

Abstract
Group 3 innate lymphoid cells (ILC3s) have emerged as master regulators of intestinal health and tissue homeostasis in mammals. Through a diverse array of cytokines and cellular interactions, ILC3s crucially orchestrate lymphoid organogenesis, promote tissue protection or regeneration, facilitate antimicrobial responses, and directly regulate adaptive immunity. Further, translational studies have found that ILC3 responses are altered in the intestine of defined patient populations with chronic infectious, inflammatory, or metabolic diseases. Therefore, it is essential to broadly understand the signals that activate, suppress, or fine-tune ILC3s in the gut. Here, we discuss recent exciting advances in this field, integrate them into our current understanding of ILC3 biology, and highlight fundamental gaps in knowledge that require additional investigation.
Key roles of ILC3s in maintaining gastrointestinal health
The gastrointestinal tract (GI tract) is an essential organ system for nutrient absorption. It is continuously exposed to dietary antigens and colonized by trillions of microbes, representing normally beneficial commensal bacteria, or the resident microbiota [1]. In addition, this organ frequently serves as an entry or infection site for many pathogens. Therefore, the intestinal immune system has to be tolerant to foreign dietary antigens and to the microbiota, while providing protection against pathogens [1]. This functional dichotomy could be why the mammalian intestine contains the largest compartment of immune system components, with numerous organized lymphoid structures and complex cellular networks. Among the different cell types, innate lymphoid cells (ILCs) represent a recently appreciated lineage that are enriched in the intestinal mucosa relative to other tissues, and which play a crucial role in regulating local immunity, inflammation and tissue homeostasis [2].
ILCs are lymphocytes that do not require or express somatically-recombined antigen-specific receptors. Based on developmental pathways, transcription factor expression and cytokine production, ILCs are classified into at least three groups [2]. Group 1 ILCs (ILC1s), including natural killer (NK) cells and non-cytotoxic ILC1s, rely on transcription factor T-bet for their development, and release the type-1 cytokine IFN-γ [3]. Group 2 ILCs (ILC2s) depend on Gata3, RORα, and Bcl11b, and generate type-2 cytokines IL-5 and IL-13. Group 3 ILCs (ILC3s), including lymphoid tissue inducer (LTi) cells and T-bet+ ILC3s, require RORγt for their development and produce type-3 cytokines IL-17 and IL-22 [3]. Given the substantial similarities to Th1, Th2, and Th17 cells, non-cytotoxic ILCs are thought to be an innate counterpart of CD4+ T helper cells, but also exhibit numerous unique and pleiotropic functions.
This review focuses on ILC3s, which are highly heterogenous in mice and humans (Box 1), and play an important role in regulating gastrointestinal health. This occurs through a number of distinct pathways, such as activating intestinal epithelial cells to produce antimicrobial peptides and eliminate pathogens, promoting the regeneration of epithelial cells to accelerate tissue repair, and regulating the homeostasis of adaptive immunity [3]. To coordinate these many functions, ILC3s have evolved a sophisticated strategy to sense the surrounding environment and promptly respond to local changes of microbes, metabolites, cytokines or neurotrophic factors, as well as systemic oscillations of the circadian rhythm. Recent advances have found abnormal ILC3 responses in the intestine of patients with chronic infections and inflammatory diseases. Understanding how ILC3 responses are regulated under healthy conditions or altered in inflammatory contexts could provoke novel strategies for preventing, treating, and curing these diseases.
Box 1: Heterogeneity of ILC3s in mice and humans.
In mice, on the basis of CCR6 and T-bet expression, ILC3s can be divided into CCR6+ (and T-bet−) ILC3s and T-bet+ ILC3s. The CCR6+ ILC3 subset includes lymphoid tissue inducer (LTi) cells that produce IL-22 and IL-17, are heterogeneous in CD4 expression, and are critical for the development of lymphoid organs, including Peyer’s patches (PP) in the small intestine prenatally. Postnatally, this subset exhibits a similar phenotype, called LTi-like cells, which predominantly localize in cryptopatches (CP) or isolated lymphoid follicles (ILF), and are essential for the formation of these lymphoid tissues [109–111]. Unlike LTi-like ILC3, T-bet+ ILC3 are diffusely localized in the lamina propria of the intestine. T-bet+ ILC3s are heterogeneous in their expression of NKp46 and are generated after birth as a result of microbiota colonization [38]. They are capable of secreting IL-22 and IFN-γ. Finally, ILC3s have the potential to lose RORγt expression and become ex-ILC3s that resemble ILC1s.
In humans, the cellular heterogeneity of ILC3s is poorly appreciated at this time, but a recent publication demonstrated that expression of NKp44, CD45RA and antigen presentation machinery distinguishes at least three subsets of ILC3s [112]. However, some of these subsets might simply represent a state of activation. Of note, human ILC3s exhibit the ability to differentiate into ex-ILC3s or ILC1s, which has been documented in several disease states, such as in the inflamed intestine of IBD patients [102, 103].
Below, we summarize our current understanding of the pathways that activate, suppress, or fine-tune ILC3 responses in the gut, while highlighting new advances as well as gaps in our knowledge that warrant further investigation.
Microbial exposure shapes ILC3 development and function in the gut
Microorganisms that colonize the intestine have a complex dialogue with ILC3s. On the one hand, ILC3s produce cytokines that modulate epithelial barrier integrity and immunity to invasive microbes [4, 5]. On the other hand, microbes crucially impact ILC3 responses either through direct stimulation or indirectly by modulating epithelial cell and myeloid cell responses [6–9]. Below, we summarize major hubs of ILC3 activation through these distinct pathways (Figure 1).
Figure 1. Activation signals of ILC3s.
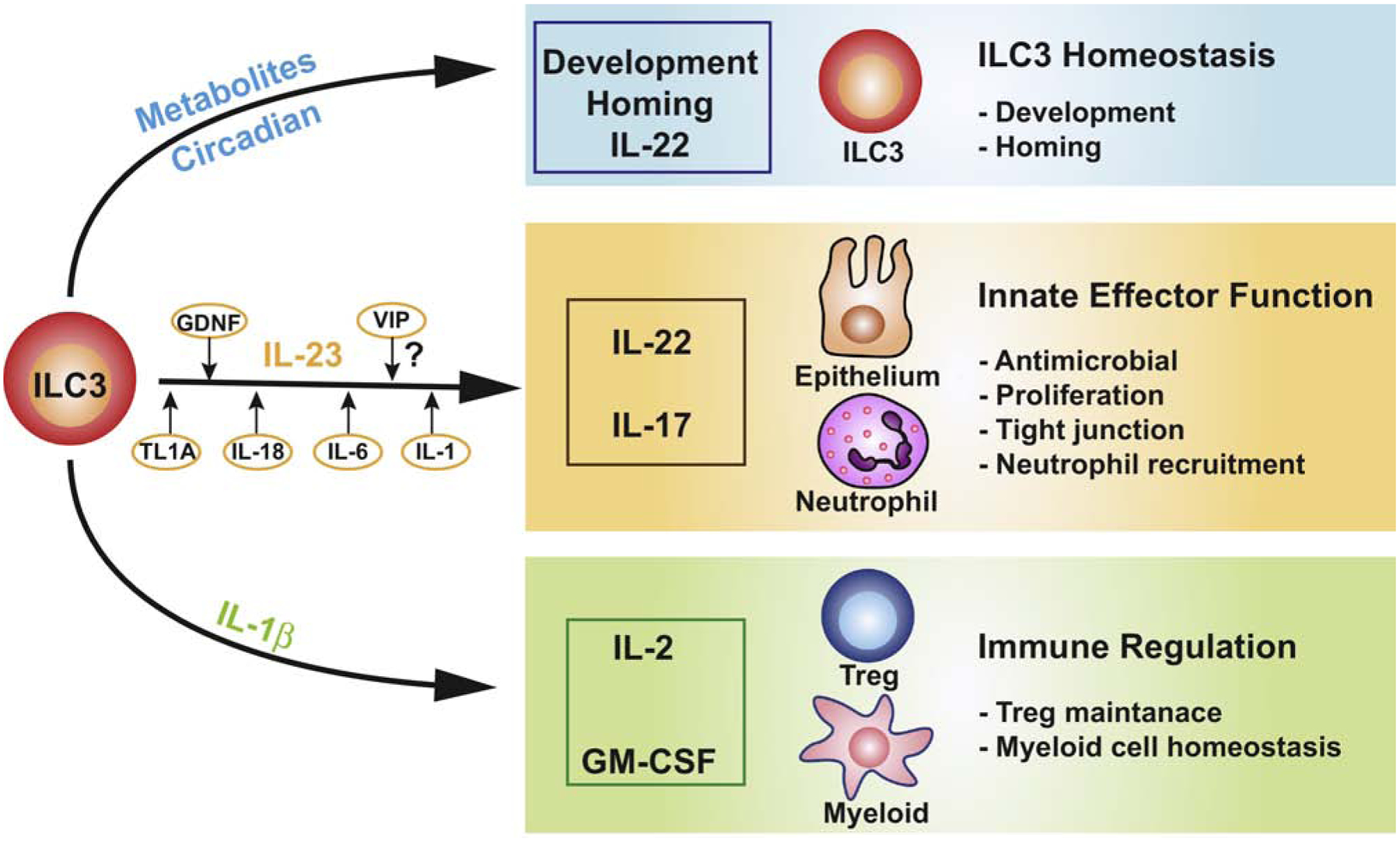
Environmental signals in the GI tract from microbiota, pathogens, neurons, and diet directly or indirectly activate ILC3s. Here, we propose that these can be grouped into three main themes. (1) Metabolites derived from microbiota or the diet (such as oxysterols, SCFAs, AhR ligands and Ras), and circadian rhythms that modulate ILC3 development, homing receptor expression, and IL-22 production, thereby maintaining ILC3 homeostasis. (2) Myeloid-derived IL-23 which serves as a master regulator of IL-22 and IL-17 production in ILC3s, enhancing the antimicrobial defense and barrier integrity of the intestine. Cytokines including TL1A, IL-18, IL-6, and IL-1, and neurotrophic factor GDNF fine-tune IL-22 production. (3) IL-1β released by macrophages, which stimulates IL-2 expression and GM-CSF production by ILC3s, orchestrating immune regulation in the intestine.
Direct activation of ILC3s by microbes
The trillions of microbiota resident in the gut produce an extensive repertoire of metabolites [10–14], which has the potential to manipulate host physiology and immune responses. In the colon of humans and mice, the main metabolites of undigested carbohydrates by bacterial fermentation are short-chain fatty acids (SCFA) [10]. SCFA, including acetic acid, propionic acid, and butyric acid, are not only energy sources, but also play an important role in the development and function of the host immune system [10]. The receptors of SCFA, such as FFAR2, FFAR3, and GPR109A, are expressed by many immune cells [10]. In the mouse colon, FFAR2 is highly enriched in ILC3s [6]. Mice fed with SCFA or an FFAR2 agonist (WO 2011/076732 A1) in drinking water, have enhanced frequencies of colonic ILC3s and IL-22 production relative to control mice, whereas ILC3 specific genetic deletion of Ffar2 (Ffar2ΔRorc) impairs the proliferation and production of IL-22 by CCR6+ ILC3s, resulting in impaired host defense to Citrobacter rodentium or tissue repair following DSS-induced intestinal injury in mice [6]. SCFA regulate IL-22 production through AKT-STAT3 and ERK-STAT3 signaling in colonic ILC3s in mice [6]. This demonstrates a direct role for microbe-derived metabolites in promoting ILC3 responses via sensing receptors. However, these responses can be nuanced and subset-, receptor- or location-dependent, given that another study demonstrated that microbiota-derived butyrate negatively regulates NKp46+ ILC3s and their cytokine production in Peyer’s Patches (PP) of the small intestine in mice [15]. In vitro butyrate stimulation decreases the number of ILC3s and IL-22 production compared to vehicle control [15]. Consistently, GPR109a agonist reduces the frequency of ILC3s in the colon, whereas the deletion of GPR109a (Gpr109a−/−) promotes higher numbers of ILC3 in the context of Rag1−/− mice compared with Gpr109a+/+ mice at steady state [16]. The precise SCFA inputs that impact subpopulations of ILC3s across different anatomical sites requires further investigation.
Tryptophan is an energy source of Lactobacilli sp., a member of the intestinal microbiota. After digestion, tryptophan metabolites are produced and act as ligands for the aryl hydrocarbon receptor (AhR) [11]. AhR is a ligand-activated transcription factor, ubiquitously expressed in lymphocytes, including ILC3s [17]. Whole body- and lineage-specific deletion of AhR (Ahr−/− and AhrΔRorc, respectively) in mice has revealed an essential role for this pathway in promoting ILC3-mediated postnatal development of isolated lymphoid follicles (ILF) and cryptopatches (CP), the development or maintenance of T-bet+ ILC3s, and, for the cytokine production in ILC3s that is required for immunity to C. rodentium [7, 14, 18–21]. AhR directly binds to the mouse Il22 locus to control cytokine production [7]. In addition to promoting host defense against pathogens, AhR-mediated IL-22 production by ILC3s promotes protective responses following DNA damage in epithelial stem cells [22]. Compared to mice with a normal diet, mice fed a diet deficient in AhR ligands exhibit impaired IL-22 production by ILC3s, and enhanced DNA damage following exposure to a carcinogen, resulting in more mutations and enhanced tumorigenesis [22]. Notably, in humans, an AhR agonist (FICZ) in combination with IL-1β, stimulate ILC3s to produce IL-22 in vitro, whereas an AhR antagonist (CH-223191) causes the differentiation of ILC3s to IFN-γ-expressing ex-ILC3s, confirming a conserved function of AhR in supporting ILC3s [21].
In addition to receptors for microbe-derived metabolites, human ILC3s also express Toll-like receptors (TLRs) which directly interact with pathogen associated molecular patterns, suggesting another potential direct recognition of microorganisms by ILC3s [23]. TLR transcripts, including those encoding TLR1, 2, 5, 6, 7, and 9 (but not 3 and 4) have been detected in human ILC3s [23]. Among them, TLR2 has the highest mRNA expression [23]. Together with IL-23 or IL-2, TLR2 ligands induce IL-22, IL-13, and IL-5 production in human ILC3s in vitro through direct activation of NF-κB and JAK signaling [23]. Microbial sensing by TLRs may be specific to human ILC3s, since a comparable expression profile has not been observed in mouse ILC3s [23].
Natural cytotoxicity receptors (NCRs), such as NKp46, NKp44, and NKp30, could be another pathway for direct recognition of microbes by ILC3s, given the fact that NCRs recognize ligands derived from viruses, bacteria, fungi, and parasites [24]. Indeed, the engagement of NKp44 triggers TNFα expression in human ILC3s [25]. When combined with IL-1, IL-7, and IL-23, NKp44 stimulation of human ILC3s synergistically promotes IL-22, GM-CSF, IL-2 and TNFα production in vitro [25]. NF-κB and NFAT activations are required for NKp44-mediated activation of ILC3s [25]. However, this may also be restricted to human ILC3s, as NKp44 is not found in mice, and the engagement of NKp46 on mouse ILC3s has no impact on cytokine production [25, 26]. Notably, the ligands that bind to NCRs are various and not fully characterized [24]; and, different ligands may bind to the same NCR [27]. Additionally, cytokines in the microenvironment may affect interactions between NCRs and their ligands [24]. The bona fide NCR ligands expressed by ILC3s and their functional impact in different microenvironments require further investigation.
Indirect activation of ILC3s through microbiota-induced IL-1β
In addition to direct regulation, microbes indirectly modulate ILC3 activation by stimulating cytokine release from myeloid cells or epithelial cells in the GI tract [8, 9]. Microbiota trigger IL-1β production by intestinal macrophages that then act on ILC3s to produce GM-CSF in mice [8]. ILC3-derived GM-CSF regulates the homeostasis of myeloid cells that subsequently support regulatory T cell (Treg) expansion and limit responses to dietary antigens in a mouse model of oral tolerance [8]. In the mouse small intestine, microbiota stimulate Myd88- and Nod2-dependent pathways to promote macrophage production of IL-1β, which drives ILC3s to produce IL-2 [9]. ILC3s are the dominant source of IL-2 in this compartment, which is required for Treg maintenance and tolerance to dietary antigens in mice [9]. Collectively, these findings implicate a novel axis where microbiota-dependent sensing by myeloid cells results in IL-1β production and subsequent activation of ILC3 to produce GM-CSF and IL-2, coordinating immune regulation throughout the GI tract [8, 9]. Consistent with this, humans with inflammatory bowel disease (IBD) exhibit reduced frequencies of IL-2 producing ILC3s in their intestine as compared with healthy controls [9]. It remains unclear what drives the dysfunction of ILC3s in this context, and this remains an active area of investigation.
Indirect activation of ILC3s by microbe-induced IL-23
ILC3s are an important source of IL-22 in the intestine [28]. IL-23 is the master regulator of IL-22 secretion in ILC3s and is produced by myeloid cells in response to microbes [17]. For example, germ-free mice have dramatically reduced frequencies of IL-22+ ILC3s in the intestine, in part due to reduced IL-23 production and impaired development of T-bet+ ILC3s, compared with conventional mice [4, 29]. Segmented filamentous bacteria (SFB) predominantly colonize the ileum of the small intestine and potently induce Th17 cell differentiation in mice [30, 31]. Recent evidence suggests a crucial role for SFB in activating ILC3s as well. Specifically, colonization of SFB increases Sca-1 and IL-22 production in ILC3s in response to IL-23 from myeloid cells in mice [32, 33]. Moreover, in Rag1-deficient mice (Rag1−/−), SFB colonization enhances IL-23 derived from CCR2+ myeloid cells and subsequent STAT3 activation in ILC3s and intestinal epithelial cells [34]. This activation of ILC3s by microbes is likely specific to anatomical locations; indeed, our group recently demonstrated a subset of microbiota that colonize the interior of intestinal lymphoid tissues, leading to a selective expansion of ILC3s in the PP and mesenteric lymph nodes (mLN), but not in the intestinal lamina propria of wild type mice [35].
Besides commensal bacteria, opportunistic microbes or pathogens influence the activation of ILC3s. Infection with C. rodentium leads to acute colitis in mice, which mimics enterohemorrhagic and enteropathogenic Escherichia coli infection in humans [36]. IL-22 is crucial for the early host defense against C. rodentium [37]. For instance, extensive studies have demonstrated that the expression of IL-22 increases in both T-bet+ and CCR6+ subsets of ILC3s during C. rodentium infection in response to IL-23 released by dendritic cells, macrophages, or CX3CR1+ phagocytes in the mouse intestine [4, 5, 38–42]. To protect against C. rodentium infection, the expression of STAT3 in ILC3s is essential, which directly binds to the Il22 locus [43]. Additionally, during C. rodentium infection, ILC3-derived lymphotoxin is required for optimal IL-22 production in ILC3s and host protection [44]. Inactivation of lymphotoxin by depletion of Ltb in ILC3s (LtbΔRorc) significantly reduces IL-22 expression in the colon of infected mice. Lymphotoxin promotes IL-22 production by binding to LTβR on dendritic cells and triggering IL-23 production [44]. Furthermore, IL-23 also upregulates the expression of other cytokines in ILC3s. Indeed, Helicobacter hepaticus colonization in mice causes the upregulation of IL-23 in the colon, which induces the activation of ILC3s to produce IL-17A and IFN-γ, leading to induction of colitis in the absence of IL-10 or adaptive immunity [45]. Salmonella enterica Typhimurium infection in mice induces IFN-γ production in T-bet+ ILC3s in response to IL-12 stimulation, which may be important in protection from this pathogen [38].
Pathways fine-tune ILC3 responses
While IL-22 production by ILC3s is dominantly driven by IL-23, there are also other critical pathways fine-tuning this response that act synergistically. Germ-free and antibiotics-treated mice exhibit reduced tumor necrosis factor-like cytokine 1A (TL1A) production by CX3CR1+ mononuclear phagocytes (MNP) relative to conventional mice [46]. MNP-derived TL1A, together with IL-23, stimulates robust IL-22 expression in both T-bet+ and CCR6+ ILC3s to protect against acute DSS-induced colitis in mice [39, 46]. However, in chronic colitis, stimulation of TL1A may also trigger the expression of the co-stimulatory molecule, OX40L, and support expansion of pro-inflammatory CD4+ T cells in vitro [46]. Additionally, activation of TL1A receptor, death receptor 3 (DR3) by an anti-DR3 agonistic antibody, induces GM-CSF production in ILC3s through the p38-MAPK pathway, leading to loss of colonic ILC3s in an IL-23-dependent manner, and exacerbation of anti-CD40-induced and DSS-induced colitis in mice [47]. Notably, increased TL1A protein amounts have been reported in colonic tissues of patients with ulcerative colitis or Crohn’s disease, and genetic variants in the TL1A coding gene, TNFSF15, have been associated with Crohn’s disease [48].
IL-6 and IL-1β are other myeloid-derived cytokines that can be induced by microbiota to impact ILC3 production of IL-22 [49–51]. Administration of IL-6 increases the production of IL-17A and IL-22 in colonic ILC3s from mice [52]. Moreover, ILC3s from human IBD patients produce more IL-17A and IL-22 when stimulated with IL-6 [52]. In addition, oral infection of mice with Toxoplasma gondii, a protozoan parasite, induces IL-22 expression in ILC3s [53]. Of note, the induction of IL-22 in this context is driven by IL-18. [53]. IL-22 expression is reduced in Il18−/− mice, and acting in synergy with IL-23, IL-18 can boost IL-22 expression in ILC3s both in vitro and in vivo in mice [53]. In a similar manner, infection with rotavirus, a double-stranded RNA virus (and the leading cause of viral gastroenteritis in infants [54]), promotes ILC3 expansion and cytokine production in the mouse small intestine [54]. Rotavirus infection increases IL-1α expression mainly in intestinal epithelial cells; and, antibody-mediated neutralization of IL-1α inhibits in vivo IL-22 production by ILC3s and enhances rotavirus replication [54]. This indicates that IL-1α released by intestinal epithelial cells is responsible for the activation of ILC3 in this context. The pathways fine-tuning ILC3 responses are specific to the context, anatomical location, and cellular interactions, and thus, although our knowledge has rapidly advanced in this area, further mechanistic investigation is warranted to fully appreciate how intestinal ILC3s are optimally regulated in health and disease.
Neurotrophic regulation of ILC3 homeostasis and function
Mucosal tissues are densely innervated, particularly in the intestine. The enteric nervous system, containing millions of neurons, is the largest part of the autonomic nervous system [55, 56]. Recent seminal studies have shown that neuropeptides and neurotransmitters produced by the nervous system or associated cell types directly regulate ILC3s in the intestine [57, 58].
Regulation of ILC3s by neuropeptides
Glial cells sense the microenvironment in the gut in a MyD88-dependent manner and release glial-derived neurotrophic factors (GDNF) [59]. ILC3s in the mouse intestine exhibit high expression of Ret, a receptor tyrosine kinase that is activated by GDNF. Mice with a deletion of Ret (RetGFP/GFP) lack PP and have reduced IL-22-expressing ILC3s in the intestine relative to Ret+/+ mice [59, 60]. Conditional deletion of Ret in ILC3s (RetΔRorc) exacerbates intestinal inflammation induced by DSS treatment or C. rodentium infection relative to control animals, while gain-of-function Ret (RetMEN2B) protects mice from DSS-induced intestinal damage and inflammation [59]. Mechanistically, activation of Ret by GDNF promotes Il22 transcription in ILC3s by triggering STAT3 and p38 MAPK/ERK-AKT signaling cascades in mice [59]. This seminal study demonstrated how ILC3s are regulated by enteric glial cells.
The enteric nervous system senses nutrient processing and uptake in the intestine. Vasoactive intestinal protein (VIP), a neuropeptide involved in coordinating feeding responses [61], is predominantly expressed by enteric neurons in mice. Notably, VIP-expressing neurons are in close proximity with CCR6+ ILC3s in the small intestine in mice, which highly express a VIP receptor Vipr2 [62]. Vipr2 agonist (BAY-559837) suppresses IL-22 production in CCR6+ ILC3s from mice both in vitro and in vivo, and conditional depletion of Vipr2 (Vipr2ΔRorc) in ILC3s increases the number of IL-22-producing CCR6+ ILC3s relative to control mice [62]. Of note, feeding induces the production of VIP in enteric neurons, and subsequently reduces IL-22+ ILC3s, subsequently permitting expansion of SFB in mice [62]. By contrast, fasting in mice inhibits SFB growth in an IL-22-dependent manner [62]. Altogether, this study demonstrated that ILC3s recognize VIP-VIPR2 signals in response to food consumption, which is essential for the control of microbiota [62]. In contrast, another study reported that in vitro stimulation with VIP of ILC3 purified from the small intestine of mice induced IL-22 production in a VIPR2-dependent manner. Additionally, injection of exogenous VIP enhanced IL-22 production in ILC3s in vivo, and Vipr2−/− mice exhibited reduced IL-22 secretion by ILC3s, concomitant with more severe inflammation following exposure to DSS, relative to wildtype mice [63].
In addition to Ret and Vipr2, ILC3s also express cholinergic receptors including Chrm1, Chrm2, Chrm4, and Chrm5 [64]. In vitro stimulation of murine ILC3s with Ach (acetylcholine), a vagus-derived neurotransmitter, activates the protective PCTR1 (protectin conjugate in tissue regeneration) biosynthetic pathway, indicating a cross-talk between ILC3 and the vagus nerve. Indeed, vagotomy in mice decreases the number of peritoneal ILC3s and delays the resolution of E. coli infection relative to sham control mice [64]. In contrast, administration of ACh-activated ILC3s restores host defense to E. coli and tissue resolution in vagotomized mice [64]. Even though recent seminal studies have revealed the potential for neurotrophic factors in regulating ILC3s, the extent of crosstalk and physiological significance of ILC3 interactions with the nervous system represents an intriguing area for future investigation.
Circadian regulation of ILC3s
Circadian rhythms control a variety of cellular and organismal processes in mammals, including metabolism, homeostasis, and behaviors [65]. Recent studies suggest that the GI tract exhibits diurnal oscillations or a circadian clock, which is involved in regulating both physiological functions of the gut including nutrient absorption, and pathogenic conditions such as microbial infections in both mouse and human [66–69]. Microbiota, epithelial cells, and immune cells in the gut undergo diurnal oscillations and an inappropriate clock system alters their function in mice [70–73]. As an essential part of intestinal immunity, ILC3s are also connected to the biological clock. Mouse ILC3s in the gut express high amounts of circadian clock genes, including Clock, Arntl, Per1, Per2, Per3, Cry1, Cry2, Nr1d1,and Nfil3 [74–76]. ILC3s exhibit circadian expression pattern of these clock regulators as well as ILC3-related transcription factors such as Rorc, Runx1, Tox, Ahr, Rora [74]. Additionally, the production of IL-17A and IL-22 in ILC3s also displays diurnal oscillations [75, 76]. Microbiota, feeding regimens, and light-dark cycles are all regulators of circadian rhythms in gut ILC3s in mice [63, 74]. Lineage-specific deletion of Arntl (ArntlΔRorc) in ILC3s reduces the number and frequency of ILC3s selectively in the mouse intestine, indicating an intrinsic circadian regulation of ILC3 homeostasis. Moreover, decreased Rorc expression and production of IL-22 and IL-17A is also observed in Arntl-deficient ILC3s relative to wildtype ILC3s in mice [74].
The causes and consequences of circadian regulation in ILC3s are multifaceted. The master clock gene regulates the migration of ILC3s to the intestinal mucosa by controlling the expression of intestinal lamina propria homing molecules in mice, independent of cellular proliferation; conversely, Arntl-deficient ILC3s (ArntlΔRorc) exhibit reduced expression of homing molecules CCR9, α4β7, and CXCR4 relative to wildtype ILC3s [74]. In addition, Arntl deletion in ILC3s leads to the hyperactivation and engagement of the proapoptotic pathways Bcl2l11, Bax and Bim, which may also result in a reduction of ILC3s in the mouse intestine [75]. Notably, the phenotype caused by depletion of Arntl in ILC3s in mice can be reversed by antibiotic treatment, suggesting that microbiota, at least partially, are driving the hyperactivation of ILC3s and induction of proapoptotic genes [75]. Similarly, depletion of another circadian gene, Nr1d1, also affects ILC3 homeostasis in the mouse gut [76]. The frequency and cell number of intestinal NCR+ ILC3s are dramatically decreased in both Nr1d1−/− mice and mice with a conditional knockout of Nr1d1 in NCR+ ILC3s (Nr1d1ΔNcr1)[74, 76], suggesting a role for Nr1d1 in the development of this ILC3 subset [76]. Further, human ILC3s isolated from the inflamed intestine of IBD patients exhibit altered expression of circadian genes relative to ILC3s from the healthy portion of the intestine across individual patients. These results suggest that disruption of normal circadian rhythms may contribute to the reduction of intestinal ILC3s observed in IBD patients [75], although further studies are required to directly test this hypothesis.
ILC3 Regulation by diet and host-derived metabolites
Over past decades, diet has emerged as an environmental factor that is related to inflammatory conditions. Dietary metabolites influence immune responses in the GI tract, the main site for nutrition absorption [77, 78].
Retinoic acid (RA) is a metabolite of vitamin A. RA concentrations are high in the intestine and mLN in humans and mice, as the dietary retinol is absorbed by the gut [79]. Besides being essential for the development of many organs, RAs play a crucial role in the homeostasis of the immune system, including that of ILC3s. For instance, maternal RAs control the differentiation of fetal LTi cells that are responsible for secondary lymphoid organ formation in mice [77]. Inhibition of RA signaling with BMS493 in pregnant female mice has led to reduced numbers of fetal LTi cells and failure of secondary lymphoid organ development in the small intestine. In the same study, RA stimulation regulated RORγt expression via direct binding of the RA receptors RAR and RXR, to the Rorc locus, which in turn supported LTi cell development [77]. RAs also promote ILC3 populations in adult mice. A vitamin A deficient-diet or inhibiting RA signaling with the pan-RAR inhibitor BMS493 significantly decreases the frequency of ILC3s, IL-22 production, and the formation of intestinal lymphoid follicles in the mouse small intestine postnatally [78, 80]. RA signaling also determines the pattern of homing receptors on ILC3s, specifically promoting CCR9 and integrin α4β7 on ILC3s and supporting the trafficking of ILC3s to the intestine in mice [81]. RA can also induce IFN-γ production and α4β7 expression in human ILC3s in vitro [82].
Human and mouse ILC3s, especially the CD4+ LTi subset of ILC3s, express GPR183 in the intestine [83, 84]. GPR183 is a receptor for oxysterols, the hydroxylated metabolites of cholesterol. Oxysterols are mainly expressed by fibroblast stromal cells in the intestine and serve as chemoattractants for ILC3s [83]. Through binding to GPR183, oxysterols direct ILC3 migration to CP and ILF in both the colon and small intestine. For example, deletion of GPR183 in ILC3s (Gpr183ΔRorc) leads to a decreased number of CP and ILF, but not PP or mLN in mice. In addition, GPR183 deficiency (Gpr183−/−) causes reduced accumulation of ILC3s in CP and ILF, but an increase in ILC3s in the mLN, relative to wildtype mice [83, 84]. GPR183 signaling in ILC3s also protects the host from C. rodentium infection in mice [84]. However, colonic inflammation can upregulate the oxysterol-GPR183 pathway, which in turn promotes intestinal inflammation in mice, which could be clinically relevant as there is a correlation between ulcerative colitis and the expression of enzymes required for oxysterol synthesis [83].
Additionally, intestinal ILC3s express high concentrations of vitamin D receptor (VDR) [85]. Indeed, a vitamin D-deficient diet results in a lower frequency of ILC3s in both the small intestine and the colon of mice relative to those on a control diet, leading to an increased susceptibility to C. rodentium infection [86]. Global deletion of VDR significantly reduces both NKp46+ and LTi-like ILC3s in the colon, while ILC3-targeted deletion of VDR (VdrΔRorc) particularly impairs the expansion of the LTi-like subset by limiting cellular proliferation [85]. IL-22 production is also decreased in VDR-deficient ILC3s in this model and the mice are more susceptible to C. rodentium infection relative to control mice [85].
Pathways suppressing ILC3 responses in the gut
At steady state, ILC3s constitutively express effector cytokines in the GI tract and play pivotal roles in maintaining the intestinal homeostasis in humans and mice [2]. However, accumulating evidence indicates that ILC3s become dysregulated and are responsible for the initiation or progression of intestinal inflammation under certain conditions, such as following H. hepaticus colonization, or following anti-CD40-induced colitis in immunodeficient mice (Rag1−/−) [45, 87, 88]. Additionally, in mouse models, excessive IL-22 by ILC3s can provoke tumorigenesis in the colon [89, 90]. Therefore, the activation of ILC3s must be effectively and tightly controlled. In the intestine, regulatory signals from epithelial cells, adaptive immune cells, and innate immunity contribute to the suppression of ILC3s (Figure 2).
Figure 2. Suppression signals of ILC3s.
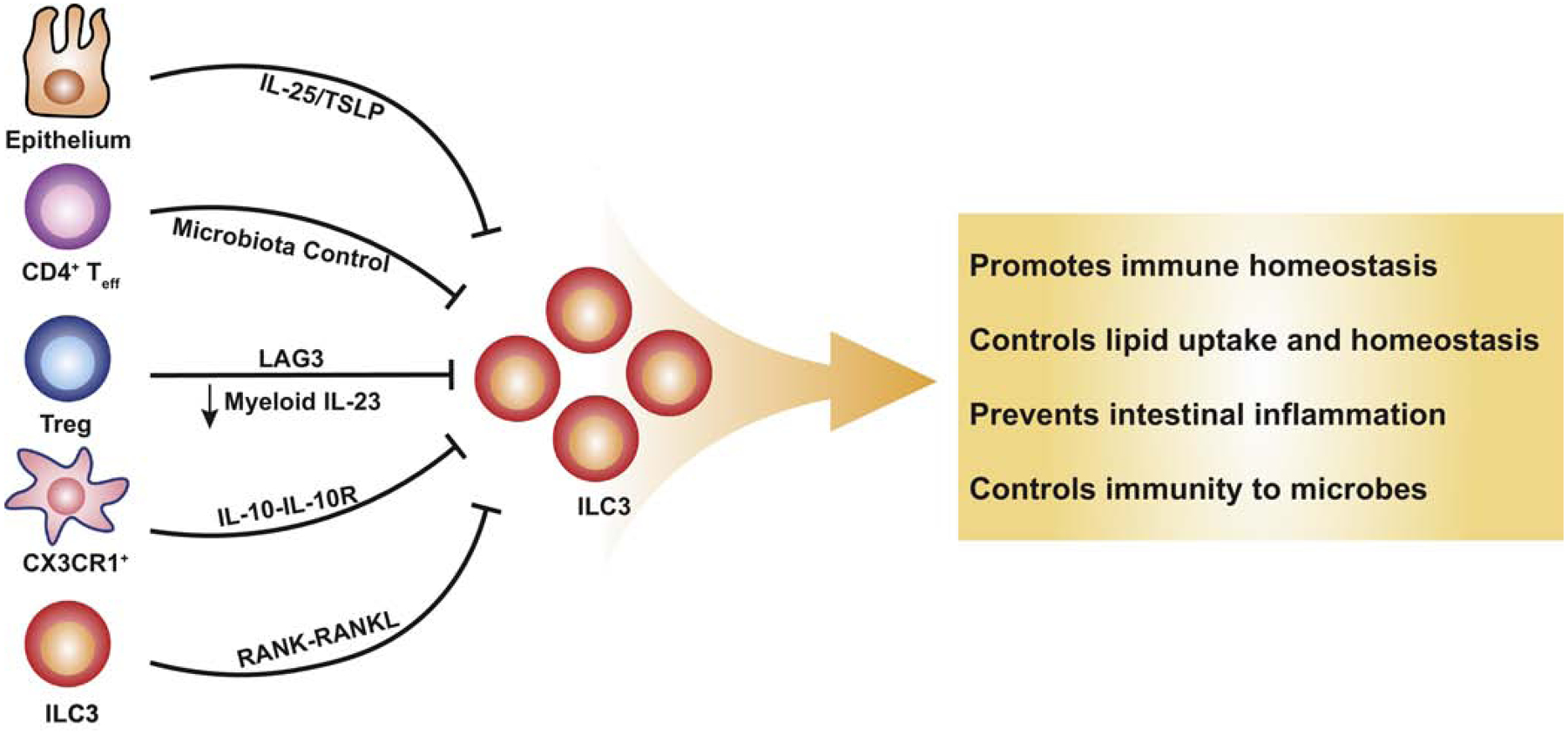
Signals derived from the epithelium, as well as a result of adaptive and innate immunity, suppress the function of ILC3s. Intestinal epithelial cells release IL-25 and TSLP indirectly inhibiting the production of IL-22 in ILC3s. Both CD4+ effector T cells and regulatory T cells are able to indirectly suppress ILC3s through distinct mechanisms. IL-10 signaling in CX3CR1+ myeloid cells indirectly repress ILC3s and RANK signaling enables self-regulation of ILC3s. Downward arrows represent suppression.
Suppression of ILC3s by intestinal epithelial cells
Intestinal epithelial cell-derived IL-25 is produced in response to microbiota colonization and negatively regulates ILC3s. Specifically, depletion of IL-25 (Il25−/−) increases IL-22 expression in ILC3s, whereas administration of IL-25 suppresses IL-22 production in mice [91]. Of note, The IL-25 receptor (IL-17RB) is not expressed by ILC3s, indicating that IL-25 indirectly inhibits ILC3s in this context. In addition, thymic stromal lymphopoietin (TSLP) expressed by intestinal epithelial cells is another negative regulator of ILC3s in mice [92]. Administration of TSLP to mice limits IL-23 induced IL-22 expression by ILC3s. Mice lacking TSLP (Tslp−/−) or antibody-mediated neutralization) protects Rag1−/− mice from C. rodentium infection. Notably, culture of ILC3s with TSLP fails to diminish IL-22 expression, indicating that similar to IL-25, TSLP also suppresses ILC3s through an indirect mechanism [92].
Negative regulation of ILC3s by adaptive immunity
A number of reports have identified crucial cross-regulation between ILC3s and T cells [93]. ILC3s in the colon suppress the expansion of CD4+ T effector cells through MHCII-dependent antigen presentation in humans and mice [94, 95]. Conversely, mice lacking T cells (Rag2−/−) exhibit substantially higher numbers of ILC3s as well as IL-22-expressing ILC3s in both the small intestine and colon [91, 96], indicating that adaptive immunity limits ILC3 activation. Reconstitution of CD4+ T cells is sufficient to normalize the number and IL-22 production of ILC3s in mice through a number of distinct mechanisms [96]. Th17 cells suppress ILC3s indirectly by decreasing SFB abundance in the small intestine, whereas Treg inhibit IL-23 production by CCR2+ myeloid cells to subsequently limit ILC3 activation in mice [34]. In addition, Treg prevent IL-23 and IL-1β production by CX3CR1+ macrophages in a LAG-3 (latent activation gene-3)-dependent manner to restrain IL-22 expression in ILC3s in the colon of mice [97]. These pathways of cross-regulation may be particularly important during the initial colonization of the intestine with microbiota wherein ILC3 activation is promoted prior to the generation of an antigen-specific CD4+ T cell response, thus allowing a fine-tuned innate and adaptive lymphocyte response that facilitates appropriate microbiota colonization and limits immune activation. Furthermore, this crosstalk is necessary to promote normal lipid uptake and homeostasis in the small intestine of mice [34]. Additional studies are warranted to fully assess these possibilities and determine their relevance to human health and disease.
Suppression of ILC3s by innate immunity
Other signals can also suppress the activation of ILC3s. IL-10 signaling in CX3CR1+ macrophages contributes to the control of ILC3 activation, and deletion of IL-10R in CX3CR1+ macrophages induces uncontrolled IL-22 expression in ILC3s in the small intestine of mice, due in part to the elevated production of IL-23 [98]. ILC3s express RANKL on their surface (a member of TNF superfamily), and deletion of RANKL in ILC3s increases the frequency of CCR6+ ILC3s in the mouse intestine, as well as IL-17A and IL-22 production stimulated by IL-23 and C. rodentium infection [99]. The expression of RORγt is also upregulated at steady state in RANKL-deficient CCR6+ ILC3s in mice [99]. RANKL directly binds to its receptor RANK which is expressed on the surface of ILC3s as well [99]. Therefore, ILC3s have the potential to cross-regulate their own activation in this context [99]. Additional pathways by which innate immunity controls ILC3s remain an additional area of active investigation.
ILC3 plasticity
Recently, a growing body of evidence supports the concept that ILC3s exhibit plasticity and have the potential to convert into other subsets of ILCs with appropriate stimuli [100]. Incubation of human ILC3s with IL-2 and IL-12 reduces IL-22 secretion, promotes the expression of IFN-γ and drives the differentiation of RORγt+ ILC3s to an ILC1 or “ex-ILC3” phenotype in vitro [51, 101]. There is also a phenotypic shift of human c-kit+ NKp44− ILC3s toward c-kit− ILC1s. In humans, this shift may be reversed by stimulation with RAs and IL-23 [102, 103]. Accordingly, evidence suggests that this ILC3 to ILC1 conversion also occurs in mice and is exacerbated in the inflamed intestine of patients with IBD [102].
Besides cytokines, transcription factors are involved in modulating ILC3 plasticity [38, 104–106]. Recently, two intermediate populations between ILC3 and ILC1 were reported in healthy humans based on flow cytometry analysis of the typical surface markers and single-cell RNA-sequencing [104]. The gene expression profiles of these intermediate populations fit into the ILC3-ILC1 spectrum. Similarly, these intermediate populations also exhibited an intermediate capacity to produce IFN-γ [104]. Moreover, the transcription factors Aiolos and T-bet cooperate to determine the fate of ILC3s. Aiolos is absent in ILC3s, but is detectable in the intermediate populations, and is also highly expressed in ILC1s [104]. T-bet has a similar expression pattern among these cell populations in human mucosal tissues. Co-expression of Aiolos and T-bet in vitro downregulates IL-22 secretion by ILC3s, but upregulates IFN-γ production in ILC3-like mouse cell line. The expression of Aiolos and T-bet are driven by TGF-β and IL-23, suggesting that environmental cytokine stimulation drives the shift of ILC3s towards ILC1s or ex-ILC3 [104]. In mice, c-Maf represses the expression of T-bet in CCR6− ILC3s by directly binding to the promoter of Tbx21 [105, 106]. Depletion of c-Maf in ILC3 increases the frequency of NKp46+ ILC3 and the expression of type 1 cytokine IFN-γ. Therefore, c-Maf acts as an essential negative regulator of T-bet+ ILC3s in mice by restraining type 1 programming and enforcing ILC3 identity [105, 106].
Finally, the expression of NKp46 on the surface of ILC3s is also reversible. Fate mapping analysis has demonstrated that a subset of NKp46− ILC3s has expressed the receptor previously [107]. Notch signaling is required to maintain the expression of NKp46, whereas TGF-β signaling in ILC3s is associated with the downregulation of NKp46 in mice. Constitutively active Tgfbr in ILC3s dramatically reduces both the percentage and the total number of NKp46+ ILC3s along the GI tract [107]. In vitro incubation of ILC3s with TGF-β also decreases the expression of NKp46 [108]. Therefore, TGF-β signaling is a negative regulator of the NKp46+ ILC3 subset. It remains unclear whether this plasticity program represents an altered state of activation, or in some contexts could be a mechanism of suppressing ILC3s, which represents an exciting opportunity to modulate ILC3s in different states of health and disease.
Concluding Remarks
While many regulatory pathways have been identified, we are only in the early stages of understanding the signals that promote, inhibit, or fine-tune ILC3s in the gut. Many questions remain unanswered (see Outstanding Questions). Some of the existing findings have caveats of being performed in immunodeficient versus immunosufficient murine hosts, which could complicate the complete interpretation of the conclusions. Similarly, not all reports utilize consistent gating strategies or appreciate the heterogeneity of ILC subsets, which is particularly lacking in humans. Therefore, it is important to reconsider all findings when interpreting new results and integrating them into a more comprehensive understanding of ILC3 biology.
Outstanding Questions Box:
Metabolites derived from gut microbes directly activate ILC3s by binding to receptors. What is the extent by which ILC3s sense microorganisms through pathogen recognition receptors directly in vivo?
A number of cytokines that activate, inhibit, or fine-tune ILC3s have been identified. What are the signaling pathways downstream of these cytokines in ILC3s that result in different outputs, and how do they cross regulate each other in synergistic, additive, or inhibitory ways?
Do metabolic pathways employed by ILC3s affect their activation? And if so, how?
The epigenetic regulation of ILC3s is at the early stages of investigation. In which ways does epigenomic regulation occur in ILC3s, and does it substantially modulate the function of these cells, or their ability to respond to microbes?
ILC3s are altered in human inflammatory diseases and cancers. How and why does the activity of ILC3s become dysregulated and can it be reversed by targeted therapies?
Murine ILC3s are highly heterogenous. What is the full heterogenicity and plasticity of human ILC3s? Do findings in mouse ILC3s fully translate to humans? Whether ILC3 subsets in humans have different activation signals, distinct functions, and separate localization, needs to be further investigated in health and disease.
Are there additional unknown functions of ILC3s beyond the well-described cellular interactions and cytokine production described in this review?
ILC3s regulate the function and response of epithelial cells and lymphocytes. Whether and how ILC3s influence the activation of cells of the myeloid lineage and other innate lymphoid cells is less understood. Neurotrophic factors have also been recently appreciated as being able to modulate the action of ILC3s by either activating or suppressing this cellular subset. How ILC3s coordinate these diverse signals to exert their function is an active area of future investigation (see outstanding questions). Further, the majority of research on ILC3s currently focuses on contexts of chemically-induced intestinal inflammation, or following bacterial colonization, while the crosstalk between ILC3s and fungi, virus, or protozoa is poorly understood. It is expected that an increased appreciation of these pathways regulating ILC3s may lead to a better understanding of human diseases where ILC3s become dysregulated. It may also lead to novel approaches that might benefit human health by targeting ILC3 responses.
Highlights Box:
ILC3s are tissue-resident lymphocytes enriched in the mammalian intestine and which crucially orchestrate immunity, inflammation and tissue homeostasis.
ILC3s are fundamentally altered in the intestine of individuals with inflammatory bowel disease, chronic infections or cancer.
Multiple cellular and molecular circuits promote ILC3 activation, including signals derived from microbiota, pathogens, neurons and dietary stimuli.
There has been recent appreciation of novel pathways that fine-tune or suppress ILC3s in the gut, such as plasticity, circadian rhythm and adaptive immunity.
Acknowledgements
We thank members of the Sonnenberg Laboratory for discussions and critical reading of the manuscript. Research in the Sonnenberg Laboratory is supported by the National Institutes of Health (R01AI143842, R01AI123368, R01AI145989, R21CA249274 and U01AI095608), the NIAID Mucosal Immunology Studies Team (MIST), the Searle Scholars Program, the American Asthma Foundation Scholar Award, an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund, a Wade F.B. Thompson/Cancer Research Institute (CRI) CLIP Investigator grant, the Meyer Cancer Center Collaborative Research Initiative, Linda and Glenn Greenberg, and the Roberts Institute for Research in IBD. G.F.S. is a CRI Lloyd J. Old STAR.
Glossary
- Gastrointestinal tract (GI tract)
organ system composed of the mouth, esophagus, stomach and intestines; absorbs energy and nutrients.
- CCR6+ ILC3
ILC3s expressing CCR6 but not T-bet or NKp46.
- DSS-induced intestinal injury
a mouse model of intestinal damage and inflammation induced by dextran sodium sulfate (DSS) in drinking water.
- NKp46+ ILC3
ILC3s expressing NKp46 and T-bet but not CCR6.
- Peyer’s patches (PP)
organized and aggregated lymphoid follicles in the small intestine.
- Cryptopatches (CP)
small clusters of lymphoid cells with predominantly lineage negative cells in the lamina propria of the intestine.
- Isolated lymphoid follicles (ILF)
organized lymphoid structures derived from cryptopatches and induced by microbiota colonization.
- Short-chain fatty acids (SCFA)
a metabolite of microbial fermentation of dietary fiber that has fewer than six carbon atoms.
- Inflammatory bowel disease (IBD)
a term for a group of intestinal disorders characterized by chronic inflammation.
- Toll-like receptors (TLR)
a class of receptors that recognize pathogen-associated molecular patterns from microorganisms.
- Pathogen-associated molecular patterns
a set of molecules from microbes that can be recognized by pattern recognition receptors to trigger immune responses.
- Natural cytotoxicity receptors (NCR)
a group of cell surface receptors predominantly expressed by NK cells.
- Regulatory T cells (Treg)
subset of T cells that mediates immunologic tolerance
- Segmented filamentous bacteria (SFB)
commensal bacteria that attach to the epithelium of the ileum and induce intestinal Th17 cells in mice.
- Opportunistic pathogens
non-pathogenic for the healthy host; becoming pathogenic when the host is compromised.
- Rag-deficient mice (Rag1−/− or Rag2−/−)
mice lacking mature T and B lymphocytes.
- Circadian rhythms
physiological processes following a daily cycle.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maloy K and Powrie F (2011) Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474 (7351), 298–306. [DOI] [PubMed] [Google Scholar]
- 2.Vivier E et al. (2018) Innate Lymphoid Cells: 10 Years On. Cell 174 (5), 1054–1066. [DOI] [PubMed] [Google Scholar]
- 3.Spits H et al. (2013) Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol 13 (2), 145–9. [DOI] [PubMed] [Google Scholar]
- 4.Satoh-Takayama N et al. (2008) Microbial Flora Drives Interleukin 22 Production in Intestinal NKp46(+) Cells that Provide Innate Mucosal Immune Defense. Immunity 29 (6), 958–970. [DOI] [PubMed] [Google Scholar]
- 5.Sonnenberg G et al. (2011) CD4(+) Lymphoid Tissue-Inducer Cells Promote Innate Immunity in the Gut. Immunity 34 (1), 122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chun E et al. (2019) Metabolite-Sensing Receptor Ffar2 Regulates Colonic Group 3 Innate Lymphoid Cells and Gut Immunity. Immunity 51 (5), 871-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiu J et al. (2012) The Aryl Hydrocarbon Receptor Regulates Gut Immunity through Modulation of Innate Lymphoid Cells. Immunity 36 (1), 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mortha A et al. (2014) Microbiota-Dependent Crosstalk Between Macrophages and ILC3 Promotes Intestinal Homeostasis. Science 343 (6178), 1477-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou L et al. (2019) Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature 568 (7752), 405-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rooks M and Garrett W (2016) Gut microbiota, metabolites and host immunity. Nature Reviews Immunology 16 (6), 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levy M et al. (2016) Metabolites: messengers between the microbiota and the immune system. Genes Dev 30 (14), 1589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samuel B et al. (2008) Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proceedings of the National Academy of Sciences of the United States of America 105 (43), 16767–16772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swann J et al. (2011) Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proceedings of the National Academy of Sciences of the United States of America 108, 4523–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zelante T et al. (2013) Tryptophan Catabolites from Microbiota Engage Aryl Hydrocarbon Receptor and Balance Mucosal Reactivity via Interleukin-22. Immunity 39 (2), 372–385. [DOI] [PubMed] [Google Scholar]
- 15.Kim S et al. (2017) Microbiota-derived butyrate suppresses group 3 innate lymphoid cells in terminal ileal Peyer’s patches. Scientific Reports 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhatt B et al. (2018) Gpr109a Limits Microbiota-Induced IL-23 Production To Constrain ILC3-Mediated Colonic Inflammation. Journal of Immunology 200 (8), 2905–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cella M et al. (2009) A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 457 (7230), 722–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiss E et al. (2011) Natural Aryl Hydrocarbon Receptor Ligands Control Organogenesis of Intestinal Lymphoid Follicles. Science 334 (6062), 1561–1565. [DOI] [PubMed] [Google Scholar]
- 19.Lee J et al. (2012) AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nature Immunology 13 (2), 144–U58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li S et al. (2016) Ikaros Inhibits Group 3 Innate Lymphoid Cell Development and Function by Suppressing the Aryl Hydrocarbon Receptor Pathway. Immunity 45 (1), 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hughes T et al. (2014) The Transcription Factor AHR Prevents the Differentiation of a Stage 3 Innate Lymphoid Cell Subset to Natural Killer Cells. Cell Reports 8 (1), 150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gronke K et al. (2019) Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature 566 (7743), 249-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crellin N et al. (2010) Regulation of Cytokine Secretion in Human CD127(+) LTi-like Innate Lymphoid Cells by Toll-like Receptor 2. Immunity 33 (5), 752–764. [DOI] [PubMed] [Google Scholar]
- 24.Barrow A et al. (2019) The Natural Cytotoxicity Receptors in Health and Disease. Frontiers in Immunology 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glatzer T et al. (2013) ROR gamma t(+) Innate Lymphoid Cells Acquire a Proinflammatory Program upon Engagement of the Activating Receptor NKp44. Immunity 38 (6), 1223–1235. [DOI] [PubMed] [Google Scholar]
- 26.Satoh-Takayama N et al. (2009) The Natural Cytotoxicity Receptor NKp46 Is Dispensable for IL-22-Mediated Innate Intestinal Immune Defense against Citrobacter rodentium. Journal of Immunology 183 (10), 6579–6587. [DOI] [PubMed] [Google Scholar]
- 27.Kruse P et al. (2014) Natural cytotoxicity receptors and their ligands. Immunology and Cell Biology 92 (3), 221–229. [DOI] [PubMed] [Google Scholar]
- 28.Takatori H et al. (2009) Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. Journal of Experimental Medicine 206 (1), 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanos S et al. (2009) ROR gamma t and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46(+) cells. Nature Immunology 10 (1), 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ivanov I et al. (2009) Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell 139 (3), 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaboriau-Routhiau V et al. (2009) The Key Role of Segmented Filamentous Bacteria in the Coordinated Maturation of Gut Helper T Cell Responses. Immunity 31 (4), 677–689. [DOI] [PubMed] [Google Scholar]
- 32.Sano T et al. (2015) An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell 163 (2), 381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Atarashi K et al. (2015) Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 163 (2), 367–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mao K et al. (2018) Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature 554 (7691), 255-+. [DOI] [PubMed] [Google Scholar]
- 35.Fung T et al. (2016) Lymphoid-Tissue-Resident Commensal Bacteria Promote Members of the IL-10 Cytokine Family to Establish Mutualism. Immunity 44 (3), 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.SCHAUER D and FALKOW S (1993) ATTACHING AND EFFACING LOCUS OF A CITROBACTER-FREUNDII BIOTYPE THAT CAUSES TRANSMISSIBLE MURINE COLONIC HYPERPLASIA. Infection and Immunity 61 (6), 2486–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng Y et al. (2008) Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nature Medicine 14 (3), 282–289. [DOI] [PubMed] [Google Scholar]
- 38.Klose C et al. (2013) A T-bet gradient controls the fate and function of CCR6(−)ROR gamma t(+) innate lymphoid cells. Nature 494 (7436), 261–265. [DOI] [PubMed] [Google Scholar]
- 39.Longman R et al. (2014) CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. Journal of Experimental Medicine 211 (8), 1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manta C et al. (2013) CX(3)CR1(+) macrophages support IL-22 production by innate lymphoid cells during infection with Citrobacter rodentium. Mucosal Immunology 6 (1), 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seo S et al. (2015) Intestinal macrophages arising from CCR2(+) monocytes control pathogen infection by activating innate lymphoid cells. Nature Communications 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang B et al. (2019) Macrophage beta 2-Integrins Regulate IL-22 by ILC3s and Protect from Lethal Citrobacter rodentium-Induced Colitis. Cell Reports 26 (6), 1614-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo X et al. (2014) Induction of Innate Lymphoid Cell-Derived Interleukin-22 by the Transcription Factor STAT3 Mediates Protection against Intestinal Infection. Immunity 40 (1), 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tumanov A et al. (2011) Lymphotoxin Controls the IL-22 Protection Pathway in Gut Innate Lymphoid Cells during Mucosal Pathogen Challenge. Cell Host & Microbe 10 (1), 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buonocore S et al. (2010) Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464 (7293), 1371–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Castellanos J et al. (2018) Microbiota-Induced TNF-like Ligand 1A Drives Group 3 Innate Lymphoid Cell-Mediated Barrier Protection and Intestinal T Cell Activation during Colitis. Immunity 49 (6), 1077-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J et al. (2019) Activation of DR3 signaling causes loss of ILC3s and exacerbates intestinal inflammation. Nature Communications 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Siakavellas S and Bamias G (2015) Tumor Necrosis Factor-like Cytokine TL1A and Its Receptors DR3 and DcR3: Important New Factors in Mucosal Homeostasis and Inflammation. Inflammatory Bowel Diseases 21 (10), 2441–2452. [DOI] [PubMed] [Google Scholar]
- 49.NICAISE P et al. (1993) INFLUENCE OF INTESTINAL BACTERIAL-FLORA ON CYTOKINE (IL-1, IL-6 AND TNF-ALPHA) PRODUCTION BY MOUSE PERITONEAL-MACROPHAGES. European Cytokine Network 4 (2), 133–138. [PubMed] [Google Scholar]
- 50.Schirmer M et al. (2016) Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell 167 (4), 1125-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cella M et al. (2010) Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc Natl Acad Sci U S A 107 (24), 10961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Powell N et al. (2015) Interleukin 6 Increases Production of Cytokines by Colonic Innate Lymphoid Cells in Mice and Patients With Chronic Intestinal Inflammation. Gastroenterology 149 (2), 456-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Munoz M et al. (2015) Interleukin-22 Induces Interleukin-18 Expression from Epithelial Cells during Intestinal Infection. Immunity 42 (2), 321–331. [DOI] [PubMed] [Google Scholar]
- 54.Hernandez P et al. (2015) Interferon-lambda and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nature Immunology 16 (7), 698-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rao M and Gershon M (2016) The bowel and beyond: the enteric nervous system in neurological disorders. Nature Reviews Gastroenterology & Hepatology 13 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chesne J et al. (2019) Neuro-immune regulation of mucosal physiology. Mucosal Immunology 12 (1), 10–20. [DOI] [PubMed] [Google Scholar]
- 57.Veiga-Fernandes H and Artis D (2018) Neuronal-immune system cross-talk in homeostasis. Science 359 (6383), 1465–1466. [DOI] [PubMed] [Google Scholar]
- 58.Quatrini L et al. (2018) Neuroendocrine regulation of innate lymphoid cells. Immunol Rev 286 (1), 120–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ibiza S et al. (2016) Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature 535 (7612), 440-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Veiga-Fernandes H et al. (2007) Tyrosine kinase receptor RET is a key regulator of Peyer’s Patch organogenesis. Nature 446 (7135), 547–551. [DOI] [PubMed] [Google Scholar]
- 61.Lelievre V et al. (2007) Gastrointestinal dysfunction in mice with a targeted mutation in the gene encoding vasoactive intestinal polypeptide: A model for the study of. intestinal ileus and Hirschsprung’s disease. Peptides 28 (9), 1688–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Talbot J et al. (2020) Feeding-dependent VIP neuron-ILC3 circuit regulates the intestinal barrier. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seillet C et al. (2019) The neuropeptide VIP confers anticipatory mucosal immunity by regulating ILC3 activity. Nat Immunol. [DOI] [PubMed] [Google Scholar]
- 64.Dalli J et al. (2017) Vagal Regulation of Group 3 Innate Lymphoid Cells and the Immunoresolvent PCTR1 Controls Infection Resolution. Immunity 46 (1), 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Asher G and Sassone-Corsi P (2015) Time for food: the intimate interplay between nutrition, metabolism, and the circadian clock. Cell 161 (1), 84–92. [DOI] [PubMed] [Google Scholar]
- 66.Hussain MM (2014) Regulation of intestinal lipid absorption by clock genes. Annu Rev Nutr 34, 357–75. [DOI] [PubMed] [Google Scholar]
- 67.Tahara Y and Shibata S (2013) Chronobiology and nutrition. Neuroscience 253, 78–88. [DOI] [PubMed] [Google Scholar]
- 68.Bellet MM et al. (2013) Circadian clock regulates the host response to Salmonella. Proc Natl Acad Sci U S A 110 (24), 9897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liang X et al. (2015) Rhythmicity of the intestinal microbiota is regulated by gender and the host circadian clock. Proc Natl Acad Sci U S A 112 (33), 10479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thaiss CA et al. (2014) Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell 159 (3), 514–29. [DOI] [PubMed] [Google Scholar]
- 71.Mukherji A et al. (2013) Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell 153 (4), 812–27. [DOI] [PubMed] [Google Scholar]
- 72.Yu X et al. (2013) TH17 cell differentiation is regulated by the circadian clock. Science 342 (6159), 727–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Y et al. (2017) The intestinal microbiota regulates body composition through NFIL. Science 357 (6354), 912–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Godinho-Silva C et al. (2019) Light-entrained and brain-tuned circadian circuits regulate ILC3s and gut homeostasis. Nature 574 (7777), 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Teng F et al. (2019) A circadian clock is essential for homeostasis of group 3 innate lymphoid cells in the gut. Sci Immunol 4 (40). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Q et al. (2019) Circadian rhythm-dependent and circadian rhythm-independent impacts of the molecular clock on type 3 innate lymphoid cells. Science Immunology 4 (40). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van de Pavert SA et al. (2014) Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature 508 (7494), 123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Spencer S et al. (2014) Adaptation of Innate Lymphoid Cells to a Micronutrient Deficiency Promotes Type 2 Barrier Immunity. Science 343 (6169), 432–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim CH (2018) Control of Innate and Adaptive Lymphocytes by the RAR-Retinoic Acid Axis. Immune Netw 18 (1), e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goverse G et al. (2016) Vitamin A Controls the Presence of ROR gamma(+) Innate Lymphoid Cells and Lymphoid Tissue in the Small Intestine. Journal of Immunology 196 (12), 5148–5155. [DOI] [PubMed] [Google Scholar]
- 81.Kim M et al. (2015) Retinoic Acid Differentially Regulates the Migration of Innate Lymphoid Cell Subsets to the Gut. Immunity 43 (1), 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ruiter B et al. (2015) Vitamins A and D have antagonistic effects on expression of effector cytokines and gut-homing integrin in human innate lymphoid cells. Clinical and Experimental Allergy 45 (7), 1214–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Emgård J et al. (2018) Oxysterol Sensing through the Receptor GPR183 Promotes the Lymphoid-Tissue-Inducing Function of Innate Lymphoid Cells and Colonic Inflammation. Immunity 48 (1), 120–132.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chu C et al. (2018) Anti-microbial Functions of Group 3 Innate Lymphoid Cells in Gut-Associated Lymphoid Tissues Are Regulated by G-Protein-Coupled Receptor 183. Cell Rep 23 (13), 3750–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.He L et al. (2019) Vitamin D/Vitamin D Receptor Signaling Is Required for Normal Development and Function of Group 3 Innate Lymphoid Cells in the Gut. Iscience 17, 119-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin Y et al. (2019) Vitamin D Is Required for ILC3 Derived IL-22 and Protection From Citrobacter rodentium Infection. Frontiers in Immunology 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Powell N et al. (2012) The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity 37 (4), 674–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Song C et al. (2015) Unique and redundant functions of NKp46(+) ILC3s in models of intestinal inflammation. Journal of Experimental Medicine 212 (11), 1869–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kirchberger S et al. (2013) Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med 210 (5), 917–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huber S et al. (2012) IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 491 (7423), 259–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sawa S et al. (2011) RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol 12 (4), 320–6. [DOI] [PubMed] [Google Scholar]
- 92.Giacomin PR et al. (2015) Epithelial-intrinsic IKKα expression regulates group 3 innate lymphoid cell responses and antibacterial immunity. J Exp Med 212 (10), 1513–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sonnenberg GF and Hepworth MR (2019) Functional interactions between innate lymphoid cells and adaptive immunity. Nat Rev Immunol 19 (10), 599–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hepworth MR et al. (2013) Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 498 (7452), 113–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hepworth MR et al. (2015) Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science 348 (6238), 1031–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Korn LL et al. (2014) Conventional CD4+ T cells regulate IL-22-producing intestinal innate lymphoid cells. Mucosal Immunol 7 (5), 1045–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bauché D et al. (2018) LAG3. Immunity 49 (2), 342–352.e5. [DOI] [PubMed] [Google Scholar]
- 98.Bernshtein B et al. (2019) IL-23-producing IL-10Rα-deficient gut macrophages elicit an IL-22-driven proinflammatory epithelial cell response. Sci Immunol 4 (36). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bando JK et al. (2018) The Tumor Necrosis Factor Superfamily Member RANKL Suppresses Effector Cytokine Production in Group 3 Innate Lymphoid Cells. Immunity 48 (6), 1208–1219.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Colonna M (2018) Innate Lymphoid Cells: Diversity, Plasticity, and Unique Functions in Immunity. Immunity 48 (6), 1104–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vonarbourg C et al. (2010) Regulated expression of nuclear receptor RORγt confers distinct functional fates to NK cell receptor-expressing RORγt(+) innate lymphocytes. Immunity 33 (5), 736–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bernink JH et al. (2013) Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol 14 (3), 221–9. [DOI] [PubMed] [Google Scholar]
- 103.Bernink JH et al. (2015) Interleukin-12 and −23 Control Plasticity of CD127(+) Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity 43 (1), 146–60. [DOI] [PubMed] [Google Scholar]
- 104.Cella M et al. (2019) Subsets of ILC3-ILC1-like cells generate a diversity spectrum of innate lymphoid cells in human mucosal tissues. Nat Immunol 20 (8), 980–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Parker ME et al. (2020) c-Maf regulates the plasticity of group 3 innate lymphoid cells by restraining the type 1 program. J Exp Med 217 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tizian C et al. (2020) c-Maf restrains T-bet-driven programming of CCR6-negative group 3 innate lymphoid cells. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Viant C et al. (2016) Transforming growth factor-β and Notch ligands act as opposing environmental cues in regulating the plasticity of type 3 innate lymphoid cells. Sci Signal 9 (426), ra46. [DOI] [PubMed] [Google Scholar]
- 108.Verrier T et al. (2016) Phenotypic and Functional Plasticity of Murine Intestinal NKp46+ Group 3 Innate Lymphoid Cells. J Immunol 196 (11), 4731–8. [DOI] [PubMed] [Google Scholar]
- 109.Mebius RE et al. (1997) Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity 7 (4), 493–504. [DOI] [PubMed] [Google Scholar]
- 110.Eberl G et al. (2004) An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol 5 (1), 64–73. [DOI] [PubMed] [Google Scholar]
- 111.Bouskra D et al. (2008) Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456 (7221), 507–10. [DOI] [PubMed] [Google Scholar]
- 112.Björklund Å et al. (2016) The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nat Immunol 17 (4), 451–60. [DOI] [PubMed] [Google Scholar]