

Highlights
-
•
A HPLC-MS/MS method for analysis of 6-O-demethylmenisporphine in biomatrices was firstly developed.
-
•
The HPLC-MS/MS method was validated as FDA guidelines.
-
•
First report on the in vivo process of 6-O-demethylmenisporphine.
-
•
The oral bioavailability of 6-O-demethylmenisporphine was up to 51.52%.
Keywords: 6-O-demethylmenisporphine, Pharmacokinetics, Tissue distribution, Excretion, Menispermi Rhizoma, HPLC-MS/MS
Abstract
6-O-demethylmenisporphine, a major active oxoisoaporphine alkaloid isolated from Menispermi Rhizoma, has been confirmed to possess significant bioactivities, including anti-cancer and anti-hypoxia effects. However, few researches on quantifying 6-O-demethylmenisporphine in biosamples have been performed. In this research, a sensitive HPLC-MS/MS approach for determining 6-O-demethylmenisporphine in various biological matrices (plasma, tissue, urine, bile and feces) of rat has been constructed. Carbamazepine was chosen as the internal standard (IS). All biosamples were prepared using a simple one-step acetonitrile precipitation. A Capcell Pak C18 column coupled with an isocratic mobile phase consisted of acetonitrile (0.1% formic acid)-water (90:10, v/v), was employed to separate 6-O-demethylmenisporphine from endogenous interferences. Peak responses were detected by multiple reaction monitoring (MRM) transitions with m/z 308.0 → 264.9 for 6-O-demethylmenisporphine and m/z 237.0 → 194.1 for IS in positive-ion mode. The approach exhibited perfect linearity over a range of 5–2000 ng/mL for plasma and 2–1000 ng/mL for various tissue, urine, bile and feces. The lower limit of quantification (LLOQ) for analyte among different biological samples ranged from 2 ng/mL to 5 ng/mL. The newly established method was simple, efficient and sensitive, which was successfully applied to investigate the absorption, distribution, and excretion of 6-O-demethylmenisporphine after oral dosing to rats. The results indicated that 6-O-demethylmenisporphine could be well absorbed into blood circulation and widely distributed in various tissues after oral dosing, the oral bioavailability was up to 51.52%. Meanwhile, it was widely metabolized in vivo and eliminated as the metabolites, the unconverted form was excreted mainly by feces route. The bioavailability, tissue distribution and excretion characteristics of 6-O-demethylmenisporphine were firstly revealed, which will provide references for further development of 6-O-demethylmenisporphine as an anti-tumor drug candidate.
1. Introduction
Traditional Chinese medicine (TCM) enjoys a long history of preventing and treating diseases. It has attracted much attention and become increasingly popular in many countries due to its unique advantages such as low prices, high safety and efficacy. Based on these benefits, more and more people have come to prefer to use TCM. Recently, some TCMs, such as Qingfeipaidu soup, Jinhuaqinggan granules, and Lianhuaqingwen capsule, etc, have been widely used to treat Corona Virus Disease 2019 (COVID-19) and achieved good clinical efficacy. Because of the complex nature of multiple components contained in TCM, it is necessary and urgent to explore the in vivo dynamic changes of bioactive single-components for elucidating curative mechanism. Menispermi Rhizoma (Chinese name: beidougen), deriving from the rhizomes of Menispermum dauricum DC. (Menispermaceae), is one of the famous TCMs with widely usage in Asia. It was used clinically to treat various diseases including lumbago, scrofula, diarrhea, dysentery and rheumatic diseases [1]. Modern pharmacological investigations displayed that Menispermi Rhizoma possesses multiple pharmacological effects like anti-oxidant [2], anti-bacterial [3], anti-inflammatory [4], [5], anti-hypoxia [6], protection against ischemia/reperfusion injury [7], [8], anti-arrhythmia [9], anti-thrombosis [10], and anti-tumor activities [11]. All these biological activities of Menispermi Rhizoma could be attributed to the active alkaloids belonging to categories bisbenzylisoquinolines, morphinanes, protoberberines and oxoisoaporphines [12]. In the past decades, many researches have mainly focused on bisbenzylisoquinoline alkaloids, whereas the researches on oxoisoaporphine alkaloids are scarce.
6-O-demethylmenisporphine is a major bioactive oxoisoaporphine alkaloid firstly isolated from Menispermi Rhizoma [13]. Oxoisoaporphine alkaloids are one type of alkaloids with unique structure, which have attracted much attention of researchers and exhibited numerous pharmacological activities including anti-tumor [14], [15], [16], anti-oxidant and anti-hypoxia [2], [6], [17], telomerase inhibitory [18], cholinesterase and β-amyloid (Aβ) inhibitory [19], [20]. 6-O-demethylmenisporphine is a typical oxoisoaporphine alkaloid with relatively high content among oxoisoaporphine alkaloids from Menispermi Rhizoma, which shows similar biological activities with oxoisoaporphine alkaloid, like anti-tumor and anti-hypoxia. It has been reported that 6-O-demethylmenisporphine displayed anti-tumor properties against diverse cancer cells such as MCF-7, H-460, HT-29 and CEM cells [16]. In addition, our previous study showed that 6-O-demethylmenisporphine could remarkably protect EAhy 926 endothelial cells from hypoxia-induced injury [6]. Based on the above significant pharmacological activities, 6-O-demethylmenisporphine could be a potential candidate drug for treating cancer.
Despite the fact that many candidate drugs exhibit obvious pharmacological activities in vitro, they might fail due to their poor bioavailability or instability in vivo. Hence, pharmacokinetic properties in vivo play a crucial role in the success of a drug candidate, which not only provide valuable information of efficacy and toxicity, but also interpret the major target sites and disposition of drug [21]. So far, the pharmacokinetic properties of 6-O-demethylmenisporphine are lacking. In order to better understand its pharmacological effects, it is crucial to establish a sensitive method for investigating the bioavailability, tissue distribution and disposition of 6-O-demethylmenisporphine.
Currently, several analytical approaches like HPLC-UV and UPLC-MS/MS have been employed to quantify 6-O-demethylmenisporphine in Menispermi Rhizoma [22], [23]. However, the quantification of 6-O-demethylmenisporphine in biological matrices (various tissue, urine, bile and feces) has not been published. Taken into full consideration of MS advantages including good separation efficiency, high sensitivity and selectivity, an efficient HPLC-MS/MS approach was established for determining 6-O-demethylmenisporphine in biological samples in our study. It was fully validated and successfully applied to explore the pharmacokinetics, bioavailability, tissue distribution and excretion patterns of 6-O-demethylmenisporphine following oral (p.o.) and intravenous (i.v.) dosing to rats. To our knowledge, it was the first report on the exploration of bioavailability, distribution, and excretion characteristics of 6-O-demethylmenisporphine. The outcome would not only lay theoretical basis for elucidating the pharmacodynamic effects, target organs and druggability, but also provide a reference for the evaluation of preclinical safety of 6-O-demethylmenisporphine.
2. Materials and methods
2.1. Materials
6-O-demethylmenisporphine was isolated from Menispermi Rhizoma in our laboratory. HPLC-DAD and HPLC-MS analysis showed that the purity percentages of 6-O-demethylmenisporphine was 98.10%. The raw material was obtained from Anguo medicine market (Hebei province, China), which was authenticated by Professor Tianxiang Li (Pharmacognosy Department of Tianjin University of Traditional Chinese Medicine). A voucher specimen (No. 20190701) has been stored in the School of Chinese Materia Medica, Tianjin University of Traditional Chinese Medicine. The separation, purification and identification methods of 6-O-demethylmenisporphine as well as its spectral data have been published by author [6]. The structure (Fig. 1 ) of 6-O-demethylmenisporphine was confirmed based on MS and NMR spectral data, as well as comparison with the reported literature data [13]. Carbamazepine with a purity of 99.00% (internal standard, batch No. 100142–201706) was obtained from China Food and Drug Inspection and Research Institute (Beijing, China). Tetrahydrofuran and formic acid of analytical grade were purchased from Sigma-Aldrich (USA). Dimethyl sulfoxide and polyethylene glycol 400 were obtained from Sinopharm Chemical Reagent Co., Ltd. (Beijing, China). Analytical grade methanol and acetonitrile were purchased from Merck (Germany). Water (HPLC-grade) was purified by SZ97 ultrapure water preparation device (Shanghai, China).
Fig. 1.

Chemical structures and product ion mass spectra of 6-O-demethylmenisporphine (A) and carbamazepine (B; IS).
2.2. HPLC-MS/MS conditions
Quantification of 6-O-demethylmenisporphine in the biological samples was performed using an Agilent 1200 series high performance liquid chromatography-tandem an Agilent 6410B triple quadrupole mass spectrometer (Agilent Technologies, USA). A Capcell Pak C18 column (150 mm × 4.6 mm, i.d., 5 μm) compatible with a column temperature of 40 °C was employed to separate 6-O-demethylmenisporphine from complex biomatrices. Injection volume was 5 μL. Elution was fulfilled with an isocratic mobile phase (delivered at 0.5 mL/min) consisting of acetonitrile (0.1% formic acid)-water (90:10, v/v). The auto-sampler was maintained at 4 °C. 6-O-demethylmenisporphine and internal standard (IS) were quantified via positive electrospray ionization interface. They were monitored by multiple reaction monitoring (MRM) mode with the transitions of m/z 308.0 → 264.9 for 6-O-demethylmenisporphine and m/z 237.0 → 194.1 for IS. The compound-dependent parameters such as collision energy and fragmentor voltage were optimized as 36 eV and 150 V for 6-O-demethylmenisporphine and 26 eV and 130 V for IS, respectively. Dwell time for each ion pair was 50 ms. High purity nitrogen served as both nebulizing and drying gas. The optimal mass spectrometric parameters were obtained as follows: the flow rate of drying gas was 11 L/min; gas temperature was kept at 300 °C; and nebulizer press and capillary voltage were maintained at 30 psi and 4 kV, respectively. Chromatograms were acquired and integrated by MassHunter workstation.
2.3. Standard and sample preparation
Primary standard solution of 6-O-demethylmenisporphine was obtained by dissolving 5 mg of 6-O-demethylmenisporphine in 3 mL of tetrahydrofuran and diluted with methanol to achieve 0.1 mg/mL. Working solutions (2–2000 ng/mL) of 6-O-demethylmenisporphine were made by stepwise dilution with methanol. Carbamazepine (IS) primary solution with a concentration of 0.1 mg/mL was prepared in a similar manner, then it was diluted with methanol to get 100 ng/mL IS working solution. All solutions were stored at 4 °C.
Standard curves of 6-O-demethylmenisporphine were achieved by evaporating 50 μL working solutions with a gentle stream of N2 at 35 °C and then adding 50 μL blank biological matrix to yield concentrations of 5.0, 10, 25, 100, 500, 1000 and 2000 ng/mL for pharmacokinetic study, 2.0, 5.0, 10, 25, 100, 500 and 1000 ng/mL for tissue distribution and excretion study, respectively. Quality control (QC) samples were prepared following the same processing procedure listed above at the concentrations of 5.0, 15, 150 and 1600 ng/mL for pharmacokinetic study and 2.0, 6.0, 80 and 800 ng/mL for tissue distribution and excretion study.
2.4. Sample pretreatment
In our research, all biological samples were frozen at −80 °C. Samples were taken out and thawed at ambient temperature before analysis. After thawing, they were processed with a simple and efficient protein precipitation procedure (PPT).
2.4.1. Preparation of plasma samples
Firstly, 20 μL IS working solution was transferred into a clean glass tube and dried with N2 (35 °C), then 50 μL of rat plasma and 20 μL of 30% formic acid (v/v) in water were added. After shaking for 1 min, the sample was deproteinized with 250 μL acetonitrile, and vortex-mixed for 3 min. Then, 270 μL of supernatant was harvested after centrifuging for 10 min at 12,000 g, which was further evaporated with N2 (35 °C). Dried residue was dissolved in 100 μL acetonitrile–water solution (9:1, v/v). The mixture was vortexed for 1 min and centrifugated for 10 min at 12,000 g. Finally, 80 μL of supernatant was pipetted into an inner lining-pipe inserted in the autosampler vial, and 5 μL aliquot was applied for HPLC-MS/MS analysis.
2.4.2. Preparation of tissue samples
After thawing the tissue samples, approximately 0.1 g tissue (get all if below 0.1 g) was put into a 2 mL Eppendorf tube and further homogenized in methanol (1:4, w/v). Then, protein precipitation was performed by mixing 50 μL of tissue homogenate with 250 μL acetonitrile. The detailed processing procedure was the same as described in “Section 2.4.1.”.
2.4.3. Preparation of urine, bile and feces samples
For bile and urine samples, 50 μL of bile or urine was taken out and processed in a similar procedure as described in “Section 2.4.1.”. For feces samples, a pestle and mortar were used to crush the fecal samples, and 0.1 g of powdered sample was homogenized with methanol (1:5, w/v). Then 50 μL of homogenized sample was processed in the same way as presented in “Section 2.4.1.”
If the concentrations of plasma and fecal homogenate exceeded the range of calibration curve in the whole process of analysis, these samples could be reanalyzed by moderately dilution with the corresponding blank biological matrix.
2.5. Method validation
The parameters of established method were validated according to the guidelines of US Food and Drug Administration (FDA) [24]. In the process of analysis of tissue samples, the validation of the method was conducted using liver as the representative tissue.
2.5.1. Specificity
Specificity was demonstrated by analyzing blank biosamples derived from six individuals, spiked biological samples at lower limit of quantification (LLOQ) concentration and actual biosamples following oral administration of 6-O-demethylmenisporphine to exclude the influences from endogenous interferences. In light of FDA guidelines, there should be no endogenous responses in the retention position of 6-O-demethylmenisporphine and IS for blank biological samples. Carryover was assessed by injecting extracted blank biological samples in six replicates after analysis of an upper limit of quantitation (ULOQ) sample, which could be neglected when the responses in the blank bio-matrices were below 20% of LLOQ.
2.5.2. Linearity and sensitivity
The linearity was tested by analyzing the calibration curve in duplicate on three consecutive days. Using weighted (1/x 2) linear regression, a calibration curve was generated from the peak area ratios of 6-O-demethylmenisporphine and IS to the corresponding concentration of 6-O-demethylmenisporphine, and the coefficient (r) of which should be above 0.99. Limit of detection (LOD), depicted as the concentration of analyte giving a signal to noise ratio of 3. LLOQ, depicted as the concentration of 6-O-demethylmenisporphine giving a S/N ratio above 10, could be evaluated by six replicate analyses with a percentage relative standard deviation (precision) below 20% and a percentage relative error (accuracy) not exceeding ± 20%.
2.5.3. Accuracy and precision
For intra-assay variations, QC samples (n = 6) at four levels (LLOQ and three QC levels) were determined on the same day. For inter-assay variations, the determination of 6-O-demethylmenisporphine QC samples (n = 6) at four levels were performed on three consecutive days. Precision, expressed as RSD% (relative standard deviation), was expected to be less than 15% while accuracy, expressed as RE% (deviation from the true value), the criteria of which was within ± 15%. For LLOQ, the RSD% and RE% should not exceed ± 20%.
2.5.4. Recovery and matrix effect
Six replicates of QC samples at three levels were analyzed to assess recovery and matrix effect of 6-O-demethylmenisporphine in various biological samples. The extraction recovery for analyte was estimated by the peak response ratios of analyte in extracted QC samples to those of it in the samples spiked post-extraction blank biological matrix at equivalent levels. The matrix effect was carried out by comparing mean peak responses of analyte in the spiked samples (post-extraction) with those of it contained in the standard solutions at the corresponding three levels. As described above, the recovery and matrix effect of carbamazepine at a concentration of 40 ng/mL were measured in a similar way as the analyte.
2.5.5. Stability
The measurements of stability were conducted to assess the stability of biological samples exposed to different storage and handling conditions. For each condition, three levels of QC samples (n = 6) were analyzed. For short-term stability, QC samples exposed to room temperature for 8 h were tested; for long-term stability, QC samples stored at –80 °C in a freezer for two weeks were determined; for freeze–thaw cycles stability, QC samples which underwent three freeze–thaw cycles were investigated. Post-preparative stability was determined by keeping extracted QC sample in an auto-sampler at 4 °C for 12 h. An acceptance criterion for all stability samples didn’t exceed ± 15%. The standard solutions stability for 6-O-demethylmenisporphine and IS were evaluated by determining the solutions which were kept at 4 °C for one month.
2.5.6. Dilution integrity
Dilution procedure might be required when the concentrations of analyte in biological matrices were higher than ULOQ. In order to validate the dilution integrity, a dilution investigation was performed with plasma sample concentration of 10 μg/mL and feces homogenate concentration of 15 μg/mL, which would be diluted 20-fold with corresponding blank biological matrix to be within the range of calibration curve (n = 6). The results of RSD% and RE% should not exceed ± 15%.
2.6. Animal experiments and sampling
SPF grade Sprague-Dawley (SD) rats (male, weighting from 200 to 240 g) were chosen as the experimental animals, which were purchased from Tianjin Aochen Laboratory Animal Co., Ltd (Tianjin, China). Rats were kept in an air-conditioned laboratory (22–26 °C, 40–60% humidity) with a 12 h dark/light cycle and acclimatized in the above conditions for 7 days. In addition, all of the experimental rats were starved overnight to exclude the dietary disturbances, yet had free access to water before administration. Animal Ethics Committee of Tianjin University of Traditional Chinese Medicine has approved the experimental protocols of this research. In the light of institutional Guidelines for Animal Care and Use, animal researches were conducted in strict accordance with standard operating procedures. According to the literature, the dosage form of 6-O-demethylmenisporphine for p.o. and i.v. administration was prepared by dissolving a certain amount of 6-O-demethylmenisporphine in the solution consisting of dimethyl sulphoxide, polyethylene glycol-400 and water at a ratio of 5:45:50 (v/v) [21].
For pharmacokinetics researches, twelve SD rats were stochasticly assigned to two groups (6 rats each group). One group was intravenously administered the 6-O-demethylmenisporphine solution at 0.1 mg/kg, the other group was orally given with 6-O-demethylmenisporphine at 1 mg/kg. For intravenous dosing, the drug was delivered into the tail vein using a disposable syringe; for oral administration, the drug was injected into stomach using a gastric lavage needle. Following oral administration (p.o. 1 mg/kg) and intravenous injection (i.v. 0.1 mg/kg), 250 μL of blood samples from eye postorbital venin were collected in the sodium heparin-coated vials at designated time points (0 (pre-dosing), 5, 10, 20, 45 min and 1, 2, 3, 4, 5, 8, 12, 24, 36, 48 h), and then immediately centrifuged (12,000 g, 10 min). Plasma (100 μL) was harvested into a new vial and frozen (−80 °C) before use.
As for the tissue distribution researches, thirty SD rats were assigned to five groups at random (n = 6 each group). The first group rats were euthanized by decapitation to obtain the blank tissue samples prior to administration. The remaining four groups were sacrificed after collecting blood through abdominal aorta at 0.75, 2.5, 10 and 24 h time points following oral dosing of 6-O-demethylmenisporphine at 1 mg/kg. Afterwards, tissue samples including spleen, liver, heart, kidney, lung, brain, stomach, muscle, small intestine and large intestine were immediately excised and flushed with cold sodium chloride solution (0.9%) so as to remove the impact of blood or chyme. Subsequently, all tissues were wiped dry with blotting paper and kept at − 80 °C.
In order to explore the excretion of orally given 6-O-demethylmenisporphine (1 mg/kg), six rats were employed for collecting urine and feces samples, which were individually fed in metabolism cages and allowed ad libitum to feed and water. Both feces and urine were harvested pre-dosing and at 0–2, 2–4, 4–6, 6–8, 8–12, 12–24 and 24–48 h after dosing. As for the bile collection, six rats were anesthetized with diethyl ether, then abdominal surgery was performed after disinfecting with 70% ethanol, bile duct catheterization was performed for collecting bile samples pre-dosing and at 0–2, 2–4, 4–6, 6–8, 8–12 and 12–24 h post-dosing. Finally, the weight of feces freeze-dried and the volumes of urine and bile for each time interval were recorded. All biosamples were frozen at − 80 °C.
2.7. Data processing
Non-compartmental pharmacokinetic analyses were carried out via the Drug and Statistics 3.2.7 program (Chinese Society of Pharmacology). Using this software, the pharmacokinetic parameters including area under the plasma concentration–time curve (AUC), elimination half-life (t 1/2), total body clearance (CLz), and mean residence time (MRT) were calculated, and the peak concentration (C max) and time-to-peak concentration (T max) were obtained directly from each individual set of data. The oral bioavailability of 6-O-demethylmenisporphine was calculated in accordance with the following formula:
3. Results and discussion
3.1. HPLC-MS/MS optimization
To get great chromatographic behavior and appropriate analysis speed, the chromatographic conditions were optimized. A Capcell Pak C18 column compatible with a slow flow rate (0.5 mL/min) was selected in this study. With the above column, the mobile phase compositions such as different types of organic phase and concentration of organic modifier, were tested to obtain the optimal chromatographic performance. It turned out that acetonitrile had more advantages over methanol in terms of shorter run time, lower background noise and higher signal intensity. Meanwhile, the sharper and more symmetrical peak occurred with organic phase containing 0.1% formic acid. Overall consideration of above conditions, an isocratic elution with acetonitrile (0.1% formic acid)-water (90:10, v/v) was chosen during the analysis.
Considering that MS conditions played a critical role in obtaining higher ion responses of 6-O-demethylmenisporphine and IS, electrospray ionization in positive and negative mode were tested. It was observed that both analyte and IS presented higher signal peak in positive ion mode due to the presence of nitrogen atom in their molecules. The precursor ions obtained for 6-O-demethylmenisporphine and IS were at m/z 308.0 and m/z 237.0, respectively. To get high selectivity and sensitivity for analysis, MRM mode was selected for quantification. And the precursor-to-product ions for MRM transitions were m/z 308.0 → 264.9 for 6-O-demethylmenisporphine and m/z 237.0 → 194.1 for IS. The typical mass spectra of 6-O-demethylmenisporphine and IS as well as their structures were presented in Fig. 1. The compound-dependent parameters like collision energy and fragmentor voltage were optimized by manual manipulation, the results were shown in “Section 2.2.”
3.2. Sample preparation
Sample processing is the key rate-limiting step of drug analysis in vivo. In our study, liquid–liquid extraction (LLE) and PPT were tried during sample processing. The results demonstrated that both PPT with acetonitrile and LLE with ethyl acetate offered a higher extraction recovery for analyte and IS. In comparison with the PPT, LLE provided cleaner extract and lower background. However, Overall consideration of high-throughput sample analysis in vivo and high sensitivity of MS, the simple and time-saving PPT approach became the final choice. The analyte is acidic due to the existence of associated phenolic hydroxyl group in the structure. Water containing different proportions of formic acid, acetic acid and hydrochloric acid were tested to improve the extraction yield of 6-O-demethylmenisporphine. As a result, formic acid can achieve the optimum recovery. Thus, 250 μL acetonitrile combined with 20 μL of 30% formic acid was used to precipitate protein and improve process efficiency. In addition, different solvents (methanol, water, methanol–water (1:1, v/v)) were investigated to homogenize the tissues during the tissue processing. As a result, methanol gave the highest extraction efficiency. Hence, all tissues were homogenized in methanol (1:4, w/v).
3.3. Selection of internal standard
A proper IS plays an important role in biopharmaceutical analysis. In this study, a series of structurally similar analogs of 6-O-demethylmenisporphine such as carbamazepine, dauriporphinoline, dauriporphine and menisporphine were tested as IS. Among them, dauriporphinoline and dauriporphine could be eluted later than the analyte, which extended the analysis time of each sample. Under the above HPLC-MS/MS conditions, it can’t separate the peaks of menisporphine and analyte. Thus, carbamazepine was finally selected as the IS for its appropriate chromatographic behavior and extraction recovery.
3.4. Method validation
3.4.1. Specificity
Fig. 2, Fig. 3 illustrate typical chromatograms of 6-O-demethylmenisporphine and IS derived from blank rat biosamples (plasma, liver homogenate, urine, bile and feces), QC samples and actual biosamples. From the above figure we can see that both analyte and IS displayed well-separated peaks with no obvious interfering peaks found in their chromatographic region (retention time: 6.1 min for 6-O-demethylmenisporphine, 3.6 min for IS).
Fig. 2.

Representative MRM chromatograms of 6-O-demethylmenisporphine (a) and carbamazepine (b; IS): (A) blank rat plasma, (B) blank plasma spiked with 6-O-demethylmenisporphine at LLOQ (5 ng/mL) and IS (40 ng/mL), (C) real plasma collected at 2 h after oral administration of 1 mg/kg 6-O-demethylmenisporphine, (D) real plasma collected at 2 h after intravenous administration of 0.1 mg/kg 6-O-demethylmenisporphine.
Fig. 3.

Representative MRM chromatograms of 6-O-demethylmenisporphine (a) and carbamazepine (b; IS): (A) blank rat liver; (B) blank liver spiked with 6-O-demethylmenisporphine at LLOQ (2 ng/mL) and IS (40 ng/mL); (C) liver sample 10 h after oral administration of 1 mg/kg 6-O-demethylmenisporphine; (D) urine sample, (E) bile sample and (F) feces sample 2–4 h after oral administration of 1 mg/kg 6-O-demethylmenisporphine.
Following the analysis of an ULOQ sample, no significant signals of analyte and IS were found in the blank rat matrix, demonstrating that carryover from late-eluting residues could be neglected.
3.4.2. Linearity and sensitivity
A summary of calibration curves in all biological samples was listed in Table 1 , all calibration curves displayed perfect linearity (r ≥ 0.99) over corresponding linearity range. The LODs for 6-O-demethylmenisporphine were 1.5, 0.6, 0.6, 0.6, 0.6 ng/mL in plasma, tissue, urine, bile and feces sample, respectively. The LLOQs of 6-O-demethylmenisporphine were as low as 5 ng/mL in plasma as well as 2 ng/mL in various organs, urine, bile and feces samples, respectively, which provide a sufficient sensitivity to meet the requirements of in vivo bioanalysis of 6-O-demethylmenisporphine.
Table 1.
Calibration curves of 6-O-demethylmenisporphine in different biological samples.
| Biosamples | Calibration curves | Correlation coefficient (r) | Linear range (ng/mL) | LLOQs (ng/mL) |
|---|---|---|---|---|
| Plasma | y = 0.1915 x + 0.0084 | 0.9932 | 5–2000 | 5 |
| Heart | y = 1.0197 x + 0.0462 | 0.9929 | 2–1000 | 2 |
| Liver | y = 0.3023 x + 0.0029 | 0.9910 | 2–1000 | 2 |
| Spleen | y = 0.4697 x + 0.0346 | 0.9926 | 2–1000 | 2 |
| Lung | y = 0.4821 x + 0.0089 | 0.9956 | 2–1000 | 2 |
| Kidney | y = 0.3693 x + 0.0853 | 0.9914 | 2–1000 | 2 |
| Brain | y = 0.7218 x + 0.0426 | 0.9904 | 2–1000 | 2 |
| Stomach | y = 1.0084 x + 0.2472 | 0.9937 | 2–1000 | 2 |
| Muscle | y = 0.4962 x + 0.0899 | 0.9940 | 2–1000 | 2 |
| Small Intestine | y = 0.5257 x + 0.0241 | 0.9910 | 2–1000 | 2 |
| Large Intestine | y = 0.6548 x + 0.0171 | 0.9900 | 2–1000 | 2 |
| Urine | y = 0.6006 x + 0.0369 | 0.9996 | 2–1000 | 2 |
| Bile | y = 0.5496 x + 0.0738 | 0.9921 | 2–1000 | 2 |
| Feces | y = 2.3760 x + 0.1622 | 0.9919 | 2–1000 | 2 |
3.4.3. Accuracy and precision
Table 2 summarizes intra-assay and inter-assay variations of 6-O-demethylmenisporphine at four concentrations (LLOQ and three QC levels) in all the tested biomatrices. The RE values of accuracy ranged from – 10.9% to 13.0%, while the RSD values of precision ranged between 3.5% and 11.1%. The above results were all in the acceptable limits, indicating that the approach exhibited satisfactory reproducibility for quantification of 6-O-demethylmenisporphine in all biosamples.
Table 2.
Precision and accuracy for HPLC-MS/MS analysis of 6-O-demethylmenisporphine in rat plasma, liver, urine, bile and feces (n = 6).
| Biosamples | QC concentration (ng/mL) | Intra-day | Inter-day | ||
|---|---|---|---|---|---|
| Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | Accuracy (RE, %) | ||
| Plasma | 5 | 8.2 | −8.5 | 9.5 | −5.6 |
| 15 | 6.2 | −2.0 | 9.1 | −2.6 | |
| 150 | 7.1 | −3.8 | 8.4 | −9.1 | |
| 1600 | 3.6 | −2.8 | 10.1 | −7.9 | |
| Liver | 2 | 3.5 | −5.4 | 7.2 | −6.4 |
| 6 | 6.3 | 10.7 | 8.2 | 5.4 | |
| 80 | 3.5 | −10.2 | 5.4 | −8.0 | |
| 800 | 5.4 | 5.5 | 8.4 | −10.9 | |
| Urine | 2 | 7.9 | −8.2 | 8.3 | −1.5 |
| 6 | 10.0 | 7.7 | 9.8 | 8.9 | |
| 80 | 6.7 | −6.1 | 7.0 | −5.0 | |
| 800 | 5.4 | −6.5 | 5.9 | −7.0 | |
| Bile | 2 | 9.0 | −4.3 | 8.5 | 5.7 |
| 6 | 8.7 | −1.9 | 11.0 | 4.1 | |
| 80 | 5.4 | −3.2 | 6.2 | −7.0 | |
| 800 | 6.8 | 5.4 | 7.6 | 11.2 | |
| Feces | 2 | 7.6 | 8.9 | 9.7 | −5.5 |
| 6 | 11.1 | −10.9 | 9.0 | −10.5 | |
| 80 | 9.5 | 9.8 | 10.3 | 12.2 | |
| 800 | 7.7 | 8.6 | 9.0 | 13.0 | |
3.4.4. Extraction recovery and matrix effect
The average recoveries for 6-O-demethylmenisporphine at three QC levels in all biosamples (Table 3 ) were within 84.5–101.3%, which the RSD values were no more than 15% (4.5–13.6%). The matrix effects for 6-O-demethylmenisporphine (Table 3) were evaluated to be ranged between 86.7% and 110.9%. The mean recovery and matrix effect of IS (40 ng/mL in all biomatrices) were within 80.1%-90.9% and 85.9%-106.2%, respectively. All data revealed that the sample processing procedure adopted gave high extraction efficiency and the influence of ion from biomatrices could be neglected.
Table 3.
Extraction recovery and matrix effect of 6-O-demethylmenisporphine and IS in rat plasma, liver, urine, bile and feces (n = 6).
| Sample matrix | Analytes | QC concentration (ng/mL) | Recovery (%) | RSD (%) | Matrix effect (%) | RSD (%) |
|---|---|---|---|---|---|---|
| Plasma | 6-O-demethylmenisporphine | 15 | 88.4 ± 7.4 | 8.4 | 110.9 ± 3.6 | 3.2 |
| 150 | 85.5 ± 6.2 | 7.3 | 102.4 ± 5.5 | 5.4 | ||
| 1600 | 93.2 ± 6.1 | 6.5 | 109.3 ± 2.5 | 2.3 | ||
| IS | 40 | 80.1 ± 4.0 | 5.0 | 90.2 ± 3.8 | 4.2 | |
| Liver | 6-O-demethylmenisporphine | 6 | 91.5 ± 7.9 | 8.6 | 100.9 ± 12.7 | 12.6 |
| 80 | 88.2 ± 7.2 | 8.2 | 101.2 ± 6.7 | 6.6 | ||
| 800 | 92.4 ± 8.5 | 9.2 | 105.3 ± 6.7 | 6.4 | ||
| IS | 40 | 88.2 ± 7.9 | 9.0 | 106.2 ± 12.1 | 11.4 | |
| Urine | 6-O-demethylmenisporphine | 6 | 91.3 ± 12.0 | 13.1 | 105.5 ± 8.4 | 7.9 |
| 80 | 94.2 ± 5.2 | 5.5 | 98.3 ± 5.9 | 6.0 | ||
| 800 | 90.1 ± 6.8 | 7.5 | 95.7 ± 7.0 | 7.3 | ||
| IS | 40 | 90.9 ± 5.4 | 5.9 | 86.1 ± 6.3 | 7.3 | |
| Bile | 6-O-demethylmenisporphine | 6 | 97.6 ± 13.3 | 13.6 | 89.0 ± 12.6 | 14.1 |
| 80 | 85.9 ± 9.7 | 11.3 | 86.7 ± 11.3 | 13.0 | ||
| 800 | 89.9 ± 7.9 | 8.8 | 88.4 ± 9.0 | 10.2 | ||
| IS | 40 | 81.6 ± 11.7 | 14.4 | 85.9 ± 6.9 | 8.0 | |
| Feces | 6-O-demethylmenisporphine | 6 | 101.3 ± 9.7 | 9.5 | 90.3 ± 10.5 | 11.7 |
| 80 | 94.3 ± 8.9 | 9.4 | 93.1 ± 11.2 | 12.0 | ||
| 800 | 84.5 ± 3.8 | 4.5 | 87.8 ± 10.0 | 11.4 | ||
| IS | 40 | 80.2 ± 8.9 | 11.1 | 87.1 ± 11.2 | 12.9 |
3.4.5. Stability
As displayed in Table 4 , the stability results in different biomatrices suggest that 6-O-demethylmenisporphine was stable under all the evaluated conditions such as room temperature for 8 h, −80 °C for two weeks, three freeze–thaw cycles as well as 4 °C for 12 h. No obvious degradation of 6-O-demethylmenisporphine occurred throughout the whole analysis. The working solutions of 6-O-demethylmenisporphine and IS kept at 4 °C were evaluated to be stable for at least one month.
Table 4.
Stability results of 6-O-demethylmenisporphine in rat plasma, liver, urine, bile and feces under different conditions (n = 3).
| Sample matrix | QC concentration (ng/mL) | At room temperature for 8 h | Frozen (-80 °C) for two weeks | Three freeze–thaw cycles | Post-preparation stability | ||||
|---|---|---|---|---|---|---|---|---|---|
| (RSD, %) | (RE, %) | (RSD, %) | (RE, %) | (RSD, %) | (RE, %) | (RSD, %) | (RE, %) | ||
| Plasma | 15 | 3.2 | 2.6 | 5.6 | 4.6 | 6.4 | 4.3 | 2.1 | −5.9 |
| 150 | 5.4 | −5.7 | 3.9 | −6.3 | 4.9 | 6.2 | 5.3 | 3.9 | |
| 1600 | 5.5 | 6.9 | 8.4 | −5.4 | 7.1 | −9.4 | 7.6 | 7.0 | |
| Liver | 6 | 5.4 | 6.5 | 2.3 | −0.6 | 2.9 | −7.9 | 6.7 | 4.3 |
| 80 | 6.7 | 5.8 | 5.7 | 3.9 | 5.0 | −3.1 | 7.2 | −4.5 | |
| 800 | 3.2 | −3.5 | 4.0 | −6.4 | 3.2 | 4.5 | 5.9 | −7.8 | |
| Urine | 6 | 6.0 | −3.2 | 8.7 | −4.9 | 6.6 | −2.2 | 3.4 | −7.6 |
| 80 | 3.2 | 4.7 | 4.1 | −3.1 | 8.7 | 2.4 | 5.6 | −8.7 | |
| 800 | 4.5 | −6.0 | 2.1 | 1.9 | 6.5 | 7.6 | 5.8 | 5.4 | |
| Bile | 6 | 7.6 | −3.7 | 6.5 | −8.0 | 4.7 | −7.6 | 3.2 | −1.9 |
| 80 | 6.5 | −5.4 | 4.1 | −4.3 | 3.8 | 2.4 | 3.9 | −3.3 | |
| 800 | 4.3 | 3.1 | 1.9 | 6.0 | 2.9 | 3.5 | 5.8 | −5.7 | |
| Feces | 6 | 3.5 | −4.6 | 5.7 | −4.9 | 3.9 | 3.1 | 4.5 | −5.4 |
| 80 | 4.6 | 3.9 | 3.7 | −1.6 | 3.1 | −3.4 | 8.7 | 2.0 | |
| 800 | 2.1 | −9.0 | 8.7 | 3.5 | 4.5 | −5.1 | 5.4 | −8.7 | |
3.4.6. Dilution integrity
1/20 dilution samples were analyzed in six replicates. The RE values were in the range of − 5.2% to 8.3% with the RSD results less than 7.8%, which revealed that plasma and feces samples with concentrations exceeding ULOQ could be reanalyzed after appropriate dilution with corresponding blank bio-matrix.
3.5. Pharmacokinetic study
In the present study, following a single intravenous injection (0.1 mg/kg) and a single intragastric administration (1.0 mg/kg) of 6-O-demethylmenisporphine, the pharmacokinetic profiles of 6-O-demethylmenisporphine in rat plasma were monitored using the new developed HPLC-MS/MS approach. Fig. 4 presents the concentration versus time changes of 6-O-demethylmenisporphine in plasma. A summary of main non-compartment pharmacokinetic parameters obtained from the results of DAS data processing are displayed in Table 5 .
Fig. 4.
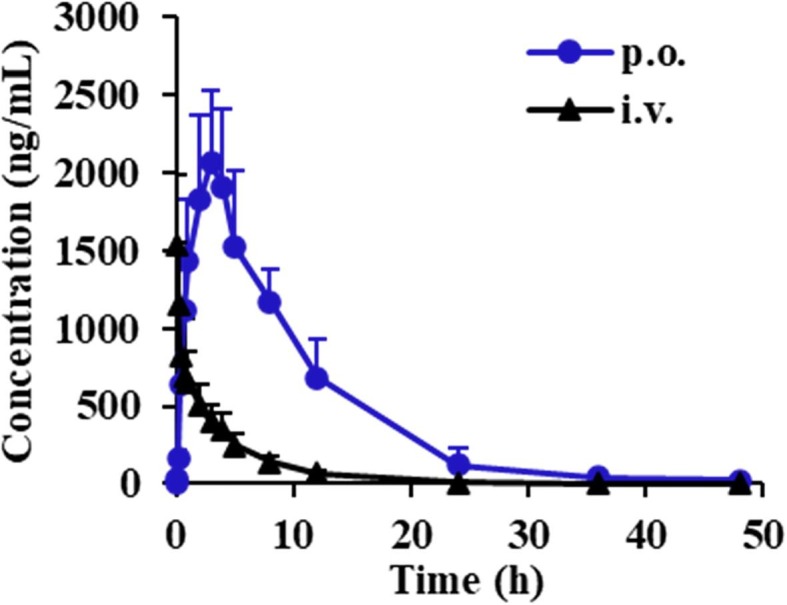
Plasma concentration–time profiles of 6-O-demethylmenisporphine in rats after given a single p.o. dose of 1.0 mg/kg and given i.v. dose of 0.1 mg/kg (n = 6).
Table 5.
Pharmacokinetic parameters of 6-O-demethylmenisporphine after a single dose of intragastric administration (1.0 mg/kg) or intravenous injection (0.1 mg/kg) in rats (n = 6).
| Parameters | Intragastric (Mean ± SD) | Intravenous (Mean ± SD) |
|---|---|---|
| Cmax (μg/L) | 2201.24 ± 412.34 | 1556.36 ± 450.21 |
| Tmax (h) | 2.80 ± 0.84 | 0.10 ± 0.03 |
| t1/2z (h) | 5.10 ± 2.91 | 4.52 ± 1.09 |
| AUC0-t (μg/L*h) | 21215.89 ± 6310.61 | 4117.86 ± 1032.26 |
| AUC0-∞ (μg/L*h) | 21506.06 ± 6017.22 | 4201.20 ± 1027.83 |
| CLz (L/h/kg) | – | 0.025 ± 0.01 |
| CLz/F (L/h/kg) | 0.05 ± 0.01 | – |
| MRT0-t (h) | 7.82 ± 2.15 | 4.71 ± 0.48 |
| MRT0-∞ (h) | 8.13 ± 1.92 | 5.28 ± 0.99 |
| Bioavailability (%) | 51.52% |
Cmax: maximum plasma concentration; Tmax: time to Cmax; t1/2: half-life; AUC0-t: area under the curve from 0 to last time; AUC0-∞: area under the curve from 0 to infinite time; CLz/F: apparent oral clearance; MRT: mean residence time.
As shown in Fig. 4, Fig. 6, 6-O-demethylmenisporphine exhibited an obvious single peak after p.o. dosing. The peak concentration (C max) was detected to be 2201.24 ± 412.34 ng/mL with the peak time at 2.80 ± 0.84 h, demonstrating that 6-O-demethylmenisporphine could be absorbed into blood circulation at a moderate speed. Based on the calculation formula in “Section 2.7″ as well as exposure levels AUC0-t (p.o. and i.v.), the oral bioavailability of 6-O-demethylmenisporphine was estimated as 51.52%, which means that 6-O-demethylmenisporphine could be well absorbed. We concluded that the relatively good oral bioavailability was closely related to physicochemical properties of 6-O-demethylmenisporphine, such as small molecular weight (below 500 Da), high lipophilicity (Log P = 2.33) and permeability. In addition, 6-O-demethylmenisporphine shows weak acidity due to the existence of phenolic hydroxyl group in molecular structure, the acidic environment of the stomach keeps 6-O-demethylmenisporphine in a free state post-dosing and further promotes the absorption of 6-O-demethylmenisporphine. In view of the elimination half-life (t 1/2) time of 5.10 ± 2.91 h and mean residence time (MRT0-∞) of 8.13 ± 1.92 h, 6-O-demethylmenisporphine was slowly eliminated after p.o. administration. The slow absorption and elimination process suggested that 6-O-demethylmenisporphine might possess a relatively long duration of action and exhibit better therapeutic effects. Thus, the compound might be developed into an oral drug.
Fig. 6.

Urinary, biliary and fecal cumulative excretion-time profiles of 6-O-demethylmenisporphine in rats after a single p.o. dose of 1.0 mg/kg (n = 6).
3.6. Tissue distribution study
Tissue distribution of 6-O-demethylmenisporphine after p.o. dosing to rats (1.0 mg/kg) was investigated by the validated HPLC-MS/MS method. The distribution tendency and concentrations in all the tested tissues at 0.75, 2.5, 10 and 24 h are shown in Fig. 5 .
Fig. 5.

Mean concentration of 6-O-demethylmenisporphine in rat tissues at 0.75, 2.5 10 and 24 h after a single p.o. dose of 1.0 mg/kg (n = 6).
It is reported that physicochemical properties of drug played a key role in the permeability from blood system to tissues [25]. As mentioned above, 6-O-demethylmenisporphine is a small molecule with high lipophilicity, which promotes its penetration through cell membrane. Hence, the results showed that 6-O-demethylmenisporphine went through rapid and widespread distribution to various organs after oral administration. The level of 6-O-demethylmenisporphine in gastrointestinal tract was higher at 0.75 h after dosing, which might be attributed to mode of oral dosing. Whereas, the highest level of 6-O-demethylmenisporphine in other tested tissues occurred at 2.5 h after dosing, which were consistent with the pharmacokinetic profiles, and then the concentration rapidly decreased within the following 2.5–24 h, suggesting that no apparent long-term accumulation was noticed in all tested tissues.
Among these tissues, the highest C max of 6-O-demethylmenisporphine was (1061.5 ± 86.7) ng/mL observed in kidney, followed by heart, lung, liver, muscle, spleen, brain orderly. The high concentration presence of 6-O-demethylmenisporphine in kidney revealed that kidney is the major target organ of 6-O-demethylmenisporphine, the specific pharmacological effect requires further research. It is reported that phenoic alkaloids (containing 6-O-demethylmenisporphine) from Menispermum dauricum have significant protective effect on myocardial injury induced by ischemia/reperfusion [7], which perhaps benefits from the accumulation of 6-O-demethylmenisporphine in the heart and brain. In addition, it was certified that 6-O-demethylmenisporphine exhibits clear anti-tumor activities against H-460 lung cancer cells [16]. In combination with relatively high level of distribution in lung tissue, 6-O-demethylmenisporphine could be a potential anti-lung cancer candidate. The distribution of 6-O-demethylmenisporphine in brain implied that it could penetrate the blood–brain barrier and play a potential therapeutic role in brain injury and nervous disorders, such as cerebral ischemia and Alzheimer’s disease. So far, oxoisoaporphine alkaloids have the dual effects of enhancing cholinergic neurotransmission and reducing AchE aggregation according to literature, which are expected to become anti-Alzheimer’s agent [26].
3.7. Excretion study
The newly established method was also used for quantifying 6-O-demethylmenisporphine in urine, bile and feces after p.o. dosing to rats (1.0 mg/kg). Fig. 6 presents the fecal, urinary and biliary cumulative excretion-time profiles of 6-O-demethylmenisporphine in rats. The data suggested that 22.6620% in total of the dose was recovered, the prototype drug accounts for 22.5641% of the dose in feces, 0.0882% in urine and 0.0097% in bile. Following oral administration, the cumulative excretion ratio of 6-O-demethylmenisporphine arrived the plateau at 12 h in feces, 24 h in urine and bile, respectively. Above results implied that 6-O-demethylmenisporphine might be widely metabolized by drug-metabolizing enzymes in vivo and mainly eliminated as the metabolites, the unconverted drug was mainly excreted in feces.
4. Conclusions
This study presented a comprehensive investigation of pharmacokinetics, bioavailability, tissue distribution and excretion of 6-O-demethylmenisporphine after dosing to rats. An efficient HPLC-MS/MS approach, combined with a time-saving sample processing procedure (PPT), was constructed and applied for rapid quantification of 6-O-demethylmenisporphine in various biomatrices, such as rat plasma, tissues, urine, bile and feces. The pharmacokinetic parameters T max (2.80 ± 0.84 h) and t 1/2 (5.10 ± 2.91 h) of 6-O-demethylmenisporphine showed that the drug could be slowly absorbed and eliminated. The oral bioavailability was up to 51.52%, which implied the considerable absorption after oral dosing. For tissue distribution, 6-O-demethylmenisporphine was quickly and extensively distributed in gastrointestinal tract, kidney, heart, lung, and muscle, etc. Meanwhile, it also could penetrate the blood–brain barrier. The excretion profiles demonstrated that the proportion of drug excreted as unconverted form was 22.66%, which was mainly excreted via fecal route. To our knowledge, it is the first time to reveal the bioavailability, tissue distribution and excretion characteristics of 6-O-demethylmenisporphine, which provide references for clarifying biological activity mechanism and druggability, and lay an experimental basis for further development of 6-O-demethylmenisporphine as an anti-tumor drug candidate.
Author statement
Each of the coauthors in our manuscript has seen and agreed with each of the changes made to this manuscript in the revision and to the way his or her name is listed.
Declaration of Competing Interest
The authors declared that there is no conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 81703690, 81803356 and 81471823), Natural Science Foundation of Tianjin (Nos.19JCQNJC12200 and 18JCYBJC29000) and Foundation of Logistics College of Chinese People’s Armed Police Forces (WHB201711, WHTD-201808).
References
- 1.Yu J., Zhu B.L., Su D., Jiang Z. Pharmacokinetic and excretion study of three alkaloids in rats using UPLC-MS/MS after oral administration of menispermi rhizoma capsules. RSC Adv. 2018;8:31633. doi: 10.1039/c8ra04084b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yi T., Jin Y.J., Jia J.J., Li X.F. Study on chemical constituents from rhizome of menispermum dauricum. J. Yanbian Univ. Nat. Sci. 2017;43:128–130. [Google Scholar]
- 3.Li J., Bi H., Du B., Sun Y., Zhang H. Study on the bacteriostatic action of Menispermi alkaloid by microcalorimetry. J. Qufu Norm. Univ. Nat. Sci. 2010;04:96–102. [Google Scholar]
- 4.Su Q., He J., Wang Z.Y., Lv L., Suo Y., Wang J.J., Zheng Z.W., Huo C.C., Li J. Intestinal anti-inflammatory effect of the rhizome extracts of Menispermum dauricum DC. on trinitrobenzene sulfonic acid induced ulcerative colitis in mice. J. Ethnopharmacol. 2016;193:12–20. doi: 10.1016/j.jep.2016.07.047. [DOI] [PubMed] [Google Scholar]
- 5.Sun D., Zhou M.G., Ying X.H., Cheng B.F., Han Y.Q., Nie Y., Hou Y.Y. Identification of nuclear factor-κB inhibitors in the folk herb Rhizoma Menispermi via bioactivity-based ultra-performance liquid chromatography/quadrupole time-of-flight mass spectrometry analysis. BMC Complem. Altern. M. 2014;14:356. doi: 10.1186/1472-6882-14-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shao J., Shi C.F., Wei J.X., Li Y.X., Guo X.J. Chemical constituents from rhizome of Menispermum dauricum and their anti-hypoxic activities. China J. Chin. Mater. Med. 2019;44:723–729. doi: 10.19540/j.cnki.cjcmm.20181121.003. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X.J., Guo L.J., Qu L., Lv Q. Protective effects of phenolic alkaloids from Menispermum dauricum on inflammatory injury following focal cerebral ischemia-reperfusion in rats. Acta Pharm. Sin. 2004;39:661–665. [PubMed] [Google Scholar]
- 8.Zhao B., Chen Y., Sun X., Zhou M., Ding J., Zhan J.J., Guo L.J. Phenolic alkaloids from Menispermum dauricum rhizome protect against brain ischemia injury via regulation of GLT-1, EAAC1 and ROS generation. Molecules. 2012;17:2725–2737. doi: 10.3390/molecules17032725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao J., Lian Y., Lu C.F., Jing L., Yuan H.T., Peng S.Q. Inhibitory effects of a bisbenzylisoquinline alkaloid dauricine on HERG potassium channels. J. Ethnopharmacol. 2012;141:685–691. doi: 10.1016/j.jep.2011.08.054. [DOI] [PubMed] [Google Scholar]
- 10.Kong X.Y., Gong P.L. Effect of phenolic alkaloids of Menispermum dauricum on thrombosis and platelet aggregation. Acta Pharm. Sin. 2005;40:916–919. [PubMed] [Google Scholar]
- 11.Wang J.Y., Sun S., Liu L., Yang W.S. Induction of apoptosis in human cervical carcinoma HeLa cells with active components of Menispermum dauricum. Genet. Mol. Res. 2014;13:3545–3552. doi: 10.4238/2014.February.13.17. [DOI] [PubMed] [Google Scholar]
- 12.Wei J.X., Fang L.L., Liang X.L., Su D., Guo X.J. A sensitive and selective UPLC-MS/MS method for simultaneous determination of 10 alkaloids from Rhizoma Menispermi in rat plasma and its application to a pharmacokinetic study. Talanta. 2015;144:662–670. doi: 10.1016/j.talanta.2015.07.023. [DOI] [PubMed] [Google Scholar]
- 13.Tan N.H., Zhao S.X., Ding L.S., Ye W.C. Oxoisoaporphines. J. Chin. Pharm. Univ. 1990;21:377–379. [Google Scholar]
- 14.Tang H., Wang X.D., Wei Y.B., Huang S.L., Huang Z.S., Tan J.H., An L.K., Wu J.Y., Chan A.S.C., Gu L.Q. Oxoisoaporphine alkaloid derivatives: Synthesis, DNA binding affinity and cytotoxicity. Eur. J. Med. Chem. 2008;43:973–980. doi: 10.1016/j.ejmech.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Yu B.W., Meng L.H., Chen J.Y., Zhou T.X., Cheng K.F., Ding J., Qin G.W. Cytotoxic oxoisoaporphine alkaloids from Menispermum dauricum. J. Nat. Prod. 2001;64:968–970. doi: 10.1021/np000532t. [DOI] [PubMed] [Google Scholar]
- 16.Cheng J.J., Tsai T.H., Lin L.C. New alkaloids and cytotoxic principles from Sinomenium acutum. Planta Med. 2012;78:1873–1877. doi: 10.1055/s-0032-1327785. [DOI] [PubMed] [Google Scholar]
- 17.Sobarzo-Sánchez E., Soto P.G., Valdés R.C., Sánchez G., Hidalgo M.E. Applied biological and physicochemical activity of isoquinoline alkaloids: oxoisoaporphine and boldine. Molecules. 2012;17:10958–10970. doi: 10.3390/molecules170910958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei Z.Z., Qin Q.P., Chen J.N., Chen Z.F. Oxoisoaporphine as Potent Telomerase Inhibitor. Molecules. 2016;21:1534. doi: 10.3390/molecules21111534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y.P., Ning F.X., Yang M.B., Li Y.C., Nie M.H., Ou T.M., Tan J.H., Huang S.L., Li D., Gu L.Q., Huang Z.S. Syntheses and characterization of novel oxoisoaporphine derivatives as dual inhibitors for cholinesterases and amyloid beta aggregation. Eur. J. Med. Chem. 2011;46:1572–1581. doi: 10.1016/j.ejmech.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Tang H., Wei Y.B., Zhang C., Ning F.X., Qiao W., Huang S.L., Ma L., Huang Z.S., Gu L.Q. Synthesis, biological evaluation and molecular modeling of oxoisoaporphine and oxoaporphine derivatives as new dual inhibitors of acetylcholinesterase/butyrylcholinesterase. Eur. J. Med. Chem. 2009;44:2523–2532. doi: 10.1016/j.ejmech.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 21.Wang P., Sun J.B., Xu J.Y., Yan Q., Gao E.Z., Qu W., Zhao Y.L., Yu Z.G. Pharmacokinetics, tissue distribution and excretion study of dictamnine, a major bioactive component from the root bark of Dictamnus dasycarpus Turcz. (Rutaceae) J. Chromatogr. B. 2013;942–943:1–8. doi: 10.1016/j.jchromb.2013.10.025. [DOI] [PubMed] [Google Scholar]
- 22.Wei J.X., Chen J., Liang X.L., Guo X.J. Microwave-assisted extraction in combination with HPLC-UV for quantitative analysis of six bioactive oxoisoaporphine alkaloids in Menispermum dauricum DC. Biomed. Chromatog. 2016;30:241–248. doi: 10.1002/bmc.3541. [DOI] [PubMed] [Google Scholar]
- 23.Wei J.X., Jiang Z., Cui Z., Guo X.J. Rapid determination of eight oxoisoaporphine alkaloids in Rhizoma Menispermi by the optimal homogenate extraction followed by UPLC-MS/MS. Anal. Bioanal. Chem. 2015;407:5535–5540. doi: 10.1007/s00216-015-8704-4. [DOI] [PubMed] [Google Scholar]
- 24.F.a.D. Administration Bioanalytical Method Validation Guidance for Industry 2018. Available online:https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf.
- 25.Leeson P. Drug discovery: chemical beauty contest. Nature. 2012;481:455–456. doi: 10.1038/481455a. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J.Y., Chen L., Sun J.B. Oxoisoaporphine alkaloids, prospective anti-Alzheimer's, anticancer and antidepression agents. ChemMedChem. 2018;13:1262–1274. doi: 10.1002/cmdc.201800196. [DOI] [PubMed] [Google Scholar]