

Abstract
Three biorthogonal click reactions, a photoinitiated thiol-yne reaction, an azide-alkyne cycloaddition, and a methyltetrazine-transcyclooctene Diels Alder, were used to independently control the presentation of several bioactive proteins to valvular interstitial cells (VICs) in hydrogel scaffolds. Tethered fibroblast growth factor (FGF-2) was found to suppress myofibroblast activation (from 48±7% to 17±6%) and promote proliferation (from 10±2% to 54±3%) at a concentration of 10 ng/mL. In the presence of the pro-fibrotic cytokine transforming growth factor-beta (TGF-β1) FGF-2 could protect the VIC fibroblast phenotype, even at much higher concentrations of TGF-β1 than that of FGF-2. With respect to the fibrocalcific VIC phenotype, TGF-β1 and bone-morphogenic protein-2 (BMP-2) were found to synergistically promote calcific nodule formation (a five-fold increase in nodules compared to TGF-β1 or BMP-2 alone). Exploiting the orthogonal click reactions, FGF-2, TGF-β1 and BMP-2 combinations were patterned into distinct regions on a hydrogel to control VIC activation and nodule formation. Cellular crosstalk between separate regions of the same scaffold was affected by the size of each region as well as the interfacial area between different regions. Collectively, these results demonstrate the versatility and robustness of a photoinitiated thiol-yne reaction to template pendant functionalities that allow for the bioconjugation of multiple proteins. This approach maintains protein bioactivity, providing an in vitro platform capable of achieving a better understanding of the complex mechanisms involved in tissue fibrosis.
Keywords: hydrogels, click chemistry, protein patterning, bio-orthogonal, valvular interstitial cells, myofibroblast
1. Introduction
Synthetic hydrogels provide a versatile platform to engineer cellular microenvironments that can recapitulate key aspects of the extracellular matrix (ECM)[1–4]. These platforms act as simplified models of the complex in vivo environment to probe singular and synergistic effects of ECM cues and their interplay with cells in a systematic and tunable manner. Recent advances in bioconjugation chemistry provide user-tunable techniques to incorporate cell signaling functionalities in the hydrogel[5,6]. However, fabrication of such material platforms necessitates versatile bioconjugation methods that are compatible with proteins, allow for spatiotemporal control, and are bioorthogonal. For example, conjugation techniques that rely on radical chemistry may damage the secondary structure of fragile proteins and render them bio-inactive[7]. Further, incorporation of multiple proteins can necessitate multiple swelling and washing steps for each successive protein immobilization, so processes that use only a single step to conjugate multiple molecules are advantageous.
Towards addressing some of these challenges, pioneering work has demonstrated patterning of singular proteins in a defined manner[5,6] or even multiple peptides[9]. For example, Deforest et al. used bioorthogonal chemistries to photoreversibly immobilize the extracellular matrix protein vitronectin within a cell-laden hydrogel[6]. Mosiewicz et al. introduced a mild enzymatic conjugation approach that could spatially pattern a single protein and maintain bioactivity to direct cell migration[8]. Wylie et al. exploited physical binding interactions to achieve simultaneous immobilization of multiple proteins in distinct patterns[9]. More recently, Grim et al. developed a reversible tethering strategy via an allyl sulfide chaintransfer agent to reversibly tether and release proteins in a hydrogel scaffold[10]. However, there is a paucity of methods that are relevant for protein introduction, especially those that allow spatiotemporal control — an aspect of the 4th dimension. Building on this paradigm, we present a mild and simple chemical approach that uses multiple bio-click reactions to spatially define multiple protein cues simultaneously within a hydrogel scaffold that is useful for cell culture. First, a photo-click reaction is used to covalently introduce multiple, pendant functional groups in a spatially controlled manner. In a second step, bioorthogonal click reactions are used to introduce proteins in the pre-patterned, templated regions to achieve spatial control of multiple bioactive proteins.
To demonstrate potential applications of this system, valvular interstitial cells (VICs) were identified as a model cell type as they receive numerous biochemical cues from the ECM to guide their function. VICs are the primary cell type that reside in the heart valve and are known to play an active role in maintenance of the valve extracellular matrix (ECM) in response to injury. Upon damage to the heart valve, these cells transition from a quiescent fibroblast phenotype into an activated myofibroblastic phenotype[11–14], which is characterized by an increase of α-smooth muscle actin (αSMA) stress fibers[15]. The abnormal and misregulated activation of VIC myofibroblasts has been found to cause tissue fibrosis[16]. Further, during the progression of fibrosis, VICs can form “nodules”, which can lead to valve calcification[17] and severe valve stenosis[18]. Biochemical cues have been found to affect VIC behaviors (e.g., proliferation, activation, calcification) in both in vivo and in vitro studies. For instance, FGF-2 has been found to promote the quiescent fibroblast phenotype as well as to induce cellular proliferation[19–21], while TGF-β1 can promote VIC transition into a myofibroblast phenotype[22–24]. Both TGF-β1 and BMP-2 are believed to play a role in VIC nodule formation[25–28]. However, these signaling pathways are complex[29] and are further convoluted by mechanical stimuli[30]. In order to further elucidate the role of these proteins on VIC activation, there is an increased desire for in vitro models that allow for the controlled presentation of matrix signals in a biologically relevant context[31].
We demonstrate a versatile hydrogel platform to study the cooperative effects of the mitogen promoter FGF-2, myofibroblast activator TGF-β1, and osteogenic effector BMP-2 over a broad experimental space on the proliferation, myofibroblast activation, and calcific nodule formation of VICs. By achieving precise spatial control over cue presentation, we aim to elucidate experimental conditions that can be translated to fibrotic and calcific valve diseases. Specifically, a series of bioorthogonal click and light-based chemistries are utilized to pattern proteins simultaneously within an poly(ethylene glycol)-based hydrogel that was developed previously to culture VICs with high viability[15]. Protein immobilization is achieved through two bioorthogonal click reactions: (i) copper-free, strain promoted azide-alkyne cycloaddition (SPAAC) and (ii) inverse electron demand Diels-Alder reaction (IEDDA). Since these reactions are orthogonal to each other, it was envisaged that these reactions could be used to immobilize dual proteins simultaneously. A photoinitiated thiol-yne reaction is used to sequentially incorporate complementary moieties for each of the two bioorthogonal pairs (e.g., azide from SPAAC, tetrazine from IEDDA) in a spatiotemporal manner through the controlled illumination of specific regions of the gel. Additionally, control over the applied light dose can be used to vary the concentration of the patterned click group into the network, thus tuning the total amount of protein immobilized in a given area. Finally, proteins tethered with a complimentary bioorthogonal pair (e.g., dibenzocyclooctyne (DBCO) from SPAAC, transcyclooctene (TCO) from IEDDA) are swelled into the hydrogel in cytocompatible media solutions and click with their complementary pre-patterned moiety in one step. This allows highly-defined spatial patterning of proteins in mild, cytocompatible solutions to recapitulate key facets of the more complex organization and presentation of multiple cues within the in vivo valve cellular microenvironment.
As a demonstration of the method, we study the synergistic influence of multiple proteins on VIC behavior in vitro. Here, we present FGF-2, TGF–β1 and BMP-2 in a hydrogel system and investigate the effects of a broad range of concentrations and combinations on VIC proliferation, myofibroblast activation, and calcific nodule formation. Experiments measure proliferation through Ki-67 nuclear expression, measure αSMA expression and examine alizarin red and alkaline phosphatase staining to quantify VIC nodule formation. Overall, this study demonstrates how material design can be leveraged to create heterogeneous and complex cell culture environments to control cell fate and function, as well as to investigate how the presentation of multiple biochemical cues can affect cellular responses.
2. Materials and Methods
2.1. Synthesis of PEG-alkyne
Briefly, 4-arm PEG amine HCl (JenKem, 5 kDa) (1.0 g, 0.2 mmol) was dissolved in dichloromethane and triethylamine (0.14 mL, 1.0 mmol) was added and stirred at room temperature (RT) for 15 mins. To the above mixture were added 4-pentynoic acid (0.16 g, 1.6 mmol), diisopropyl carbodiimide (DIC) (0.25 mL, 1.6 mmol) and dimethyl aminopyridine (DMAP) (0.02 g, 0.2 mmol) at 0°C. The mixture was then stirred at RT for 24 hrs. The reaction mixture was then precipitated in diethyl ether (45.0 mL), centrifuged and dried under reduced pressure. The precipitate was dissolved in distilled water and dialyzed in 2L DI H2O for 24 hrs replacing water after 12 hrs. The dialyzed solution was then lyophilized to obtain the final macromer as colorless powder.
2.2. Synthesis of PEG-azide
Briefly, 4-arm PEG (Jenkem, 5 kDa) (0.18 g, 0.04 mmol) was dissolved in dichloromethane (DCM) and pyridine at 0°C. Methanesulfonyl chloride (20 molar equivalents to PEG) was dissolved in DCM and added dropwise to the reaction mixture. The solution was stirred overnight, washed with aqueous sodium bicarbonate, dried with magnesium sulfate, and concentrated under reduced pressure. The resultant activated PEG was dissolved in dimethylformamide (DMF) along with five-fold equivalents of sodium azide. The solution was heated to 80°C and stirred overnight under argon. The product was filtered, concentrated under reduced pressure, and precipitated in diethyl ether (45.0 mL). The precipitate was then dissolved in distilled water and dialyzed in 2L DI H2O for 24 hrs replacing water after 12 hrs. The dialyzed solution was then lyophilized to obtain the final macromer as colorless powder.
2.3. Synthesis of disulfide azide
To a solution of 5-azido pentanoic acid (Bachem, 0.86 g, 6.66 mmol) in DMF was added cystamine-hydrochloride (0.5 g, 2.22 mmol) followed by triethylamine (0.45 g, 4.44 mmol) and then the mixture was stirred for 10 mins. To the above mixture was added diisopropyl carbodiimide (DIC) (0.84 g, 6.66 mmol) and dimethyl aminopyridine (DMAP) (54 mg, 0.44 mmol) at 0°C and the mixture was stirred at room temperature for 24 hrs. The reaction mixture was diluted with water and extracted with ethyl acetate multiple times. The combined organic extracts were dried over sodium sulphate and then evaporated under reduced pressure. The crude concentrate was then purified by silica gel column chromatography using ethyl acetate and hexane as the mobile phase to obtain the final product as a colorless powder.
2.4. Synthesis of disulfide methyltetrazine
To a solution of methyltetrazine-PEG-NHS ester (Click Chemistry Tools, 100 mg, 0.19 mmol) in DMSO was added cystamine-hydrochloride (21 mg, 0.09 mmol) followed by triethylamine (33 μL, 0.23 mmol) and the reaction mixture was stirred for an hour. The above mixture was then diluted with distilled water and dialyzed (using dialysis membrane with 500D M.wt cut-off) for 24 hrs replacing water 4 times to remove small molecular reagents and unreacted starting material. The dialyzed solution was then lyophilized to obtain the disulphide containing tetrazine dimer.
2.5. Functionalization of proteins
Transcyclooctene-BSA-Fluorescein was synthesized by adding 5μL of transcyclooctene-NHS ester (Click Chemistry Tools, 20mM) in DMSO to an aqueous solution of BSA-Fluorescein (Life Technologies, 100.0 μL, 10mg/mL). The reaction took place at room temperature while on an orbital shaker for 1 hour. The TCO-BSA-Fluorescein was then stored at −20°C until use.
Dibenzocylcooctyne-BSA-Rhodamine was synthesized by adding 5μL of DBCO NHS ester (Click Chemistry Tools 20mM) in DMSO to an aqueous solution of BSA-Rhodamine (Life Technologies, 100.0 μL, 10mg/mL). The reaction took place at room temperature while on an orbital shaker for 1 hour. The DBCO-BSA-Rhodamine was then stored at −20°C until use.
DBCO-FGF-2 was synthesized by adding 3.9 μL of DBCO-NHS ester (Click Chemistry Tools 2 mM) in DMSO to an aqueous solution of FGF-2 (Sigma-Aldrich, 500 μL, 100 μg/mL). The reaction took place at room temperature while on an orbital shaker for 1 hour. The DBCO-FGF-2 was then stored at −20°C until use.
DBCO-BMP-2 was synthesized by adding 5.1 μL of TCO-NHS ester (Click Chemistry Tools 200 μM) in DMSO to an aqueous solution of BMP-2 (Sigma-Aldrich, 500 μL, 20 μg/mL). The reaction took place at room temperature while on an orbital shaker for 1 hour. The DBCO-FGF-2 was then stored at −20°C until use.
TCO-TGF-β1 was synthesized by adding 1.1 μL of TCO-NHS ester (Click Chemistry Tools 200 μM) in DMSO to an aqueous solution of TGF-β1 (R&D Systems, 500 μL, 2 μg/mL). The reaction took place at room temperature while on an orbital shaker for 1 hour. The TCO-TGF-β1 was then stored at −20°C until use.
2.6. Hydrogel Formation
To form a 100 μm thick hydrogel substrate, 6.0 μL of 200 mg/mL 4-arm, 10k molecular weight PEG-alkyne solution, 4.4 μL of 100 mg/mL 4-arm, 5k molecular weight PEG-azide solution, 2.2 μL of 25 mM Azide-RGD solution and 0.5 μL of 320 mM copper sulfate were combined. 0.5 μL of 3.2 M sodium ascorbate was added rapidly and the solution was transferred in between a sigmacote treated glass slide and azide-functionalized cover glass (fabricated as reported previously[10]). After 6 minutes, the resultant gel was removed and soaked in a saturated ammonium chloride solution with mild shaking for 16 hours with at least three buffer changes, to remove remaining copper.
2.7. Rheology measurements of hydrogels
The moduli of hydrogels with different crosslinker concentrations were measured on a Discovery Hybrid Rheometer (TA Instruments) at room temperature. Optically thin hydrogels with a thickness of 100 μm were formed in situ between a Quartz bottom plate and an 8 mm diameter stainless steel upper plate. The gel network evolution was monitored using a dynamic time sweep at 1% strain. Young’s modulus was calculated with E = 2 * (1 + v) * G’, where a Poisson’s ratio (v) of 0.5 was assumed[32]. Young’s modulus will be reported to indicate the modulus of the hydrogels for all subsequent discussions.
2.8. Photopatterning of hydrogel
10μL of either disulfide containing click group (azide or methyltetrazine) (50mM stock) was dissolved in 67.5μL DMSO with 10μL of 50mM tris(2-carboxyethyl) Phosphine (TCEP). After 15 minutes, 0.15 μL of the photoinitiator Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) (67mM) was added to the mixture. Hydrogel substrates were incubated in the patterning solution for 4 hours before UV exposure. A photomask with lithographic patterns and a collimated UV light source (Omicure) were then used to generate patterns on the hydrogel substrates (methods as previous reported[30]. After UV exposure, hydrogel substrates were washed with a 1:1 DMSO:H2O solution for 3 hours with three buffer changes. Hydrogel substrates were then sterilized by a 1-hr incubation with 5% Isopropanol and two 1-hr washes with sterile PBS at 37°C under gentle shaking. Labeled proteins were diluted in sterilized PBS and incubated with hydrogel substrates overnight at 4°C for protein immobilization. Hydrogels were then washed 3-times (1-hour each) with sterile PBS to remove untethered proteins before cell seeding.
2.9. VIC isolation and culture
Primary valvular interstitial cells (VICs) were isolated from fresh male porcine hearts (Hormel) as previously described[19,33]. Briefly, porcine aortic valve leaflets were excised from the hearts, rinsed in Earle’s Balanced Salt Solution (Life Technologies) with 1% penicillin-streptomycin (Life Technologies) and 0.5 mg/mL fungizone (Life Technologies) and subsequently incubated in a collagenase solution (250 units/mL, Worthington) for 30 min at 37 °C. Endothelial cells were removed by vortexing and centrifugation, followed by another incubation in collagenase solution for 60 min at 37 °C. Filtration of the solution with a 100 μm cell strainer was conducted to separate VICs from the remaining extracellular matrix. Next, VICs were pelleted by centrifugation and then re-suspended in growth media (Media 199 (DMEM, Gibco Life Technology #11150–059) supplemented with 15% fetal bovine serum (FBS, Life Technologies), 2% penicillin-streptomycin, and 0.5 mg/mL fungizone). Fibroblast growth factor 2 (FGF-2) and transforming growth factor β 1 (TGF-β1) were supplemented as stated (FGF-2, Sigma-Aldrich and TGF-β1, R&D Systems). The isolated VICs were expanded on tissue culture polystyrene (TCPS) until 80% confluence and frozen down in FBS containing 20% DMSO and stored in liquid nitrogen as Passage 1 (P1). P2 to P4 VICs were generated by expanding the P1 stock in growth media (DMEM supplemented with 15% FBS, 50 U/ml penicillin, 50 mg/ml streptomycin and 1 mg/ml fungizone), and P2 to P4 VICs were used in all the reported experiments. VICs were cultured on TCPS in growth media at 5 × 105 cells/cm2 for two days prior to seeding on hydrogels at 40,000 cells/cm2 in media with a serum level of 1% FBS for all conditions except for osteogenic differentiation medium conditions(15% FBS)[34]. Hydrogel samples were then transferred to a new plate with fresh media 24 h post cell seeding. For myofibroblast activation studies, VICs were cultured for 2 days in 1% FBS medium before fixation, while for calcific nodule formation, VICs were for culture for 5 days in 1% FBS medium for before fixation for immunostaining and 7 days in 15% FBS osteogenic differentiation medium before fixation and histological staining (Alizarin Red & ALP staining).
2.10. Immunostaining
VICs were fixed in 4% paraformaldehyde for 45 mins at room temperature, rinsed in PBS twice, and then permeabilized using 0.1% TritonX-100 in PBS for 1 h. Next, samples were blocked in PBS with 5% bovine serum albumin (BSA) for 1h at room temperature to minimize non-specific protein binding. Anti-KI67 (1:250, rabbit, Abcam) and anti-αSMA (1:200, mouse, Abcam) primary antibodies in 5% BSA were applied to samples and incubated for 1h at room temperature with gentle shaking. Primary antibodies were removed by rinsing in PBST (0.5 wt% Tween-20 in PBS) for 10 min. Samples were then incubated at room temperature with secondary antibodies (1:1000, goat anti-rabbit AlexaFluor 488; goat anti-mouse AlexaFluor 647, invitrogen), phalloidin (1:300, Sigma Aldrich) and DAPI (1 mg/mL; Sigma) in PBS with 1% BSA. After 1 h, the secondary antibody solution was removed and the samples were rinsed three times for 10 min with PBST. All immunostained samples were stored in PBS at 4°C until imaging (Operetta; Perkin Elmer).
2.11. Western Blots
Chemiluminescence western blot techniques were used to determine α-SMA protein expression levels. Protein expression values are relative to total protein (Ponceau stain) staining. Polyvinylidene difluoride (PVDF) membranes (Bio-Rad) were used and probed for primary α-SMA antibody (Abcam) diluted 1:2000 in blocking solution (5% milk in 0.1% Tween TBS (TBST)) at 4C overnight. Membranes were washed with TBST and incubated with a secondary goat-anti-mouse horseradish peroxidase conjugated antibody (Jackson ImmunoResearch) at 1:10000 dilution in blocking solution for 1h at RT. Protein chemiluminescent signal was detected using luminol-based enhanced chemiluminescence (ECL) horseradish peroxidase (HRP) substrate (ThermoFisher) and an ImageQuant LAS 4000 detector.
2.12. Histology Staining
Alizarin red staining was performed with alizarin red solution kit (Millipore). VICs were fixed in 10% formalin for 15 mins at room temperature, rinsed in PBS twice for 10 mins. Alizarin red solution was filtered with 0.45 μm filter before addition. After 5 mins of incubation with alizarin red solution at room temperature and protected from light, excess dye was removed and the hydrogels were washed with DI H20 three times for 15 mins before imagining with brightfield microscope (Nikon CFI60). Alkaline phosphatase (ALP) staining was performed with alkaline phosphatase detection kit (Millipore). ALP detection solution was prepared by mixing Fast Red Violet (FRV) with Naphthol AS-BI phosphate solution and water in a 2:1:1 ratio (FRV:Naphthol:water). VICs culture on hydrogel substrates were fixed in 4% paraformaldehyde for 2 mins at room temperature and rinsed with 1x TBST buffer (20 mM Tris-HCl, pH 7.4, 0.15M NaCl, 0.05% Tween-20). 1mL ALP staining solution was added to each well in a 12-well plate, followed by 15 mins incubation protected from light at room temperature. Gels were rinsed with 1x PBST and stored in PBS before imaging.
2.13. Statistical analysis
For each experiment, at least three biological replicates were performed using different pools of male porcine hearts with two technical replicates in each biological replicate. Each pool of male porcine VICs consists of at least 10 porcine hearts. For cell analyses, at least 200 cells were assessed per condition per technical replicate. Data were compared using one-way ANOVAs and Bonferroni post-tests in Prism 7 (GraphPad Software, Inc) unless otherwise stated. Data is presented as mean ± standard deviation.
3. Results
3.1. Control of VIC activation via solubly presented cytokines
Hydrogel substrates with tunable mechanical properties designed for primary valvular interstitial cell (VIC) culture were formed by reacting 4-arm poly(ethylene glycol) (PEG) azide and alkyne macromers via a Copper click mechanism (Figure 1A). As hydrogel substrates stiffness can regulate VIC activation[30,35], three different hydrogel formulations were designed with different crosslinking densities. As shown in Figure S1A, hydrogel substrates with Young’s moduli of 1.8±0.2 kPa (soft), 7.8±0.3 kPa (intermediate), 33.1±0.5 kPa (stiff) were fabricated. Significant α–smooth muscle actin (αSMA) activation differences were observed for VICs cultured on soft, intermediate and stiff substrates, of 18±6%, 48±7%, 89±9%, respectively. The intermediate hydrogel substrate was chosen to study the effect of biochemical cues on VIC activation, not only because VICs showed a mixed population of fibroblast and myofibroblast on these substrates, but also because 7.8 kPa is similar to the stiffness of healthy heart valve tissue[30,36]. VIC activation can then be influenced via the introduction of exogenous cytokines to culture media. Fibroblast growth factor 2 (FGF-2) was introduced to promote a quiescent VIC phenotype, as quantified by the decreased expression of αSMA stress fibers (Figure 1B). FGF-2 treatment resulted in a significant decrease of VIC activation from 48±7% to 19±3% at 10ng/mL, while no decrease was observed with treatments of 0.1 and 1ng/mL (Figure 1B).
Figure 1. VIC activation is regulated by biochemical cues.
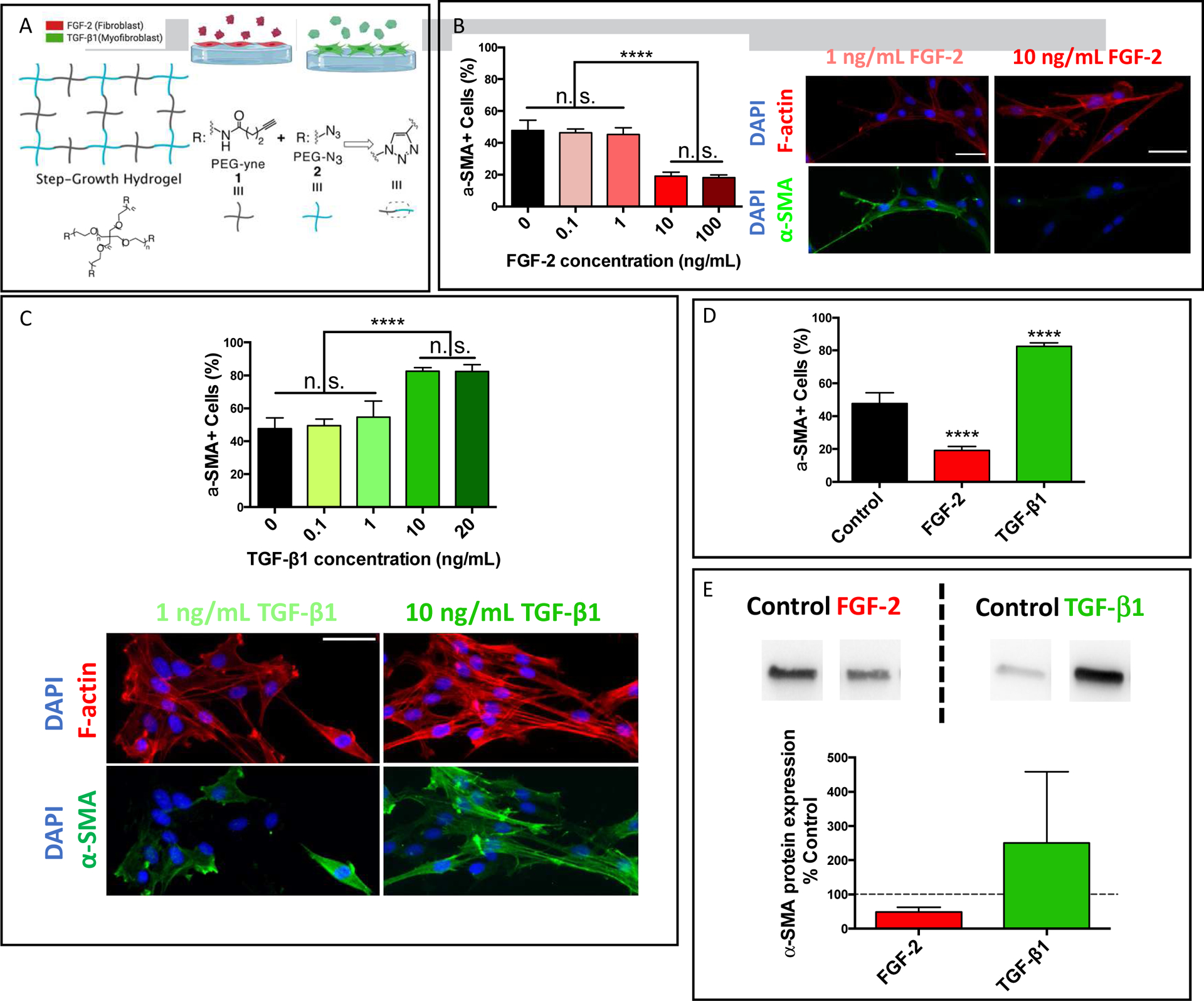
A) Schematic of hydrogel substrates formed by copolymerizing PEG-Azide and PEG-Alkyne macromers via a copper catalyzed click reaction. B) Dose curve of solubly delivered FGF-2 on VIC activation cultured on hydrogel substrates with a Young’s modulus, E = 7.8±0.3 kPa. Representative immunostaining images of αSMA stress fibers (green) and F-actin (red) for VICs treated with either 1 or 10 ng/mL of FGF-2. C) Dose curve of solubly presented TGF-β1 on VIC activation on hydrogel substrates with a Young’s modulus, E = 7.8±0.3 kPa. Representative immunostaining images of αSMA stress fibers (green) and F-actin (red) for VICs treated with either 1 or 10 ng/mL of TGF-β1. D) FGF-2 and TGF-β1 effects, both at 10 ng/mL, on VIC phenotypic changes as assessed by the percentage of αSMA stress fiber positive cells. E) FGF-2 and TGF-β1, both at 10 ng/mL, effects on VIC αSMA protein expression as analyzed via western blot. Values are plotted as mean ± standard deviation. *: p<0.05, ****: p<0.0001, based on one-way ANOVA. n = 3 with duplicates, more than 200 VICs were analyzed per sample. Scale bar = 50 μm.
Transforming growth factor β 1 (TGF-β1) was then chosen as a pro-fibrotic cue to promote the activated myofibroblast phenotype. VIC activation increased from 48±7% to 83±2% with treatment of TGF-β1 at a concentration of 10ng/mL (Figure 1C). Comparing these conditions, three distinct cell populations were observed: a mixed population of fibroblast and myofibroblast in untreated conditions, a myofibroblast dominant population with TGF-β1 treatment, and a fibroblast dominant population in FGF-2 treated conditions (Figure 1D). These distinct populations were further confirmed via western blot analysis of αSMA protein expression, demonstrating a significant decrease in expression in FGF-2 conditions and an increase in TGF-β1 conditions (Figure 1E).
3.2. Control of VIC phenotype with tethered biochemical cues
The effects of these cues on VIC activation after tethering into a hydrogel platform was then investigated. Hydrogels were synthesized off-stoichiometry with excess of 4-arm PEG alkyne, so that it maintained excess ‘yne’ functionalities throughout the network to enable subsequent thiol-yne photo patterning. The two orthogonal click pairs (azide / dibenzocclooctyne (DBCO) and methyltetrazine / transcyclooctene (TCO)) were chosen due to their orthogonality, as well as that the tethered groups (azide and methyltetrazine) are unaffected by the photoinitiated thiol-yne reaction. Proteins functionalized with the complementary moieties (DBCO and TCO, correspondingly) were then swelled in and tethered into the hydrogel scaffold. First, hydrogels were formed and ubiquitously photopatterned with either azide or methyltetrazine moieties (Figure 2A). FGF-2 and TGF-β1 were then modified with the corresponding click pairs (DBCO and TCO, respectively), a process that did not significantly alter their bioactivity (Figure S2). These proteins were then swollen into the networks individually to tether them ubiquitously throughout the hydrogels. A Smad3-GFP reporter cell line[10,37] was used to confirm TGF-β1 signaling on the TGF-β1 patterned gels. Cells on TGF-β1 patterned gels had two-fold higher levels of nuclear localized Smad-3, indicating TGF-β1 signaling was activated, while cells on non-patterned gels had mostly cytoplasmic Smad-3 (Figure 2B). Similarly, to confirm preserved bioactivity of tethered FGF-2, cellular proliferation was assessed, as the mitogenic properties of this cues have been previously shown[19]. A five-fold increase (from 10±2% to 54±3%) in the proliferative marker Ki-67 was observed on FGF-2 patterned gels compared to non-patterned conditions (Figure 2C). TGF-β1 tethering did not influence cell proliferation compared to VICs on unmodified gels (Figure S3). Myofibroblast activation was then quantified for VICs on gels patterned with either FGF-2 or TGF-β1. A significant increase (from 48±7% to 82±3%) in αSMA positive cells was observed on TGF-β1 gels, while there was a significant decrease (from 48±7% to 17±6%) of activated cells on FGF-2 gels (Figure 2D). These values were similar when compared to the effects of solubly delivered cues (Figure 1D&2D). This was further confirmed via αSMA protein expression analysis, where αSMA expression was decreased for cells on FGF-2 patterned gels and increased for cells on TGF-β1 gels (Figure 2D & Figure S4).
Figure 2. Full length proteins remain bioactive after immobilization.

A) Schematic of the method for full-length proteins conjugation into hydrogel scaffold via a photo-initiated thiol-yne reaction and two orthogonal click reactions, SPAAC and IEDDA. B) The bioactivity of TGF-β1 (10 ng/mL) after tethering was confirmed by a Smad3 nuclear localization in a mouse embryonic fibroblast (MEF) reporter cell line, as shown in representative immunostaining images of Smad (green) and nucleus (blue), and relative nuclear GFP intensity quantification. C) FGF-2 remains bioactive after immobilization, upregulating VIC proliferation by five-fold at 10 ng/mL, as quantified by percentage of Ki67 positive cells. Representative immunostaining images for cytoskeletal F-actin (red) and Ki67 (purple). D) Tethered FGF-2 and TGF-β1 regulate VIC phenotypic changes and αSMA protein expression at 10 ng/mL. Representative immunostaining images of F-actin (red), and αSMA stress fibers (green) of VICs in response to FGF-2 and TGF-β1 tethered to the substrate. Representative images of western blot for αSMA protein and quantification of intensity values with respect to control samples. Values are plotted as mean ± standard deviation. ****: p<0.0001, based on one-way ANOVA. n = 3 with duplicates, more than 200 cells were analyzed per sample. Scale bar = 50 μm.
3.3. Incorporation of multiple bioactive cues into a single hydrogel network
Multiple proteins were then tethered into single networks to control cell fate and to study the interactions between biochemical cues. Hydrogel substrates were first homogeneously patterned with a pendant azide moiety. After removing unreacted azide moieties, a second patterning step was performed to ubiquitously incorporate the methyltetrazine group. Sequentially, two proteins functionalized with either DBCO or TCO moiety were swelled in and tethered into the scaffold homogeneously. Here, both FGF-2 and TGF-β1 were tethered into hydrogel networks (Figure 3A). FGF-2 (10 ng/mL) was able to protect VICs from the pro-fibrotic effects of soluble TGF-β1 (10ng/mL), reducing VIC activation from 83±2% to 16±5% (Figure 3B). As TGF-β1 concentration was increased to 20 ng/mL, VIC activation significantly increased to 29±6%, but levels remained significantly lower than conditions with 20 ng/mL TGF-β1 alone (82±3% activation) (Figure 3B & Figure 1D). This protective effect was also observed on gels with tethered TGF-β1 where the introduction of FGF-2 resulted in VIC deactivation from 83±2% to 56±7%, even at FGF-2 concentrations ten-fold lower than that of TGF-β1 (Figure 3B, Figure 1D). VIC response to tethered proteins was comparable with that when treated with equivalent concentrations of the soluble cues (Figure S5).
Figure 3. Multiple proteins are presented simultaneously to study their interactive effect on VIC cellular responses.

A) Schematic of the method for multiple full-length protein conjugation into a single hydrogel scaffold via orthogonal click reactions, SPAAC and IEDDA. B) FGF-2 has a protective effect on VICs maintaining their fibroblast phenotype against TGF-β1 myofibroblast activation as observed by measuring αSMA positive cells. C) TGF-β1 and BMP-2 (both at 10 ng/mL) have synergistic effects on VIC calcific nodule formation, as shown in representative brightfield images of VICs in response to multiple tethered proteins and quantification of nodule number per area. Values are plotted as mean ± standard deviation. **: p<0.01, ****: p<0.0001, based on one-way ANOVA. n = 3 with duplicates, more than 200 VICs were analyzed per sample. Scale bar = 200 μm.
The combined effect of multiple biochemical cues on VIC nodule formation, another important VIC cellular response related to valve calcification, was investigated. In order to study VIC nodule formation, stiff hydrogel substrates (E ~ 33.1±0.5 kPa) were chosen to promote VIC nodule formation as well as to mimic the stiffness of diseased heart valve tissue[36]. TGF-β1 and bone morphogenesis protein-2 (BMP-2) were introduced to VIC culture, due to the pro-myofibroblast-activation potential of TGF-β1 and the osteogenic potential of BMP-2. BMP-2 was functionalized with DBCO without significantly altering its bioactivity (Figure S6). A synergistic effect of TGF-β1 and BMP-2 was observed, where only VICs on gels patterned with both proteins demonstrated significant nodule formation (1530±380 nodules/cm2) compared to those on unpatterned gels (30±90 nodules/cm2) or those patterned solely with TGF-β1 (180±250 nodules/cm2) or BMP-2 (270±290 nodules/cm2) (Figure 3C). Histological staining demonstrated significant increases in calcium phosphate (Alizarin Red) and osteogenic differentiation (alkaline phosphatase) in the dual patterned gels compared to the other conditions (Figure 3D).
3.4. Spatial control over bioactive cues in a single gel
Distinct spatial patterns were created within individual PEG hydrogels using a photomask with lithographic patterns (Figure 4A). By controlling the regions of UV exposure during each step of photo-patterning, azide and tetrazine moieties were immobilized into distinct regions. After the spatial immobilization of azide and tetrazine, a solution of both DBCO and TCO modified proteins was added to the scaffold. Fluorescently labeled BSA was first chosen as a model to demonstrate spatial control over protein patterning and spatial fidelity. BSA tethering was achieved with high specificity in a variety of patterns (Figure 4B). Light exposure could also be varied to control protein incorporation (Figure 4B). A series of circle patterns was chosen for further experiments. FGF-2 and TGF-β1 were patterned in this approach to control VIC activation in a single hydrogel network. VICs in the FGF-2 patterned region demonstrated a deactivated, quiescent fibroblast phenotype (28±9% activation), while those located in the TGF-β1 patterned region had an activated, myofibroblast phenotype (79±9% activation) (Figure 4C). These conditions demonstrated similar levels of VIC activation to those cultured on gels with a single patterned cue (Figure 4D & Figure 2D).
Figure 4. Spatial control over protein presentation and their effects on VIC myofibroblast activation.

A) Schematic of the method to conjugate multiple full-length proteins incorporated into a single hydrogel scaffold with precise spatial control by photo-initiated thiol-yne reaction and two orthogonal click reactions, SPAAC and IEDDA. B) BSA was used as model protein to demonstrate pattern orthogonality and fidelity. Light exposure is controlled to alter protein immobilization as shown by the linear correlation between relative intensity of the pattern and light dose. The light intensity at 365 nm was kept constant at 10 mW/cm2 and gels were exposed for up to 100s with 20s intervals. C) Illustration of spatially patterned FGF-2 and TGF-β1 at 10 ng/mL, as well as representative images of VICs immunostained for nucleus (blue, DAPI), Cell Mask (red, cytoplasmic stain) and αSMA (green) near the interface of the two patterns. D) Quantification of VIC activation on FGF-2 and TGF-β1 patterned regions, validating FGF-2 anti-fibrotic effects and TGF-β1 pro-fibrotic effects. Values are plotted as mean ± standard deviation. ****: p<0.0001, based on one-way ANOVA. n = 3 with duplicates, more than 200 VICs were analyzed per sample. Scale bar = 50 μm.
3.5. Control over cell-cell interaction on spatially patterned hydrogel substrates
Hydrogels with several pattern area sizes and varied interface perimeter were then designed to control VIC cell-cell interaction on individual hydrogel substrates. As the size of the TGF-β1 patterned region increased by 4-fold, VIC activation in the FGF-2 region increased from 28±9% to 44±6% (Figure 5A). This demonstrates that an increase in the size of the myofibroblast population can lead to increased activation of the neighboring VICs, even when exposed to the anti-fibrotic cue FGF-2.
Figure 5. Spatial control over the presentation of multiple proteins to understand the effects of cell-cell interactions on VIC activation.

A) Illustration of pattern designs for TGF-β1 (10 ng/mL, activating) and FGF-2 (10 ng/mL, deactivating) presentation and quantification of VIC activation in different regions. Increasing the size of the TGF-β1 pattern induces higher VIC activation on FGF-2 patterned region. B) Illustration of different patterns and regions of interest, as well as quantification of VIC activation in these different regions based on distance from the TGF-β1 patterned region. VICs in the mid section of the hydrogels with the larger TGF-β1 pattern have increased VIC activation with respect to those in the outer section. C) Illustration of pattern designs with identical TGF-β1 pattern sizes, but different interfacial areas with the FGF-2 regions. Quantification of VIC activation in each of the different regions. Increased interfacial area between the TGF-β1 and FGF-2 regions results in significant increase in VIC activation on the FGF-2 regions. D) Representative images of VIC activation around regions interface on different pattern designs. Immunostained images for nucleus (blue, DAPI), Cell Mask (red, cytoplasmic stain) and αSMA stress fibers (green). Values are plotted as mean ± standard deviation. *: p<0.05, **: p<0.01, ***: p<0.001, ****: p<0.0001, based on one-way ANOVA. n = 3 with duplicates, more than 200 VICs were analyzed per sample. Scale bar = 50 μm.
To further elucidate the mechanism behind this phenomenon, VIC activation on the FGF-2 region was analyzed with respect to the distance from the TGF-β1 region. As shown in Figure 5B, VIC activation on the hydrogel substrate with an enlarged TGF-β1 pattern was significantly higher when closer to the TGF-β1 pattern (“mid region”) (49±4%) compared to VICs farther away (“outer region”) (41±4%,). The TGF-β1 pattern was then varied to create conditions with an identical pattern size, but different degrees of interface between the different patterned zones. While VIC activation within the TGF-β1 patterns was consistent between conditions (~80%), activation increased from 44±6% to 60±5% in the FGF-2 region as the TGF-β1 pattern was split into smaller patterns (Figure 5C).
Similar effects of cell-cell interaction were also observed on VIC nodule formation. VICs formed significantly more nodules on TGF-β1/BMP-2 dual patterned region (1380±480 nodules/cm2) compared to TGF-β1 region (320±280 nodules/cm2), as shown by the representative images around the interface in Figure 6C. There was an increase in nodule number formation (320±280 nodules/cm2 to 600±420 nodules/cm2) in the TGF-β1 region by enlarging the size of the dual protein (TGF-β1/BMP-2) region by four-fold (Figure 6A). Similarly, an increase in calcification (Alizarin Red) and osteogenic markers (ALP) was observed in conditions with larger dual-protein patterned region. Pattern designs with the same surface area but different interfacial regions (as those used previously in Figure 5) were then used. No significant changes in nodule density was observed as the interface between the two regions was increased (Figure 6A).
Figure 6. Spatial control over the presentation of multiple proteins to understand the effects of cell-cell interaction on VIC nodule formation.

A) Illustration of pattern designs for TGF-β1 (10 ng/mL) and TGF-β1/BMP-2 (both 10 ng/mL) presentation and quantification of VIC nodule numbers in different regions. Larger TGF-β1/BMP-2 pattern induces more VIC nodule formation on the TGF-β1 only region. B) Illustration of different patterns designs for FGF-2 (10 ng/mL) and TGF-β1 /BMP-2 (both 10 ng/mL) presentation and quantification of VIC nodule numbers on hydrogel substrate with different pattern designs. There is no signficant difference in VIC activation between the 4 FGF-2 patterns. C) Representative images of VIC nodule formation by immunostaining for nucleus (blue, DAPI) and cytoplasmic marker Cell Mask (red) around the interfacial area between the different regions of interest. D) Representative brightfield images of VIC nodule formation via histological stainings, alizarin red and ALP, in different regions of interest. Values are plotted as mean ± standard deviation. *: p<0.05, ***: p<0.001, ****: p<0.0001, based on one-way ANOVA. n = 3 with duplicates. Scale bar = 50 μm.
Finally, three proteins were patterned into a single hydrogel substrate simultaneously in order to achieve spatial control of VIC nodule formation, as well as to further elucidate the effects of VIC cell-cell interactions on a single hydrogel. FGF-2 was patterned in the outer region of gels with an inner region containing TGF-β1 and BMP-2. While nodule formation was observed outside the dual-patterned regions of previous gels (Figure 5), the inclusion of FGF-2 prevented significant nodule formations outside the TGF-β1 /BMP-2 region (Figure 6B & 6D). Furthermore, as the TGF-β1 /BMP-2 pattern size was increased, there was no increase in nodule formation in the FGF-2 region. Similarly, when altering the interface perimeter of the patterns, no significant change in VIC nodule formation was observed (Figure 6B). Thus, multiple bioactive proteins could be independently patterned to precisely and spatially control VIC nodule formation, demonstrating the potential applications of this hydrogel system in tissue engineering and regenerative medicine, especially in vitro models to study valve disease and allow for drug screening.
4. Discussion
The ability to create heterogeneous cell culture environments has been of particular interests to the field of biomaterials of late. The cellular niche involves numerous different signaling moieties, and the ability to recreate this fully in vitro has proven challenging. Spatial control over the presentation of multiple full-length proteins is particularly difficult. Several strategies have been developed, where either multiple protein mimetic peptides[38] or single proteins are incorporated into a hydrogel scaffold[10]. Recent systems have developed this concept further, demonstrating how ligand-receptor interactions can be exploited to achieve simultaneous immobilization of multiple proteins in distinct patterns[9]. In this manuscript, several orthogonal click reactions were leveraged to spatially control the incorporation of multiple proteins simultaneously.
This platform allowed for the investigation of the interactions between multiple proteins in cell culture. While tethered FGF-2 demonstrated an anti-fibrotic effect on cultured VICs, it was also able to protect cells against the pro-fibrotic influence of TGF-β1. VICs on FGF-2 patterned gels required twice the concentration of TGF-β1 to cause a significant increase in activation, and remained less activated (compared to TGF-β1 treatment alone) even as the TGF-β1 concentration was raised to ten-fold that of FGF-2. This aligns with current literature that has hypothesized that FGF-2 has a role in maintaining VIC quiescence in a healthy valve, and acts to reverse VIC activation as a part of injury resolution[19].
This protein patterning approach allowed for control of cell fate within distinct zones in a single hydrogel substrate. VICs are known to exhibit two distinct phenotypes: a quiescent fibroblast state and an activated myofibroblast state[16]. Current efforts are being taken to understand the factors that are involved in the chronic or irreversible activation of these cells in the heart valve[39]. The creation of distinct biochemical signaling zones in a single hydrogel culture environment provides a unique opportunity to investigate the interaction of these cell types. Upon the introduction of a small TGF-β1 pattern into FGF-2 containing gels, there was little observed interaction between the cell types (i.e., cells in the FGF-2 region were not significantly more activated compared to cells on gels patterned with FGF-2 alone). However, as the TGF-β1 pattern diameter was increased from 3mm to 6mm, the protective effect of FGF-2 was diminished. In the 6mm TGF-β1 patterned gels, VICs on FGF-2 regions were 44±6% activated (Figure 5A), similar to the activation percentage on unmodified gels (Figure 1A). This parallels with a common consideration of aortic valve fibrosis, where a positive feedback loop (as myofibroblast populations increase, their influence on quiescent fibroblasts also increases) can accelerate fibrosis[11,40]. This effect can be a result from soluble factors secreted by activated VICs (such as TGF-β)[11,40], inducing VICs on the FGF-2 region to activate; via cell-cell contact or a combination of both. This effect was further exacerbated as the boundary between FGF-2 and TGF-β1 patterned regions was increased while maintaining the same pattern surface area as previous patterns. VIC activation on FGF-2 regions increased significantly from 44±6% to 60±5% when 9 smaller TGF-β1 patterns were used rather than a single large pattern (Figure 5B). These findings indicate that cell-cell contact could play a role in VIC crosstalk, which could be useful for discovering potential medical treatments for early stage fibrosis.
This platform was also useful in investigating the biochemical signaling that causes VIC nodule formation and calcification. Spatial control over nodule formation in hydrogel culture can be potentially useful in the fields of drug screening, disease modeling, and regenerative medicine. The role of specific cytokines in calcification of the aortic valve, a process known as aortic valve stenosis, has been debated. Both TGF-β1 and BMP-2 have been identified as possibly having roles in this pathway[25–27]. It was observed that the simultaneous presentation of both cues had a synergistic effect on VIC nodule formation and calcification. Similar to the previous results with TGF-β1 and FGF-2, nodule formation increased in the single TGF-β1 pattern as the dual protein pattern size was increased. However, when FGF-2 was patterned in the region surrounding the TGF-β1/BMP-2 pattern, this effect was no longer observed. This result could indicate that FGF-2 also has a protective role against calcification, rather than only fibrosis, and that misregulation of this process may contribute to valve calcification.[20] In this study, the interfacial area between VICs exposed to TGF-β1/BMP-2 and FGF-2 was controlled to investigate if a critical size of activation exists that leads to nodule formation, as this is a characteristic of fibrocalcific heart valve disease. Looking forward, the material design developed in this submission might further be applied to investigate further how VIC nodule nucleation occurs and how the calcification process begins. In a broader context, this system can be utilized to recapitulate complex biological interfaces and to assess cellular dynamics in response to these biochemical stimuli.
This dual protein patterning approach may prove beneficial for the design of complex cell culture environments in vitro. While this platform can recreate complex biochemical environments in a 2D manner, the translation to a 3D environment can be used to ask these complex biological questions. However, when using this approach for encapsulate cells (i.e., a 3D system), the cells would be exposed to the bioactive proteins during the patterning steps. System design must then be considered to select conditions where the initial dosing (both exposure time and concentration) do not convolute the effects of the cues on cell behavior. Further studies investigating the simultaneous patterning of multiple different proteins with high spatial resolution can be used to ask biological questions related to synergistic interactions of multiple cytokines using a simple material design.
5. Conclusions
As more is learned about the complexities of the cellular niche, more versatile cell culture scaffolds are needed to properly study cell function and disease. In this study, an orthogonal multi-protein patterning approach was designed to achieve precise and spatial control over three proteins in a hydrogel scaffold. This hydrogel system was used to study the interactive effects of multiple proteins on VIC myofibroblast activation and calcific nodule formation. Simultaneous patterning of these proteins revealed both synergies, as in the case of nodule formation, and protective effects, with FGF-2 shielding cells against pro-fibrotic cues. Furthermore, this system demonstrated how cell-cell interactions in complex biochemical environments can alter cellular behavior, as well as how varied biochemical presentation can affect this. Finally, it was shown how materials can be designed to precisely control cellular behaviors, which is of importance to tissue engineering and regenerative medicine. These findings are relevant to understanding the aortic valve niche, as well as for designing materials to model other complex cellular environments.
Supplementary Material
Acknowledgements
The authors would like to thank Megan E. Schroeder for help with VIC isolation. The authors would also like to acknowledge support from the National Institutes of Health (Grant R01 HL132353).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Sharing
The raw data used in this study are available from the corresponding author upon reasonable request.
References
- [1].Tibbitt MW, Anseth KS, Biotechnol. Bioeng 2009, 103, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Discher DE, Mooney DJ, Zandstra PW, Science (80-. ) 2009, 324, 1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Burdick JA, Murphy WL, Nat. Commun 2012, 3, 1269. [DOI] [PubMed] [Google Scholar]
- [4].Annabi N, Tamayol A, Uquillas JA, Akbari M, Bertassoni LE, Cha C, Camci-Unal G, Dokmeci MR, Peppas NA, Khademhosseini A, Adv. Mater 2014, 26, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].C. A. DeForest, Anseth KS, Nat. Chem 2011, 3, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].C. A. DeForest, Tirrell DA, Nat. Mater 2015, 14, 523. [DOI] [PubMed] [Google Scholar]
- [7].Floyd RA, Carney JM, Ann. Neurol 1992. [DOI] [PubMed] [Google Scholar]
- [8].Mosiewicz KA, Kolb L, van der Vlies AJ, Martino MM, Lienemann PS, Hubbell JA, Ehrbar M, Lutolf MP, Nat. Mater 2013, 12, 1072. [DOI] [PubMed] [Google Scholar]
- [9].Wylie RG, Ahsan S, Aizawa Y, Maxwell KL, Morshead CM, Shoichet MS, Nat. Mater 2011, 10, 799. [DOI] [PubMed] [Google Scholar]
- [10].Grim JC, Brown TE, Aguado BA, Chapnick DA, Viert AL, Liu X, Anseth KS, ACS Cent. Sci 2018, 4, 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Liu AC, Joag VR, Gotlieb AI, Am. J. Pathol 2007, 171, 1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yip CYY, Simmons CA, Cardiovasc. Pathol 2011, 20, 177. [DOI] [PubMed] [Google Scholar]
- [13].Usprech J, Chen WLK, Simmons CA, Wiley Interdiscip. Rev. Syst. Biol. Med 2016, 8, 169. [DOI] [PubMed] [Google Scholar]
- [14].Durbin AD, Gotlieb AI, Cardiovasc. Pathol 2002, 11, 69. [DOI] [PubMed] [Google Scholar]
- [15].Mabry KM, Lawrence RL, Anseth KS, Biomaterials 2015, 49, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rabkin-Aikawa E, Farber M, Aikawa M, Schoen FJ, J. Heart Valve Dis 2004, 13, 841. [PubMed] [Google Scholar]
- [17].Merryman WD, Schoen FJ, Curr. Cardiol. Rep 2013, 15, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schoen FJ, Circulation 2008, 118, 1864. [DOI] [PubMed] [Google Scholar]
- [19].Gonzalez Rodriguez A, Schroeder ME, Walker CJ, Anseth KS, APL Bioeng 2018, 2, 046104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cushing MC, Mariner PD, Liao J-T, Sims EA, Anseth KS, FASEB J 2008, 22, 1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Boilly B, Vercoutter-Edouart AS, Hondermarck H, Nurcombe V, Le Bourhis X, Cytokine Growth Factor Rev 2000, 11, 295. [DOI] [PubMed] [Google Scholar]
- [22].Khan R, Sheppard R, Immunology 2006, 118, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hagler MA, Hadley TM, Zhang H, Mehra K, Roos CM, Schaff HV, Suri RM, Miller JD, Cardiovasc. Res 2013, 99, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liu AC, Gotlieb AI, Am. J. Pathol 2008, 173, 1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fukui N, Ikeda Y, Ohnuki T, Hikita A, Tanaka S, Yamane S, Suzuki R, Sandell LJ, Ochi T, J. Biol. Chem 2006, 281, 27229. [DOI] [PubMed] [Google Scholar]
- [26].Lim J, Ehsanipour A, Hsu JJ, Lu J, Pedego T, Wu A, Walthers CM, Demer LL, Seidlits SK, Tintut Y, Am. J. Pathol 2016, 186, 2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xu S, Liu AC, Gotlieb AI, Cardiovasc. Pathol 2010, 19, 236. [DOI] [PubMed] [Google Scholar]
- [28].Jian B, Narula N, Li Q, Mohler ER, Levy RJ, Ann. Thorac. Surg 2003, 75, 457. [DOI] [PubMed] [Google Scholar]
- [29].Monroe MN, Nikonowicz RC, Grande-Allen KJ, Acta Biomater 2019, 97, 420. [DOI] [PubMed] [Google Scholar]
- [30].Ma H, Killaars AR, DelRio FW, Yang C, Anseth KS, Biomaterials 2017, 131, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Neves SC, Moroni L, Barrias CC, Granja PL, Trends Biotechnol 2019. [DOI] [PubMed] [Google Scholar]
- [32].Hiemenz PC, Lodge T, Polymer chemistry; CRC Press, 2007. [Google Scholar]
- [33].Johnson CM, Hanson MN, Helgeson SC, J. Mol. Cell. Cardiol 1987, 19, 1185. [DOI] [PubMed] [Google Scholar]
- [34].Duan B, Yin Z, Hockaday Kang L, Magin RL, Butcher JT, Acta Biomater 2016, 36, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Quinlan AMT, Billiar KL, J. Biomed. Mater. Res . Part A 2012, 100A, n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang H, Tibbitt MW, Langer SJ, Leinwand LA, Anseth KS, Proc. Natl. Acad. Sci 2013, 110, 19336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Erickson RA, Liu X, Mol. Biol. Cell 2009, 20, 1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yu LMY, Kazazian K, Shoichet MS, J. Biomed. Mater. Res . Part A 2007, 82A, 243. [DOI] [PubMed] [Google Scholar]
- [39].Lu L, Guo J, Hua Y, Huang K, Magaye R, Cornell J, Kelly DJ, Reid C, Liew D, Zhou Y, Chen A, Xiao W, Fu Q, Wang BH, Clin. Exp. Pharmacol. Physiol 2017, 44, 55. [DOI] [PubMed] [Google Scholar]
- [40].Farrar EJ, Pramil V, Richards JM, Mosher CZ, Butcher JT, Biomaterials 2016, 105, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.