

SUMMARY
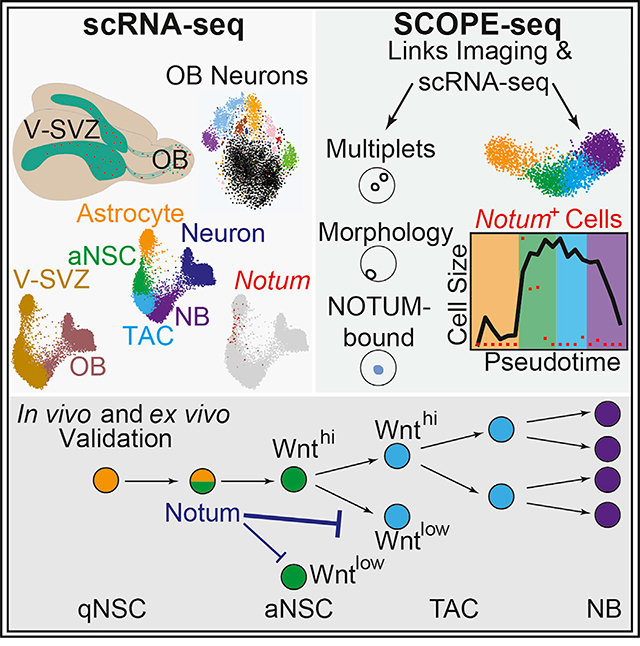
In the adult ventricular-subventricular zone (V-SVZ), neural stem cells (NSCs) generate new olfactory bulb (OB) neurons and glia throughout life. To map adult neuronal lineage progression, we profiled >56,000 V-SVZ and OB cells by single-cell RNA sequencing (scRNA-seq). Our analyses reveal the molecular diversity of OB neurons, including fate-mapped neurons, lineage progression dynamics, and an NSC intermediate enriched for Notum, which encodes a secreted WNT antagonist. SCOPE-seq technology, which links live-cell imaging with scRNA-seq, uncovers cell-size transitions during NSC differentiation and preferential NOTUM binding to proliferating neuronal precursors. Consistently, application of NOTUM protein in slice cultures and pharmacological inhibition of NOTUM in slice cultures and in vivo demonstrated that NOTUM negatively regulates V-SVZ proliferation. Timely, context-dependent neurogenesis demands adaptive signaling among neighboring progenitors. Our findings highlight a critical regulatory state during NSC activation marked by NOTUM, which attenuates WNT-stimulated proliferation in NSC progeny.
In Brief
Mizrak et al. combine single-cell RNA-seq, SCOPE-seq, and genetic fate mapping to study adult olfactory bulb neurogenesis from the ventricular-subventricular zone (V-SVZ). They reveal the key molecular and cellular changes during neuronal lineage differentiation and highlight an activating NSC intermediate marked by Notum, an extracellular signal that controls V-SVZ proliferation.
Graphical Abstract
INTRODUCTION
Neurogenesis persists in two principal regions of the adult mouse brain: the subgranular zone of the dentate gyrus and the ventricular-subventricular zone (V-SVZ) located in the walls of the lateral ventricles (Doetsch et al., 1999; Fiorelli et al., 2015; Mirzadeh et al., 2008). The V-SVZ contains both quiescent neural stem cells (qNSCs) and actively dividing neural stem cells (aNSCs) (Codega et al., 2014; Mich et al., 2014), with intrinsic regional identities defined during embryogenesis (Fuentealba et al., 2015), generating different olfactory bulb (OB) interneuron subtypes or glia depending on their location (Brill et al., 2009; Delgado and Lim, 2015; Kohwi et al., 2005; López-Juárez et al., 2013; Merkle et al., 2014, 2007; Mizrak et al., 2019; Ortega et al., 2013; Zweifel et al., 2018). V-SVZ qNSCs are specialized astrocytes with radial morphology (reviewed in Kriegstein and Alvarez-Buylla, 2009; Mirzadeh et al., 2008), and once activated, many of them divide symmetrically, resulting in their depletion (Obernier et al., 2018). aNSCs give rise to transit-amplifying cells (TACs), which generate neuroblasts (NBs). NBs migrate via the rostral migratory stream (RMS) to the OB (Figure 1A), where they terminally differentiate into interneurons. V-SVZ NSC-to-OB interneuron differentiation requires cells with a broad distribution of developmental states; however, the functional and cellular identities of key intermediate states and the gene expression programs pertinent to these transitions are unknown. Although recent efforts defined OB neuron subtypes (Eng et al., 2019; Tepe et al., 2018), a comprehensive characterization of the cellular states during V-SVZ to OB lineage progression and a classification of V-SVZ NSC-derived OB neurons are lacking and require the combination of single-cell RNA sequencing (scRNA-seq) with genetic fate mapping.
Figure 1. Fate-Mapping Experiments and OB Neuron Subtypes.

(A) Top: schema of adult mouse brain (left) showing the migration of V-SVZ neuronal precursors (red) to the OB. Coronal schema (right) highlighting the main OB layers (gl, glomerular layer; gr, granule cell layer; mi, mitral cell layer; epl, external plexiform layer; ipl, internal plexiform layer), anterior olfactory nucleus (aoni), and RMS, which terminates in the subependymal zone (RMS/SEZ). Bottom: four- and six-week-old GCERT2 and NESFLPO animals were injected with tamoxifen for three consecutive days and assayed four weeks after the last injection.
(B) Violin plots showing log2 (counts per million [CPM] + 1) expression values of neuron subtype markers (false discovery rate [FDR] < 0.05). Black and red circles indicate cell numbers and fraction of TdTom+ cells, respectively, in each cluster.
(C) Top: t-distributed stochastic neighbor embedding (t-SNE) of major OB neurons colored separately. Bottom: ISH images showing spatial distribution of subtype markers and their scaled expression on t-SNE.
(D) Coronal images showing the co-localization of endogenous TdTom and main OB neuron subtype markers. The corresponding OB layer was indicated on each image. Scale bars: 20 μm.
Intercellular communication in the V-SVZ is essential for proper adult neurogenesis but has not been fully explored at the single-cell level. γ-aminobutyric acid (GABA) released by NBs inhibits NSC proliferation (Alfonso et al., 2012; Liu et al., 2005), whereas contact-mediated NOTCH signaling between NSCs and TACs regulates NSC quiescence (Kawaguchi et al., 2013). WNT signaling plays a crucial role in orchestrating proliferation and differentiation of adult V-SVZ NSCs (Adachi et al., 2007; Azim et al., 2014; Ortega et al., 2013; Qu et al., 2010; Zywitza et al., 2018). Although these studies provide insights into the significance of WNT signaling in V-SVZ, they do not reveal the key WNT pathway regulators that modulate these interactions. In particular, WNT signaling can be negatively regulated by multiple families of secreted proteins that modulate WNT ligand activity (reviewed in Driehuis and Clevers, 2017). Therefore, these secreted negative regulators could facilitate adaptive signaling between NSCs and their progeny to cope with sustained WNT ligand exposure. However, the key secreted negative regulators of WNT signaling crucial for adult neurogenesis and their cellular origins and target cells are unknown.
Several scRNA-seq platforms (Klein et al., 2015; Macosko et al., 2015; Yuan and Sims, 2016) have been applied to address cellular heterogeneity and temporal gene expression changes in various stem cell contexts, including the V-SVZ (Mizrak et al., 2019; Zywitza et al., 2018); however, these studies lacked OB single-cell expression profiling and fate-mapping reporters and thus could not link V-SVZ NSCs to their progeny in the OB. Importantly, current high-throughput methods are unable to link live-cell imaging and scRNA-seq data from the same cell and cannot measure, for example, morphological features of individual cells. This is particularly problematic in applications of these technologies to stem cells, in which an intracellular reporter or a surface marker is often used to identify cells. In this study, we applied large-scale scRNA-seq of fate-mapped cells and an improved version of single-cell optical phenotyping and expression sequencing (SCOPE-seq) (Yuan et al., 2018) to directly link live-cell imaging with scRNA-seq and revealed the cellular dynamics of adult neurogenesis. We uncovered a rare intermediate NSC population enriched for Notum, which we demonstrate to be an important mediator of V-SVZ intercellular signaling.
RESULTS
Large-Scale Single-Cell Profiling of Adult Mouse OB with Fate-Mapping Reporters
We profiled >56,000 cells from matched V-SVZs and OBs of animals carrying either the well-established hGFAP::CreERT2; Rosa26LSL-TdTomato (GCERT2) (Ganat et al., 2006; Mich et al., 2014; Obernier et al., 2018) or the newer V-SVZ-enriched rat-Nes::FLPOER; Rosa26FSF-TdTomato (NESFLPO) fate-mapping reporter alleles (Lao et al., 2012; Petrova et al., 2013; Wojcinski et al., 2017) (Figure 1A; Figure S1A). The animals received tamoxifen at postnatal day (P) 28 or P42 for three consecutive days and were sacrificed at P56 or P70. Postnatal neurogenesis declines dramatically in 4-month-old adult mice (Daynac et al., 2016), and gradual temporal changes in subtype-specific OB neurogenesis were previously reported (Batista-Brito et al., 2008; Lemasson et al., 2005). Therefore, experiments were performed at two early time points with higher neuron output to avoid biasing our analysis toward particular OB neuron subtypes. NESFLPO specifically labels cells along the walls of the lateral ventricles, which generate TdTomato+ (TdTom+) OB cells four weeks after tamoxifen induction (Figure S1B). We identified ten major OB cell populations, including NBs and neurons (Levine et al., 2015; Shekhar et al., 2016) (Figure S1C). Morphological classification of OB neurons depends on overlapping markers (reviewed in Nagayama et al., 2014), whereas our subclustering of the >4,500 OB neurons revealed nine molecularly distinct subtypes (Figure 1B; Table S1). TdTom expression was detected in multiple neuron subtypes (64 TdTom+ neurons) (Figure S1D), with ≥50% of TdTom+ neurons in the abundant Neuron1 and Neuron2 (34 TdTom+ neurons), whereas the fraction of TdTom+ neurons was significantly enriched in Neuron3 (Figure 1B; Table S1). Gad1 and Gad2 expression was widespread in OB neurons, supporting their GABAergic identity with weaker expression in NBs (Lledo et al., 2008; Nagayama et al., 2014) (Figure 1B).
Next, we surveyed the Allen Mouse Brain Atlas In Situ Hybridization (ISH) database for subtype-specific markers (Table S1) to examine their spatial distribution in the multilayered OB. Using combinations of markers per subtype, we confirmed enrichment of Neuron1, Neuron2, and Neuron8 subtypes in the granule cell layer, whereas Neuron4, Neuron5, and Neuron6 markers showed enrichment in the glomerular layer (Figure 1C; Figure S1E). Neuron7 and Neuron9 markers were predominantly expressed in the anterior olfactory nucleus and external plexiform layer, respectively, and Neuron3 markers were enriched in the mitral cell layer and the external plexiform-glomerular layer boundary (Figure 1C). Consistent with scRNA-seq, immunostainings demonstrated TdTom co-localization with neuron subtype markers in their predetermined positions, validating fate-mapping reporter expression in the sparse OB neuron subtypes (Figure 1D).
Dissection of Neuronal Lineage Progression Revealed Gene Expression Features and Key Cellular Intermediates
To address the complexity of adult neuronal lineage progression and regulation, we generated force-directed visualizations (Weinreb et al., 2018) of neuronal lineage trajectories from V-SVZ astrocytes to OB neurons using both the NESFLPO and the GCERT2 datasets (12,334 and 7,903 cells, respectively). The resulting trajectories revealed that V-SVZ and OB NBs co-cluster, and lineage progression is constricted by two developmental transitions (Figure 2A; Table S2). To identify gene signatures associated with neuronal differentiation stages, we factorized the data with single-cell hierarchical Poisson factorization (scHPF) (Levitin et al., 2019) and projected cell scores from different factors onto the lineage trajectories. For each gene expression program or factor, scHPF computes a score for each cell and gene that indicates the strength of its association with a factor. Using scHPF, we identified signatures of two intermediate populations corresponding to factors 2 and 5, in addition to known gene expression programs (Figure 2B; Figure S2A; Table S2). In the OB, migrating NBs with high cell scores for factor 5 were marked by Prokr2 and Htr3a, genes for which mouse mutants impair NB migration to the OB (García-González et al., 2017; Wen et al., 2019). Notum, which encodes a secreted WNT inhibitor, was the highest-ranked gene in factor 2 and marked the V-SVZ transition between astrocyte and aNSC clusters. Factor 2 was also enriched for Ascl1 and Sfrp1 that encodes another secreted WNT inhibitor, distinctly separated from factor 1, and enriched for astrocyte genes such as Gfap, Slc1a3, and Cldn10 (Figure 2B).
Figure 2. Dissection of Neuronal Lineage Progression and Intermediate States.

(A) Force-directed neuronal lineage trajectories comprising V-SVZ astrocytes, aNSCs, TACs, V-SVZ NBs, OB NBs, and neurons in both NESFLPO and GCERT2. Each cell type and/or sample is colored separately.
(B) Cell scores for six scHPF factors are projected on force-directed lineage trajectories. Two high-scoring genes are highlighted for each factor. Genes marking the transition states are in red.
(C) Binomial enrichment of genes in astrocyte and aNSC clusters compared with the remaining neuronal lineage.
(D) Log2 (CPM+1) expression values of Cldn10, Notum, Ascl1, and Mcm2 in all GCERT2 (7,903 cells), NESFLPO (12,334 cells), and TdTom+ (349 cells) neuronal lineage cells isolated from GCERT2 and NESFLPO. Cells without marker detection are in gray.
(E) Representative enriched and depleted pathways (FDR < 0.01) in Notum+ neuronal lineage cells (NES, normalized enrichment score).
(F) Phenotypic volume analysis of neuronal lineage clusters (mean ± standard error of the mean).
(G) Average expression of the top five most specific OB neuron subtype markers (FDR < 0.01) (Table S1) in cell clusters.
To identify markers of the qNSC-aNSC transition, we analyzed genes enriched in astrocyte and aNSC clusters compared with the rest of the lineage, which revealed Notum as a top marker of the intermediate cells (Figures 2C and 2D). Notum expression preceded Ascl1 expression enriched by factor 2 in the neuronal lineage, suggesting additional heterogeneity (Figure 2D). To investigate the heterogeneity among the intermediate cells, we calculated the pointwise mutual information (PMI), a measure of co-expression, for all gene pairs. As expected, genes enriched by factor 2 had high PMI but also formed significantly distinct co-expression modules (Figure S2B), indicating multistep differentiation in the intermediate population and/or heterogeneity in NSC activation. Importantly, we observed Notum enrichment in V-SVZ endothelial cells (Figure S2C), consistent with previous reports of NOTUM expression in brain endothelial cells (Zhang et al., 2014). Next, we identified canonical pathways enriched in Notum+ cells in the neuronal lineage (Table S3) by gene set enrichment analysis (GSEA) (Kharchenko et al., 2014; Subramanian et al., 2005). The G1/S transition gene set was significantly upregulated, suggesting an advanced cell-cycle state in Notum+ NSCs (Figure 2E).
V-SVZ qNSCs are regionally heterogeneous, generating distinct OB interneurons depending on their location (reviewed in Chaker et al., 2016; Obernier and Alvarez-Buylla, 2019). We identified several main OB neuron subtypes expressing the fate-mapping reporter (Figures 1B–1D); however, it is unknown whether gene expression heterogeneity is preserved along neuronal lineage progression. To test this, we calculated the phenotypic volume of each cluster in the trajectory, a metric for cellular heterogeneity (Azizi et al., 2018). Astrocytes formed the most heterogeneous cluster (Figure 2F), partly because of the inclusion of qNSCs and niche astrocytes in this cluster, whereas aNSC and TACs were more uniform. We observed an increase in cellular heterogeneity in NBs and OB neurons (Figure 2F). Next, we plotted the expression of top markers of neuron subtypes with a higher fraction of TdTom+ cells (Figure 1B; Table S1). Interestingly, these subtype-specific gene signatures varied in the apparent timing of emergence during the lineage progression. The expression of Neuron2 and Neuron3 markers was particularly enriched in OB NBs, compared with NSCs and TACs (Figure 2G), suggesting an activation of a terminal differentiation program at the NB stage, whereas we did not detect traces of Neuron9 markers in NBs as expected or Neuron7 markers located in anterior olfactory nucleus (Figure 1C). Newborn neurons were previously reported in the anterior olfactory nucleus (Shapiro et al., 2009) and other olfactory processing areas (Feliciano et al., 2015); however, the migration and the maturation status of these cells is not fully clear, and our data suggest a lack of connection between V-SVZ and OB NBs and Neuron7.
SCOPE-Seq Profiling Reveals Cell-Size Dynamics along Neuronal Lineage Progression and a Bias of NOTUM toward Proliferating Cells
We next sought to determine the spatial arrangement of NOTUM-expressing cells in the V-SVZ. Immunostainings using a Nestin reporter mouse that expresses CFP under the control of the rat Nestin promoter (Nestin-CFP) (Mignone et al., 2004; Wojcinski et al., 2017) revealed strong NOTUM labeling, particularly in the ventral V-SVZ (Figure 3A; Figure S2D), consistent with Notum enrichment in that region (Mizrak et al., 2019). We also observed NOTUM labeling away from the ventricular surface, likely in endothelial cells, which are strongly enriched in Notum expression compared with other brain cell types (Zhang et al., 2014), and peripheral endothelial cells (Munji et al., 2019). Whole-mount staining of the V-SVZ in Nestin-CFP animals (Mirzadeh et al., 2008, 2010) showed NOTUM labeling on the ventricular surface with partial co-localization with GFAP, which marks many V-SVZ qNSCs and some aNSCs (Codega et al., 2014; Mich et al., 2014; Pastrana et al., 2009) (Figure 3B). To further characterize V-SVZ neuronal lineage progression and the Notum+ intermediate, we performed SCOPE-seq profiling of >2,500 V-SVZ neuronal lineage cells. SCOPE-seq (Yuan et al., 2018) uses optically decodable mRNA capture beads to link imaging of dissociated single cells (e.g., cell size, surface staining, and multiplet detection) captured in microwells with their scRNA-seq profiles (Figure 3C; Table S4). The neuronal lineage trajectory generated from SCOPE-seq after multiplet removal recapitulated the lineage progression order and Notum expression in the intermediate population (Figure 3C; Table S1). Changes in cell size among V-SVZ neuronal progenitors have been reported in long-term cultured (Costa et al., 2011) and dissociated V-SVZ cells (Codega et al., 2014). When optically decoded cells in the neuronal lineage (737/2,888 cells) were analyzed (Gunderson et al., 2004; Yuan et al., 2018), we observed a significant increase in cell size at the early aNSC stage (Figure 3D). This imaging-based intermediate stage clearly overlapped with Notum-expressing cells (Figure 3D). Another significant shift in cell size was observed in the early-to-late NB transition with a decrease in late NB size, likely because of cell-cycle exit. Given the enrichment of G1/S transition gene signatures in Notum+ cells, the SCOPE-seq data demonstrate dynamic cell-size control during V-SVZ NSC differentiation, in which Notum marks activating NSCs with an intermediate cell size between the cells in astrocyte and those in aNSC clusters.
Figure 3. SCOPE-Seq Profiling of V-SVZ Neuronal Lineage Cells.

(A) Coronal image of ventral V-SVZ showing NOTUM, Nestin-CFP, and GFAP labeling. Nestin-CFP expression was detected using anti-GFP staining. Nestin-CFP reliably labels most ventricle-contacting cells, including NSCs and ependymal cells. Scale bars: 20 μm.
(B) Whole-mount staining showing NOTUM labeling in ventricle-contacting cells (lateral wall). Cyan arrowhead, NOTUM+GFAP+ cells; white, NOTUM−GFAP+ cells; yellow, NOTUM+GFAP− cells. Scale bars: 20 μm.
(C) Top: workflow for SCOPE-seq using optically barcoded mRNA capture beads. Following barcoded cDNA synthesis, optical demultiplexing identifies the barcode on each bead. Scale bar: 20 μm. Bottom: pseudotemporal ordering of V-SVZ neuronal lineage cells, and Notum marking the astrocyte-aNSC transition. Astrocyte, orange; aNSC, green; TAC, blue; NB, purple.
(D) Average size of pseudotemporally ordered neuronal lineage cells and the fraction of Notum+ cells in each pseudotime bin. Shading color corresponds to the stages of differentiation labeled in (C).
(E) Gene signatures of NOTUM-bound cells showing significant enrichment in aNSC and TAC.
(F) Box plot of GSEA data showing the enrichment or depletion of WNT signaling pathway in individual cells and the quantification of enrichment scores in each cluster (unpaired t test, p < 0.001).
As a secreted WNT antagonist, NOTUM is active in the extracellular space (Zhang et al., 2015), it but can also be retained at the cell surface via binding to glycosaminoglycans on glypicans (Kakugawa et al., 2015). We used SCOPE-seq to identify NOTUM-bound optically decoded cells (45/737 cells) in the neuronal lineage based on anti-NOTUM surface labeling (Figure S3A). These cells showed enrichment of aNSC and TAC cluster gene signatures compared with astrocytes and NBs (Figure 3E). This result was consistent with the flow cytometric quantification showing stronger NOTUM surface labeling on cells with higher epidermal growth factor (EGF)-ligand binding affinity (Codega et al., 2014) compared with Nestin-CFP+ EGF binding− cells, containing V-SVZ qNSCs (unpaired t test, p < 0.01) (Figure S3B). GSEA showed significant enrichment of WNT signaling activity in aNSCs and TACs (Figure 3F), suggesting actively dividing V-SVZ progenitors as potential targets of secreted WNT inhibitors, including NOTUM.
Modulation of NOTUM Activity Causes Aberrant Adult Neurogenesis
Given the context-dependent roles of WNT signaling in the V-SVZ (Adachi et al., 2007; Azim et al., 2014; Ortega et al., 2013; Qu et al., 2010; Zywitza et al., 2018), we next examined the function of NOTUM in V-SVZ regulation. We modulated NOTUM activity in the V-SVZ using either a selective small molecule inhibitor (ABC99) (Suciu et al., 2018) or a recombinant mouse NOTUM protein. We first investigated the effects of these perturbations ex vivo in organotypic slice cultures from 7- to 8-week-old Nestin-CFP reporter mice (Mignone et al., 2004; Wojcinski et al., 2017) treated with vehicle (saline), recombinant mouse NOTUM protein, ABC99, or the inactive control compound ABC101 (Figure 4A). After five days of continuous treatment in serum-free media supplemented with EGF, slices were treated with EdU and fixed (Figure 4A). We observed a significant increase in proliferating (EdU+) V-SVZ progenitors in the ABC99-treated slices (Figures 4B, 4C, and 4E), whereas addition of NOTUM protein suppressed V-SVZ proliferation. ABC99 treatment resulted in a dramatic expansion of the ventral V-SVZ with significant spreading of EdU+ cells toward brain parenchyma (Figures 4B, 4D, and 4E). It is possible that the absence of proper migratory cues in organotypic slice cultures caused the ventral V-SVZ progenitors to expand as a group toward the brain parenchyma in ABC99-treated slices rather than migrating along the RMS as they would in vivo. In summary, these experiments demonstrate a direct inhibitory effect of NOTUM on V-SVZ proliferation, particularly in the ventral V-SVZ, in which NOTUM levels are enriched (Figure 3A) (Mizrak et al., 2019).
Figure 4. Modulation of NOTUM Activity Causes Aberrant Adult Neurogenesis.

(A) Left: schema of organotypic slice cultures. Slices were treated with vehicle (DMSO), recombinant mouse NOTUM protein (rmNOTUM), ABC101 (inactive compound), or ABC99 (NOTUM inhibitor) for five consecutive days. LV, lateral ventricle; LW, lateral wall; SW, septal wall; CC, corpus callosum.
(B) Coronal schema of the V-SVZ showing the location of the images in (E), (I), and (J).
(C) Quantification of EdU+ proliferating cells per V-SVZ area (in square millimeters) in brain slices treated with vehicle, NOTUM protein, ABC101, and ABC99 (n = 4 replicates, mean ± standard deviation [SD], p < 0.01, unpaired t test).
(D) Density plot showing the distance of EdU+ cells within each sample from the midline of the lateral ventricle. ABC99-treated slices were significantly disorganized and contained many EdU+ V-SVZ cells, which migrated away from the ventricle (p < 0.01, Kolmogorov-Smirnov test).
(E) Rostro-caudal level matched images of the control or the inhibitor-treated brain slices. Scale bars: 100 μm.
(F) Schema showing i.p. injection regimen. D–2 denotes the first injection day, whereas D0 indicates the last injection day.
(G) Normalized anti-GFP staining intensity in each quantified cell population.
(H) Quantification of GFAP+EdU−, GFAP+EdU+, GFAP−EdU+, and DCX+ cells per V-SVZ area (in square millimeters), showing a significant increase in NSC progeny (p < 0.01, unpaired t test), but not in GFAP+EdU−, and GFAP+EdU+ NSCs in ABC99-injected animals at 1 dpi (n = 3 replicates, mean ± SD, p < 0.01, unpaired t test).
(I) Representative level-matched images from ventral V-SVZ (B), showing the increase in DCX+ cells in ABC99-treated animals. Scale bars: 20 μm.
(J) GFAP and EdU stainings of level-matched mid-V-SVZ (B) showing the increase in EdU+ cells in ABC99-treated animals. Scale bars: 20 μm.
Next, ABC99 or ABC101 was injected intraperitoneally (i.p.) into Nestin-CFP mice for three consecutive days to assess the effects of NOTUM inhibition in vivo. Animals were sacrificed one day after the last injection (1 dpi) (Figure 4F). Short-term systemic ABC99 administration showed no obvious adverse effects on animal health, as previously reported (Pentinmikko et al., 2019). To measure NOTUM activity and ensure that systemic treatment inhibits NOTUM activity in the V-SVZ, we used mass spectrometry-based activity-based protein profiling (MS-ABPP) (reviewed in Cravatt et al., 2008) and developed a targeted liquid chromatography-tandem mass spectrometry (LC-MS/MS) method to detect NOTUM-derived peptides. We detected significant NOTUM activity in the V-SVZ (Table S5) and a 40%–50% inhibition of V-SVZ NOTUM activity after ABC99 injections (Figure S3C). We confirmed WNT pathway activation in the V-SVZ following NOTUM inhibition via qRT-PCR of Axin2 in microdissected V-SVZ tissue from ABC99- or ABC101-injected controls at 1 dpi (unpaired t test, p < 0.05) (Figure S3D). Thus, ABC99 is able to reach to brain and inhibit WNT signaling without an adverse effect.
To understand the effects of NOTUM inhibition in vivo, we quantified different NSC populations and their progeny following ABC99 and ABC101 treatments. Immunostaining of different rostro-caudal levels of the V-SVZ (Figure S3E) revealed a robust increase in EdU+ progenitors, as well as doublecortin-expressing (DCX+) NBs in the ABC99-treated animals at 1 dpi compared with ABC101-treated mice (Figures 4B and 4H–4J). In addition to the labeling of ventricle-contacting NSCs and ependymal cells (Figure 3A), we found that the staining intensity for Nestin-CFP (anti-GFP staining) was graded, with stronger labeling in GFAP+ NSCs (p < 0.01, unpaired t test), and significantly weaker expression in NBs, reflecting decreasing perdurance of CFP in progeny of Nestin-expressing NSCs (Figure 4G). We did not detect significant change in the abundance of GFAP+EdU− NSCs used as a proxy for qNSCs following ABC99 treatment. Although we observed a significant increase in GFAP−EdU+ intermediate progenitors (Figure 4H), the abundance of GFAP+EdU+ NSCs was not significantly different between the two conditions, suggesting a more localized impact of NOTUM inhibition on transit-amplifying NSC progeny. The increase in NBs following ABC99 treatment was noticeably higher in the ventral V-SVZ, in which NOTUM expression is enriched (Figure 3A), and resulted in pockets of irregular NB expansion (Figures 4B and 4I). NB expansion upon Notum inhibition is likely, partly due to the increase in the number of intermediate progenitors, because they are upstream of NBs in the V-SVZ hierarchy (reviewed in Chaker et al., 2016; Ponti et al., 2013) (Figure 2; Figure S3G). However, we do not rule out the possibilities that NOTUM inhibition may affect V-SVZ NB migration or cause premature NB production with defective migratory properties, leading to their increased accumulation. Altogether, consistent with the slice culture findings and SCOPE-seq data, our in vivo data show that NOTUM-mediated inhibition of WNT signaling is more specific to proliferating neuronal progenitors and indicates NOTUM prevents their overexpansion in the V-SVZ.
DISCUSSION
Regulation of lineage progression from adult NSCs to OB neurons is achieved by stringent molecular checkpoints to ensure accurate cell production and to prevent excessive progenitor activation that could lead to stem cell depletion or pathogenic tissue overgrowth. The mouse adult V-SVZ is continuously exposed to proliferative WNT ligands in cerebrospinal fluid (Lehtinen et al., 2011) and the V-SVZ niche (Figure S3F). Most adult V-SVZ NSCs divide symmetrically (Obernier et al., 2018), making them prone to depletion. Therefore, molecular mechanisms must be in place to regulate proliferative signals in the niche (e.g., WNT ligands) to ensure robust neurogenesis. We used scRNA-seq and SCOPE-seq to identify a negative regulator of WNT signaling, Notum, as a marker of an NSC intermediate. We demonstrated that NOTUM levels must be tightly controlled to maintain normal neurogenesis in the adult V-SVZ. We propose that as poised qNSCs are activating, they release NOTUM to inhibit proliferation of nearby NSC progeny to attenuate their expansion (Figure S3G), thus possibly providing a favorable environment for their daughter cells.
Our analysis also revealed substantial changes in cell size at astrocyte-aNSC and early NB-to-late NB transitions (Figure 3D). Coordination of cell cycle and volume is essential to maintain cellular homeostasis. In particular, progressions through G1/S and G2/M phases are size dependent (Jones et al., 2017). It will be important to investigate molecular mechanisms underlying the cell-size changes in NSCs, such as nutritional state, translational activation (Llorens-Bobadilla et al., 2015), and circadian clock regulation, as well as their reversibility in the intermediate populations, and to determine the critical size thresholds for NSC activation and cell-cycle exit.
We defined nine molecularly distinct OB neuron subtypes and validated fate-mapping reporter expression in several subtypes. Analysis of neuronal trajectories revealed a convergence of heterogeneous astrocytes to more uniform aNSCs and TACs, and late branching during NB to neuron differentiation. These dynamics suggest cell-intrinsic programming, such as epigenetic heterogeneity among aNSCs and TACs. For example, diverse sets of transcription factors expressed in regionally distinct qNSCs (reviewed in Chaker et al., 2016) may establish epigenetic memory that is maintained in aNSCs and TACs and later contributes to a specification event at the NB stage. Future epigenomic characterization of V-SVZ NSCs and their progeny may address this complexity.
We also used an improved version of SCOPE-seq, which enables direct imaging of protein surface binding via detection of fluorescently labeled cells, thus avoiding flow sorting, which can significantly reduce cell quality and RNA content. SCOPE-seq can also be used to detect cells expressing intracellular reporters, such as lineage tracing markers, while simultaneously providing morphological insight, such as cell size. Developmental trajectories of different germinal regions consisting of cells with a broad distribution of imaging features (e.g., cell size, shape, intracellular reporter expression, and surface marker expression detected with antibodies) can be greatly enhanced using SCOPE-seq to identify different stem cell states.
Elucidating the mediators of intercellular communication in stem cell niches is essential to better understanding the mechanisms of stem cell regulation and to developing strategies for tissue regeneration. In this study, we identified an intermediate NSC population using scRNA-seq to which we attribute a key functional role. Transitioning stem cells represent the dynamic nature of the V-SVZ niche. Because they are primed to generate multiple progenitors, their communication with the NSC progeny already in the niche is critical for efficient neurogenesis. Similar strategies may also be employed by stem cell intermediates in other adult tissues, ensuring on-demand cell replenishment.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-GFP | Nacalai Tesque | Cat#04404-84; RRID: AB_10013361 |
| Chicken polyclonal anti-GFAP | Abcam | Cat#ab4674; RRID: AB_304558 |
| Chicken polyclonal anti-TH | Millipore | Cat#ab9702; RRID: AB_570923 |
| Goat polyclonal anti-DCX | Santa Cruz Biotechnology | Cat#sc-8066; RRID: AB_2088494 |
| Rabbit polyclonal anti-CALB2 | Millipore Sigma | Cat#ab5054; RRID: AB_2068506 |
| Rabbit polyclonal anti-EOMES | Millipore Sigma | Cat#ab2283; RRID: AB_10806889 |
| Mouse monoclonal anti-PVALB | Millipore Sigma | Cat#mab1572; RRID: AB_2174013 |
| Rabbit polyclonal anti-DOT1L | Abcam | Cat#ab64077; RRID: AB_2095412 |
| Mouse monoclonal anti-RELN | Millipore Sigma | Cat#mab5364; RRID: AB_2179313 |
| Rabbit polyclonal anti-NOTUM | Sigma | Cat#abs762 |
| Rat monoclonal anti- mouse CD14 PECy7 | BioLegend | Cat#1223315; RRID: AB_10641133 |
| Mouse seroblock FcR | BioRad | Cat#Buf041A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Papain | Worthington | Cat#LS003119 |
| PIPES | Sigma | Cat#P1851 |
| Ovomucoid | Worthington | Cat#LS003087 |
| Percoll | Sigma | Cat#P1644 |
| Calcein AM | Invitrogen | Cat#C3100MP |
| 2-Mercaptoethanol | Fisher Scientific | Cat#BP176-100 |
| Mouse recombinant EGF | Thermofisher | Cat# PMG8041 |
| Alexa Fluor 647 EGF | Thermofisher | E35351 |
| Recombinant Mouse Notum Protein | R&D Systems | Cat # 9150-NO-050 |
| N2 supplement | Life Sciences | 17502048 |
| B27 supplement | Life Sciences | 12587-010 |
| ABC99 | Suciu et al., 2018 | The Cravatt Lab |
| ABC101 | Suciu et al., 2018 | The Cravatt Lab |
| Critical Commercial Assays | ||
| Nextera XT DNA Library Preparation Kit | Illumina | Cat#FC-131-1024 |
| Qubit dsDNA HS Assay Kit | ThermoFisher | Cat#Q32854 |
| High Sensitivity DNA chips kit | Agilent Technologies | Cat#5067-4626 |
| NextSeq 500/550 High Output v2 kit (75 cycles) | Illumina | Cat#FC-404-2005 |
| SOLAμ™ SPE Plates | Thermofisher | Cat#60209-001 |
| EASY-Spray column | Thermofisher | Part# ES800 |
| DC™ Protein Assay Kit II | BioRad | Cat#5000112 |
| Millicell Cell Culture Insert | Millipore Sigma | Cat# PICM0RG50 |
| Deposited Data | ||
| Raw and processed data | This manuscript | GEO: GSE134918 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 00664 |
| Mouse: B6.Cg-Tg(GFAP-cre/ERT2)505Fmv/J | The Jackson Laboratory | JAX: 012849 |
| Mouse: B6;129S6-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze | The Jackson Laboratory | JAX: 007908 |
| Mouse: ratNes::FLPOER | The Joyner lab | Wojcinski et al., 2017 |
| Mouse: Rosa26FRT-STOP-FRT-TdTomato | The Jackson Laboratory | JAX: 021875 |
| Mouse: Nestin-CFP | The Joyner lab | Wojcinski et al., 2017 |
| Oligonucleotides | ||
| Template Switching Oligonucleotide AAGCAGTGGTATCAACGCAGAGTGAATrGrGrG | Macosko et al., 2015 | IDT |
| Gapdh primer F 5′- CCAAGGTGTCCGTCGTGGATCT-3′ | https://pga.mgh.harvard.edu/primerbank/ | IDT |
| Gapdh primer R 5′- GTTGAAGTCGCAGGAGACAACC-3′ | https://pga.mgh.harvard.edu/primerbank/ | IDT |
| Axin2 primer F 5′-TGACTCTCCTTCCAGATCCCA-3′ | https://pga.mgh.harvard.edu/primerbank/ | IDT |
| Axin2 primer R 5′-TGCCCACACTAGGCTGACA-3′ | https://pga.mgh.harvard.edu/primerbank/ | IDT |
| Software and Algorithms | ||
| Phenograph | Levine et al., 2015 | https://github.com/jacoblevine/PhenoGraph |
| Force-directed graphs | Weinreb et al., 2018 | https://github.com/AllonKleinLab/SPRING/ |
| t- distributed stochastic neighbor embedding (t-SNE) | Van der Maaten and Hinton, 2008 | https://lvdmaaten.github.io/tsne/ |
| Rstudio | R Studio Team (2015) | https://www.rstudio.com |
| Gene Set Enrichment Analysis | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| SCDE | Kharchenko et al., 2014 | http://hms-dbmi.github.io/scde/ |
| scHPF | Levitin et al., 2019 | https://github.com/simslab/scHPF |
| Skyline | https://skyline.ms/project/home/begin.view?/ | |
| Other | ||
| DNase I | Worthington | Cat#LS002139 |
| Drop-seq beads | ChemGenes | Cat# MACOSKO-2011-10 |
| SCOPE-seq beads | ChemGenes | Custom Beads |
| Maxima H Minus Reverse Transcriptase | ThermoFisher | Cat#EP0752 |
| Exo-I | New England Biolabs | Cat#M0293L |
| SUPERaseIN | ThermoFisher | Cat#AM2696 |
| KAPA HotStart ReadyMix | Kapabiosystems | Cat#KK2602 |
| Ampure XP beads | Beckman Coulter | Cat#A63881 |
| Buffer TCL | QIAGEN | Cat#1031576 |
| miRNeasy RNA isolation kit | QIAGEN | Cat#217004 |
| iScript cDNA preparation kit | BioRad | Cat#1708890 |
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter A. Sims (pas2182@cumc.columbia.edu).
Materials Availability
SCOPE-seq beads are a custom synthesis product from Chemgenes. This study did not generate any other new unique reagents or mouse lines.
Data and Code Availability
The accession number for the sequencing data and count matrices reported in this paper is GEO: GSE134918. Data analysis code used for scRNA-seq is available at https://github.com/simslab.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All experiments were performed according to protocols approved by IACUC at the Columbia University and the Memorial Sloan Kettering Cancer Center. Animals were given access to food and water ad libitum and were housed on a 12 hr light/dark cycle, and sacrificed at the same time of day. The following mouse lines were used for the experiments: Nestin-CFP (Mignone et al., 2004; Wojcinski et al., 2017), B6.Cg-Tg(GFAP-cre/ERT2)505Fmv/J mice (Stock no: 012849, The Jackson Laboratories) (Ganat et al., 2006), Nestin-FlpoER (Lao et al., 2012; Petrova et al., 2013; Wojcinski et al., 2017), Rosa26LSL-TdTomato (Ai14, Stock: 007909, The Jackson Laboratories), Rosa26FRT-STOP-FRT-TdTomato derived from Ai65 (Stock no: 021875, The Jackson Laboratories), and C57BL/6J (Stock no: 00664, The Jackson Laboratories). For TdTomato induction, four- or six-weeks old mice were injected with 100 mg/kg of tamoxifen (Sigma) intraperitoneally (IP) for three consecutive days, and sacrificed four weeks after the last injection. ABC99 and ABC101 were injected at 10mg/kg for three consecutive days (Related to Figure 4). Subcutaneous EdU (50 mg/kg) injections were done one hour before the animals were sacrificed. For in vivo and ex vivo immunostainings, equal numbers of male and female mice were used for each condition. Matched V-SVZ and OB scRNA-seq experiments were performed on the following animals at the indicated time-points: male P56 V-SVZ NESFLPO (1 mouse); male P70 V-SVZ NESFLPO (1 mouse); male P56 OB NESFLPO (1 mouse); male P70 OB NESFLPO (1 mouse); male P56 V-SVZ GCERT2 (1 mouse); female P56 V-SVZ GCERT2 (1 mouse); male P70 V-SVZ GCERT2 (1 mouse); female P70 V-SVZ GCERT2 (1 mouse); male P56 OB GCERT2 (1 mouse); female P56 OB GCERT2 (1 mouse); male P70 OB GCERT2 (1 mouse); female P70 OB GCERT2 (1 mouse) (Related to Figure S1). SCOPE-seq experiments were performed on male C57BL/6J animals (2 mice) (Related to Figure 3).
METHOD DETAILS
Single cell RNA-sequencing sample preparation
Whole-mounts of the lateral and septal walls of the V-SVZ were dissected as previously described (Mirzadeh et al., 2010; Mizrak et al., 2019), and combined prior to dissociation. Olfactory bulb (OB) was also removed to profile V-SVZ and OB from the same animal. For scRNA-seq experiments, sex and age of each animal, and the number of single cells analyzed are indicated in the Figure S1. Dissected whole mounts were dissociated as described previously (Mizrak et al., 2019). Briefly, minced V-SVZ pieces were digested with papain (Worthington, 6 mg per sample, 10 min at 37°C) in PIPES solution (120 mM NaCl, 5 mM KCl, 20 mM PIPES (Sigma), 0.45% glucose, 1× Antibiotic/Antimycotic (GIBCO), and phenol red (Sigma) in water; pH adjusted to 7.6). After trituration to single cells in DMEM/F12 containing ovomucoid (Worthington, 0.7 mg/ml) and DNase (Worthington, 0.5 mg/ml), the cell suspension was layered on top of 22% Percoll (Sigma) and centrifuged for 10 mins at 4°C without brakes to remove debris and myelin. The single cell suspension was washed by two rounds of centrifugation (1300 rpm at 4°C) and resuspended in 1× Tris Buffered Saline. Cells were stained with Calcein AM live stain dye (Fisher, 1:500) on ice for 30 minutes, and were passed through a 40 μm cell strainer (Fisher) to remove any cell clumps before loading.
Single cell library preparation and sequencing
Single cell capture and reverse transcription (RT) were performed as previously described (Yuan and Sims, 2016). Cells were loaded in the microwell devices at a concentration of 400–1000 cells per μl. Cell capture, lysis (2-Mercaptoethanol: Fisher Scientific, Buffer TCL: QIAGEN), and reverse transcription (Maxima H Minus Reverse Transcriptase: ThermoFisher, SUPERaseIN: Thermofisher, Template switch oligo: IDT) were all performed on 150K microwell devices (microwell size: x: 50 μm, y: 50 μm, z: 58 μm) controlled by an automated microfluidics system. The devices were scanned on a fluorescence microscope (Eclipse Ti-U, Nikon) during the RNA capture step to check for lysis efficiency. RT reactions were performed on Drop-seq beads (MACOSKO-2011–10, ChemGenes) also captured in the microwells. Beads were collected from the devices and pooled for cDNA amplification step. Following exonuclease I treatment (New England Biolabs), cDNA was amplified by PCR for 14 cycles (KAPA HotStart ReadyMix, Kapabiosystems), and used as input for Nextera tagmentation reactions. cDNA and library quality was assessed using both Qubit and Bioanalyzer (Agilent Technologies). All DNA purification steps were carried out using Ampure XP beads (Beckman). High-quality samples were sequenced on a NextSeq 500 sequencer using a 75 cycle High Output Kit (Illumina, 21 cycle read 1, 63 cycle read 2, and 8 cycle index read).
Immunohistochemistry, Microscopy, and Image Analysis
8–10 week-old animals were anesthetized, and transcardially perfused with PBS followed by 4% PFA and brains were post-fixed in 4% PFA for 24 hours. After fixation, brains were treated with 30% sucrose until they are embedded in OCT (Tissue-Tek) for cryosectioning. Sections were prepared at 18 μm on cryostat (Leica, CM3050S). Slice cultures were fixed in 4% PFA at end point, and stained at floating sections. Health of the slices was determined based on EdU (Click-IT) incorporation. For immunofluorescent analysis, sections (cryo and slice) were washed with 1X PBS and blocked with blocking buffer (5% BSA in PBS with 0.1% (or 0.3% for slice cultures) Triton-X). Primary antibodies were provided in blocking buffer overnight at 4°C. Washes were performed in PBS with 0.1% Triton-X (or 0.3% for slice cultures). Alexa Fluor conjugated secondary antibodies (Life sciences was supplemented 1/500 in blocking for 1–2 hours at room temperature. After the final washes, EdU staining was performed using Click-IT by manufacturer’s protocol. Hoechst 33258 or DAPI (Invitrogen) was used as counter stain. Slides were mounted using Fluorogel Mounting medium (Electron Microscopy Sciences). The following antibodies were used: rat anti-GFP (1:1000, Nacalai Tesque 04404–84), chicken anti-GFAP (1:1000, Abcam ab4674), rabbit anti-NOTUM (1:500, Sigma abs762), goat anti-DCX (1:500, Santa Cruz sc-8066), chicken anti-TH (1:400, Millipore ab9702), mouse anti-RELN (1:200, Millipore mab5364), rabbit anti-DOT1L (1:500, ab64077), rabbit anti-EOMES (1:500, ab2283), rabbit anti-CALB2 (1:200, Millipore ab5054), and mouse anti-PVALB (1:500, mab1572).
Images were taken using a Zeiss LSM 880 Confocal Microscope or a DM6000 Leica fluorescent microscope. For quantifications of in vivo stainings, we analyzed sections at three different rostral/caudal levels (from Bregma +1.45 mm to Bregma 0.0 mm) from three replicates (Figure S3E). The quantifications from these three levels were averaged to find the total V-SVZ percentages per replicate. Cells in the entire dorsal-ventral extent of V-SVZ were quantified using Fiji software (Schindelin et al., 2012). The identical quantification method was applied to all stainings, replicates and conditions using rostro-caudal level matched sections. Organotypic slice culture quantifications were performed on four V-SVZ replicates. Two-way statistical comparisons were conducted using two-tailed unpaired t test on three or four biological replicates, which is similar to sample sizes employed in the field. All quantification data was presented as mean+/−standard deviation. For anti-GFP staining intensity measurements, GFP staining intensity for each cell nucleus was measured using high magnification images, and normalized to the average GFP staining intensity of all Nestin-CFP+ cells in the same section.
Quantitative RT-PCR and Flow Cytometry
ABC99 or ABC101 were injected at 10mg/kg for three consecutive days and the animals were sacrificed at 1 dpi. RNA was isolated from microdissected V-SVZ lateral walls using miRNeasy RNA isolation kit (QIAGEN). cDNA was prepared using iScript cDNA preparation kit (BioRad). Following qRT-PCR (Applied BioSystems, Step One) using primers against Axin2, relative gene expression levels were quantified using ddCT method. Gapdh was used as the housekeeping control to normalize the gene expression. The following primers were used:
Axin2 F 5′-TGACTCTCCTTCCAGATCCCA-3′
Axin2 R 5′-TGCCCACACTAGGCTGACA-3′
Gapdh F 5′- CCAAGGTGTCCGTCGTGGATCT-3′
Gapdh R 5′- GTTGAAGTCGCAGGAGACAACC-3′
For flow cytometry, V-SVZ lateral walls from 8–10 weeks old NestinCFP animals were dissected, and the single cell suspensions were prepared as described above. Following the Percoll gradient, cells were washed by centrifugation (1300 rpm at 4°C). All staining and washes were performed on ice in 1% BSA, 0.1% Glucose HBSS. After each staining step, cells were washed twice by centrifugation (1300 rpm at 4°C). Cells were first incubated in mouse seroblock FcR (1:100, BioRad buf041A) for 10 minutes to avoid nonspecific binding of the antibodies. Cells were then incubated for 15 minutes in anti-NOTUM (1:50), followed by Alexa Fluor 555 secondary antibody staining (1:2000). Finally, cells were stained for 15 minutes with PE-Cy7 conjugated rat anti-CD14 (1:50, Biolegend 123315), and Alexa Fluor 647 conjugated EGF (1:250, Thermofisher E35351). At high concentrations, NOTUM antibody non-specifically interacts with Fc receptors on macrophage, monocyte, microglia, lymphocytes, which can be efficiently blocked with the mouse seroblock FcR incubation (nearly 20-fold depletion in CD14+ NOTUM+ cells). After the final washes, cells were passed through a 40 μm cell strainer (Fisher) to remove any cell clumps. BD FACS Fortessa was used for flow cytometric analysis, and the data was analyzed using FlowJo. All gates were set using single-color control samples.
V-SVZ organotypic Slice Cultures
7–8 week old NestinCFP/+ animals were euthanized and the brains were dissected into fresh directly into cold HBSS and then embedded in 4% low melting agarose in 1X PBS. Brains were mounted on the chucks and live 250–300um thick coronal sections were obtained using vibratome (Leica). Sections were collected into Neurobasal Media (Invitrogen) supplemented with N2 and B27 Supplements and 20ng/mL EGF (Invitrogen). Functionally validated recombinant mouse NOTUM protein was acquired from R&D Systems (Cat # 9150-NO-050). Sections used for this study spanned the forebrain regions (Bregma +1.4mm to +1mm) Sections were carefully transferred over the membrane inserts (Millipore, Millicell cell culture inserts) that are placed over 1mL of media. Alternating sections were treated either with ABC99 or ABC101. Media was changed every day, and 10 ul of media was dropped on each slice to keep them moist. Slices were cultured at 37°C with 5% CO2. Prior to fixation at end point, cultures were pulsed with EdU (2.5 μg/mL) for five hours. Health of the slices was determined based on clear EdU (Click-IT) incorporation.
SCOPE-seq sample and library preparation, and sequencing
Single cell suspensions of V-SVZ cells from male C57BL/6J animals were prepared as described above. Following the Percoll gradient, cells were washed in 1% BSA, 0.1% Glucose HBSS by centrifugation (1300 rpm at 4°C). Cells were first incubated in anti-NOTUM (1:50) for 15 minutes on ice, washed twice by centrifugation (1300 rpm at 4°C), and incubated in Alexa Fluor 647 secondary antibody (1:2000) for 15 minutes on ice. Cells were then washed twice, and stained with Calcein AM live stain dye (Fisher, 1:500) in 1X TBS on ice for 30 minutes. The resulting cell suspension was passed through a 40 μm cell strainer, and loaded in a microwell device (> 130K microwells) at a concentration of 1000 cells per μl. Untrapped cells were flushed out with 1× TBS, and the device was scanned under an inverted automated Nikon Eclipse Ti2 microscope in two fluorescence channels and the bright field channel in ~35 minutes. Optically barcoded (OBC) beads (Chemgenes) were then loaded in the microwell device, and untrapped beads were flushed out with 1× TBS. The microwell device containing the cells and the beads was connected to the computer-controlled reagent and temperature delivery system to perform on-chip RT as described above. The device was scanned during RNA capture to assess the lysis efficiency. After the post-RT washes were completed, the device was disconnected from the automated reagent delivery system. On-chip Exo I treatment was performed at 37°C for 45 minutes followed by TE/TW buffer wash (10 mM Tris pH 8.0, 1 mM EDTA, 0.01% Tween-20). The device was then connected to the automated optical demultiplexing system for reagent delivery.
Optical demultiplexing was performed as previously described with several modifications (Yuan et al., 2018). Each optically decodable mRNA capture bead is conjugated to an oligonucleotide with two, 8-base sequences, each of which is selected from a different set of 96 sequences (Table S4). The barcodes are synthesized combinatorially using solid-phase oligonucleotide synthesis (Chemgenes). Together, the two sequences form the cell-identifying barcode for a given bead, and the complete set of cell-identifying barcodes includes 96×96 = 9,216 possible barcodes. Further multiplexing capacity is achieved by preparing sequencing libraries separately from beads deposited in different sub-arrays of the same device (Yuan et al., 2018). The oligonucleotides also contain interspersed random sequences that form a unique molecular identifier (UMI) and are terminated with poly(dT) to facilitate mRNA capture by hybridization to the poly(A) tail.
To identify the two sequences on each bead that form the cell-identifying barcode, we use sequential fluorescence hybridization. In each cycle of fluorescence hybridization, we introduce a set of 8-base, Cy5-labeled oligonucleotides to probe the first sequence on each bead and a set of 8-base, Cy3-labeled oligonucleotides to probe the second sequence. The probes are pooled such that all 96 sequences in each set can be identified with eight cycles of sequential fluorescence hybridization (Gunderson et al., 2004) (Table S4). Each cycle consists of a background scan and probe scan (two fluorescence channels; Cy3 and Cy5, and bright field), and probe hybridization and stripping (2× SSC buffer with tween-20 and 150mM NaOH melting solution). Background scans showed near absolute probe stripping efficiency. Each pool of hybridization mixture consists of Cy3- and Cy5-labeled oligonucleotides complementary to OBCs (96×96 possible combinations). After the optical demultiplexing workflow, all wells were sealed with perfluorinated oil (Sigma, F3556), the device with separated regions was cut in to 15 pieces with razor blades. Beads from each region were placed in a microcentruge tube containing 100% ethanol, and collected by vortexing, sonication, and centrifugation. By preparing and indexing libraries separately from 15 different device regions, we effectively expand our barcoding capacity to 96×96×15 = 138,240 barcodes. The beads were then washed with TE/SDS (10 mM Tris-HCl, 1 mM EDTA, 0.5% SDS) once, TE/TW twice, and nuclease-free water. cDNA was amplified by PCR for 16 cycles (KAPA HotStart ReadyMix, Kapabiosystems), and used as input for Nextera tagmentation reactions. Library preparation was performed as described above resulting in 15 sequencing libraries. These libraries were pooled, and sequenced on a NextSeq 500 sequencer (Illumina) using a 75 cycle High Output Kit (26 cycle read 1, 58 cycle read 2, and 8 cycle index read).
Mass Spectrometry Based Activity-Based Protein Profiling
To prepare V-SVZ proteome, ABC99 or ABC101 were injected at 10mg/kg for three consecutive days and the animals were sacrificed 4 hours after the last injection. Whole-mounts of the lateral wall of V-SVZ were dissected (Mirzadeh et al., 2010) and snap frozen. Frozen samples from each mouse were homogenized using probe sonication in 300 μL of cold DPBS. Protein concentrations were determined using Bio-Rad DC protein assay. Due to the low protein yields, four SVZ proteomes from each group (ABC101 or ABC99-treated) were combined into a single sample and split into two 1.0 mL samples at a protein concentration of 2.7 mg/mL. Tissue homogenates (1.0 mL, 2.7 mg/mL) were treated with FP-biotin (10 μL, 1 mM in DMSO) to a final concentration of 10 μM and incubated for 1 hour at 37°C with gentle rotation. To remove unreacted probe, protein was precipitated by addition of a cold acetone:methanol (9:1) mixture (8 mL) and incubated at −20°C freezer overnight. The samples were centrifuged at 4,700 × g for 15 min at 4°C resulting in the formation of a protein pellet at the bottom of the tube. The supernatant was removed by inversion and the pellet was allowed to air dry for 10 min at room temperature. The protein pellet was dissolved in 0.2 mL of a denaturing solution containing urea (8.0 M), sodium dodecyl sulfate (SDS) (1%), and 1,4-dithiothreitol (DTT) (200 mM) in DPBS. Following complete dissolution, the samples were incubated for 30 min at 37°C and then treated with iodoacetamide (20 μL, 500 mM in DPBS) for an additional 30 min at room temperature. The samples were diluted with 0.25% SDS in DPBS (1.4 mL) and incubated with DPBS-washed streptavidin agarose beads (60 μL) for 1 h at room temperature. The streptavidin beads were then sequentially washed with 0.25% SDS (10 × 0.8 mL), DPBS (10 × 1.1 mL), and distilled water (10 × 1.1 mL). The washed beads were transferred to a clean vessel and trypsin (1.6 μg in 100 μl of 2 M urea in DPBS) was added. Samples were incubated overnight at room temperature with agitation. The beads were removed from the digestion using filtration and washed with DPBS (100 μL). The combined filtrates were then acidified by addition of formic acid (20 μL of 100% formic acid) and desalted using SOLAμ™ SPE Plates (HRP 2 mg / 1 mL). Samples were dried by centrifugal evaporation and stored at −80°C until analysis.
Dry peptide samples were reconstituted in water containing 0.1% formic acid (20 μL) and 10 μL were injected onto an EASY-Spray column (15 cm × 75 μm ID, PepMap C18, 3 μm particles, 100 Å pore size, Thermo Fisher Scientific) using a Dionex RSLCnano LC (Thermo Fisher Scientific). Peptides were separated over a 30 min gradient of 0 to 40% acetonitrile (0.1% formic acid) and analyzed on an Orbitrap Fusion Lumos (Thermo Fisher Scientific) operated using a parallel reaction monitoring (PRM) method for three distinct NOTUM peptides and a single peptide from three other common serine hydrolase targets: ABHD6, FAAH and MGLL. Selected ions were isolated and fragmented by high energy collision dissociation (HCD) and fragments were detected in the Orbitrap at 15,000 or 30,000 resolution. Sequences, targets and targeting parameters can be found in Table S5 (PRM Parameters).
QUANTIFICATION AND STATISTICAL ANALYSIS
Single cell sequencing data processing and clustering
The sequencing reads were demultiplexed, aligned, and quantified as described previously (Mizrak et al., 2019). Briefly, read 2 was trimmed to remove 3′ polyA tails (> 7 A’s), and fragments with fewer than 24 remaining nucleotides were discarded. Trimmed reads were aligned to the Mus musculus genome and transcriptome annotation (GRCm38, Gencode annotation vM10) using STAR v.2.5.0 with parameters −sjdbOverhang 65–twopassMode Basic–outSAMtype BAM Unsorted (Dobin et al., 2013). Only reads with unique, strand-specific alignments to exons were kept for further analysis. TdTomato allele was originally designed as tdTomato::WPRE::polyA. As the sequencing data is 3′ biased, we used the WPRE sequence with flanking sequences to detect TdTomato expression in cells. WPRE is denoted as WRPE in the expression matrices.
We extracted 12-nt cell barcodes (CBs) and 8-nt unique molecular identifies (UMIs) from read 1. Degenerate CBs containing either ‘N’s or more than four consecutive ‘G’s were discarded. Synthesis errors, which can result in truncated 11-nt CBs, were corrected similarly to a previously reported method (Shekhar et al., 2016). Briefly, we identified all CBs with at least twenty apparent molecules and for which greater than 90% of UMI-terminal nucleotides were ‘T’. These putative truncated CBs were corrected by removing their last nucleotide. This twelfth nucleotide became the new first nucleotide of corresponding UMIs, which were also trimmed of their last (‘T’) base. We modified our protocol for commercial Drop-seq beads from Chemgenes with a modified structure. A ‘V’ base was added to the end of the UMI upstream of the poly(T) tail for quality-control purposes. Our analytical pipeline was adjusted to recognize truncated CBs by the presence of excessive ‘T’ bases one nucleotide downstream of the position used in the previous pipeline. All reads with the same CB, UMI, and gene mapping were collapsed to represent a single molecule. To correct for sequencing errors in UMI’s, we further collapsed UMI’s that were within Hamming distance 1 of another UMI with the same barcode and gene. To correct for sequencing errors in cell barcodes, we then collapsed CBs that were within Hamming distance one of another barcode, had at least 20 unique UMI-gene pairs a piece, and had at least 75% overlap of their UMI-gene pairs. Finally, we repeated UMI error correction and collapse using the error-corrected CBs. The remaining barcode-UMI-gene triplets were used to generate a digital gene expression matrix. The number of cells included in each sample was determined based on the inflection point in the cumulative histograms for molecules as previously described (Macosko et al., 2015; Yuan and Sims, 2016).
For clustering, we modified our method (Levitin et al., 2019) to select genes detected in fewer cells than expected given their apparent expression level. Briefly, for variable gene selection only, we normalized the molecular counts for each cell to sum to 1. Genes were then ordered by their normalized expression values. For each gene g, we computed a dropout score as follows:
where fg is the fraction of cells in the dataset that expressed g, and fmax,g is the maximum fg in a rolling window of 25 genes centered on g. To cluster and visualize the data, we computed a cell by cell Spearman’s correlation matrix using the marker genes identified above. Using this matrix, we constructed a k-nearest neighbors graph (k = 20), which we then used as input to Louvain clustering with Phenograph (Levine et al., 2015). After clustering, we identified genes most specific to each cluster using a binomial test (Shekhar et al., 2016). The same similarity matrix, transformed into a distance matrix by subtracting its values from 1, was used as input to tSNE for visualization. V-SVZ and OB samples from different genotypes (GCERT2 and NESFLPO) were clustered separately. The neuron sub-clustering was performed on OB neurons only, and the clusters with overlapping binomially specific markers were merged. Astrocyte, aNSC, TAC and NB clusters from V-SVZ, and NB and Neuron clusters from OB were merged and re-clustered (k = 20), and the resulting k-nearest neighbors graphs were visualized using force-directed graphs to examine their lineage relationship (Weinreb et al., 2018). Doublets, marked by significant enrichment of canonical markers for more than one cell type (e.g., Astrocyte-Oligodendrocytes, Astrocyte-Neuron) were removed from any analysis. Finally, any contaminating cells (e.g., a small cluster of endothelial cells in the larger neuron cluster) formed separate clusters in the sub-clustering analysis, and were removed from the subsequent analyses. The following numbers of total and TdTom+ cells were analyzed in each scRNA-seq sample (TdTom+ cell number/total cell number): male P56 V-SVZ NESFLPO = 44/2657; male P70 V-SVZ NESFLPO = 264/13821; male P56 OB NESFLPO = 64/6249; male P70 OB NESFLPO = 129/9479; male P56 V-SVZ GCERT2 = 16/2032; female P56 V-SVZ GCERT2 = 56/3928; male P70 V-SVZ GCERT2 = 118/4139; female P70 V-SVZ GCERT2 = 34/3189; male P56 OB GCERT2 = 80/1704; female P56 OB GCERT2 = 105/3010; male P70 OB GCERT2 = 88/4935; female P70 OB GCERT2 = 31/1371.
Single cell hierarchical poisson factorization (scHPF), Pointwise Mutual Information (PMI), phenotypic volume, differential expression and Gene Set Enrichment (GSEA) analyses
scHPF detects latent factors explaining the discrete expression patterns in the scRNaseq datasets (Levitin et al., 2019). Each gene and cell have a score for each factor demonstrating the gene’s contribution to the factor, and the factor’s contribution to the detected expression in the cell respectively. scHPF was applied to the merged neuronal lineage cells from GCERT2 and NESFLPO using mouse protein coding genes. To select the optimal number of factors, first we ran scHPF for different number of factors, K. For each value of K, we calculated the maximum pairwise overlap of the 100 highest-scoring genes in each factor, and considered overlap significant if p < 0.05 by a hypergeometric test. We picked the model with maximum K (p > 0.05) and identified the factors with consistent expression patterns both in GCERT2 and NESFLPO, which resulted in eleven factors (Table S2).
To assess co-expression and mutual exclusivity of genes, we first identified the significantly expressed genes (detected in more than 30 cells) in the GCERT2 neuronal lineage. For each pair genes, we computed the log-transformed ratio of the joint probability of detecting the two genes in the same cell to the product of their marginal detection probabilities (pointwise mutual information):
Positive values of R indicate co-expression, and negative values indicate mutual exclusivity beyond what would be expected by chance. The results for the top factor 2 genes (Table S2) were summarized in Figure S2B. Significant clusters of these genes were isolated using R function pvclust(), which uses bootstrap resampling to compute p values (method.dist = “euclidean,”method.hclust = “average,” nboot = 1000; alpha = 0.95).
Phenotypic volume in Figure 2F was computed as previously described (Azizi et al., 2018). Briefly, we merged the scRNA-seq count matrices for neuronal lineage cells from the V-SVZ and OB of both the GCERT2 and NESFLPO reporter mice, and applied the Phenograph-based clustering method described above to identify clusters of astrocytes, aNSCs, TACs, neuroblasts, and mature neurons. We transformed the matrix of molecular counts to log2(counts per thousand+1), including only genes with a > 500 total counts in the dataset. We then computed phenotypic volumes for sub-matrices comprised of astrocyte, aSNC, TAC, SVZ-neuroblasts, OB-neuroblasts, or mature neurons as the product of positive eigenvalues of the gene expression covariance matrix for each of the six populations.
To test the enrichment of WNT signaling pathway (Gene Set M5493-Molecular Signatures Database, Broad Institute) in the SCOPE-seq dataset, we computed the expression of each gene in each cell as the z-scored log2(counts per million + 1) and produced a ranked list for each cell. GSEA (GSEA v.2.2.2, https://www.gsea-msigdb.org/gsea/login.jsp) was conducted (Subramanian et al., 2005) for the WNT pathway gene set against each ranked list using the Java implementation using the following command:
java -cp gsea2–2.2.2.jar -Xmx2512m xtools.gsea.GseaPreranked -scoring_scheme classic –setmin 10 –setmax 1000 –nperm 1000
Single-cell differential expression (SCDE) analysis was used to identify differentially expressed genes in Notum+ cells (Kharchenko et al., 2014). To identify canonical pathways (C2 canonical pathways collection-Broad Institute) enriched or depleted in these cells, SCDE output was pre-ranked based on effect size, and the resulting list was inputted to GSEA using the parameters described above. The results were summarized in Table S3.
SCOPE-seq imaging and sequencing data analysis
Image analysis was performed using ImageJ. To identify the OBC sequence, each bead was identified in the bright field channel (setAutoThreshold(“Default”); setOption (“BlackBackground,” false); run(“Convert to Mask”)), and Cy3 and Cy5 channel intensities were measured in these regions of interest (ROIs). OBC calling was performed using a bead-by-bead algorithm (Gunderson et al., 2004). Cell ROIs were extracted from the cell loading scan using the calcein signal, the intensity measurements were performed for two fluorescence channels (calcein AM and anti-NOTUM fluorescence). The coordinates for the center of the wells and the well ROIs were extracted from the bright field channel (Auto Local Threshold, method = Bernsen, radius = 30, parameter_1 = 40, parameter_2 = 0). Cells were assigned to the center of their wells, the wells containing more than one cell were marked as multiplets.
Both the calcein AM and anti-NOTUM fluorescence intensity distributions were bimodal for microwells containing individual cells (Figure S3A). We thresholded the anti-NOTUM and calcein AM fluorescence at the intensity between the two modes of each intensity histogram with the fewest cells to identify a set calcein AM+/NOTUM+ and calcein AM+/NOTUM− cells for further analysis. We discarded cells for which the same cell-identifying barcode was associated with more than one cell in the same sub-array of the device.
To analyze the scRNA-seq data from SCOPE-Seq, we first extract the cell-identifying barcode and UMI from Read 1. The sequences in the two sets that are combined to form each cell-identifying barcode have a Hamming distance of at least three for all sequence pairs within each set. Therefore, we correct at most one substitution error in each of the two sequences should one arise from sequencing, oligonucleotide synthesis, or other sources. We only keep reads with a complete cell-identifying barcode. Next, we align the reads from Read 2 using the STAR aligner and keep strand-specific alignments to exons as described above after removing poly(A) tails > 7 bases long from the 3′ end of each read. Finally, we use the UMIs to estimate the number of molecules associated with each gene in each cell as described above. Clustering and visualization of the scRNA-seq profiles resulting from SCOPE-seq was carried out as described above for conventional scRNA-seq. In addition, using the imaging data obtained in the SCOPE-seq experiment, we excluded cell multiplets.
To associate imaging features with pseudotime in the neuronal lineage trajectory, we first extracted neuronal lineage cells from the SCOPE-seq data based on Phenograph clustering of the entire dataset. This included all clusters enriched in markers of astrocytes, aNSCs, TACs, and NBs. After marker selection using the drop-out curve for the neuronal lineage cells as described above, we computed a similarity matrix of pairwise Spearman correlation coefficients from which we constructed a k-nearest neighbors graph (k = 20) which we projected in two dimensions as a force-directed graph using the spring_layout function in the networkx Python module. We then fit a polynomial to the resulting two-dimensional projection, which we interpret as the average trajectory. We then assign each cell to a position along this average trajectory by determining its minimum distance from the polynomial curve. Finally, we grouped the cells into 20 bins of equal size along the polynomial trajectory to produce Figure 3D where we plot the average area (based on calcein AM fluorescence) of the cells in each bin and the fraction of Notum mRNA+ cells in each bin.
To associate NOTUM surface staining with gene expression signatures of the four major subpopulations in the neuronal lineage (astrocyte, aNSC, TAC, and NB), we first computed the pointwise mutual information (PMI) between NOTUM surface staining from SCOPE-seq imaging (a categorical where a cell is either positive or negative) and expression of each gene from scRNA-seq (a second categorical where a gene is either detected or not in a given cell):
Here, P(NOTUM,g) is the joint probability that a cell is both positive for NOTUM surface staining and positive for detection of the mRNA encoding gene g, P(NOTUM) is the probability that a cell is positive for NOTUM surface staining, and P(g) is the probability that a cell is positive for detection of the mRNA encoding gene g. Therefore, if a gene co-occurs with NOTUM surface staining more than expected by chance, RNOTUM,g is positive. To identify genes that were significantly associated with NOTUM surface staining, we used a permutation test, shuffling the identities of NOTUM surface positive cells 1,000 times and correcting the resulting p values for multiple comparisons using the Benjamini-Hochberg procedure as implemented in the Python module statsmodels. We created a gene set comprised of genes detected in greater than 20 cells with pcorr < 0.05. We then created a ranked list of genes for each of the four neuronal lineage clusters using −/+log(p) where p is the p value from the binomial test used to identify genes specific to each cluster that is described above. This parameter is positive for genes that are positively enriched in a given cluster and negative for genes that are depleted. Finally, we used GSEA to compute normalized enrichment scores for the gene set comprised of NOTUM surface staining-associated genes in the ranked list for each of the four clusters using the GSEA parameters described above. The results are summarized in Figure 3E.
Activity-Based Protein Profiling Data Analysis
Raw data files were uploaded analyzed in Skyline (v4.2.0.19009) to determine the relative abundance of each serine hydrolase peptide in vehicle-treated samples relative to ABC99-treated samples. Peptide quantification was performed by calculating the sum of the peak areas corresponding to 6 fragment ions from each peptide (Table S5). To determine protein activity relative to the controls, an area ratio was calculated for each fragment ion, dividing the peak area of an ABC99-treated sample by the mean peak area of the ABC101-control samples. The relative protein activity was found from the slope of the regression line obtained by plotting the treated ratios against mean control ratios. The peptides and fragment ions were pre-selected from in-house reference spectral libraries acquired in data-dependent acquisition mode to identify authentic spectra for each peptide. Product ions used for Skyline quantification appear in Table S5.
Supplementary Material
Highlights.
Neurogenesis studied in ventricular-subventricular zone (V-SVZ) and olfactory bulb (OB)
scRNA-seq reveals lineage progression from V-SVZ to OB and a Notum+ NSC transition
SCOPE-seq identifies a Notum+ NSC intermediate based on cell size and NOTUM targets
NOTUM prevents uncontrolled expansion of neuronal progenitors in the V-SVZ
ACKNOWLEDGMENTS
A.L.J. was supported by R01NS092096 from NIH/NINDS. N.S.B. was supported by postdoctoral fellowship C32599GG from NYSTEM/USA and K99NS112605–01 from NIH/NINDS. P.A.S. was funded by R33CA202827 from NIH/NCI and R44HG010003 from NIH/NHGRI.
Footnotes
DECLARATION OF INTERESTS
Columbia University has filed patent applications on SCOPE-seq, and P.A.S. and J.Y. are listed as inventors.
REFERENCES
- Adachi K, Mirzadeh Z, Sakaguchi M, Yamashita T, Nikolcheva T, Gotoh Y, Peltz G, Gong L, Kawase T, Alvarez-Buylla A, et al. (2007). Beta-catenin signaling promotes proliferation of progenitor cells in the adult mouse subventricular zone. Stem Cells 25, 2827–2836. [DOI] [PubMed] [Google Scholar]
- Alfonso J, Le Magueresse C, Zuccotti A, Khodosevich K, and Monyer H (2012). Diazepam binding inhibitor promotes progenitor proliferation in the postnatal SVZ by reducing GABA signaling. Cell Stem Cell 10, 76–87. [DOI] [PubMed] [Google Scholar]
- Azim K, Fischer B, Hurtado-Chong A, Draganova K, Cantú C, Zemke M, Sommer L, Butt A, and Raineteau O (2014). Persistent Wnt/β-catenin signaling determines dorsalization of the postnatal subventricular zone and neural stem cell specification into oligodendrocytes and glutamatergic neurons. Stem Cells 32, 1301–1312. [DOI] [PubMed] [Google Scholar]
- Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, et al. (2018). Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 174, 1293–1308.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista-Brito R, Close J, Machold R, and Fishell G (2008). The distinct temporal origins of olfactory bulb interneuron subtypes. J. Neurosci. 28, 3966–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brill MS, Ninkovic J, Winpenny E, Hodge RD, Ozen I, Yang R, Lepier A, Gascón S, Erdelyi F, Szabo G, et al. (2009). Adult generation of glutamatergic olfactory bulb interneurons. Nat. Neurosci. 12, 1524–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaker Z, Codega P, and Doetsch F (2016). A mosaic world: puzzles revealed by adult neural stem cell heterogeneity. Wiley Interdiscip. Rev. Dev. Biol. 5, 640–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codega P, Silva-Vargas V, Paul A, Maldonado-Soto AR, Deleo AM, Pastrana E, and Doetsch F (2014). Prospective identification and purification of quiescent adult neural stem cells from their in vivo niche. Neuron 82, 545–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa MR, Ortega F, Brill MS, Beckervordersandforth R, Petrone C, Schroeder T, Götz M, and Berninger B (2011). Continuous live imaging of adult neural stem cell division and lineage progression in vitro. Development 138, 1057–1068. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Wright AT, and Kozarich JW (2008). Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 77, 383–414. [DOI] [PubMed] [Google Scholar]
- Daynac M, Morizur L, Chicheportiche A, Mouthon MA, and Boussin FD (2016). Age-related neurogenesis decline in the subventricular zone is associated with specific cell cycle regulation changes in activated neural stem cells. Sci. Rep. 6, 21505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado RN, and Lim DA (2015). Embryonic Nkx2.1-expressing neural precursor cells contribute to the regional heterogeneity of adult V-SVZ neural stem cells. Dev. Biol. 407, 265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Caillé I, Lim DA, García-Verdugo JM, and Alvarez-Buylla A (1999). Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97, 703–716. [DOI] [PubMed] [Google Scholar]
- Driehuis E, and Clevers H (2017). WNT signalling events near the cell membrane and their pharmacological targeting for the treatment of cancer. Br. J. Pharmacol. 174, 4547–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, Yun J, Cronin C, Karp C, Yuan GC, and Cai L (2019). Transcriptome-scale superresolved imaging in tissues by RNA seqFISH. Nature 568, 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliciano DM, Bordey A, and Bonfanti L (2015). Noncanonical Sites of Adult Neurogenesis in the Mammalian Brain. Cold Spring Harb. Perspect. Biol. 7, a018846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorelli R, Azim K, Fischer B, and Raineteau O (2015). Adding a spatial dimension to postnatal ventricular-subventricular zone neurogenesis. Development 142, 2109–2120. [DOI] [PubMed] [Google Scholar]
- Fuentealba LC, Rompani SB, Parraguez JI, Obernier K, Romero R, Cepko CL, and Alvarez-Buylla A (2015). Embryonic Origin of Postnatal Neural Stem Cells. Cell 161, 1644–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganat YM, Silbereis J, Cave C, Ngu H, Anderson GM, Ohkubo Y, Ment LR, and Vaccarino FM (2006). Early postnatal astroglial cells produce multilineage precursors and neural stem cells in vivo. J. Neurosci. 26, 8609–8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-González D, Khodosevich K, Watanabe Y, Rollenhagen A, Lübke JHR, and Monyer H (2017). Serotonergic Projections Govern Postnatal Neuroblast Migration. Neuron 94, 534–549.e9. [DOI] [PubMed] [Google Scholar]
- Gunderson KL, Kruglyak S, Graige MS, Garcia F, Kermani BG, Zhao C, Che D, Dickinson T, Wickham E, Bierle J, et al. (2004). Decoding randomly ordered DNA arrays. Genome Res. 14, 870–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AR, Forero-Vargas M, Withers SP, Smith RS, Traas J, Dewitte W, and Murray JAH (2017). Cell-size dependent progression of the cell cycle creates homeostasis and flexibility of plant cell size. Nat. Commun. 8, 15060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakugawa S, Langton PF, Zebisch M, Howell S, Chang TH, Liu Y, Feizi T, Bineva G, O’Reilly N, Snijders AP, et al. (2015). Notum deacylates Wnt proteins to suppress signalling activity. Nature 519, 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi D, Furutachi S, Kawai H, Hozumi K, and Gotoh Y (2013). Dll1 maintains quiescence of adult neural stem cells and segregates asymmetrically during mitosis. Nat. Commun. 4, 1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Silberstein L, and Scadden DT (2014). Bayesian approach to single-cell differential expression analysis. Nat. Methods 11, 740–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, and Kirschner MW (2015). Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohwi M, Osumi N, Rubenstein JL, and Alvarez-Buylla A (2005). Pax6 is required for making specific subpopulations of granule and periglomerular neurons in the olfactory bulb. J. Neurosci. 25, 6997–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, and Alvarez-Buylla A (2009). The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32, 149–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao Z, Raju GP, Bai CB, and Joyner AL (2012). MASTR: a technique for mosaic mutant analysis with spatial and temporal control of recombination using conditional floxed alleles in mice. Cell Rep. 2, 386–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Zappaterra MW, Chen X, Yang YJ, Hill AD, Lun M, Maynard T, Gonzalez D, Kim S, Ye P, et al. (2011). The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron 69, 893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasson M, Saghatelyan A, Olivo-Marin JC, and Lledo PM (2005). Neonatal and adult neurogenesis provide two distinct populations of newborn neurons to the mouse olfactory bulb. J. Neurosci. 25, 6816–6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amir ED, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, et al. (2015). Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162, 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitin HM, Yuan J, Cheng YL, Ruiz FJ, Bush EC, Bruce JN, Canoll P, Iavarone A, Lasorella A, Blei DM, and Sims PA (2019). De novo gene signature identification from single-cell RNA-seq with hierarchical Poisson factorization. Mol. Syst. Biol. 15, e8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang Q, Haydar TF, and Bordey A (2005). Nonsynaptic GABA signaling in postnatal subventricular zone controls proliferation of GFAP-expressing progenitors. Nat. Neurosci. 8, 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lledo PM, Merkle FT, and Alvarez-Buylla A (2008). Origin and function of olfactory bulb interneuron diversity. Trends Neurosci. 31, 392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens-Bobadilla E, Zhao S, Baser A, Saiz-Castro G, Zwadlo K, and Martin-Villalba A (2015). Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell 17, 329–340. [DOI] [PubMed] [Google Scholar]
- López-Juárez A, Howard J, Ullom K, Howard L, Grande A, Pardo A, Waclaw R, Sun YY, Yang D, Kuan CY, et al. (2013). Gsx2 controls region-specific activation of neural stem cells and injury-induced neurogenesis in the adult subventricular zone. Genes Dev. 27, 1272–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkle FT, Mirzadeh Z, and Alvarez-Buylla A (2007). Mosaic organization of neural stem cells in the adult brain. Science 317, 381–384. [DOI] [PubMed] [Google Scholar]
- Merkle FT, Fuentealba LC, Sanders TA, Magno L, Kessaris N, and Alvarez-Buylla A (2014). Adult neural stem cells in distinct microdomains generate previously unknown interneuron types. Nat. Neurosci. 17, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mich JK, Signer RA, Nakada D, Pineda A, Burgess RJ, Vue TY, Johnson JE, and Morrison SJ (2014). Prospective identification of functionally distinct stem cells and neurosphere-initiating cells in adult mouse forebrain. eLife 3, e02669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignone JL, Kukekov V, Chiang AS, Steindler D, and Enikolopov G (2004). Neural stem and progenitor cells in nestin-GFP transgenic mice. J. Comp. Neurol. 469, 311–324. [DOI] [PubMed] [Google Scholar]
- Mirzadeh Z, Merkle FT, Soriano-Navarro M, Garcia-Verdugo JM, and Alvarez-Buylla A (2008). Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell 3, 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadeh Z, Doetsch F, Sawamoto K, Wichterle H, and Alvarez-Buylla A (2010). The subventricular zone en-face: wholemount staining and ependymal flow. J. Vis. Exp. 39, 1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrak D, Levitin HM, Delgado AC, Crotet V, Yuan J, Chaker Z, Silva-Vargas V, Sims PA, and Doetsch F (2019). Single-Cell Analysis of Regional Differences in Adult V-SVZ Neural Stem Cell Lineages. Cell Rep. 26, 394–406.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munji RN, Soung AL, Weiner GA, Sohet F, Semple BD, Trivedi A, Gimlin K, Kotoda M, Korai M, Aydin S, et al. (2019). Profiling the mouse brain endothelial transcriptome in health and disease models reveals a core blood-brain barrier dysfunction module. Nat. Neurosci. 22, 1892–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayama S, Homma R, and Imamura F (2014). Neuronal organization of olfactory bulb circuits. Front. Neural Circuits 8, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obernier K, and Alvarez-Buylla A (2019). Neural stem cells: origin, heterogeneity and regulation in the adult mammalian brain. Development 146, dev156059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obernier K, Cebrian-Silla A, Thomson M, Parraguez JI, Anderson R, Guinto C, Rodas Rodriguez J, Garcia-Verdugo J-M, and Alvarez-Buylla A (2018). Adult Neurogenesis Is Sustained by Symmetric Self-Renewal and Differentiation. Cell Stem Cell 22, 221–234.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega F, Gascón S, Masserdotti G, Deshpande A, Simon C, Fischer J, Dimou L, Chichung Lie D, Schroeder T, and Berninger B (2013). Oligodendrogliogenic and neurogenic adult subependymal zone neural stem cells constitute distinct lineages and exhibit differential responsiveness to Wnt signalling. Nat. Cell Biol. 15, 602–613. [DOI] [PubMed] [Google Scholar]
- Pastrana E, Cheng LC, and Doetsch F (2009). Simultaneous prospective purification of adult subventricular zone neural stem cells and their progeny. Proc. Natl. Acad. Sci. USA 106, 6387–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentinmikko N, Iqbal S, Mana M, Andersson S, Cognetta AB 3rd, Suciu RM, Roper J, Luopajärvi K, Markelin E, Gopalakrishnan S, et al. (2019). Notum produced by Paneth cells attenuates regeneration of aged intestinal epithelium. Nature 571, 398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova R, Garcia AD, and Joyner AL (2013). Titration of GLI3 repressor activity by sonic hedgehog signaling is critical for maintaining multiple adult neural stem cell and astrocyte functions. J. Neurosci. 33, 17490–17505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponti G, Obernier K, Guinto C, Jose L, Bonfanti L, and Alvarez-Buylla A (2013). Cell cycle and lineage progression of neural progenitors in the ventricular-subventricular zones of adult mice. Proc. Natl. Acad. Sci. USA 110, E1045–E1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Q, Sun G, Li W, Yang S, Ye P, Zhao C, Yu RT, Gage FH, Evans RM, and Shi Y (2010). Orphan nuclear receptor TLX activates Wnt/beta-catenin signalling to stimulate neural stem cell proliferation and self-renewal. Nat. Cell Biol. 12, 31–40, S31–S39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro LA, Ng K, Zhou QY, and Ribak CE (2009). Subventricular zone-derived, newly generated neurons populate several olfactory and limbic forebrain regions. Epilepsy Behav. 14 (Suppl 1), 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, et al. (2016). Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell 166, 1308–1323.e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suciu RM, Cognetta AB 3rd, Potter ZE, and Cravatt BF (2018). Selective Irreversible Inhibitors of the Wnt-Deacylating Enzyme NOTUM Developed by Activity-Based Protein Profiling. ACS Med. Chem. Lett. 9, 563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepe B, Hill MC, Pekarek BT, Hunt PJ, Martin TJ, Martin JF, and Arenkiel BR (2018). Single-Cell RNA-Seq of Mouse Olfactory Bulb Reveals Cellular Heterogeneity and Activity-Dependent Molecular Census of Adult-Born Neurons. Cell Rep. 25, 2689–2703.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Maaten L, and Hinton G (2008). Visualizing Data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605. [Google Scholar]
- Weinreb C, Wolock S, and Klein AM (2018). SPRING: a kinetic interface for visualizing high dimensional single-cell expression data. Bioinformatics 34, 1246–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Zhang Z, Li Z, Liu G, Tao G, Song X, Xu Z, Shang Z, Guo T, Su Z, et al. (2019). The PROK2/PROKR2 signaling pathway is required for the migration of most olfactory bulb interneurons. J. Comp. Neurol. 527, 2931–2947. [DOI] [PubMed] [Google Scholar]
- Wojcinski A, Lawton AK, Bayin NS, Lao Z, Stephen DN, and Joyner AL (2017). Cerebellar granule cell replenishment postinjury by adaptive reprogramming of Nestin+ progenitors. Nat. Neurosci. 20, 1361–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, and Sims PA (2016). An Automated Microwell Platform for Large-Scale Single Cell RNA-Seq. Sci. Rep. 6, 33883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Sheng J, and Sims PA (2018). SCOPE-Seq: a scalable technology for linking live cell imaging and single-cell RNA sequencing. Genome Biol. 19, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cheong SM, Amado NG, Reis AH, MacDonald BT, Zebisch M, Jones EY, Abreu JG, and He X (2015). Notum is required for neural and head induction via Wnt deacylation, oxidation, and inactivation. Dev. Cell 32, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel S, Marcy G, Lo Guidice Q, Li D, Heinrich C, Azim K, and Raineteau O (2018). HOPX Defines Heterogeneity of Postnatal Subventricular Zone Neural Stem Cells. Stem Cell Reports 11, 770–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zywitza V, Misios A, Bunatyan L, Willnow TE, and Rajewsky N (2018). Single-Cell Transcriptomics Characterizes Cell Types in the Subventricular Zone and Uncovers Molecular Defects Impairing Adult Neurogenesis. Cell Rep. 25, 2457–2469.e8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the sequencing data and count matrices reported in this paper is GEO: GSE134918. Data analysis code used for scRNA-seq is available at https://github.com/simslab.