

Abstract
Engineering polyketide synthases (PKS) to produce new metabolites requires an understanding of catalytic points of failure during substrate processing. Growing evidence indicates the thioesterase (TE) domain as a significant bottleneck within engineered PKS systems. We created a series of hybrid PKS modules bearing exchanged TE domains from heterologous pathways and challenged them with both native and non-native polyketide substrates. Reactions pairing wildtype PKS modules with non-native substrates primarily resulted in poor conversions to anticipated macrolactones. Likewise, product formation with native substrates and hybrid PKS modules bearing non-cognate TE domains was severely reduced. In contrast, non-native substrates were converted by most hybrid modules containing a substrate compatible TE, directly implicating this domain as the major catalytic gatekeeper and highlighting its value as a target for protein engineering to improve analog production in PKS pathways.
Keywords: Polyketides, Biosynthesis, Biocatalysis, Natural products, PKS Engineering
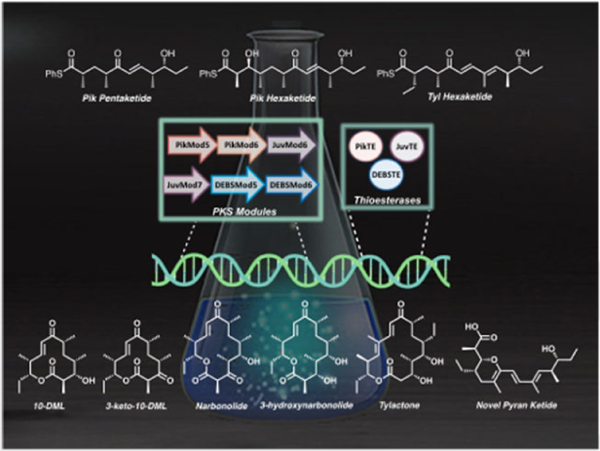
Graphical Abstract
Improved catalysis with engineered polyketide synthases: Pairing wild-type polyketide synthases with non-native substrates largely failed to produce the anticipated products. A series of hybrid modules bearing heterologous thioesterase domains were generated and employed to alleviate the observed catalytic bottleneck, resulting in the efficient processing of non-native substrates and an unexpected path to product diversity.
Engineered polyketide synthases (PKSs) are often found to be highly inefficient, displaying significant decreases in product yield or failing to generate the anticipated metabolite entirely compared to a wild type module against its native substrate.[1] Determining the point(s)-of-failure in these engineered pathways remains challenging due to the dependence on downstream domains to successfully process intermediates into mature, detectable small molecules.[2] In contrast, targeted in vitro reactions in which isolated PKS modules process full-length synthetic substrates provides information about decreased catalytic function within a PKS module. Rigorous characterization of the products generated in engineered PKS reactions is crucial to understanding and optimizing catalysis and metabolite production in these systems. Previous in vitro experiments have demonstrated that PKS modules can process non-native substrates in some cases and have delineated obstacles that limit engineering of PKS systems.[3] The present study focuses on how substitution of the thioesterase (TE) domain within a PKS module effects its ability to process native and non-native substrates. Herein, TE domains are shown to be a primary point of catalytic inefficiency in the processing of non-native substrates. We demonstrate that engineered modules containing a heterologous substrate-matched TE domain generally restore polyketide production.
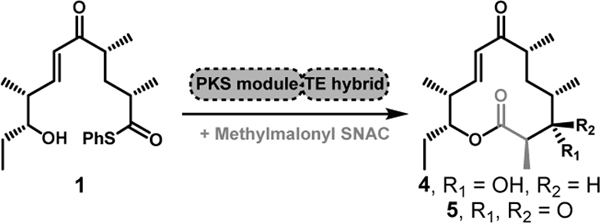
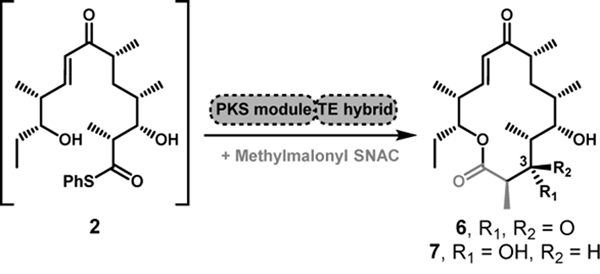
Initial experiments attempted to convert full-length synthetic polyketide intermediates (1–3, Figure 1A)[4] into the corresponding macrolactones (4–8) using the terminal two modules from the pikromycin (Pik) and erythromycin (DEBS) systems, and the penultimate module from the juvenimicin (Juv) pathway. These modules share similar domain architecture and ability to process advanced polyketide substrates into their respective macrolactone natural products (4, 6, 9–10, Figure 1B). Notably, the presence or absence of a ketoreductase (KR) domain within a PKS module influences if the final macrolactone contains a 3-hydroxy (4, 7) or 3-keto (5, 6) functionality. The use of penultimate modules for cyclization required the incorporation of a TE in place of C-terminal docking domains, modifications that have been designed previously for Pik Module 5 (Mod5) and DEBS Mod5.[5]
Figure 1.
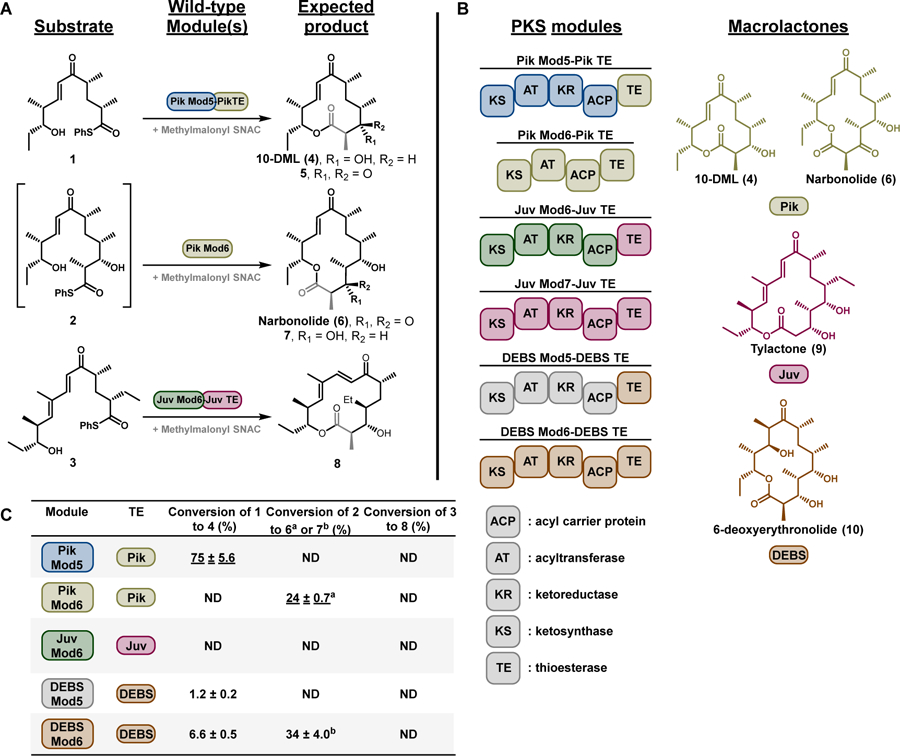
(a) Reaction scheme of PKS modules processing their native full-length synthetic substrates 14a, 24b, 34c to the corresponding macrolactones. Substrate 2 is generated by in situ photolysis of the 2-nitrobenzyloxymethyl ether (NBOM) protected native hexaketide.4b (b) Domain architecture of the PKS modules used in this study and the native macrolactone products from each respective biosynthetic pathway. (c) Table of percent conversion for reactions with substrates 1–3. The product yield for each PKS module processing its native substrate is underlined. Conversion to each macrolactone was monitored by HPLC with data represented as the mean ± standard deviation where n = 3. ND = not detected.
Wild-type PKS modules (Figure 1A) efficiently processed native substrates in vitro into corresponding macrolactones (Figure 1C, underlined), while modules presented with non-native substrates typically showed a severe decrease in product formation. The Pik Mod5-Pik TE hybrid protein efficiently processed native substrate (1) to 10-dml (4) with 75% conversion; however, when DEBS Mod5-DEBS TE and DEBS Mod6-DEBS TE were reacted with 1 as a non-native substrate, poor conversions of 1.2 and 6.6% were observed, respectively. Likewise, reactions with 2 followed a similar trend. Consistent with our previous report[4b], Pik Mod6-Pik TE efficiently processes 2 to narbonolide (6). Although DEBS Mod6-DEBS TE produced substantial levels of 3-hydroxy-narbonolide 7 (due to the presence of a KR in the DEBS Mod6 domain architecture), all other non-native reaction pairings failed to produce appreciable levels of either macrolactone 6 or 7 from 2. Furthermore, all reactions containing 3 failed to generate the predicted product (8).
This analysis of PKS modules with native and non-native substrates reinforces the challenge of designing and engineering PKS modules for synthetic biology applications.[6] Recently, we have shown[7] that TE modifications have a significant effect on the catalytic processing of non-native substrates by PKS modules. We concluded that the decreased yields from reactions containing non-native substrates (Figure 1C) are at least partially due to ineffective processing within the TE domain.
To determine if the decreased yields originate from compatibility issues between the polyketide intermediates and the TE domain in each terminal module, we generated a series of PKS fusion proteins where the native TE domain was exchanged for a corresponding domain from a heterologous system (see Supporting Information). Modules were chosen from the functionally related Pik, DEBS, and Juv biosynthetic pathways which generate macrolactones that vary in size from 12- to 16-membered rings (Figure 1B). All modules tested possess the core ketosynthase (KS), acyl transferase (AT), and acyl carrier protein (ACP) domains critical for extension of a polyketide with an additional acyl unit, and all except Pik Mod6 contain a ketoreductase (KR) domain, which reduces the post-extension β-keto group to a hydroxyl (i.e., Figure 1A, 4 versus 5). This provided hybrid PKS modules with either a cognate TE from its own biosynthetic pathway or non-cognate TE domain from the other two pathways (Figure 1, Table 1). We then characterized the effect of the non-cognate TE on the ability of the hybrid PKS modules to process native and non-native substrates.
Table 1.
Evaluation of PKS TE hybrid modules with 1, the native substrate of Pik Mod5.[a]
 | |||
|---|---|---|---|
| Module | TE | Conversion to 4 (%) | Conversion to 5 (%) |
 |
 |
1.2 ±0.2 | trace |
 |
ND | ND | |
 |
21 ± 07 | trace | |
 |
|
6.6 ± 0.5 | ND |
|
ND | ND | |
|
17 ± 0.5 | 6.6 ± 0.1 | |
 |
|
2.3 ± 0.4 | trace |
|
ND | ND | |
|
41 ± 2.8 | trace | |
 |
|
2.9 ± 0.2 | trace |
|
ND | ND | |
|
75 ± 5.6 | trace | |
Reactions where the TE domain is paired with the substrate are underlined. Conversion to 4 or 5 was monitored by HPLC with data represented as the mean ± standard deviation where n = 3. Trace=detected by LC-MS but below the detection limit of HPLC. ND= not detected.
Our initial expansion tested the flexibility of the hybrid TE modules using 1 as the substrate (Table 1). While all the hybrid modules share the same domain configuration (KS-AT-KR-ACP-TE) and are expected to produce 10-dml (4) from 1, 3-keto-10-dml (5) has also been reported as a result of domain-skipping.[3] HPLC quantification of the reaction products compared to authentic standards of 4 and 5[7b] provided new insights into two key questions: First, can PKS module-TE domain hybrids from different type I PKS pathways function in an engineered context, and second, how does a match between the TE domain and the incoming linear (acyl-ACP) intermediate affect the efficiency of product formation? In all reactions where 1 was a non-native substrate (e.g. DEBS Mod5, DEBS Mod6, and Juv Mod6 with 1), we achieved significant conversions to 10-dml (4, Table 1) when the substrate (1) matched the TE domain (Pik TE, underlined yields). Our results demonstrate that the KS-AT-ACP-KR domains of these PKS modules maintain a suitable level of catalytic function when the downstream TE domain (Pik TE) is correctly matched to the incoming unnatural intermediate (1). This conclusion is supported by the 21, 17, 41, and 75% conversions from the reactions containing, DEBS Mod5-Pik TE, DEBS Mod6-Pik TE, Juv Mod6-Pik TE, Pik Mod5-Pik TE, respectively (Table 1). These results also indicate that the DEBS and Juv TE domains are unable to efficiently generate 12-membered macrolactones as the percent conversions of hybrid modules containing these TE domains were low, even when the substrate (1) was native to the PKS module (Pik Mod5). The observation that pairing of the TE domain to the native polyketide substrate considerably enhances product yield reinforces the value of assessing the innate substrate specificity of TE domains in the context of engineered PKSs. Although attenuated yields may also be attributed to one or more of the other PKS catalytic domains (e.g. KS, AT, KR, ACP), the TE has been identified as a central feature for effective off-loading and cyclization of polyketides.[7] Selection or modification of a TE within a module can therefore increase product yields and processing of non-native substrates.
We next expanded our studies to assess substrate flexibility by probing hybrid TE modules for the ability to accept and process Pik hexaketide 2[4b] into 14-membered macrolactones using HPLC quantification against known standards (Table 2). Similar to the results using 1, substitution of the cognate Pik TE domain in Pik Mod6 with either the DEBS or Juv TE domains led to a severe decrease in the ability of Pik Mod6 to convert 2 into macrolactone 6. However, when reacted with DEBS Mod6, 2 could be converted into 6 and 7. Due to the domain architecture of DEBS Mod6, the expected product is C-3-OH macrolactone 7. Compound 7 is produced with the DEBS and Juv TE fusion proteins, but with the Pik TE, C-3-keto macrolactone 6 is favored. This result suggests a preference of the various TE domains for functionality at the C-3 position that matches their native chain elongation intermediate when offloading 14-membered rings. Thus, the hybrid DEBS Mod6-Pik TE produces predominantly narbonolide (6), suggesting that this hybrid mediates skipping of the KR domain [5a, 8], in preference for macrolactonization with the keto group at C-3, as dictated by the match between the TE and substrate oxidation state (Table 2).
Table 2.
Evaluation of PKS TE hybrid modules with 2.[a]
 | |||
|---|---|---|---|
| Module | TE | Conversion to 6 (%) | Conversion to 7 (%) |
 |
|
trace | 34 ± 4.0 |
|
ND | 9.6 ± 1.0 | |
|
22 ± 0.7 | 3.5 ± 0.1 | |
 |
|
ND | trace |
|
ND | ND | |
|
trace | ND | |
 |
|
trace | ND |
|
ND | ND | |
|
24 ± 0.7 | ND | |
Substrate 2 is generated by in situ photolysis of the 2-nitrobenzyloxymethyl ether (NBOM) protected native hexaketide.4b Conversion to 6 or 7 was monitored by HPLC with data represented as the mean ± standard deviation where n = 3. Trace = detected by LC-MS but below the detection limit of HPLC. ND = not detected.
These data show that the ability of a TE domain to effectively cyclize an advanced polyketide intermediate depends on the functionality present in the linear polyketide chain (Figure 2). The native linear seco-acid product of Pik Mod6 is unreduced (i.e. keto group) at the C-3 position and its cognate TE displays a preference for this ACP-bound substrate. Surprisingly, when substituted onto the DEBS Mod6 as the DEBS Mod6-Pik TE hybrid, the Pik TE retains its preference for the 3-keto ACP-tethered intermediate and selectively catalyzes cyclization to 6. This result suggests that the binding affinity of the unreduced ACP-tethered intermediate is higher toward the non-cognate Pik TE rather than to its own natural KR domain, thus leading to release of 6 instead of KR-mediated reduction of the β-keto group at C-3 and release of 7. This ability of the Pik TE to strongly interact with C-3 keto intermediates is also observed in the reaction of 1 with DEBS Mod6-Pik TE leading to 5 (Table 1).
Figure 2.
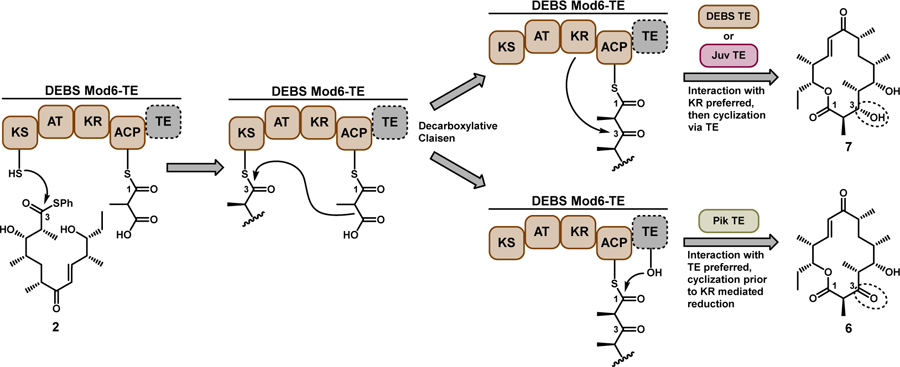
Biocatalytic mechanism of the processing of 2 by DEBS Mod6. Individual PKS-TE fusion proteins produce divergent products apparently due to innate affinities between the β-keto thioester and the TE versus KR.
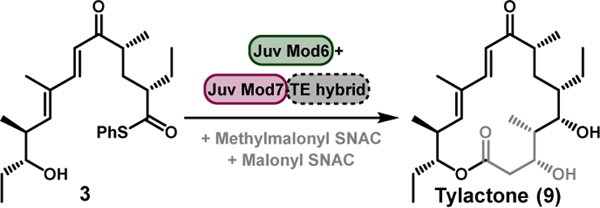
Next, we tested the ability of hybrid TEs to retain activity when coupled with an upstream module, an interaction critical to late-stage PKS engineering as it ensures that a hybrid protein will function correctly in the context of other modules. Reactions were performed using 3[4c] in its native context with Juv Mod6, which must then pass an elongated intermediate to Juv Mod7-TE hybrids for an additional extension and cyclization (Table 3). LC-MS analysis of these reactions revealed that DEBS, Juv, and Pik TE domains all possessed the ability to generate a 16-membered macrolactone when fused to Juv Mod7. HPLC quantification of tylactone (9) production demonstrated Juv Mod6 + Juv Mod7-Juv TE to be the most efficient (34%), followed closely by Juv Mod7-DEBS TE (30%). This result not only shows that PKS hybrid TE modules are catalytically competent when used with upstream modules, but also further supports selectivity in the TE for 3-hydroxy or 3-keto functionality.
Table 3.
Reaction of 3 with Juv Mod6 + Juv Mod7 TE hybrids.[a]
 | ||
|---|---|---|
| Module | TE | Conversion to 9 (%) |
 |
|
30 ± 0.8 |
|
34 ± 3.3 | |
|
trace | |
Conversion to 9 was monitored by HPLC with data represented as the mean ± standard deviation where n = 3. Trace=detected by LC-MS but below the detection limit of HPLC.
Having identified the crucial role of the TE domain in engineered PKS systems, we sought to harness hybrid TE modules to produce unnatural macrolactones. Accordingly, we utilized the Juv Mod6 TE hybrids and screened with substrate 3 (the natural substrate for this module), to probe for the ability to generate the predicted 14-membered diene macrolactone 8 (Scheme 1). Analytical experiments assessed by LC-MS initially indicated that a product corresponding to the correct mass was formed only when Juv Mod6 was paired with the Pik TE domain. To confirm this initial result, we performed a 0.1 mmol scale reaction and purified the product by preparatory HPLC. Surprisingly, the isolated product was a pyran containing seco-acid 13 (Scheme 1) as opposed to the expected macrolactone 8. The historical reliance on in vivo reactions and assessing fermentation extracts by liquid chromatography-mass spectrometry (LC-MS) analysis of the culture medium has limited the ability to characterize new structures.[2a, 2b] LC-MS analysis can provide ambiguous data with isomeric molecules generating identical mass spectra, precluding definitive structural assignment. Importantly, pyran 13 and the anticipated macrolactone 8 share the same exact mass and were ultimately distinguished by a full complement of NMR studies (see Supporting Information).
Scheme 1.
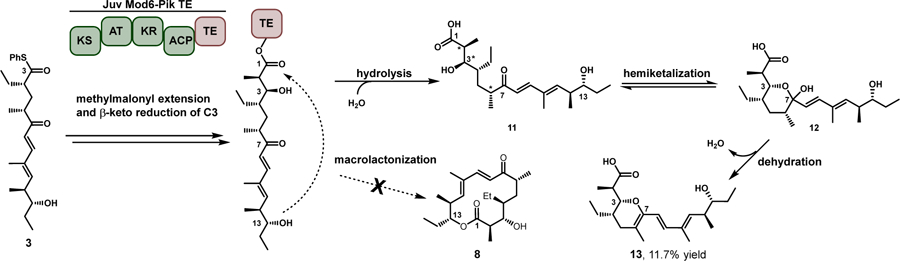
Reaction of Juv Mod6-Pik TE with Tyl hexaketide 3 results in pyran 13, which is identical in mass to macrolactone 8. This outcome was only achieved via extension of Tyl hexaketide through Juv Mod6-Pik TE and hydrolytic offloading to the linear seco acid 11, which can rapidly interconvert to the hemiketal containing product 12. Finally, the hemiketal is spontaneously dehydrated to produce the observed pyran 13. Stereochemistry displayed is that of the native product 11 and subsequent hemiketalization (12). *represent likely points of inverted stereochemistry seen in the isolated product.
Initial efforts aimed at evaluating the origin of this new dehydrated pyran functionality centered on discerning whether it resulted directly from TE mediated catalysis, or by offloading the linear acid with subsequent spontaneous hemiketalization and dehydration. Addressing this issue was approached using two methods: first a serine to alanine TE inactivating mutant was generated in Juv Mod6-Pik TE (Juv Mod6-Pik TES148A) to decouple the activity of the TE domain from that of the module. Second, a rapid time course analysis was conducted using LC-MS analysis to monitor the release of seco-acid and/or hemiketal products as well as formation of the dehydrated pyran moiety. Initial analytical reactions with Juv Mod6-PikTES148A failed to produce recognizable seco-acid/hemiketal products.
Next, we evaluated the time course experiments with the goal of understanding the origin of the dehydrated pyran (see Supporting Information). These efforts yielded emergence of a substance corresponding to either the seco-acid and/or hemiketal upon incubation for 1–2 hours. Semi-preparative scale up and characterization of this compound to determine its identity as a linear acid, hemiketal or a mixture of the two proved challenging, however, a fraction consisting of a mixture of four isomers was obtained. Efforts to separate these molecules by subsequent rounds of purification proved futile indicating the mixture is likely interconverting. NMR characterization yielded nearly complete assignment of the four compounds, which include three linear chain stereoisomers (sites of epimerization are unclear) as well as a single hemiketal constituent 12. In order to confirm that this mixture could spontaneously dehydrate, the initial mixture was subjected to buffered conditions with and without Juv Mod6-Pik TE. Monitoring the product profile over time demonstrated partial conversion to the dehydrated pyran 13 irrespective of Juv Mod6-Pik TE enzyme. These results indicate that the extended hexaketide is likely offloaded from the PKS module as the seco-acid, which rapidly interconverts between the hemiketal and the seco-acid, and subsequently dehydrates to generate the final isolated product 13 (Scheme 1).
The work described herein provides further insight into the substrate flexibility of type I modular PKS TE domains and their application in PKS engineering. The TE domain is a critical catalytic gatekeeper for the processing of unnatural substrates into macrolactones and other new molecules, and partially explains previous studies where non-native substrates failed to be processed by engineered PKS modules.[5a] To explore the extent of this limitation and overcome it, we generated a series of type I PKS modules fused with TE domains from three related biosynthetic pathways and assessed each for catalytic activity with select polyketide substrates to emulate the final catalytic steps in engineered pathways. Although the TE interactions are not the only factor involved in polyketide assembly and offloading (e.g. Juv Mod6 hybrids fail to process 2, Table 2), we achieved robust catalysis of non-native substrates through PKS modules when the TE domain was matched with the substrate from its native PKS. In addition to explaining the attenuated yields of polyketides obtained from engineered PKSs described in previous studies,[7] the current work generates further experimental support for the critical role of the TE domain in the processing of unnatural intermediates. These results are in accord with allied efforts performed in fungal iterative PKSs, which identified the TE domain as a key gatekeeper in the production of fungal polyketides.[9]
Furthermore, our results indicate that identity of the TE domain may also alter the sequence of catalytic events that occurs in engineered PKS modules. This possibility is highlighted by the product divergence when DEBS Mod6 TE hybrids process the unnatural substrate 2, giving either 6 (3-keto) or 7 (3-hydroxy) macrolactones predominantly (Figure 1). Recent work from Kalkreuter et al. also noted the gatekeeper role of the KR by observing that assembly of unnatural polyketide intermediates resulted in a propensity to skip the PKS module reductive step, setting keto functionality at the respective C-atom.[10] Future structural and protein engineering studies will focus on delineating the role of the TE domain in the processing of unnatural substrates. By exploring how non-native TE domains can contribute to diminished product formation observed with combinatorial biosynthesis and directed pathway engineering strategies, we will be able to more effectively design bacterial type I PKS pathways for enhanced production of polyketide natural product analogs, a long-standing goal in secondary metabolite biosynthesis and synthetic biology.[1, 3, 11]
Supplementary Material
Acknowledgements
We gratefully acknowledge NIH grant R35 GM118101, and the Hans W. Vahlteich Professorship (D.H.S.). This research was partially supported by Rackham Merit Predoctoral Fellowships (D.A.H. and A.A.K.), T32-CA009676 (A.A.K. training fellowship), and an American Foundation for Pharmaceutical Education Predoctoral Fellowship (D.A.H.). We thank Dr. Zachary Litman for critical reading of this manuscript.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Weissman KJ, Nat. Prod. Rep 2016, 33, 203–230. [DOI] [PubMed] [Google Scholar]
- [2].a) McDaniel R, Thamchaipenet A, Gustafsson C, Fu H, Betlach M, Betlach M, Ashley G, Proc. Natl. Acad. Sci. U. S. A 1999, 96, 1846–1851; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xue Q, Ashley G, Hutchinson CR, Santi DV, Proc. Natl. Acad. Sci. U. S. A 1999, 96, 11740–11745; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jacobsen JR, Keatinge-Clay AT, Cane DE, Khosla C, Bioorg. Med. Chem 1998, 6, 1171–1177; [DOI] [PubMed] [Google Scholar]; d) Leaf T, Cadapan L, Carreras C, Regentin R, Ou S, Woo E, Ashley G, Licari P, Biotechnol. Prog 2000, 16, 553–556; [DOI] [PubMed] [Google Scholar]; e) Kinoshita K, Williard PG, Khosla C, Cane DE, J. Am. Chem. Soc 2001, 123, 2495–2502; [DOI] [PubMed] [Google Scholar]; f) Harvey CJ, Puglisi JD, Pande VS, Cane DE, Khosla C, J. Am. Chem. Soc 2012, 134, 12259–12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kittendorf JD, Sherman DH, Bioorg. Med. Chem 2009, 17, 2137–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Hansen DA, Rath CM, Eisman EB, Narayan AR, Kittendorf JD, Mortison JD, Yoon YJ, Sherman DH, J. Am. Chem. Soc 2013, 135, 11232–11238; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hansen DA, Koch AA, Sherman DH, J. Am. Chem. Soc 2015, 137, 3735–3738; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lowell AN, DeMars MD, Slocum ST, Yu F, Anand K, Chemler JA, Korakavi N, Priessnitz JK, Park SR, Koch AA, Schultz PJ, Sherman DH, J. Am. Chem. Soc 2017, 139, 7913–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Mortison JD, Kittendorf JD, Sherman DH, J. Am. Chem. Soc 2009, 131, 15784–15793; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Watanabe K, Wang CCC, Boddy CN, Cane DE, Khosla C, J. Biol. Chem 2003, 278, 42020–42026; [DOI] [PubMed] [Google Scholar]; c) Yin Y, Lu H, Khosla C, Cane DE, J. Am. Chem. Soc 2003, 125, 5671–5676. [DOI] [PubMed] [Google Scholar]
- [6].Kalkreuter E, Williams GJ, Curr. Opin. Microbiol 2018, 45, 140–148. [DOI] [PubMed] [Google Scholar]
- [7].a) Hansen DA, Koch AA, Sherman DH, J. Am. Chem. Soc 2017, 139, 13450–13455; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Koch AA, Hansen DA, Shende VV, Furan LR, Houk KN, Jiménez-Osés G, Sherman DH, J. Am. Chem. Soc 2017, 139, 13456–13465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chemler JA, Tripathi A, Hansen DA, O’Neil-Johnson M, Williams RB, Starks C, Park SR, Sherman DH, J. Am. Chem. Soc 2015, 137, 10603–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Xu Y, Zhou T, Zhang S, Xuan L-J, Zhan J, Molnár I, J. Am. Chem. Soc 2013, 135, 10783–10791; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xu Y, Zhou T, Zhang S, Espinosa-Artiles P, Wang L, Zhang W, Lin M, Gunatilaka AAL, Zhan J, Molnár I, Proc. Natl. Acad. Sci. U. S. A 2014, 111, 12354–12359; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Newman AG, Vagstad AL, Belecki K, Scheerer JR, Townsend CA, Chem. Commun. (Camb.) 2012, 48, 11772–11774; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Newman AG, Vagstad AL, Storm PA, Townsend CA, J. Am. Chem. Soc 2014, 136, 7348–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kalkreuter E, CroweTipton JM, Lowell AN, Sherman DH, Williams GJ, J. Am. Chem. Soc 2019, 141, 1961–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Kittendorf JD, Sherman DH, Curr. Opin. Biotechnol 2006, 17, 597–605; [DOI] [PubMed] [Google Scholar]; b Wong FT, Khosla C, Curr. Opin. Chem. Biol 2012, 16, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.