

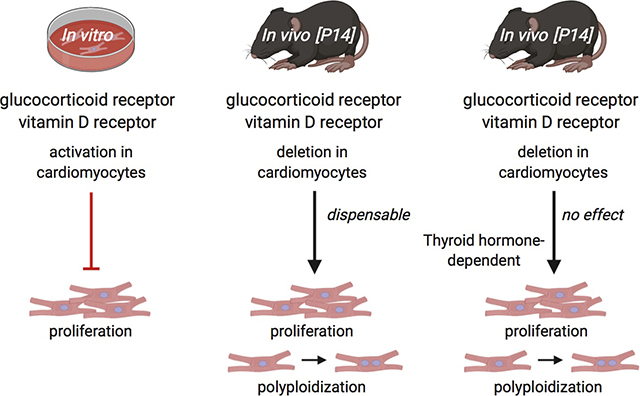
Abstract
Cardiomyocyte (CM) proliferative potential varies considerably across species. While lower vertebrates and neonatal mammals retain robust capacities for CM proliferation, adult mammalian CMs lose proliferative potential due to cell-cycle withdrawal and polyploidization, failing to mount a proliferative response to regenerate lost CMs after cardiac injury. The decline of murine CM proliferative potential occurs in the neonatal period when the endocrine system undergoes drastic changes for adaptation to extrauterine life. We recently demonstrated that thyroid hormone (TH) signaling functions as a primary factor driving CM proliferative potential loss in vertebrates. Whether other hormonal pathways govern this process remains largely unexplored. Here we showed that agonists of glucocorticoid receptor (GR) and vitamin D receptor (VDR) suppressed neonatal CM proliferation. We next examined CM nucleation and proliferation in neonatal mutant mice lacking GR or VDR specifically in CMs, but we observed no difference between mutant and control littermates at postnatal day 14. Additionally, we generated compound mutant mice that lack GR or VDR and express dominant-negative TH receptor alpha in their CMs, and similarly observed no increase in CM proliferative potential compared to dominant-negative TH receptor alpha mice alone. Thus, although GR and VDR activation is sufficient to inhibit CM proliferation, they seem to be dispensable for neonatal CM cell-cycle exit and polyploidization in vivo. In addition, given the recent report that VDR activation in zebrafish promotes CM proliferation and tissue regeneration, our results suggest distinct roles of VDR in zebrafish and rodent CM cell-cycle regulation.
Graphical Abstract
1. Introduction
Cardiovascular disease is the leading killer in the United States, mostly due to heart failure after myocardial infarction (MI) [1,2]. After ischemic injury like MI induces CM death in the hearts of adult mammals, lost CMs are not replenished and fibrotic tissue permanently replaces previously functional cardiac muscle. However, lower vertebrates like zebrafish (Danio rerio), newts (Notophthalmus viridescens), and axolotls (Ambystoma mexicanum) display robust CM proliferation and myocardial regeneration after cardiac injury [2–4]. Intriguingly, neonatal mammals also transiently possess considerable CM proliferative potential. Ischemic heart injury induced in newborn mice at P0 or P1 resolves as fibrosis-free, fully-regenerated cardiac muscle by 21 days post-injury, mediated by existing CMs that proliferate to reconstitute the lost myocardium [5,6]. This transient CM proliferative potential is lost in mice after the first week as neonatal CMs binucleate and permanently exit the cell cycle [7,8]. Because the regeneration-competent hearts of lower vertebrates and neonatal mice consist predominantly of mononuclear CMs while the non-regenerative hearts of adult mice consist primarily of binuclear CMs, this developmental binucleation is implicated as a critical inhibitor of CM proliferative potential [4,9–11]. However, the underlying molecular processes that drive neonatal rodent CM binucleation and cell cycle withdrawal are not fully understood.
Recent evidence indicates that thyroid hormones (THs) play a crucial role in driving CM binucleation and suppressing CM proliferative potential in vertebrates [12]. TH signaling is a critical regulator of metabolism and thermogenesis conserved across vertebrates and is particularly active in endotherms [13]. THs enhance CM contractility in vivo and rise soon after birth in neonatal mice, coinciding with the closure of the CM proliferative window [14,15]. Serum TH levels are also substantially lower in newts and zebrafish than in non-regenerative mammals [16,17]. Neonatal mice dosed with propylthiouracil (PTU) – a potent inhibitor of TH synthesis – through postnatal day 14 (P14) show significantly increased mononuclear CM percentage and CM proliferation [12]. These mononuclear CMs and CM proliferation phenotypes are also observed in mutant mice in which TH signaling is specifically inactivated in CMs [12]. These mutant mouse hearts also display enhanced recovery after MI as indicated by cardiac contractile functions and fibrosis one month post-surgery. Conversely, exogenous TH suppresses CM proliferation and cardiac regeneration in adult zebrafish. These data strongly suggest an inhibitory role played by TH in the developmental control of CM proliferative potential during the neonatal window.
In addition to TH, other hormones such as glucocorticoids and vitamin D have been shown to suppress CM proliferation in culture. Glucocorticoids are stress-associated steroid hormones produced by the adrenal glands that act on nearly all organs in the body via binding to the glucocorticoid receptor (GR) [18–20]. Specifically, glucocorticoids like dexamethasone are established cell cycle regulators known to repress the cell cycle primarily through the GR, which acts as a transcription factor after glucocorticoid binding [21,22]. It has been reported that GR activation inhibits neonatal rat CM proliferation, promotes neonatal rat CM hypertrophy, and increases CM binucleation through epigenetic repression of Cyclin D2 gene [23–25]. Furthermore, adult zebrafish heart regeneration is impaired by either GR agonist exposure [26] or crowding-induced stress through GR activation [27].
Vitamin D is a steroid hormone precursor that is hydroxylated sequentially in the liver and kidneys into ercalcitriol (D2) or calcitriol (D3). These active hormones can bind to vitamin D receptors (VDRs) and regulate downstream gene expression in target cells [28]. Alfacalcidol (Alfa) is a vitamin D analog that forms calcitriol directly after hydroxylation in the liver [29]. Anti-proliferative effects of vitamin D analogs have been reported in mammalian cells generally [30] as well as mammalian CMs and CM-derived cell lines specifically [31,32]. Notably, VDR signaling may have distinct regulatory roles across vertebrates. Unlike in mammals, vitamin D analogs alfacalcidol and calcipotriene significantly increase CM proliferation in embryonic zebrafish, whereas VDR-inhibitor PS121912 significantly decreases CM proliferation [33]. Alfacalcidol also stimulates CM proliferation in adult zebrafish during heart regeneration, while VDR suppression decreases CM proliferation during regeneration [33].
Intriguingly, CM-specific deletion of either GR or VDR promotes cardiac hypertrophy [34–36]. This cardiac enlargement phenotype together with the documented functions to inhibit CM proliferation in vitro led us to investigate the possibility that GR and VDR signaling alters CM proliferative potential during the neonatal window in vivo either alone or in combination with TH signaling activation.
2. MATERIALS & METHODS
2.1. Animals
Mouse protocols were conducted in accordance with the Institutional Animal Care and Use Committee (IACUC) of the University of California, San Francisco. CD-1 (Charles River), Myh6-Cre (20), TRαAMI/AMI (here named ThraDN/DN) (12), Nr3c1f/f (here named Grf/f) (8), and Vdrf/f (9) mouse lines were maintained according to the University of California, San Francisco institutional guidelines. All mice were between 1 and 14 days old when analyzed for in vivo experiments, including cardiomyocyte nucleation and proliferation analyses. Subcutaneous (subQ) injections and MI surgery were performed in accordance with IACUC guidelines. Primary cardiomyocytes for in vitro experiments were cultured from P1 and P7 CD-1 neonatal mice of mixed sex. For all experiments, both male and female mice were used and no gender difference was observed.
2.2. Reagents
The following reagents were used: rabbit anti-PCM1 (1:2000) (Sigma HPA023370), rabbit anti-PCM1 (H262) (1:200) (Santa Cruz Biotechnology SC-67204), Alexa Fluor 488 donkey anti-rabbit IgG (Molecular Probes 1874771) (1:500), rat anti Ki-67 monoclonal antibody (SolA15), mouse anti-cTnT (1:400)(Thermo Fisher Scientific MS295P1), Mouse anti-phospho Histone 3 antibody (1:500) (Millipore Mill-05–806), rabbit anti-Aurora B (1:100)(Abcam GR3194983–1),Southern Biotech Dapi-Fluoromount-G Clear Mounting Media (Southern Biotech 0100–20), WGA CF633 conjugates (1:200), 5-Ethynyl-2-deoxyuridine (Santa Cruz Biotechnology SC-284628), Click-it EdU imaging kit (ThermoFisher Scientific C10337), Collagenase Type II (Worthington LS004177), Ethyl carbamate (Alfa Aesar AAA44804–18), calcitriol (Sigma-Aldrich 32222–06-3), alfacalcidol (Sigma-Aldrich L91201381), dexamethasone (TCI America TCI-D1961–1G), hydrocortisone (Alfa Aesar AAA16292–03), corticosterone (Enzo 89149–746), sodium chloride (Sigma-Aldrich S9888–10KG), mifepristone (VWR 89162), PS121912 (obtained from University of Wisconsin-Milwaukee), potassium chloride (Sigma-Aldrich P9541–1KG), monopotassium phosphate (Fisher 5028048), sodium phosphate dibasic heptahydrate (Fisher S25837), magnesium sulfate heptahydrate (FisherS25414), HEPES (Sigma-Aldrich H4034–100g), sodium bicarbonate (EMD EM-SX0320–1), taurine (Sigma-Aldrich T8691–100G), biacetyl monoxime (Sigma-Aldrich B0753–100G), glucose (Amresco 97061–164), EGTA (Amresco 0732–288), protease XIV (P5147–100MG), fetal bovine serum (JRS CCFAP004), calcium chloride (Amresco 97061–904), primocin (Invivogen NC9141851), DMEM (CCF CCFAA005), TRIzol reagent (Thermo Fisher Scientific), SYBR Select Master Mix (Applied Biosystems, 4472908), iScript RT Supermix (Bio-Rad 1708841).
2.3. Methods
2.3.1. Generating mouse lines
Mice heterozygous for the Tg(Myh6-Cre)2182Mds transgene (here referred to as “Myh6-Cre”) were bred with mice homozygous for the Nr3c1f/f allele (here named Grf/f) to generate Myh6-Cre;Grf/+ mice. These Myh6-Cre;Grf/+ mice were then bred with Grf/f mice to generate mutant Myh6-Cre;Grf/f mice and littermate controls of the following genotypes: Grf/+, Grf/f, and Myh6-cre;Grf/+. Similarly, mice heterozygous for Myh6-Cre were bred with mice homozygous for the Vdrf/f allele to generate Myh6-Cre;Vdrf/+ mice. These Myh6-Cre;Vdrf/+ mice were then bred with Vdrf/f mice to generate mutant Myh6-Cre;Vdrf/f mice and littermate controls of the following genotypes: Vdrf/+, Vdrf/f, and Myh6-Cre;Vdrf/+.
In parallel, mice heterozygous for Myh6-Cre were bred with mice homozygous for the ThraDN allele to generate Myh6-Cre;ThraDN/+ mice that were subsequently bred with either mice homozygous for the Grf/f allele or mice homozygous for the Vdrf/f allele to generate both Myh6-Cre;ThraDN/+;Grf/+ mice or Myh6-Cre;ThraDN/+;Vdrf/+ mice, respectively. The Myh6-Cre;ThraDN/+;Grf/+ mice were bred with Grf/f mice to generate experimental Myh6-Cre;ThraDN/+;Grf/f mice and control Myh6-Cre;ThraDN/+;Grf/+ mice. The Myh6-Cre;ThraDN/+;Vdrf/+ mice were bred with Vdrf/f mice to generate experimental Myh6-Cre;ThraDN/+;Vdrf/f mice and control Myh6-Cre;ThraDN/+;Vdrf/+ mice.
2.3.2. Analysis of cardiomyocyte nucleation
Ventricular tissues were fixed in 3.7% paraformaldehyde for 48 hours followed by incubation in 50% w/v potassium hydroxide solution overnight. After a brief wash with PBS, tissues were gently crushed to release dissociated cardiomyocytes. Cells were further washed with PBS three times, deposited on slides, and then allowed to dry out completely. (Note: CMs could be stained for cTnT after the last PBS wash by suspension in primary antibody solution overnight and subsequent suspension of the CMs in secondary antibody solution for 120 minutes, prior to plating on glass slides.) Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI). To determine cardiomyocyte nucleation, images of spotted cardiomyocytes were analyzed in ImageJ. The number of mononuclear, binuclear, and polynuclear cardiomyocytes was determined manually using the Count Tool. At least 130 cardiomyocytes per each sample were analyzed.
2.3.3. Neonatal cardiomyocyte isolation and culture
Freshly dissected ventricles from P1 or P7 CD-1 strain neonatal mice extracted and perfused with Perfusion Buffer (12 mM sodium chloride, 1.5 mM potassium chloride, 60 μM monopotassium phosphate, 60 μM sodium phosphate dibasic heptahydrate, 120 μM magnesium sulfate heptahydrate, 1mM HEPES, 4.6 mM sodium bicarbonate, 30 mM taurine, 10 mM biacetyl monoxime, 5.5 mM glucose, and 400 μM EGTA). Cardiomyocytes were serially isolated in Digestion Buffer (2 mg/mL collagenase II and 500 μg/mL protease XIV in perfusion buffer) for 2h at 37°C during agitation. Isolated cardiomyocytes were cultured in Plating Media (10% FBS and 100 μg/mL primocin in DMEM) and plated on a 96-well cell culture plate at a density of ~25 thousand cells/well.
2.3.4. Quantitative PCR
24 hours after plating P1 CMs, old Plating Media was changed and 5 μM EdU Plating Media containing each drug of interest at the requisite concentration was added to each treatment well. (Control wells received Plating Media with 5 μM EdU but no dissolved drugs.) 48 hours after drug addition, Plating Media was removed and each well was immediately washed with 100 μL PBS before RNA was immediately isolated using TRIzol Reagent (Thermo Fisher Scientific) according to manufacturer’s protocols. 100 ng RNA was used to synthesize cDNA using iScript RT Supermix (Bio-Rad 1708841) according to manufacturer’s protocols. Quantitative PCR was performed using the SYBR Select Master Mix (Applied Biosystems, 4472908) and the 7900HT Fast Real-Time PCR system (Applied Biosystems). DNA content was measured via qPCR targeting GR targets Pam [37] and Cacna1c [38] and VDR target Vdr [32]. Relative CM DNA content was measured via normalization against CM housekeeping gene, Actb using the following primers: Pam forward = 5’- CAGAACTATCCCAGAAGAGGC-3’; Pam reverse = 5’- TTCTGTTTCTTTGTGATGCCCA-3’; Cacna1c forward = 5’- ATGAAAACACGAGGATGTACGTT-3’; Cacna1c reverse = 5’- ACTGACGGTAGAGATGGTTGC-3’; Vdr forward =5’- ACCCTGGTGACTTTGACCG-3’; Vdr reverse = 5’- GGCAATCTCCATTGAAGGGG-3’; Actb forward =5’- AGTGTGACGTTGACATCCGT-3’; Actb reverse = 5’- TGCTAGGAGCCAGAGCAGTA-3’.
2.3.5. Neonatal cardiomyocyte in vitro immunohistochemistry
24 hours (48 hours for P7 CMs) after plating, old Plating Media was changed and 5 μM EdU Plating Media containing each drug of interest at the requisite concentration was added to each treatment well. (Control wells received Plating Media with 5 μM EdU but no dissolved drugs.) 48 hours (72 hours for P7 CMs) after drug addition, Plating Media was removed and each well was immediately washed with 100 μL PBS, fixed with 3.7% PFA for 15 minutes at room temperature, permeabilized in 0.2% Triton X-100 in PBS (PBST), blocked in 5% normal donkey serum (NDS) in PBST for 1 hour at room temperature, and incubated with primary antibodies in PBST overnight at 4°C. After primary antibody incubation, sections were incubated in their corresponding secondary antibody for 2 hours at room temperature, and (if appropriate) the Click-it EdU imaging kit was used to visualize EdU via conjugation to sulfo-Cyanine 5-azide dye (Lumiprobe A3330). In all samples, nuclei were visualized by staining with DAPI.
2.3.6. Uninjured neonatal CM proliferation analysis
CD-1 littermate hearts were harvested at P1, P4, P7, and P10 for proliferation analysis.
2.3.7. Uninjured neonatal injections
From birth until P7, CD-1 littermates were subcutaneously injected daily with either 5 μg dexamethasone (in 0.9% saline) per gram body weight, 1 ng alfacalcidol (in 0.9% saline) per gram body weight, or saline control. All hearts were harvested for analysis on P7.
2.3.8. Neonatal myocardial infarction (MI) and subsequent injections
MI injury was induced in P1 CD-1 mice via ligation of the LAD coronary artery while the animal was anesthetized on ice. After surgery, the chest wall and skin were sutured closed and animals were allowed to recover. Between P1 and P8, they were subcutaneously injected daily with either 5 μg dexamethasone (in 0.9% saline) per gram body weight, 5 μg mifepristone (in 0.9% saline) per gram body weight, 1 ng alfacalcidol (in 0.9% saline) per gram body weight, 1 ng PS121912 (in 0.9% saline) per gram body weight, or saline control. All hearts were harvested for analysis on P8.
2.3.9. In vivo cardiomyocyte immunohistochemistry
Experimental mice were anesthetized by injection of 20% ethyl carbamate in 1X PBS, their hearts were freshly excised, soaked briefly in 30% sucrose, and then embedded in O.C.T. Compound (Tissue Tek, cat#4583) and flash frozen on a metal block cooled by liquid nitrogen. Embedded samples were then sectioned with a Leica CM3050S to 5 μm thickness. Tissue sections were then fixed in 3.7% paraformaldehyde for 15 min at room temperature, permeabilized in 0.2% PBST, blocked in 5% NDS in PBST for 1 hour at room temperature, and incubated with primary antibodies in PBST overnight at 4°C. After primary antibody incubation, sections were incubated in their corresponding secondary antibody for 2 hours at room temperature and mounted in DAPI to visualize nuclei.
2.3.10. Statistical analysis
The number of samples per each experimental condition is listed in the description of the corresponding figure legend. Statistical significance was determined using the one-way ANOVA test (Fig. 1), and Student’s T-test for the rest of the figures.
Figure 1. Glucocorticoid receptor (GR) and vitamin D receptor (VDR) agonists suppress P1 mouse cardiomyocyte (CM) proliferation in vitro.

(A) Primary CMs (PCM1+ nuclei within cTnT+ cell bodies) cultured from P1 neonatal mouse hearts in the presence of 100 nM hydrocortisone, 100 nM corticosterone, or 100 nM dexamethasone incorporated 5-fold less EdU over 48 hr than CMs cultured without any GR agonist (DMSO). (B) Similarly, primary P1 neonatal CMs cultured with 1 μM calcitriol and 1 μM alfacalcidol incorporated 2-fold less EdU than those cultured with no VDR agonist (DMSO). Values are reported as mean ± SEM (n=3 hearts in A; n=5 hearts in B). NS, not significant. *p < 0.05, **p < 0.01. Scale bars: 25 μm.
3. RESULTS
3.1. Glucocorticoid and vitamin D receptor activation inhibits neonatal mouse cardiomyocyte proliferation in vitro
We first examined if GR and VDR activation regulates neonatal CM proliferation in vitro. Primary CMs were isolated from P1 mice and cultured with one of three GR agonists for 48 hours: hydrocortisone, corticosterone, or dexamethasone. Proliferating CMs were positive for cTnT (gray), PCM-1 rings (red), and EdU incorporation (green) (Fig. 1). CM proliferation is inhibited by 100 nM hydrocortisone (0.30 ± 0.03%), corticosterone (0.15 ± 0.1%), and dexamethasone (0.07 ± 0.09%) as compared to CMs cultured without any GR agonists (1.22 ± 0.44%) (Fig. 1A). We also cultured P1 CMs with two VDR agonists: calcitriol and alfacalcidol. Neonatal CM proliferation was inhibited by 1 μM calcitriol (0.42 ± 0.15%) and alfacalcidol (0.35 ± 0.05%) relative to CMs cultured without any VDR agonists (0.87 ± 0.10%) (Fig. 1B). These anti-proliferative effects of GR and VDR agonists were also observed using phospho-histone H3 (pHH3) as the marker for CM proliferation (Fig. S2A). Furthermore, in P1 CMs dexamethasone treatment upregulated GR target genes Pam 2.4-fold and Cacna1c 3.5-fold while alfacalcidol increased the expression of a VDR target gene Vdr 2.3-fold (Fig. S6A & B). This in vitro suppression of neonatal CM proliferation led us to investigate the in vivo contribution of GR and VDR to cardiomyocyte proliferative potential during the neonatal window.
3.2. Glucocorticoid and vitamin D receptors in cardiomyocytes are dispensable for the regulation of neonatal mouse cardiomyocyte proliferation and binucleation in vivo
To elucidate the physiological role of GR in regulating neonatal CM proliferative potential in vivo, we generated Myh6-Cre;Grf/f knockout mice in which CM-specific CRE recombinase (Myh6-Cre) deletes the Gr gene (also named as Nuclear Receptor Subfamily 3 Group C member 1, Nr3c1), resulting in CM-specific loss of GR signaling (Fig. 2A). We examined heart phenotypes at P14 when most mouse CMs have completed binucleation and withdrawn from the cell cycle [7]. Interestingly, unlike our in vitro primary CMs, no changes of CM proliferative activity in vivo were observed at P14 (Fig. 2B–D). Myh6-Cre;Grf/f knockout mice did not show an increase in the heart weight to body weight ratio (6.21 ± 0.16) compared to littermate controls (6.16 ± 0.18) (Fig. 2B). Additionally, P14 Myh6-Cre;Grf/f hearts were comparable to littermate control hearts in terms of mononuclear CM percentage (13.6 ± 1.1% vs 13.7 ± 2.0%) (Fig. 2C and S5). Analysis of CM proliferation using the Ki67 marker also showed no increase in Ki67+ CMs in mutant hearts (1.23 ± 0.55% vs 1.09 ± 0.23%) (Fig. 2D). No difference was observed between control and mutant hearts when CM mitosis (positive for pHH3) and cytokinesis (Aurora B kinase at the cleavage furrow) were analyzed (Fig. S3A and C). Altogether, we did not observe that CM-specific loss of GR affects CM proliferative potential during this window.
Figure 2. CM-specific loss of glucocorticoid receptor (GR) signaling does not enhance CM proliferative potential in vivo.

(A) Schematic for generating Myh6-Cre;Grf/f mice with CM-specific deletion of floxed endogenous Gr and assessing CM proliferative potential at P14. Littermate Grf/f mice served as control animals. (B) Mononuclear CM percentage did not differ between Myh6-Cre;Grf/f mice and littermate controls at P14. (C) Heart weight (mg) to body weight (g) ratio (HW:BW) did not differ between Myh6-Cre;Grf/f mice and littermate controls at P14. (D) No difference in the number of Ki67+ CMs was observed between Myh6-Cre;Grf/f mice and littermate controls at P14. White arrows indicate Ki67+ CM nuclei. Values are reported as mean ± SEM (n=3 hearts). NS, not significant. *p < 0.05. Scale bars: 100 μm.
To investigate the contribution of VDR to the control of neonatal CM proliferative potential in vivo, we generated mice with CM-specific loss of VDR (Myh6-Cre;Vdrf/f) (Fig. 3A). As with GR deletion, no changes in CM proliferative activity were observed at P14 (Fig. 3B–D). The heart weight to body weight ratio did not significantly differ between Myh6-Cre;Vdrf/f knockout mice compared to littermate controls (Fig. 3B). Additionally, P14 Myh6-Cre;Vdrf/f hearts did not significantly differ from littermate control hearts in terms of mononuclear CM percentage (15.8 ± 3.2% vs 14.0 ± 1.0%), Ki67+ CM proliferation (0.93 ± 0.29% vs 0.61 ± 0.11%) (Fig. 3C & D and S5), CM mitosis or cytokinesis (Fig. S3A and C). As with GR loss, VDR loss alone did not affect neonatal CM proliferative potential in vivo.
Figure 3. CM-specific loss of Vitamin D receptor (VDR) signaling does not enhance CM proliferative potential in vivo.

(A) Schematic for generating Myh6-Cre;Vdrf/f mice with CM-specific deletion of floxed endogenous VDR and assessing CM proliferative potential at P14. Littermate controls included phenotypically-identical genotypes: Vdrf/f and Myh6-Cre;Vdrf/+. (B) Mononuclear CM percentage did not differ between Myh6-Cre;Vdrf/f mice and littermate controls at P14. (C) Heart weight to body weight ratio (HW:BW) did not differ between Myh6-Cre;Vdrf/f mice and littermate controls at P14. (D) No difference in the number of Ki67+ CMs was observed between Myh6-Cre;Vdrf/f mice and littermate controls at P14. White arrows indicate Ki67+ CM nuclei. Values are reported as mean ± SEM (n=3). NS, not significant. *P < 0.05. Scale bars: 100 μm.
3.3. Loss of glucocorticoid or vitamin D receptor does not enhance cardiomyocyte proliferation potential in mice with deficiency in thyroid hormone signaling in vivo
It is plausible that the potential inhibitory effect of GR and VDR activation on CM proliferation in vivo is masked by other major regulators. One such candidate is TH receptor. We recently showed that TH signaling activation suppresses CM proliferative potential [12], and mice with deficiency in TH receptor activation in CMs show enhanced CM proliferation and have ~30% mononuclear CMs at P14. However, the majority of CMs in the TH receptor deficient heart still become binucleated and arrested in the cell-cycle, implicating the existence of other pathways that contribute to CM binucleation and cell-cycle withdrawal in the neonatal period. We next investigated whether loss of GR or VDR signaling affect neonatal CM proliferative potential in combination with loss of TH signaling. We bred Myh6-Cre;ThraDN/+;Grf/f mice that express dominant negative TH receptor alpha (THRADN) and delete Gr specifically in CMs (Fig. 4A). The heart weight to body weight ratio for Myh6-Cre;ThraDN/+;Grf/f (7.28 ± 0.05) did not significantly differ from littermate Myh6-Cre;ThraDN/+;Grf/+ controls (7.61 ± 0.55) (Fig. 4B). Additionally, compound mutant hearts did not display significant increases in CM proliferation (3.5 ± 0.7% vs 3.2 ± 0.7%) or CM nucleation (32.7 ± 8.7% vs 33.4 ± 6.1%) compared to littermate Myh6-Cre;ThraDN/+;Grf/+ controls at P14 (Fig. 4C & D and S5). No difference was observed between control and mutant hearts when CM mitosis and cytokinesis were examined (Fig. S3A and C). Thus, we did not observe any significant effect of GR loss on top of TH signaling suppression on in vivo neonatal CM proliferative potential.
Figure 4. Loss of glucocorticoid receptor (GR) does not enhance cardiomyocyte (CM) proliferative potential in mice with CM-specific deficiency of thyroid hormone signaling.

(A) Schematic for generating Myh6-Cre;ThraDN/+;Grf/f mice with CM-specific deletion of floxed endogenous Gr and CM-restricted expression of a dominant negative (DN) thyroid hormone receptor alpha (Thra). (B) Mononuclear CM percentage did not differ between Myh6-Cre;ThraDN/+;Grf/f mice and Myh6-Cre;ThraDN/+;Grf/+ controls at P14. (C) Heart weight to body weight ratio (HW:BW) did not differ between Myh6-Cre;ThraDN/+;Grf/f mice and Myh6-Cre;ThraDN/+;Grf/+ controls at P14. (D) No difference in the number of Ki67+ CMs was observed between Myh6-Cre;ThraDN/+;Grf/f mice and Myh6-Cre;ThraDN/+;Grf/+ controls at P14. Yellow arrowheads indicate mononuclear CMs. White arrowheads indicate Ki67+ CM nuclei. Values are reported as mean ± SEM (n=3 animals). NS, not significant. *p < 0.05. Scale bars: 100 μm.
Finally, we bred Myh6-Cre;ThraDN/+;Vdrf/f mice and observed no significant difference in the heart weight to body weight ratio (6.83 ± 0.14 vs 7.23 ± 0.83), CM nucleation (24.4 ± 2.3% vs 26.9 ± 3.4%), or CM proliferation (3.0 ± 0.3 vs 3.9 ± 1.0%) compared to Myh6-Cre;ThraDN/+;Vdrf/+ littermate controls at P14 (Fig. 5A–D and S5). Both mutant and control mice have similar levels of CM mitosis and cytokinesis (Fig. S3A and C). As such, we did not observe a significant effect of VDR loss on neonatal CM proliferative potential on top of TH signaling suppression. Overall, our results show that although GR and VDR activation in cultured P1 CMs is sufficient to inhibit CM proliferation, they seem to be dispensable for CM cell-cycle exit and binucleation in vivo between P0 and P14.
Figure 5. Loss of vitamin D receptor (VDR) does not enhance cardiomyocyte (CM) proliferative potential in mice with CM-specific deficiency of thyroid hormone signaling.

(A) Schematic for generating Myh6-Cre;ThraDN/+;Vdrf/f mice with CM-specific deletion of floxed endogenous VDR and CM-restricted expression of a dominant negative (DN) thyroid hormone receptor alpha (Thra). (B) Mononuclear CM percentage did not differ between Myh6-Cre;ThraDN/+;Vdrf/f mice and Myh6-Cre;ThraDN/+;Vdrf/+ controls at P14. (C) Heart weight to body weight ratio (HW:BW) did not differ between Myh6-Cre;ThraDN/+;Vdrf/f mice and Myh6-Cre;ThraDN/+;Vdrf/+ controls at P14. (D) No difference in the number of Ki67+ CMs was observed between Myh6-Cre;ThraDN/+;Vdrf/f mice and Myh6-Cre;ThraDN/+;Vdrf/+ controls at P14. Yellow arrows indicate mononuclear CMs. White arrows indicate Ki67+ CM nuclei. Values are reported as mean ± SEM. (n=3) NS, not significant. *p < 0.05. Scale bars: 100 μm.
4. DISCUSSION
Mammalian cardiomyocytes undergo postnatal cell-cycle exit and polyploidization when they lose regenerative potential [7,9,39–41]. The intrinsic and extrinsic molecular drivers of this process have been under intensive studies [4,42–46]. Our observation that both GR and VDR agonists inhibit proliferation of primary neonatal mouse CMs in vitro (Fig. 1, Fig. S2A) is consistent with previous reports in rodent CM culture and rodent cardiac myocyte cell lines [23,24,31,32,47]. GR activation inhibits neonatal rat CM proliferation and increases CM binucleation [23,24]. VDR activation reduces expression of both c-myc and PCNA protein levels, and inhibits cell proliferation in neonatal rat CM culture [47]. At the concentrations of VDR and GR agonists in our study, we did not observe obvious toxicity that leads to gross CM cell loss (shown in the images of Fig. 1) or changes in CM beating frequency. Furthermore, our observations that P1 CMs upregulate GR- and VDR-responsive genes upon GR and VDR agonist treatment (Fig. S6A & B) and that GR and VDR antagonists can mitigate the anti-proliferative effects of their agonists (Fig. S2B) further support that activation of these receptors themselves inhibits neonatal CM proliferation.
Interestingly, while P1 and P7 CMs have similar levels of baseline proliferation in vivo (Fig. S4A), their proliferative potential in response to VDR activation differs. We observed that unlike in P1 CMs, activation of VDR signaling did not affect proliferation in cultured P7 CMs (Fig. S1). It is possible that the ~60% downregulation of Vdr in the heart between birth and P7 may explain this differential response [48] (Fig. S6D). In contrast to our finding, calcitriol was previously reported to induce robust proliferation of zebrafish CMs and cultured P7 mouse CMs [33]. The differential response of P7 CMs to VDR activation in different groups is intriguing and merits further investigation.
Unlike our observations in cultured CMs, our in vivo analyses did not reveal a significant role for either receptor in regulating neonatal CM proliferative potential loss: neither CM nucleation nor CM proliferation was influenced by either GR or VDR loss in P14 mice (Fig. 2, Fig. 3, Fig. S7). However, injecting neonatal mice with exogenous dexamethasone and alfacalcidol from birth to P7 did reduce in vivo CM proliferation at P7, suggesting that GR and VDR can suppress CM proliferation in vivo if activated beyond physiological levels (Fig. S4B). As such, while GR and VDR signaling are sufficient to inhibit CM proliferation in culture, we did not observe their regulatory influence on CM proliferative potential to be determinative in vivo at physiological levels in the neonate. Moreover, we did not observe cardiomyocyte hypertrophy of these aforementioned mutant mice at P14, suggesting that the hypertrophic phenotypes described in both mutant mice [34,35] may develop later in the postnatal life (Fig. S3B). Altogether, our data indicate that while agonist treatment both in vitro and in vivo can inhibit CM proliferation, GR and VDR are not physiological regulators of CM proliferation at endogenous glucocorticoid and vitamin D levels in the neonatal time window.
To further explore the contribution of GR and VDR to in vivo CM proliferation in the context of cardiac injury, we induced MI with coronary artery ligation in P1 mice and injected them daily with either saline, GR-agonist dexamethasone (Dex), GR-antagonist mifepristone (Mif), VDR-agonist alfacalcidol (Alfa), or VDR-antagonist PS121912 (PS) until 7 days post-injury, which is the global CM proliferative peak after cardiac injury [49]. Dex-injected mice had 80% fewer proliferating CMs than saline-injected controls (Fig. S4C). CM proliferation in none of the other conditions differed significantly from controls. These results suggest that GR activation is sufficient but not necessary to suppress CM proliferation after injury and that VDR activation is neither sufficient nor necessary to affect CM proliferation after injury.
A possible explanation between our differential observations in vitro and in vivo may be that GR and VDR signaling are not activated during the neonatal window we examined. Previously-published mouse transcriptome data supports this: neither GR targets Pam and Cacna1c nor VDR target Vdr are upregulated in the mouse heart at P14 [50] (Fig. S6E–G). Similarly, previously-published RNAseq data shows downregulation of both Pam and Vdr in the neonatal heart [48] (Fig. S6C & D). Additionally, the possibility remains that there are other in vivo factors not present in vitro whose regulatory influence over CM proliferative potential is strong enough to mask any contributions from either GR or VDR signaling. Taken together, these data suggest neither GR nor VDR are strongly activated at P14 and do not contribute substantially to the neonatal loss of CM proliferative potential.
As previously stated, one such in vivo factor that exerts a stronger effect on CM proliferative potential than GR or VDR may be thyroid hormone (TH). We previously established a potent inhibitory role for TH signaling over CM proliferative potential [12]. Still, it remained possible that GR or VDR signaling may alter the observed effects of loss of TH signaling on CM proliferative potential. When we analyzed P14 mice both expressing ThraDN and lacking either GR or VDR in their CMs, no significant effects of either GR or VDR loss on CM proliferative potential were observed (Fig. 4, Fig. 5, Fig. S3A & C, Fig. S7). Thus, the regulatory influence of TH over CM proliferative potential is strong enough to overpower any contributions from either GR or VDR at physiological levels. Further investigation is needed to explore the molecular and endocrine factors that act in parallel to and downstream of thyroid hormone to establish vertebrate cardiac regenerative potential.
In summation, our results suggest that while GR and VDR signaling suppress neonatal CM proliferation in vitro, they do not function as major triggers of neonatal CM cell-cycle withdrawal and binucleation in vivo. Future investigation into other signaling factors that may contribute to postnatal CM cell-cycle exit alongside TH signaling is warranted.
Supplementary Material
Highlights.
Stimulation of the glucocorticoid and vitamin D receptors suppress neonatal mouse cardiomyocyte proliferation in vitro
Glucocorticoid and vitamin D receptors in cardiomyocytes are dispensable for the regulation of neonatal mouse cardiomyocyte proliferation and polyploidization in vivo
Loss of glucocorticoid or vitamin D receptors in cardiomyocytes does not enhance in vivo cardiomyocyte proliferative potential in mice with cardiomyocyte-specific thyroid hormone signaling deficiency
ACKNOWLEDGEMENTS
We thank A. Arnold for sharing PS121912, Alexander Amram for substantial contributions to proliferation analysis of CMs both in vitro and in vivo, Rachel B. Bigley and Kentaro Hirose for assistance in breeding and genotyping the mutant mice in our experiments. The figure in the graphic abstract was made in ©BioRender - biorender.com.
Funding: This work is supported by a NIGMS IMSD fellowship, Hillblom fellowship, and NIH F31 predoctoral fellowship (to S.C.), a UCSF-IRACDA postdoctoral fellowship (to A.P.), NIH (R01HL13845) Pathway to Independence Award (R00HL114738), Edward Mallinckrodt Jr. Foundation, March of Dimes Basil O’Conner Scholar Award, American Heart Association Beginning Grant-in-Aid, American Federation for Aging Research, Life Sciences Research Foundation, Program for Breakthrough Biomedical Research, UCSF Eli and Edythe Broad Center of Regeneration Medicine and Stem Cell Research Seed Grant, UCSF Academic Senate Committee on Research, REAC Award (Harris Fund), Department of Defense, and Cardiovascular Research Institute (to G.N.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics-2015 update : A report from the American Heart Association. Circulation 2015; 131: e29–39. [DOI] [PubMed] [Google Scholar]
- 2.González-Rosa JM, Burns CE, Burns CG. Zebrafish heart regeneration: 15 years of discoveries. Regeneration 2017; 4: 105–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Witman N, Murtuza B, Davis B, Arner A, Morrison JI. Recapitulation of developmental cardiogenesis governs the morphological and functional regeneration of adult newt hearts following injury. Dev. Biol. 2011; 354: 67–76. [DOI] [PubMed] [Google Scholar]
- 4.Vivien CJ, Hudson JE, Porrello ER. Evolution, comparative biology and ontogeny of vertebrate heart regeneration. npj Regen. Med. 2016; 1: 16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jopling C, Sleep E, Raya M, Marti M, Raya A, Belmonte JCI, et al. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 2010; 464: 606–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porrello ER, Mahmoud AI, Simpson E, Hill J, James A, Olson EN, et al. Transient Regenerative Potential of the Neonatal Mouse Heart. Science 2011; 331: 1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am. J. Physiol. 1996; 271: H2183–H2189. [DOI] [PubMed] [Google Scholar]
- 8.Cao T, Liccardo D, LaCanna R, Zhang X, Lu R, Chen X, et al. Fatty acid oxidation promotes cardiomyocyte proliferation rate but does not change cardiomyocyte number in infant mice. Front. Cell Dev. Biol. 2019; 7: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mortem IP, Hearts H, Adler C, Costabel U. Myocardial DNA and Cell Number Under the Influence of Cytostafics. 1980; 125: 109–125. [DOI] [PubMed] [Google Scholar]
- 10.Brodsky VY, Chernyaev AL, Vasilyeva IA. Variability of the cardiomyocyte ploidy in normal human hearts. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1992; 61: 289–294. [DOI] [PubMed] [Google Scholar]
- 11.Haubner BJ, Scheider J, Schweigmann U, Schuetz T, Dichtl W, Velik-Salchner C, et al. Functional Recovery of a Human Neonatal Heart after Severe Myocardial Infarction. Circ. Res. 2016; 118: 216–221. [DOI] [PubMed] [Google Scholar]
- 12.Hirose K, Payumo AY, Cutie S, Hoang A, Zhang H, Guyot R, et al. Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science 2019; 364: 184–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buffenstein R, Woodley R, Thomadakis C, Daly TJ, Gray DA. Cold-induced changes in thyroid function in a poikilothermic mammal, the naked mole-rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001; 280: R149–55. [DOI] [PubMed] [Google Scholar]
- 14.Li M, Iismaa SE, Naqvi N, Nicks A, Husain A, Graham RM, et al. Thyroid hormone action in postnatal heart development. Stem Cell Res. 2014; 13: 582–591. [DOI] [PubMed] [Google Scholar]
- 15.Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 2012; 14: 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liversage RA, Korneluk RG. Serum levels of thyroid hormone during forelimb regeneration in the adult newt, Notophthalmus viridescens. J. Exp. Zool. 1978; 206: 223–228. [Google Scholar]
- 17.Chang J, Wang M, Gui W, Zhao Y, Yu L, Zhu G, et al. Changes in Thyroid Hormone Levels during Zebrafish Development. Zoolog. Sci. 2012; 29: 181–184. [DOI] [PubMed] [Google Scholar]
- 18.Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013; 132: 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans RM. The Steroid and Thyroid Hormone Receptor Superfamily on JSTOR. Science 1988; 240: 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rog-Zielinska EA, Thompson A, Kenyon CJ, Brownstein DG, Moran CM, Richardson J, et al. Glucocorticoid receptor is required for foetal heart maturation. Hum. Mol. Genet. 2013; 22: 3269–3282. [DOI] [PubMed] [Google Scholar]
- 21.Crochemore C, Michaelidis TM, Fischer D, Loeffler JP, Almeida OFX. Enhancement of p53 activity and inhibition of neural cell proliferation by glucocorticoid receptor activation. FASEB J. 2002; 16: 761–770. [DOI] [PubMed] [Google Scholar]
- 22.Smith E, Redman RA, Logg CR, Coetzee GA, Kashara N, Frenkel B, et al. Glucocorticoids Inhibit Developmental Stage-specific Osteoblast Cell Cycle. J. Biol. Chem. 2000; 275: 19992–20001. [DOI] [PubMed] [Google Scholar]
- 23.Gay MS, Li Y, Xiong F, Lin T, Zhang L. Dexamethasone treatment of newborn rats decreases cardiomyocyte endowment in the developing heart through epigenetic modifications. PLoS One 2015; 10: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gay MS, Dasgupta C, Li Y, Kanna A, Zhang L. Dexamethasone Induces Cardiomyocyte Terminal Differentiation via Epigenetic Repression of Cyclin D2 Gene. J. Pharmacol. Exp. Ther. 2016; 358: 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitehurst RM, Zhang M, Bhattacharjee A, Li M. Dexamethasone induced hypertrophy in rat neonatal cardiac myocytes involves an elevated L-type Ca2+ current. J. Mol. Cell. Cardiol. 1999; 31: 1551–1558. [DOI] [PubMed] [Google Scholar]
- 26.Huang WC, Yang CC, Chen IH, Liu YM, Chang SJ, Chuang YJ, et al. Treatment of Glucocorticoids Inhibited Early Immune Responses and Impaired Cardiac Repair in Adult Zebrafish. PLoS One 2013; 8: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jazwinska A, Sallin P. Acute stress is detrimental to heart regeneration in zebrafish. Open Biol. 2016; 6: 160012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bikle DD. Vitamin D metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014; 21: 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ringe JD, Schacht E. Improving the outcome of established therapies for osteoporosis by adding the active D-hormone analog alfacalcidol. Rheumatol. Int. 2017; 28: 103–111. [DOI] [PubMed] [Google Scholar]
- 30.Samuel S, Sitrin MD. Vitamin D’s role in cell proliferation and differentiation. Nutr. Rev. 2008; 66: S116–124. [DOI] [PubMed] [Google Scholar]
- 31.Hlaing SM, Garcia LA, Contreras JR, Norris KC, Ferrini MG, Artaza JN, et al. 1,25-Vitamin D3 promotes cardiac differentiation through modulation of the WNT signaling pathway. J. Mol. Endocrinol. 2014; 53: 303–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J. Steroid Biochem. Mol. Biol. 2007; 103: 533–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han Y, Chen A, Umansky KB, Oonk KA, Choi WE, Dickson AL, et al. Vitamin D Stimulates Cardiomyocyte Proliferation and Controls Organ Size and Regeneration in Zebrafish. Dev. Cell 2019; 48: 853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen S, Law CS, Grigsby CL, Olsen K, Hong TT, Zhang Y, et al. Cardiomyocyte-specific deletion of the vitamin D receptor gene results in cardiac hypertrophy. Circulation 2011; 124: 1838–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oakley RH, Ren R, Cruz-Topete D, Bird GS, Myers PH, Boyle MC, et al. Essential role of stress hormone signaling in cardiomyocytes for the prevention of heart disease. Proc. Natl. Acad. Sci. 2013; 110: 17035–17040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cruz-Topete D, Oakley RH, Carroll NG, He B, Myers PH, Xu X, et al. Deletion of the Cardiomyocyte Glucocorticoid Receptor Leads to Sexually Dimorphic Changes in Cardiac Gene Expression and Progression to Heart Failure. J. Am. Heart Assoc. 2019; 8: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clerch LB, Baras AS, DeCarlo Massaro G, Hoffman EP, Massaro D. DNA microarray analysis of neonatal mouse lung connects regulation of KDR with dexamethasone-induced inhibition of alveolar formation. Am. J. Physiol. - Lung Cell. Mol. Physiol. 2004; 286: L411–419. [DOI] [PubMed] [Google Scholar]
- 38.Kosmidis G, Bellin M, Ribero MC, Van Meer B, Oostward DWV, Passier R, et al. Altered calcium handling and increased contraction force in human embryonic stem cell derived cardiomyocytes following short term dexamethasone exposure. Biochem. Biophys. Res. Commun. 2015; 467: 998–1005. [DOI] [PubMed] [Google Scholar]
- 39.Bergmann O, Zdunek S, Felker A, Jovinge S, Salehpour M, Alkass K, et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015; 161: 1566–1575. [DOI] [PubMed] [Google Scholar]
- 40.Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am. J. Physiol. 1997; 272: H220–H226. [DOI] [PubMed] [Google Scholar]
- 41.Alkass K, Panula J, Westman M, Wu T, Bergmann O. No Evidence for Cardiomyocyte Number Expansion in Preadolescent Mice. Cell 2015; 163: 1026–1036. [DOI] [PubMed] [Google Scholar]
- 42.Senyo SE, Lee RT, Kühn B. Cardiac regeneration based on mechanisms of cardiomyocyte proliferation and differentiation. Stem Cell Res. 2014; 13: 532–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xin M, Olson EN, Bassel-Duby R. Mending broken hearts: Cardiac development as a basis for adult heart regeneration and repair. Nature Reviews Molecular Cell Biology 2013; 14: 529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiang MSW, Kikuchi K. Endogenous Mechanisms of Cardiac Regeneration. International Review of Cell and Molecular Biology 2016; 326: 67–131. [DOI] [PubMed] [Google Scholar]
- 45.Foglia MJ, Poss KD. Building and re-building the heart by cardiomyocyte proliferation. Development 2016; 143: 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Øvrebø JI, Edgar BA. Polyploidy in tissue homeostasis and regeneration. Development 2018; 145: dev156034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Connell TD, Berry JE, Jarvis AK, Somerman MJ, Simpson RU. 1,25-Dihydroxyvitamin D3 regulation of cardiac myocyte proliferation and hypertrophy. Am. J. Physiol. Circ. Physiol. 1997; 272: H1751–H1758. [DOI] [PubMed] [Google Scholar]
- 48.O’Meara CC, Wamstad JA, Gladstone RA, Fomovsky GM, Butty VL, Shrikumar A, et al. Transcriptional reversion of cardiac myocyte fate during mammalian cardiac regeneration. Circ. Res. 2015; 116: 804–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Porrello ER, Mahmoud AI, Simpson E, Hill JA, James A, Olson EN, et al. Transient Regenerative Potential of the Neonatal Mouse Heart. Science 2011; 331: 1078–1080 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cardoso-Moreira M, Halbert J, Valloton D, Velten B, Chen C, Shao Y, et al. Gene expression across mammalian organ development. Nature 2019; 571: 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.