

Abstract
Guanine radicals are important reactive intermediates in DNA damage. Hydroxyl radical (HO•) has long been believed to react with 2’-deoxyguanosine (dG) generating 2’-deoxyguanosin-N1-yl radical (dG(N1-H)•) via addition to the nucleobase π-system and subsequent dehydration. This basic tenet was challenged by an alternative mechanism, in which the major reaction of HO• with dG was proposed to involve hydrogen atom abstraction from the N2-amine. The 2’-deoxyguanosin-N2-yl radical (dG(N2-H)•) formed was proposed to rapidly tautomerize to dG(N1-H)•. We report the first independent generation of dG(N2-H)• in high yield via photolysis of 1. dG(N2-H)• is directly observed upon nanosecond laser flash photolysis (LFP) of 1. The absorption spectrum of dG(N2-H)• is corroborated by DFT studies, and anti- and syn-dG(N2-H)• are resolved for the first time. The LFP experiments showed no evidence for tautomerization of dG(N2-H)• to dG(N1-H)• within hundreds of microseconds. This observation suggests that the generation of dG(N1-H)• via dG(N2-H)• following hydrogen atom abstraction from dG is unlikely to be a major pathway when HO• reacts with dG.
Keywords: DNA damage, guanine oxidation, radical reactivity, laser flash photolysis, oxidative stress
GRAPHICAL ABSTRACT
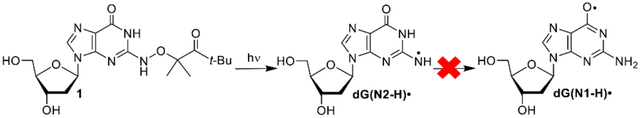
dG(N2-H)• and dG(N1-H)• are major reactive species produced upon guanine oxidation. dG(N2-H)• was independently generated from 1. dG(N2-H)• reacts significantly more slowly with thiol reducing agents than alkyl radicals. Contrary to previous reports, there is no evidence that dG(N2-H)• rearranges to dG(N1-H)• on the sub-millisecond timescale
Introduction
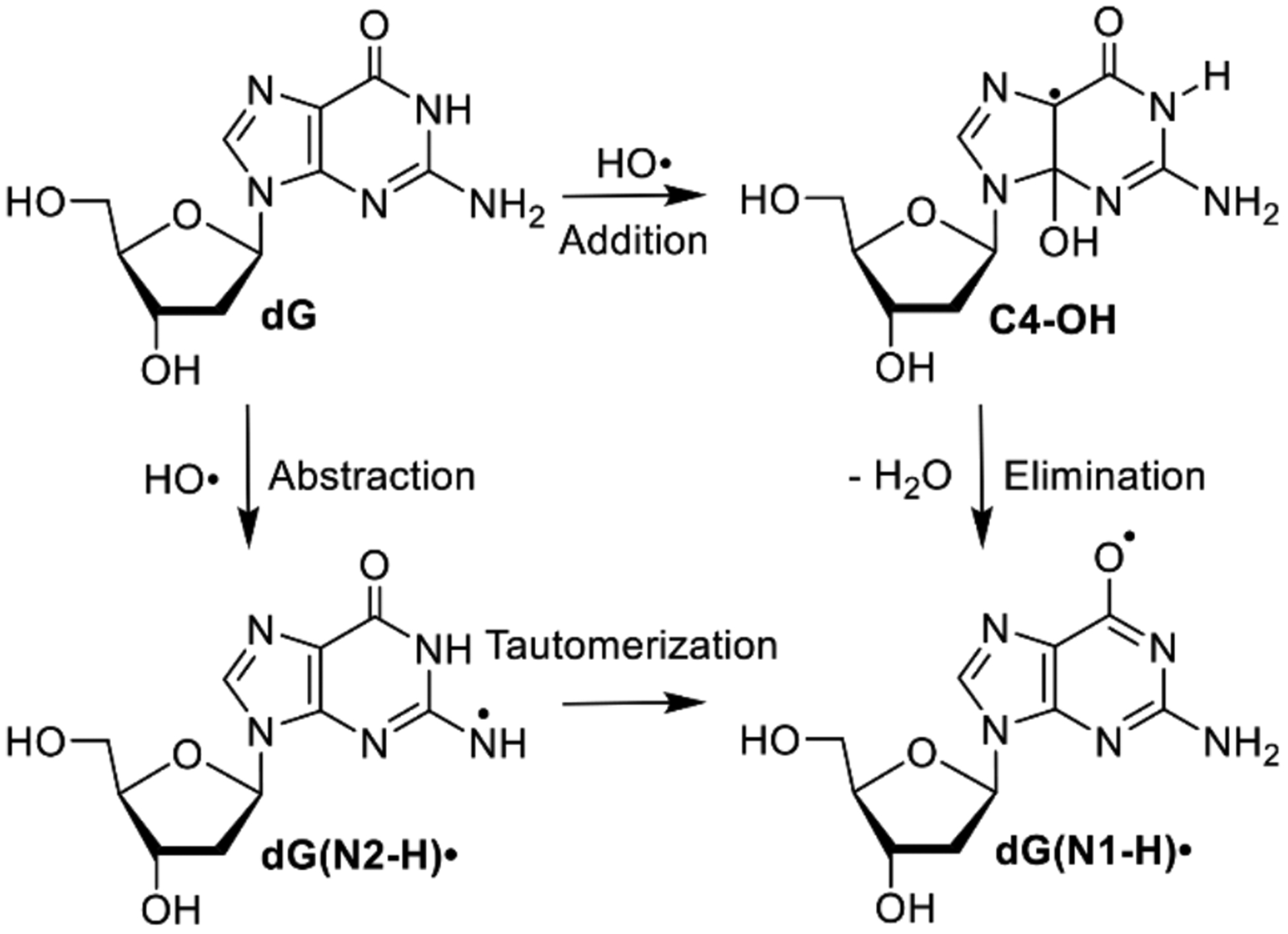
Nucleic acid oxidation is central to human health. For instance, it is a factor in aging and in the development of cancer.[1] DNA is also the molecular target for a variety of cancer treatments. Many of these methods involve one electron oxidation of DNA, with ionizing radiation being the most common.[2] Ionizing radiation damages DNA directly by ionizing DNA and indirectly by ionizing water, which generates hydroxyl radical (HO•), a highly reactive DNA damaging species.[3] 2’-Deoxyguanosine (dG) is the most readily oxidized of the 4 native nucleosides and is also a primary contributor to electron transfer in one-electron oxidized DNA.[4] Consequently, the reactive intermediates produced upon dG oxidation, and their reactivity, have been the focus of important theoretical studies and experimental investigations for the past 30 years.[5] Pulse radiolysis has been used extensively to characterize the early, rapid dG oxidation events that are complete on the sub-millisecond timescale.[5c] dG(N1-H)• is the major and thermodynamically most stable intermediate generated by reaction of dG with HO• (Scheme 1).[6] However, the mechanism(s) by which dG(N1-H)• is formed is controversial and was recently proposed to arise by tautomerization of dG(N2-H)• (Scheme 1).[7] We have resolved this problem by using near-UV photolysis of a synthetic precursor to dG(N2-H)• in conjunction with time-resolved spectroscopy, time-dependent DFT calculations and product studies.
Scheme 1.
Generation of guanine radicals by hydroxyl radical (HO•).
It is widely accepted that HO• reacts with dG yielding dG(N1-H)• via an addition-elimination mechanism. HO• adds to C4, C5 and C8 atoms of guanine. C4-OH (Scheme 1) is proposed to be the major product, accounting for 60–70% of the reactions.[6a] Computational studies indicate that upon barrierless addition of HO•, C4-OH produces dG(N1-H)• via loss of hydroxide to form an ion pair, followed by N1-deprotonation.[5b] Calculations predict that ion pair formation encounters ~6.5 kcal/mol barrier and is the rate determining step. N1-deprotonation within the ion pair is kinetically and thermodynamically favored (>7 kcal/mol) over the N2-position. The calculated energy difference between the radicals is basis set dependent, but dG(N1-H)• is 2–4 kcal/mol more stable than dG(N2-H)•.[8]
The HO• pathway to dG(N1-H)• through C4-OH was challenged by a series of pulse radiolysis experiments carried out on various guanine derivatives.[7] The authors posited that HO• preferentially abstracts the N2-hydrogen atom, instead of adding to the p-bond to generate C4-OH (Scheme 1). Initially formed dG(N2-H)• was proposed to rearrange to the more stable dG(N1-H)• with a < 30 μs half-life at 298 K and an activation energy of ~5.5 kcal/mol. The feasibility of the hydrogen atom abstraction of this alternative mechanism was corroborated by DFT calculations.[5b] Pulse radiolysis, in conjunction with spectroscopic detection (and other measurements), is a powerful approach for studying reactive intermediate chemistry. However, one limitation is that multiple reactive intermediates can be produced. This can potentially complicate analysis, particularly if reactive intermediates have overlapping spectral properties. To simplify the examination of purine radicals and resolve mechanistic conflicts in the aforementioned studies, we designed a photochemical precursor (1) that generates a single purine intermediate, dG(N2-H)•. We thoroughly examined the reactivity of this radical by product analysis following UV-photolysis of 1 and time-resolved LFP experiments that corroborate each other. In addition to refuting the sub-millisecond tautomerization of dG(N2-H)•, we also distinguished the anti- and syn-conformers of dG(N2-H)• for the first time.
Results and Discussion
Design and synthesis of a photochemical precursor (1) for dG(N2-H)•.
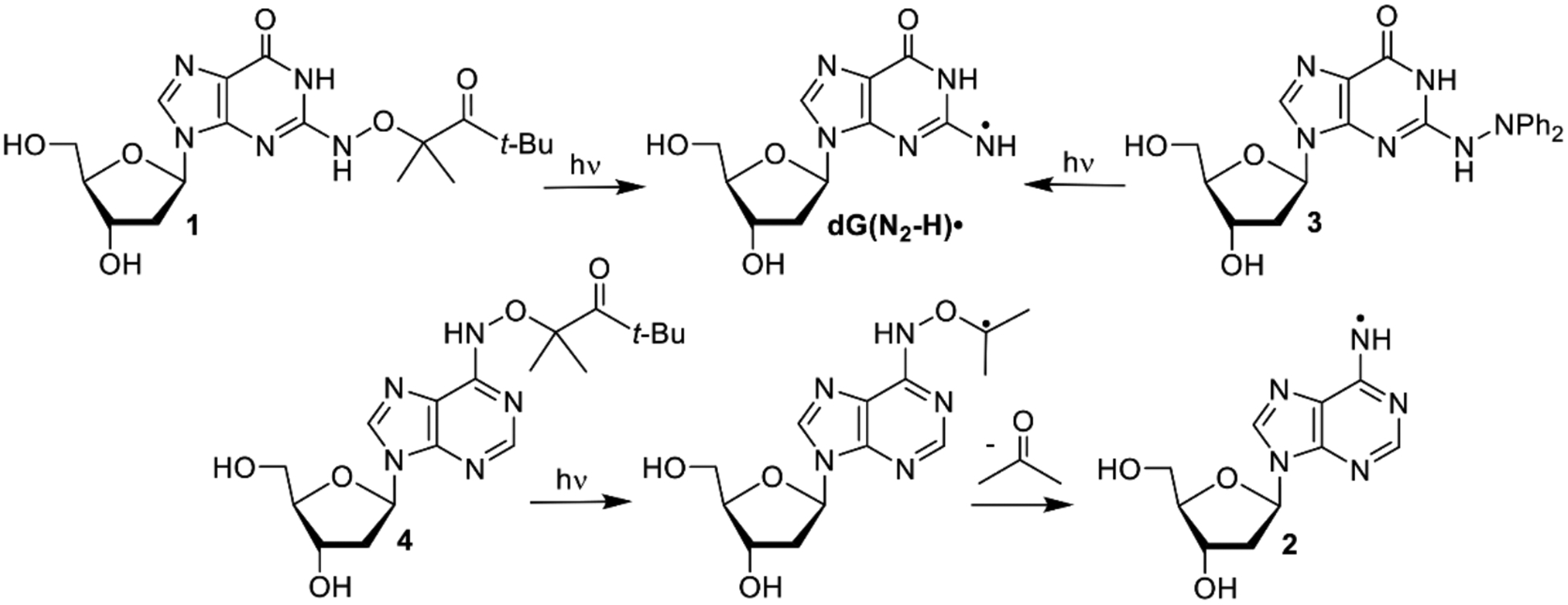
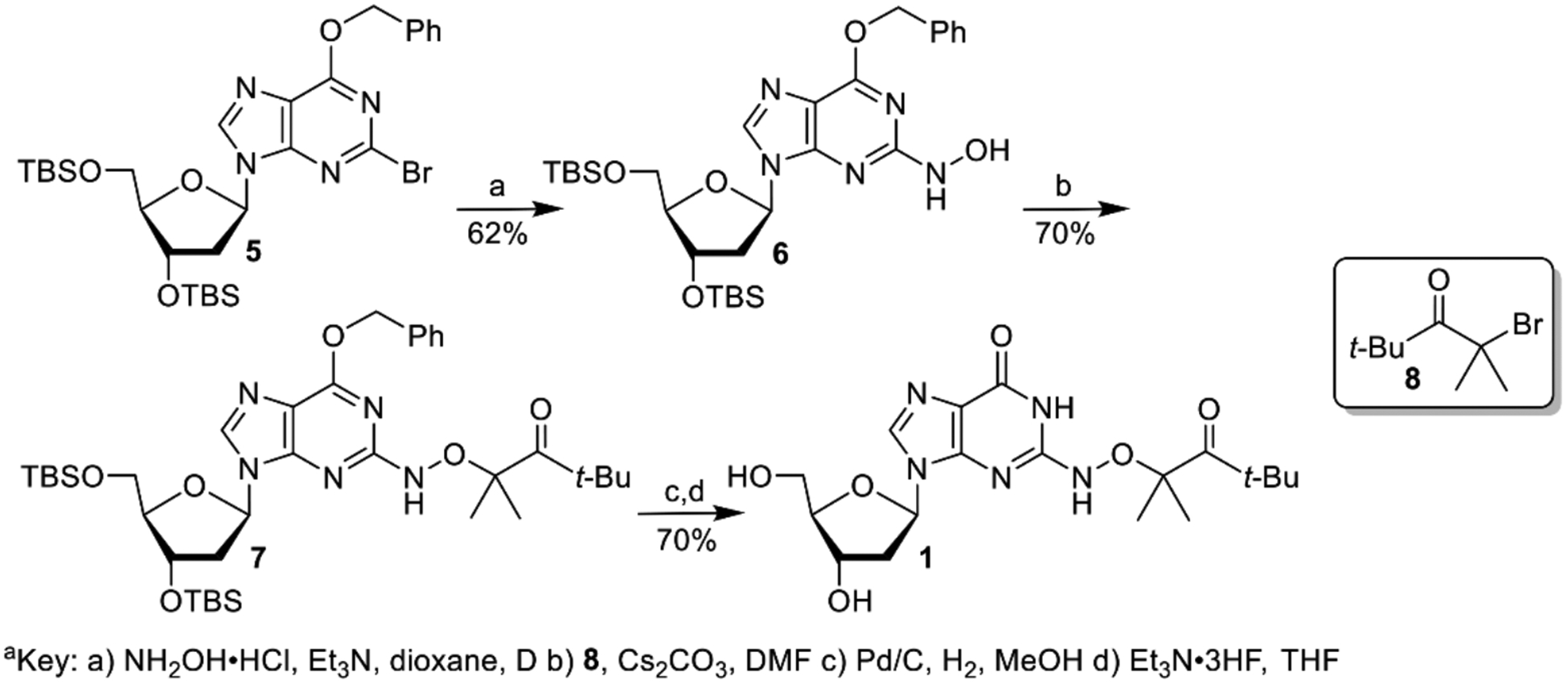
We previously generated dG(N2-H)• and 2’-deoxyadenosin-N6-yl radical (2) from the corresponding diphenyl hydrazines (e.g. 3, Scheme 2).[9] However, photochemical conversion of 3 to dG(N2-H)• is too inefficient for laser flash photolysis examination of dG(N2-H)•. More recently, we reported on a method for generating 2’-deoxyadenosin-N6-yl radical (2) from a ketone precursor (4, Scheme 2).[10] Upon photolysis, 4 undergoes Norrish Type I photocleavage followed by rapid b-fragmentation. Using acetone as triplet photosensitizer greatly accelerated the conversion of 4 and allowed us to obtain the spectrum of 2. However, photosensitization of 3 by ketones was not attempted, because photodissociation of tetraphenylhydrazine occurs from the excited singlet.[11] Furthermore, we anticipated that the ketones, which photo oxidize 8-oxodGuo, would do the same to the more readily oxidizable 3.[9, 12] We rationalized that 1 would yield dG(N2-H)• via the analogous cascade of reactions that 4 undergoes upon photolysis, and could also be sensitized by ketones (Scheme 2). The synthetic approach to.1 was strongly influenced by that of 4 and started from previously reported 5 (Scheme 3).[10a, 13] Substitution of the bromide in 5 by hydroxylamine (6) was slower than the analogous reaction in the synthesis of 4 due to the increased electron density of guanine. The formation of 6 required higher temperature than that for the dA analogue, and dioxane was substituted for THF, because of its higher boiling point. Introduction of the hindered ketone 8 using previously reported conditions, in which NaH was used as base led to the formation of an undesired product. Subsequently, the substitution was successfully carried out using Cs2CO3 as base. The desired ketone (1) was then obtained via standard debenzylation and desilylation conditions.
Scheme 2.
Photochemical generation of purine radicals from synthetic precursors.
Scheme 3.
Synthesis of dG(N2-H)• precursor 1.[a]
Photochemical generation of dG(N2-H)• and product studies.
With an eye on utilizing 1 as a source of dG(N2-H)• in DNA, we carried out photolyses in Pyrex vessels using lamps whose maximum output is at 350 nm. Although the λmax for 1 occurs in a far shorter region (λmax = 260 nm, ε = 1.22 × 104 M−1s−1 in H2O) than where these lamps emit, the absorption band tails above 300 nm (Figure S5). The quantum yield for disappearance of 1 under these conditions, measured using 2-hydroxy-2-methylpropiophenone as an actinometer, was similar (F = 1.8 ´ 10−3) to that of 4 (F = 1.5 ´ 10−3).[10a, 14] However, the weaker absorbance of 1 above 300 nm resulted in less efficient photochemical conversion than 4, such that only ~10% of the ketone was consumed following 8 h direct irradiation. Inspired by the sensitization used during the photolysis of 4, we used acetone (2% v/v,150 mM) to sensitize the reaction and achieved increased conversion to 62.4 ± 0.9% after 8 h in the presence of PhSH as reducing agent. The yield of dG and mass balance were high when Fe2+ or PhSH were used as a reducing agent (Table 1). In contrast to 2, bmercaptoethanol (BME) also effectively trapped dG(N2-H)• producing dG.[10a] We attribute this difference to the fact that guanine is more electron-rich than adenine. The corresponding nitrogen radical (dG(N2-H)•) is less electrophilic than 2, and encounters lower energy barriers when reacting with electronegative thiol hydrogen atom donors. These observations are consistent with our hypothesis regarding the polarity matching between hydrogen atom donors and neutral purine radicals.[10a] Purine electronic properties also manifest themselves when photolysis is carried out in the absence of a reducing agent. Under these conditions the mass balance and dG yield decrease almost two-fold but the conversion rate of 1 is approximately twice as high as when a reducing agent added (Figure S6). This is consistent with the proposal that the radical precursor can serve as a reducing reagent for dG(N2-H)•, a pathway that will be significant in laser flash photolysis (LFP) experiments described below.
Table 1.
Product studies upon anaerobic photolysis of 1 in the presence of acetone (2% v/v) as sensitizer.[a]
| Reducing Agent (mM) | % Yield dG[b] | % Mass Balance[b] |
|---|---|---|
| Fe2+ (10) | 85 ± 2 | 90 ± 1 |
| PhSH (10) | 89 ± 3 | 93 ± 1 |
| BME (10) | 87 ± 2 | 91 ± 2 |
| None[c] | 55 ± 3 | 65 ± 1 |
| None[d] | 35 ± 2 | 44 ± 3 |
[1] = 0.1 mM.
Average ± std. dev. of 3 experiments.
Anaerobic photolysis.
Aerobic photolysis
Although acetone photosensitizes 1, comparison to the sensitizer’s effect on 4 indicated that photochemical conversion would be too low for LFP studies.[10a] Consequently, we considered photolysis in the presence of other photosensitizers. Acetophenone was a promising candidate due to its efficient intersystem crossing, long triplet lifetime, and relatively high triplet energy.[15] Anaerobic photolyses were carried out in the presence of acetophenone (1% v/v, 86 mM), and the consumption of the precursor was significantly accelerated. The sensitization efficiency is proportional to the percentage of acetonitrile in phosphate buffer (Table 2). The source of this solvent effect is uncertain but it is unlikely that it is due to the decreased energy of π, π* excited triplet state of acetophenone in more polar solvent mixtures.[16] In the presence of 100 mM BME, the photolysis of 1 quantitatively yielded dG, indicating that sensitization was not detrimental to the fidelity of the photochemistry.
Table 2.
Effect of acetonitrile on the conversion of 1 (0.1 mM) within 30 min in the presence of acetophenone (1% v/v).
aAverage ± std. dev. of 3 experiments.
Acetonitrile in phosphate buffer (10 mM, pH = 7.2).
Acetonitrile in water.
dG(N2-H)• characterization by laser flash photolysis and DFT studies.
In light of the observation that photosensitized photolysis of 1 is a high fidelity source of dG(N2-H)•, this system was used to directly observe the latter via transient absorption spectroscopy. Rich transient features are observed upon nanosecond pulses (355 nm) of solutions of 1 (1 mM) and acetophenone (30 mM) in aqueous buffer (pH 7.0)/acetonitrile (1:1, v:v). Following 355 nm excitation, the triplet acetophenone absorption band at ~340 nm is immediately observed.[17] This transient decays within 4 μs (Figure 1a, S7), and is accompanied by a build-up of a strong absorption band with maxima at 610 nm and 650 nm. We attribute these observations to photosensitization of 1 by triplet acetophenone and the resulting reactions that give rise to dG(N2-H)•. The timescale for the growth of dG(N2-H)• is consistent with laser flash photolysis and computational studies on the formation of 2 via b-fragmentation following Norrish Type I photocleavage of 4 (Scheme 2).[10a] The transient feature centered at 610 nm is consistent with the previously reported absorption of N1-MedG(N2-H)• from N1-MedG.[5c, 7d, 18] The red-shifted peak at 650 nm has not been reported in relevant studies.
Figure 1.
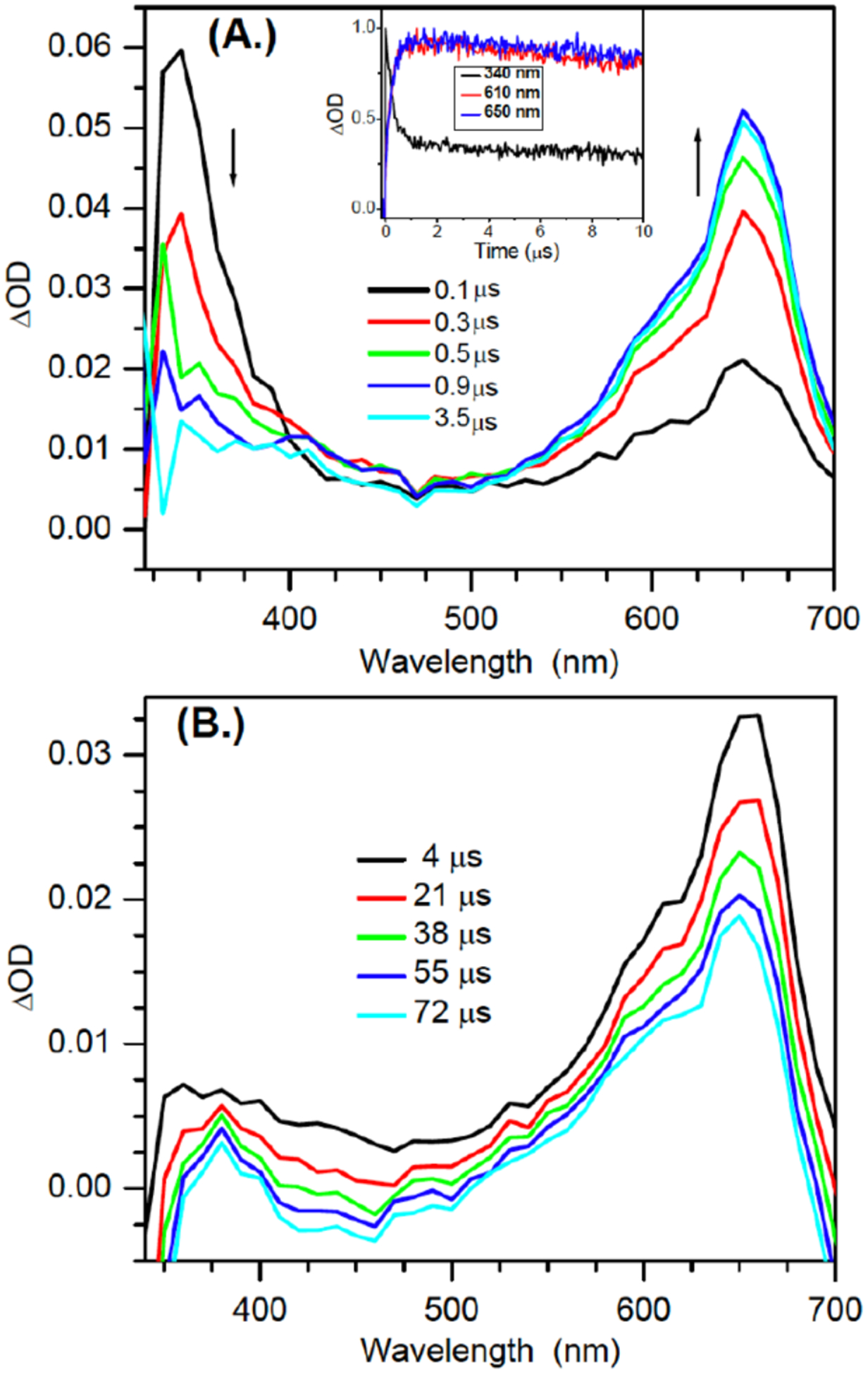
Transient UV-vis absorption spectra of 1 (1 mM) and acetophenone (30 mM) in aqueous buffer (pH 7.0) /acetonitrile (1:1, v:v) upon 355 nm laser flash photolysis in anaerobic conditions (A.) within the first 4 μs (B.) 100 μs. Inset: Normalized early time kinetics traces for the transients at 340 nm, 610 nm, 650 nm.
A variety of possible molecules responsible for the peak at 650 nm were considered. The assignment of this peak to the triplet excited state of 1, or the aminoxy alkyl radical intermediate 10 resulting from Norrish Type I cleavage analogous to that produced from 4 were ruled out by calculations. DFT calculations on 10 and the triplet excited state of 9 (the analogue of 1 lacking a 2’-deoxyribose, Figure 2) indicate that neither absorbs above 600 nm (Table S2). The observation that the growth and decay kinetics of the 650 nm peak are essentially identical to that of the 610 nm peak (Figure 1), suggested that they belong to very similar species. Given that product studies indicate that 1 produces dG(N2-H)• with high fidelity, we postulated that the peaks at 610 and 650 nm belong to different conformational isomers of dG(N2-H)• in which the remaining N2 hydrogen is either syn or anti with respect to the guanine N3 atom (Figure 2). (The naming convention is that utilized by Sevilla.[19]) The optical spectra (Figure 3a) for these two conformers in analogues lacking the deoxyribose ring (syn-, anti-Gua(N2-H)•) were calculated by TDDFT-B3LYP/6–311++G(d,p) in vacuum and under PCM. To compare with experiments, the TDDFT calculated absorption maxima for guanine radicals usually require being red-shifted by 40 – 70 nm.[6b] The calculated spectra for each Gua(N2-H)• conformer features an intense absorption band above 600 nm and weaker band in the UV (Figure 3a). The calculations reproduce the main feature of the experimental spectra for dG(N2-H)•. In addition, it is found that the λmax of the main absorption band > 600 nm is different for these two conformers (Table S2). Under PCM solvation model, the calculated lmax (after adding 60 nm) for the syn-conformer (632 nm) is red-shifted relative to the anti-conformer (616 nm). In vacuum, the calculated λmax (after adding 40 nm) for syn-Gua(N2-H)• is 652 nm and that for the anti-conformer is 612 nm. The theoretically predicted absorption wavelengths for the two conformers match the experimental spectrum obtained from the photolysis of 1 (Figure 1b), indicating that the peak with λmax = 650 nm is ascribable to syn-dG(N2-H)•, which is partially resolved from anti-dG(N2-H)• λmax = 610 nm. Experiments in G-quadruplex DNA corroborate the predicted spectral dependence upon dG(N2-H)• conformation.[20] When dG(N2-H)• is produced from the deprotonation of dG•+ in G-quadruplex structure, the observed spectrum centered at ~600 nm is consistent with anti-dG(N2-H)•. Selective formation of anti-dG(N2-H)• in the G-quadruplex is attributed to preferential syn-N2 deprotonation, which does not disrupt Hoogsteen hydrogen bonding.[20] The predicted conformational dependence of guanine radical λmax is also consistent with calculations on N9-methyl guanine radicals.[20–21]
Figure 2.
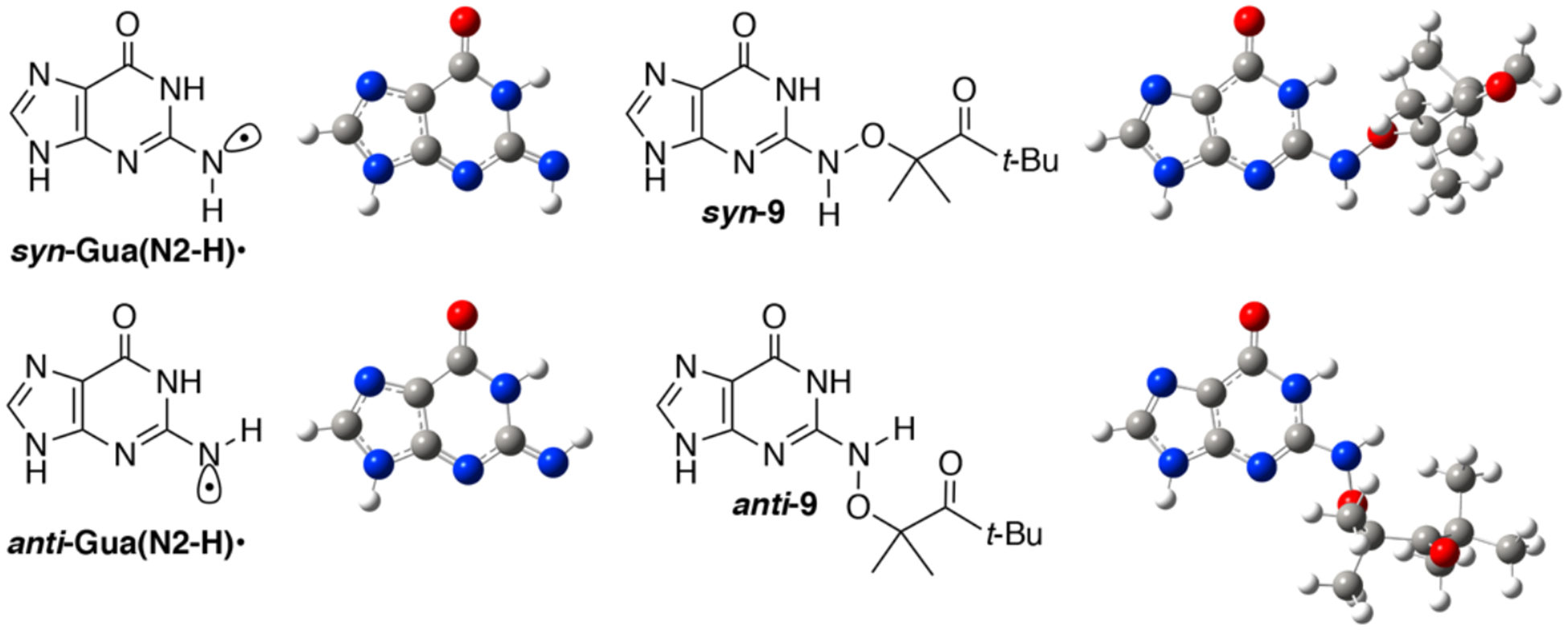
The B3LYP/6–311++G(d,p)//PCM optimized geometries of syn- and anti-Gua(N2-H)•, as well as syn- and anti-9 (the analogues for precursor 1 lacking of the deoxyribose ring).
Figure 3.
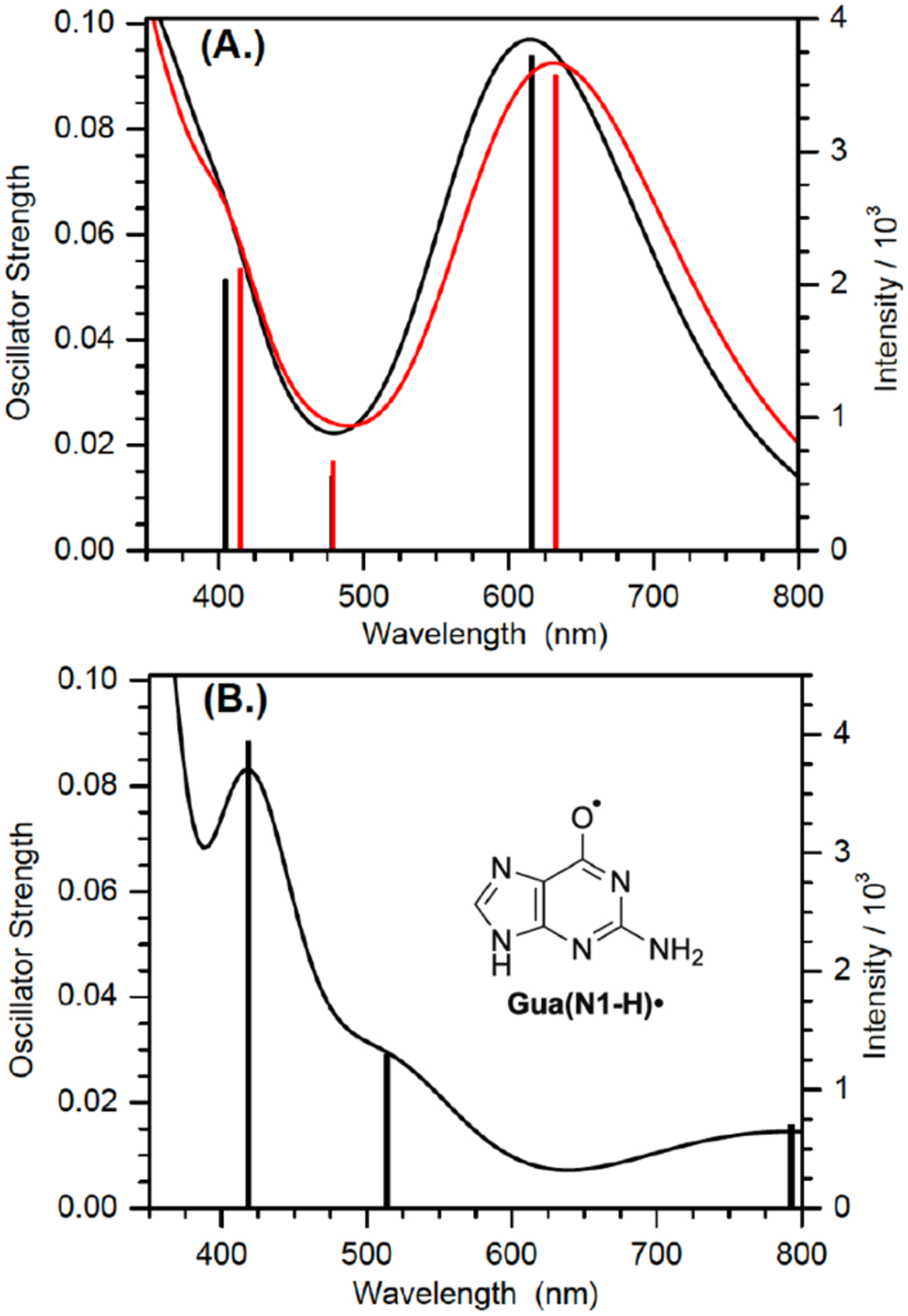
TDDFT-B3LYP /6–311++G(d,p) //PCM calculated absorption spectra of guanine radicals in water after red-shifting by 60 nm (A.): anti- (black) and syn- (red) Gua(N2-H)•; (B.) Gua(N1-H)•. Lengths of sticks correspond to the relative oscillator strengths of electronic transitions. Each electronic transition is convoluted using a Gaussian function with half width at half maximum (HWHM) of 0.33 eV.
Our calculations based on the DFT/B3LYP/6–311++G(d,p)//PCM method show that syn-Gua(N2-H)• is ~ 0.1 eV (~ 2.3 kcal/mol) lower in energy than the anti-conformer, which is in general agreement with the literature (3.0 kcal/mol at the level of B3LYP/6–31G(d)//PCM).[19] In the presence of five explicit waters and under PCM solvation model (Figure S8), the B3LYP/6–311++G(d,p) calculated energy difference between the syn- and anti-Gua(N2-H)• is further reduced to 0.064 eV (~ 1.5 kcal/mol). The small energy difference means that the two conformers of dG(N2-H)• may coexist in aqueous solution. Previously, N1-MedG(N2-H)• was produced by deprotonation of the corresponding radical cation.[5c, 7d, 18] Given the small energy difference of the two conformers and low energy barrier for deprotonation (~ 4.75 kcal/mol), the anti-and syn-conformational isomers are expected to form in approximately equal amounts from this intermediate.[22] The dominant absorption peaks (> 600 nm) of anti-dG(N2-H)• and syn-dG(N2-H)• are predicted to overlap significantly with comparable intensity (Figure 3a). Consequently, it is expected that the two peaks of the anti- and syn- dG(N2-H)• may not be resolved in the spectrum containing both conformers in approximately equal amounts. The spectra of N1-MedG(N2-H)• produced by one-electron oxidation and deprotonation, where only a single broad peak centered at ~ 630 nm are consistent with this.[5c, 7d, 18]
In contrast to the generation of N1-MedG(N2-H)• via deprotonation of N1-MedG•+, the conformation dG(N2-H)• generated via sensitized photolysis of 1 is controlled by the conformation of the precursor, which also exists in anti- and syn-conformations (Figure 2). B3LYP/6–311++G(d,p)//PCM calculations of the analogue lacking a 2’-deoxyribose indicate that syn-8 is more stable than anti-8 by 0.21 eV (~4.83 kcal/mol). The more abundant syn-1 is expected to result in a greater amount of syn-dG(N2-H)• than anti-dG(N2-H)•. Moreover, DFT calculations at DFT/B3LYP/6–311++G(d,p)//PCM level showed that the energy barrier for the transition between syn-dG(N2-H)• and anti-dG(N2-H)• is 0.89 eV (20.5 kcal/mol) (Figure S9). This significant energy barrier suggests that the interconverison between the two conformers is kinetically infeasible on the LFP experiment timescale. These results are consistent with a recent report by Sevilla in which rotation of the N2-H group in N1-MedG(N2-H)• from 0 to 60° with respect to the purine ring also indicated a high rotational barrier.[6b] Consequently, the significant rotational barrier and the preference for generating syn-dG(N2-H)• by 1 results in a partially resolved spectrum of the two conformers. (Figure 1). The transient spectrum for dG(N2-H)• with maximum signal (4 μs) is reproduced by combining two broad peaks with λmax at ~ 610 and 650 nm (Figure 4). Dividing these two peak areas by their respective molar absorption coefficients (estimated using the calculated oscillator strengths in Table S2) suggests that the observed spectrum is the result of an ~ 2:1 mixture of syn-dG(N2-H)• relative to anti-dG(N2-H)•.
Figure 4.
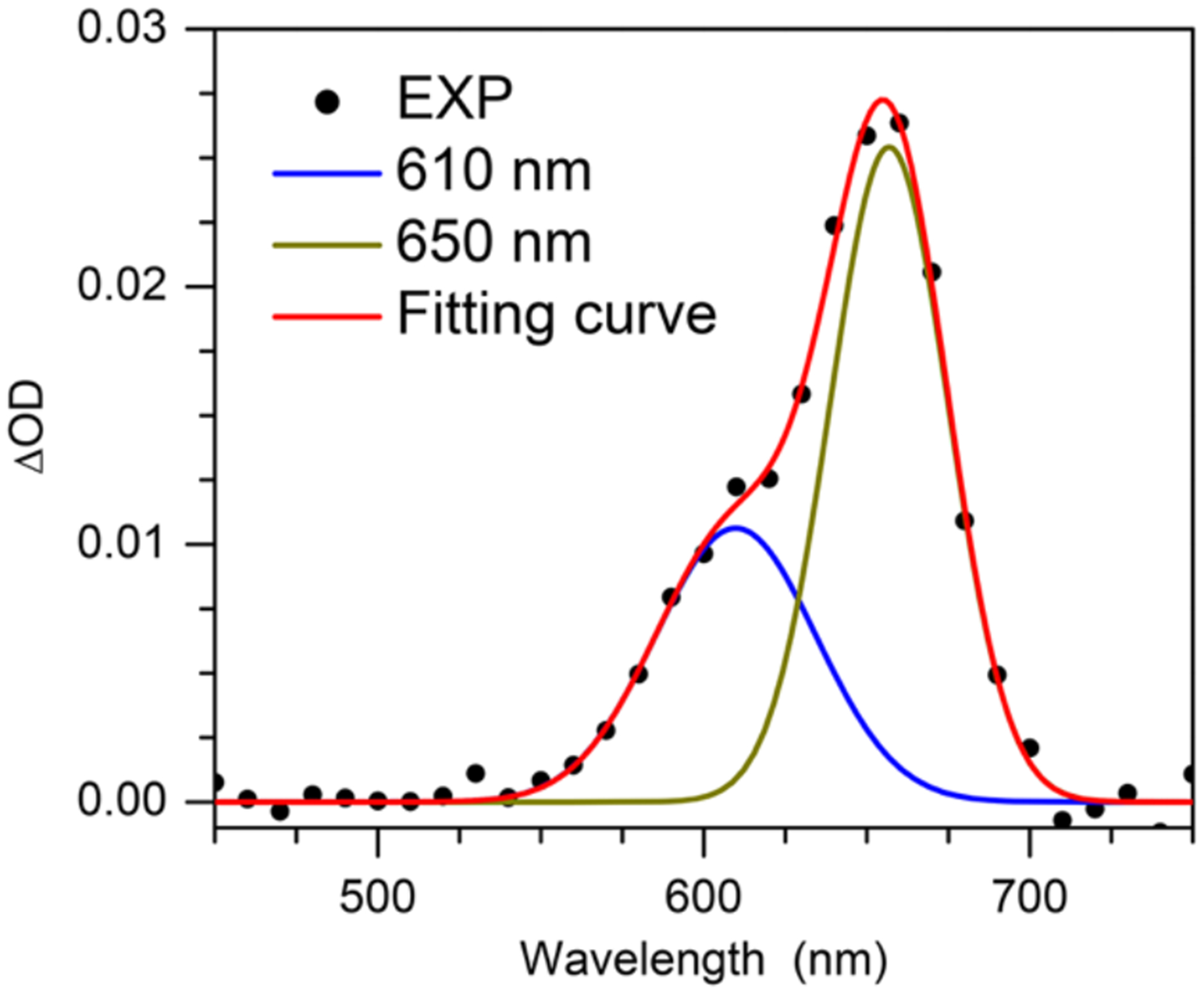
Experimental (black dots) transient UV-vis absorption spectrum at 4 μs of dG(N2-H)• with the fitted spectrum (red line), composed of two individual Gaussian bands peaking at ~ 610 (blue line) and ~ 650 nm (dark yellow line).
The decays of the 610 and 650 nm bands produced upon sensitized photolysis of 1 (1 mM) in the absence of additional reducing agent were fitted to double-exponentials and exhibited comparable lifetimes (Figure 5, Table 3), corroborating the proposal that the bands are attributable to syn- and anti-dG(N2-H)•. The shorter lifetime decay constants change little with the addition of reducing agent, but are dependent on the concentration of the radical precursor. We attribute this decay pathway to the reduction of dG(N2-H)• by its precursor (1), a process that was detected in product studies (Table 1). This bimolecular process is more prominent in the laser flash photolysis experiments, which are carried out at significantly higher concentrations of 1 and greater photon fluxes. In contrast, the slower decay process is affected by the addition of glutathione (GSH) or BME. Attributing the change in the lifetimes of the slower decay constants for the transient at 610 nm and 650 nm to reduction of dG(N2-H)• by the thiols indicates that BME (~2.7 – 2.9 × 104 M−1s−1) reacts approximately 10-fold more slowly with the nitrogen-centered radical than does GSH (~3.0 – 3.2 × 105 M−1s−1). These rate constants are considerably slower than what would be expected for reaction with a carbon-centered radical.[23] However, they are consistent with reactions of other nitrogen-centered radicals that are conjugated to electron accepting substituents, such as a purine ring (e.g. 2).[10a, 24] These radicals and dG(N2-H)• are electron deficient and kinetically mismatched for reaction with thiols.
Figure 5.
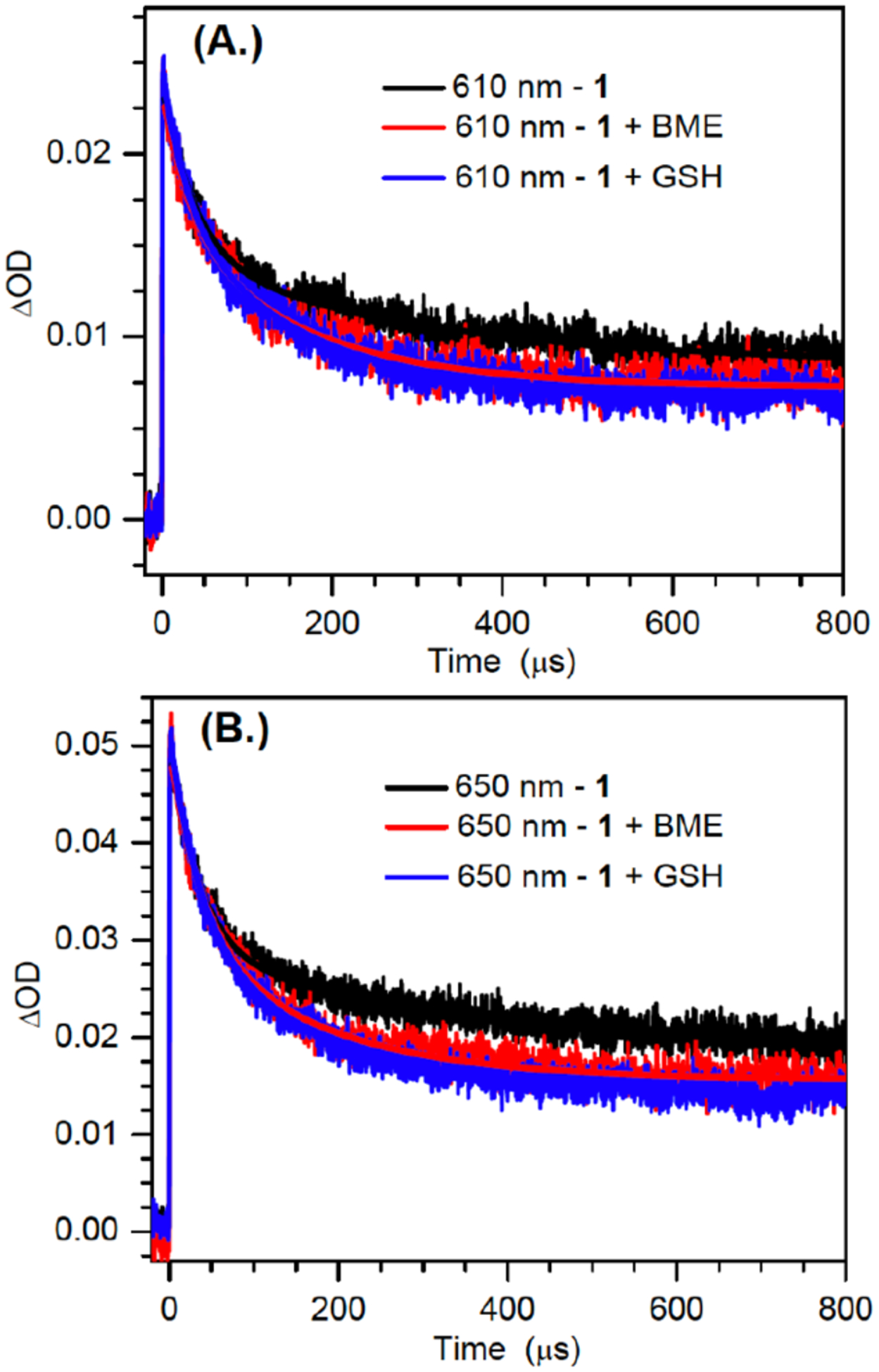
Decay kinetics traces and biexponential fitting for the 610 nm (a); and 650 nm (b) bands obtained from the photosensitized photolysis of 1 (1mM) in the absence and presence of thiols of BME (100 mM) and GSH (10 mM).
Table 3.
Double exponential fitting results for the decay kinetics at 610 nm and 650 nm in the absence and presence of thiols.
| Reducing agent (mM) | 610 nm τ1 (μs) | 610 nm τ2 (μs) | 650 nm τ1 (μs) | 650 nm τ2 (μs) |
|---|---|---|---|---|
| None | 37.5 ±1.1 | 298.9 ± 4.7 | 34.0 ±1.0 | 322.4 ± 4.7 |
| BME (100) | 34.9 ± 2.7 | 161.2 ± 6.6 | 38.3±1.3 | 166.5 ± 2.6 |
| GSH (10) | 30.4 ± 1.9 | 151.5 ± 4.2 | 35.1± 1.9 | 165.5 ± 4.4 |
dG(N2-H)• also features a less intense absorption band at ~ 370 nm, which is evident upon diminution of the transient absorption of triplet acetophenone after 4 μs. Subsequently, the band at 370 nm decays at comparable rates as those at 610 nm and 650 nm (Figure S10).[7] This observation is inconsistent with the proposed tautomerization of dG(N2-H)• to dG(N1-H)•. Pulse radiolysis experiments indicate that dG(N1-H)• absorbs strongly in this region.[5c] TDDFT-B3LYP/6–31++G(d,p)//PCM calculations indicate that dG(N1-H)• should absorb more strongly in this region than dG(N2-H)• (Figure 3, Table S2). These spectral features for the two radicals are affirmed by other computational studies.[6b] Furthermore, radiolysis studies showed that dG(N1-H)• decays relatively slowly with a lifetime of ~0.07 s.[25] Consequently, if dG(N2-H)• tautomerized to dG(N1-H)• with the reported first-order rate constant of 2.3 × 104 s−1 (t½ < 30 μs), the absorption in the 370 nm region would have maintained its intensity or even increased slightly during the time that the longer wavelength bands for dG(N2-H)• decay (Figure 1, 3).[7d].[6b] Finally, the observation that the intense absorption features of dG(N2-H)• at 610 nm and 650 nm exist for hundreds of microseconds, provide additional evidence against rapid tautomerization (Figure S11).
Conclusion
On account of their being common intermediates in DNA damage, the structure and reactivity of purine radicals have garnered significant interest. UV-photolysis of appropriately designed precursors is a common approach for generating homogeneous solutions of these and other DNA radicals.[26] The generation and reactivity of dG(N2-H)• has been a contentious issue, in part because it has only been produced using radiolysis, which may not produce homogeneous solutions of the radical. To address this issue, we developed a photochemical precursor (1) that produces dG(N2-H)• in high yield, as evidenced by product studies. Photosensitization by acetophenone enabled using 1 as a high fidelity source of dG(N2-H)• in laser flash photolysis experiments, where the distinct spectral features of anti- and syn-dG(N2-H)• are resolved . LFP affirmed product studies, showing that dG(N2-H)• is reduced by precursor 1. dG(N2-H)• also reacts with thiols, albeit significantly more slowly than carbon-centered radicals.
Importantly, photochemical generation of dG(N2-H)• from 1 enabled us to address the proposed microsecond timescale tautomerization of dG(N2-H)• to dG(N1-H)•. Chatgilialoglu, Steenken and Sevilla had independently reported spectra that they attributed to dG(N2-H)• or N1-MedG(N2-H)• upon radiolysis of a variety of guanosine derivatives.[5c, 7, 18] The observed absorption spectra following irradiation of N1-methylated substrates under different conditions were in good agreement, exhibiting defined absorption bands with λmax between 610 and 630 nm.[5c, 7b, 18] These spectra were very different than that observed following generation of dG•+ near neutral pH where a transient exhibiting λmax ~370 nm and a weaker absorption band at ~500 nm were attributed to dG(N1-H)•.[5c] These well-defined spectra were also in contrast to those reported following reaction of 8-bromoguanosine with solvated electron, and either dG or guanosine (G) with HO•.[7b–d] The authors ascribed the broad transients that extend from ~500 – 650 nm to dG(N2-H)•. In addition, the authors attributed the first order decay (t½ <30 μs) of absorption at 620 nm to tautomerization of dG(N2-H)• to dG(N1-H)•, despite the lack of sufficient spectroscopic evidence for the growth of the latter[7a],[7c] and the contradiction with the high barrier of 18.68 kcal/mol for the tautomerization.[8] LFP generation of dG(N2-H)• from 1 unambiguously shows that the radical does not tautomerize to dG(N1-H)• on even the hundreds of microseconds timescale, an observation that is consistent with recent experiments in G-quadruplexes.[20] Given these two independent reports that refute purine radical tautomerization, one must also question whether hydroxyl radical generates substantial quantities of dG(N1-H)• by abstracting a hydrogen atom from the N2-amino group of 2’-deoxyguanosine and subsequent tautomerization.[7c, 7d] The differing conclusions drawn from various radiolysis experiments may be attributable to differences in precursors, concentrations, doses and dose rates, as well as the inherent lack of chemical specificity when high energy species such as hydroxyl radical are used to generate reactive intermediates. UV-photolysis of a designed precursor (1) to dG(N2-H)• is not limited in this way. We conclude that it is unlikely that hydroxyl radical reacts directly with 2’-deoxyguanosine to yield dG(N2-H)•, and that this radical does not readily tautomerize to the more stable dG(N1-H)•.
Supplementary Material
Acknowledgements
We are grateful for support from the National Institute of General Medical Sciences (GM-054996 and GM-131736). H.S. thanks the funding support from the National Natural Science Foundation of China (21933005, 21727803 and 21425313). We thank Amit Adhikary and Michael Sevilla for helpful discussions.
References
- [1].a) Dizdaroglu M, Mutat. Res. Rev. Mutagen 2015, 763, 212–245; [DOI] [PubMed] [Google Scholar]; b) Cadet J, Davies KJA, Medeiros MHG, Di Mascio P, Wagner JR, Free Rad. Biol. Med 2017, 107, 13–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Dizdaroglu M, Jaruga P, Free Rad. Res 2012, 46, 382–419; [DOI] [PubMed] [Google Scholar]; b) Cadet J, Wagner JR, Cold Spring Harbor Perspect. Biol 2013, 5, A012559/012551-A012559/012518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].in Free-Radical-Induced DNA Damage and Its Repair: A Chemical Perspective (Ed.: von Sonntag C), Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, pp. 335–355. [Google Scholar]
- [4].a) Steenken S, Jovanovic SV, J. Am. Chem. Soc 1997, 119, 617–618; [Google Scholar]; b) Thapa B, Schlegel HB, J. Phys. Chem. A 2015, 119, 5134–5144; [DOI] [PubMed] [Google Scholar]; c) Genereux JC, Barton JK, Chem. Rev 2010, 110, 1642–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Steenken S, Chem. Rev 1989, 89, 503–520; [Google Scholar]; b) Kumar A, Pottiboyina V, Sevilla MD, J. Phys. Chem. B 2011, 115, 15129–15137; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Candeias LP, Steenken S, J. Am. Chem. Soc 1989, 111, 1094–1099. [Google Scholar]
- [6].a) Candeias LP, Steenken S, Chem. - Eur. J 2000, 6, 475–484; [DOI] [PubMed] [Google Scholar]; b) Kumar A, Sevilla MD, J. Phys. Chem. A 2019, 123, 3098–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Chatgilialoglu C, Caminal C, Altieri A, Vougioukalakis GC, Mulazzani QG, Gimisis T, Guerra M, J. Am. Chem. Soc 2006, 128, 13796–13805; [DOI] [PubMed] [Google Scholar]; b) Chatgilialoglu C, Caminal C, Guerra M, Mulazzani QG, Angew. Chem. Int. Ed 2005, 44, 6030–6032; [DOI] [PubMed] [Google Scholar]; c) Chatgilialoglu C, D’Angelantonio M, Guerra M, Kaloudis P, Mulazzani QG, Angew. Chem. Int. Ed 2009, 48, 2214–2217; [DOI] [PubMed] [Google Scholar]; d) Chatgilialoglu C, Angelantonio MD, Kciuk G, Bobrowski K, Chem. Res. Toxicol 2011, 24, 2200–2206. [DOI] [PubMed] [Google Scholar]
- [8].Adhikary A, Kumar A, Becker D, Sevilla MD, J. Phys. Chem. B 2006, 110, 24171–24180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zheng L, Lin L, Qu K, Adhikary A, Sevilla MD, Greenberg MM, Org. Lett 2017, 19, 6444–6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Zheng L, Griesser M, Pratt DA, Greenberg MM, J. Org. Chem 2017, 82, 3571–3580; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zheng L, Greenberg MM, J. Am. Chem. Soc 2017, 139, 17751–17754; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sun H, Zheng L, Yang K, Greenberg MM, J. Am. Chem. Soc 2019, 141, 10154–10158; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zheng L, Greenberg MM, J. Am. Chem. Soc 2018, 140, 6400–6407; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Sun H, Zheng L, Greenberg MM, J. Am. Chem. Soc 2018, 140, 11308–11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Anderson RW, Hochstrasser RM, J. Phys. Chem 1976, 80, 2155–2159. [Google Scholar]
- [12].Adam W, Saha-Möller CR, Schönberger A, J. Am. Chem. Soc 1996, 118, 9233–9238. [Google Scholar]
- [13].Harwood EA, Hopkins PB, Sigurdsson ST, J. Org. Chem 2000, 65, 2959–2964. [DOI] [PubMed] [Google Scholar]
- [14].Scaiano JC, Stamplecoskie KG, Hallett-Tapley GL, Chem. Comm 2012, 48, 4798–4808. [DOI] [PubMed] [Google Scholar]
- [15].a) Lamola AA, J. Chem. Phys 1967, 47, 4810–4816; [DOI] [PubMed] [Google Scholar]; b) Yang N-C, McClure DS, Murov S, Houser JJ, Dusenbery R, J. Am. Chem. Soc 1967, 89, 5466–5468; [Google Scholar]; c) Lamola AA, Hammond GS, J. Chem. Phys 1965, 43, 2129–2135. [Google Scholar]
- [16].a) Narra S, Shigeto S, J. Phys. Chem. B 2015, 119, 3808–3814; [DOI] [PubMed] [Google Scholar]; b) Hayes WP, Timmons CJ, Spectrochimica Acta 1965, 21, 529–541. [Google Scholar]
- [17].Lutz H, Breheret E, Lindqvist L, J. Phys. Chem 1973, 77, 1758–1762. [Google Scholar]
- [18].Adhikary A, Kumar A, Munafo SA, Khanduri D, Sevilla MD, Phys. Chem. Chem. Phys 2010, 12, 5353–5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Adhikary A, Kumar A, Becker D, Sevilla MD, J. Phys. Chem. B 2006, 110, 24171–24180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Banyasz A, Martínez-Fernández L, Balty C, Perron M, Douki T, Improta R, Markovitsi D, J. Am. Chem. Soc 2017, 139, 10561–10568. [DOI] [PubMed] [Google Scholar]
- [21].Martínez Fernández L, Cerezo J, Asha H, Santoro F, Coriani S, Improta R, ChemPhotoChem 2019, 3, 846–855. [Google Scholar]
- [22].Wu L, Jie J, Liu K, Su H, Acta Chimica Sinica 2014, 72, 1182–1186. [Google Scholar]
- [23].Newcomb M, Tetrahedron 1993, 49, 1151–1176. [Google Scholar]
- [24].Horner JH, Musa OM, Bouvier A, Newcomb M, J. Am. Chem. Soc 1998, 120, 7738–7748. [Google Scholar]
- [25].Rokhlenko Y, Geacintov NE, Shafirovich V, J. Am. Chem. Soc 2012, 134, 4955–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Greenberg MM, Adv. Phys. Org. Chem 2016, 50, 119–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.