

Abstract
Head and neck cancer is a highly genetic and metabolic heterogeneous collection of malignancies of the lip, oral cavity, salivary glands, pharynx, esophagus, paranasal sinuses, and larynx with five-year survival rates ranging from 12% to 93%. Patients with head and neck cancer typically present with advanced stage III, IVa, or IVb disease and are treated with comprehensive modality including chemotherapy, radiotherapy, and surgery. Despite advancements in treatment modality and technique, noisome recurrence, invasiveness, and resistance as well as posttreatment complications severely influence survival rate and quality of life. Thus, new therapeutic strategies are urgently needed that offer enhanced efficacy with less toxicity. ROS in cancer cells plays a vital role in regulating cell death, DNA repair, stemness maintenance, metabolic reprogramming, and tumor microenvironment, all of which have been implicated in resistance to chemo-/radiotherapy of head and neck cancer. Adjusting ROS generation and elimination to reverse the resistance of cancer cells without impairing normal cells show great hope in improving the therapeutic efficacy of chemo-/radiotherapy of head and neck cancer. In the current review, we discuss the pivotal and targetable redox-regulating system including superoxide dismutases (SODs), tripeptide glutathione (GSH), thioredoxin (Trxs), peroxiredoxins (PRXs), nuclear factor erythroid 2-related factor 2/Kelch-like ECH-associated protein 1 (Nrf2/keap1), and mitochondria electron transporter chain (ETC) complexes and their roles in regulating ROS levels and their clinical significance implicated in chemo-/radiotherapy of head and neck cancer. We also summarize several old drugs (referred to as the non-anti-cancer drugs used in other diseases for a long time) and small molecular compounds as well as natural herbs which effectively modulate cellular ROS of head and neck cancer to synergize the efficacy of conventional chemo-/radiotherapy. Emerging interdisciplinary techniques including photodynamic, nanoparticle system, and Bio-Electro-Magnetic-Energy-Regulation (BEMER) therapy are promising measures to broaden the potency of ROS modulation for the benefit of chemo-/radiotherapy in head and neck cancer.
1. Introduction
Head and neck cancer (HNC) is the seventh most frequently occurring malignancy worldwide in 2018 (accounting for 4.9% of all cancer sites) [1]. It is reported that lip, oral cavity, and pharyngeal cancers could be responsible for the 529,500 new cancer cases (accounting for about 3.8% of all cancer cases) and the 292,300 cancer-related deaths (accounting for about 3.6% of all cancer deaths) in 2012 globally, and the incidence is predicted to increase by 62% to 856,000 cases in 2035 [2]. Due to the tenacious resistance of cancer cells to therapy, the five-year survival rate has not been significantly improved during past decade [3]. Commonly used radiation and chemotherapy drugs affect the prognosis of HNC through reactive oxygen species (ROS) regulation directly and indirectly [4]. The balance of cellular ROS is levered by ROS generators including mitochondrial ROS, NADPH oxidases, and other enzymes and ROS eliminators such as superoxide dismutases (SODs), tripeptide glutathione (GSH), and nuclear factor erythroid 2-related factor 2/Kelch-like ECH-associated protein 1 (Nrf2/Keap1) [5]. ROS has been implicated in cancer initiation, formation, and development as well as therapy resistance [6]. In spite of some inspiring clinical trials concerning ROS modulation in comprehensive treatment of HNC, the personalized treatments call for multiple therapeutic strategies. During the past years, genetic or pharmaceutic methods for modulating ROS in HNC are showing great preclinical and clinical significance in the combined modality of chemo-/radiotherapy. Ongoing researches from other groups and our own are making efforts in modulating the cellular ROS level to enhance the efficacy of chemo-/radiotherapy and to decrease side effects and toxicity without compromising therapeutic efficacy in the treatment of HNC.
2. The Epidemiology of Head and Neck Cancer and Leading Therapeutic Challenges
Head and neck cancer incorporates multiple organs from complex anatomical topographies which include the lip (C00), oral cavity (C02-06), salivary glands (C07-08), oropharynx (C01, C09-C10), nasopharynx (C11), hypopharynx (C12-14), esophagus (C15), paranasal sinuses (C30-31), and larynx (C32) [1, 2, 7–9] (Figure 1(a)). About 85-90% of HNC are squamous carcinoma that originated from epithelial cells (HNSCC) [9, 10]. There are more than 800,000 new cases and 500,000 deaths of esophageal, lip, oral cavity, and nasopharyngeal cancers worldwide [11, 12]. In 2020, there are 84,070 estimated new cases and 30,670 estimated deaths of HNC in the United States. The oral cavity and pharynx cancers rank first among the new cases of HNC, while they rank eighth (4%) among all cancer sites in males. The esophageal cancers top the list of HNC mortality [13]. In general, males are more inclined to suffer from HNC [1]. Advancing age is a disadvantage to HNC prognosis. HPV status of HNC influences the therapeutic outcome; HPV-positive HNC are associated with a better response to chemotherapy and radiotherapy even with stage IV disease [8, 14]. The five-year survival rates of HNC range from 12% to 93% from among different ages, gender, educational levels, race, and geographical locations as well as different cancer sites, pathological grades, and received therapy [2, 3, 12, 15, 16].
Figure 1.
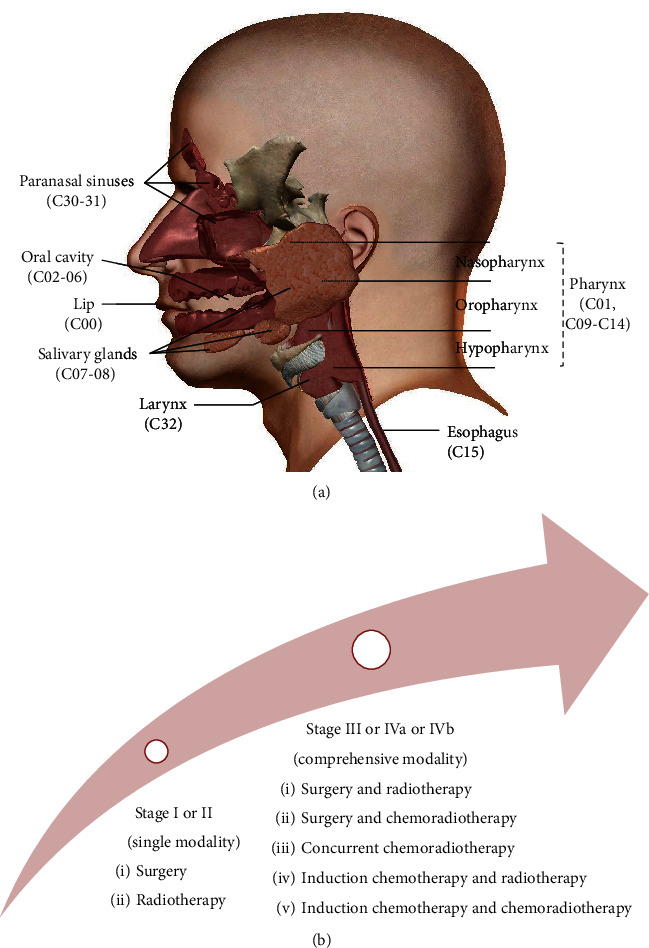
Anatomical sites and treatment of HNC. (a) Head and neck cancers incorporate multiple anatomical regions concerning the lip (C00), oral cavity (C02-06), salivary glands (C07-08), oropharynx (C01, C09-C10), nasopharynx (C11), hypopharynx (C12-14), esophagus (C15), paranasal sinuses (C30-31), and larynx (C32). International Classification of Diseases 10th revision, website: http://www.who.int/classifications/icd/icdonlineversions/en/. (b) HNC patients with early stages (stages I and II) are recommended for single modality including surgery or radiotherapy. Comprehensive modality including surgery, radiotherapy, and chemotherapy is guided for advanced cases (stages III, IVa, and IVb). Note. NCCN Clinical Practice Guidelines in Oncology: Head and Neck Cancers, website: https://www.nccn.org.
Due to the special anatomical position, HNC are prone to exert a negative impact on language, respiration, eating, swallowing, and digestion. Besides, rapid blood supply and lymphatic regurgitation render HNC to be inclined to cervical lymph node metastasis [14]. The treatment strategy depends on individual factors concerned with the site of the cancer and tumor/node/metastasis (TNM) stage, as well as patient preference [14, 16]. In general, HNC at an early stage (TNM: I and II) are well controlled after surgery or radiotherapy. HNC at an advanced stage (TNM: III, IVa, and IVb) are locally invasive and accompanied by metastasis of cervical lymph nodes. It is difficult to completely remove the cancer. They call for comprehensive treatment of surgery, radiotherapy, and chemotherapy to reduce the original lesion or control the postoperative period [17–19] (Figure 1(b)). Unfortunately, two-thirds of patients with HNC are advanced cases (T3-T4 and/or cervical adenopathy) when they are first examined, at which point they have lost the optimum time for surgery [20]. Cisplatin- (CDDP-) based chemotherapy and adjuvant radiotherapy are still the first-line treatment options for advanced patients [14]. In spite of the advancement of diagnosis and treatment modality, such as minimally invasive transoral surgery, intensity-modulated radiotherapy (IMRT), gene-targeting drugs (anti-EGFR therapy), and immunotherapy (anti-PD-1 therapy) of HNC, the long-term survival rate of patients is not substantially improved [21]. Disappointingly, more than 50% of patients develop recurrence in either local or distant sites within two years of treatment [22, 23]. Recurrence and posttherapy complications (marrow depression, immune suppression, muscle fibrosis, renal toxicity, mucosal damage, salivary gland secretion disorders, mandibular fractures, and necrosis) severely affect the quality of life and lead to a high morbidity of HNC patients [8, 24]. Resistance to treatment is correlated with recurrence and morbidity. Thus, developing new treatment strategies to surmount recurrence and complications is vital for improving the long-term survival and quality of life of patients with HNC [25, 26]. Cancer cells are prone to increase oxidative stress and switch the metabolism pattern to aerobic glycolysis called the Warburg effect [27–29]. Targeting these unique biochemical alterations in cancer cells might be a feasible strategy to prevent therapy resistance and ameliorate the prognosis [30].
3. Redox Adaptation in Cancer Cells and Its Implicated Modulation in Chemo-/Radiotherapy of HNC
Reactive oxygen species (ROS) is a term that denotes a series of intermediate products produced during the oxidative metabolism of cells, including two-electron (nonradical) ROS such as hydrogen peroxide (H2O2), organic hydroperoxides (ROOH), singlet molecular oxygen (1O2), hypochlorous acid (HOCl), hypobromous acid (HOBr), and ozone (O3); free radical ROS include the superoxide anion radical (O2−), the hydroxyl radical (·OH), the peroxyl radical (ROO·), and the alkoxyl radical (RO·) [5]. Mitochondrial electron transport chain (ETC) complex [31] and nicotinamide adenine dinucleotide phosphate oxidases (NOXs) [32] are the major endogenous sources of ROS. To protect lipids, proteins, and nucleic acids from indiscriminate damage induced by free radicals, cells arrange a complex network of antioxidant systems to maintain genomic stability and proper cellular function [6]. SODs and GSH are the predominant antioxidant systems [30] (Figure 2). Other ROS generators including cytochrome p450, lipoxygenase, and xanthine oxidase and scavengers such as catalase (CAT), peroxiredoxins (PRXs), glutathione peroxidases (GPXs), vitamin C, and vitamin E closely participate in the redox system [6, 33]. Nrf2/Keap1 complex regulates redox hemostasis by sensing oxidative stress and then activating downstream antioxidant elements such as glutathione-S-transferases (GST), NAD(P)H:quinone oxidoreductase (NQO1), PRXs, GPXs, and CAT [34–36]. Other redox-sensitive transcription factors such as nuclear factor-κB (NF-κB), p53, and hypoxia inducible factor 1 (HIF-1) lead to elevation of ROS-eliminating enzymes like SOD and GSH, activating survival factors such as myeloid cell leukaemia-1 (Mcl-1) and B-cell lymphoma-2 (Bcl-2), and inhibition of cell death factors [30].
Figure 2.
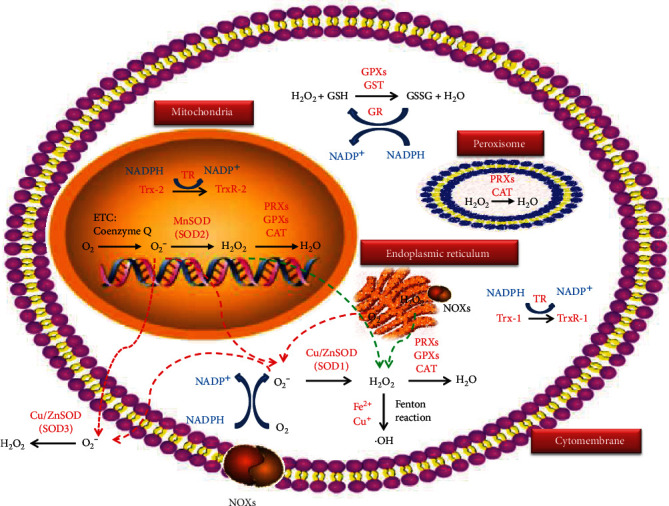
ROS sources and antioxidant systems. Mitochondrial respiration ETC and the membrane-bond NOX complexes are the two major ROS resources. Leakage of electrons from ETC is mediated by coenzyme Q and produces O2− through O2. There are three isoforms of SODs to defend oxidation. Cu/Zn SOD (SOD1) in the cytoplasm, MnSOD (SOD2) in the mitochondria, and Cu/Zn SOD (SOD3) in the extracellular matrix can rapidly convert O2− to H2O2. NOXs catalyze the generation of O2− from O2 and NADPH. H2O2 is converted to toxic ·OH by a metal (Fe2+ or Cu+) catalyst through the Fenton reaction. H2O2 can be converted into H2O by PRXs, GPXs, and CAT. Besides, Trxs (the cytoplasmic Trx-1 and the mitochondrial Trx-2) can reduce oxidized PRXs. Trxs themselves are also reduced to TrxR by TR using NADPH as an electron donor. GPXs oxidize reduced GSH to GSSH. GSSH is reduced back to GSH by GR accompanied by an electron from NADPH. Note. ETC: electron transport chain; NOXs: NADPH oxidase; SODs: superoxide dismutases; H2O2: hydrogen peroxide; NADPH: nicotinamide adenine dinucleotide phosphate; ·OH: hydroxyl radicals; PRXs: peroxiredoxins; GPXs: glutathione peroxidases; CAT: catalase; Trx: thioredoxin (oxidized); Trx-R: thioredoxin (reduced); TR: thioredoxin reductase; GSH: tripeptide glutathione (reduced); GSSH: glutathione disulfide (oxidized); GR: glutathione. Green dotted lines denote H2O2 diffusion. Red dotted lines denote O2− diffusion.
In normal cells, redox balance is well orchestrated via antioxidant defense systems. Once exposed to continuous exogenous stimuli such as radiation and carcinogens and endogenous oncogene activation such as H-Ras, the normal cells fail to leverage the redox balance, thus forming cancer cells [37]. To adapt to the oxidative stress, the initiated cancer cells will tactfully enhance the antioxidant enzymes accordingly. Consequently, both the ROS level and ROS-scavenging enzymes are increased to benefit cancer cell survival, metastasis, and even resistance [6, 38]. In other words, ROS represents a double-edged sword [39]. Basal levels of ROS can maintain the homeostasis of normal cells; chronic and low levels of ROS promote cell mitosis and increase genomic instability to induce the occurrence and progression of tumors; moderate concentrations of ROS cause temporary or permanent cell cycle arrest and may induce cell differentiation [39]; acute and high concentrations of ROS damage macromolecules and thus induce apoptosis, necrosis, and ferroptosis [40]. Therefore, the high concentration of ROS in cancer cells and the defects of their antioxidant damage defense system render cancer cells more susceptible to ROS modulation. In the case of the same concentration of ROS, cancer cells first undergo apoptosis while normal cells can tolerate it [41–43]. Adjusting intracellular ROS levels to efficiently kill cancer cells and reduce the side effect of chemo-/radiotherapy is currently considered as the fundamental means of cancer treatment [30, 40, 44] (Figure 3).
Figure 3.
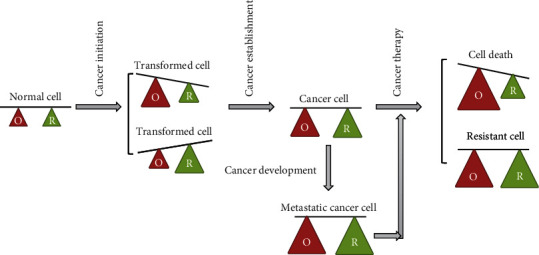
Redox adaptation in cancer formation, development, and therapy. Cellular redox homeostasis is maintained by ROS generation and elimination balance in normal cells. Once continuous exogenous stimulus and endogenous oncogene activation disrupt the balance, either a high level of ROS is produced or antioxidants are excessively enhanced, and cancer cells are hence formed. In order to survive oxidative stress, these cancer cells regain redox homeostasis via multiple mechanisms such as increasing ROS-scavenging enzymes. During the development of cancer and even during the process of therapy resistance, the cancer cells gradually enhance both ROS level and antioxidant enzymes. Thus, abrogating the adaptation mechanisms by increasing the ROS level beyond a threshold that is incompatible for cellular survival and attenuating antioxidant defense systems can be an attractive strategy to kill cancer cells and thus reverse resistance and limit cancer progression. Note. O: oxidative status; R: reducing status.
During chemotherapy and/or radiotherapy in HNC, frequent resistance and accompanying side effects are the head-scratching puzzles. Despite the development of gene-targeted drugs such as bortezomib, sorafenib, and cetuximab for the treatment of HNC, evasion of therapy remains the main obstacle to cure [21]. The implicated regulation of ROS is of great significance for cancer treatment, because commonly used radiation and chemotherapy drugs affect the prognosis of HNC through ROS regulation directly and indirectly [4] (Figure 4). Physiological mechanisms which mediate the chemotherapy efficacy by ROS are as follows: (1) cell death regulation [45–48], (2) deoxyribonucleic acid (DNA) damage repair [49–51], (3) drug metabolism [30, 52, 53], (4) tumor microenvironment [54, 55], and (5) cancer stem cell (CSC) characteristics [56]. In radiation biology, an “oxygen effect” is an important phenomenon which refers to the enhanced killing effect in the presence of oxic conditions. Irradiation exposure can induce mitochondrial-dependent ROS generation [57]. ROS-modulated DNA damage repair [58–61], cell death regulation [62–65], tumor microenvironment [66, 67], and CSC characteristics [67, 68] greatly affect the radiotherapy efficiency. Among these biological factors, cell death and DNA damage are the most common aspects regulated by cellular redox status.
Figure 4.
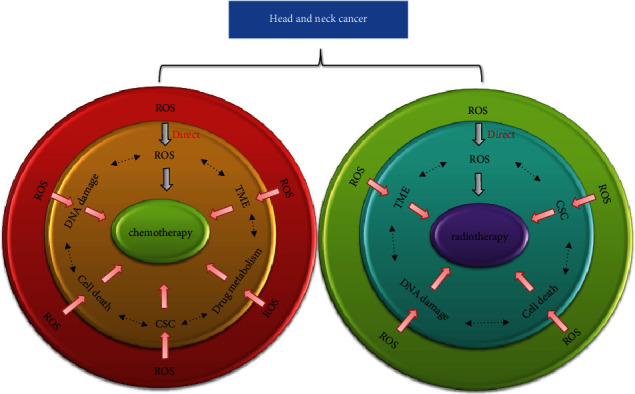
ROS is implicated in the modulation in the chemo-/radiotherapy of HNC. ROS can directly and indirectly affect the efficiency of chemotherapy drugs such as cisplatin and 5-Fu and/or radiation therapy in HNC. A direct effect is seen in terms of ROS-induced lethal genetic damage. Indirect mechanisms include cell death regulation such as apoptosis and autophagy, DNA damage repair, drug metabolism, cancer stem cell (CSC) characteristics, and tumor microenvironment (TME) which are modulated by ROS in the chemotherapy of HNC. Radiotherapy exerts its function through induction of DNA damage within the cell. Except for drug metabolism, other mechanisms are all involved in ROS-mediated radiotherapy efficacy in HNC. Because of the dual role of ROS, the complex modulation network can adapt towards the killing effect of cancer cells or readapting the therapy stimuli. Generally, low and chronic ROS may call for more antioxidant stress defense to protect cancer cells, while high and acute ROS may kill cancer cells with no margin for adaptation. Note. ROS: reactive oxygen species; HNC: head and neck cancer; 5-Fu: 5-flurouracil; DNA: deoxyribonucleic acid; CSC: cancer stem cell; TME: tumor microenvironment.
Currently, it is recognized that CSC presenting self-renewal and pluripotent differentiation capabilities are more inclined to obtain heterogeneous, aggressive, and resistant phenotypes [69, 70]. Especially in poorly vascularized hypoxic tumor niches, CSC characteristics can be well maintained with a high level of ROS-eliminating enzymes, drug resistance transporter proteins, DNA repair enzymes, and antiapoptotic proteins such as Bcl-2 [71, 72]. A lower ROS concentration is found in CSC-enriched populations from irradiated head and neck cancers, compared with nontumorigenic cells [73]. With prolonged exposure to low oxygen levels, CSC cells may undergo epithelial-to-mesenchymal transition (EMT) and acquire the ability to invade and metastasise to local lymph nodes and distant organs [71]. ROS have been implicated in EMT via the activation of EMT-inducing transcription factors including Snail/Slug, ZEB1/2, Twist1/2, HIF-1, and Dlx-2 by modulating upstream signaling pathways such as epidermal growth factor (EGF), Wnt/β-catenin, transforming growth factor-β (TGF-β), mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinse/protein kinase B (PI3K/Akt), Hedgehog, and Notch [68, 74, 75]. Moreover, EMT is closely linked to CSC and the metabolic alteration of cancer cells to avoid hostile environments [76, 77]. Tumor cell-derived low level of ROS inhibits caveolin-1 expression in cancer-associated fibroblasts (CAFs) which is implicated in the stabilization and nuclear accumulation of EMT-inducing transcription factor HIF-1 [78, 79]. The tightly oxygen-regulated subunit HIF-1α effectively induces angiogenic genes such as VEGF [80] and shifts glucose metabolism from aerobic respiration to anaerobic glycolysis via transactivation of glucose transporter GLUT-1 and lactate dehydrogenase (LDH) [81, 82]. An enhanced HIF-1α level has been observed in the CSC subpopulation of HNSCC [67] and linked to poor prognosis and resistance to chemotherapy and radiotherapy [83, 84]. Pharmacological depletion of ROS scavengers reduces the colony-forming capacity of CSC and then increases the radiosensitivity of HNC [73]. Moreover, the capacity of cellular ROS to sensitize the chemo-/radiotherapy of cancer cells depends on the basal level of ROS in such cells. Below a certain threshold, ROS can facilitate survival, but if a certain limit is broken through, cells will die due to intolerance [40]. Adjusting the appropriate ROS level can synergize conventional therapy while reducing the dosage of chemotherapeutic drugs and/or radiation in the clinical condition and thereby alleviating the potential side effects.
In view of the strong reactivity, short life, and opposing roles of ROS, specific quantification and localization of ROS are an important cornerstone for a thorough understanding of its role in cancer initiation, development, and therapy. There are small molecule probes and gene-encoded probes designed to detect whole-cell ROS and mitochondrial ROS. The advantages and disadvantages of these probes are listed in Table 1. Only by clearly understanding the characteristics and defects of these probes can we obtain the accurate research outcomes concerning cellular stress response and therapeutic dose. Besides, methods designed for real-time monitoring of the kinetic changes in the cellular redox state in vivo may further facilitate a comprehensive understanding of the mechanisms of redox biology [85].
Table 1.
The advantages and disadvantages of several ROS probes.
| Name | Advantages | Disadvantages | Reference |
|---|---|---|---|
| DCFH-DA | Convenient to use | Photosensitivity and autoxidation; not specified to detect H2O2; oxidized by cytochrome c | [86, 87] |
| DHE | Convenient to use; specified to detect O2− | Produces two products with similar fluorescence characteristics which need to be resolved by HPLC and other means; photosensitivity and autoxidation | [88] |
| DHR | Convenient to use; specified to detect ONOO− | Intermediates can be reduced by mercaptan and vitamin C; autoxidation | [89] |
| FlAmBE | Convenient to use; stable fluorescence | Not specified to detect ONOO−; high background fluorescence | [90] |
| HKSOX-1/1r | Specified to detect superoxide; stable fluorescence; specified to detect O2−; insensitive to low pH | Not clear | [91] |
| MitoSOX | TPP group localized in mitochondria; convenient to use; specified to detect O2− | Interferes with mitochondrial metabolism; mitochondrial membrane; potential-dependent location; produces two products with similar fluorescence characteristics which need to be resolved by HPLC; photosensitivity and autoxidation | [92] |
| MitoPY1 | TPP group localized in mitochondria; convenient to use; stable fluorescence | Mitochondrial membrane potential-dependent location; not specified to detect ONOO−; high background fluorescence | [93] |
| MitoAR/HR | Rhodamine group localized in mitochondria; convenient to use; specified to detect ·OH/HClO | Mitochondrial membrane potential-dependent location | [94] |
| HKSOX-1m | TPP group localized in mitochondria; specified to detect O2−; stable fluorescence; insensitive to low pH | Mitochondrial membrane potential-dependent location | [91] |
| FRR2 | Rhodamine group localized in mitochondria; convenient to use; reversible real-time detection; stable fluorescence | Nonspecific; mitochondrial membrane potential-dependent location | [95] |
| Pep1-NP | Cationic styrene localized in mitochondria; convenient to use; specified to detect H2O2; stable fluorescence | Not clear | [96] |
| Hyper | Highly specific to H2O2; reversible real-time detection; stable fluorescence; MLS group localized in subcellular structure; independent of membrane potential | pH sensitive; limitation of cell transfection efficiency | [97] |
| RoGFP2-Orp1 | Highly specific to H2O2; reversible real-time detection; stable fluorescence; MLS group localized in subcellular structure; independent of membrane potential; pH insensitivity | Limitation of cell transfection efficiency | [98] |
Note. DCFH-DA: 2,′7′-dichlorofluorescein diacetate; H2O2: hydrogen peroxide; DHE: dihydroethidium; O2−: superoxide anion radical; DHR: dihydrorhodamine; ONOO-: peroxynitrite anion; FlAmBE: boric acid ester derivative; HKSOX-1/1r/1m: novel O2- probes using carboxy tetrafluorofluorescein as fluorescence group (HKSOX-1/1r for cellular retention, HKSOX-1m for mitochondria-targeting); pH: potential of hydrogen; MitoSOX: DHE for mitochondria-targeting; TPP: triphenyl-phosphine; HPLC: high-performance liquid chromatography; MitoPY1: FlAmBE for mitochondria-targeting; MitoAR/HR: DHR for mitochondria-targeting; ·OH: hydroxyl radical; HClO: hypochlorous acid; FRR2: a novel DHR probe; Pep1-NP: a novel boric acid probe targeting mitochondria; Hyper: a genetic probe specific for H2O2; RoGFP2-Orp1: redox-sensitive green fluorescent proteins 2; MLS: mitochondrial localization sequences.
4. Modulate ROS Generation and Elimination to Improve the Efficacy of Chemo-/Radiotherapy in HNC
Once cancer cells are exposed to chemotherapy, radiation, and other treatments, the readaption of the redox status is launched. This in turn provides us a platform to modulate ROS scavengers and generators in order to improve the efficacy of chemo-/radiotherapy.
4.1. Targeting the SOD Antioxidant System in HNC
Superoxide dismutases (SODs) are the main antioxidants which can rapidly and specifically convert O2− to hydrogen peroxide (H2O2). Three isoforms of SODs are found in mammals: SOD1 (Cu/ZnSOD) in the cytoplasm, SOD2 (MnSOD) in the mitochondria, and SOD3 (Cu/ZnSOD) in the extracellular matrix [6]. Noteworthy, the homotetramer SOD2 (MnSOD) which is the most researched SOD in cancer is found exclusively in the mitochondrial matrix [99]. MnSOD acts as a double-edged sword in cancer development [100]. Some researches show that the expression level of MnSOD is decreased compared with normal tissues in breast cancer, pancreatic cancer, and ovarian cancer [101–103]. On the contrary, other researches reveal the higher expression of MnSOD in the malignant progression of gastric cancer, lung cancer, and esophageal cancer [104–106]. During radiation, MnSOD plays a vital role modulating cellular redox balance towards the good and bad sides known as radiosensitization and redioresistance [107]. This dual effect may be ascribed to differences in the expression and/or activity of other antioxidant enzymes like GSH/GSSH, thioredoxins, and catalases in different types of cancers.
SOD mimics such as MnTnBuOE-2-PyP5+ (BMX-001) and Mn (II) pentaaza macrocycle (GC4419) possessing high SOD-like activity show great hope in multiple clinical applications [108]. Ashcraf et al. found that MnTnBuOE can alleviate mucositis (manifested as xerostomia and fibrosis in salivary glands) induced by radiation in non-tumor-bearing female C57BL/6 mice with a dose-modifying factor of 0.77. Human pharyngeal squamous carcinoma cell FaDu xenograft nude mice treated with a combination of RT and MnBuOE showed greater radiosensitivity than a single RT group. The dose adjustment factor is analyzed as 1.3 [109]. Another report from this team discovered that lower doses of MnBuOE mitigated cisplatin-induced oral ulcer formation, bleeding, and furfuration in the radiation area. MnBuOE did not meddle with RT/cisplatin-regulated neoplasm growth [110]. BMX-001 is undergoing phase I clinical trials concerning its safety and pharmacokinetic and radiation protection in conditions of locally advanced head and neck cancer (clinical trial number: NCT02990468).
A randomized, double-blind phase IIb clinical trial of the effects of GC4419 on radiation-induced mucositis in patients with head and neck cancer was completed on 29 August 2019. 223 patients with HNC from 44 institutions who were planning to receive definitive or postoperative IMRT plus cisplatin were randomly allocated into the 30 mg GC4419, 90 mg GC4419, and placebo groups. The outcomes are inspiring. Compared with the placebo group, 90 mg GC4419 treatment showed a decreasing incidence, duration, and severity of oral mucositis induced by 60-72 Gy IMRT (at least two oral mucosal sites) and concurrent cisplatin. No significant toxicity specified or enhanced by GC4419 in IMRT plus cisplatin treatment was observed. A phase III clinical trial (clinical trial number: NCT03689712) to investigate the effects of GC4419 on radiation-induced oral mucositis in patients with head and neck cancer is currently in progress [111].
4.2. Targeting the GSH Antioxidant System in HNC
Tripeptide Glutathione (GSH) is an important intracellular antioxidant that powerfully transfers hydrogen peroxide to H2O and plays a role in the detoxification of many peroxides and electrophilic compounds [112]. Cysteine-glutamate antiporter (System xc−; xCT) encoded by SLC7A11 acts as cysteine importer to the cellular ROS which is essential for GSH biosynthesis [113]. Glutamate-cysteine ligase (GCL) synthesizes substrate cysteine, glycine, and glutamate to GSH [6]. That is to say, cysteine availability and GCL activity determine the synthesis of GSH. GPXs and GST oxidize reduced GSH to glutathione disulfide (GSSG). GSSG can be reduced by glutathione reductase (GR) back to GSH [114]. Meanwhile, nicotinamide adenine dinucleotide phosphate (NADPH) serves as an electron donor [115]. The ratio of reduced and oxidized glutathione (GSH : GSSG) is a representative indicator of cell antioxidant capacity. The imbalance in the synthesis and conversion of GSH is widely implicated in Parkinson's disease [116], cystic fibrosis [117], skin whitening [118], diabetes [119], and schizophrenia [120] as well as cancer [112, 121].
Increased GSH has long been considered as an accomplice in the progression and multidrug resistance of cancer [122–126]. GSH depletion obtained by the irreversible GCL inhibitor BSO is the most commonly used method and is associated with many chemotherapy drugs. However, previous phase I clinical trials concerning the anticancer effect of GSH inhibitor buthionine sulfoximine (BSO) were unsatisfactory [127, 128]. Shortcomings such as a short half-life and nonselective GSH depletion on normal cells limited its clinical application. Over the past two decades, BSO stood at a standstill and did not proceed to Phase II clinical trials. Based on this, researchers carried out a large amount of work with respect to GSH analogues [129] or a combination treatment with other antioxidant systems [130]. Key elements such as GST and xCT in the GSH synthesis process are also excavated to solve chemoresistance [125]. Telcyta (TLK-286), a GSH analogue, has completed the phase II/III clinical trials concerning its treatment efficacy combined with cisplatin, carboplatin, doxorubicin, paclitaxel, and docetaxel in several types of locally advanced or metastatic or refractory resistant cancers (https://www.clinicaltrials.gov/). However, HNC are not covered in these trials. The clinical application of TLK-286 in HNC is hence not suggested in the latest NCCN and ASCO guidelines [17, 19].
There are some preclinical researches in the matter of the GSH antioxidant system in HNC. The combination of BSO and the thioredoxin reductase (TrxR) inhibitor auranofin can synergistically sensitize erlotinib-induced cell death of HNC in vitro and in vivo [131]. On the other hand, the BSO and auranofin combination can simultaneously activate the Nrf2-antioxidant response element pathway which may lead to suboptimal GSH and Trx inhibition in resistant HNC. Thus, inhibition of Nrf2 is proven to make the anticancer effect of BSO and auranofin back to the optimum for HNC [130].
Ethacrynic acid (ECA), a GST inhibitor, was designed to be a methoxy poly(ethylene glycol)-poly(lactide)-disulfide bond-methacrynic acid (MPEG-PLA-SS-ECA) nanoparticle drug carrier, which encapsulates pingyangmycin (PYM) or carboplatin (CBP) separately. The PYM- and CBP-resistant oral squamous cell carcinoma cell lines SCC15/PYM and SCC15/CBP were established to examine the reversal effect of drug resistance by the MPEG-PLA-SS-ECA/PYM and MPEG-PLA-SS-ECA/CBP nanoparticle. The resistant factor values of MPEG-PLA-SS-ECA/PYM and MPEG-PLA-SS-ECA/CBP nanoparticles in SCC15/CBP and SCC15/PYM cells were 1.51 and 1.24. Effective inhibition of GST by nanoparticles shows great hope in reversing PYM and CBP drug resistance in oral cancer [132]. These findings are expected to proceed to further clinical trials.
4.3. Targeting the Trx Antioxidant System in HNC
The thioredoxin (Trx) system is a disulfide reductase system widely existing in many species from prokaryotes to mammals. It is composed of Trx, thioredoxin reductase (TrxR), coenzyme α-NADPH, and Trx-interacting protein (TXNIP) [133]. The predominant location is the cytoplasm containing Trx-1 and TrxR-1, and the subordinate location is mitochondria containing Trx2 and TrxR-2 [134]. Trx with a conserved redox catalytic site (-Cys-Gly-Pro-Cys-) can affect multiple biological functions such as intracellular redox regulation, DNA synthesis, selenium metabolism, cell growth regulation, and apoptosis [135]. TrxR is the only known enzyme capable of reducing Trx, which regulates the protein's thiol/disulfide bond balance by disulfide reductase activity. The dynamic balance between TrxR reduction ability and oxidative stress is the key factor to ensure body homeostasis [130, 136]. Elevated levels of Trx system proteins (Trx-1, TrxR-1, Trx-2, and TrxR-2) and decreased levels of TNXIP protein are involved in various cancers [137–140]. A similar phenomenon was discovered in oral cancers [141–143] and esophageal adenocarcinoma [144]. Moreover, Kaplan-Meier's analysis revealed that the expression level of Trx was significantly related with a lower 5-year survival rate in patients with tongue squamous cell carcinoma [141]. The expression level of TrxR-1 in HPV− cells is much higher than in HPV+ cells in HNSCC. This leads to intrinsic resistance to radiation in HPV− cells [145]. Trx inhibitors such as 1-methylpropyl 2-imidazolyl disulfide (PX-12), 4-benzothiazole-substituted quinol (PMX464), and suberoylanilide hydroxamic acid (SAHA) exert anticancer activity by ROS generation, cell cycle arrest, and apoptosis induction via MAPK signaling pathways [136]. SAHA can synergize the killing effect of bortezomib in EBV-positive nasopharyngeal carcinoma (NPC) HK1-EBV, HONE1-EBV, HA, and C666-1 cell lines. In vivo, bortezomib and SAHA effectively induced apoptosis and inhibited the growth of NPC xenografts in nude mice. ROS generation and subsequent induction of apoptosis indicated by elevated levels of cleaved caspases 3, 7, and 9 and cleaved PARP are the key mechanisms for this synergistic effect [146].
4.4. Targeting the PRX Antioxidant System in HNC
Peroxiredoxins (PRXs) are a family of 22-27 kDa non-selenium-dependent glutathione peroxidases that catalyze the reduction of H2O2 and peroxynitrite (ONOO−). There are six subtypes of Prxs (Prx I-VI) found in mammals [147]. PRXs participate in the occurrence and development of tumors by regulating the level of redox inside and outside the mitochondria [148]. Prx1 was observed to be significantly increased in ESCC clinical tissue samples [149]. Activation of the mTOR/p70S6K pathway is involved in Prx1-promoted tumorigenesis [150]. Another study discovered that Prx II was greatly augmented in patients who failed to respond to chemotherapy or radiation therapy. And in head and neck cancer UMSCC-11A cells, the expression level of Prx II was elevated after 3 Gy radiation or treatment of cisplatin (5 mg/ml) and 5-flurouracil (5-Fu) (2.5 mg/ml). The antisense of PrxII could be sensitized to radiation or chemotherapy inducing apoptosis in UMSCC-11A cells [151]. In a recent study, the expression level of Prx6 was analyzed by immunohistochemistry in 95 ESCC samples and 26 paired adjacent normal tissues. Prx6 was upregulated in ESCC tissues and correlated with the elevated proliferation markers such as Ki67, PCNA, and CyclinD1. Silencing Prx6 greatly inhibited the proliferation of Eca-109 and TE-1, while the overexpression of Prx6 facilitated the migration and invasion of Eca-109 and TE-1 via elevating the Akt and Erk1/2 signaling pathway. Moreover, the downregulation of Prx6 synergizes the apoptosis induced by 8 Gy X-ray irradiation. These findings are further validated in the ESCC xenograft mode in vivo. Inhibition of Prx6 shows a novel therapeutic strategy for radiosensitization in ESCC [152].
4.5. Targeting the Nrf2/Keap1 Antioxidant System in HNC
Nrf2 and Keap1 are the major proteins that coordinate the induction and transcription of various antioxidant enzymes [34]. Under normal physiological conditions, Nrf2 binds to the Keap1/CUL3/RBX1 E3-ubiquitin ligase complex in large amounts and degrades rapidly in the cytoplasm. When the oxide accumulates, Nrf2 and Keap1 dissociate and transfer or bind to antioxidant enzymes in the promoter region of detoxification phase II enzymes, such as NQO1, GST, glutathione peroxidase (Gpx), peroxidase I, glutathione ligase, glutathione, epoxide hydrolase, and heme oxygenase (HO-1). These enzymes can protect the body from active substances (such as ROS) and some toxic substances [34, 35]. A large number of studies have shown that Nrf2 is related to the occurrence of metabolic disorders and cancer initiation, and these are well reviewed by Cuadrado et al. and Rojo de la Vega et al. [153, 154]
Nrf2 gene (NFE2LE) mutations are a mechanism of Nrf2 activation which has been correlated with poor survival [155]. Besides, a high frequency (60%) of DNA level inactivation to the Nrf2 inhibitor Keap1/CUL3/RBX1 E3-ubiquitin ligase complex is related to HNSCC. And this complex disruption is unique to HNSCC. The median survival rate was decreased when the altered complex increased. Nrf2 activation is an underlying prognostic indicator in HNSCC [156].
A recent retrospective study concerning Nrf2 was conducted in 183 patients with confirmed stage I to VI HNSCC. A higher level of Nrf2 was associated with a poorer overall survival (median OS: 45.5 months versus 60 months). This is further validated through the Cancer Genome Atlas (TCGA) database. The OS for Nrf2high versus Nrf2low is 40 months versus 90 months, and disease-free survival (DFS) in the Nrf2high group is 64 months compared with 100 months in the Nrf2low group. Nrf2 expression was significantly higher in cisplatin-resistant and nonresponder patients than good responders. HO-1, the Nrf2-targeted gene, was also elevated in cisplatin-resistant HNNC patients. Knockdown of Nrf2 reversed the sphere-forming efficiency that marks the cancer stem cell characteristics in FaDu cells [157]. Inhibition of Nrf2 by artesunate leading to a reversal of the ferroptosis resistance in cisplatin-resistant HNC cells has been reported [158]. These findings hint at some clues for the targeted therapy of the Nrf2/Keap1 system and complimentary strategy towards drug resistance.
4.6. Targeting the ETC Complexes in HNC
In cancer cells, mitochondria electron transporter chain (ETC) complexes become more active to produce ATP and ROS which induce drug resistance via ATP-driven multidrug efflux pumps. Elevated ROS promote certain antioxidant systems to attain redox balance. Therefore, disturbing ETC complexes show great potential for tackling drug resistance. On one hand is the consumption of as much ATP as possible, while on the other hand ROS levels are increased facilitating cellular apoptosis [40]. Proteomic expression profiling reveals reduction of COX7A2 (cytochrome c oxidase subunit 7A2), a subunit of ETC complex IV, which is related to patients with esophageal adenocarcinoma who respond to cisplatin plus 5-Fu therapy. Silencing of COX7A2 in OE19 cells leads to an abnormal cup-shaped structure of the mitochondria observed by electron microscopy. The combination treatment of cisplatin/5-Fu after silencing COX7A2 significantly inhibits OE19 cell proliferation [159].
5. Repurpose Old Drugs Modulating ROS for a New Life
The so-called “new use of old drugs” refers to the non-anti-cancer drugs that have been used for a long time in clinical practice. These drugs are applied to new fields because of their anticancer effects. By this, not only is the safety of drugs ensured, but the long cycle of new drug development and screening is also avoided (Table 2).
Table 2.
Old drugs modulating ROS as an adjuvant agent in the chemo-/radiotherapy of HNC.
| Drug | Site | Experimental model | Effective dose | Cotherapy | ROS detection | Biological effects | Mechanisms | Reference |
|---|---|---|---|---|---|---|---|---|
| Sulfasalazine | Larynx | In vitro (HN3, HN4, and HN9; HN3-cisR, HN4-cisR, and HN9-cisR cells) In vivo (HN9-cisR xenograft nude mice) |
In vitro (1 mM) In vivo (250 mg/kg daily) |
+Cisplatin In vitro (20 μM) In vivo (5 mg/kg weekly) |
DCFH-DA flow cytometry | Synergistic effect | ↑ROS, ↓GSH, ↓xCT, ↑γH2AX | [162] |
|
| ||||||||
| DCA | Larynx | In vitro (HN2, 3, 4, 5, 9, and 10; SNU-1041, 1066, and 1076; HN4-cisR and HN9-cisR cells) In vivo (HN4-cisR and HN9-cisR xenograft nude mice) |
In vitro (15-30 mM) In vivo (0.5 g/l once per week) |
+Cisplatin In vitro (10-30 μM) In vivo (5 mg/kg once per week) |
DCFH-DA+MitoSOX flow cytometry and confocal microscopy | Synergistic effect: enhances apoptosis | ↑mROS, ↓ΔΨm, ↓PDK2, ↑p21, ↓pPDHE1α, ↑c-PARP, ↑PUMA, ↑CC3 | [165] |
|
| ||||||||
| Melatonin | Oral cavity | In vitro (Cal-27, SCC-9 cell) |
1.5 mM | +Radiation (8 Gy) | DCFH-DA spectrofluorometer | Synergistic effects: enhance apoptosis and lethal autophagy | ↑GSSG/GSH, ↑Bax/Bcl-2, ↓NIX, ↑ATG12-ATG5 | [173] |
|
| ||||||||
| Melatonin | Oral cavity | In vitro (Cal-27, SCC-9 cell) |
1.5 mM | +Cisplatin (10 μM) | DCFH-DA spectrofluorometer | Synergistic effects: enhance apoptosis and lethal autophagy | ↑GSSG/GSH, ↑Bax/Bcl-2, ↑NIX, ↑ATG12-ATG5 | [173] |
|
| ||||||||
| Thioridazine | Larynx | In vitro (AMC-HN4 cell) |
10 μM | +Carboplatin | DCFH-DA+MitoSOX flow cytometry and fluorescence microscope | Synergistic effect: enhances apoptosis | ↑ROS, ↓PSMA5, ↑Nrf2, ↓c-FLIP, ↓Mcl-1, ↑c-PARP, ↑CC3 | [180] |
|
| ||||||||
| Aspirin | Larynx | In vitro (HN3, 4, and 9; HN3R, 4R, and 9R cells) In vivo (HN9R xenograft nude mice) |
In vitro (5-10 mM) In vivo (10 mg/kg daily) |
+Sorafenib In vitro (5-10 μM) In vivo (10 mg/kg daily) |
DCFH-DA flow cytometry | Synergistic effect | ↑ROS, ↓xCT, ↓GSH, ↑c-PARP, ↓p65, ↓Mcl-1 | [183] |
|
| ||||||||
| Aspirin | Larynx | In vitro (HN3, 4, and 9; HN3R, 4R, and 9R cells) In vivo (HN9R xenograft nude mice) |
In vitro (5-10 mM) In vivo (10 mg/kg daily) |
+Cisplatin In vitro (10 μM) In vivo (5 mg/kg weekly) |
DCFH-DA flow cytometry | Synergistic effect | ↓xCT, ↓GSH, ↑c-PARP, ↓p65, ↓Mcl-1, ↑p-p53 | [183] |
|
| ||||||||
| Salinomycin | Nasopharynx | In vitro (CNE-1, CNE-2, SUNE1, 6-10B, 5-8F, SUNE1R cell) |
2 μM | +Radiation (4 Gy) | DCFH-DA flow cytometry | Synergistic effect: enhances apoptosis | ↑ROS, ↓Nrf2, ↓survivin | [186] |
| Metformin | HNSCC | In vitro (HN30, HN31 cell) Clinical samples |
2.5 mM | +Radiation (4 Gy) | DCFH-DA flow cytometry | Synergistic effect: induces senescence | ↑ROS, ↓ME2, ↑p21, ↑NADP/NADPH, ↑SA-β-gal | [192] |
Note. mM: millimole; μM: micromole; DCFH-DA: 2′,7′-dichlorofluorescein diacetate; ROS: reactive oxygen species; GSH: glutathione; GSSG: oxidized glutathione; xCT: cysteine-glutamate antiporter; γH2AX: H2A histone family member X; DCA: dichloroacetic acid; mROS: mitochondrial reactive oxygen species; ΔΨm: mitochondrial membrane potential; PDK2: pyruvate dehydrogenase kinase 2; p21: protein 21; PDHE1α: pyruvate dehydrogenase E1-α; c-PARP: cleaved poly-ADP ribose polymerase; PUMA: p53 upregulated modulator of apoptosis; CC3: cleaved caspase 3; Bcl-2: B-cell lymphoma-2; Bax: Bcl-2-associated X protein; NIX: adenovirus E1B 19 kDa interacting protein 3-like; ATG: autophagy related; PSMA5: proteasome subunit alpha 5; Nrf2: nuclear factor E2-related factor 2; c-FLIP: cellular FLICE-like inhibitory protein; Mcl-1: myeloid cell leukaemia-1; p65: protein 65; p-p53: phosphorylated protein 53; ME2: malic enzyme 2; NADP: nicotinamide adenine dinucleotide phosphate; NADPH: nicotinamide adenine dinucleotide phosphate oxidase; SA-β-gal: senescence-associated β-galactosidase.
5.1. Sulfasalazine
Sulfasalazine is an anti-inflammatory drug that has been applied to treat inflammatory bowel disease and rheumatoid arthritis for decades [160]. Recent studies show that sulfasalazine, a nonsubstrate xCT inhibitor, can efficiently kill cancer cells. Sulfasalazine can eliminate cellular detoxification by GSH depletion and enhance the anticancer effect by upregulating ferroptosis in HNC [161]. In HNC cisplatin-resistant HN3-cisR, HN4-cisR, and HN9-cisR cells, 1 mM sulfasalazine can enhance cisplatin-induced cell death in terms of a significant decrease of GSH. Pretreatment of N-acetylcysteine (NAC) can block this effect. In HN9-cisR xenograft nude mice, a combination of sulfasalazine with cisplatin showed greater inhibition of tumor growth than either single group [162]. Thus, the synergy of sulfasalazine with conventional chemotherapeutic agents is promising in the treatment of advanced and resistant HNC.
5.2. Dichloroacetic Acid
Dichloroacetic acid (DCA), an inhibitor of pyruvate dehydrogenase kinase, has been approved by the FDA for treating a rare hereditary lactate metabolism disorder [163]. During the past decade, DCA has been repurposed for enhancing cancer therapy efficacy by overcoming resistance to chemotherapeutic drugs [164]. Even so, DCA has rarely been checked in HNC. Downregulation of PDK2 by DCA switches bioenergetics towards mitochondrial oxidative phosphorylation which leads to an increase in mitochondrial reactive oxygen species (mROS) in the larynx cancer cisplatin-resistant cell lines AMC-HN4-cisR and HN9-cisR, thus sensitizing a cisplatin effect in vitro and in vivo [165]. DCA-induced apoptosis by the inhibition of PDK1 in HNSCC cells can be further enhanced by cetuximab-mediated downregulation of ASCT2, which is a glutamine transporter [166]. One issue should be dealt with caution when the use of DCA in cancer treatment is concerned. Long-term exposure to DCA may shift normal cells such as immune cells to a greater oxidative metabolism in which the condition of normal physiology function is disturbed [164].
5.3. Melatonin
Melatonin, N-acetyl-5-methoxytryptamine, is a compound containing an indole ring approved by FDA as a raw material for dietary supplements. In China, the use of melatonin as a raw material for health foods is allowed, requiring a purity of more than 99.5% and a recommended daily dosage of 1-3 mg limited to improving sleep (product standard: GB/T5009.170-2003; http://www.nhc.gov.cn). During recent years, melatonin has been found to possess anti-inflammatory, antioxidant, and anticancer activities [167–171]. The melatonin gel [172] has completed a phase II clinical trial (EudraCT number: 2015-001534-13) in 80 patients with HNC. The results showed that melatonin can protect oral mucosa against the side effects of radiotherapy. Fernandez-Gil et al. have researched an enhancing cytotoxic role of melatonin combined with rapamycin in HNSCC cells. Moreover, they found that a high concentration of melatonin could sensitize HNC cells to CDDP and irradiation by enhancing the mitochondrial ROS and then inducing apoptosis and lethal autophagy [173]. A combined melatonin and irradiation treatment decreased the mitophagic marker NIX, while a combined melatonin and cisplatin treatment increased NIX [173]. This is perhaps due to different ROS levels enhanced by each kind of combination. Even so, melatonin shows great hope in combination with radiotherapy or chemotherapy for better therapeutic efficiency.
5.4. Thioridazine
Thioridazine was approved for use in the United States in 1978 and was indicated for the therapy of acute and chronic psychosis. A high concentration of thioridazine administration is prone to cause prolongation of the QTc interval and increase sudden death risk [174–176]. However, a low concentration of thioridazine is reported to induce apoptosis, inhibit angiogenesis and metastasis, and overcome drug resistance in cancer treatment [177–179]. A combination of thioridazine with carboplatin significantly induced mitochondrial apoptosis and downregulated apoptosis-related proteins c-FLIP and Mcl-2 which can be reversed by knockdown of PSMA5, a proteasome subunit. Besides, a combined treatment with carboplatin and thioridazine could induce ROS production and activate Nrf2 translocation as well as antioxidant response elements within 1 h in HNC AMC-HN4 cells. ROS scavengers (NAC, trolox, and glutathione-ethyl-ester) inhibited Nrf2 translocation and PSMA5 expression. Mitochondrial ROS have a critical role in carboplatin plus thioridazine-induced apoptosis. Moreover, a combination of thioridazine and carboplatin did not induce cell death in normal human mesangial and umbilical vein cells. Thus, a low concentration of thioridazine is a promising adjuvant agent in carboplatin-resistant HNC [180].
5.5. Acetylsalicylic Acid
Acetylsalicylic acid (aspirin), a nonsteroidal anti-inflammatory drug, has been used for relieving inflammation and preventing cardiovascular events [181]. It is reported that aspirin can inhibit tumor growth and metastasis [182]. A low concentration of aspirin (1-3 mM) can synergize 1-3 μM sorafenib to more cell death in HNC cisplatin-resistant HN3R, HN4R, and HN9R cells. The combination of aspirin and sorafenib significantly decreased GSH level and elevated total ROS levels in cisplatin-resistant HNC cells. This effect can be revered by the antioxidant trolox. Furthermore, aspirin and sorafenib could synergize cisplatin-induced cytotoxicity in resistant HNC cells. In HN9R xenograft nude mice, the effect of aspirin plus sorafenib on cisplatin has been confirmed and that this trip-combination greatly suppressed tumor growth without affecting the weight of mice. Aspirin is promising in synergizing sorafenib alone or combined with sorafenib to synergize cisplatin in anticancer therapeutics of HNC [183].
5.6. Salinomycin
Salinomycin is a carboxypolyether potassium ionophore antibiotic isolated from the fermentation broth of Streptomyces albus by Miyazaki et al. in the process of screening for new antibiotics in 1974 [184]. Salinomycin has been widely used in the prevention and control of coccidiosis in poultry animals in the past. In 2009, Gupta et al. conducted a high-throughput screening of more than 16,000 chemicals and found that salinomycin can selectively kill breast cancer stem cells, and its killing effect is 100 times more than that of the clinical first-line chemotherapy drug paclitaxel [185]. The nasopharyngeal carcinoma radioresistant SUNE1R cells expressed higher Nrf2 compared to parental SUNE1 cells. Salinomycin can restore the radiosensitivity of SUNE1R cells by inducing apoptosis which is mediated via Nrf2 inhibition and ROS generation [186]. In view of these effects, salinomycin is perhaps a promising adjuvant agent to modulate ROS for enhancing the radiosensitivity of HNC. However, more in vivo experiments concerning its efficacy and toxicity should be further carried out.
5.7. Metformin
Metformin has been approved to treat type 2 diabetes since 1957 in Europe [187]. Due to lactic acidosis, metformin was taken off the US market; however, later it has been proven safe and effective in controlling glucose levels and was reapplied in 1995 [188, 189]. In 2005, metformin was used to reduce the incidence of cancer in patients with diabetes [190]. Since then, metformin has been vastly explored in the anticancer field. Metformin has been reported to inhibit the proliferation and viability of HNSCC cells via an AMPK-dependent manner [191]. In another research, metformin could suppress both HNSCC HN30 (wtp53) and HN31 (p53 with 2 missense mutations) cells via the downregulation of malic enzyme 2 (ME2) driven by ROS generation. Noteworthy, metformin exerted a more efficient inhibitory effect in HN31 cells which are resistant to radiation [192]. This provided an AMPK-independent manner for metformin to enhance the radiation effect against resistant HNSCC.
6. Exploit Novel Small Molecular Compounds Targeting ROS
Small molecular compounds composed of several or dozens of atoms have always been commonly used in clinical medicine due to their many advantages, such as a definite curative effect, less adverse effects, and smaller molecular weight, which are easily absorbed [193]. It is also one of the hot spots in the field of medicinal chemistry drug development. Based on the intensive implication of ROS in cancer treatment, here we reviewed several novel compounds modulating ROS as a potential adjuvant therapy of HNC (Table 3).
Table 3.
Small molecular compounds modulating ROS in chemo-/radiotherapy of HNC.
| Compound | Site | Experimental model | Effective dose | Cotherapy | ROS detection | Biological effect | Mechanisms | Reference |
|---|---|---|---|---|---|---|---|---|
| CHW09 | Oral cavity | In vitro (Ca9-22, CAL 27 cancer cell, and normal gingival fibroblast HGF-1 cell) |
10 μg/ml | +Radiation (12 Gy) |
DCFH-DA flow cytometry | Synergistic effects | ↑ROS, ↑CC3, ↑CC8, ↑c-PARP, ↑8-oxodG, ↑γH2AX | [195] |
|
| ||||||||
| Oxamate | Nasopharynx | In vitro (CNE-1, CNE-2 cell) In vivo (CNE-1 xenograft nude mice) |
In vitro (20, 50, 100 mM) In vivo (750 mg/kg daily for 3 weeks) |
+Radiation (9.9 Gy) |
DCFH-DA flow cytometry | Synergistic effect: enhances apoptosis and G2/M arrest | ↑ROS, ↓ATP, ↓CDK1/cyclin B1, ↓Bcl-2, ↑Bax, ↑CC3 | [197] |
|
| ||||||||
| D-Allose | Tongue | In vitro (HSC-3 cell) |
25 mM | +Radiation (4 Gy) |
DCFH-DA fluorescence microscopy | Synergistic effect: enhances apoptosis | ↑ROS, ↑TXNIP, ↓TRX | [201] |
|
| ||||||||
| D-Allose | Tongue | In vitro (HSC-3 cell) In vivo (HSC-3 xenograft nude mice) |
In vitro (10 mM) In vivo (500 mM 5 times/week for 3 weeks) |
+Docetaxel In vitro (0.1 ng/ml) In vivo (12 mg/kg on day 0 and day 7) |
DCFH-DA fluorescence microscopy | Synergistic effect: enhances apoptosis G2/M arrest | ↑ROS, ↑TXNIP, ↓TRX | [202] |
|
| ||||||||
| SAHA | Nasopharynx | In vitro (HK1-EBV, HONE1-EBV, HA, C666-1, NP460, HK2 cell) In vivo (C666-1, HONE1, HA xenograft nude mice) |
In vitro (5 μM) In vivo (50 mg/kg 5 days per week for 4 weeks) |
+Bortezomib In vitro (30 nM) In vivo (60 μg/kg) |
DCFH-DA flow cytometry | Synergistic effect: enhances apoptosis | ↑ROS, ↑c-PARP, ↑CC3, ↑CC7, ↑CC9 | [146] |
|
| ||||||||
| NaB | Esophagus | In vitro (KYSE-150, KYSE-150R cells) |
0.5, 1 μM | +Radiation (5 Gy) | DCFH-DA flow cytometry | Synergistic effect: enhances apoptosis, G2/M arrest, and DNA damage | ↑ROS, ↓Bmi-1, ↑p21, ↓DNA-PKcs, ↓NBS1, ↓Rad51, ↑γH2AX | [65] |
|
| ||||||||
| TSA | Esophagus | In vitro (KYSE-150, KYSE-150R cells) |
5, 10 mM | +Radiation (5 Gy) | DCFH-DA flow cytometry | Synergistic effect enhances apoptosis, G2/M arrest, and DNA damage | ↑ROS, ↓Bmi-1, ↑p21, ↓DNA-PKcs, ↓NBS1, ↓Rad51, ↑γH2AX | [65] |
Note. ROS: reactive oxygen species; DCFH-DA: 2′,7′-dichlorofluorescein diacetate; CHW09: sulfonyl chromen-4-ones; SAHA: vorinostat; NaB: sodium butyrate; TSA: hydroxamic acid trichostatin A; c-PARP: poly-ADP ribose polymerase; CC3: cleaved caspase 3; CC7: cleaved caspase 7; CC8: cleaved caspase 8; CC9: cleaved caspase 9; 8-oxodG: 8-oxo-2′-deoxyguanosine; γH2AX: H2A histone family member X; NQO1: NAD(P)H:quinone oxidoreductase 1; Bcl-2: B-cell lymphoma-2; Bax: Bcl-2-associated X protein; ATP: adenosine-triphosphate; CDK1: cyclin-dependent kinase 1; c-PARP: cleaved PARP; Bmi-1: B-lymphoma Mo-MLV insertion region 1; p21: protein 21; DNA-PKcs: DNA-dependent protein kinase, catalytic unit; NBS1: Nijmegen breakage syndrome 1; RAD51: radioresistance protein 51; TXNIP: Trx-interacting protein.
6.1. CHW09
Chromones are oxygen-containing heterocyclic compounds that possess anti-inflammatory and anticancer abilities. A sulfonyl substituent is installed on the chrome-4-one skeleton. This synthesized compound is named CHW09. In vitro, CHW09 can efficiently kill oral cancer cells Ca9-22 and CAL 27 with a mild decrease in viability in the normal human gingival fibroblast cell HGF-1. Cellular ROS and mitochondrial superoxide were both induced, and subsequent apoptosis and DNA damage were enhanced after the treatment of CHW09. The high-stress status renders cancer cells more sensitive to ROS-generating agents [194]. A combination of 10 μg/ml CHW09 and 12 Gy radiation synergistically inhibits proliferation and induces apoptosis of Ca9-22 and CAL 27 [195]. However, the animal experiments are not available now.
6.2. Oxamate
Lactate dehydrogenase (LDH) is a major glycolytic enzyme which catalyzes the transformation of pyruvate to lactate. As the Warburg effect commonly exists in cancer cells with elevated glucose consumption and aerobic glycolysis, the LDH expression is increased at the same time in various types of cancer [196]. Oxamate, a LDH competitive inhibitor, provides an attractive chance to develop a novel cancer therapeutic strategy. In nasopharyngeal carcinoma CNE-1 and CNE-2 cells, oxamate efficiently synergizes radiation by upregulating ROS level and subsequent G2/M arrest and apoptosis. Besides, inhibition of LDH disturbed energy metabolism and significantly decreased ATP production. An in vivo experiment further validated the synergizing effect of oxamate in radiation [197]. Even so, the small size and high polarity of oxamate limit its catalytic activity and permeability. Several N-alkyl-oxamates are synthesized [198]. Further experiments are imperative concerning their inhibitory effects on LDH and anticancer actions.
6.3. D-Allose
D-Allose is a rare aldohexose with many physiological functions including lowering blood lipid and blood glucose concentrations, scavenging free radicals in the body, and reducing ischemia-reperfusion injury and anticancer effects [199]. It is noteworthy that D-allose can inhibit carcinogenesis under oxidative stress and can induce the expression of TXNIP which inhibits the proliferation of HNC cells [200]. Hoshikawa et al. reported the radiosensitizing effect of D-allose on HNC HSC-3 cells using a 3D culture method. A combination of D-allose and radiotherapy had better effects than the two alone. The radiation enhancement rate reached 1.61 and 2.11 after 10 mM and 25 mM allose treatment, respectively. The radiation treatment alone could not increase the expression of the mRNA level of TXNIP, while allose combined with radiation treatment could significantly increase the expression of TXNIP which can significantly induce the generation of cellular ROS and the occurrence of apoptosis [201]. Besides, D-allose can synergize docetaxel-induced apoptosis by increasing TXNIP and ROS in vitro and in vivo [202]. Most importantly, allose has no known side effects [203], so the combined use of allose and radiation or docetaxel may become a new treatment strategy for HNC [204].
6.4. Histone Deacetylase Inhibitors
Tumorigenesis and progression are the result of the interaction of heredity and epigenetics. As an important epigenetic modification, histone deacetylation plays an important role in the occurrence and development of a tumor [205]. The abnormal expression of histone deacetylase (HDAC) in normal tissues and cells will promote the development of a tumor, and it is related to the proliferation and apoptosis, angiogenesis, metastasis, and drug resistance of tumor cells, and becomes a new target of tumor treatment [206–209]. HDAC inhibitors such as vorinostat (SAHA), romidepsin, belinostat, and panobinostat have been approved by FDA as anticancer drugs (https://www.fda.gov). More combination modalities concerning SAHA with conventional chemotherapy drugs are undergoing preclinical researches [146, 209, 210]. SAHA can synergize bortezomib-induced apoptosis via the upregulation of ROS in nasopharyngeal carcinoma cells. Further in vivo experiments confirmed this effect [146]. Sodium butyrate (NaB) and hydroxamic acid trichostatin A (TSA) are another two HDAC inhibitors that sensitize radiation by downregulating Bmi-1 and then increasing ROS generation and impairing DNA repair in esophageal squamous cell carcinoma radioresistant KYSE-150R cells. HDAC inhibitors as anticancer drugs complementary to chemo-/radiotherapy show a great potential [211].
7. Natural Herbs Effectively Modulating ROS Are Important Drug Candidates
Natural herbs combined with surgery and chemo-/radiotherapy show a certain effect in clinical cancer treatment. Mechanistically, the imbalance between ROS generation and elimination in cancer provides an opportunity for natural herbs. Generally, ROS upregulation synergizes conventional chemo-/radiotherapy, while the downregulation of ROS may protect normal tissue from side effects. Here, we reviewed several natural herbs modulating ROS in the comprehensive treatment of HNC (Table 4).
Table 4.
Natural products modulating ROS in chemo-/radiotherapy of HNC.
| Category | Herb | Site | Experimental model | Effective dose | Cotherapy | ROS detection | Biological effect | Mechanisms | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Flavonoids | Quercetin | Larynx | In vitro (Hep2 cell) |
40 μM | +Cisplatin (2.5 μg/ml) |
— | Synergistic effects | ↓Cu/Zn SOD, ↓p-AKT, ↑p-JNK, ↑c-FOS, ↓Bcl-2, ↓Bcl-xL, ↓survivin, ↑Bax, ↑cytochrome c, ↑caspase-3, ↑caspase-9, ↓NOS, ↓HSP70, ↓Ki-67, ↓telomerase | [216] |
| Naringin | Esophagus | In vitro (YM1 cancer stem cell) In vivo (YM1 xenograft mouse) |
In vitro (354 μM) In vivo (50 mg/kg) |
+Doxorubicin | — | Reduce side effect, restore the antioxidant defense system | ↑SOD | [217] | |
| AIF | Esophagus | In vitro (Eca109, KYSE-30 cells) In vivo (Eca109 xenograft nude mice) |
In vitro (5 μM) In vivo (20 mg/kg/day) |
+Radiation In vitro (6 Gy) In vivo (2 Gy/min on days 10 and 30) |
DCFH-DA confocal microscope | Synergistic effects: enhance apoptosis G2/M arrest | ↑ROS, ↓Nrf2, ↓HO-1, ↓NQO1, γ↑H2AX | [218] | |
| Wogonin | Larynx | In vitro (HN2, -HN3, -HN4, -HN5, and -HN9; SNU-1041, SNU-1066, and SNU-1076; HN4-cisR and HN9-cisR cells; normal cell: HOK-1, HOF, and HEK) In vivo (HN4-cisR or HN9-cisR xenograft nude mice) |
50 mg/kg | +Cisplatin | DCFH-DA flow cytometry | Synergistic effects: enhance apoptosis | ↑ROS, ↓GSH, ↓Nrf2, ↓GST, ↑p53, ↑p-JNK, ↑c-PARP, ↑PUMA | [219] | |
|
| |||||||||
| Polyphenols | Curcumin | Oral cavity | In vitro (normal cell SGNs and cancer cell PE/CA-PJ15) In vivo (male adult Wistar rats) |
In vitro (3.37, 6.75 μM) In vivo (200 mg/kg) |
+Cisplatin | — | Otoprotective effect: antioxidant activity Synergistic effects: prooxidant and anti-inflammatory |
Protective mechanisms: ↑Nrf2, ↑HO-1, ↓p53, ↓NF-κB Synergistic mechanisms: ↓Nrf2 (nuclear), ↓NF-κB, ↓pSTAT-3, ↑p53 |
[223] |
| Curcumin | Pharynx | In vitro (HPV-cells: FaDu, SQ20B, JHU-022, HEK-001, and MSK-Leuk1; HPV+ cells: UPCI-SCC090 and UPCI-SCC154) In vivo (FaDu xenograft nude mice) |
In vitro (10 μM) In vivo (10 μM) |
+Radiation In vitro (0, 2, 4, 6 Gy) In vivo (0, 2, 4, 6 Gy) |
— | Synergistic effects: inhibition of antioxidant defense system | ↓TxnRd1 | [145] | |
| FA | Oral cavity | In vitro (normal cell SGNs and cancer cell PE/CA-PJ15) In vivo (male adult Wistar rats) |
In vitro (100-600 μM) In vivo (600 mg/kg) |
+Cisplatin | — | Otoprotective effect: antioxidant activity Synergistic effects: prooxidant at lower concentrations (100-600 μM) Antagonistic effect: antioxidant at higher concentrations (>600 μM) |
Protective mechanisms: ↑Nrf2, ↑HO-1, ↓P53 Synergistic mechanisms: ↓Nrf2 (nuclear), ↓pSTAT-3 Antagonistic mechanisms: ↑Nrf2 (nuclear), ↑pSTAT-3 |
[223] | |
| DPP-23 | Larynx | In vitro (HN3, HN3-cisR, HN4, HN4-cisR, HN9, HN9-cisR, HOK-1 cells) In vivo (HN9-cisR xenograft nude mice) |
In vitro (2-40 μM) In vivo (10 mg/kg) |
+Cisplatin In vitro (10 μM) In vivo (5 mg/kg) |
DCFH-DA flow cytometry | Synergistic effects: inhibition of antioxidant defense system and activation of apoptosis | ↑ROS, ↓GSH, ↓Nrf2, ↓HO-1, ↑p53, ↑c-PARP, ↑p21 | [224] | |
| EGCG | Oral cavity | In vitro (normal cell: NHOK; cancer cell: HSC-2) |
50-100 μM | +Doxorubicin 0.625-5 μM | DCFH-DA fluorescence microscope | Chemoprotective effect | ↓ROS | [225] | |
| TA | Oral cavity | In vitro (normal cell: NHOK; cancer cell: HSC-2) |
12.5-50 μM | +Doxorubicin 0.625-5 μM | DCFH-DA fluorescence microscope | Chemoprotective effect | ↓ROS | [225] | |
| Epicatechin | Oral cavity | In vitro (oral fibroblast cells) In vivo (Zebrafish) |
In vitro (50 μM) In vivo (200 μM) |
+Radiation In vitro (20 Gy) In vivo (20 Gy) |
DCFH-DA flow cytometry | Radioprotective effect: reduce apoptosis and restore MMP | ↓ROS, ↓p38, ↓p-JNK, ↓CC3 | [226] | |
| Epicatechin | Oral cavity | In vitro (human keratinocyte HaCaT cell) In vivo (Sprague-Dawley rats) |
In vitro (100 μM) In vivo (2 mM/day after radiation for 23 days) |
+Radiation In vitro (20 Gy) In vivo (30 Gy) |
DCFH-DA flow cytometry | Radioprotective effect | ↓ROS, ↓p-JNK, ↓p38, ↓CC3, ↓NOX3 | [227] | |
|
| |||||||||
| Quinones | Plumbagin | Tongue | In vitro (CAL27 cell, cisplatin-resistant cell line CAL27/CDDP) In vivo (CAL27/CDDP xenograft nude mice) |
In vitro (5 μM) In vivo (3 mg/kg every two days) |
+Cisplatin In vitro (16.7 μM) In vivo (4 mg/kg every three days) |
DCFH-DA+MitoSOX fluorescence microscope | Synergistic effects: enhance apoptosis and autophagy | ↑ROS, ↓Bcl-2, ↑Bax, ↑CC3, ↑Beclin-1, ↓p62, ↑LC-II/LC-I, ↓p-AKT, ↓p-mTOR, ↑p-JNK | [238] |
| β-Lapachone | Head and neck | In vitro (FaDu, Detroit 562, SqCC/Y1, UMSCC-10A) In vivo (SqCC/Y1 xenograft SCID-NOD mice clinical samples) |
In vitro (2.5 μM) In vivo (10 mg/kg every other day) |
+Radiation In vitro (2 Gy) In vivo (10 Gy) |
DCFH-DA flow cytometry | Synergistic effects: enhance apoptosis and NDA damage | ↓NQO1, ↑ROS, ↓Bcl-2, ↓ATP, ↑γH2AX | [233] | |
|
| |||||||||
| Terpenoids | Oridonin | Larynx | In vitro (Hep-2 and Tu212 cells) In vivo (Hep-2 xenograft nude mice) |
In vitro (12, 24, and 36 μM) In vivo (20 mg/kg) |
+Cetuximab In vitro (10 μg/ml) In vivo (1 mg/mice) |
DCFH-DA flow cytometry | Synergistic effects: enhance apoptosis and G2/M arrest | ↑ROS, ↑CC8, ↑CC3, ↑c-PARP, ↑p21, ↑Fas, ↑FADD, ↑FasL, ↓ICAD, ↓cyclin B1, ↑p-cdc2, ↑p-cdc25c, ↓NAC, ↓CAT, ↑p-JNK, ↓p-EGFR | [244] |
|
| |||||||||
| Ginsenosides | Ro | Esophagus | In vitro (ECA-109, TE-1 cell) |
50 μM | +5-Fluorouracil 100 μg/ml | — | Synergistic effects: enhance DNA repair and inhibit autophagic flux | ↑ESR2, ↑NCF1, ↑ATG-7, ↑CC3, ↑CC9, ↑c-PARP, ↑p62, ↓LC3BII/LC3BI, ↑CHEK1 | [248] |
| KRG | Oral cavity | In vitro (normal keratinocyte HaLa cell, cancer SCC25, SCC1484 cell) In vivo (zebrafish) |
In vitro (10-100 μg/ml) In vivo (30 μg/ml) |
+Radiation In vitro (8 Gy) In vivo (20 Gy) |
DCFH-DA flow cytometry | Radioprotective effect | ↓ROS, ↓ATM, ↓p-p53, ↓p-JNK, ↓p-p38, ↓CC3 | [249] | |
Note. ROS: reactive oxygen species; DCFH-DA: 2′,7′-dichlorofluorescein diacetate; SOD: superoxide dismutase; AKT: protein kinase B; p-AKT: phosphorylated AKT; JNK: c-Jun N-terminal kinase; p-JNK: phosphorylated-JNK; c-FOS: cellular oncogene fos; Bcl-2: B-cell lymphoma-2; Bcl-xL: B-cell lymphoma-extra large; Bax: Bcl-2-associated X protein; NOS: nitric oxide synthase; HSP70: heat shock protein 70; AIF: alpinumisoflavone; Nrf2: nuclear factor (erythroid-derived 2)-like 2 transcription factor; HO-1: heme oxygenase; NQO1: NAD(P)H:quinone oxidoreductase 1; γH2AX: H2A histone family member X; GSH: glutathione; GST: glutathione-S-transferases; p53: protein 53; c-PARP: cleaved poly-ADP ribose polymerase; PUMA: p53 upregulated modulator of apoptosis; NF-κB: nuclear factor kappa-B; STAT: signal transducer and activator of transcription; TxnRd1: thioredoxin reductase 1; FA: ferulic acid; p21: protein 21; EGCG: epigallocatechin gallate; TA: tannic acid; CC3: cleaved caspase 3; NOX3: triphosphopyridine nucleotide oxidase 3; p62: sequestosome-1; LC3-II: light chain 3 II; LC3-I: light chain 3 I; p-mTOR: phosphorylated mammalian target of rapamycin; CC8: cleaved caspase 8; FADD: Fas-associated death domain; FasL: Fas ligand; ICAD: caspase-3-activated DNase inhibitor; p-cdc2: phosphorylated cdc2; p-cdc25c: phosphorylated cdc25c; NAC: N-acetyl cysteine; CAT: catalase; EGFR: epidermal growth factor receptor; p-EGFR: phosphorylated EGFR; Ro: ginsenoside Ro; ESR2: estrogen receptor 2; NCF1: neutrophil cytosolic factor 1; ATG-7: autophagy-related 7; CC9: cleaved caspase 9; CHEK1: checkpoint kinase 1; KRG: Korean red ginseng; p-p53: phosphorylated p53.
7.1. Flavonoids
Flavonoids, a group of important naturally occurring compounds found in several edible vegetables, fruits, and medicinal plants, are structured by connecting two benzene rings with phenolic hydroxyl groups through the central three-carbon chain (C6-C3-C6) [212]. It is reported that flavonoids can be used to protect the cardiovascular system, lower diabetes risk, cure neurodegenerative disorders, restore cognition after stroke, and suppress cancer progression [213–215]. Although flavonoids do not seem to be potent enough to be used as a monotherapy in the treatment of cancers, these compounds have been suggested to render considerable clinical benefits when applied in combination with radiotherapy or chemotherapy. Quercetin can synergize cisplatin-induced mitochondrial apoptosis via downregulating Cu/Zn SOD which leads to elevated ROS in larynx cancer Hep2 cells [216]. Naringin has a protective role in doxorubicin-induced toxicity towards normal tissues without sacrificing its anticancer effect by elevating SOD and total antioxidant capacity against the esophageal cancer stem cell YM1 in xenograft nude mice [217]. Alpinumisoflavone (AIF) could significantly increase the radiosensitivity of esophageal squamous cell carcinoma (ESCC) indicated by enhanced apoptosis, DNA damage, and cell cycle arrest which are mechanically achieved by ROS generation and Nrf2 antioxidant system inhibition both in vitro and in vivo [218]. Wogonin, isolated from the root of Scutellaria baicalensis Georgi, could selectively kill HNC cells by upregulating intracellular ROS with no obvious cytotoxic effect against normal oral keratinocytes, oral fibroblasts, and skin keratinocytes. Mechanically, wogonin induces HNC cell death via JNK and PARP activation resulting from the inhibition of Nrf2-GSTP1. Combined wogonin synergizes cisplatin-induced cell death of cisplatin-resistant HNC HN4R and HN9R cells by enhanced ROS in vitro and in vivo. These findings show great hope in the chemosensitivity potential of wogonin in advanced HNC [219].
7.2. Polyphenols
Curcumin is a hydrophobic phenol isolated from Curcuma longa and possesses a variety of pharmacological effects including antidiabetic, antiamyloid, antidepressant, antibacterial, cardioprotective, anti-inflammatory, antioxidant, and anticancer properties [220]. Multiple animal and human studies prove that curcumin is nontoxic even at high doses [221]. Curcumin can inhibit the effects of prosurvival and antiapoptotic elements such as NF-κB and reduce the radiation adaptation in order to enhance the radiation-induced killing effect in various cancer cells [222]. A higher expression of TxnRd1 leads to intrinsic resistance to radiation in HPV− cells. Curcumin can effectively downregulate TxnRd1 and then sensitize HPV− cells to radiation [145]. In a very recent research, curcumin and ferulic acid (FA) both show an antioxidant ability by upregulating the Nrf2/HO-1 pathway for protecting the cochlea after cisplatin treatment without sacrificing the anticancer therapeutic effect in the human oral squamous carcinoma cell line PE/CA-PJ15 and in animal models. One thing to mention is that FA exhibits a biphasic response wherein at lower concentrations it exerts an oxidant function and at higher concentrations it promotes an antioxidant function for chemoresistance. Judging from this, curcumin seems the optimum regimen for effective treatment [223]. A novel synthetic polyphenol conjugate, (E)-3-(3,5-dimethoxyphenyl)-1-(2-methoxyphenyl) prop-2-en-1-one (DPP-23), can efficiently kill cisplatin-resistant HN3-cisR, HN4-cisR, and HN9-cisR cells without harming normal HOK-1 cells. DPP-23 inhibits Nrf2 antioxidant systems and activates p53 expression, thus boosting an increase in cisplatin-mediated apoptosis in vitro and in vivo [224]. Epigallocatechin gallate (EGCG) and tannic acid (TA) could mitigate doxorubicin-induced keratinocyte toxicity without impairing the anticancer effect of doxorubicin at a certain concentrations. An additive cellular ROS increase was not observed after combination treatment of doxorubicin with either 50 μM EGCG or 50 μM TA in oral keratinocyte cells [225]. Epicatechin can protect normal oral fibroblasts from radiation via downregulating ROS and subsequent apoptosis. This is also validated in zebrafish. Epicatechin inhibits JNK and p38 signaling pathways but not the ERK pathway during this physiological process [226]. Another study also confirmed a radioprotective role of epicatechin in human keratinocyte HaCaT cells and in Sprague-Dawley rats via ROS regulation and JNK and p38 pathway alterations [227].
7.3. Naphthoquinones
β-Lapachone (3,4-dihydro-2,2-dimethyl-2H-naphthol (1,2-b) pyran-5,6-dione (C15H14O3)) is a natural naphthoquinone, originally an isomer of lapacho, obtained from the bark of the purple Ipe in South America. Various studies have demonstrated that β-lapachone can induce cell death in solid cancers including esophageal and oral cancers [228–230]. ARQ 761, a β-lapachone analogue, has completed a phase I clinical trial (clinical trial number: NCT01502800) in advanced solid tumors. Outcomes show that ARQ 761 possesses a modest single-agent activity. The most common adverse effect is anemia [231]. Several derivatives have been developed throughout the years. β-Lapachone and its 3-iodine derivatives (3-I-α-lapachone and 3-I-β-lapachone) efficiently kill OSCC HSC-3 cells by enhancing ROS and inducing G2/M arrest, DNA fragmentation, and mitochondria-dependent apoptosis. These results are synchronized in an in vivo study, and the toxicology towards normal tissue is slight [232]. In another multifaceted study, NQO1 is highly expressed in HNC clinical tissue samples, and β-lapachone can synergize radiation to enhance apoptosis and DNA damage by inhibiting NQO1 in HNC FaDu, Detroit 562, SqCC/Y1, and UMSCC-10A cells and also in SqCC/Y1 xenograft nude mice [233]. Thus, the combination treatment of β-lapachone and radiotherapy for QNO1+ HNC patients shall be further tested in clinical trials.
Plumbagin (5-hydroxy-2-methyl-1, 4-naphthoquinone (C11H8O3)), isolated from Plumbago zeylanica L., Juglans regia, Juglans cinerea, and Juglans nigra, exerts antibacterial, antifungal, antiatherosclerosis, and anticancer effects [234]. Our research group has devoted to research its anticancer properties in HNC in recent years. Plumbagin can induce ROS, G2/M arrest, apoptosis, and autophagy in addition to reversing Epithelial-Mesenchymal Transitions (EMT) and cancer stem cell characteristics via inhibiting PI3K/Akt/mTOR, GLUT-1, MAPK, and Nrf2 signaling pathways of oral squamous cell carcinoma (OSCC) in vitro and in vivo [235–237]. In our very recent research, the cisplatin-resistant cell line CAL27/CDDP is applied to verify the chemosensitivity of plumbagin in cisplatin treatment. Outcomes show that plumbagin can efficiently synergize cisplatin-induced apoptosis via upregulating cellular ROS and mitochondrial hydrogen peroxide. Autophagy is also induced by plumbagin and cisplatin, while it is hard to determine its definite anticancer or protective role. Besides, these effects can all be reversed by antioxidant NAC. In CAL27/CDDP xenograft nude mice, we are glad to observe that the combination of plumbagin and cisplatin can significantly reduce tumor volume without affecting the weight of the mice [238]. In order to prompt the clinical utility of plumbagin, we also carried out stable isotope labeling with amino acids in cell culture (SILAC) quantitative proteomics technology to fully reveal the possible molecular targets of plumbagin on OSCC [236]. More well-designed experiments are going on to determine plumbagin's anticancer effect in chemo-/radiotherapy in HNC.
7.4. Terpenoids
Oridonin is a natural bioactive diterpenoid isolated from Rabdosia rubescens, which has been a widely used herb in traditional Chinese medicine [239]. Oridonin shows great anticancer potential with low adverse effect [240]. In human laryngeal squamous cell carcinoma (LSCC) Hep-2 cells, oridonin can induce G2/M phase arrest and apoptosis by targeting caspase 9 to enhance ROS production [240, 241]. Hep-2 is a cell line characterized by high EGFR expression. Hence, the inhibition of EGFR with tyrphostin AG1478 can augment oridonin-induced intrinsic and extrinsic apoptosis via ROS generation in Hep-2 cells [242]. Oridonin is reported to synergize cetuximab. The setting is that cetuximab exhibits unsatisfactory efficacy as a single agent in HNSCC patients [243]. The combined treatment with oridonin and cetuximab could induce Fas-dependent apoptosis and G2/M arrest through triggering ROS generation in LSCC Hep-2 and Tu212 cells. EGFR and JNK signaling pathways are involved in these biological effects. In vivo experiments validate the combined anticancer effect of oridonin and cetuximab [244]. Thus, oridonin is a promising drug targeting ROS in combination with cetuximab in resistant cases.
7.5. Ginsenosides
Ginsenosides, the main active ingredient of ginseng, have significant anticancer activity by inhibiting cell proliferation, promoting apoptosis, inducing cell cycle arrest of cancer cells, inhibiting tumor angiogenesis, and synergizing with chemo-/radiotherapy [245]. Besides, ginsenosides can activate the body's immunity through different ways to fight against cancer [246]. Some kinds of ginsenosides are undergoing clinical trials [247]. Ro, a kind of ginsenoside monomer, can activate estrogen receptor 2 (ESR2), which leads to the activation of neutrophil cytosolic factor 1 (NCF1), a subunit of NADPH oxidase, and then leads to the elevation of ROS production. It is reported that Ro can synergize the killing effect of 5-fluorouracilin by upregulating ROS and subsequently inhibiting protective autophagy in esophageal cancer ECA-109 and TE-1 cells. NAC, an antioxidant, substantially reversed Ro-mediated autophagy inhibition in ECA-109 and TE-1 cells and reversed cell death enhanced by the combination of Ro and 5-fluorouracilin [248]. Korean red ginseng (KRG) whose effective constituent is ginsenoside shows great potential in radiosensitivity in oral cancer SCC25 and SCC1484 cells and radioprotection in normal keratinocyte HaCaT cells. Radiation can induce cell death in HaCaT cells by increasing intracellular ROS and membrane damage. When radiation is combined with KRG, the injury of HaCaT cells was greatly alleviated accompanied by ROS elimination and downregulation of p38 and JNK signaling pathways. This protective effect was verified in a zebrafish embryo toxicity model. These findings show that KRG can potentially be used as a protective drug against radiation-induced oral mucositis without impairing the killing effect of cancer cells [249].
8. Emerging Interdisciplinary Techniques Broaden the Potency of ROS in Chemo-/Radiotherapy in HNC
8.1. Application of Photodynamic Therapy (PDT)
Photodynamic therapy (PDT) is a recognized treatment for incurable head and neck cancer [250, 251]. PDT may be particularly useful for the treatment of early unresectable lesions and remission of locally recurrent esophageal cancer [252], resulting in prolonged survival [253]. Besides, the application of PDT will not affect treatment options for future relapses or second primary disease [254]. Conventional PDT starts with the administration of a photosensitizer (PS), which is excited by locally applied light after 2-4 days. The activated PS subsequently converts oxygen to ROS that can damage DNA, proteins, and lipids, ultimately resulting in cell death [255]. However, side effects of conventional PDT (using hydrophobic PS) are common, including damage to normal surrounding tissues and skin phototoxicity. Hence, there is a trend that associates PDT with other chemotherapeutic agents to reduce tumor resistance and improve the efficacy of treatment [256]. Targeted PDT with cetuximab-IRDye700DX conjugates is currently being tested in patients diagnosed with advanced stage HNSCC (clinical trial number: NCT02422979). The first results of this trial indicate that patients responded well to this therapy, while experiencing limited side effects [257]. The following table lists some recent researches concerning combinations of PDT with chemotherapy drugs which show a synergistic anticancer effect and are expected in future clinical trials (Table 5).
Table 5.
Combination treatment of PDT with chemotherapy drugs in HNC.
| Site | Model | Photosensitizer/laser irradiation | Cotherapy | ROS detection | Effect | Mechanisms | Reference |
|---|---|---|---|---|---|---|---|
| Larynx | In vitro (AMC-HN3 cell) In vivo (AMC-HN3 xenograft nude mice |
Radachlorin (0.9 J/cm2) | +Carboplatin | DCFH-DA confocal microscope, flow cytometry | Synergistic effects: reduce side effect | ↑cytochrome c ↓EGFR, ↑ROS | [258] |
|
| |||||||
| Larynx | In vitro (Hep-2 cell) |
mTHPC (2 J/cm2) | +Cisplatin (5 μM) | — | Synergistic effects | ↓Bcl-2, ↓PD-L1 ↓ATG-7, ↓LC3-II/LC3-I | [259] |
|
| |||||||
| Oral cavity | In vitro (BHY cell) |
mTHPC (1.8 J/cm2) | +Oxaliplatin (0.1-100 μM) | DCFH-DA flow cytometry | Synergistic effects | ↑ROS, ↑S-phase arrest | [260] |
|
| |||||||
| Esophagus | In vitro (KYSE-70 cell) |
mTHPC (1.8 J/cm2) | +Cisplatin (0.01-50 μM) | DCFH-DA flow cytometry | Synergistic effects | ↑ROS | [260] |
|
| |||||||
| Head and neck | In vitro (cisplatin-resistant SQ20B and JSQ3 cell and cisplatin-sensitive HNSCC135 and SCC61 cell) In vivo (SQ20B xenograft nude mice) |
Pyrolipid (54 J/cm2) | +Cisplatin (0.5 mg/kg) | — | Synergistic effects: enhance apoptosis | ↑IL-6, ↑TNF-α, ↑IFN-γ | [261] |
Notes. DCFH-DA: 2′,7′-dichlorofluorescein diacetate; mTHPC: meta-Tetra (hydroxyphenyl) chlorin; EGFR: epidermal growth factor receptor; Bcl-2: B-cell lymphoma-2; ROS: reactive oxygen species; LC3: microtubule-associated protein light chain 3; ATG-7: autophagy-related 7; TNF-α: tumor necrosis factor-α; IL-6: interleukin-6; IFN-γ: interferon-γ.
8.2. Application of the Nanoparticle System Based on ROS Modulation
Nanomedicine is an emerging form of treatment that is dedicated to alternative drug delivery and improved therapeutic efficacy, while reducing harmful side effects on normal tissues. New nanoparticle systems with highly flexible and rapid drug design and production capabilities can be created, which can be designed based on the genetic characteristics of tumors; therefore, making the drug selection for individual patient treatment and overcoming multiple forms of multidrug resistance look promising and open up new prospects for cancer treatment [262–268]. The effect of metal nanoparticles on ROS concentration has been shown to play a role in both radiosensitization and radioprotection. Many studies have investigated the effects of nanoparticle structure and surface functionalization on nanoparticle absorption and radiosensitization during radiotherapy [269].
In OSCC cells, RPTD/HP nanoparticles were effectively internalized and showed effective effects on cell growth inhibition and apoptosis induction after laser irradiation. In OSCC tumor-bearing mice, RPTD/HP nanoparticles show excellent tumor-targeting ability and significantly inhibit tumor growth through a variety of mechanisms after local laser irradiation [270].
Cur-NPs induce apoptosis and inhibit cell growth in human oral cancer cisplatin-resistant CAL27 cells, but they have no cytotoxicity on normal human gingival fibroblast cells and normal keratinocyte cells. The results show that Cur-NPs trigger apoptotic cell death by regulating the function of MDR1 and the production of ROS. The activation of caspase 9 and caspase 3 associated with intrinsic signaling pathways is its main pharmacological effect. Cur-NPs are expected to become a new drug against cisplatin-resistant human oral cancer [271].
8.3. BEMER Therapy Possesses Great Potential in Radiosensitization
Application of low-dose electromagnetic fields (EMF) in the regulation of cellular processes is a complementary therapeutic method. EMF therapy can effectively normalize the tissue microcirculation, thus sensitizing cancer therapy [272–277]. A unique system is the Bio-Electro-Magnetic-Energy-Regulation (BEMER) which employs a low-frequency pulsed magnetic field (maximum 35 μT) with a series of half-wave-like sinusoidal intensity transformation. The BEMER system facilitates an increase in blood vessel and microcirculation to improve organ blood flow, nutrient supply, and removal of metabolites. BEMER treatment showed an obvious radiosensitizing effect in a time-dependent manner by deriving a high ROS level and increasing the number of DNA double-strand breaks (DSBs) in the HNC cell line UTSCC15 which were cultured in a 3D laminin-rich extracellular matrix [278].
9. Conclusions and Perspectives
The estimated new cases of HNC have gradually increased during the past ten years [7, 13, 279]. The five-year survival rate of HNC has not been significantly improved especially in esophageal cancer which is as low as 12% [3]. Two-thirds of the patients with HNC are advanced cases when they are first examined [20]. Comprehensive modality is required for advanced HNC covering surgery, radiotherapy, and chemotherapy. However, response failures and severe side effects still affect the prognosis and quality of life of patients. Methods that simultaneously increase the therapeutic response of cancer cells and protect normal tissues are needed to ameliorate the treatment outcome.
The cellular redox status greatly affects the chemo-/radiotherapy efficiency directly or indirectly by multiple biological events such as cell death induction [45–47], DNA damage repair [45–47], stemness maintenance [68], metabolic reprogramming [81, 82], and tumor microenvironment modification [78]. In the past decade, researchers around the world have carried out numerous researches dedicated to enhancing and adjuvanting the effect of chemo-/radiotherapy by finely adjusting the cellular redox of HNC. These research findings contribute to extend the molecular mechanisms and orchestrate therapeutic strategies to overcome resistance and reduce the side effects, and finally improve long-term outcomes for HNC.
Increasing efficacy may be obtained by combining agents with established conventional treatments. In this review paper, we list several “old drugs” and novel small molecular compounds which efficiently modulate cellular or mitochondrial ROS levels to synergize chemo-/radiotherapy. The mechanisms in terms of energy metabolism, cell death, and DNA damage are investigated. Redox-related signaling pathways such as Nrf2/Keap1, MAPKs, p53, NF-κB, and STAT3 have been discovered to be involved in the process of chemo-/radiosensitization at different degrees using multiple cell and animal models.
In view of the dual role of ROS at different cell stages and the fact that the basic level is different for different types of cells, it should require considerable caution when adjusting ROS. In general, the success of cancer treatments by inducing oxidative damage or disrupting antioxidant systems to suspend the cancer progression of redox homeostasis should be tailored according to tumor stage and pathological pattern, antioxidant levels in the microenvironment of the tumor, and the endogenous antioxidant capacity [6]. Oxidation-reduction screening may include the expression rates of different oxidoreductase enzymes and the comparison of antioxidant enzyme expression between tumor tissues and normal tissues [280]. Prominent detection kits by simple sampling methods concerning the redox status of patients with HNC may bring benefits. This approach can be used to better scheme the treatment for each patient and maximize the effectiveness of the treatment to annihilate the cancerous tissue and reduce adverse harm to normal tissue.
Besides, the overexpression of EGFR was discovered in 80-100% of HNC patients [281]. Inhibition of EGFR strengthens the apoptosis induction of ROS-generating agents in HNC. In the context of EGFR, the downregulation of the glutamine transporter ASCT2 can sensitize HNSCC cells to combination therapy with radiation, cetuximab, or cisplatin to induce higher ROS and then evoke more apoptosis [166, 282]. Ptoxin, a new immunotoxin, was obtained by fusing a novel EGFR-targeted antibody into pseudomonas exotoxin A. Ptoxin can effectively boost ROS via inhibiting the Nrf2-Keap1 antioxidant pathway, thus inducing apoptosis in EGFR+ esophageal cancer cells [283]. DpdtbA, a dithiocarbamate derivative, can effectively inhibit p53/EGFR/AK and produce ROS via inactivating and downregulating SOD which lead to the occurrence of apoptosis [284]. In the future, the combination of Ptoxin and/or DpdtbA with chemotherapy or radiotherapy shows great potential against the most deadly esophageal cancer.
Noteworthy is the emergence of natural herbs that are considered to be putative chemo-/radiosensitizers. Due to the drug resistance of small molecule-targeted inhibitors, researchers are currently committed to developing “double target” or “multitarget” agents. The value of natural herbs is their capability of exerting their anticancer ability via multiple targets which may be developed as effective and ideal drugs to improve cancer treatment especially under the complex circumstance of redox. Defects such as poor solution, low bioavailability, and inefficient extraction of natural herbs limit their future application. Several approaches including the development of synthetic analogues, the use of nanoparticles and other efficient delivery agents to improve bioavailability, and the employment of phospholipid complexes to increase solubility have shown promise in overcoming these challenges [285, 286]. Interdisciplinary techniques such as PDT, nanoparticle transfer system, and the BEMER system show great potential in personalized ROS modulation at a large scale in future combination therapy. More mechanistic studies and randomized controlled trials are required to confirm the benefits.
Acknowledgments
This work and publication costs were financially supported by the National Natural Science Foundation of China (grant number 81860480), the Youth Science Fund Project of the Jiangxi Provincial Science and Technology Department (grant number 20181BAB215022), and the Young Teachers Research and Development Fund Project of Nanchang University (grant number 4209-16100009-PY201818).
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R. L., Torre L. A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Shield K. D., Ferlay J., Jemal A., et al. The global incidence of lip, oral cavity, and pharyngeal cancers by subsite in 2012. CA: A Cancer Journal for Clinicians. 2017;67(1):51–64. doi: 10.3322/caac.21384. [DOI] [PubMed] [Google Scholar]
- 3.Arnold M., Laversanne M., Brown L. M., Devesa S. S., Bray F. Predicting the future burden of esophageal cancer by histological subtype: international trends in incidence up to 2030. American Journal of Gastroenterology. 2017;112(8):1247–1255. doi: 10.1038/ajg.2017.155. [DOI] [PubMed] [Google Scholar]
- 4.Srinivas U. S., Tan B. W. Q., Vellayappan B. A., Jeyasekharan A. D. ROS and the DNA damage response in cancer. Redox Biology. 2019;25, article 101084 doi: 10.1016/j.redox.2018.101084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sies H., Jones D. P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nature Reviews Molecular Cell Biology. 2020;21(7):363–383. doi: 10.1038/s41580-020-0230-3. [DOI] [PubMed] [Google Scholar]
- 6.Chio I. I. C., Tuveson D. A. ROS in cancer: the burning question. Trends in Molecular Medicine. 2017;23(5):411–429. doi: 10.1016/j.molmed.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siegel R. L., Miller K. D., Jemal A. Cancer statistics, 2019. CA: A Cancer Journal for Clinicians. 2018;69(1):7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 8.Cohen E. E. W., LaMonte S. J., Erb N. L., et al. American Cancer Society head and neck cancer survivorship care guideline. CA: A Cancer Journal for Clinicians. 2016;66(3):203–239. doi: 10.3322/caac.21343. [DOI] [PubMed] [Google Scholar]
- 9.Mehanna H., Paleri V., West C. M. L., Nutting C. Head and neck cancer—part 1: epidemiology, presentation, and prevention. BMJ. 2010;341(sep20 1, article c4684) doi: 10.1136/bmj.c4684. [DOI] [PubMed] [Google Scholar]
- 10.Baxi S., Fury M., Ganly I., Rao S., Pfister D. G. Ten years of progress in head and neck cancers. Journal of the National Comprehensive Cancer Network. 2012;10(7):806–810. doi: 10.6004/jnccn.2012.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jemal A., Bray F., Center M. M., Ferlay J., Ward E., Forman D. Global cancer statistics. CA: A Cancer Journal for Clinicians. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 12.Torre L. A., Bray F., Siegel R. L., Ferlay J., Lortet-Tieulent J., Jemal A. Global cancer statistics, 2012. CA: A Cancer Journal for Clinicians. 2015;65(2):87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 13.Siegel R. L., Miller K. D., Jemal A. Cancer statistics, 2020. CA: A Cancer Journal for Clinicians. 2020;70(1):7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 14.Chow L. Q. M. Longo D. L., editor. Head and neck cancer. New England Journal of Medicine. 2020;382(1):60–72. doi: 10.1056/NEJMra1715715. [DOI] [PubMed] [Google Scholar]
- 15.Chen W., Zheng R., Baade P. D., et al. Cancer statistics in China, 2015. CA: A Cancer Journal for Clinicians. 2016;66(2):115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 16.Mehanna H., West C. M. L., Nutting C., Paleri V. Head and neck cancer—part 2: treatment and prognostic factors. BMJ. 2010;341(sep28 2, article c4690) doi: 10.1136/bmj.c4690. [DOI] [PubMed] [Google Scholar]
- 17.Koyfman S. A., Ismaila N., Crook D., et al. Management of the neck in squamous cell carcinoma of the oral cavity and oropharynx: ASCO clinical practice guideline. Journal of Clinical Oncology. 2019;37(20):1753–1774. doi: 10.1200/JCO.18.01921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colevas A. D., Yom S. S., Pfister D. G., et al. NCCN guidelines insights: head and neck cancers, version 1.2018. Journal of the National Comprehensive Cancer Network. 2018;16(5):479–490. doi: 10.6004/jnccn.2018.0026. [DOI] [PubMed] [Google Scholar]
- 19.Adelstein D., Gillison M. L., Pfister D. G., et al. NCCN guidelines insights: head and neck cancers, version 2.2017. Journal of the National Comprehensive Cancer Network. 2017;15(6):761–770. doi: 10.6004/jnccn.2017.0101. [DOI] [PubMed] [Google Scholar]
- 20.Cognetti D. M., Weber R. S., Lai S. Y. Head and neck cancer: an evolving treatment paradigm. Cancer. 2008;113(S7) Supplement 7:1911–1932. doi: 10.1002/cncr.23654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cramer J. D., Burtness B., Le Q. T., Ferris R. L. The changing therapeutic landscape of head and neck cancer. Nature Reviews Clinical Oncology. 2019;16(11):669–683. doi: 10.1038/s41571-019-0227-z. [DOI] [PubMed] [Google Scholar]
- 22.Argiris A., Karamouzis M. V., Raben D., Ferris R. L. Head and neck cancer. The Lancet. 2008;371(9625):1695–1709. doi: 10.1016/S0140-6736(08)60728-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leemans C. R., Braakhuis B. J. M., Brakenhoff R. H. The molecular biology of head and neck cancer. Nature Reviews Cancer. 2011;11(1):9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 24.Nekhlyudov L., Lacchetti C., Davis N. B., et al. Head and neck cancer survivorship care guideline: American Society of Clinical Oncology clinical practice guideline endorsement of the American Cancer Society Guideline. Journal of Clinical Oncology. 2017;35(14):1606–1621. doi: 10.1200/JCO.2016.71.8478. [DOI] [PubMed] [Google Scholar]
- 25.Suh Y., Amelio I., Urbano T. G., Tavassoli M. Clinical update on cancer: molecular oncology of head and neck cancer. Cell Death & Disease. 2014;5(1, article e1018) doi: 10.1038/cddis.2013.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zibelman M., Mehra R. Overview of current treatment options and investigational targeted therapies for locally advanced squamous cell carcinoma of the head and neck. American Journal of Oncology. 2016;39(4):396–406. doi: 10.1097/COC.0000000000000283. [DOI] [PubMed] [Google Scholar]
- 27.Wilkie M. D., Anaam E. A., Lau A. S., et al. TP53 mutations in head and neck cancer cells determine the Warburg phenotypic switch creating metabolic vulnerabilities and therapeutic opportunities for stratified therapies. Cancer Letters. 2020;478:107–121. doi: 10.1016/j.canlet.2020.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koppenol W. H., Bounds P. L., Dang C. V. Otto Warburg's contributions to current concepts of cancer metabolism. Nature Reviews Cancer. 2011;11(5):325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 29.Icard P., Shulman S., Farhat D., Steyaert J. M., Alifano M., Lincet H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resistance Updates. 2018;38:1–11. doi: 10.1016/j.drup.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 30.Trachootham D., Alexandre J., Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nature Reviews Drug Discovery. 2009;8(7):579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 31.Zhao R. Z., Jiang S., Zhang L., Yu Z. B. Mitochondrial electron transport chain, ROS generation and uncoupling (review) International Journal of Molecular Medicine. 2019;44(1):3–15. doi: 10.3892/ijmm.2019.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bedard K., Krause K. H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 33.Glorieux C., Calderon P. B. Catalase, a remarkable enzyme: targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biological Chemistry. 2017;398(10):1095–1108. doi: 10.1515/hsz-2017-0131. [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto M., Kensler T. W., Motohashi H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiological Reviews. 2018;98(3):1169–1203. doi: 10.1152/physrev.00023.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annual Review of Pharmacology and Toxicology. 2013;53(1):401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaramillo M. C., Zhang D. D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Development. 2013;27(20):2179–2191. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irani K., Xia Y., Zweier J. L., et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275(5306):1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 38.Sosa V., Moliné T., Somoza R., Paciucci R., Kondoh H., LLeonart M. E. Oxidative stress and cancer: an overview. Ageing Research Reviews. 2013;12(1):376–390. doi: 10.1016/j.arr.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 39.Emanuele S., D’Anneo A., Calvaruso G., Cernigliaro C., Giuliano M., Lauricella M. The double-edged sword profile of redox signaling: oxidative events as molecular switches in the balance between cell physiology and cancer. Chemical Research in Toxicology. 2018;31(4):201–210. doi: 10.1021/acs.chemrestox.7b00311. [DOI] [PubMed] [Google Scholar]
- 40.Cui Q., Wang J. Q., Assaraf Y. G., et al. Modulating ROS to overcome multidrug resistance in cancer. Drug Resistance Updates. 2018;41:1–25. doi: 10.1016/j.drup.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 41.Kong Q., Beel J. A., Lillehei K. O. A threshold concept for cancer therapy. Medical Hypotheses. 2000;55(1):29–35. doi: 10.1054/mehy.1999.0982. [DOI] [PubMed] [Google Scholar]
- 42.Pelicano H., Carney D., Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resistance Updates. 2004;7(2):97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 43.Fry F., Jacob C. Sensor/effector drug design with potential relevance to cancer. Current Pharmaceutical Design. 2006;12(34):4479–4499. doi: 10.2174/138161206779010512. [DOI] [PubMed] [Google Scholar]
- 44.de Sá Junior P. L., Câmara D. A. D., Porcacchia A. S., et al. The roles of ROS in cancer heterogeneity and therapy. Oxidative Medicine and Cellular Longevity. 2017;2017:12. doi: 10.1155/2017/2467940.2467940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen B., Rao X., House M. G., Nephew K. P., Cullen K. J., Guo Z. GPx3 promoter hypermethylation is a frequent event in human cancer and is associated with tumorigenesis and chemotherapy response. Cancer Letters. 2011;309(1):37–45. doi: 10.1016/j.canlet.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 46.Shin D., Kim E. H., Lee J., Roh J. L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radical Biology and Medicine. 2018;129:454–462. doi: 10.1016/j.freeradbiomed.2018.10.426. [DOI] [PubMed] [Google Scholar]
- 47.Narita N., Ito Y., Takabayashi T., et al. Suppression of SESN1 reduces cisplatin and hyperthermia resistance through increasing reactive oxygen species (ROS) in human maxillary cancer cells. International Journal of Hyperthermia. 2018;35(1):269–278. doi: 10.1080/02656736.2018.1496282. [DOI] [PubMed] [Google Scholar]
- 48.Xue D. F., Pan S. T., Huang G., Qiu J. X. ROS enhances the cytotoxicity of cisplatin by inducing apoptosis and autophagy in tongue squamous cell carcinoma cells. International Journal of Biochemistry & Cell Biology. 2020;122, article 105732 doi: 10.1016/j.biocel.2020.105732. [DOI] [PubMed] [Google Scholar]
- 49.Bauman J. E., Austin M. C., Schmidt R., et al. ERCC1 is a prognostic biomarker in locally advanced head and neck cancer: results from a randomised, phase II trial. British Journal of Cancer. 2013;109(8):2096–2105. doi: 10.1038/bjc.2013.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Banerjee R., Russo N., Liu M., et al. TRIP13 promotes error-prone nonhomologous end joining and induces chemoresistance in head and neck cancer. Nature Communications. 2014;5(1) doi: 10.1038/ncomms5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shen B., Huang D., Ramsey A. J., et al. PD-L1 and MRN synergy in platinum-based chemoresistance of head and neck squamous cell carcinoma. British Journal of Cancer. 2020;122(5):640–647. doi: 10.1038/s41416-019-0697-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krzyzanowski D., Bartosz G., Grzelak A. Collateral sensitivity: ABCG2-overexpressing cells are more vulnerable to oxidative stress. Free Radical Biology and Medicine. 2014;76:47–52. doi: 10.1016/j.freeradbiomed.2014.07.020. [DOI] [PubMed] [Google Scholar]
- 53.Karthikeyan S., Hoti S. L., Nazeer Y., Hegde H. V. Glaucarubinone sensitizes KB cells to paclitaxel by inhibiting ABC transporters via ROS-dependent and p53-mediated activation of apoptotic signaling pathways. Oncotarget. 2106;7(27):42353–42373. doi: 10.18632/oncotarget.9865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Senthebane D. A., Rowe A., Thomford N. E., et al. The role of tumor microenvironment in chemoresistance: to survive, keep your enemies closer. International Journal of Molecular Sciences. 2017;18(7):p. 1586. doi: 10.3390/ijms18071586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu H., Jiang W., Wang Q., Hang L., Wang Y., Wang Y. ROS-sensitive biomimetic nanocarriers modulate tumor hypoxia for synergistic photodynamic chemotherapy. Biomaterials Science. 2019;7(9):3706–3716. doi: 10.1039/C9BM00634F. [DOI] [PubMed] [Google Scholar]
- 56.Ferreira J. A., Peixoto A., Neves M., et al. Mechanisms of cisplatin resistance and targeting of cancer stem cells: adding glycosylation to the equation. Drug Resistance Updates. 2016;24:34–54. doi: 10.1016/j.drup.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 57.Quintiliani M. Modification of radiation sensitivity: the oxygen effect. International Journal of Radiation Oncology • Biology • Physics. 1979;5(7):1069–1076. doi: 10.1016/0360-3016(79)90621-7. [DOI] [PubMed] [Google Scholar]
- 58.Hutchinson M.-K. N. D., Mierzwa M., D’Silva N. J. Radiation resistance in head and neck squamous cell carcinoma: dire need for an appropriate sensitizer. Oncogene. 2020;39(18):3638–3649. doi: 10.1038/s41388-020-1250-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schötz U., Balzer V., Brandt F.-W., et al. Dual PI3K/mTOR inhibitor NVP-BEZ235 enhances radiosensitivity of head and neck squamous cell carcinoma (HNSCC) cell lines due to suppressed double-strand break (DSB) repair by non-homologous end joining. Cancers. 2020;12(2):p. 467. doi: 10.3390/cancers12020467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xiong W., Zhao J., Yu H., et al. Elevated expression of AKR1C3 increases resistance of cancer cells to ionizing radiation via modulation of oxidative stress. PLoS One. 2014;9(11, article e111911) doi: 10.1371/journal.pone.0111911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang H., Yue J., Jiang Z., et al. CAF-secreted CXCL1 conferred radioresistance by regulating DNA damage response in a ROS-dependent manner in esophageal squamous cell carcinoma. Cell Death & Disease. 2017;8(5, article e2790) doi: 10.1038/cddis.2017.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Digomann D., Linge A., Dubrovska A. SLC3A2/CD98hc, autophagy and tumor radioresistance: a link confirmed. Autophagy. 2019;15(10):1850–1851. doi: 10.1080/15548627.2019.1639302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kruger M., Pabst A. M., Al-Nawas B., Horke S., Moergel M. Paraoxonase-2 (PON2) protects oral squamous cell cancer cells against irradiation-induced apoptosis. Journal of Cancer Research and Clinical Oncology. 2015;141(10):1757–1766. doi: 10.1007/s00432-015-1941-2. [DOI] [PubMed] [Google Scholar]
- 64.Fitzgerald A. L., Osman A. A., Xie T.-X., et al. Reactive oxygen species and p21Waf1/Cip1 are both essential for p53-mediated senescence of head and neck cancer cells. Cell Death & Disease. 2015;6(3, article e1678) doi: 10.1038/cddis.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dong Q., Sharma S., Liu H., et al. HDAC inhibitors reverse acquired radio resistance of KYSE-150R esophageal carcinoma cells by modulating Bmi-1 expression. Toxicology Letters. 2014;224(1):121–129. doi: 10.1016/j.toxlet.2013.10.014. [DOI] [PubMed] [Google Scholar]
- 66.Nicolay N. H., Wiedenmann N., Mix M., et al. Correlative analyses between tissue-based hypoxia biomarkers and hypoxia PET imaging in head and neck cancer patients during radiochemotherapy-results from a prospective trial. European Journal of Nuclear Medicine and Molecular Imaging. 2020;47(5):1046–1055. doi: 10.1007/s00259-019-04598-9. [DOI] [PubMed] [Google Scholar]
- 67.Wozny A. S., Lauret A., Battiston-Montagne P., et al. Differential pattern of HIF-1α expression in HNSCC cancer stem cells after carbon ion or photon irradiation: one molecular explanation of the oxygen effect. British Journal of Cancer. 2017;116(10):1340–1349. doi: 10.1038/bjc.2017.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wozny A. S., Vares G., Alphonse G., et al. ROS production and distribution: a new paradigm to explain the differential effects of X-ray and carbon ion irradiation on cancer stem cell migration and invasion. Cancers. 2019;11(4):p. 468. doi: 10.3390/cancers11040468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reid P., Marcu L. G., Olver I., Moghaddasi L., Staudacher A. H., Bezak E. Diversity of cancer stem cells in head and neck carcinomas: the role of HPV in cancer stem cell heterogeneity, plasticity and treatment response. Radiotherapy and Oncology. 2019;135:1–12. doi: 10.1016/j.radonc.2019.02.016. [DOI] [PubMed] [Google Scholar]
- 70.Hu J., Mirshahidi S., Simental A., et al. Cancer stem cell self-renewal as a therapeutic target in human oral cancer. Oncogene. 2019;38(27):5440–5456. doi: 10.1038/s41388-019-0800-z. [DOI] [PubMed] [Google Scholar]
- 71.Najafi M., Farhood B., Mortezaee K., Kharazinejad E., Majidpoor J., Ahadi R. Hypoxia in solid tumors: a key promoter of cancer stem cell (CSC) resistance. Journal of Cancer Research and Clinical Oncology. 2020;146(1):19–31. doi: 10.1007/s00432-019-03080-1. [DOI] [PubMed] [Google Scholar]
- 72.Garcia-Mayea Y., Mir C., Masson F., Paciucci R., LLeonart M. E. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Seminars in Cancer Biology. 2020;60:166–180. doi: 10.1016/j.semcancer.2019.07.022. [DOI] [PubMed] [Google Scholar]
- 73.Diehn M., Cho R. W., Lobo N. A., et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee S. Y., Ju M. K., Jeon H. M., et al. Oncogenic metabolism acts as a prerequisite step for induction of cancer metastasis and cancer stem cell phenotype. Oxidative Medicine and Cellular Longevity. 2018;2018:28. doi: 10.1155/2018/1027453.1027453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Alamoud K. A., Kukuruzinska M. A. Emerging insights into Wnt/β-catenin signaling in head and neck cancer. Journal of Dental Research. 2018;97(6):665–673. doi: 10.1177/0022034518771923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen L., Li Y.-C., Wu L., et al. TRAF 6 regulates tumour metastasis through EMT and CSC phenotypes in head and neck squamous cell carcinoma. Journal of Cellular and Molecular Medicine. 2017;22(2):1337–1349. doi: 10.1111/jcmm.13439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kang H., Kim H., Lee S., Youn H., Youn B. Role of metabolic reprogramming in epithelial-mesenchymal transition (EMT) International Journal of Molecular Sciences. 2019;20(8):p. 2042. doi: 10.3390/ijms20082042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fiori M. E., Di Franco S., Villanova L., Bianca P., Stassi G., De Maria R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Molecular Cancer. 2019;18(1) doi: 10.1186/s12943-019-0994-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee S. Y., Jeong E. K., Ju M. K., et al. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Molecular Cancer. 2017;16(1) doi: 10.1186/s12943-016-0577-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fu J., Zhang J., Gong Y., Testa C. L., Klein-Szanto A. J. Regulation of HIF-1 alpha by the proprotein convertases furin and PC7 in human squamous carcinoma cells. Molecular Carcinogenesis. 2015;54(9):698–706. doi: 10.1002/mc.22131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Meijer T. W. H., Kaanders J. H. A. M., Span P. N., Bussink J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clinical Cancer Research. 2012;18(20):5585–5594. doi: 10.1158/1078-0432.CCR-12-0858. [DOI] [PubMed] [Google Scholar]
- 82.Lu H., Li X., Luo Z., Liu J., Fan Z. Cetuximab reverses the Warburg effect by inhibiting HIF-1-regulated LDH-A. Molecular Cancer Therapeutics. 2013;12(10):2187–2199. doi: 10.1158/1535-7163.MCT-12-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu S. L., Li Y. J., Liao K., et al. 2-Methoxyestradiol inhibits the proliferation and migration and reduces the radioresistance of nasopharyngeal carcinoma CNE-2 stem cells via NF-κB/HIF-1 signaling pathway inactivation and EMT reversal. Oncology Reports. 2017;37(2):793–802. doi: 10.3892/or.2016.5319. [DOI] [PubMed] [Google Scholar]
- 84.Warfel N. A., El-Deiry W. S. HIF-1 signaling in drug resistance to chemotherapy. Current Medicinal Chemistry. 2014;21(26):3021–3028. doi: 10.2174/0929867321666140414101056. [DOI] [PubMed] [Google Scholar]
- 85.Schwarzländer M., Dick T. P., Meyer A. J., Morgan B. Dissecting redox biology using fluorescent protein sensors. Antioxidants & Redox Signaling. 2016;24(13):680–712. doi: 10.1089/ars.2015.6266. [DOI] [PubMed] [Google Scholar]
- 86.Hempel S. L., Buettner G. R., O’Malley Y. Q., Wessels D. A., Flaherty D. M. Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: comparison with 2′,7′-dichlorodihydrofluorescein diacetate, 5(and 6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate, and dihydrorhodamine 123. Free Radical Biology Medicine. 1999;27(1-2):146–159. doi: 10.1016/S0891-5849(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 87.Zhang X., Gao F. Imaging mitochondrial reactive oxygen species with fluorescent probes: current applications and challenges. Free Radical Research. 2015;49(4):374–382. doi: 10.3109/10715762.2015.1014813. [DOI] [PubMed] [Google Scholar]
- 88.Robinson K. M., Janes M. S., Pehar M., et al. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(41):15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wardman P. Methods to measure the reactivity of peroxynitrite-derived oxidants toward reduced fluoresceins and rhodamines. Methods in Enzymology. 2008;441:261–282. doi: 10.1016/S0076-6879(08)01214-7. [DOI] [PubMed] [Google Scholar]
- 90.Lippert A. R., Van de Bittner G. C., Chang C. J. Boronate oxidation as a bioorthogonal reaction approach for studying the chemistry of hydrogen peroxide in living systems. Accounts of Chemical Research. 2011;44(9):793–804. doi: 10.1021/ar200126t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hu J. J., Wong N. K., Ye S., et al. Fluorescent probe HKSOX-1 for imaging and detection of endogenous superoxide in live cells and in vivo. Journal of the American Chemical Society. 2015;137(21):6837–6843. doi: 10.1021/jacs.5b01881. [DOI] [PubMed] [Google Scholar]
- 92.Roelofs B. A., Ge S. X., Studlack P. E., Polster B. M. Low micromolar concentrations of the superoxide probe MitoSOX uncouple neural mitochondria and inhibit complex IV. Free Radical Biology Medicine. 2015;86:250–258. doi: 10.1016/j.freeradbiomed.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dickinson B. C., Chang C. J. A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. Journal of the American Chemical Society. 2008;130(30):9638–9639. doi: 10.1021/ja802355u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Koide Y., Urano Y., Kenmoku S., Kojima H., Nagano T. Design and synthesis of fluorescent probes for selective detection of highly reactive oxygen species in mitochondria of living cells. Journal of the American Chemical Society. 2007;129(34):10324–10325. doi: 10.1021/ja073220m. [DOI] [PubMed] [Google Scholar]
- 95.Kaur A., Haghighatbin M. A., Hogan C. F., New E. J. A FRET-based ratiometric redox probe for detecting oxidative stress by confocal microscopy, FLIM and flow cytometry. Chemical Communications. 2015;51(52):10510–10513. doi: 10.1039/C5CC03394B. [DOI] [PubMed] [Google Scholar]
- 96.Wen Y., Liu K., Yang H., et al. Mitochondria-directed fluorescent probe for the detection of hydrogen peroxide near mitochondrial DNA. Analytical Chemistry. 2015;87(20):10579–10584. doi: 10.1021/acs.analchem.5b03326. [DOI] [PubMed] [Google Scholar]
- 97.Belousov V. V., Fradkov A. F., Lukyanov K. A., et al. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nature Methods. 2006;3(4):281–286. doi: 10.1038/nmeth866. [DOI] [PubMed] [Google Scholar]
- 98.Albrecht S. C., Barata A. G., Grosshans J., Teleman A. A., Dick T. P. In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metabolism. 2011;14(6):819–829. doi: 10.1016/j.cmet.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 99.Borgstahl G. E. O., Parge H. E., Hickey M. J., Beyer W. F., Hallewell R. A., Tainer J. A. The structure of human mitochondrial manganese superoxide dismutase reveals a novel tetrameric interface of two 4-helix bundles. Cell. 1992;71(1):107–118. doi: 10.1016/0092-8674(92)90270-m. [DOI] [PubMed] [Google Scholar]
- 100.Che M., Wang R., Li X., Wang H. Y., Zheng X. F. S. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discovery Today. 2016;21(1):143–149. doi: 10.1016/j.drudis.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chuang T. C., Liu J. Y., Lin C. T., et al. Human manganese superoxide dismutase suppresses HER2/neu-mediated breast cancer malignancy. FEBS Letters. 2007;581(23):4443–4449. doi: 10.1016/j.febslet.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 102.Cullen J. J., Weydert C., Hinkhouse M. M., et al. The role of manganese superoxide dismutase in the growth of pancreatic adenocarcinoma. Cancer Research. 2003;63(6):1297–1303. [PubMed] [Google Scholar]
- 103.Hu Y., Rosen D. G., Zhou Y., et al. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. Journal of Biological Chemistry. 2005;280(47):39485–39492. doi: 10.1074/jbc.M503296200. [DOI] [PubMed] [Google Scholar]
- 104.Miranda A., Janssen L., Bosman C. B., et al. Superoxide dismutases in gastric and esophageal cancer and the prognostic impact in gastric cancer. Clinical Cancer Research. 2000;6(8):3183–3192. [PubMed] [Google Scholar]
- 105.Chung-man Ho J., Zheng S., Comhair S. A., Farver C., Erzurum S. C. Differential expression of manganese superoxide dismutase and catalase in lung cancer. Cancer Research. 2001;61(23):8578–8585. [PubMed] [Google Scholar]
- 106.Hu H., Luo M.-l., Du X.-l., et al. Up-regulated manganese superoxide dismutase expression increases apoptosis resistance in human esophageal squamous cell carcinomas. Chinese Medical Journal. 2007;120(23):2092–2098. doi: 10.1097/00029330-200712010-00006. [DOI] [PubMed] [Google Scholar]
- 107.Holley A. K., Miao L., Clair D. K. S., Clair W. H. S. Redox-modulated phenomena and radiation therapy: the central role of superoxide dismutases. Antioxidants & Redox Signaling. 2014;20(10):1567–1589. doi: 10.1089/ars.2012.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Batinic-Haberle I., Tovmasyan A., Spasojevic I. Mn porphyrin-based redox-active drugs: differential effects as cancer therapeutics and protectors of normal tissue against oxidative injury. Antioxidants & Redox Signaling. 2018;29(16):1691–1724. doi: 10.1089/ars.2017.7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ashcraft K. A., Boss M. K., Tovmasyan A., et al. Novel manganese-porphyrin superoxide dismutase-mimetic widens the therapeutic margin in a preclinical head and neck cancer model. International Journal of Radiation Oncology • Biology • Physics. 2015;93(4):892–900. doi: 10.1016/j.ijrobp.2015.07.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Birer S. R., Lee C. T., Choudhury K. R., et al. Inhibition of the continuum of radiation-induced normal tissue injury by a redox-active Mn porphyrin. Radiation Research. 2017;188(1):94–104. doi: 10.1667/RR14757.1.S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Anderson C. M., Lee C. M., Saunders D. P., et al. Phase IIb, randomized, double-blind trial of GC4419 versus placebo to reduce severe oral mucositis due to concurrent radiotherapy and cisplatin for head and neck cancer. Journal of Clinical Oncology. 2019;37(34):3256–3265. doi: 10.1200/JCO.19.01507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Harris I. S., Treloar A. E., Inoue S., et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27(2):p. 314. doi: 10.1016/j.ccell.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 113.Shin C.-S., Mishra P., Watrous J. D., et al. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nature Communications. 2017;8(1) doi: 10.1038/ncomms15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Couto N., Wood J., Barber J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radical Biology and Medicine. 2016;95:27–42. doi: 10.1016/j.freeradbiomed.2016.02.028. [DOI] [PubMed] [Google Scholar]
- 115.Mannervik B. The enzymes of glutathione metabolism: an overview. Biochemical Society Transactions. 1987;15(4):717–718. doi: 10.1042/bst0150717. [DOI] [PubMed] [Google Scholar]
- 116.Spina M. B., Cohen G. Dopamine turnover and glutathione oxidation: implications for Parkinson disease. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(4):1398–1400. doi: 10.1073/pnas.86.4.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Day B. J. Glutathione: a radical treatment for cystic fibrosis lung disease? Chest. 2005;127(1):12–14. doi: 10.1378/chest.83.5.39S. [DOI] [PubMed] [Google Scholar]
- 118.Sonthalia S., Daulatabad D., Sarkar R. Glutathione as a skin whitening agent: facts, myths, evidence and controversies. Indian Journal of Dermatology, Venereology Leprology. 2016;82(3):262–272. doi: 10.4103/0378-6323.179088. [DOI] [PubMed] [Google Scholar]
- 119.Conn J. W., Louis L. H., Johnston M. W. Alleviation of experimental diabetes in man by administration of reduced glutathione (GSH): metabolic implications. Science. 1949;109(2829):279–280. doi: 10.1126/science.109.2829.279. [DOI] [PubMed] [Google Scholar]
- 120.Rodríguez-Santiago B., Brunet A., Sobrino B., et al. Association of common copy number variants at the glutathione S-transferase genes and rare novel genomic changes with schizophrenia. Molecular Psychiatry. 2010;15(10):1023–1033. doi: 10.1038/mp.2009.53. [DOI] [PubMed] [Google Scholar]
- 121.Chatterjee S., Chakrabarti S., Sengupta B., et al. Prevalence of CYP1A1 and GST polymorphisms in the population of northeastern India and susceptibility of oral cancer. Oncology Research Featuring Preclinical and Clinical Cancer Therapeutics. 2009;17(9):397–403. doi: 10.3727/096504009788912499. [DOI] [PubMed] [Google Scholar]
- 122.Lim J. K. M., Delaidelli A., Minaker S. W., et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proceedings of the National Academy of Sciences of the United States of America. 2019;116(19):9433–9442. doi: 10.1073/pnas.1821323116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Silva M. M., Rocha C. R. R., Kinker G. S., Pelegrini A. L., Menck C. F. M. The balance between NRF2/GSH antioxidant mediated pathway and DNA repair modulates cisplatin resistance in lung cancer cells. Scientific Reports. 2019;9(1) doi: 10.1038/s41598-019-54065-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kou L., Sun R., Xiao S., et al. Ambidextrous approach to disrupt redox balance in tumor cells with increased ROS production and decreased GSH synthesis for cancer therapy. ACS Applied Materials & Interfaces. 2019;11(30):26722–26730. doi: 10.1021/acsami.9b09784. [DOI] [PubMed] [Google Scholar]
- 125.Traverso N., Ricciarelli R., Nitti M., et al. Role of glutathione in cancer progression and chemoresistance. Oxidative Medicine and Cellular Longevity. 2013;2013:10. doi: 10.1155/2013/972913.972913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.McLellan L. I., Wolf C. R. Glutathione and glutathione-dependent enzymes in cancer drug resistance. Drug Resistance Updates. 1999;2(3):153–164. doi: 10.1054/drup.1999.0083. [DOI] [PubMed] [Google Scholar]
- 127.O'Dwyer P. J., Hamilton T. C., LaCreta F. P., et al. Phase I trial of buthionine sulfoximine in combination with melphalan in patients with cancer. Journal of Clinical Oncology. 1996;14(1):249–256. doi: 10.1200/JCO.1996.14.1.249. [DOI] [PubMed] [Google Scholar]
- 128.Bailey H. H., Ripple G., Tutsch K. D., et al. Phase I study of continuous-infusion L-S, R-buthionine sulfoximine with intravenous melphalan. Journal of the National Cancer Institute. 1997;89(23):1789–1796. doi: 10.1093/jnci/89.23.1789. [DOI] [PubMed] [Google Scholar]
- 129.Wu J. H., Batist G. Glutathione and glutathione analogues; therapeutic potentials. Biochimica et Biophysica Acta - General Subjects. 2013;1830(5):3350–3353. doi: 10.1016/j.bbagen.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 130.Roh J. L., Jang H., Kim E. H., Shin D. Targeting of the glutathione, thioredoxin, and Nrf2 antioxidant systems in head and neck cancer. Antioxidants & Redox Signaling. 2017;27(2):106–114. doi: 10.1089/ars.2016.6841. [DOI] [PubMed] [Google Scholar]
- 131.Sobhakumari A., Love-Homan L., Fletcher E. V. M., et al. Choi J., editor. Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS One. 2012;7(10):p. e48175. doi: 10.1371/journal.pone.0048175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Han B., Wang Y., Wang L., Shang Z., Wang S., Pei J. Preparation of GST inhibitor nanoparticle drug delivery system and its reversal effect on the multidrug resistance in oral carcinoma. Nanomaterials. 2015;5(4):1571–1587. doi: 10.3390/nano5041571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ren X., Zou L., Lu J., Holmgren A. Selenocysteine in mammalian thioredoxin reductase and application of ebselen as a therapeutic. Free Radical Biology and Medicine. 2018;127:238–247. doi: 10.1016/j.freeradbiomed.2018.05.081. [DOI] [PubMed] [Google Scholar]
- 134.Holmgren A. Thioredoxin. Annual Review of Biochemistry. 1985;54(1):237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 135.Tinkov A. A., Bjørklund G., Skalny A. V., et al. The role of the thioredoxin/thioredoxin reductase system in the metabolic syndrome: towards a possible prognostic marker? Cellular and Molecular Life Sciences. 2018;75(9):1567–1586. doi: 10.1007/s00018-018-2745-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jia J. J., Geng W. S., Wang Z. Q., Chen L., Zeng X. S. The role of thioredoxin system in cancer: strategy for cancer therapy. Cancer Chemotherapy and Pharmacology. 2019;84(3):453–470. doi: 10.1007/s00280-019-03869-4. [DOI] [PubMed] [Google Scholar]
- 137.Samaranayake G. J., Troccoli C. I., Huynh M., et al. Thioredoxin-1 protects against androgen receptor-induced redox vulnerability in castration-resistant prostate cancer. Nature Communications. 2017;8(1):p. 1204. doi: 10.1038/s41467-017-01269-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bhatia M., McGrath K. L., Di Trapani G., et al. The thioredoxin system in breast cancer cell invasion and migration. Redox Biology. 2016;8:68–78. doi: 10.1016/j.redox.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kaimul A. M., Nakamura H., Masutani H., Yodoi J. Thioredoxin and thioredoxin-binding protein-2 in cancer and metabolic syndrome. Free Radical Biology and Medicine. 2007;43(6):861–868. doi: 10.1016/j.freeradbiomed.2007.05.032. [DOI] [PubMed] [Google Scholar]
- 140.Lin F., Zhang P., Zuo Z., et al. Thioredoxin-1 promotes colorectal cancer invasion and metastasis through crosstalk with S100P. Cancer Letters. 2017;401:1–10. doi: 10.1016/j.canlet.2017.04.036. [DOI] [PubMed] [Google Scholar]
- 141.Zhu X., Huang C., Peng B. Overexpression of thioredoxin system proteins predicts poor prognosis in patients with squamous cell carcinoma of the tongue. Oral Oncology. 2011;47(7):609–614. doi: 10.1016/j.oraloncology.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 142.Iwasawa S., Yamano Y., Takiguchi Y., Tanzawa H., Tatsumi K., Uzawa K. Upregulation of thioredoxin reductase 1 in human oral squamous cell carcinoma. Oncology Reports. 2011;25(3):637–644. doi: 10.3892/or.2010.1131. [DOI] [PubMed] [Google Scholar]
- 143.Banerjee S., Mukherjee S., Mitra S., Singhal P. Altered expression of mitochondrial antioxidants in oral squamous cell carcinoma. Journal of Oral Science. 2017;59(3):439–446. doi: 10.2334/josnusd.16-0655. [DOI] [PubMed] [Google Scholar]
- 144.Feingold P. L., Surman D. R., Brown K., et al. Induction of thioredoxin-interacting protein by a histone deacetylase inhibitor, entinostat, is associated with DNA damage and apoptosis in esophageal adenocarcinoma. Molecular Cancer Therapeutics. 2018;17(9):2013–2023. doi: 10.1158/1535-7163.MCT-17-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tuttle S., Hertan L., Daurio N., et al. The chemopreventive and clinically used agent curcumin sensitizes HPV− but not HPV+ HNSCC to ionizing radiation, in vitro and in a mouse orthotopic model. Cancer Biology & Therapy. 2014;13(7):575–584. doi: 10.4161/cbt.19772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hui K. F., Lam B. H. W., Ho D. N., Tsao S. W., Chiang A. K. S. Bortezomib and SAHA synergistically induce ROS-driven caspase-dependent apoptosis of nasopharyngeal carcinoma and block replication of Epstein-Barr virus. Molecular Cancer Therapeutics. 2013;12(5):747–758. doi: 10.1158/1535-7163.MCT-12-0811. [DOI] [PubMed] [Google Scholar]
- 147.Rhee S. G., Kil I. S. Multiple functions and regulation of mammalian peroxiredoxins. Annual Review of Biochemistry. 2017;86(1):749–775. doi: 10.1146/annurev-biochem-060815-014431. [DOI] [PubMed] [Google Scholar]
- 148.Ismail T., Kim Y., Lee H., Lee D. S., Lee H. S. Interplay between mitochondrial peroxiredoxins and ROS in cancer development and progression. International Journal of Molecular Sciences. 2019;20(18):p. 4407. doi: 10.3390/ijms20184407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ren P., Ye H., Dai L., et al. Peroxiredoxin 1 is a tumor-associated antigen in esophageal squamous cell carcinoma. Oncology Reports. 2013;30(5):2297–2303. doi: 10.3892/or.2013.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gong F., Hou G., Liu H., Zhang M. Peroxiredoxin 1 promotes tumorigenesis through regulating the activity of mTOR/p70S6K pathway in esophageal squamous cell carcinoma. Medical Oncology. 2015;32(2):p. 455. doi: 10.1007/s12032-014-0455-0. [DOI] [PubMed] [Google Scholar]
- 151.Park S. H., Chung Y. M., Lee Y. S., et al. Antisense of human peroxiredoxin II enhances radiation-induced cell death. Clinical Cancer Research. 2000;6(12):4915–4920. [PubMed] [Google Scholar]
- 152.He Y., Xu W., Xiao Y., et al. Overexpression of peroxiredoxin 6 (PRDX6) promotes the aggressive phenotypes of esophageal squamous cell carcinoma. Journal of Cancer. 2018;9(21):3939–3949. doi: 10.7150/jca.26041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cuadrado A., Rojo A. I., Wells G., et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nature Reviews Drug Discovery. 2019;18(4):295–317. doi: 10.1038/s41573-018-0008-x. [DOI] [PubMed] [Google Scholar]
- 154.Rojo de la Vega M., Chapman E., Zhang D. D. NRF2 and the hallmarks of cancer. Cancer cell. 2018;34(1):21–43. doi: 10.1016/j.ccell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Stransky N., Egloff A. M., Tward A. D., et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Martinez V. D., Vucic E. A., Thu K. L., Pikor L. A., Lam S., Lam W. L. Disruption of KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex components by multiple genetic mechanisms: association with poor prognosis in head and neck cancer. Head Neck. 2015;37(5):727–734. doi: 10.1002/hed.23663. [DOI] [PubMed] [Google Scholar]
- 157.Noman A. S. M., Parag R. R., Rashid M. I., et al. Widespread expression of Sonic hedgehog (Shh) and Nrf 2 in patients treated with cisplatin predicts outcome in resected tumors and are potential therapeutic targets for HPV-negative head and neck cancer. Therapeutic Advances in Medical Oncology. 2020;12 doi: 10.1177/1758835920911229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Roh J. L., Kim E. H., Jang H., Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biology. 2017;11:254–262. doi: 10.1016/j.redox.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Aichler M., Elsner M., Ludyga N., et al. Clinical response to chemotherapy in oesophageal adenocarcinoma patients is linked to defects in mitochondria. Journal of Pathology. 2013;230(4):410–419. doi: 10.1002/path.4199. [DOI] [PubMed] [Google Scholar]
- 160.Goldman P., Peppercorn M. A. Drug therapy: sulfasalazine. New England Journal of Medicine. 1975;293(1):20–23. doi: 10.1056/NEJM197507032930105. [DOI] [PubMed] [Google Scholar]
- 161.Shin D., Lee J., You J. H., Kim D., Roh J. L. Dihydrolipoamide dehydrogenase regulates cystine deprivation-induced ferroptosis in head and neck cancer. Redox Biology. 2020;30, article 101418 doi: 10.1016/j.redox.2019.101418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Roh J. L., Kim E. H., Jang H. J., Park J. Y., Shin D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Letters. 2016;381(1):96–103. doi: 10.1016/j.canlet.2016.07.035. [DOI] [PubMed] [Google Scholar]
- 163.Naito E., Kuroda Y., Toshima K., et al. Effect of sodium dichloroacetate on human pyruvate metabolism. Brain & Development. 1989;11(3):195–197. doi: 10.1016/S0387-7604(89)80098-1. [DOI] [PubMed] [Google Scholar]
- 164.Kankotia S., Stacpoole P. W. Dichloroacetate and cancer: new home for an orphan drug? Biochimica et Biophysica Acta. 2014;1846(2):617–629. doi: 10.1016/j.bbcan.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 165.Roh J. L., Park J. Y., Kim E. H., Jang H. J., Kwon M. Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer. Cancer Letters. 2016;371(1):20–29. doi: 10.1016/j.canlet.2015.11.023. [DOI] [PubMed] [Google Scholar]
- 166.Lu H., Li X., Lu Y., Qiu S., Fan Z. ASCT2 (SLC1A5) is an EGFR-associated protein that can be co-targeted by cetuximab to sensitize cancer cells to ROS-induced apoptosis. Cancer Letters. 2016;381(1):23–30. doi: 10.1016/j.canlet.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zakki S. A., Muhammad J. S., Li J. L., et al. Melatonin triggers the anticancer potential of phenylarsine oxide via induction of apoptosis through ROS generation and JNK activation. Metallomics. 2020;12(3):396–407. doi: 10.1039/C9MT00238C. [DOI] [PubMed] [Google Scholar]
- 168.NaveenKumar S. K., Hemshekhar M., Kemparaju K., Girish K. S. Hemin-induced platelet activation and ferroptosis is mediated through ROS- driven proteasomal activity and inflammasome activation: Protection by Melatonin. Biochimica et Biophysica Acta-Molecular Basis of Disease. 2019;1865(9):2303–2316. doi: 10.1016/j.bbadis.2019.05.009. [DOI] [PubMed] [Google Scholar]
- 169.Zou Z. W., Liu T., Li Y., et al. Melatonin suppresses thyroid cancer growth and overcomes radioresistance _via_ inhibition of p65 phosphorylation and induction of ROS. Redox Biology. 2018;16:226–236. doi: 10.1016/j.redox.2018.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lin Y. W., Lee L. M., Lee W. J., et al. Melatonin inhibits MMP-9 transactivation and renal cell carcinoma metastasis by suppressing Akt-MAPKs pathway and NF-κB DNA-binding activity. Melatonin inhibits MMP-9 transactivation and renal cell carcinoma metastasis by suppressing Akt-MAPKs pathway and NF-κB DNA-binding activity. Journal of Pineal Research. 2016;60(3):277–290. doi: 10.1111/jpi.12308. [DOI] [PubMed] [Google Scholar]
- 171.Ju H. Q., Li H., Tian T., et al. Melatonin overcomes gemcitabine resistance in pancreatic ductal adenocarcinoma by abrogating nuclear factor-κB activation. Journal of Pineal Research. 2016;60(1):27–38. doi: 10.1111/jpi.12285. [DOI] [PubMed] [Google Scholar]
- 172.Moneim A. A., Guerra-Librero A., Florido J., et al. Oral mucositis: melatonin gel an effective new treatment. International Journal of Molecular Sciences. 2017;18(5, article E1003) doi: 10.3390/ijms18051003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fernandez-Gil B. I., Guerra-Librero A., Shen Y.-Q., et al. Melatonin enhances cisplatin and radiation cytotoxicity in head and neck squamous cell carcinoma by stimulating mitochondrial ROS generation, apoptosis, and autophagy. Oxidative Medicine and Cellular Longevity. 2019;2019:12. doi: 10.1155/2019/7187128.7187128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fenton M., Rathbone J., Reilly J., Sultana A. Thioridazine for schizophrenia. Cochrane Database of Systematic Reviews. 2007;1(3, article CD001944) doi: 10.1002/14651858.CD001944.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Thanacoody R. H., Daly A. K., Reilly J. G., Ferrier I. N., Thomas S. H. Factors affecting drug concentrations and QT interval during thioridazine therapy, Clinical Pharmacology Therapeutics. 2007;82(5):555–565. doi: 10.1038/sj.clpt.6100195. [DOI] [PubMed] [Google Scholar]
- 176.Bisset A. F. Discontinuation of thioridazine. Risks must be balanced. BMJ. 2002;325(7370):967–968. doi: 10.1136/bmj.325.7370.967/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nagel D., Spranger S., Vincendeau M., et al. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell. 2012;22(6):825–837. doi: 10.1016/j.ccr.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 178.Lu M., Li J., Luo Z., et al. Roles of dopamine receptors and their antagonist thioridazine in hepatoma metastasis. OncoTargets and Therapy. 2015;8:1543–1552. doi: 10.2147/OTT.S77373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Spengler G., Molnar J., Viveiros M., Amaral L. Thioridazine induces apoptosis of multidrug-resistant mouse lymphoma cells transfected with the human ABCB1 and inhibits the expression of P-glycoprotein. Anticancer Research. 2011;31(12):4201–4205. [PubMed] [Google Scholar]
- 180.Seo S. U., Cho H. K., Min K. J., et al. Thioridazine enhances sensitivity to carboplatin in human head and neck cancer cells through downregulation of c-FLIP and Mcl-1 expression. Cell Death & Disease. 2017;8(2, article e2599) doi: 10.1038/cddis.2017.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Torpy J. M., Livingston E. H. JAMA patient page. Aspirin therapy. JAMA. 2013;309(15):p. 1645. doi: 10.1001/jama.2013.3866. [DOI] [PubMed] [Google Scholar]
- 182.Liao D., Zhong L., Duan T., et al. Aspirin suppresses the growth and metastasis of osteosarcoma through the NF-κB pathway. Clinical Cancer Research. 2015;21(23):5349–5359. doi: 10.1158/1078-0432.CCR-15-0198. [DOI] [PubMed] [Google Scholar]
- 183.Roh J. L., Kim E. H., Jang H., Shin D. Aspirin plus sorafenib potentiates cisplatin cytotoxicity in resistant head and neck cancer cells through xCT inhibition. Free Radical Biology and Medicine. 2017;104:1–9. doi: 10.1016/j.freeradbiomed.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 184.Miyazaki Y., Shibuya M., Sugawara H., et al. Salinomycin, a new polyether antibiotic. Journal of Antibiotics. 1974;27(11):814–821. doi: 10.7164/antibiotics.27.814. [DOI] [PubMed] [Google Scholar]
- 185.Gupta P. B., Onder T. T., Jiang G., et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138(4):645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhang G., Wang W., Yao C., Ren J., Zhang S., Han M. Salinomycin overcomes radioresistance in nasopharyngeal carcinoma cells by inhibiting Nrf2 level and promoting ROS generation. Biomedicine & Pharmacotherapy. 2017;91:147–154. doi: 10.1016/j.biopha.2017.04.095. [DOI] [PubMed] [Google Scholar]
- 187.Bailey C. J. Metformin: historical overview. Diabetologia. 2017;60(9):1566–1576. doi: 10.1007/s00125-017-4318-z. [DOI] [PubMed] [Google Scholar]
- 188.Flory J., Lipska K. Metformin in 2019. JAMA. 2019;321(19):1926–1927. doi: 10.1001/jama.2019.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Pernicova I., Korbonits M. Metformin--mode of action and clinical implications for diabetes and cancer. Nature Reviews Endocrinology. 2014;10(3):143–156. doi: 10.1038/nrendo.2013.256. [DOI] [PubMed] [Google Scholar]
- 190.Evans J. M. M., Donnelly L. A., Emslie-Smith A. M., Alessi D. R., Morris A. D. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330(7503):1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sikka A., Kaur M., Agarwal C., Deep G., Agarwal R. Metformin suppresses growth of human head and neck squamous cell carcinoma via global inhibition of protein translation. Cell Cycle. 2014;11(7):1374–1382. doi: 10.4161/cc.19798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Woo S. H., Yang L. P., Chuang H. C., et al. Down-regulation of malic enzyme 1 and 2: sensitizing head and neck squamous cell carcinoma cells to therapy-induced senescence. Head Neck. 2016;38(Supplement 1):E934–E940. doi: 10.1002/hed.24129. [DOI] [PubMed] [Google Scholar]
- 193.Roskoski R. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacological Research. 2015;100:1–23. doi: 10.1016/j.phrs.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 194.Tang J. Y., Wu C. Y., Shu C. W., Wang S. C., Chang M. Y., Chang H. W. A novel sulfonyl chromen-4-ones (CHW09) preferentially kills oral cancer cells showing apoptosis, oxidative stress, and DNA damage. Environmental Toxicology. 2018;33(11):1195–1203. doi: 10.1002/tox.22625. [DOI] [PubMed] [Google Scholar]
- 195.Tang J. Y., Shu C. W., Wang C. L., et al. Sulfonyl chromen-4-ones (CHW09) shows an additive effect to inhibit cell growth of X-ray irradiated oral cancer cells, involving apoptosis and ROS generation. International Journal of Radiation Biology. 2019;95(9):1226–1235. doi: 10.1080/09553002.2019.1625490. [DOI] [PubMed] [Google Scholar]
- 196.Abdel-Wahab A. F., Mahmoud W., Al-Harizy R. M. Targeting glucose metabolism to suppress cancer progression: prospective of anti-glycolytic cancer therapy. Pharmacological Research. 2019;150, article 104511 doi: 10.1016/j.phrs.2019.104511. [DOI] [PubMed] [Google Scholar]
- 197.Zhai X., Yang Y., Wan J., Zhu R., Wu Y. Inhibition of LDH-A by oxamate induces G2/M arrest, apoptosis and increases radiosensitivity in nasopharyngeal carcinoma cells. Oncology Reports. 2013;30(6):2983–2991. doi: 10.3892/or.2013.2735. [DOI] [PubMed] [Google Scholar]
- 198.Laganá G., Barreca D., Calderaro A., Bellocco E. Lactate dehydrogenase inhibition: biochemical relevance and therapeutical potential. Current Medicinal Chemistry. 2019;26(18):3242–3252. doi: 10.2174/0929867324666170209103444. [DOI] [PubMed] [Google Scholar]
- 199.Chen Z., Chen J., Zhang W., Zhang T., Guang C., Mu W. Recent research on the physiological functions, applications, and biotechnological production of D-allose. Applied Microbiology Biotechnology. 2018;102(10):4269–4278. doi: 10.1007/s00253-018-8916-6. [DOI] [PubMed] [Google Scholar]
- 200.Hoshikawa H., Mori T., Mori N. In vitro and in vivo effects of D-allose: up-regulation of thioredoxin-interacting protein in head and neck cancer cells. Annals of Otology, Rhinology & Laryngology. 2010;119(8):567–571. doi: 10.1177/000348941011900810. [DOI] [PubMed] [Google Scholar]
- 201.Hoshikawa H., Indo K., Mori T., Mori N. Enhancement of the radiation effects by D-allose in head and neck cancer cells. Cancer Letters. 2011;306(1):60–66. doi: 10.1016/j.canlet.2011.02.032. [DOI] [PubMed] [Google Scholar]
- 202.Indo K., Hoshikawa H., Kamitori K., et al. Effects of D-allose in combination with docetaxel in human head and neck cancer cells. International Journal of Oncology. 2014;45(5):2044–2050. doi: 10.3892/ijo.2014.2590. [DOI] [PubMed] [Google Scholar]
- 203.Iga Y., Nakamichi K., Shirai Y., Matsuo T. Acute and sub-chronic toxicity of D-allose in rats. Bioscience, Biotechnology, and Biochemistry. 2014;74(7):1476–1478. doi: 10.1271/bbb.100121. [DOI] [PubMed] [Google Scholar]
- 204.Hoshikawa H., Kamitori K., Indo K., et al. Combined treatment with D-allose, docetaxel and radiation inhibits the tumor growth in an in vivo model of head and neck cancer. Oncology Letters. 2018;15(3):3422–3428. doi: 10.3892/ol.2018.7787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hesham H. M., Lasheen D. S., Abouzid K. A. M. Chimeric HDAC inhibitors: comprehensive review on the HDAC-based strategies developed to combat cancer. Medicinal Research Reviews. 2018;38(6):2058–2109. doi: 10.1002/med.21505. [DOI] [PubMed] [Google Scholar]
- 206.New M., Olzscha H., La Thangue N. B. HDAC inhibitor-based therapies: can we interpret the code? Molecular Oncology. 2012;6(6):637–656. doi: 10.1016/j.molonc.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Jang B., Shin J. A., Kim Y. S., et al. Growth-suppressive effect of suberoylanilide hydroxamic acid (SAHA) on human oral cancer cells. Cellular Oncology. 2016;39(1):79–87. doi: 10.1007/s13402-015-0255-3. [DOI] [PubMed] [Google Scholar]
- 208.Grabarska A., Łuszczki J. J., Nowosadzka E., et al. Histone deacetylase inhibitor SAHA as potential targeted therapy agent for larynx cancer cells. Journal of Cancer. 2017;8(1):19–28. doi: 10.7150/jca.16655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Citro S., Bellini A., Miccolo C., Ghiani L., Carey T. E., Chiocca S. Synergistic antitumour activity of HDAC inhibitor SAHA and EGFR inhibitor gefitinib in head and neck cancer: a key role for ΔNp63α. British Journal of Cancer. 2019;120(6):658–667. doi: 10.1038/s41416-019-0394-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Suzuki M., Endo M., Shinohara F., Echigo S., Rikiishi H. Enhancement of cisplatin cytotoxicity by SAHA involves endoplasmic reticulum stress-mediated apoptosis in oral squamous cell carcinoma cells. Cancer Chemotherapy and Pharmacology. 2009;64(6):1115–1122. doi: 10.1007/s00280-009-0969-x. [DOI] [PubMed] [Google Scholar]
- 211.West A. C., Johnstone R. W. New and emerging HDAC inhibitors for cancer treatment. Journal of Clinical Investigation. 2014;124(1):30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wang T. Y., Li Q., Bi K. S. Bioactive flavonoids in medicinal plants: structure, activity and biological fate. Asian Journal of Pharmaceutical Sciences. 2018;13(1):12–23. doi: 10.1016/j.ajps.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Spencer J. P. E. The impact of flavonoids on memory: physiological and molecular considerations. Chemical Society Reviews. 2009;38(4):1152–1161. doi: 10.1039/b800422f. [DOI] [PubMed] [Google Scholar]
- 214.Pan M. H., Ho C. T. Chemopreventive effects of natural dietary compounds on cancer development. Chemical Society Reviews. 2008;37(11):2558–2574. doi: 10.1039/b801558a. [DOI] [PubMed] [Google Scholar]
- 215.Heiss C., Keen C. L., Kelm M. Flavanols and cardiovascular disease prevention. European Heart Journal. 2010;31(21):2583–2592. doi: 10.1093/eurheartj/ehq332. [DOI] [PubMed] [Google Scholar]
- 216.Sharma H., Sen S., Singh N. Molecular pathways in the chemosensitization of cisplatin by quercetin in human head and neck cancer. Cancer Biology & Therapy. 2014;4(9):949–955. doi: 10.4161/cbt.4.9.1908. [DOI] [PubMed] [Google Scholar]
- 217.Tajaldini M., Samadi F., Khosravi A., Ghasemnejad A., Asadi J. Protective and anticancer effects of orange peel extract and naringin in doxorubicin treated esophageal cancer stem cell xenograft tumor mouse model. Biomedicine & Pharmacotherapy. 2020;121, article 109594 doi: 10.1016/j.biopha.2019.109594. [DOI] [PubMed] [Google Scholar]
- 218.Zhang B., Fan X., Wang Z., Zhu W., Li J. Alpinumisoflavone radiosensitizes esophageal squamous cell carcinoma through inducing apoptosis and cell cycle arrest. Biomedicine & Pharmacotherapy. 2017;95:199–206. doi: 10.1016/j.biopha.2017.08.048. [DOI] [PubMed] [Google Scholar]
- 219.Kim E. H., Jang H., Shin D., Baek S. H., Roh J. L. Targeting Nrf2 with wogonin overcomes cisplatin resistance in head and neck cancer. Apoptosis. 2016;21(11):1265–1278. doi: 10.1007/s10495-016-1284-8. [DOI] [PubMed] [Google Scholar]
- 220.Stohs S. J., Chen O., Ray S. D., Ji J., Bucci L. R., Preuss H. G. Highly bioavailable forms of curcumin and promising avenues for curcumin-based research and application: a review. Molecules. 2020;25(6):p. 1397. doi: 10.3390/molecules25061397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Epstein J., Sanderson I. R., Macdonald T. T. Curcumin as a therapeutic agent: the evidence from in vitro, animal and human studies. British Journal of Nutrition. 2010;103(11):1545–1557. doi: 10.1017/S0007114509993667. [DOI] [PubMed] [Google Scholar]
- 222.Sak K. Radiosensitizing potential of curcumin in different cancer models. Nutrition Cancer. 2019;2019:1–14. doi: 10.1080/01635581.2019.1681480. [DOI] [PubMed] [Google Scholar]
- 223.Paciello F., Fetoni A. R., Mezzogori D., et al. The dual role of curcumin and ferulic acid in counteracting chemoresistance and cisplatin-induced ototoxicity. Scientific Reports. 2020;10(1):p. 1063. doi: 10.1038/s41598-020-57965-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kim E. H., Jang H., Roh J. L. A novel polyphenol conjugate sensitizes cisplatin-resistant head and neck cancer cells to cisplatin via Nrf2 inhibition. Molecular Cancer Therapeutics. 2016;15(11):2620–2629. doi: 10.1158/1535-7163.MCT-16-0332. [DOI] [PubMed] [Google Scholar]
- 225.Sheng H., Ogawa T., Niwano Y., Sasaki K., Tachibana K. Effects of polyphenols on doxorubicin-induced oral keratinocyte cytotoxicity and anticancer potency against oral cancer cells. Journal of Oral Pathology & Medicine. 2018;47(4):368–374. doi: 10.1111/jop.12685. [DOI] [PubMed] [Google Scholar]
- 226.Shin H. A., Shin Y. S., Kang S. U., et al. Radioprotective effect of epicatechin in cultured human fibroblasts and zebrafish. Journal of Radiattion Research. 2014;55(1):32–40. doi: 10.1093/jrr/rrt085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Shin Y. S., Shin H. A., Kang S. U., et al. Effect of epicatechin against radiation-induced oral mucositis: in vitro and in vivo study. PLoS One. 2013;8(7):p. e69151. doi: 10.1371/journal.pone.0069151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Jeon Y.-J., Bang W., Shin J.-C., et al. Downregulation of Sp1 is involved in β-lapachone-induced cell cycle arrest and apoptosis in oral squamous cell carcinoma. International Journal of Oncology. 2015;46(6):2606–2612. doi: 10.3892/ijo.2015.2972. [DOI] [PubMed] [Google Scholar]
- 229.Sunassee S. N., Veale C. G. L., Shunmoogam-Gounden N., et al. Cytotoxicity of lapachol, β-lapachone and related synthetic 1,4-naphthoquinones against oesophageal cancer cells. European Journal of Medicinal Chemistry. 2013;62:98–110. doi: 10.1016/j.ejmech.2012.12.048. [DOI] [PubMed] [Google Scholar]
- 230.Ferraz da Costa D. C., Rangel L. P., da Cunha Martins-Dinis M. M. D., da Silva Ferretti G. D., Ferreira V. F., Silva J. L. Anticancer potential of resveratrol, β-lapachone and their analogues. Molecules. 2020;25(4):p. 893. doi: 10.3390/molecules25040893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Gerber D. E., Beg M. S., Fattah F., et al. Phase 1 study of ARQ 761, a β-lapachone analogue that promotes NQO1-mediated programmed cancer cell necrosis. British Journal of Cancer. 2018;119(8):928–936. doi: 10.1038/s41416-018-0278-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Dias R. B., de Araújo T. B. S., de Freitas R. D., et al. β-Lapachone and its iodine derivatives cause cell cycle arrest at G2/M phase and reactive oxygen species-mediated apoptosis in human oral squamous cell carcinoma cells. Free Radical Biology Medicine. 2018;126:87–100. doi: 10.1016/j.freeradbiomed.2018.07.022. [DOI] [PubMed] [Google Scholar]
- 233.Li L. S., Reddy S., Lin Z. H., et al. NQO1-mediated tumor-selective lethality and radiosensitization for head and neck cancer. Molecular Cancer Therapeutics. 2016;15(7):1757–1767. doi: 10.1158/1535-7163.MCT-15-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Tripathi S. K., Panda M., Biswal B. K. Emerging role of plumbagin: cytotoxic potential and pharmaceutical relevance towards cancer therapy. Food and Chemical Toxicology. 2019;125:566–582. doi: 10.1016/j.fct.2019.01.018. [DOI] [PubMed] [Google Scholar]
- 235.Zhou S.-F., Pan S.-T., Qin Y., et al. Plumbagin induces G2/M arrest, apoptosis, and autophagy via p 38 MAPK- and PI3K/Akt/mTOR-mediated pathways in human tongue squamous cell carcinoma cells. Drug Design Development and Therapy. 2015;9:1601–1626. doi: 10.2147/DDDT.S76057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Zhou S.-F., Pan S.-T., Qin Y., et al. Plumbagin suppresses epithelial to mesenchymal transition and stemness via inhibiting Nrf2-mediated signaling pathway in human tongue squamous cell carcinoma cells. Drug Design Development and Therapy. 2015;9:5511–5551. doi: 10.2147/dddt.s89621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Na S., Zhang J., Zhou X., et al. Plumbagin-mediating GLUT1 suppresses the growth of human tongue squamous cell carcinoma. Oral Diseases. 2018;24(6):920–929. doi: 10.1111/odi.12799. [DOI] [PubMed] [Google Scholar]
- 238.Xue D., Pan S.-T., Zhou X., et al. Plumbagin enhances the anticancer efficacy of cisplatin by increasing intracellular ROS in human tongue squamous cell carcinoma. Oxidative Medicine and Cellular Longevity. 2020;2020:21. doi: 10.1155/2020/5649174.5649174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lv F., Xu X. J. Studies on the chemical constituents of Rabdosia rubescens. Zhong Yao Cai. 2008;31(9):1340–1343. [PubMed] [Google Scholar]
- 240.Kang N., Cao S.-J., Zhou Y., et al. Inhibition of caspase-9 by oridonin, a diterpenoid isolated from Rabdosia rubescens, augments apoptosis in human laryngeal cancer cells. International Journal of Oncology. 2015;47(6):2045–2056. doi: 10.3892/ijo.2015.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Kang N., Zhang J. H., Qiu F., et al. Induction of G(2)/M phase arrest and apoptosis by oridonin in human laryngeal carcinoma cells. Journal of Natural Products. 2010;73(6):1058–1063. doi: 10.1021/np9008199. [DOI] [PubMed] [Google Scholar]
- 242.Kang N., Zhang J. H., Qiu F., Tashiro S., Onodera S., Ikejima T. Inhibition of EGFR signaling augments oridonin-induced apoptosis in human laryngeal cancer cells via enhancing oxidative stress coincident with activation of both the intrinsic and extrinsic apoptotic pathways. Cancer Letters. 2010;294(2):147–158. doi: 10.1016/j.canlet.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 243.Bernier J. Cetuximab in the treatment of head and neck cancer. Expert Review of Anticancer Therapy. 2014;6(11):1539–1552. doi: 10.1586/14737140.6.11.1539. [DOI] [PubMed] [Google Scholar]
- 244.Cao S., Xia M., Mao Y., et al. Combined oridonin with cetuximab treatment shows synergistic anticancer effects on laryngeal squamous cell carcinoma: involvement of inhibition of EGFR and activation of reactive oxygen species-mediated JNK pathway. International Journal of Oncology. 2016;49(5):2075–2087. doi: 10.3892/ijo.2016.3696. [DOI] [PubMed] [Google Scholar]
- 245.Liu Z. Q. Chemical insights into ginseng as a resource for natural antioxidants. Chemical Reviews. 2012;112(6):3329–3355. doi: 10.1021/cr100174k. [DOI] [PubMed] [Google Scholar]
- 246.Zhang Y., Qiu Z., Qiu Y., Su T., Qu P., Jia A. Functional regulation of ginsenosides on myeloid immunosuppressive cells in the tumor microenvironment. Integrative Cancer Therapies. 2019;18 doi: 10.1177/1534735419886655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Yu S. E., Mwesige B., Yi Y. S., Yoo B. C. Ginsenosides: the need to move forward from bench to clinical trials. Journal of Ginseng Research. 2019;43(3):361–367. doi: 10.1016/j.jgr.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Zheng K., Li Y., Wang S., et al. Inhibition of autophagosome-lysosome fusion by ginsenoside Ro via the ESR2-NCF1-ROS pathway sensitizes esophageal cancer cells to 5-fluorouracil-induced cell death via the CHEK1-mediated DNA damage checkpoint. Autophagy. 2016;12(9):1593–1613. doi: 10.1080/15548627.2016.1192751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Chang J. W., Park K. H., Hwang H. S., Shin Y. S., Oh Y. T., Kim C. H. Protective effects of Korean red ginseng against radiation-induced apoptosis in human HaCaT keratinocytes. Journal of Radiation Research. 2014;55(2):245–256. doi: 10.1093/jrr/rrt109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Civantos F. Photodynamic therapy for head and neck lesions in the subtropics. Journal of the National Comprehensive Cancer Network. 2012;10(Supplement 2):S-65–S-68. doi: 10.6004/jnccn.2012.0179. [DOI] [PubMed] [Google Scholar]
- 251.Indrasari S. R., Timmermans A. J., Wildeman M. A., et al. Remarkable response to photodynamic therapy in residual T4N0M0 nasopharyngeal carcinoma: a case report. Photodiagnosis Photodynamic Therapy. 2012;9(4):319–320. doi: 10.1016/j.pdpdt.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 252.Moghissi K. Where does photodynamic therapy fit in the esophageal cancer treatment jigsaw puzzle? Journal of the National Comprehensive Cancer Network. 2012;10(Supplement 2):S-52–S-55. doi: 10.6004/jnccn.2012.0176. [DOI] [PubMed] [Google Scholar]
- 253.Lindenmann J., Matzi V., Neuboeck N., et al. Individualized, multimodal palliative treatment of inoperable esophageal cancer: clinical impact of photodynamic therapy resulting in prolonged survival. Lasers in Surgery Medicine. 2012;44(3):189–198. doi: 10.1002/lsm.22006. [DOI] [PubMed] [Google Scholar]
- 254.Nyst H. J., Tan I. B., Stewart F. A., Balm A. J. Is photodynamic therapy a good alternative to surgery and radiotherapy in the treatment of head and neck cancer? Photodiagnosis Photodynamic Therapy. 2009;6(1):3–11. doi: 10.1016/j.pdpdt.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 255.Zhou Z., Song J., Nie L., Chen X. Reactive oxygen species generating systems meeting challenges of photodynamic cancer therapy. Chemical Society Reviews. 2016;45(23):6597–6626. doi: 10.1039/C6CS00271D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Rosin F. C. P., Teixeira M. G., Pelissari C., Correa L. Photodynamic therapy mediated by 5-aminolevulinic acid promotes the upregulation and modifies the intracellular expression of surveillance proteins in oral squamous cell carcinoma. Photochemistry and Photobiology. 2018;95(2):635–643. doi: 10.1111/php.13029. [DOI] [PubMed] [Google Scholar]
- 257.Cognetti D., Curry J. M., Gillenwater A. M., et al. A phase 2a, multicenter, open-label study of RM-1929 photoimmunotherapy in patients with recurrent head and neck cancer. International Journal of Radiation Oncology • Biology • Physics. 2018;100(5) doi: 10.1016/j.ijrobp.2017.12.158. [DOI] [Google Scholar]
- 258.Hwang H., Biswas R., Chung P. S., Ahn J. C. Modulation of EGFR and ROS induced cytochrome c release by combination of photodynamic therapy and carboplatin in human cultured head and neck cancer cells and tumor xenograft in nude mice. Journal of Photochemistry and Photobiology B: Biology. 2013;128:70–77. doi: 10.1016/j.jphotobiol.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 259.Xue K., Wang Y. N., Zhao X., Zhang H. X., Yu D., Jin C. S. Synergistic effect of meta-tetra (hydroxyphenyl)chlorin-based photodynamic therapy followed by cisplatin on malignant Hep-2 cells. OncoTargets and Therapy. 2019;12:5525–5536. doi: 10.2147/OTT.S198422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Lange C., Bednarski P. J. Evaluation for synergistic effects by combinations of photodynamic therapy (PDT) with temoporfin (mTHPC) and Pt (II) complexes carboplatin, cisplatin or oxaliplatin in a set of five human cancer cell lines. International Journal of Molecular Sciences. 2018;19(10) doi: 10.3390/ijms19103183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.He C., Liu D., Lin W. Self-assembled core-shell nanoparticles for combined chemotherapy and photodynamic therapy of resistant head and neck cancers. ACS Nano. 2015;9(1):991–1003. doi: 10.1021/nn506963h. [DOI] [PubMed] [Google Scholar]
- 262.Markman J. L., Rekechenetskiy A., Holler E., Ljubimova J. Y. Nanomedicine therapeutic approaches to overcome cancer drug resistance. Advanced Drug Delivery Reviews. 2013;65(13-14):1866–1879. doi: 10.1016/j.addr.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Mariadoss A. V. A., Vinayagam R., Senthilkumar V., et al. Phloretin loaded chitosan nanoparticles augments the pH-dependent mitochondrial-mediated intrinsic apoptosis in human oral cancer cells. International Journal of Biological Macromolecules. 2019;130:997–1008. doi: 10.1016/j.ijbiomac.2019.03.031. [DOI] [PubMed] [Google Scholar]
- 264.Zhang P., Wang P., Yan L., Liu L. Synthesis of gold nanoparticles with Solanum xanthocarpum extract and their in vitro anticancer potential on nasopharyngeal carcinoma cells. International Journal of Nanomedicine. 2018;13:7047–7059. doi: 10.2147/IJN.S180138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Wang Y., Zhang Y., Guo Y., et al. Synthesis of zinc oxide nanoparticles from Marsdenia tenacissima inhibits the cell proliferation and induces apoptosis in laryngeal cancer cells (Hep-2) Journal of Photochemistry and Photobiology B: Biology. 2019;201, article 111624 doi: 10.1016/j.jphotobiol.2019.111624. [DOI] [PubMed] [Google Scholar]
- 266.Sánchez-Rodríguez C., Palao-Suay R., Rodrigáñez L., et al. α-Tocopheryl succinate-based polymeric nanoparticles for the treatment of head and neck squamous cell carcinoma. Biomolecules. 2018;8(3) doi: 10.3390/biom8030097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Jin H., Zhu T., Huang X., et al. ROS-responsive nanoparticles based on amphiphilic hyperbranched polyphosphoester for drug delivery: light-triggered size-reducing and enhanced tumor penetration. Biomaterials. 2019;211:68–80. doi: 10.1016/j.biomaterials.2019.04.029. [DOI] [PubMed] [Google Scholar]
- 268.Satapathy S. R., Nayak A., Siddharth S., Das S., Nayak D., Kundu C. N. Metallic gold and bioactive quinacrine hybrid nanoparticles inhibit oral cancer stem cell and angiogenesis by deregulating inflammatory cytokines in p53 dependent manner. Nanomedicine. 2018;14(3):883–896. doi: 10.1016/j.nano.2018.01.007. [DOI] [PubMed] [Google Scholar]
- 269.Howard D., Sebastian S., Le Q. V., Thierry B., Kempson I. Chemical mechanisms of nanoparticle radiosensitization and radioprotection: a review of structure-function relationships influencing reactive oxygen species. International Journal of Molecular Sciences. 2020;21(2) doi: 10.3390/ijms21020579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Shi S., Zhang L., Zhu M., et al. Reactive oxygen species-responsive nanoparticles based on PEGlated prodrug for targeted treatment of oral tongue squamous cell carcinoma by combining photodynamic therapy and chemotherapy. ACS Applied Materials & Interfaces. 2018;10(35):29260–29272. doi: 10.1021/acsami.8b08269. [DOI] [PubMed] [Google Scholar]
- 271.Chang P.-Y., Peng S.-F., Lee C.-Y., et al. Curcumin-loaded nanoparticles induce apoptotic cell death through regulation of the function of MDR1 and reactive oxygen species in cisplatin-resistant CAR human oral cancer cells. International Journal of Oncology. 2013;43(4):1141–1150. doi: 10.3892/ijo.2013.2050. [DOI] [PubMed] [Google Scholar]
- 272.Francisco A.-C., Mar S.-A., Irene C., et al. Could radiotherapy effectiveness be enhanced by electromagnetic field treatment? International Journal of Molecular Sciences. 2013;14(7):14974–14995. doi: 10.3390/ijms140714974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Sanie-Jahromi F., Saadat M. Different profiles of the mRNA levels of DNA repair genes in MCF-7 and SH-SY5Y cells after treatment with combination of cisplatin, 50-Hz electromagnetic field and bleomycin. Biomedecine & Pharmacotherapy. 2017;94:564–568. doi: 10.1016/j.biopha.2017.07.115. [DOI] [PubMed] [Google Scholar]
- 274.Akbarnejad Z., Eskandary H., Dini L., et al. Cytotoxicity of temozolomide on human glioblastoma cells is enhanced by the concomitant exposure to an extremely low-frequency electromagnetic field (100 Hz, 100 G) Biomedecine & Pharmacotherapy. 2017;92:254–264. doi: 10.1016/j.biopha.2017.05.050. [DOI] [PubMed] [Google Scholar]
- 275.Vadalà M., Morales-Medina J. C., Vallelunga A., Palmieri B., Laurino C., Iannitti T. Mechanisms and therapeutic effectiveness of pulsed electromagnetic field therapy in oncology. Cancer Medicine. 2016;5(11):3128–3139. doi: 10.1002/cam4.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Sanie-Jahromi F., Saadat I., Saadat M. Effects of extremely low frequency electromagnetic field and cisplatin on mRNA levels of some DNA repair genes. Life Sciences. 2016;166:41–45. doi: 10.1016/j.lfs.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 277.Baharara J., Hosseini N., Farzin T. R. Extremely low frequency electromagnetic field sensitizes cisplatin-resistant human ovarian adenocarcinoma cells via P 53 activation. Cytotechnology. 2016;68(4):1403–1413. doi: 10.1007/s10616-015-9900-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Storch K., Dickreuter E., Artati A., Adamski J., Cordes N. BEMER electromagnetic field therapy reduces cancer cell radioresistance by enhanced ROS formation and induced DNA damage. PLoS One. 2016;11(12, article e0167931) doi: 10.1371/journal.pone.0167931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Jemal A., Siegel R., Xu J., Ward E. Cancer statistics, 2010. CA: A Cancer Journal for Clinicians. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 280.Mu J., Lin J., Huang P., Chen X. Development of endogenous enzyme-responsive nanomaterials for theranostics. Chemical Society Reviews. 2018;47(15):5554–5573. doi: 10.1039/C7CS00663B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Shin D. M., Ro J. Y., Hong W. K., Hittelman W. N. Dysregulation of epidermal growth factor receptor expression in premalignant lesions during head and neck tumorigenesis. Cancer Research. 1994;54(12):3153–3159. [PubMed] [Google Scholar]
- 282.Tao X., Lu Y., Qiu S., Wang Y., Qin J., Fan Z. AP1G1 is involved in cetuximab-mediated downregulation of ASCT2-EGFR complex and sensitization of human head and neck squamous cell carcinoma cells to ROS- induced apoptosis. Cancer Letters. 2017;408:33–42. doi: 10.1016/j.canlet.2017.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Yang Y., Tian Z., Ding Y., et al. EGFR-Targeted Immunotoxin Exerts Antitumor Effects on Esophageal Cancers by Increasing ROS Accumulation and Inducing Apoptosis via Inhibition of the Nrf2-Keap1 Pathway. Journal of Immunology Research. 2018;2018:10. doi: 10.1155/2018/1090287.1090287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Wang Z., Li C., Li Y., et al. DpdtbA-Induced Growth Inhibition in Human Esophageal Cancer Cells Involved Inactivation of the p53/EGFR/AKT Pathway. Oxidative Medicine and Cellular Longevity. 2019;2019:14. doi: 10.1155/2019/5414670.5414670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Crooker K., Aliani R., Ananth M., Arnold L., Anant S., Thomas S. M. A review of promising natural chemopreventive agents for head and neck cancer. Cancer Prevention Research. 2018;11(8):441–450. doi: 10.1158/1940-6207.CAPR-17-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Mohan A., Narayanan S., Sethuraman S., Krishnan U. M. Novel resveratrol and 5-fluorouracil coencapsulated in PEGylated nanoliposomes improve chemotherapeutic efficacy of combination against head and neck squamous cell carcinoma. Biomed Research International. 2014;2014:14. doi: 10.1155/2014/424239.424239 [DOI] [PMC free article] [PubMed] [Google Scholar]