and blocking NFκB nuclear translocation. Eur. J. Pharmacol. 2011;653:8–15. doi: 10.1016/j.ejphar.2010.11.026. [DOI] [PubMed] [Google Scholar]
- Lin C.-H., Yu M.-C., Tung W.-H., Chen T.-T., Yu C.-C., Weng C.-M., Tsai Y.-J., Bai K.-J., Hong C.-Y., Chien M.-H., Chen B.-C. Connective tissue growth factor induces collagen I expression in human lung fibroblasts through the Rac1/MLK3/JNK/AP-1 pathway. Biochim. Biophys. Acta. 2013;1833:2823–2833. doi: 10.1016/j.bbamcr.2013.07.016. [DOI] [PubMed] [Google Scholar]
- Liu P., Miao K., Zhang L., Mou Y., Xu Y., Xiong W., Yu J., Wang Y. Curdione ameliorates bleomycin-induced pulmonary fibrosis by repressing TGF-β-induced fibroblast to myofibroblast differentiation. Respir. Res. 2020;21:58. doi: 10.1186/s12931-020-1300-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., Liu, B., Zhang, G.-q., Zou, J.-f., Zou, M.-l., Cheng, Z.-s., 2018. Calpain inhibition attenuates bleomycin-induced pulmonary fibrosis via switching the development of epithelial-mesenchymal transition. Naunyn Schmiedebergs Arch. Pharmacol.391, 695–704. [DOI] [PMC free article] [PubMed]
- Maher T.M., Strek M.E. Antifibrotic therapy for idiopathic pulmonary fibrosis: time to treat. Respir. Res. 2019;20:205. doi: 10.1186/s12931-019-1161-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins, A.D., Oliveira, P.F., Alves, M.G., 2019. Assessment of Sertoli Cell Proliferation by 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide and Sulforhodamine B Assays. Curr. Protocol Toxicol.81, e85. [DOI] [PubMed]
- Mehrabadi M.E., Salemi Z., Babaie S., Panahi M. Effect of Biochanin A on Retina Levels of Vascular Endothelial Growth Factor, Tumor Necrosis Factor-Alpha and Interleukin-1Beta in Rats With Streptozotocin-Induced Diabetes. Can. J. Diabetes. 2018;42:639–644. doi: 10.1016/j.jcjd.2018.03.008. [DOI] [PubMed] [Google Scholar]
- Ming X., Ding M., Zhai B., Xiao L., Piao T., Liu M. Biochanin A inhibits lipopolysaccharide-induced inflammation in human umbilical vein endothelial cells. Life Sci. 2015;136:36–41. doi: 10.1016/j.lfs.2015.06.015. [DOI] [PubMed] [Google Scholar]
- Molina-Molina M., Machahua-Huamani C., Vicens-Zygmunt V., Llatjós R., Escobar I., Sala-Llinas E., Luburich-Hernaiz P., Dorca J., Montes-Worboys A. Anti-fibrotic effects of pirfenidone and rapamycin in primary IPF fibroblasts and human alveolar epithelial cells. BMC Pulm. Med. 2018;18:63. doi: 10.1186/s12890-018-0626-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Molina, M., Machahua-Huamani, C., Vicens-Zygmunt, V., Llatjós, R., Escobar, I., Sala-Llinas, E., Luburich-Hernaiz, P., Dorca, J., Montes-Worboys, A., 2018b. Anti-fibrotic effects of pirfenidone and rapamycin in primary IPF fibroblasts and human alveolar epithelial cells. BMC Pulm. Med.18, 63. [DOI] [PMC free article] [PubMed]
- Moon Y.J., Sagawa K., Frederick K., Zhang S., Morris M.E. Pharmacokinetics and bioavailability of the isoflavone biochanin A in rats. AAPS J.8. 2006:E433–E442. doi: 10.1208/aapsj080351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakas, A., Heldin, C.H., 2016. Mechanisms of TGFβ-Induced Epithelial-Mesenchymal Transition. J. Clin. Med.5. [DOI] [PMC free article] [PubMed]
- Oh J.S., Cho I.A., Kang K.R., You J.S., Yu S.J., Lee G.J., Seo Y.S., Kim C.S., Kim D.K., Kim S.G., Seo Y.W., Im H.J., Kim J.S. Biochanin-A antagonizes the interleukin-1β-induced catabolic inflammation through the modulation of NFκB cellular signaling in primary rat chondrocytes. Biochem. Biophys. Res. Commun. 2016;477:723–730. doi: 10.1016/j.bbrc.2016.06.126. [DOI] [PubMed] [Google Scholar]
- Pieretti A.C., Ahmed A.M., Roberts J.D., Jr., Kelleher C.M. A novel in vitro model to study alveologenesis. Am. J. Respir. Cell Mol. Biol. 2014;50:459–469. doi: 10.1165/rcmb.2013-0056OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raheja S., Girdhar A., Lather V., Pandita D. Biochanin A: a phytoestrogen with therapeutic potential. Trends Food Sci. Technol. 2018;79:55–66. [Google Scholar]
- Richeldi L., Collard H.R., Jones M.G. Idiopathic pulmonary fibrosis. The Lancet. 2017;389:1941–1952. doi: 10.1016/S0140-6736(17)30866-8. [DOI] [PubMed] [Google Scholar]
- Robbe, A., Tassin, A., Carpentier, J., Declèves, A.E., Mekinda Ngono, Z.L., Nonclercq, D., Legrand, A., 2015. Intratracheal Bleomycin Aerosolization: the Best Route of Administration for a Scalable and Homogeneous Pulmonary Fibrosis Rat Model?Biomed. Res. Int.2015, 198418. [DOI] [PMC free article] [PubMed]
- Sadri, H., Goodarzi, M.T., Salemi, Z., Seifi, M., 2017. Antioxidant Effects of Biochanin A in Streptozotocin Induced Diabetic Rats. Brazil. Arch. Biol. Tech.60.
- Salton, F., Volpe, M.C., Confalonieri, M., 2019. Epithelial⁻Mesenchymal Transition in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Medicina (Kaunas)55. [DOI] [PMC free article] [PubMed]
- Sarfraz A., Javeed M., Shah M.A., Hussain G., Shafiq N., Sarfraz I., Riaz A., Sadiqa A., Zara R., Zafar S., Kanwal L., Sarker S.D., Rasul A. Biochanin A: a novel bioactive multifunctional compound from nature. Sci. Total Environ. 2020;722 doi: 10.1016/j.scitotenv.2020.137907. [DOI] [PubMed] [Google Scholar]
- Saxena M., Balaji S.A., Deshpande N., Ranganathan S., Pillai D.M., Hindupur S.K., Rangarajan A. AMP-activated protein kinase promotes epithelial-mesenchymal transition in cancer cells through Twist1 upregulation. J. Cell Sci. 2018;131 doi: 10.1242/jcs.208314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnolo, P., Balestro, E., Aliberti, S., Cocconcelli, E., Biondini, D., Casa, G.D., Sverzellati, N., Maher, T.M., 2020. Pulmonary fibrosis secondary to COVID-19: a call to arms?The Lancet Respir. Med.. [DOI] [PMC free article] [PubMed]
- Sun Q., Wu Y., Zhao F., Wang J. Maresin 1 inhibits transforming growth factor-β1-induced proliferation, migration and differentiation in human lung fibroblasts. Mol. Med. Rep. 2017;16:1523–1529. doi: 10.3892/mmr.2017.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szliszka E., Czuba Z.P., Mertas A., Paradysz A., Krol W. Urologic Oncology: Seminars and Original Investigations. Elsevier; 2013. The dietary isoflavone biochanin-A sensitizes prostate cancer cells to TRAIL-induced apoptosis; pp. 331–342. [DOI] [PubMed] [Google Scholar]
- Tan J.W., Tham C.L., Israf D.A., Lee S.H., Kim M.K. Neuroprotective effects of biochanin A against glutamate-induced cytotoxicity in PC12 cells via apoptosis inhibition. Neurochem. Res. 2013;38:512–518. doi: 10.1007/s11064-012-0943-6. [DOI] [PubMed] [Google Scholar]
- Tjin G., White E.S., Faiz A., Sicard D., Tschumperlin D.J., Mahar A., Kable E.P., Burgess J.K. Lysyl oxidases regulate fibrillar collagen remodelling in idiopathic pulmonary fibrosis. Dis. Model. Mech. 2017;10:1301–1312. doi: 10.1242/dmm.030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Li J.J., Chen Y.M. Biochanin A extirpates the epithelial-mesenchymal transition in a human lung cancer. Exp. Ther. Med. 2018;15:2830–2836. doi: 10.3892/etm.2018.5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Li J.J., Chen Y.M. Biochanin A extirpates the epithelial-mesenchymal transition in a human lung cancer. Exp. Ther. Med. 2018;15:2830–2836. doi: 10.3892/etm.2018.5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L., Song F., Li H., Li Y., Li J., He Q.-Y., Zhang D., Wang F., Zhang M., Zhao H., Feng T., Zhao Y.-Y., Wang S.-W. Submicron emulsion of cinnamaldehyde ameliorates bleomycin-induced idiopathic pulmonary fibrosis via inhibition of inflammation, oxidative stress and epithelial-mesenchymal transition. Biomed. Pharmacother. 2018;102:765–771. doi: 10.1016/j.biopha.2018.03.145. [DOI] [PubMed] [Google Scholar]
- Yang J.-Y., Tao L.-J., Liu B., You X.-Y., Zhang C.-F., Xie H.-F., Li R.-S. Wedelolactone Attenuates Pulmonary Fibrosis Partly Through Activating AMPK and Regulating Raf-MAPKs Signaling Pathway. Front. Pharmacol. 2019;10 doi: 10.3389/fphar.2019.00151. 151-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C., Zhang P., Lou L., Wang Y. Perspectives Regarding the Role of Biochanin A in Humans. Front. Pharmacol. 2019;10 doi: 10.3389/fphar.2019.00793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaghloul M.S., Abdel-Salam R.A., Said E., Suddek G.M., Salem H.A.-R. Attenuation of Bleomycin-induced pulmonary fibrosis in rats by flavocoxid treatment. Egypt. J. Basic Appl. Sci.4. 2017:256–263. [Google Scholar]
- Zhang Y., Chen W.A. Biochanin A inhibits lipopolysaccharide-induced inflammatory cytokines and mediators production in BV2 microglia. Neurochem. Res. 2015;40:165–171. doi: 10.1007/s11064-014-1480-2. [DOI] [PubMed] [Google Scholar]
- Zhou G., Sun X., Qin Q., Lv J., Cai Y., Wang M., Mu R., Lan H.Y., Wang Q.W. Loss of Smad7 Promotes Inflammation in Rheumatoid Arthritis. Front. Immunol. 2018;9:2537. doi: 10.3389/fimmu.2018.02537. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.