

Abstract
PURPOSE OF REVIEW:
This article discusses the clinical presentation, evaluation, and management of the patient with optic neuritis. Initial emphasis is placed on clinical history, examination, diagnostic testing, and medical decision making, while subsequent focus is placed on examining specific inflammatory optic neuropathies. Clinical clues, examination findings, neuroimaging, and laboratory testing that differentiate autoimmune, granulomatous, demyelinating, infectious, and paraneoplastic causes of optic neuritis are assessed, and current treatments are evaluated.
RECENT FINDINGS:
Advances in technology and immunology have enhanced our understanding of the pathologies driving inflammatory optic nerve injury. Clinicians are now able to interrogate optic nerve structure and function during inflammatory injury, rapidly identify disease-relevant autoimmune targets, and deliver timely therapeutics to improve visual outcomes.
SUMMARY:
Optic neuritis is a common clinical manifestation of central nervous system inflammation. Depending on the etiology, visual prognosis and the risk for recurrent injury may vary. Rapid and accurate diagnosis of optic neuritis may be critical for limiting vision loss, future neurologic disability, and organ damage. This article will aid neurologists in formulating a systematic approach to patients with optic neuritis.
INTRODUCTION
Optic neuritis, or inflammation of the optic nerves, is a frequent cause of acute optic nerve injury in children and adults. While optic neuritis is frequently associated with multiple sclerosis (MS), the causes of optic neuritis are protean. As a result, the prognosis and treatment of optic neuritis will vary depending upon the etiology, the duration and severity of vision loss, prior injury, and the success of prior treatment. Optimal care of patients with optic neuritis therefore depends on rapid recognition, appropriate diagnostic studies, and early institution of effective therapies.
Multiple causes of optic nerve inflammation exist: autoimmunity, infection, granulomatous disease, paraneoplastic disorders, and demyelination. Rapid determination of the etiology of optic neuritis is important for implementing timely and appropriate treatment. In addition, understanding the cause of optic neuritis informs on visual prognosis, illuminates future health risks, and directs additional evaluations and treatments. Differentiating between various causes of optic neuritis, however, often requires a multifaceted evaluation that extends beyond a clinical history and neuro-ophthalmologic examination. Visual field perimetry, optical coherence tomography (OCT), MRI, serologic testing, and CSF analysis may help to focus the differential diagnosis or identify an alternative diagnosis. Therefore, an initial overview of the clinical presentation, examination findings, evaluation, and treatment of the patient with optic neuritis is warranted.
KEY POINTS.
The classic presentation of optic neuritis associated with multiple sclerosis is unilateral, moderate, painful vision loss with an afferent pupillary defect and normal fundus examination. Bilateral vision loss, lack of pain, and severe loss of vision should raise concern for an alternative inflammatory optic neuropathy.
Neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein (MOG)-IgG optic neuritis cause severe vision loss and are more frequently bilateral. MOG-IgG optic neuritis frequently causes significant optic disc edema.
EVALUATING AND TREATING THE PATIENT WITH OPTIC NEURITIS
The evaluation of the patient with optic neuritis begins with a careful history and examination that provides the framework for guiding and interpreting further laboratory, imaging, and visual testing. The following sections provide a road map for the evaluation of the patient with optic neuritis, highlighting how history, examination, visual function, OCT, and neuroimaging may be used to hone the differential diagnosis and focus therapy.
Presentation and Examination
Optic neuritis characteristically presents as acute, unilateral, painful vision loss. In the Optic Neuritis Treatment Trial,1 95% of patients showed unilateral vision loss and 92% had associated retroorbital pain that frequently worsened with eye movement. Some inflammatory and infectious causes of inflammatory optic neuropathy, however, present with subacute visual decline and variable levels of eye discomfort (TABLE 3–1). Therefore, patients with chronic vision loss and the absence of eye pain should raise suspicion for an alternative cause of optic neuropathy or vision loss. Bilateral optic neuritis is more common in children and in adults who are seropositive for myelin oligodendrocyte glycoprotein IgG (MOG-IgG) or anti-aquaporin-4 (AQP4) IgG.2–4
TABLE 3–1.
Demographics and Clinical Presentation of Optic Neuritis
| Diagnosis | Demographics | Onset of Vision Loss | Pain | Eye Findings | Associated Systemic/Neurologic Disease |
|---|---|---|---|---|---|
| Multiple sclerosis | Young adults, female predominance | Acute or subacute | Yes | Normal or mild disc edemaa | Signs or symptoms of multiple sclerosis |
| Neuromyelitis optica (NMO) spectrum disorder | Older adults, strong female predominance | Acute or subacute | Yes | Normal or mild disc edemaa | Longitudinally extensive transverse myelitis, area postrema syndrome, SIADH |
| MOG-IgG | Pediatric and adult, no sex predilection | Acute or subacute | Yes | Frequent disc edema; often moderate or severea | Myelitis, ADEM |
| Seronegative (AON, RION, CRION) | Young adults, female predominance | Acute or subacute | Yes | Normal or mild edema; rare uveitisa | Autoimmune serology, steroid dependence |
| Granulomatous (sarcoidosis, granulomatosis with polyangiitis) | All age groups, sarcoidosis more common in African and Caribbean ethnicity, granulomatosis with polyangiitis peaks at older age | Subacute | Variable | Normal or edematous disc Sarcoid: uveitis, vitreitis, periphlebitis, episcleritis Granulomatosis with polyangiitis: scleritis, conjunctivitis, uveitis, vitreitis, vasculitis, orbital inflammation |
Sarcoidosis: hilar adenopathy, lung fibrosis, cardiac symptoms, erythema nodosum Granulomatosis with polyangiitis: sinus disease, otitis, nasal ulcers, glomerulonephritis, systemic vasculitis, pleural effusion |
| Autoimmune (Sjögren syndrome, systemic lupus erythematosus) | Young adults, strong female predominance | Acute or subacute | Variable | Normal or edematousa Sjögren syndrome: dry eye |
Sjögren syndrome: xerostomia, dental disease, pancreatitis Systemic lupus erythematosus: malaise, arthritis, malar rash, renal disease, thrombosis |
| GFAP-IgG | Early to late adulthood, no sex predilection | Not reported | No | Disc edema, normal intracranial pressure | Encephalitis, meningoencephalitis, myelitis, seizure |
| Paraneoplastic (CRMP-5) | Older adults, no sex predilection | Subacute | No | Disc edema, vitreitis, retinal vascular leak | Malignancy |
| Neuroretinitis | No age predilection, no sex predilection | Subacute | No | Disc edema with macular star | Viral prodrome, infection, catscratch disease |
| Syphilis | Concurrent HIV, high-risk behavior | Acute, subacute or chronic | Variable | Frequent severe disc edema, neuroretinitis, episcleritis, uveitis | Meningitis, encephalitis, cranial nerve palsies |
| Lyme disease | Lyme-endemic region | Acute or subacute | Rarely reported | Disc edema, neuroretinitis | Myalgia, arthralgia, erythema migrans |
| Tuberculosis | Tuberculosis-endemic region, Immunocompromise | Acute or subacute | Infrequent | Normal or edema, uveitis, orbital apex syndrome | Pulmonary disease, meningitis, lymphadenopathy |
| Viral infection | Viral-endemic regions, zoster reactivation, immunocompromise | Acute or subacute | Variable | Normal or edema, retinal necrosis, chorioretinitis | Zoster, fever, rash, lymphadenopathy, immunocompromise |
ADEM = acute disseminated encephalomyelitis; AON = autoimmune optic neuropathy; CRION = chronic relapsing inflammatory optic neuropathy; CRMP-5 = collapsin response mediator protein-5; GFAP-IgG = glial fibrillary acidic protein immunoglobulin G; HIV = human immunodeficiency virus; MOG-IgG = myelin oligodendrocyte glycoprotein immunoglobulin G; RION = relapsing isolated optic neuritis; SIADH = syndrome of inappropriate secretion of antidiuretic hormone.
Optic disc pallor if recurrent disease.
Examination of the patient with optic neuritis typically reveals visual acuity loss, visual field loss, color vision deficits, and an afferent pupillary defect in the affected eye. The absence of an afferent pupillary defect should always raise diagnostic concern unless the patient has bilateral involvement or a history of optic neuropathy in the fellow eye. The extent of visual acuity loss may vary significantly. In idiopathic optic neuritis and optic neuritis associated with MS, high-contrast visual acuity loss is moderate, with the majority of patients having acuity better than 20/200.5 Conversely, optic neuritis associated with neuromyelitis optica spectrum disorder (NMOSD) or MOG-IgG often presents with severe vision loss worse than 20/400.6,7 The severity of vision loss associated with infectious, granulomatous, and paraneoplastic optic neuropathy varies based on the extent and duration of disease. In idiopathic optic neuritis, the funduscopic examination is typically normal, with less than 25% of patients presenting with disc edema. Significant disc inflammation, disc hemorrhages, or ocular inflammation should raise concern for infection, granulomatous inflammation, or optic neuritis associated with MOG-IgG. TABLE 3–1 presents common clinical symptoms and examination findings for inflammatory optic neuropathies.
Visual Testing: Perimetry, Evoked Potentials, Optical Coherence Tomography
Visual evoked potentials provide a sensitive test of the axonal transmission along the optic nerve. While an abnormal P100 latency on visual evoked potential testing confirms the presence of an optic neuropathy, visual evoked potential testing will not differentiate the cause of inflammation nor inform on visual prognosis. Visual evoked potentials, however, may prove useful in confirming subtle cases of optic neuritis. Furthermore, a significant increase in P100 latency without a drop in amplitude is consistent with a demyelinating optic neuropathy, while a drop in amplitude suggests concurrent axonal injury.
Visual field defects due to optic neuritis vary considerably. Therefore, the pattern of visual field loss is not specific for any subtype of optic neuritis. Diffuse or central visual field loss is the most frequent pattern observed in acute idiopathic optic neuritis and MS optic neuritis8; altitudinal field loss may be more frequent in NMOSD optic neuritis than MS optic neuritis.9,10
OCT is a noninvasive imaging technology capable of identifying subtle optic nerve and retinal pathology. OCT frequently identifies peripapillary retinal nerve fiber layer thickening in acute optic neuritis that evolves into focal retinal nerve fiber layer and macular thinning.11 Unfortunately, initial studies have not identified any correlation between acute retinal nerve fiber layer changes and visual outcomes or treatment response. OCT angiography is a new adaptation of OCT technology that provides high-resolution information on retinal blood vessels and may provide novel diagnostic and prognostic information in patients with optic neuritis. OCT angiography reveals decreased vessel density in the peripapillary retina and macula following optic neuritis.12 In NMOSD, reduced peripapillary and parafoveal vessel density is observed independent of a history of optic neuritis and appears to correlate with visual function.13 In complex cases, OCT may be helpful to document associated retinal abnormalities or identify alternative diagnoses such as chorioretinitis, central serous chorioretinopathy, and acute macular neuroretinopathy. TABLE 3–2 summarizes the results of paraclinical testing that may impact the diagnosis of optic neuritis in the acute setting.
TABLE 3–2.
Ophthalmic Diagnostics in Optic Neuritis
| Diagnosis | Visual Evoked Potentials | Visual Fields | Optical Coherence Tomography |
|---|---|---|---|
| Multiple sclerosis | P100 latency prolonged with normal or mildly reduced amplitude | Diffuse field loss, central scotoma | Acute peripapillary retinal nerve fiber layer (RNFL) thickening with subsequent peripapillary RNFL and GC+IPL thinning |
| Neuromyelitis optica (NMO) spectrum disorder | P100 latency prolonged with mildly reduced amplitude, reduced amplitude with normal latency absent response | Total loss, central, quadrant, altitudinal | Severe peripapillary RNFL thinning |
| MOG-IgG | P100 latency prolonged with normal or mildly reduced amplitude | Not reported | Peripapillary RNFL thinning and GC+IPL thinning, worsens with recurrence |
| Seronegative (AON, RION, CRION) | P100 latency prolonged with mildly reduced amplitude, reduced amplitude with normal latency (CRION) | Central scotoma, constriction, altitudinal (CRION) | Severe peripapillary RNFL thinning worsening with recurrent disease, microcystic macular edema |
| Granulomatous (sarcoid, granulomatosis with polyangiitis) | P100 latency prolonged with normal or mildly reduced amplitude | Central scotomas, occasional hemianopic and altitudinal defects | Peripapillary RNFL thickening, retinal and subretinal fluid, choroidal nodules (sarcoid) |
| Autoimmune (Sjögren syndrome, systemic lupus erythematosus) | P100 latency prolonged with normal or mildly reduced amplitude | Variable | Peripapillary RNFL thinning, choroidopathy (systemic lupus erythematosus) |
| GFAP-IgG | Normal | Arcuate defects, enlarged blind spot, diffuse loss | Peripapillary RNFL thickening |
| Paraneoplastic (CRMP-5) | P100 latency prolonged; electroretinogram may be abnormal | Arcuate defects, constriction; enlarged blind spot, paracentral scotoma, diffuse loss | Peripapillary RNFL thickening, retinal hyperreflective material |
| Neuroretinitis | P100 latency normal or modestly prolonged; electroretinogram normal | Central or centrocecal scotoma | Peripapillary RNFL thickening, outer retinal fluid or hyperreflective material |
| Syphilis | P100 latency prolonged | Variable | Peripapillary RNFL thickening, choroidopathy, retinal or subretinal fluid |
| Lyme disease | P100 latency prolonged with normal or mildly reduced amplitude; latency may be normal in neuroretinitis | Central or centrocecal scotoma | Peripapillary RNFL thickening, outer retinal fluid or hyperreflective material if neuroretinitis |
| Tuberculosis | Not reported | Variable, but enlarged blind spot and central scotoma are most common | Peripapillary RNFL thickening if disc edema, rare choroidal lesions |
| Viral infection | Not reported | Variable | Retinal thinning, cystic fluid, hyperreflective lesions with retinitis and necrosis, outer and inner retinal hyperreflective material with chorioretinitis |
AON = autoimmune optic neuropathy; CRION = chronic relapsing inflammatory optic neuropathy; CRMP-5 = collapsin response mediator protein-5; GC+IPL = ganglion cell plus inner plexiform layer thickness; GFAP-IgG = glial fibrillary acidic protein immunoglobulin G; MOG-IgG = myelin oligodendrocyte glycoprotein immunoglobulin G; RION = relapsing isolated optic neuritis.
KEY POINTS.
Ophthalmic testing is not generally helpful in differentiating acute optic neuropathies. Visual evoked potentials may help to detect subtle optic nerve injury when clinical examination findings are uncertain.
Optical coherence tomography may be useful in detecting subtle retinal pathology or documenting the extent of prior injury in cases of recurrent optic neuritis.
MRI of the orbits is the most sensitive diagnostic test (90%) for optic neuritis; however, a normal orbital MRI scan does not exclude optic neuritis.
The pattern of inflammation of the optic nerve on MRI may provide diagnostic information. NMOSD optic neuritis more often affects the optic chiasm, intracranial optic nerve, and optic tracts; MOG-IgG optic neuritis frequently inflames the intraorbital optic nerve and optic nerve sheath. Both disorders may be bilateral with longitudinally extensive lesions.
Antinuclear autoantibodies are observed in many patients with optic neuritis; however, they are much less frequent in multiple sclerosis-associated optic neuritis.
Magnetic Resonance Imaging
MRI is an exquisitely sensitive tool for the detection of optic neuritis. MRI of the orbits with fat suppression and gadolinium enhancement detects acute optic neuritis lesions in 95% of affected individuals within 20 days of vision loss14; T2-weighted images with fat suppression and short tau inversion recovery (STIR) detect lesions in up to 89% of acute optic neuritis cases with abnormalities persisting for as long as 6 weeks in 92% of cases.15,16 In addition, the distribution and appearance of optic nerve, orbital, brain, and meningeal inflammation associated with acute optic neuritis may help to differentiate between autoimmune, infectious, and granulomatous inflammation (TABLE 3–3). For example, bilateral optic neuritis is more common in NMOSD and MOG-IgG disease than in MS. Lesions involving the optic chiasm and optic tract are highly suggestive of optic neuritis associated with NMOSD. Longitudinally extensive lesions of the retrobulbar optic nerve are commonly observed in both NMOSD and MOG-IgG disease, more so with MOG-IgG disease (FIGURE 3–1).4,17 Perineural optic nerve enhancement (optic perineuritis) is frequent with MOG-IgG-associated optic neuritis (FIGURE 3–1)6; however, in certain clinical circumstances, syphilis, tuberculosis, sarcoidosis, and granulomatosis with polyangiitis (previously known as Wegener granulomatosis) should also be considered.
TABLE 3–3.
MRI Findings in Optic Neuritis
| Diagnosis | Optic Nerve Imaging | Orbital Imaging | Brain and Spinal Cord Imaging |
|---|---|---|---|
| Multiple sclerosis | Unilateral, retrobulbar and canalicular, short anterior segmental lesions, optic nerve enhancement | Negative | Periventricular ovoid lesions, subcortical and juxtacortical lesions |
| Neuromyelitis optica (NMO) spectrum disorder | May be bilateral, often intracranial involving chiasm and optic tract, often longitudinally extensive, optic nerve enhancement | Negative | Longitudinally extensive transverse myelitis, posterior fossa and periaqueductal gray lesions, hypothalamic lesions |
| MOG-IgG | Frequently bilateral and retrobulbar, often longitudinally extensive, optic nerve and perineural sheath enhancement | Enhancement of perineural orbital tissue | Myelitis (thoracolumbar and conus predominance), lesions of deep gray nuclei |
| Seronegative (AON, RION, CRION) | Retrobulbar, optic nerve enhancement, occasional nerve swelling | Normal | Normal |
| Granulomatous (sarcoidosis, granulomatosis with polyangiitis) | Commonly unilateral, frequent combined optic nerve and sheath enhancement | Sarcoid: orbital apex inflammation Granulomatosis with polyangiitis: orbital cellulitis, orbital mass, orbital pseudotumor |
Sarcoidosis: periventricular lesions, leptomeningeal lesions, pituitary and hypothalamic lesions Granulomatosis with polyangiitis: pachymeningitis, nonspecific gray and white matter lesions due to vasculitis |
| Autoimmune (Sjogren syndrome, systemic lupus erythematosus) | Retrobulbar, optic nerve enhancement | Normal | Systemic lupus erythematosus: infarcts and dural thrombosis |
| GFAP-IgG | Normal | Normal | Linear radial perivascular enhancement, meningitis, myelitis, leptomeningeal and ependymal enhancement |
| Paraneoplastic (CRMP-5) | Bilateral, optic nerve enhancement | Normal | Cerebellar atrophy, mesial temporal lesions, cerebellar lesions, myelitis (may be longitudinally extensive) |
| Neuroretinitis | Normal, occasional high T2 signal or enhancement in proximal optic nerve | Normal | Normal |
| Syphilis | Optic nerve and perineural sheath enhancement | Occasional enhancement of orbital fat | Leptomeningeal enhancement, encephalitis, myelitis, infarct |
| Lyme disease | Retrobulbar, optic nerve enhancement | Normal | Cranial nerve enhancement, periventricular and subcortical lesions |
| Tuberculosis | Retrobulbar, optic nerve and sheath enhancement | Orbital tuberculoma, dacryoadenitis | Leptomeningeal enhancement, ependymitis, tuberculoma |
| Viral infection | Retrobulbar, optic nerve enhancement | Normal | Variable depending on pathogen |
AON = autoimmune optic neuropathy; CRION = chronic relapsing inflammatory optic neuropathy; CRMP-5 = collapsin response mediator protein-5; GFAP-IgG = glial fibrillary acidic protein immunoglobulin G; MOG-IgG = myelin oligodendrocyte glycoprotein immunoglobulin G; MRI = magnetic resonance imaging; RION = relapsing isolated optic neuritis.
FIGURE 3–1.
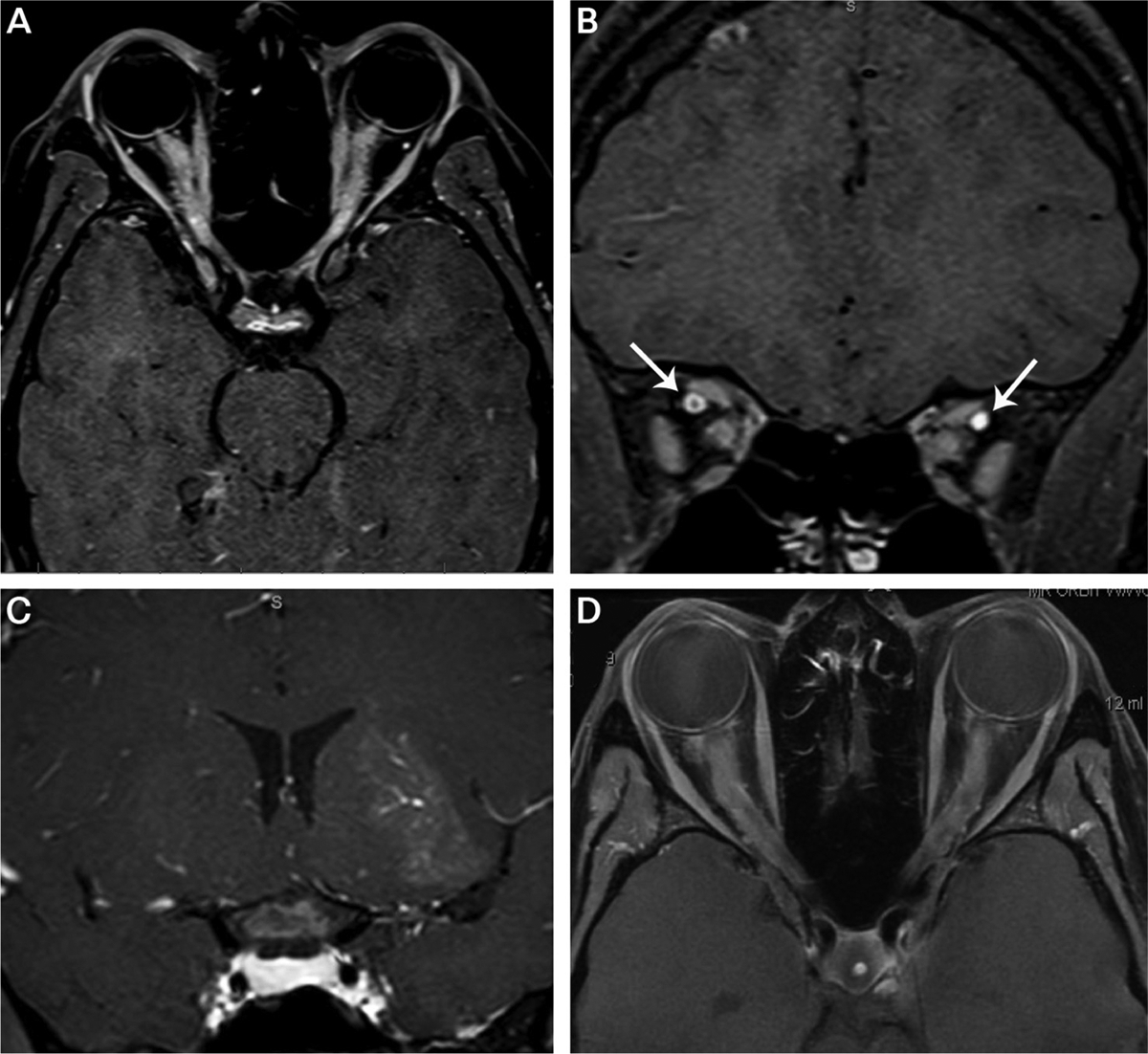
Fat-suppressed postcontrast T1-weighted orbital imaging of optic neuritis. A, Axial image shows bilateral longitudinally extensive lesions involving the orbital and intracanalicular optic nerves in a patient with myelin oligodendrocyte glycoprotein antibody (MOG-IgG) optic neuritis. Note the enhancement of the optic discs suggestive of disc edema. B, Coronal image of optic nerves in panel A showing both sheath (right eye) and nerve (left eye) enhancement. Gadolinium contrast fills both superior orbital veins (arrows). C, Coronal image showing an enlarged, enhancing optic chiasm in a patient with neuromyelitis optica (NMO) spectrum disorder-associated optic neuritis. Additional cloudlike enhancement is seen in the left thalamus/basal ganglia due to NMO inflammation. D, Bilateral enhancing lesions of the orbital and intracanalicular optic nerves in a patient with chronic relapsing immune-mediated optic neuropathy.
The presence and pattern of brain and spinal cord MRI lesions also may provide valuable diagnostic clues. T2-hyperintense and gadolinium-enhancing lesions in multiple regions of the brain and/or spinal cord may be highly suggestive or diagnostic of MS,18 whereas periependymal, fornix, and hypothalamic lesions may favor NMOSD.19 Prior or concurrent longitudinally extensive spinal cord lesions would indicate a high likelihood of NMOSD or MOG-IgG encephalomyelitis; more frequent involvement of the lower cord and conus is seen with MOG-IgG disease.20 Optic neuritis with glial fibrillary acidic protein autoantibodies (GFAP-IgG) may be with a unique pattern of linear perivascular enhancement.21 Meningeal enhancement and thickening might focus the evaluation toward granulomatous disease or infection.
Serology and CSF Analysis
Autoimmune serology and CSF analysis may yield critical clues that either focus the differential diagnosis or clarify an underlying etiology. While not specific for any cause of optic neuritis, antinuclear autoantibodies (ANA) are more common in patients with NMOSD or MOG-IgG optic neuritis than in those with MS.22,23 Patients with optic neuritis with NMOSD and GFAP-IgG may demonstrate serum and CSF antineuronal autoantibodies that are commonly reported in autoimmune encephalopathy panels.21,24 A mild CSF pleocytosis is frequently observed in patients with acute optic neuritis; however, extensive pleocytosis (>100 cells/mm3) is observed more often in patients with MOG-IgG. Typically, pleocytosis of less than 50 cells/mm3 is noted in cases of MS-associated optic neuritis. Eosinophils and polymorphonuclear cells in the CSF are suggestive of NMOSD. Oligoclonal bands and intrathecal IgG synthesis, hallmarks of optic neuritis associated with MS, are uncommon in NMOSD and MOG-IgG-related optic neuritis and rarely persist.22,25 GFAP-IgG may be restricted to the CSF in affected patients.21 In contrast, most AQP4-IgG is produced in the periphery, and testing for CSF AQP4-IgG is neither sensitive or cost-effective.26 If suspicion exists for infection or granulomatous disease, serum and CSF should be evaluated for syphilis, Lyme bacillus, and angiotensin-converting enzyme (ACE). Additional serologic testing for Bartonella henselae should be considered in cases of neuroretinitis in which optic disc edema is accompanied by a macular star of exudates located in a radial pattern around the fovea; serologic testing for cytoplasmic antineutrophil cytoplasmic antibodies (c-ANCA) should be included in optic neuritis cases under consideration for granulomatosis with polyangiitis (formerly known as Wegener granulomatosis).
KEY POINTS.
CSF pleocytosis may be highest in MOG-IgG optic neuritis, whereas CSF eosinophils are suggestive of NMOSD.
Oligoclonal bands should suggest multiple sclerosis-associated optic neuritis, especially if they persist.
Aquaporin-4 IgG is rarely, if ever, isolated to the CSF.
Treatment
Administration of high doses of corticosteroids is the standard treatment for acute optic neuritis. In the Optic Neuritis Treatment Trial, IV methylprednisolone (1000 mg/d for 3 days), followed by oral prednisone (1 mg/kg/d for 11 days) accelerated visual recovery but failed to improve functional outcomes.1 Subsequent studies in patients with relapsing MS or optic neuritis have demonstrated that doses of corticosteroid equivalent to 1000 mg IV methylprednisolone, administered IV or orally, provides an equivalent therapeutic effect of accelerated recovery.27,28 IM or subcutaneous adrenocorticotropic hormone (ACTH) is also approved for the treatment of acute optic neuritis and provides an alternative option for enhancing corticosteroid signaling. Lower doses of oral prednisone (1 mg/kg/d or less) should be avoided in cases of idiopathic optic neuritis as an increased risk of relapse exists.1 Chronic treatment with low-dose oral prednisone, however, is important for the treatment of sarcoid optic neuritis and recurrent optic neuritis due to chronic relapsing inflammatory optic neuropathy.29,30 In the Optic Neuritis Treatment Trial, treatment with IV methylprednisolone was reported to delay conversion to MS in the first 2 years.31 A similar finding, however, was not observed in the original Optic Neuritis Treatment Trial dataset,1 likely resulting from the reclassification of study participants.32 Indeed, independent studies evaluating the effects of IV methylprednisolone on relapse rates in patients with MS have failed to observe a similar effect.33,34
IV immunoglobulin (IVIg) and plasma exchange have been evaluated in patients with optic neuritis that is refractory to high-dose corticosteroid treatment. IVIg (2 g/kg) failed to improve contrast sensitivity or visual function in patients with acute optic neuritis or MS with refractory vision loss.35,36 Treatment response may have been limited because of the delayed administration of IVIg in both studies. In contrast, plasma exchange has resulted in improved visual outcomes in patients with corticosteroid-refractory optic neuritis and NMOSD optic neuritis.7,37 While the frequency of responders varied, the majority of patients with optic neuritis treated with plasma exchange had improvement in their visual function. Increased response to plasma exchange has been associated with male sex, lower baseline disability, rapid initiation of treatment, and shorter relapse duration.38,39 While the optimal use and timing of plasma exchange in patients with optic neuritis has yet to be defined, the natural history of poor visual recovery in NMOSD and recurrent optic neuritis argues that plasma exchange should be considered as a first-line treatment in certain clinical circumstances, as discussed later in this article. Therapeutic apheresis, using immunoadsorption, offers an alternative to plasma exchange outside of the United States and has been reported to benefit steroid-refractory optic neuritis.40
If infection is prominent in the differential diagnosis of a patient with optic neuritis, it is prudent to begin appropriate antibiotic therapy as soon as possible. Symptomatic therapy with corticosteroids may be initiated concurrently unless otherwise contraindicated. Antibiotic or antiviral therapies may be tailored or discontinued based on diagnostic imaging, serology, cultures, or CSF analysis. For optic neuritis associated with Bartonella infection, the utility of antibiotic therapy remains unclear; however, significant vision loss, systemic infection, and immunocompromised status should bolster consideration for antibiotics.41
THE SPECTRUM OF OPTIC NEURITIS
The differential diagnosis of optic neuritis has undergone a significant transformation over the past decade. An expanding spectrum of autoimmune, paraneoplastic, and idiopathic inflammatory disorders have been associated with acute optic neuritis, and their visual prognosis, response to treatment, and risk for recurrence vary substantially. The following sections address the diagnosis and treatment of specific inflammatory and infectious causes of optic neuritis.
KEY POINTS.
High-dose IV methylprednisolone (1000 mg/d IV for 3 days) is effective at improving the speed of recovery of optic neuritis. Small studies have demonstrated that the type of steroid and mode of delivery (oral versus IV) are likely inconsequential. Lower dosages of oral prednisone (1 mg/kg) are contraindicated for acute optic neuritis treatment because of a higher risk of relapse.
Plasma exchange may be useful in treating steroid-resistant optic neuritis, severe optic neuritis due to NMOSD, and recurrent optic neuritis at risk for poor recovery. Time to administration of plasma exchange may be critical to treatment success.
Optic neuritis is the initial presentation of multiple sclerosis in 25% of individuals. The presence of enhancing and nonenhancing brain MRI lesions meeting dissemination in space criteria by the 2017 McDonald criteria is diagnostic of multiple sclerosis. If no enhancing lesions are present, oligoclonal bands may provide dissemination in time criteria according to the 2017 McDonald criteria.
Optic Neuritis Associated With Multiple Sclerosis
Optic neuritis affects roughly 70% of patients with MS and is the presenting symptom in approximately 25% of individuals. Women are affected twice as often as men, and whites dominate the racial distribution. Optic neuritis is frequently unilateral and painful and progresses over 1 to 2 weeks; bilateral optic neuritis and severe vision loss (count fingers or worse) are uncommon. Optic disc edema is infrequent (35%) (FIGURE 3–2); hemorrhagic disc edema (5.6%), retinal exudates (1.8%), and vitreal cells (3.3%) are rare and impart a reduced risk for MS.5,42 OCT of the peripapillary retinal nerve fiber layer has demonstrated that mild segmental swelling of the nerve fiber layer (>110% of normal thickness) is present in 82% of affected eyes and persists in 58% of eyes at 1 month.11 Peripapillary and macular OCT show relatively rapid and concurrent thinning of the peripapillary retinal nerve fiber layer and macular ganglion cell plus inner plexiform layer. The change in ganglion cell plus inner plexiform layer thickness in the first month may predict poor visual acuity after 6 months.43 At 1 month, visual acuity of 20/50 or less, contrast sensitivity less than 1.0 log units, and visual field mean deviation of −15 dB or lower also predict moderate to severe vision loss at 6 months.44 T1 gadolinium enhancement is evident in 95% of acute MS-associated optic neuritis lesions within 20 days of vision loss14; lesions are typically short and anterior when compared to NMOSD- and MOG-associated optic neuritis.4,45 The location and length of enhancement of the optic nerve lesion on MRI do not correlate with visual recovery. Brain MRI and CSF analysis are important for determining MS risk in patients with idiopathic optic neuritis using the 2017 McDonald criteria.18 The presence and pattern of enhancing and T2-weighted brain MRI lesions may be diagnostic of MS or provide dissemination in space criteria. In addition, the presence of CSF oligoclonal bands now fulfills dissemination in time criteria for patients with optic neuritis who meet clinical or MRI criteria for dissemination in space.
FIGURE 3–2.
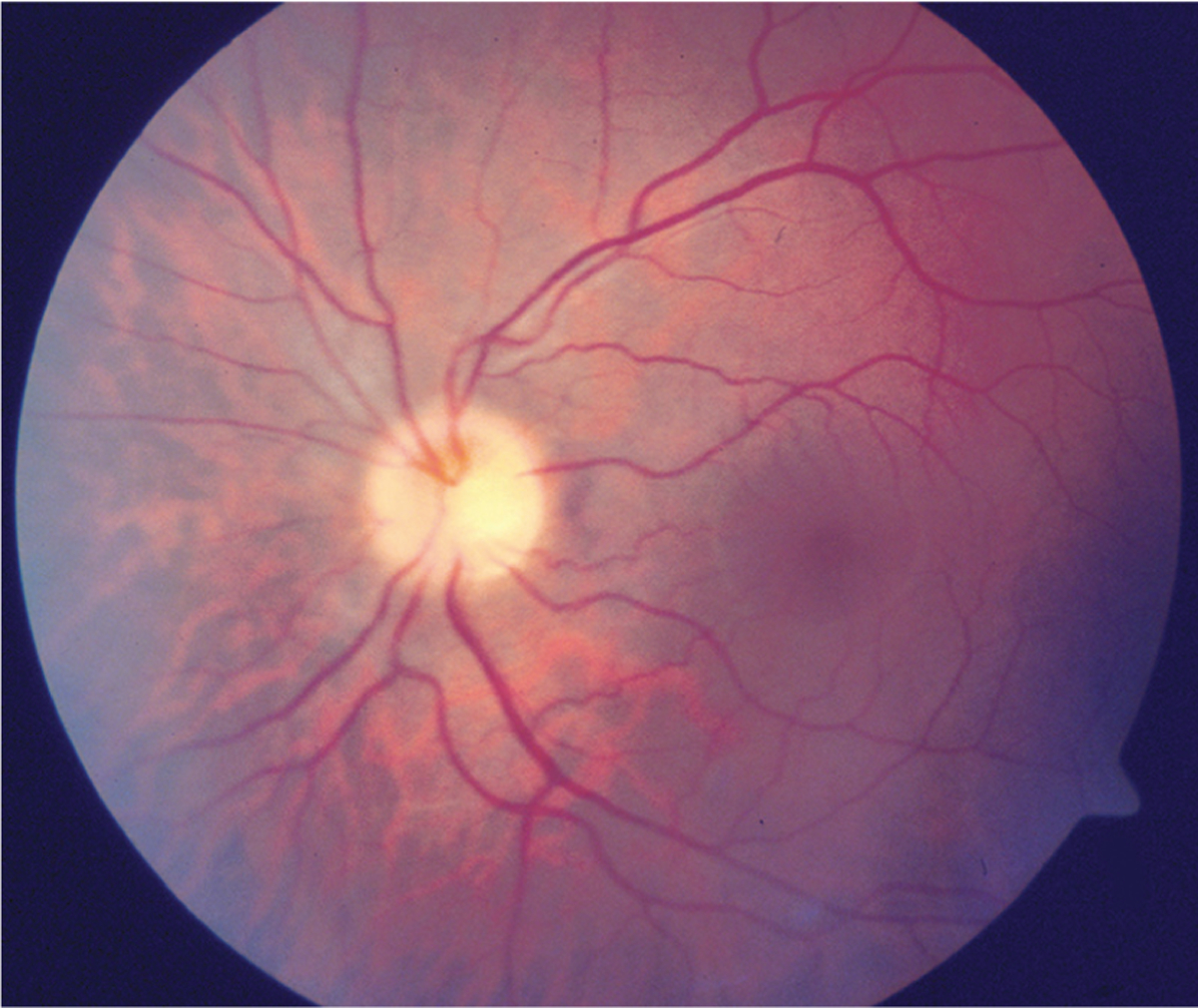
Mild optic disc edema associated with multiple sclerosis.
Visual recovery from MS-associated optic neuritis is generally good. In the Optic Neuritis Treatment Trial, the median acuity at recovery was 20/16. Visual acuity returned to 20/40 or better in 91% of subjects with 20/200 or worse vision at entry, independent of treatment with corticosteroids. As noted previously, treatment with IV methylprednisolone hastened the speed of visual recovery in the Optic Neuritis Treatment Trial but did not influence long-term outcomes. Patients with MS-associated optic neuritis who have poor visual recovery following treatment with high-dose corticosteroids often respond to plasma exchange.37 The optimal timing for institution of plasma exchange after IV methylprednisolone, however, remains to be determined. A phase 2 clinical trial recently evaluated phenytoin for neuroprotection in acute optic neuritis. Phenytoin treatment significantly reduced peripapillary retinal nerve fiber layer loss at 6 months but had no effect on visual outcomes.46
Optic Neuritis Associated With Neuromyelitis Optica Spectrum Disorder
NMOSD is a central nervous system (CNS) inflammatory disorder that frequently involves the optic nerves and spinal cord. According to the International Consensus criteria,47 NMOSD is classified as either seropositive or seronegative based on the presence of serum AQP4-IgG. Approximately 80% of affected individuals are seropositive for AQP4-IgG, and the presence of a core clinical presentation (optic neuritis, acute myelitis, area postrema syndrome, acute brainstem syndrome, symptomatic narcolepsy, or acute diencephalic syndrome with NMOSD-typical MRI lesions, or symptomatic cerebral syndrome with NMOSD-typical brain lesions) and serum AQP4-IgG is diagnostic of disease.47 NMOSD-associated optic neuritis may also be diagnosed in AQP4-IgG seronegative individuals who have concurrent or prior core clinical presentations that meet additional criteria.47 Optic nerve MRI criteria for NMOSD-associated optic neuritis requires a T2-hyperintense or T1-weighted gadolinium-enhancing lesion extending over half of the optic nerve length or involving the optic chiasm. Optic nerve head edema is rare, as with MS-associated optic neuritis. Optic neuritis associated with NMOSD is seen more often in women and nonwhites. It is typically severe (mean acuity ≤20/400),7 and bilateral optic neuritis occurs in up to 20% of cases.48 Retinal nerve fiber layer loss is typically more severe following NMOSD-associated optic neuritis.49,50
Complete response to high-dose corticosteroids is much less common than in MS-associated optic neuritis, occurring in only 36%.48 Retrospective studies comparing IV methylprednisolone to plasma exchange have documented improved visual acuity and visual fields using plasma exchange (CASE 3–1).7,37 A short interval (≤5 days) between optic neuritis onset and the institution of plasma exchange is associated with an increased probability of complete improvement.39 Therefore, patients with acute optic neuritis suspicious for NMOSD may benefit from initiation of plasma exchange before confirmation of AQP4-IgG seropositivity. MRI and CSF findings raising concern for NMOSD include longitudinally extensive or bilateral optic nerve lesions, inflammation of the optic chiasm or optic tracts, periependymal brain lesions, polymorphonuclear CSF pleocytosis, or CSF eosinophils. Patients with seropositive NMOSD are at high risk for relapse, and initiation of immunosuppressive therapy is therefore recommended.47 Differentiating MS-associated optic neuritis from NMOSD-associated optic neuritis is critical as some immunomodulatory therapies approved for MS may exacerbate NMOSD disease activity.51–53
Optic Neuritis Associated With Myelin Oligodendrocyte Glycoprotein Autoantibodies
Serum autoantibodies against MOG (MOG-IgG) identify a cohort of patients with a strong predilection for isolated and recurrent optic neuritis.2,3 MOG-IgG-associated optic neuritis is slightly more common in females (57%), displays no distinct ethnic predilection, and is frequently bilateral (37% to 44%) (CASE 3–2).3,6 The age of onset may vary considerably (range: 2 to 79 years); adults with disease onset between 20 and 45 years of age commonly present with unilateral optic neuritis, while older patients more often present with bilateral optic neuritis. Simultaneous optic neuritis and transverse myelitis mimicking NMOSD has been reported in up to 25% of cohorts.3,6 MOG-IgG disease commonly presents as acute disseminated encephalomyelitis (ADEM) or recurrent optic neuritis in children.3,54
Vision loss due to MOG-IgG-associated optic neuritis is typically severe (mean: count fingers vision), painful (86%), and associated with optic disc edema (86%) (FIGURE 3–5).6 Common MRI findings include longitudinally extensive (more than half the length) optic nerve lesions (80%) and perineural enhancement (50%). MOG-IgG optic neuritis is frequently steroid responsive and steroid dependent.55 Although severe vision loss has been reported, it is typically due to repeated attacks of optic neuritis; the risk of severe visual impairment from a single event of optic neuritis is low.6 IV methylprednisolone is therefore the treatment of choice in cases of acute MOG-IgG optic neuritis. If IV methylprednisolone has failed to improve vision with prior events or MOG-IgG optic neuritis is recurring in a previously affected eye, then a combination of plasma exchange and IV methylprednisolone should be considered. MOG-IgG optic neuritis frequently recurs; 80% of patients have two or more attacks over a median time of 2.9 years.6 Long-term immunosuppressants used to prevent MOG-IgG optic neuritis include corticosteroids, azathioprine, mycophenolate mofetil, and rituximab. The optimal preventive therapy, however, has yet to be determined.3,54
FIGURE 3–5.
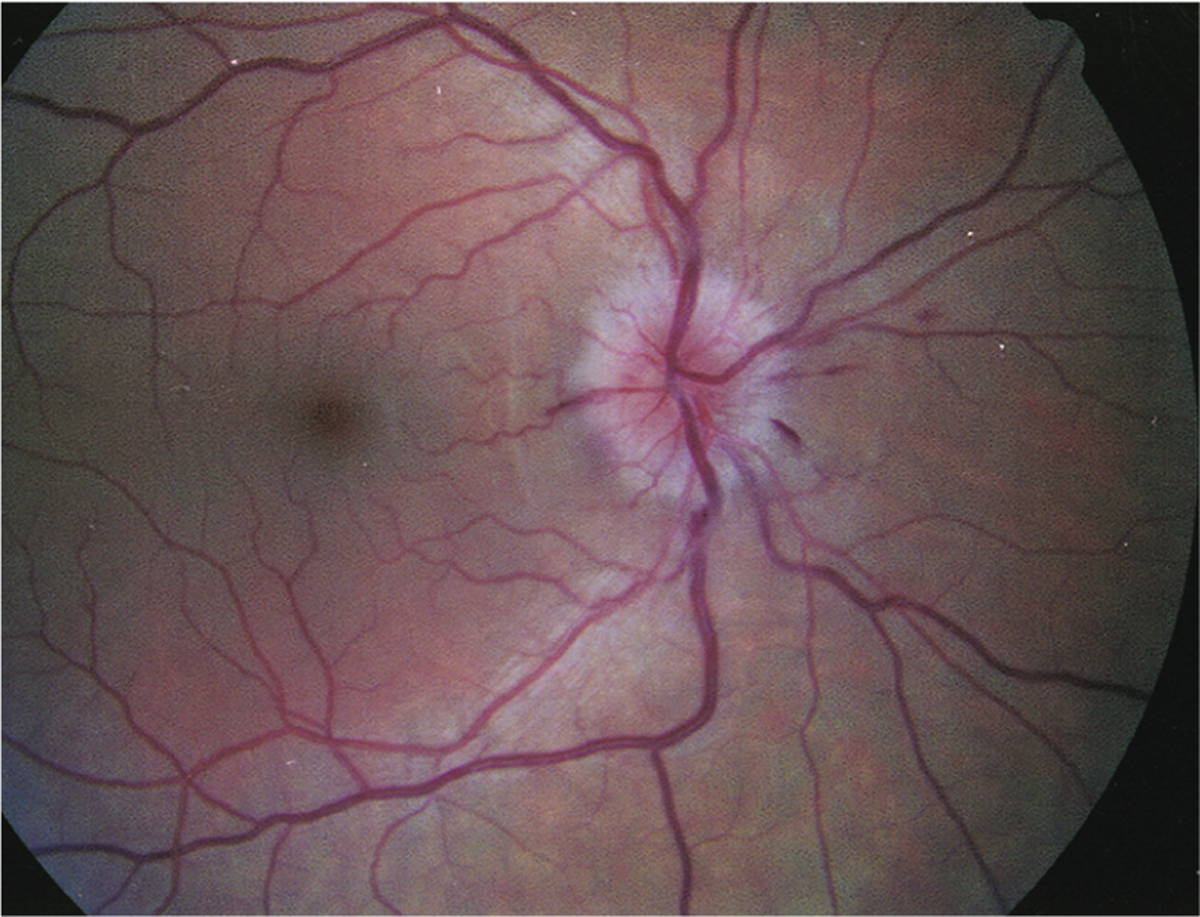
Optic disc edema and retinal inflammation in optic neuritis. Optic disc edema with nerve fiber layer (splinter) hemorrhages due to myelin oligodendrocyte glycoprotein (MOG)-IgG optic neuritis.
KEY POINTS.
It is important to differentiate optic neuritis due to NMOSD from that due to multiple sclerosis. The prognosis for visual recovery is poorer for NMOSD optic neuritis, and the risk for recurrence is high.
NMOSD optic neuritis should prompt consideration for early plasma exchange.
Treatments for multiple sclerosis, such as interferon beta, fingolimod, and natalizumab, have been documented to exacerbate NMOSD disease activity.
MOG-IgG disease frequently causes recurrent optic neuritis. Bilateral optic neuritis, longitudinal optic nerve lesions, optic nerve sheath enhancement, and steroid responsiveness are important clinical and radiologic clues.
GFAP-IgG encephalomyelitis is commonly associated with optic nerve papillitis. As a result, disc edema is prominent, but vision loss is rare.
Perivascular radial enhancement on MRI is highly suggestive of GFAP-IgG disease. GFAP-IgG may be isolated to the CSF in a large fraction of patients.
Optic Neuritis Associated With Glial Fibrillary Acidic Protein Autoantibodies
Serum and CSF autoantibodies against GFAP-IgG have been recently identified in patients with inflammatory meningitis, encephalitis, and myelitis. Roughly 40% of patients with GFAP-IgG meningoencephalitis demonstrated inflammatory optic disc edema on examination.21,56 The optic disc edema was bilateral and symmetric, and the CSF opening pressure was normal in all patients except for two individuals who showed a mildly elevated intracranial pressure (<300 mm H2O). Additional findings consistent with inflammatory papillitis included mild vitreitis, venular leakage on fluorescein angiography, and resolution with high-dose corticosteroids. Inflammatory lesions of the retrobulbar optic nerve were not reported.21,56 Many affected individuals showed a characteristic radial perivascular enhancement on brain MRI. Visual acuity was minimally affected; however, some patients showed nerve fiber layer bundle defects and optic disc atrophy after clinical resolution. The combination of meningoencephalitis, bilateral papillitis, and normal MRI of the optic nerves, with or without radial perivascular enhancement on brain MRI, should prompt testing for serum and CSF GFAP-IgG. Initial treatment of GFAP-IgG papillitis is with IV methylprednisolone followed by high-dose oral corticosteroids.21,56
Recurrent Idiopathic Optic Neuritis
Autoimmune optic neuropathy, chronic relapsing inflammatory optic neuropathy, and relapsing isolated optic neuritis are terms used in the literature to describe cases of recurrent, seronegative optic neuritis. Autoimmune optic neuropathy was initially described by Dutton and colleagues57 in patients with recurrent events of steroid-responsive optic neuritis. Visual acuity loss may range from mild to severe; pain is infrequent, and the fundus examination often reveals mild edema. Laboratory and imaging tests are typically unrevealing, although positive ANA and anticardiolipin antibodies have been reported in some series. Skin biopsy has revealed evidence of histopathologic vasculitis in roughly 25% of patients and immunoglobulin, immune complex, or complement deposition in most others.58 Relapsing isolated optic neuritis is used to describe patients with spontaneous and isolated attacks of nonprogressive, unilateral, or bilateral optic neuritis.59 Patients with relapsing isolated optic neuritis are predominantly female with moderate levels of vision loss (average: 20/80). Systemic illness is infrequent (14%), and patients infrequently display ANA seropositivity (15%). CSF pleocytosis is minimal, and oligoclonal bands are uncommon (19%).
CASE 3–1.
A 35-year-old woman presented with acute painful loss of vision in the right eye that began 1 month earlier. The vision loss progressed over 2 days and reached light perception. She received 3 days of high-dose IV methylprednisolone 2 weeks earlier, and her vision improved but remained very blurry. She denied any additional neurologic or ophthalmic problems. She had experienced a similar episode of painful right eye vision loss 5 years ago, from which she recovered after IV methylprednisolone.
On examination, visual acuity was 20/500 right eye and 20/20 left eye with a right afferent pupillary defect. There was central vision loss in the right eye and dense visual field suppression. The right optic nerve was pale; the left was normal. Optical coherence tomography (OCT) of the peripapillary retinal nerve fiber layer showed severe thinning in the right eye (FIGURE 3–3a). MRI of her orbits showed longitudinally extensive T1 gadolinium enhancement of the intraorbital right optic nerve (FIGURE 3–3b) and T2-hyperintense signal in the intracranial right optic nerve (FIGURE 3–3c).
COMMENT
Recurrent severe optic neuritis is concerning for neuromyelitis optica spectrum disorder (NMOSD)-associated optic neuritis. Suspicion for NMOSD optic neuritis is raised by the significant retinal nerve fiber layer thinning in the previously affected right eye. The prognosis for visual recovery is poor given the severe vision loss 1 month after disease onset and the lack of response to IV steroids. Serologic testing returned positive for aquaporin-4 antibodies (titer: 1:10,000). The patient was admitted to the hospital and received 5 consecutive days of plasma exchange. Vision returned to 20/30 in the right eye, and she was started on azathioprine for prophylactic therapy.
FIGURE 3–3.
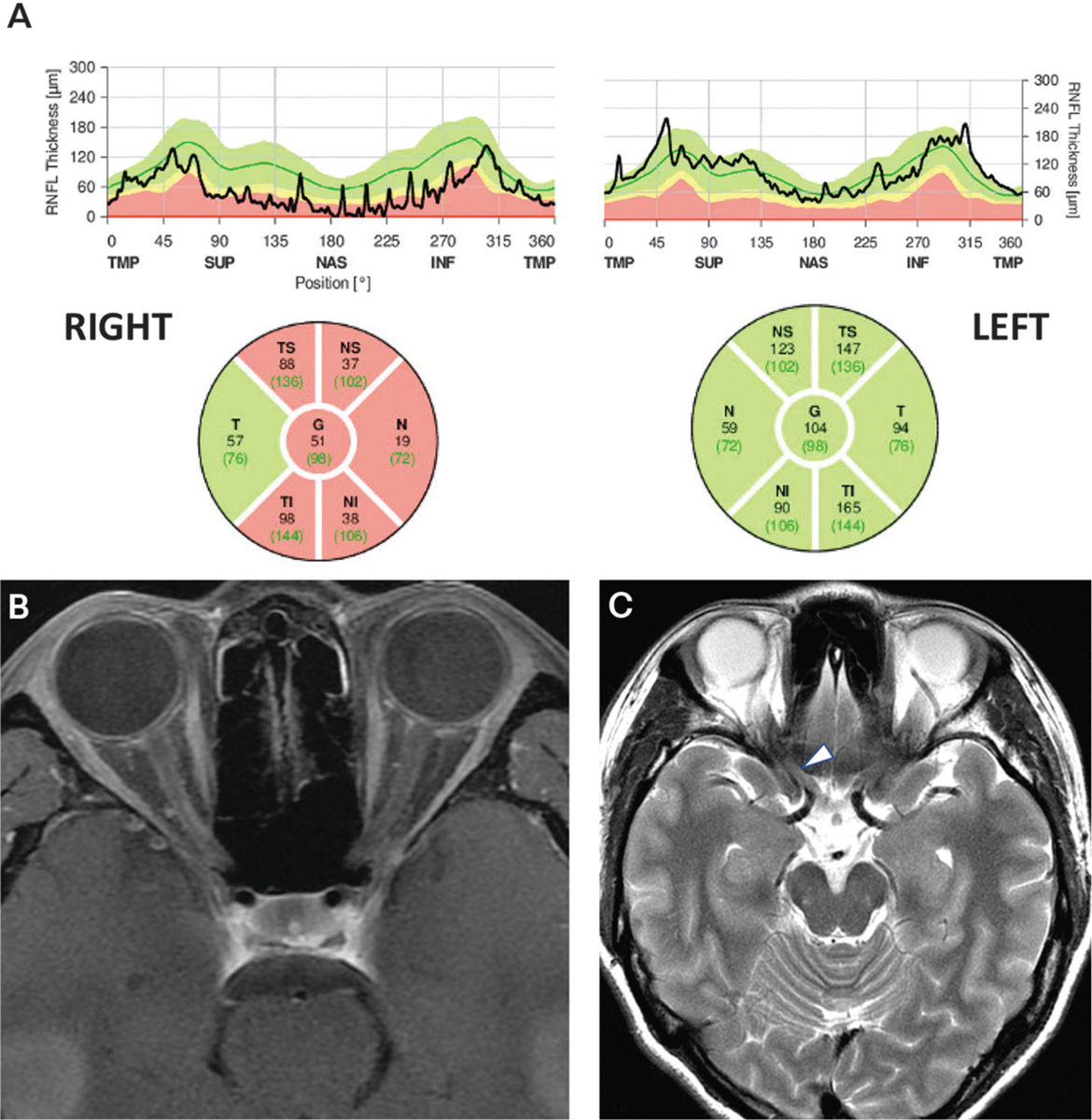
Findings of the patient in CASE 3–1. A, Spectral domain optical coherence tomography of the peripapillary retinal nerve fiber layer showing severe thinning on the right (mean right: 51 microns; mean left: 104 microns). B, Axial fat-suppressed postcontrast T1-weighted MRI showing a subtle longitudinally extensive enhancing lesion of right orbital nerve and sheath. C, Axial T2-weighted MRI showing subtle signal abnormality (arrowhead) in the posterior right optic nerve.
Generally, response to IV methylprednisolone is excellent, with 70% of patients with relapsing isolated optic neuritis recovering to better than 20/25 acuity. Relapse following steroid withdrawal is uncommon (10%) in patients with relapsing isolated optic neuritis.
In contrast, chronic relapsing inflammatory optic neuropathy is used to describe patients with recurrent episodes of isolated unilateral or bilateral optic neuritis who demonstrate progressive vision loss between attacks.60 Chronic relapsing inflammatory optic neuropathy was initially defined as a steroid-dependent condition; however, later publications have noted only that patients with chronic relapsing inflammatory optic neuropathy require immunosuppression for prevention of optic neuritis relapses.30 Indeed, patients with chronic relapsing inflammatory optic neuropathy demonstrate a threefold increase (31% versus 10%) in risk of relapse compared to patients with relapsing isolated optic neuritis following discontinuation of corticosteroids.59 Patients with chronic relapsing inflammatory optic neuropathy are predominantly female and ethnically diverse. Visual acuity loss is generally severe, and the average visual acuity on recovery is worse than in relapsing isolated optic neuritis (20/40 versus 20/25).30,59,60 The reasons for worse visual outcome are likely multifactorial: interattack progression, attack frequency, and the timing of acute treatment and chronic immunosuppression. Similar to patients with relapsing isolated optic neuritis, patients with chronic relapsing inflammatory optic neuropathy are rarely ANA seropositive, lack concurrent systemic disease, and rarely produce CSF oligoclonal bands.30,59,60 Acute attacks of chronic relapsing inflammatory optic neuropathy are usually responsive to IV methylprednisolone; however, chronic immunosuppression is generally required. Immunosuppressive agents include prednisone, azathioprine, mycophenolate mofetil, cyclophosphamide, and IVIg.30
CASE 3–2.
An 18-year-old man with a history of recurrent optic neuritis presented with acute, painful bilateral vision loss. The eye pain worsened with eye movement, and vision declined to its nadir over 5 days. He had experienced his last episode of optic neuritis 6 months earlier and was currently on B-cell depletion therapy with rituximab. He denied any prior or concurrent neurologic problems.
On examination, visual acuity was 20/40 right eye and 20/30 left eye. He had central field loss in both eyes, sluggish pupils, and no afferent pupillary defect. Optic disc pallor was seen bilaterally.
Aquaporin-4 IgG, antinuclear antibodies, and antineutrophil cytoplasmic antibodies were negative. MRI showed bilateral optic nerve T2 signal hyperintensity and swelling (FIGURES 3–4a and 3–4b); optical coherence tomography showed minimal peripapillary retinal nerve fiber layer thinning in both eyes (FIGURE 3–4c).
COMMENT
Recurrent bilateral optic neuritis in a young man with longitudinally extensive optic nerve inflammation with sheath involvement is highly suggestive of myelin oligodendrocyte glycoprotein (MOG)-IgG optic neuritis. MOG-IgG testing by cell-binding assay was positive in this patient. He was treated with 5 days of IV methylprednisolone and had prompt return of normal vision. Because of recurrent disease activity on rituximab, the patient was switched to mycophenolate mofetil and has remained relapse-free.
FIGURE 3–4.
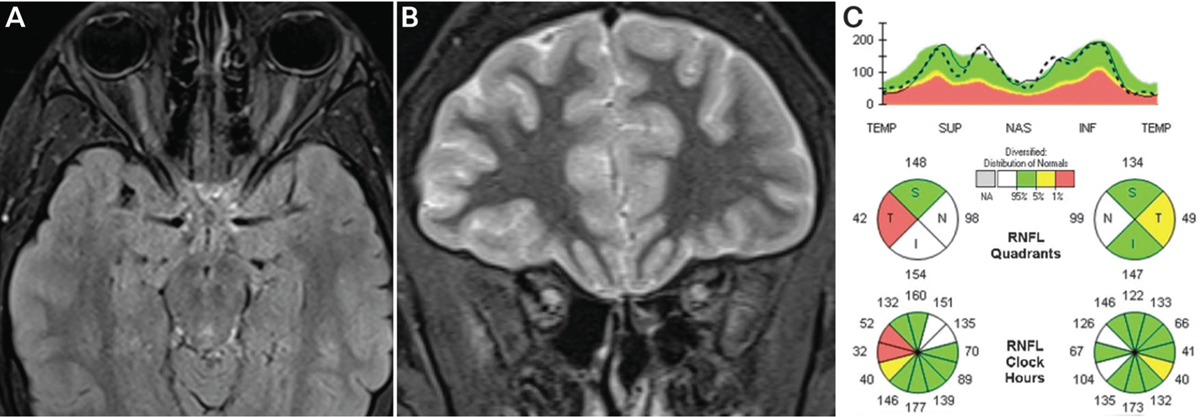
Findings of the patient in CASE 3–2. A, Axial fat-saturated T2-weighted turbo inversion recovery magnitude images showing enlarged optic nerves with long T2-hyperintense orbital lesions. B, Coronal short tau inversion recovery (STIR) images showing T2-hyperintense enlarged optic nerves. C, Spectral domain optical coherence tomography of the peripapillary retinal nerve fiber layer showing normal mean thickness in both eyes despite two prior episodes of optic neuritis. Mild segmental retinal nerve fiber layer thickening is seen in the right eye due to edema as well as focal temporal thinning due to prior episodes of optic neuritis, together leading to the normal mean thickness (pseudo-normal mean thickness). The patient had no clinical evidence of disc edema on examination.
Since autoimmune optic neuropathy, relapsing isolated optic neuritis, and chronic relapsing inflammatory optic neuropathy are diagnoses of exclusion, prior evaluations should include MRI of the brain and optic nerves, lumbar puncture, and serologic testing for AQP4-IgG and MOG-IgG. A single small case study reported a low incidence (5%) of NMOSD among chronic relapsing inflammatory optic neuropathy cases61; however, the incidence of MOG-IgG disease among chronic relapsing inflammatory optic neuropathy cases remains unknown. It is likely that advances in antigen identification will lead to more definitive categorization of patients currently diagnosed with autoimmune optic neuropathy, relapsing isolated optic neuritis, or chronic relapsing inflammatory optic neuropathy.
KEY POINT.
Autoimmune optic neuropathy, relapsing isolated optic neuritis, and chronic relapsing inflammatory optic neuropathy are idiopathic seronegative optic neuropathies characterized by their responsiveness to or dependency on steroid immunosuppression. They are currently a diagnosis of exclusion for patients with recurrent optic neuritis seronegative for AQP4-IgG and MOG-IgG.
OPTIC NEURITIS ASSOCIATED WITH SYSTEMIC IMMUNE DISORDERS
Acute optic neuritis may occur in the background of systemic or neurologic immune disorders. Occasionally, optic neuritis may be the presenting symptom of a systemic immune process. The following sections review the features of optic neuritis associated with Sjögren syndrome, systemic lupus erythematosus, and paraneoplastic disease.
Sjögren Syndrome
Sjögren syndrome is a systemic immune disorder characterized by destructive inflammation of salivary and lacrimal glands resulting in keratoconjunctivitis and xerostomia (sicca syndrome). Sjögren syndrome may present in isolation with sicca syndrome (primary Sjögren syndrome) or in association with other autoimmune connective tissue disorders (secondary Sjögren syndrome). Primary Sjögren syndrome is commonly diagnosed using the American-European Consensus Group criteria, which include ocular and oral clinical symptoms and signs, histopathology, and serology.62 A simpler classification proposed by the American College of Rheumatology is based on serology, ocular surface staining, and histopathology; it shows comparable sensitivity and specificity.63 Sjögren syndrome commonly presents in middle age and predominantly affects women. CNS involvement is infrequent (approximately 5%), and optic neuritis is present in a small percentage (4%) of these cases.64 Affected individuals are commonly treated with corticosteroids, and improvement in vision is variable; recurrent optic neuritis may occur in one-third of patients. Sjögren syndrome has been reported in conjunction with NMOSD; therefore, it is important to check for AQP4 autoantibodies in patients with primary or secondary Sjögren syndrome and optic neuritis, particularly if optic neuritis is associated with myelitis or brainstem inflammation. Despite their association, Sjögren syndrome and NMOSD are considered to be independent, overlapping autoimmune conditions; AQP4 autoantibodies are observed at a similar frequency in patients with NMOSD with and without Sjögren syndrome, and the neurologic presentations of AQP4-IgG seropositive patients with Sjögren syndrome are not distinct from those observed in NMOSD.65
Systemic Lupus Erythematosus
Isolated optic neuritis is an infrequent manifestation of systemic lupus erythematosus (SLE). In a 3-year prospective study of 370 patients with SLE without a history of neurologic involvement, only 16 patients (4.3%) had CNS events; optic neuritis comprised two of 23 events (8.7%).66 Optic neuritis in patients with SLE is typically severe, and recovery is often incomplete. In a small series of patients with SLE with optic neuritis, almost 40% had 20/200 or worse acuity at recovery and 37.5% had a recurrent event.67 Some patients with acute SLE optic neuritis respond to high-dose corticosteroids; the response to plasma exchange is not reported. For patients with recurrent disease, successful control has been reported with steroid-sparing immunosuppressants, such as cyclophosphamide, azathioprine, methotrexate, and cyclosporine. As noted for Sjögren syndrome, AQP4-IgG seropositivity is common in patients with SLE who present with NMOSD, longitudinally extensive transverse myelitis, and recurrent optic neuritis. Therefore, it is prudent to check patients with SLE with acute or recurrent optic neuritis for AQP4-IgG and treat vision loss aggressively. Routine testing for AQP4-IgG in patients with SLE with CNS manifestations that are nonsyndromic for NMOSD, however, is unlikely to be informative.65,68 In some cases of SLE, optic disc edema reflects an anterior ischemic optic neuropathy related to vasculitis or antiphospholipid disorder, imparting a worse prognosis for visual recovery.
Paraneoplastic Optic Neuritis
Paraneoplastic optic neuritis is associated with an autoimmune response against CRMP-5/CV-2.69 Small cell carcinoma is the most frequently associated tumor; however, prostate cancer, renal cell carcinoma, lung adenocarcinoma, and thymoma have been reported.69,70 Vision loss is typically subacute, progressive, and bilateral within weeks to months. Pain is unusual. Fundus examination shows optic disc edema typically with nerve fiber layer hemorrhages. Vitreitis is a striking feature. Vascular leakage secondary to peripheral retinal inflammation is also observed. CRMP-5 paraneoplastic optic neuritis rarely occurs in isolation; however, isolated CRMP-5 perioptic neuritis suggestive of MOG-IgG-associated optic neuritis has been reported.71 Central and peripheral neurologic symptoms reported with CRMP-5 paraneoplastic optic neuritis include ataxia, nystagmus, dementia, polyneuropathy, and myelitis. When concurrent myelitis is present, CRMP-5 paraneoplastic disease may mimic NMOSD.70 For more information on the neuro-ophthalmic complications of paraneoplastic disease, refer to the article “Paraneoplastic Syndromes in Neuro-ophthalmology” by Lynn Gordon, MD, PhD, and Marc Dinkin, MD,72 in this issue of Continuum.
KEY POINTS.
Isolated optic neuritis associated with systemic lupus erythematosus or Sjögren syndrome is
rare. Patients diagnosed with systemic lupus erythematosus or Sjögren syndrome and optic neuritis should be tested for AQP4-IgG.
Patients with systemic lupus erythematosus or Sjögren syndrome with optic neuritis who are seropositive for AQP4-IgG are at higher risk for poor visual recovery than patients with systemic lupus erythematosus or Sjögren syndrome without AQP4-IgG.
Paraneoplastic optic neuritis associated with collapsin response mediator protein-5 (CRMP-5) autoantibodies may mimic idiopathic optic neuritis. Bilateral, asynchronous optic neuritis with prominent vitreitis and retinal leakage in an older adult should raise clinical concern.
CRMP-5 optic neuritis is frequently accompanied by central or peripheral neurologic injury. The presence of transverse myelitis may mimic NMOSD.
Sarcoid optic neuropathy may be extremely difficult to diagnose in the absence of ocular inflammation or systemic disease.
OPTIC NEURITIS DUE TO GRANULOMATOUS DISEASE
Ophthalmic complications are frequently observed in patients with granulomatous disease, and involvement of the optic nerve may result in substantial visual morbidity. The following sections review optic neuritis and ophthalmic manifestations of sarcoidosis and granulomatosis with polyangiitis.
Sarcoidosis
Sarcoidosis frequently presents with ophthalmic complications and is proportionally more common in African and Caribbean ethnic groups.73 Anterior uveitis, vitreitis, periphlebitis, and keratoconjunctivitis are common findings. Optic neuropathy and involvement of the CNS are relatively infrequent and occur in up to 10% of patients.29 Optic neuropathy from sarcoidosis commonly presents as optic neuritis, although compressive and infiltrative injuries to the optic nerve have been described. In a large 2016 case series of sarcoid optic neuropathy,29 82% of patients presented with subacute optic neuritis evolving over days; roughly half were unilateral. Bilateral involvement was typically asynchronous. Similar to other case series,74 vision loss was significant, with median acuity ranging from 20/400 in unilateral disease to 20/150 in bilateral disease. Central field loss was the most common pattern of visual field loss; however, other hemianopic and altitudinal patterns were also observed. MRI frequently showed swelling and enhancement of the affected optic nerve with rare cases of optic perineuritis (5%) or chiasmitis (2.5%).29 In cases of sarcoid optic neuritis, additional inflammation in the orbital apex and CNS were occasionally observed.29,74
Roughly one-third of patients with sarcoid optic neuritis demonstrate concurrent intraocular inflammation at the time of presentation.60 Some published series have found a higher percentage of patients with ocular inflammation; however, the presentations were not limited to optic neuritis. Types of ocular inflammation include granulomatous anterior uveitis, panuveitis, vitreitis, nerve fiber layer infarcts, macular exudates, retinal vasculitis with its characteristic perivascular infiltrates known as “candle wax droppings” (FIGURE 3–6), andepiscleritis.75 Serum ACE is elevated in roughly 50% of patients at the time of optic neuropathy; however, some series have reported lower numbers of patients who were positive.74 Abnormal CSF is uncommon (CASE 3–3). When abnormal, CSF typically shows a lymphocytic pleocytosis with elevated CSF protein. Oligoclonal bands are noted in approximately 15% of patients. In cases of neurosarcoidosis, ACE levels in the CSF may be tested. The reported sensitivity and specificity of CSF ACE are 66.7% and 67.3%, respectively, when using a cutoff value of 2.76 The diagnosis of sarcoid optic neuritis should be confirmed with pathology showing noncaseating granulomas; chest CT, gallium scan, and fludeoxyglucose positron emission tomography (FDG-PET) often prove useful to identify inflamed tissue for sampling.73
FIGURE 3–6.
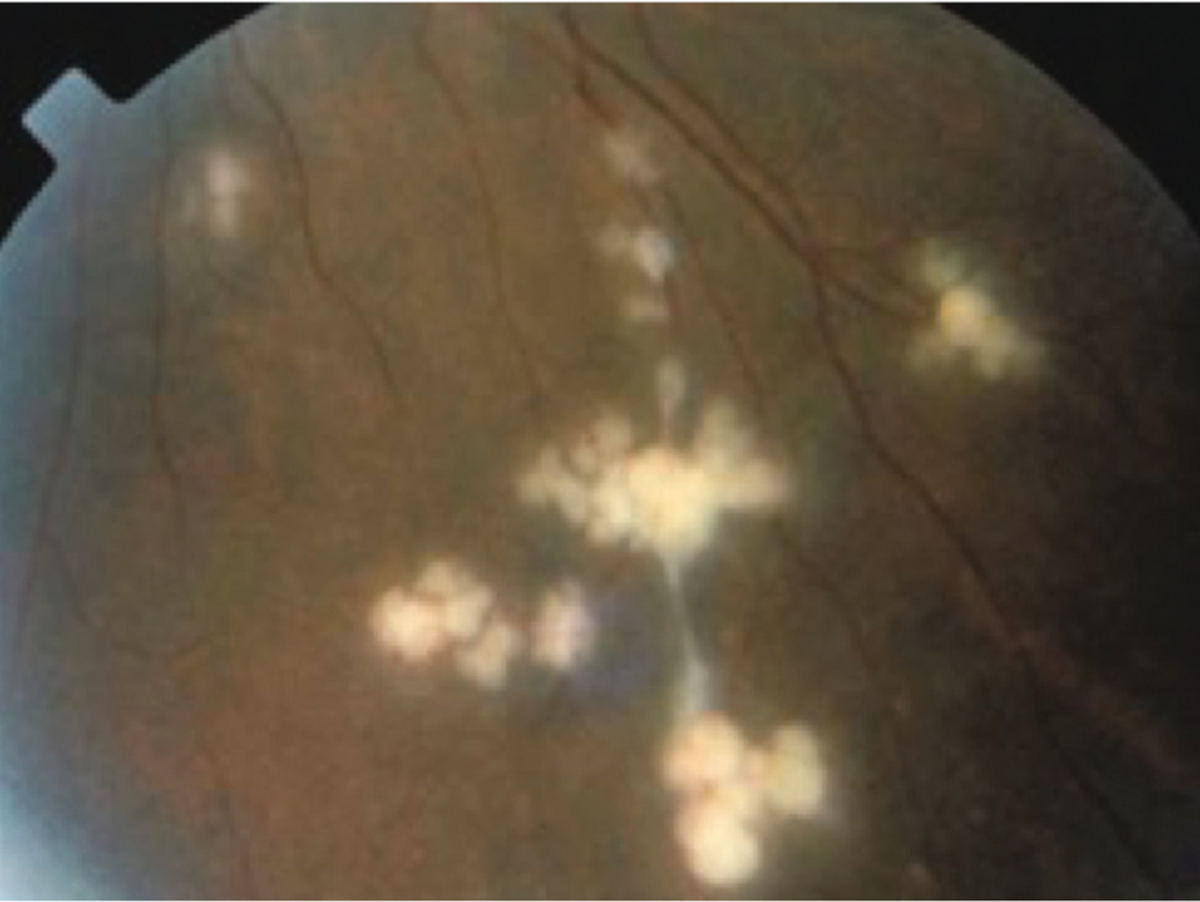
Perivascular infiltrates (candle wax droppings) due to sarcoidosis.
Since sarcoidosis typically demonstrates multiorgan involvement, treatment of sarcoid optic neuritis will often require long-term immunosuppression. Oral corticosteroids (0.5 mg/kg/d to 1.0 mg/kg/d) are the most common approach to initial therapy, although an initial administration of 3 to 5 days of IV methylprednisolone (1 g/d) may be warranted based on the acuity and severity of vision loss.29,73,74 Prolonged immunosuppression with steroid-sparing agents such as azathioprine, methotrexate, tumor necrosis factor-α inhibitors, and mycophenolate mofetil may be necessary to address breakthrough disease or steroid dependence. Final visual acuity varies significantly, and improvement does not correlate with initial MRI findings.29
CASE 3–3.
A 41-year-old man presented with 3 months of subacute, painless vision loss in the left eye. The vision loss was maximal by 2 weeks and had not recovered. He denied any additional neurologic or eye symptoms.
On examination, his visual acuity was 20/15 right eye and 20/50 left eye, and color vision was diminished in the left eye. He had superotemporal field loss in the left eye and a left afferent pupillary defect. The left optic disc was pale (FIGURE 3–7a), and the retina was normal. MRI of the orbits showed diffuse enhancement of the left optic nerve from the globe to the chiasm (FIGURE 3–7b).
IV steroids had no effect on his vision. CSF analysis showed no pleocytosis, normal protein, normal CSF angiotensin-converting enzyme level, and no oligoclonal bands. Serologic testing for antinuclear antibody, angiotensin-converting enzyme level, aquaporin-4 IgG, and myelin oligodendrocyte glycoprotein (MOG) IgG was negative.
Repeat MRI 3 months later demonstrated no change, and his vision worsened. CT of the chest showed hilar adenopathy. Mediastinal lymph node biopsy showed non-necrotizing granulomas diagnostic of sarcoidosis (FIGURES 3–7c and 3–7d). Chronic oral prednisone improved his vision to 20/30.
COMMENT
Subacute painless vision loss is an unusual presentation for optic neuritis associated with longitudinally extensive optic nerve lesions (ie, neuromyelitis optica (NMO) spectrum disorder or MOG-IgG optic neuritis). Progressive decline in this patient and persistent enhancement of the optic nerve months after presentation prompted additional investigation for alternative inflammatory disorders. Because of the common pulmonary involvement in sarcoidosis, a chest CT was performed and hilar adenopathy was identified. Hilar lymph node biopsy was diagnostic of sarcoidosis, and the patient improved with chronic steroid therapy.
FIGURE 3–7.
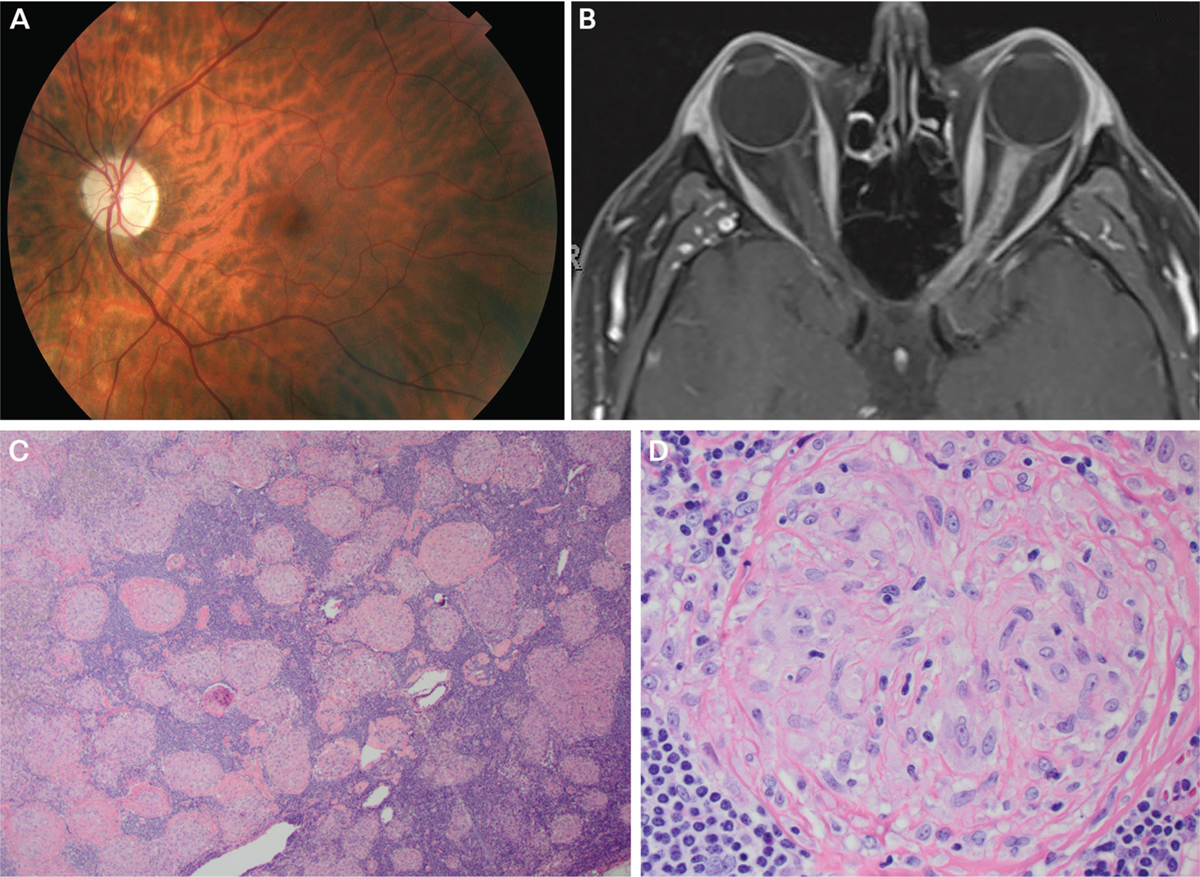
Findings of the patient in CASE 3–3. A, Fundus photography of the left eye revealing optic disc pallor. B, Axial fat-suppressed postcontrast T1-weighted MRI showing longitudinally extensive enhancement of left optic nerve. C, Low-magnification image of hilar lymph node biopsy stained with hematoxylin and eosin (H&E) showing replacement of the normal lymph node architecture by multiple small, well-defined, non-necrotizing granulomas of relatively uniform sizes and shapes that coalesce with variable degrees of fibrosis. D, High-magnification image of granuloma showing cytologic features of the epithelioid histiocytes.
Panels C and D courtesy of Jeffrey Schowinsky, MD.
Granulomatosis With Polyangiitis
Granulomatosis with polyangiitis (previously known as Wegener granulomatosis) is a multisystem small vessel granulomatous vasculitis that commonly presents with sinonasal, respiratory, and renal disease.77 Approximately 30% of patients with granulomatosis with polyangiitis are reported to have ocular involvement,78 and, in rare cases, the disease may be limited to the eye.79 Granulomatosis with polyangiitis may occur across all age groups, with a peak incidence between 64 and 75 years of age. Granulomatosis with polyangiitis has no sex predilection, and the disease can be seen in all racial and ethnic groups. The upper respiratory tract is most frequently involved; however, lower respiratory involvement with cough, dyspnea, and hemoptysis is common. Renal involvement may not be evident at disease onset, but glomerulonephritis usually develops within the first 2 years of disease. CNS involvement occurs in one-third of patients, and signs include peripheral neuropathy, cranial neuropathy, seizure, stroke, and ophthalmoplegia.80
The optic nerve is infrequently involved in granulomatosis with polyangiitis, and both retrobulbar optic neuritis and optic perineuritis have been reported.81,82 Loss of visual acuity is generally severe; however, improvement in vision has been reported with corticosteroids and immunosuppression in a limited number of cases. Compressive and ischemic optic neuropathies have also been described.78 Additional ophthalmic and neuro-ophthalmic features of granulomatosis with polyangiitis include ulcerative conjunctivitis, dacryoadenitis, orbital cellulitis, uveitis, vitreitis, vasculitis, chorioretinal ischemia, central retinal vein occlusion, and diplopia. Ocular manifestations, such as necrotizing scleritis, are indicators of both increased morbidity and mortality.78 The diagnosis of granulomatosis with polyangiitis may be made with objective evidence of two or more features of disease in the kidneys, lungs, and nasal sinuses.77 With systemic disease, 80% to 90% of patients will have positive c-ANCA. The vast majority of c-ANCA autoantibodies are directed against proteinase-3, and the remainder are against myeloperoxidase. Because of high mortality risk, treatment regimens typically combine high-dose corticosteroids with immunosuppressants such as cyclophosphamide, methotrexate, rituximab, and tumor necrosis factor-α inhibitors.77,78
INFECTIOUS OPTIC NEURITIS
Direct infection of the optic nerve is a rare cause of optic nerve inflammation. Infectious optic neuritis, however, is important to identify as rapid initiation of appropriate antimicrobial, antiviral, antifungal, or antiprotozoal agents may be important for preventing further vision loss, facilitating visual recovery, or treating concurrent systemic or CNS disease. Optic neuritis occurring in conjunction with fever, meningitis, cranial nerve palsies, and encephalitis should always raise concern for an infectious cause. A detailed history regarding infectious risk factors and travel history may help to focus on probable agents. On examination, concurrent papillitis may result in severe optic disc edema with a macular star, and concurrent ocular inflammation may generate signs of uveitis, retinitis, and chorioretinitis. A complete review of infectious optic neuritides is beyond the scope of the current review; however, the following sections focus on some common presentations and infectious agents.
Neuroretinitis
Infectious optic neuritis frequently results in neuroretinitis, a term that describes optic disc edema in combination with a starlike pattern of lipid exudate in the macula and fovea (FIGURE 3–8). Multiple bacterial, spirochetal, viral, fungal, and protozoal infections of the optic nerve may result in neuroretinitis, including Bartonella, Rickettsia, spirochetes, Coxsackievirus B, HIV, histoplasmosis, and toxoplasmosis.83 Although infectious agents are common causes of neuroretinitis, many cases are idiopathic since any cause of significant leakage from the optic disc may result in outer retinal exudates. Neuroretinitis has been reported with sarcoidosis, Behçet disease, and severe ischemic optic neuropathy and following severe papilledema. Neuroretinitis is commonly preceded by a febrile illness, and additional symptoms such as arthralgia, headache, rash, and lymphadenopathy further support an infectious etiology in many cases. Vision loss is usually painless, and central or centrocecal field loss is most common.83,84 The macular exudates (macular star) may not be evident for 2 to 6 weeks after initial presentation and may take several months to resolve. OCT may demonstrate subretinal fluid, retinal thickening, or exudates in the outer plexiform layer before they are evident on fundus examination.85 In most cases, vision loss spontaneously resolves; however, visual recovery in recurrent disease is less common.84
FIGURE 3–8.
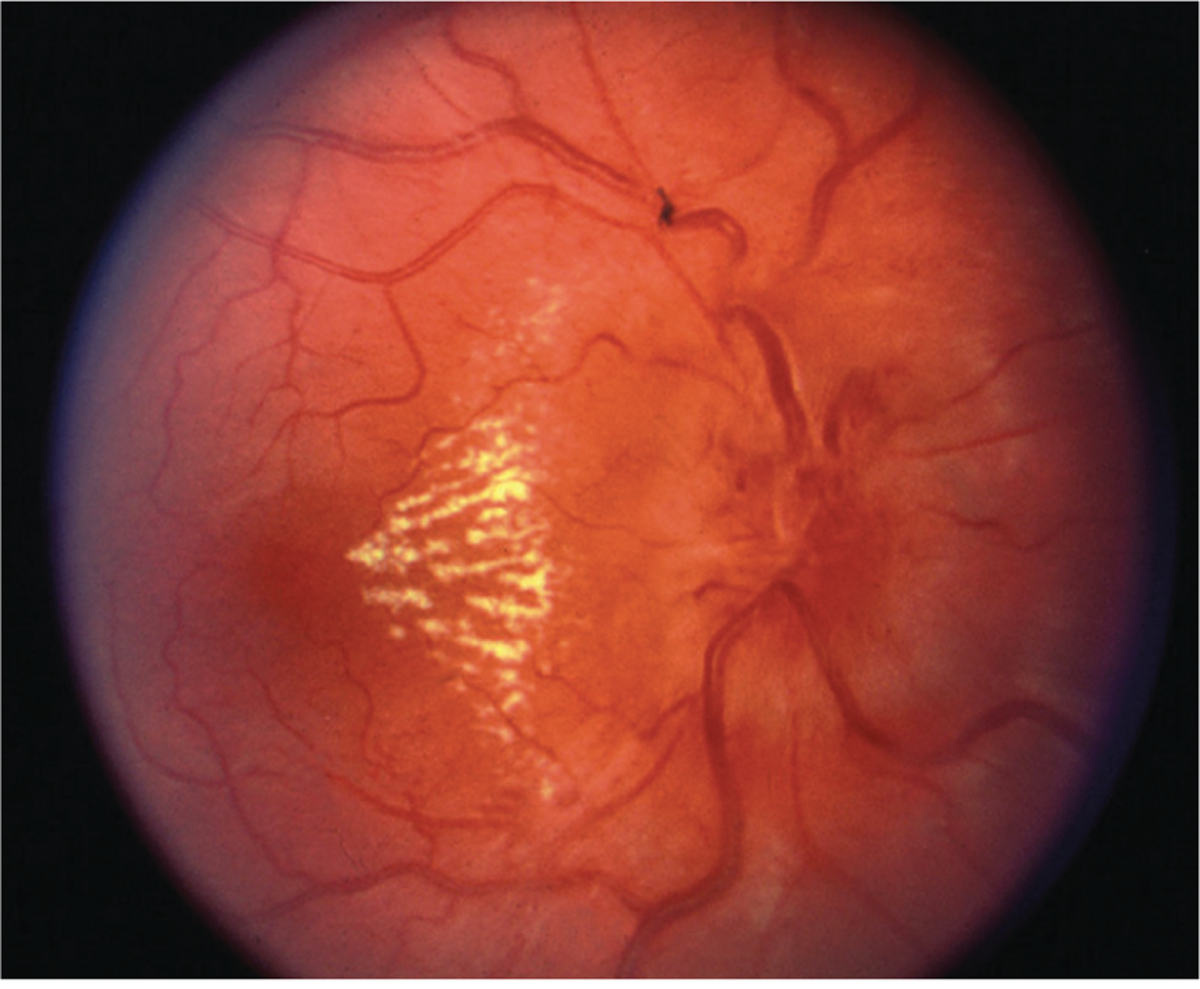
Optic disc edema with partial macular star (macular fan) due to neuroretinitis. Note the dilated venules due to venous compression from disc edema.
The most common cause of neuroretinitis is catscratch disease resulting from B. henselae infection. The majority of cases are transmitted by cat scratch or bite; however, transmission by dog has been documented. Vision loss may be mild or severe, and MRI of the optic nerves may show enhancement of the optic disc and proximal nerve.86 Serum antibodies to Bartonella confirm exposure. The benefit of antibiotic therapy for Bartonella neuroretinitis is unclear.41 Antibiotic treatment may not be prudent except for cases with systemic infection, severe vision loss, or immunocompromise.
KEY POINTS.
Angiotensin-converting enzyme levels in the serum and CSF are notoriously insensitive for neurosarcoidosis. When suspicious, CT chest, gallium scan, or fludeoxyglucose positron emission tomography are recommended for identifying involved tissue amenable to biopsy.
While multiple infectious agents have been associated with neuroretinitis, many cases are idiopathic. Exposure to common infectious causes should be evaluated. When the infectious workup is negative, alternative noninflammatory causes of optic disc edema with a macular star should be considered.
Lyme Disease
Infection with the spirochete Borrelia burgdorferi may result in CNS infection and either primary or secondary optic nerve injury. Primary infection of the optic nerve may result in optic neuritis, neuroretinitis, papillitis, or ischemic optic neuropathy.87,88 Secondary injury may result from papilledema due to elevated intracranial pressure from Lyme meningitis. In Lyme optic neuritis, vision loss is typically preceded by signs of systemic infection: headache, myalgia, and arthralgia. Vision loss is typically painless, varies from mild to severe (20/30 to count fingers vision), and is often bilateral or consecutive. Disc edema is common. Lyme optic neuritis may show optic nerve enhancement on orbital MRI and nonspecific white matter lesions on brain MRI.87 CSF will demonstrate a lymphocytic pleocytosis with intrathecal synthesis of Borrelia-specific antibodies in the vast majority of patients.89 Borrelia-specific IgM and IgG antibodies are usually detected in the blood; however, equivocal and negative tests during early disease should prompt retesting during the convalescent phase. Treatment is typically with doxycycline for CNS involvement; however, parenteral ceftriaxone is often recommended for late-stage disease.90
CASE 3–4.
A 42-year-old man presented with 2 weeks of splotchy vision in the right eye. He had noticed transient loss of vision in the right eye with postural changes. He denied any eye pain but reported fever and body rash 5 weeks prior.
On examination, vision was 20/15 in both eyes with superotemporal field loss in the right eye and a right afferent pupillary defect. Fundus examination showed significant right optic nerve edema with associated nerve fiber layer hemorrhages (FIGURE 3–9). He had increased lower extremity tone, hyperreflexia, and bilateral extensor plantar responses. MRI of the orbits showed mild T2-hyperintense signal in the right optic nerve without gadolinium enhancement. Slit-lamp examination showed iritis.
COMMENT
Visual field loss, transient visual obscurations, and significant disc edema with hemorrhages and ocular inflammation are atypical for idiopathic optic neuritis. While myelin oligodendrocyte glycoprotein (MOG) IgG commonly causes significant disc edema, ocular inflammation is not observed with MOG-IgG optic neuritis and MRI typically shows significant nerve enhancement. This patient’s prior fever and rash, ocular inflammation, and disc edema with hemorrhage suggest an infectious cause. The patient acknowledged high-risk behavior for HIV and tested positive for HIV and syphilis. He was treated with a 14-day course of IV penicillin G, and the disc edema and visual field loss resolved over the next 3 months.
FIGURE 3–9.
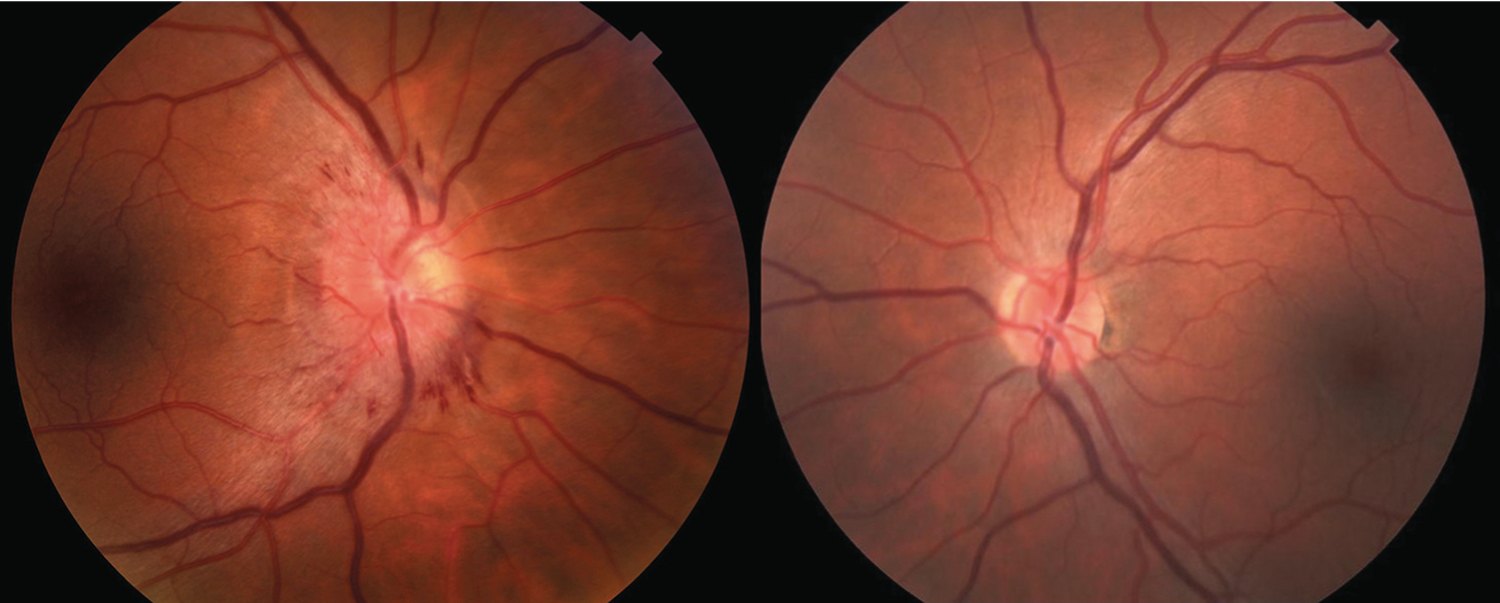
Funduscopy of the patient in CASE 3–4. Fundus photos demonstrate severe disc edema with nerve fiber layer hemorrhages in the right eye. The left optic nerve is normal. The diagnosis was optic neuritis due to syphilis.
KEY POINTS.
Optic neuritis with disc edema and cranial neuropathies should be investigated for Lyme disease in endemic areas.
Syphilitic optic neuritis is often associated with ocular inflammation. Optic disc edema, when present, is usually prominent. A detailed social history identifying high-risk behavior for HIV should be performed in suspicious cases.
Syphilis
Infection with the spirochete Treponema pallidum may progress to involve the CNS in secondary and tertiary stages of disease. Syphilitic optic neuritis is clinical evidence of neurosyphilis independent of any additional neurologic findings. Vision loss may be unilateral or bilateral; pain is uncommon. Papillitis and disc swelling are frequently observed, and the fundus examination may eventually demonstrate a macular star.91 Associated ocular inflammation is common.92 MRI of the orbits often shows perineuritis,93 in which enhancement is seen within the optic nerve sheath, but inflammation of the nerve may also be observed (CASE 3–4).94 Additional brain MRI abnormalities display a spectrum of meningovascular, inflammatory, and degenerative pathology.95 CSF typically shows a lymphocytic pleocytosis with elevated protein and intrathecal IgG synthesis. Testing for antitreponemal antibodies (eg, fluorescent treponemal antibody absorption) should be performed in the blood and CSF. Lipid antigen testing (Venereal Disease Research Laboratory [VDRL]) may be a better reflection of disease activity but has low diagnostic sensitivity in the CSF. HIV testing should be performed in patients suspected of having neurosyphilis, and clinicians should be mindful that false-negative tests for antitreponemal antibodies are common with concomitant HIV infection.91 Vision loss is usually responsive to IV antibiotics. Treatment is generally with benzathine penicillin G; however, long-term IV aqueous penicillin G is generally recommended for syphilitic optic neuritis because of its outstanding CNS penetration.91
Viral Infections
Acute viral infection is an uncommon cause of isolated optic neuritis. Herpes simplex virus (HSV), varicella-zoster virus (VZV), cytomegalovirus (CMV), Epstein-Barr virus (EBV), HIV, West Nile virus, dengue fever, mumps, measles, and rubella infections have been implicated in cases of acute optic neuritis, many with concurrent retinal, brain, or ocular inflammation.96 HSV optic neuritis may occur concurrently with or following HSV encephalitis or acute retinal necrosis. HSV optic neuritis is usually responsive to acyclovir; however, any additional benefit to the use of concurrent methylprednisolone is uncertain. VZV papillitis has been reported during primary VZV infection (chickenpox) and secondary reactivation (zoster). Vision loss is usually bilateral with primary infection and unilateral following zoster ophthalmicus. Onset after zoster may be delayed by weeks. The role of corticosteroids and antivirals again remains uncertain. Visual recovery is generally good but is limited in cases associated with posterior outer retinal necrosis.
CMV- and EBV-associated optic neuritides are rare. CMV papillitis is typically associated with CMV retinitis in patients who are immunocompromised. EBV optic neuritis is generally bilateral and retrobulbar, but papillitis, chiasmitis, and neuroretinitis cases have been reported. Most cases of EBV-associated optic neuritis respond to corticosteroids. Primary HIV infection of the optic nerve is rare but, in some cases, may be the initial manifestation of disease. Presentations include retrobulbar optic neuritis, papillitis, and neuroretinitis. Mosquito-borne arboviruses and flaviviruses (West Nile virus, dengue fever, chikungunya virus, Rift Valley fever) may cause acute optic neuritis often in association with chorioretinitis and other signs of ocular inflammation. Signs of systemic infection are common, and visual prognosis is generally good except for dengue fever. Rare cases of optic neuritis have been reported with acute mumps, rubella, and measles virus infections; vision generally recovers with corticosteroid treatment.
KEY POINTS.
Optic neuritis due to direct viral infection is rare. Clinical and examination clues include recent zoster ophthalmicus, encephalitis, immunosuppression, risk for mosquito-borne illness, or associated retinitis or chorioretinitis.
Optic neuritis from tuberculosis is often associated with uveitis and orbital apex syndrome. MRI brain findings include leptomeningeal enhancement, ependymitis, abscess, infarct, encephalitis, and tubercles.
Tuberculosis
CNS infection with Mycobacterium tuberculosis is an infrequent cause of inflammatory optic neuropathy. Affected individuals typically reside in or have traveled to an endemic area. The most common presentation of optic neuritis associated with tuberculosis is papillitis.97 Neuroretinitis and retrobulbar optic neuritis are less common. Optic nerve involvement is frequently accompanied by posterior uveitis or panuveitis; the orbital apex syndrome is also frequently observed. The onset of vision loss may be acute or insidious, with roughly half of the patients displaying a chronic course. Loss of visual acuity at presentation may vary from mild (20/30) to severe (<20/200), and the most common pattern of visual field loss is an enlarged blind spot. Vision loss from CNS tuberculosis may also arise from tubercular meningitis resulting in hydrocephalus, optic chiasmatic arachnoiditis, and compression of either the optic nerve or chiasm due to tuberculomas.96 Tuberculous optic perineuritis may be evident on orbital imaging.98
MRI of the brain may show a constellation of pathology ranging from leptomeningeal enhancement to abscesses and tuberculomas.99 CSF will typically show a chronic mixed pleocytosis with elevated protein and low glucose.100 CSF polymerase chain reaction (PCR) for M. tuberculosis is a rapid and sensitive method for diagnostic confirmation.100 Antituberculosis medications are often effective in improving vision, with roughly 75% of patients reaching 20/40 vision or better. Davis and colleagues97 did not report any benefit to visual recovery with the additional use of corticosteroids.
CONCLUSION
Optic neuritis may result from a diverse number of inflammatory and infectious conditions. Subacute, painful vision loss accompanied by an afferent pupillary defect should routinely raise concern for optic neuritis. Nevertheless, vision loss may sometimes be chronic and painless, especially with granulomatous inflammation or certain infections. Idiopathic optic neuritis associated with MS is often self-limited and has a strong chance of recovery. Inflammatory optic neuropathies associated with MOG-IgG disease and NMOSD, however, are typically more severe and often recur without prophylactic therapy. Importantly, NMOSD optic neuritis has a lower probability of visual recovery, and early intervention with plasma exchange may enhance visual recovery.
Moving forward, optimal visual recovery from acute optic neuritis may require treatment decisions before a definitive serologic, molecular, or histopathologic diagnosis. Therefore, clinicians must be aware of pertinent fundus, neuroimaging, and laboratory data that favor various inflammatory or infectious etiologies. Aggressive anti-inflammatory therapy coupled with targeted immunosuppression and/or antibiotics is likely to become the standard for minimizing short- and long-term vision loss from optic neuritis.
RELATIONSHIP DISCLOSURE:
Dr Bennett serves on the editorial boards of the Journal of Neuro-ophthalmology, Multiple Sclerosis, and Neurology: Neuroimmunology & Neuroinflammation and as a consultant for AbbVie Inc; Alexion; Chugai Pharmaceutical Co, Ltd; Clene Nanomedicine; EMD Serono, Inc; Equillium, Inc; Frequency Therapeutics; Genentech, Inc; MedImmune; and Sanofi Genzyme. Dr Bennett has received research/grant support from EMD Serono, Inc; the Guthy-Jackson Charitable Foundation; Mallinckrodt Pharmaceuticals; the National Eye Institute (R01EY022936); the National Institute of Allergy and Infectious Diseases (UM1AI110498); and Novartis AG. Dr Bennett receives publishing royalties from UpToDate, Inc, and has received personal compensation for serving as a medicolegal consultant on medical cases involving neuroinflammatory and neuro-ophthalmologic disorders.
Footnotes
UNLABELED USE OF PRODUCTS/INVESTIGATIONAL USE DISCLOSURE:
Dr Bennett discusses the unlabeled/investigational use of plasma exchange and apheresis for the treatment of optic neuritis.
REFERENCES
- 1.Beck RW, Cleary PA, Anderson MM, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med 1992;326(9): 581–588. doi: 10.1056/NEJM199202273260901. [DOI] [PubMed] [Google Scholar]
- 2.Jarius S, Paul F, Aktas O, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation 2018;15(1):134. doi: 10.1186/s12974-018-1144-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jurynczyk M, Messina S, Woodhall MR, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain 2017;140(12): 3128–3138. doi: 10.1093/brain/awx276. [DOI] [PubMed] [Google Scholar]
- 4.Ramanathan S, Prelog K, Barnes EH, et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Scler 2016;22(4):470–482. doi: 10.1177/1352458515593406. [DOI] [PubMed] [Google Scholar]
- 5.The clinical profile of optic neuritis. Experience of the Optic Neuritis Treatment Trial. Optic Neuritis Study Group. Arch Ophthalmol 1991; 109(12):1673–1678. [DOI] [PubMed] [Google Scholar]
- 6.Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. Myelin oligodendrocyte glycoprotein antibody-positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol 2018;195:8–15. doi: 10.1016/j.ajo.2018.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merle H, Olindo S, Jeannin S, et al. Treatment of optic neuritis by plasma exchange (add-on) in neuromyelitis optica. Arch Ophthalmol 2012;130(7): 858–862. doi: 10.1001/archophthalmol.2012.1126. [DOI] [PubMed] [Google Scholar]
- 8.Keltner JL, Johnson CA, Spurr JO, Beck RW. Baseline visual field profile of optic neuritis. The experience of the optic neuritis treatment trial. Optic Neuritis Study Group. Arch Ophthalmol 1993;111(2):231–234. doi: 10.1001/archopht.1993.01090020085029. [DOI] [PubMed] [Google Scholar]
- 9.Merle H, Olindo S, Jeannin S, et al. Visual field characteristics in neuromyelitis optica in absence of and after one episode of optic neuritis. Clin Ophthalmol 2013;7:1145–1153. doi: 10.2147/OPTH.S43894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakajima H, Hosokawa T, Sugino M, et al. Visual field defects of optic neuritis in neuromyelitis optica compared with multiple sclerosis. BMC Neurol 2010;10:45. doi: 10.1186/1471-2377-10-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kupersmith MJ, Mandel G, Anderson S, et al. Baseline, one and three month changes in the peripapillary retinal nerve fiber layer in acute optic neuritis: relation to baseline vision and MRI. J Neurol Sci 2011;308(1–2):117–123. doi: 10.1016/j.jns.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 12.Higashiyama T, Nishida Y, Ohji M. Optical coherence tomography angiography in eyes with good visual acuity recovery after treatment for optic neuritis. PLoS One 2017;12(2):e0172168. doi: 10.1371/journal.pone.0172168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y, Zhou L, ZhangBao J, et al. Peripapillary and parafoveal vascular network assessment by optical coherence tomography angiography in aquaporin-4 antibody-positive neuromyelitis optica spectrum disorders. Br J Ophthalmol 2019;103(6):789–796. doi: 10.1136/bjophthalmol-2018-312231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kupersmith MJ, Alban T, Zeiffer B, Lefton D. Contrast-enhanced MRI in acute optic neuritis: relationship to visual performance. Brain 2002; 125(pt 4):812–822. doi: 10.1093/brain/awf087. [DOI] [PubMed] [Google Scholar]
- 15.Kapoor R, Miller DH, Jones SJ, et al. Effects of intravenous methylprednisolone on outcome in MRI-based prognostic subgroups in acute optic neuritis. Neurology 1998;50(1):230–237. doi: 10.1212/WNL.50.1.230. [DOI] [PubMed] [Google Scholar]
- 16.Miller DH, Newton MR, van der Poel JC, et al. Magnetic resonance imaging of the optic nerve in optic neuritis. Neurology 1988;38(2):175–179. doi: 10.1212/WNL.38.2.175. [DOI] [PubMed] [Google Scholar]
- 17.Akaishi T, Sato DK, Nakashima I, et al. MRI and retinal abnormalities in isolated optic neuritis with myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies: a comparative study. J Neurol Neurosurg Psychiatry 2016;87(4): 446–448. doi: 10.1136/jnnp-2014-310206. [DOI] [PubMed] [Google Scholar]
- 18.Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2018;17(2): 162–173. doi: 10.1016/S1474-4422(17)30470-2. [DOI] [PubMed] [Google Scholar]
- 19.Pittock SJ, Weinshenker BG, Lucchinetti CF, et al. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol 2006;63(7):964–968. doi: 10.1001/archneur.63.7.964. [DOI] [PubMed] [Google Scholar]
- 20.Sato DK, Callegaro D, Lana-Peixoto MA, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 2014;82(6):474–481. doi: 10.1212/WNL.0000000000000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flanagan EP, Hinson SR, Lennon VA, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients. Ann Neurol 2017;81(2): 298–309. doi: 10.1002/ana.24881. [DOI] [PubMed] [Google Scholar]
- 22.Jarius S, Ruprecht K, Kleiter I, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation 2016;13(1):280. doi: 10.1186/s12974-016-0718-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pittock SJ, Lennon VA, de Seze J, et al. Neuromyelitis optica and non organ-specific autoimmunity. Arch Neurol 2008;65(1):78–83. doi: 10.1001/archneurol.2007.17. [DOI] [PubMed] [Google Scholar]
- 24.McKeon A, Lennon VA, Jacob A, et al. Coexistence of myasthenia gravis and serological markers of neurological autoimmunity in neuromyelitis optica. Muscle Nerve 2009;39(1): 87–90. doi: 10.1002/mus.21197. [DOI] [PubMed] [Google Scholar]
- 25.Jarius S, Paul F, Franciotta D, et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci 2011;306(1–2):82–90. doi: 10.1016/j.jns.2011.03.038. [DOI] [PubMed] [Google Scholar]
- 26.Majed M, Fryer JP, McKeon A, et al. Clinical utility of testing AQP4-IgG in CSF: Guidance for physicians. Neurol Neuroimmunol Neuroinflamm 2016;3(3):e231. doi: 10.1212/NXI.0000000000000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alam SM, Kyriakides T, Lawden M, Newman PK. Methylprednisolone in multiple sclerosis: a comparison of oral with intravenous therapy at equivalent high dose. J Neurol Neurosurg Psychiatry 1993;56(11):1219–1220. doi: 10.1136/jnnp.56.11.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morrow SA, Fraser JA, Day C, et al. Effect of treating acute optic neuritis with bioequivalent oral vs intravenous corticosteroids: a randomized clinical trial. JAMA Neurol 2018;75(6):690–696. doi: 10.1001/jamaneurol.2018.0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kidd DP, Burton BJ, Graham EM, Plant GT. Optic neuropathy associated with systemic sarcoidosis. Neurol Neuroimmunol Neuroinflamm 2016;3(5): e270. doi: 10.1212/NXI.0000000000000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petzold A, Plant GT. Chronic relapsing inflammatory optic neuropathy: a systematic review of 122 cases reported. J Neurol 2014;261(1):17–26. doi: 10.1007/s00415-013-6957-4. [DOI] [PubMed] [Google Scholar]
- 31.Beck RW, Cleary PA, Trobe JD, et al. The effect of corticosteroids for acute optic neuritis on the subsequent development of multiple sclerosis. The Optic Neuritis Study Group. N Engl J Med 1993;329(24):1764–1769. doi: 10.1056/NEJM199312093292403. [DOI] [PubMed] [Google Scholar]
- 32.Goodin DS. Perils and pitfalls in the interpretation of clinical trials: a reflection on the recent experience in multiple sclerosis. Neuroepidemiology 1999;18(2):53–63. doi: 10.1159/000069408. [DOI] [PubMed] [Google Scholar]
- 33.Sharrack B, Hughes RA, Morris RW, et al. The effect of oral and intravenous methylprednisolone treatment on subsequent relapse rate in multiple sclerosis. J Neurol Sci 2000;173(1):73–77. doi: 10.1016/S0022-510X(99)00304-4. [DOI] [PubMed] [Google Scholar]
- 34.Zivadinov R, Rudick RA, De Masi R, et al. Effects of IV methylprednisolone on brain atrophy in relapsing-remitting MS. Neurology 2001;57(7): 1239–1247. doi: 10.1212/WNL.57.7.1239. [DOI] [PubMed] [Google Scholar]
- 35.Noseworthy JH, O’Brien PC, Petterson TM, et al. A randomized trial of intravenous immunoglobulin in inflammatory demyelinating optic neuritis. Neurology 2001;56(11):1514–1522. doi: 10.1212/WNL.56.11.1514. [DOI] [PubMed] [Google Scholar]
- 36.Roed HG, Langkilde A, Sellebjerg F, et al. A double-blind, randomized trial of IV immunoglobulin treatment in acute optic neuritis. Neurology 2005;64(5):804–810. doi: 10.1212/01.WNL.0000152873.82631.B3. [DOI] [PubMed] [Google Scholar]
- 37.Deschamps R, Gueguen A, Parquet N, et al. Plasma exchange response in 34 patients with severe optic neuritis. J Neurol 2016;263(5): 883–887. doi: 10.1007/s00415-016-8073-8. [DOI] [PubMed] [Google Scholar]
- 38.Aungsumart S, Apiwattanakul M. Clinical outcomes and predictive factors related to good outcomes in plasma exchange in severe attack of NMOSD and long extensive transverse myelitis: case series and review of the literature. Mult Scler Relat Disord 2017;13:93–97. doi: 10.1016/j.msard.2017.02.015. [DOI] [PubMed] [Google Scholar]
- 39.Bonnan M, Valentino R, Debeugny S, et al. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry 2018;89(4):346–351. doi: 10.1136/jnnp-2017-316286. [DOI] [PubMed] [Google Scholar]
- 40.Koziolek MJ, Tampe D, Bähr M, et al. Immunoadsorption therapy in patients with multiple sclerosis with steroid-refractory optical neuritis. J Neuroinflammation 2012;9:80. doi: 10.1186/1742-2094-9-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhatti MT, Lee MS. Should patients with bartonella neuroretinitis receive treatment? J Neuroophthalmol 2014;34(4):412–416. doi: 10.1097/WNO.0000000000000166. [DOI] [PubMed] [Google Scholar]
- 42.Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final optic neuritis treatment trial follow-up. Arch Neurol 2008; 65(6):727–732. doi: 10.1001/archneur.65.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gabilondo I, Martínez-Lapiscina EH, Fraga-Pumar E, et al. Dynamics of retinal injury after acute optic neuritis. Ann Neurol 2015;77(3):517–528. doi: 10.1002/ana.24351. [DOI] [PubMed] [Google Scholar]
- 44.Kupersmith MJ, Gal RL, Beck RW, et al. Visual function at baseline and 1 month in acute optic neuritis: predictors of visual outcome. Neurology 2007;69(6):508–514. doi: 10.1212/01.wnl.0000267272.60714.42. [DOI] [PubMed] [Google Scholar]
- 45.Mealy MA, Whetstone A, Orman G, et al. Longitudinally extensive optic neuritis as an MRI biomarker distinguishes neuromyelitis optica from multiple sclerosis. J Neurol Sci 2015;355(1–2): 59–63. doi: 10.1016/j.jns.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raftopoulos R, Hickman SJ, Toosy A, et al. Phenytoin for neuroprotection in patients with acute optic neuritis: a randomised, placebo-controlled, phase 2 trial. Lancet Neurol 2016;15(3): 259–269. doi: 10.1016/S1474-4422(16)00004-1. [DOI] [PubMed] [Google Scholar]
- 47.Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85(2):177–189. doi: 10.1212/WNL.0000000000001729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol 2016;79(2): 206–216. doi: 10.1002/ana.24554. [DOI] [PubMed] [Google Scholar]
- 49.Naismith RT, Tutlam NT, Xu J, et al. Optical coherence tomography differs in neuromyelitis optica compared with multiple sclerosis. Neurology 2009;72(12):1077–1082. doi: 10.1212/01.wnl.0000345042.53843.d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ratchford JN, Quigg ME, Conger A, et al. Optical coherence tomography helps differentiate neuromyelitis optica and MS optic neuropathies. Neurology 2009;73(4):302–308. doi: 10.1212/WNL.0b013e3181af78b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Min JH1, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler 2012;18(1): 113–115. doi: 10.1177/1352458511431973. [DOI] [PubMed] [Google Scholar]
- 52.Kleiter I, Hellwig K, Berthele A, et al. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol 2012;69(2):239–245. doi: 10.1001/archneurol.2011.216. [DOI] [PubMed] [Google Scholar]
- 53.Kim SH, Kim W, Li XF, et al. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Mult Scler 2012;18(10): 1480–1483. [DOI] [PubMed] [Google Scholar]
- 54.Hacohen Y, Wong YY, Lechner C, et al. Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody-associated disease. JAMA Neurol 2018; 75(4):478–487. doi: 10.1001/jamaneurol.2017.4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramanathan S, Reddel SW, Henderson A, et al. Antibodies to myelin oligodendrocyte glycoprotein in bilateral and recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm 2014;1(4):e40. doi: 10.1212/NXI.0000000000000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen JJ, Aksamit AJ, McKeon A, et al. Optic disc edema in glial fibrillary acidic protein autoantibody-positive meningoencephalitis. J Neuroophthalmol 2018;38(3):276–281. doi: 10.1097/WNO.0000000000000593. [DOI] [PubMed] [Google Scholar]
- 57.Dutton JJ, Burde RM, Klingele TG. Autoimmune retrobulbar optic neuritis. Am J Ophthalmol 1982; 94(1):11–17. doi: 10.1016/0002-9394(82)90184-2. [DOI] [PubMed] [Google Scholar]
- 58.Frohman L, Dellatorre K, Turbin R, Bielory L. Clinical characteristics, diagnostic criteria and therapeutic outcomes in autoimmune optic neuropathy. Br J Ophthalmol 2009;93(12): 1660–1666. doi: 10.1136/bjo.2009.159350. [DOI] [PubMed] [Google Scholar]
- 59.Benoilid A, Tilikete C, Collongues N, et al. Relapsing optic neuritis: a multicentre study of 62 patients. Mult Scler 2014;20(7):848–853. doi: 10.1177/1352458513510223. [DOI] [PubMed] [Google Scholar]
- 60.Kidd D, Burton B, Plant GT, Graham EM. Chronic relapsing inflammatory optic neuropathy (CRION). Brain 2003;126(pt 2):276–284. doi: 10.1093/brain/awg045. [DOI] [PubMed] [Google Scholar]
- 61.Petzold A, Pittock S, Lennon V, et al. Neuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritis. J Neurol Neurosurg Psychiatry 2010; 81(1):109–111. doi: 10.1136/jnnp.2008.146894. [DOI] [PubMed] [Google Scholar]
- 62.Vitali C, Bombardieri S, Jonsson R, et al. Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis 2002;61(6):554–558. doi: 10.1016/B978-0-12-803604-4.00004-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shiboski SC, Shiboski CH, Criswell L, et al. American College of Rheumatology classification criteria for Sjögren’s syndrome: a data-driven, expert consensus approach in the Sjögren’s International Collaborative Clinical Alliance cohort. Arthritis Care Res (Hoboken) 2012;64(4): 475–487. doi: 10.1002/acr.21591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Massara A, Bonazza S, Castellino G, et al. Central nervous system involvement in Sjögren’s syndrome: unusual, but not unremarkable-clinical, serological characteristics and outcomes in a large cohort of Italian patients. Rheumatology (Oxford) 2010;49(8):1540–1549. doi: 10.1093/rheumatology/keq111. [DOI] [PubMed] [Google Scholar]
- 65.Jarius S, Jacobi C, de Seze J, et al. Frequency and syndrome specificity of antibodies to aquaporin-4 in neurological patients with rheumatic disorders. Mult Scler 2011;17(9): 1067–1073. doi: 10.1177/1352458511403958. [DOI] [PubMed] [Google Scholar]
- 66.Kampylafka EI, Alexopoulos H, Kosmidis ML, et al. Incidence and prevalence of major central nervous system involvement in systemic lupus erythematosus: a 3-year prospective study of 370 patients. PLoS One 2013;8(2):e55843. doi: 10.1371/journal.pone.0055843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin YC, Wang AG, Yen MY. Systemic lupus erythematosus-associated optic neuritis: clinical experience and literature review. Acta Ophthalmol 2009;87(2):204–210. doi: 10.1111/j.1755-3768.2008.01193.x. [DOI] [PubMed] [Google Scholar]
- 68.Asgari N, Jarius S, Laustrup H, et al. Aquaporin-4-autoimmunity in patients with systemic lupus erythematosus: a predominantly population-based study. Mult Scler 2018;24(3):331–339. doi: 10.1177/1352458517699791. [DOI] [PubMed] [Google Scholar]
- 69.Cross SA, Salomao DR, Parisi JE, et al. Paraneoplastic autoimmune optic neuritis with retinitis defined by CRMP-5-IgG. Ann Neurol 2003;54(1):38–50. doi: 10.1002/ana.10587. [DOI] [PubMed] [Google Scholar]
- 70.Jarius S, Wandinger KP, Borowski K, et al. Antibodies to CV2/CRMP5 in neuromyelitis optica-like disease: case report and review of the literature. Clin Neurol Neurosurg 2012;114(4): 331–335. doi: 10.1016/j.clineuro.2011.10.048. [DOI] [PubMed] [Google Scholar]
- 71.Igarashi N, Sawamura H, Kaburaki T, Aihara M. Anti-collapsing response-mediating protein-5 antibody-positive paraneoplastic perioptic neuritis without typical neurological symptoms. Neuroophthalmology 2017;41(1):24–29. doi: 10.1080/01658107.2016.1241283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gordon L, Dinkin M. Paraneoplastic syndromes in neuro-ophthalmology. Continuum (Minneap Minn) 2019;25(5, Neuro-ophthalmology):1401–1421. [DOI] [PubMed] [Google Scholar]
- 73.Baughman RP, Weiss KL, Golnik KC. Neuro-ophthalmic sarcoidosis. Eye Brain 2012;4:13–25. doi: 10.2147/EB.S29401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koczman JJ, Rouleau J, Gaunt M, et al. Neuro-ophthalmic sarcoidosis: the University of Iowa experience. Semin Ophthalmol 2008;23(3): 157–168. doi: 10.1080/08820530802007382. [DOI] [PubMed] [Google Scholar]
- 75.Caplan L, Corbett J, Goodwin J, et al. Neuro-ophthalmologic signs in the angiitic form of neurosarcoidosis. Neurology 1983;33(9): 1130–1135. doi: 10.1212/WNL.33.9.1130. [DOI] [PubMed] [Google Scholar]
- 76.Bridel C, Courvoisier DS, Vuilleumier N, Lalive PH. Cerebrospinal fluid angiotensin-converting enzyme for diagnosis of neurosarcoidosis. J Neuroimmunol 2015;285:1–3. doi: 10.1016/j.jneuroim.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 77.Leavitt RY, Fauci AS, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum 1990;33(8):1101–1107. doi: 10.1002/art.1780330807. [DOI] [PubMed] [Google Scholar]
- 78.Kubaisi B, Abu Samra K, Foster CS. Granulomatosis with polyangiitis (Wegener’s disease): an updated review of ocular disease manifestations. Intractable Rare Dis Res 2016;5(2):61–69. doi: 10.5582/irdr.2016.01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ahmed M, Niffenegger JH, Jakobiec FA, et al. Diagnosis of limited ophthalmic Wegener granulomatosis: distinctive pathologic features with ANCA test confirmation. Int Ophthalmol 2008;28(1):35–46. doi: 10.1007/s10792-007-9109-y. [DOI] [PubMed] [Google Scholar]
- 80.Nishino H, Rubino FA, DeRemee RA, et al. Neurological involvement in Wegener’s granulomatosis: an analysis of 324 consecutive patients at the Mayo Clinic. Ann Neurol 1993;33(1): 4–9. doi: 10.1002/ana.410330103. [DOI] [PubMed] [Google Scholar]
- 81.Niskopoulou M, Du Toit N. Optic neuritis as a feature of Wegener’s granulomatosis. Eye 2002; 16(3):320–321. doi: 10.1038/sj/eye/6700096. [DOI] [PubMed] [Google Scholar]
- 82.Takazawa T, Ikeda K, Nagaoka T, et al. Wegener granulomatosis-associated optic perineuritis. Orbit 2014;33(1):13–16. doi: 10.3109/01676830.2013.841716. [DOI] [PubMed] [Google Scholar]
- 83.Abdelhakim A, Rasool N. Neuroretinitis: a review. Curr Opin Ophthalmol 2018;29(6):514–519. doi: 10.1097/ICU.0000000000000527. [DOI] [PubMed] [Google Scholar]
- 84.Purvin V, Sundaram S, Kawasaki A. Neuroretinitis: review of the literature and new observations. J Neuroophthalmol 2011;31(1):58–68. doi: 10.1097/WNO.0b013e31820cf78a. [DOI] [PubMed] [Google Scholar]
- 85.Finger ML, Borruat FX. Dynamics of intraretinal fluid accumulation evidenced by SD-OCT in a case of cat scratch neuroretinitis. Eye (Lond) 2014;28(6):770–771. doi: 10.1038/eye.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schmalfuss IM, Dean CW, Sistrom C, Bhatti MT. Optic neuropathy secondary to cat scratch disease: distinguishing MR imaging features from other types of optic neuropathies. AJNR Am J Neuroradiol 2005;26(6):1310–1316. [PMC free article] [PubMed] [Google Scholar]
- 87.Sibony P, Halperin J, Coyle PK, Patel K. Reactive Lyme serology in optic neuritis. J Neuroophthalmol 2005;25(2):71–82. doi: 10.1097/01.WNO.0000166060.35366.70. [DOI] [PubMed] [Google Scholar]
- 88.Träisk F, Lindquist L. Optic nerve involvement in Lyme disease. Curr Opin Ophthalmol 2012;23(6): 485–490. doi: 10.1097/ICU.0b013e328358b1eb. [DOI] [PubMed] [Google Scholar]
- 89.Djukic M, Schmidt-Samoa C, Lange P, et al. Cerebrospinal fluid findings in adults with acute Lyme neuroborreliosis. J Neurol 2012;259(4): 630–636. doi: 10.1007/s00415-011-6221-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stanek G, Wormser GP, Gray J, Strle F. Lyme borreliosis. Lancet 2012;379(9814):461–473. doi: 10.1016/S0140-6736(11)60103-7. [DOI] [PubMed] [Google Scholar]
- 91.Smith GT, Goldmeier D, Migdal C. Neurosyphilis with optic neuritis: an update. Postgrad Med J 2006;82(963):36–39. doi: 10.1136/pgmj.2004.020875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Northey LC, Skalicky SE, Gurbaxani A, McCluskey PJ. Syphilitic uveitis and optic neuritis in Sydney, Australia. Br J Ophthalmol 2015;99(9):1215–1219. doi: 10.1136/bjophthalmol-2014-306168. [DOI] [PubMed] [Google Scholar]
- 93.Parker SE, Pula JH. Neurosyphilis presenting as asymptomatic optic perineuritis. Case Rep Ophthalmol Med 2012;2012:621872. doi: 10.1155/2012/621872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Frohman L, Wolansky L. Magnetic resonance imaging of syphilitic optic neuritis/perineuritis. J Neuroophthalmol 1997;17(1):57–59. [PubMed] [Google Scholar]
- 95.Nagappa M, Sinha S, Taly AB, et al. Neurosyphilis: MRI features and their phenotypic correlation in a cohort of 35 patients from a tertiary care university hospital. Neuroradiology 2013;55(4): 379–388. doi: 10.1007/s00234-012-1017-9. [DOI] [PubMed] [Google Scholar]
- 96.Kahloun R, Abroug N, Ksiaa I, et al. Infectious optic neuropathies: a clinical update. Eye Brain 2015;7:59–81. doi: 10.2147/EB.S69173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Davis EJ, Rathinam SR, Okada AA, et al. Clinical spectrum of tuberculous optic neuropathy. J Ophthalmic Inflamm Infect 2012;2(4):183–189. doi: 10.1007/s12348-012-0079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ryu WY, Kim JS. Optic perineuritis simultaneously associated with active pulmonary tuberculosis without intraocular tuberculosis. Int J Ophthalmol 2017;10(9):1477–1478. doi: 10.18240/ijo.2017.09.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Trivedi R, Saksena S, Gupta RK. Magnetic resonance imaging in central nervous system tuberculosis. Indian J Radiol Imaging 2009;19(4): 256–265. doi: 10.4103/0971-3026.57205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rock RB, Olin M, Baker CA, et al. Central nervous system tuberculosis: pathogenesis and clinical aspects. Clin Microbiol Rev 2008;21(2):243–261, table of contents. doi: 10.1128/CMR.00042-07. [DOI] [PMC free article] [PubMed] [Google Scholar]