

Abstract
Background:
Kinase fusions are rare and poorly characterized in breast cancer (BC). We aimed to characterize kinase fusions within a large cohort of advanced BC.
Patients and methods:
A total of 4854 patients with BC were analyzed by Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) targeted DNAseq and MSK-Fusion targeted RNAseq during the study time period.
Results:
Twenty-seven of 4854 (0.6%) patients harbored fusions: 11 FGFR (five FGFR2, three FGFR3, three FGFR1), five BRAF, four NTRK1, two RET, two ROS1, one ALK, one ERBB2, and one MET. A history of endocrine therapy was present in 15 (56%) of fusion-positive BC; eight of the 15 cases had available pre-treatment samples, of which six were fusion-negative. None of the fusion-positive BC samples harbored ESR1 hotspot mutations. Two patients with acquired LMNA-NTRK1 fusions and metastatic disease received larotrectinib and demonstrated clinical benefit.
Conclusion:
Kinase fusions in BC are extremely rare, and appear to be enriched in hormone-resistant, metastatic carcinomas and mutually exclusive with ESR1 mutations. The present study expands the spectrum of genetic alterations activating mitogen-activated protein kinase (MAPK) signaling that can substitute for ESR1 mutations in this setting. Molecular testing at progression after endocrine therapy should include fusion testing, particularly in the absence of ESR1 hotspot alterations, in an effort to identify additional therapeutic options which may provide substantial clinical benefit.
Keywords: breast cancer, endocrine therapy, kinase fusion, larotrectinib, resistance
INTRODUCTION
Approximately 70% of breast cancers (BC) express estrogen receptor (ER), which serves as a predictive biomarker for antiestrogen therapy. Resistance to endocrine therapy (ET) occurs in metastases, frequently the result of activating ESR1 alterations.1–4 Apart from alterations that reactivate ER, the RAS-mitogen-activated protein kinase (MAPK) pathway and MYC alterations are enriched in and predicted to mediate endocrine resistance through activation of parallel oncogenic signaling pathways.5 Still, there remain many uncharacterized causes of acquired endocrine resistance and limited insight into rational treatment approaches for such cancers.
Given the recent finding that ER-positive BC frequently express fusion proteins6 and the targetability of kinase fusions, we investigated the features of kinase fusions in BC including their emergence as a mechanism of acquired resistance to ET.
METHODS
Case selection and molecular testing
Approval was obtained from our institutional review board, and this study was conducted in accordance with US Common Rule. BC accessioned for DNA-based next generation sequencing (NGS) using Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT)7 and/or RNA-bascd targeted NGS with a custom Archer-based panel, MSK-Fusion8–10 between 1 January 2014 and 30 September 2019 were assessed for kinase fusions. The custom Archer-based MSK-Fusion panel8 used covers fusions involving the kinase domains of the following genes: ALK, BRAF, EGFR, ERBB2, ERBB4, FGFR1, FGFR2, FGFR3, KIT, MET, NTRK1, NTRK2, NTRK3, RET, and ROSl. The MSK-Fusion panel included ESR1 as of October 2018. Absence of fusions in pre-treatment material was confirmed by MSK-IMPACT, MSK-Fusion, break-apart FISH from ZytoVision (Bremerhaven, Germany) and/or Pan-Trk immunohistochemistry (IHC) with Abeam EPR17341 (Cambridge, MA) depending upon available material. Secretory carcinomas were excluded because the fusion incidence is already known.11
IHC testing for ER and progesterone receptor (PgR) and IHC and/or FISH for human epidermal growth factor receptor 2 (HER2) were carried out and reported according to the American Society of Clinical Oncology/College of American Pathologists guideline recommendations12,13 using a Food and Drug Administration-approved method. Pan-Trk IHC was also carried out if indicated.14
MSK-IMPACT, MSK-Fusion, break-apart FISH, IHC staining for ER, PgR, HER2 and pan-Trk and HER2 FISH are clinically validated assays that were carried out in Clinical Laboratory Improvement Amendments (CLIA)-accredited laboratories.
Reagents
Larotrectinib was purchased from AdooQ Bioscience, Irvine, CA. Ceritinib and fulvestrant were purchased from Sell-eckchem, Houston, TX. NTRK1 (30697), ALK (3633), phosphorylated AKT (4060), pan-AKT (2965), phosphorylated ERK1/2 (4370), ERK1/2 (4695), phospho-PLCγ (8713), PLCγ (5690), ERα (8644), PgR (8757), β-tubulin (2146), and glyc-eraldehyde-3-phosphate dehydrogenase (GAPDH) (2118) primary antibodies were obtained from Cell Signaling Technology, Danvers, MA.
Cell culture
Cell lines were maintained in a 37°C incubator with 5% C02 in humidified atmosphere. MCF7, T47D, and MCF10A cells were obtained from the American Type Culture Collection. MCF7 and T47D cells were grown in Dulbecco’s Modified Eagle Medium (DMEM)/F12 and RPMI-1640, respectively, supplemented with 10% fetal bovine serum (FBS) or 10% charcoal stripped bovine serum (Gibco, Waltham, MA), 100 μg/ml penicillin, 100 mg/ml streptomycin (Corning, Corning, NY), and 4 mM glutamine. MCF10A cells were cultured in DMEM/F12 supplemented with 5% horse serum (Gibco), 20 ng/ml EGF (Sigma-Aldrich, St. Louis, MO), 10 μg/ml insulin (Sigma-Aldrich), 0.5 μg/ml hydrocortisone (Sigma-Aldrich), 0.1 μg/ml cholera toxin (Sigma-Aldrich), 100 μg/ml penicillin, and 100 mg/ml streptomycin (Corning). For cell proliferation assay, 200 or 500 cells were seeded in each well of 96-well plates, and proliferation was measured with resazurin reagent (Fisher Scientific, Waltham, MA).
Generation of breast cell lines expressing kinase fusions
The LMNA-NTRK1 fusion expressing lentiviral vectors were generated by cloning coding sequences from LMNA exons 1–10 and NTRK1 exons 11–17 into pLenti lentiviral vectors. The coding sequences from EML4 exons 1–6 and ALK exons 20–29 were cloned into pLenti vectors to express EML4-ALK fusion protein. Lentivirus was prepared in HEK293T cells with pLenti empty vector or the plasmid with kinase fusion insert, and then used to transduce breast cell lines. Transduced cells were selected in medium supplemented with 1 mg/ml Geneticin for 2 weeks before being used in experiments.
Western blot
Protein samples were extracted from cultured cells with radioimmunoprecipitation assay (RIPA) buffer (Thermo Scientific) and quantified by the bicinchoninic acid (BCA) assay. Protein fractionated on NuPAGE 4–12% Bis-Tris gels (Life Technologies, Waltham, MA) was transferred on to nitrocellulose membranes. All primary antibodies were used with 1 : 1000 dilution, except GAPDH antibody, which was used at 1 : 5000 dilution in Tris-buffered saline, 0.1% Tween 20 containing 5% bovine serum albumin and 0.02% sodium azide. Peroxidase-conjugated goat anti-rabbit immunoglobulin G secondary antibody (Sigma-Aldrich) was used at 1 : 10000 dilution in TBST containing 2.5% skim milk. The blot was visualized by chemiluminescence.
Animal studies
Athymic nu/nu BALB/c female mice aged 6–8 weeks were obtained from Harlan Laboratories, Inc., Indianapolis, IN and maintained in pressurized ventilated caging. This study was carried out in compliance with institutional guidelines under an Institutional Animal Care and Use Committee (IACUC) approved protocol (MSKCC#12-10-016). MCF7 xenograft tumors expressing empty vector or LMNA-NTRK1 fusion were established in nude mice by subcutaneously implanting 0.18 mg sustained release 17β-estradiol pellets with a 10 g trocar into one flank followed by injecting 1 × 107 cells suspended 1 : 1 (volume) with reconstituted basement membrane (Matrigel, Collaborative Research, Bedford, MA) on the opposite side 10 days afterwards. When tumors reached a size of ~150 mm3, the 17β-estradiol pellets were removed from the mice. The mice bearing tumors expressing LMNA-NTRK1 fusion were randomly divided into two groups and treated with vehicle or 200 mg/kg of larotrectinib via gavage once daily. The mice bearing MCF7 tumors expressing the empty vector were treated with vehicle as control. Tumor dimensions were measured with Vernier calipers and tumor volumes calculated [π/6 × larger diameter × (smaller diameter)2]. In this study, there was no blinding of the investigator as randomization of animals was done. Based upon our previous work measuring the variability in size and growth of MCF7 xenografts, we estimated 10 mice/group would allow us to detect tumor size differences of >200 mm3.
RESULTS
Prevalence and clinicopathologic features
Of 4854 patients with BC analyzed via MSK-IMPACT targeted DNAseq and subsequent MSK-Fusion targeted RNAseq when indicated (10%, 495/4854) during the study time period, 27 patients (0.6%) harbored fusions (Table 1). These fusions included 11 FGFR fusions (five FGFR2, three FGFR3, three FGFR1), five BRAF fusions, four NTRK1 fusions, two RET fusions, two ROS1 fusions, one ALK fusion, one ERBB2 fusion, and one MET fusion. The median age at diagnosis for the fusion-positive cohort was 49 years with a range of 28–76 years. The clinicopathologic features are summarized in Table 2. Of the 27 BC with fusions, 21 were ductal, five lobular, and one mixed ductal and lobular. Kinase fusions were detected in 19 metastatic tumors (70%) and eight primary tumors (30%). The majority of metastatic tumors were previously exposed to ET (n = 15/19, 79%) at the time of fusion detection. In contrast, none of the primary tumors with kinase fusions had a history of ET before diagnosis. Of the four metastatic tumors with no prior ET, three were ER-negative and one was ER-positive, but the patient had declined adjuvant ET. A change in the tumor’s receptor status from ER-positive in the primary to ER-negative in the metastasis was observed in 26% (5/19) of cases, with the metastasis showing fusions in NTRK1 (n = 1), FGFR1 (n = 1), FGFR3 (n = 1), and BRAF (n = 2).
Table 1.
Kinase fusions and clinicopathologic features
| CBioPortal ID | 5′ gene | 3′ gene | NGS platform | Age (years) | Prior endocrine therapy | P versus M | Subtype | ER/PgR/HER2 |
|---|---|---|---|---|---|---|---|---|
| P-0025349 | CCDC6 exon 1 | RET exon 12 | I, F | 43 | No | P | Lobular | pos/pos/neg |
| P-0021788 | FGFR2 exon 17 | VPS26A exon 2 | I, F | 45 | Tamoxifen, anastrozole | M | Ductal | pos/pos/neg |
| P-0022337 | FGFR2 exon 17 | TBC1D4 exon 2 | I, F | 45 | No | M | Ductal | pos/pos/neg |
| P-0024183 | FGFR2 exon 17 | PLXNA2 exon 2 | I, F | 43 | No | P | Ductal | neg/neg/neg |
| P-0031242 | FGFR2 exon 17 | TACC2 exon 17 | I, F | 44 | No | M | Ductal | neg/neg/neg |
| P-0022569 | FGFR3 exon 17 | TACC3 exon 8 | I, F | 47 | No | P | Ductal | neg/pos/neg |
| P-0021893 | GTF2E2 exon 2 | FGFR1 exon 10 | I, F | 52 | Anastrozole | M | Ductal | neg/neg/neg |
| P-0031476 | HDAC2 exon 1 | ROS1 exon 34 | I, F | 34 | Letrozole, fulvestrant | M | Ductal | pos/neg/neg |
| P-0015494 | IGF1R exon 15 | ROS1 exon 34 | I, F | 55 | No | P | Ductal | neg/neg/neg |
| P-0021127 | KIAA1549 exon 10 | BRAF exon 9 | I, F | 54 | Anastrozole | M | Lobular | neg/neg/neg |
| P-0023994 | LMNA exon 10 | NTRK1 exon 11 | I, F | 43 | Anastrozole | M | Ductal | neg/neg/neg |
| P-0026614 | SPECC1L exon 9 | RET exon 12 | I, F | 56 | Anastrozole, fulvestrant | M | Lobular | pos/neg/neg |
| P-0029981 | SPOCK1 exon 6 | BRAF exon 10 | I, F | 41 | Tamoxifen | M | Ductal | pos/neg/amp |
| P-0034117 | KIAA1549 exon 12 | BRAF exon 10 | Ia | 70 | No | M | Ductal | neg/neg/neg |
| P-0000104 | ZBTB7B exon 4 | NTRK1 exon 12 | I, F | 49 | Exemestane, fulvestrant | M | Lobular | pos/neg/neg |
| P-0004477 | AGK exon2 | BRAF exon 8 | I, F | 76 | Letrozole | M | Lobular | pos/neg/neg |
| P-0006787 | ST7 exon 1 | MET exon 11 | Ia | 55 | Tamoxifen, arimidex | M | Ductal | pos/pos/neg |
| P-0024463 | FGFR3 exon 15 | TACC3 exon 10 | I, F | 50 | No | P | Ductal | neg/neg/neg |
| P-0013299 | EML4 exon 6 | ALK exon 20 | I, F | 60 | Letrozole, fulvestrant | M | Ductal | pos/neg/neg |
| P-0006601 | MINDY4 exon 5 | BRAF exon 9 | I, F | 52 | Tamoxifen, aromasin, fulvestrant | M | Ductal | neg/neg/neg |
| P-0044467 | SCP2 exon 13 | NTRK1 exon 11 | I, F | 49 | Tamoxifen, letrozole | M | Ductal | pos/neg/neg |
| P-0027759 | FGFR2 exon 17 | CCDC170 exon 10 | I, F | 58 | No | P | Ductal | neg/neg/neg |
| P-0039478 | SMIM19 exon 1 | FGFR1 exon 7 | I, F | 34 | No | P | Ductal | pos/pos/neg |
| P-0042695 | ERBB2 exon 25 | BCAR4 exon 3 | I, F | 52 | No | M | Ductal | neg/neg/neg |
| P-0042785 | GSK3B exon 1 | FGFR1 exon 2 | I, F | 28 | No | P | Ductal | neg/neg/neg |
| P-0006660 | FGFR3 exon 17 | TACC exon 7 | I, F | 51 | Letrozole, fulvestrant | M | Mixed | neg/neg/neg |
| P-0046560 | LMNA exon 12 | NTRK1 exon 12 | I, F | 48 | Letrozole, fulvestrant, tamoxifen | M | Ductal | pos/neg/neg |
ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; MSK-IMPACT, Memorial Sloan Kettering-tegrated Mutation Profiling of Actionable Cancer Targets; NGS, next generation sequencing; PgR, progesterone receptor.
I = MSK-IMPACT, F = MSK-Fusion.
No material available for MSK-Fusion.
Table 2.
Clinicopathologic characteristics of kinase fusion positive breast cancer samples
| Clinicopathologic characteristics | Number (%) |
|---|---|
| Age at breast cancer diagnosis, median (range), years | 49 (28–76) |
| Histologic subtype (primary tumor) | |
| Invasive ductal carcinoma | 21 (78%) |
| Invasive lobular carcinoma | 5 (18%) |
| Invasive mixed ductal/lobular carcinoma | 1 (4%) |
| Tumor tested | |
| Primary | 8 (30%) |
| Metastasis | 19 (70%) |
| Receptor status (n = 27) | |
| ER-positive | 13 (48%) |
| PgR-positive | 6 (22%) |
| HER2-positive | 1 (4%) |
| Triple-negative | 13 (48%) |
| Endocrine therapy (ET), metastatic tumors (n = 19) | |
| ER-positive metastasis, post-ET | 10 |
| ER-positive metastasis, no ET | 1 |
| ER-negative metastasis, post-ET | 5 |
| ER-negative metastasis, no ET | 3 |
| Primary versus metastatic receptor status, post-ET (n = 19) | |
| pos>pos | 11 (58%) |
| pos>neg | 5 (26%) |
| neg>neg | 3 (16%) |
| Frequency of selected molecular alterations* | |
| PIK3CA mutation, P = 0.05 | 15 (56%) |
| AKT1 mutation, P = 0.65 | 2 (8%) |
| ESR1 mutation, P = 0.16 | 0 |
| ERBB2 mutation, P = 1.00 | 1 (4%)a |
| CDH1 mutation, P = 0.40 | 5 (19%) |
| ERBB2 amplification, P = 0.24 | 1 (4%)a |
ER, estrogen receptor; ET, endocrine therapy; HER2, human epidermal growth factor receptor 2; PgR, progesterone receptor.
ERBB2 mutation and amplification detected in same post-ET case (P-0029981).
P value (Fisher’s exact test with two-tailed P value): frequency of molecular alterations in fusion positive versus fusion negative breast cancer.
Of the 15 patients with fusions in metastases and previous history of ET, eight had pre-treatment material available for fusion testing. In six of the eight cases, fusions were not detected in the pre-treatment sample, suggesting that these fusions arose as a mechanism of acquired resistance to ET. All patients with acquired kinase fusions and NGS testing on more than one lesion demonstrated that pre-and post-ET lesions were clonally related (Figure 1). The only patient (P-0031476) with a kinase fusion present in the primary tumor and metastasis who then received ET demonstrated progression of disease on letrozole. Figure 1 illustrates the course of the six patients with acquired kinase fusions.
Figure 1. Treatment summaries and molecular findings in patients with acquired kinase fusions.
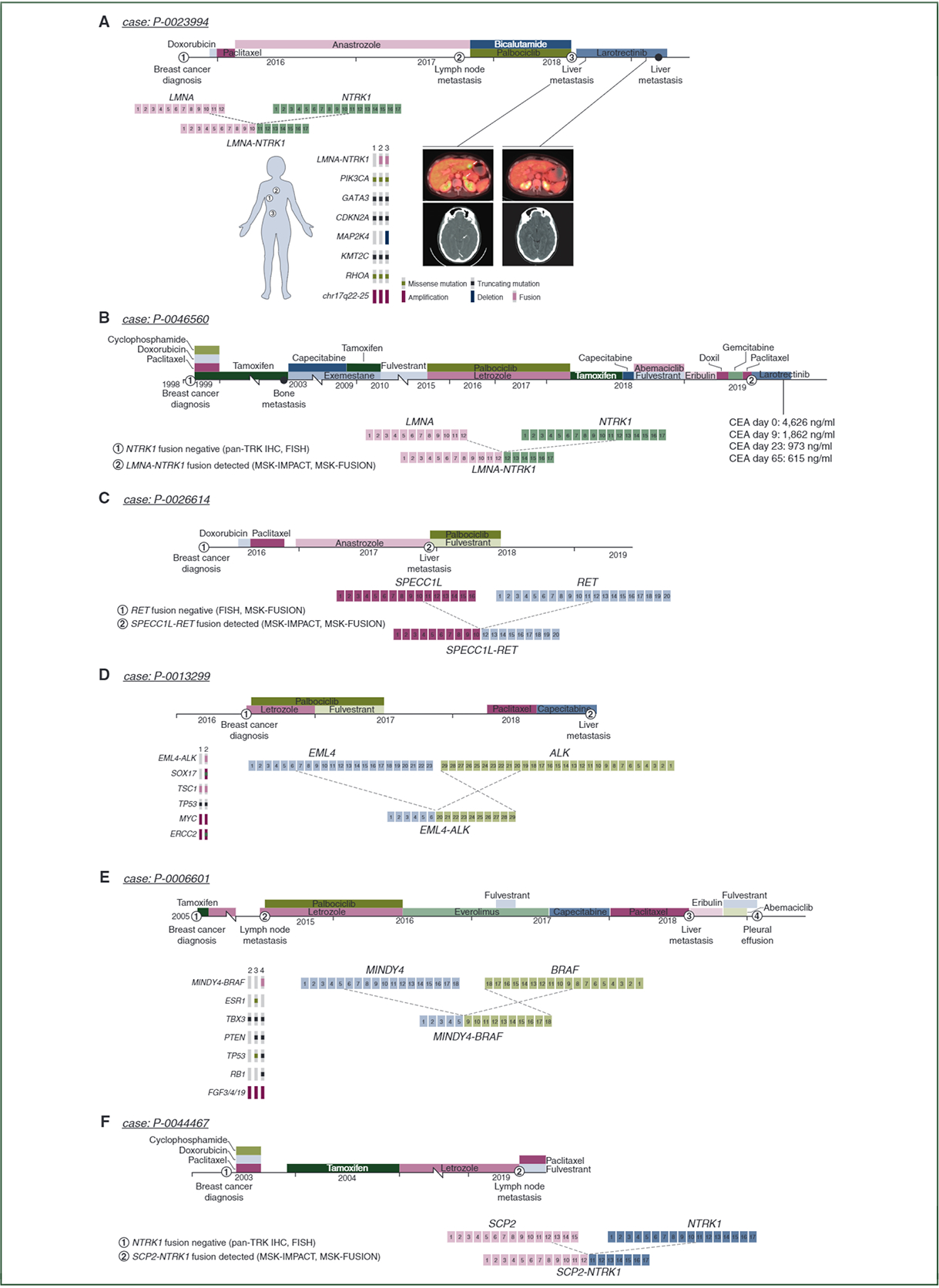
Six patients acquired kinase fusions after endocrine therapy. The first patient demonstrated an LMNA-NTRK1 fusion in the aortic lymph nodes and liver after anastrozole therapy. The second patient demonstrated an acquired LMNA-NTRK1 fusion after tamoxifen and fulvestrant. The third patient demonstrated a SPECC1L-RETfusion after anastrozole therapy. The fourth and fifth patients demonstrated acquired EML4-ALK and MINDY4-BRAF fusions, both after fulvestrant. The sixth patient demonstrated an acquired SCP2-NTRK1 fusion after tamoxifen and letrozole. The first, fourth, and fifth cases had DNA sequencing both before and at the time of the development of kinase fusions, and all three cases demonstrated shared mutations demonstrating clonal relatedness between tumor foci.
IHC, immunohistochemistry; MSK-IMPACT, Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets; TRK, receptor tyrosine kinase.
The first patient (P-0023994) with an acquired kinase fusion (Figure 1A) was diagnosed with ER-positive, PgR-positive, HER2-negative invasive ductal carcinoma with regional lymph node metastases. After adjuvant chemotherapy and ET, the patient developed multiple distant metastases (aortic lymph nodes, bone, liver, adrenal glands, and brain). This patient demonstrated a new LMNA-NTRK1 fusion by MSK-IMPACT and MSK-Fusion in the aortic lymph nodes with diffuse pan-Trk IHC staining. The absence of the fusion in the pre-treatment sample was confirmed by MSK-IMPACT, MSK-Fusion, and a negative pan-Trk IHC stain. Larotrectinib therapy was initiated and the patient demonstrated a complete response, including the brain lesions. A second patient (P-0046560, Figure 1B) with an acquired LMNA-NTRK1 fusion detected in a post-ET liver metastasis sample via MSK-IMPACT and MSK-Fusion was also originally ER-positive and PgR-positive while negative for fusions by pan-Trk IHC and NTRK1 FISH on the pre-treatment primary tumor sample. This patient received larotrectinib monotherapy and demonstrated a biochemical response: pre-larotrectinib carcinoembryonic antigen levels of 4626 ng/ml reduced to 1862 ng/ml after 9 days of treatment, 973 ng/ml after 23 days of treatment, and 615 ng/ml after 65 days of treatment. Radiologic assessment of the target lesion is planned. The four remaining patients (Figure 1C–F) all demonstrated ER-positive primary lesions followed by metastases detected on or after ET with a new kinase fusion confirmed by MSK-IMPACT and MSK-Fusion (SPECC1L-RET, EML4-ALK, MINDY4-BRAF, SCP2-NTRK1). These four patients were not eligible for kinase inhibitors during the study period. Pre-treatment samples for these patients were negative for fusions by MSK-Fusion and FISH (SPECC1L-RET), MSK-IMPACT (EML4-ALK and MINDY4-BRAF), and pan-Trk IHC and FISH (SCP2-NTRK1). None of the patients with acquired fusions demonstrated an ESR1 hotspot mutation by MSK-IMPACT in a lesion harboring a kinase fusion.
Molecular profiles of kinase fusion-positive BC.
Because MAPK alterations are enriched in endocrine resistant BC,5 we explored the frequency of concurrent previously described MAPK alterations in kinase fusion-positive BC. Of the 27 fusion-positive BC, one harbored both ERBB2 amplification and an in-frame ERBB2 kinase domain exon 20 insertion (P-0029981) while none of the remaining cases harbored other MAPK pathway alterations such as NF1, KRAS, NRAS, HRAS, ERBB3, MAP2K1, or BRAF mutations, or EGFR amplifications.
The frequencies of common molecular alterations in BC are listed in Table 2. The fusion-positive BC do not show mutual exclusivity with these alterations. None of the kinase fusion driven BC harbored ESR1 hotspot mutations in the same sample in which a fusion was detected.
Functional studies: LMNA-NTRK1 fusion promotes hormone-independent growth.
To investigate the functional impact of the LMNA-NTRK1 fusion identified in patient P-0023994, the coding sequence of the LMNA-NTRK1 fusion was cloned into the lentiviral vector to express the fusion protein in ER-positive MCF7 cells (Figure 2A). While little proliferative advantage was observed in the fusion-expressing cells cultured in FBS-supplemented media, the LMNA-NTRK1 fusion protein promoted hormone-independent growth in media supplemented with charcoal-stripped serum that mimics the low-estrogen condition of aromatase inhibition (Figure 2B). As the fusion protein contains the kinase domain of NTRK1, the downstream proliferative signaling pathways, ERK, AKT, and PLCγ were activated in MCF7 cells expressing the LMNA-NTRK1 fusion (Figure 2A). Inhibition of TrkA activity by larotrectinib, a pan-Trk inhibitor, repressed the activation of downstream pathways (Figure 2A) and abolished hormone-independent proliferation in the fusion-expressing cells cultured in charcoal-stripped serum (CSS)-supplemented media (Figure 2C). Long-term expression of the NTRK1 fusion was also associated with reduction of PgR expression, consistent with a suppressed ER signaling pathway (Figure 2A). In ER-negative, non-transformed MCF10A cells, expression of the LMNA-NTRK1 fusion activated ERK signaling and promoted EGF-independent growth (supplementary Figure SI, available at Annals of Oncology online), further demonstrating its oncogenic potential in breast-derived cells.
Figure 2. LMNA-NTRK1 fusion promotes hormone-independent growth and confers resistance to endocrine therapy.
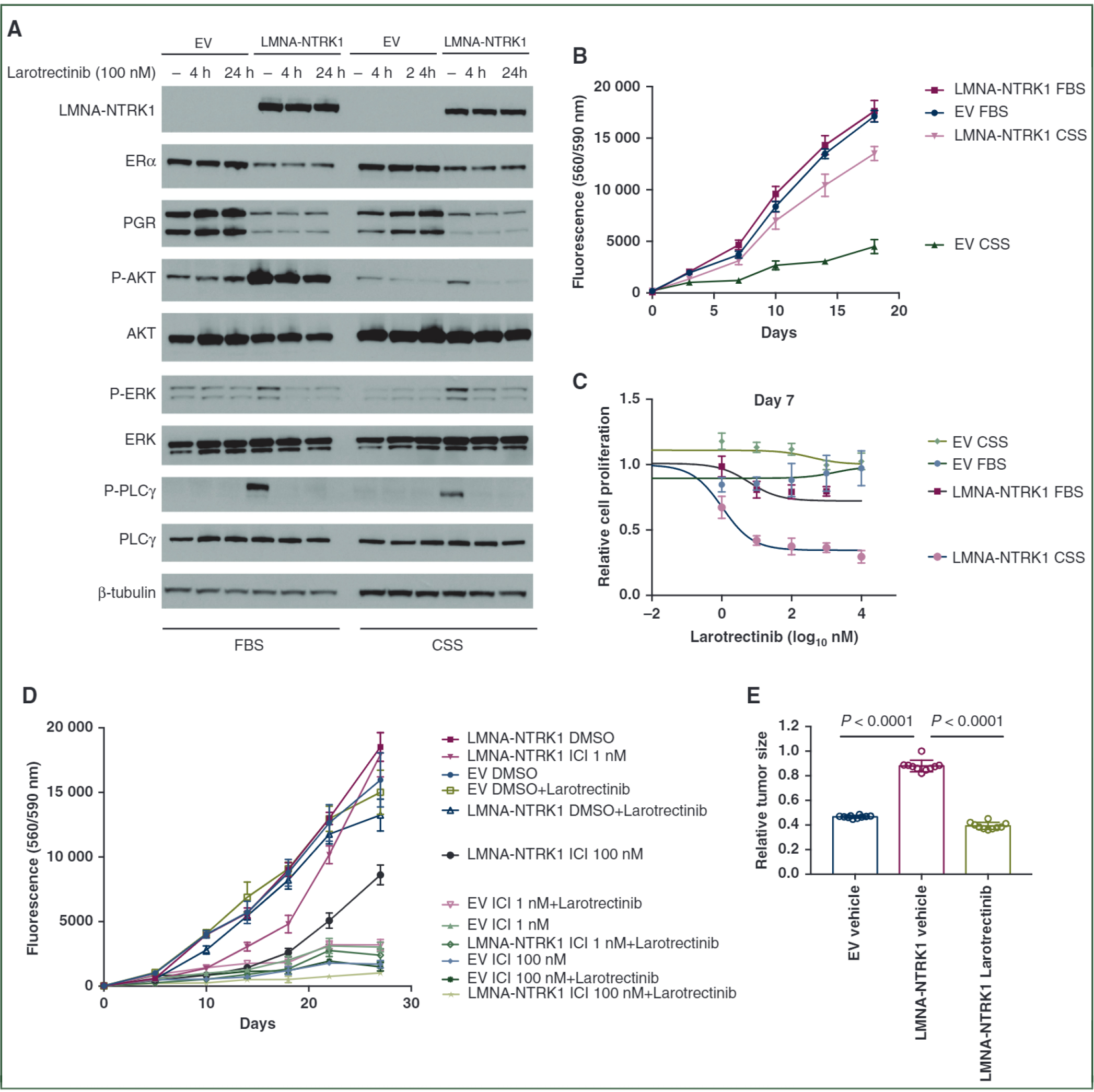
(A) Western blot analysis of ERα, PgR and phosphorylation of AKT, ERK and PLCγ in MCF7 cells expressing LMNA-NTRK1 fusion that were cultured with either 10% FBS or 10% CSS and treated with 100 nM larotrectinib for 4 h or 24 h. (B) LMNA-NTRK1 fusion promoted hormone-independent growth of MCF7 cells. MCF7 cells with empty vector (EV) or LMNA-NTRK1 fusion were seeded into 96-well plates in DMEM/F12 medium supplemented with either 10% FBS or 10% CSS, and cell proliferation was assayed with resazurin. (C) Proliferative inhibition of MCF7 cells expressing LMNA-NTRK1 fusion by larotrectinib. MCF7 cells with EV or LMNA-NTRK1 fusion were treated with various doses of larotrectinib in medium supplemented with 10% FBS or 10% CSS. (D) LMNA-NTRK1 fusion confers resistance to fulvestrant. MCF7 cells with EV or LMNA-NTRK1 fusion seeded into 96-well plates were treated with various doses of fulvestrant (ICI) and/or 100 nM larotrectinib. (E) LMNA-NTRKl-expressing MCF7 xenografts after estrogen deprivation. MCF7 xenograft tumors expressing LMNA-NTRK1 fusion were established in nude mice. Then the estradiol pellets were removed, and the mice were treated with either vehicle or 200 mg/kg of larotrectinib via gavage once daily for 2 weeks. The mice bearing MCF7 tumors expressing the EV were treated with vehicle as control. The relative tumor volume compared with the volume measured before treatment of each group ± standard deviation (n = 10 mice/group) is shown with t-test P values.
CSS, charcoal-stripped bovine serum; DMEM, Dulbecco’s Modified Eagle Medium; DMSO, dimethyl sulfoxide; ERα, estrogen receptor alpha; FBS, fetal bovine serum; PgR, progesterone receptor.
To further demonstrate the capacity of the LMNA-NTRK1 fusion in promoting ET resistance, MCF7 cells were treated with the ER antagonist fulvestrant. The fusion-expressing cells showed significant growth advantage over the control cells in the presence of fulvestrant, and furthermore this augmented growth was eliminated by co-treatment of larotrectinib (Figure 2D).
We also examined xenografts from MCF7 cells engineered to express the LMNA-NTRK1 fusion or control vector. Upon estradiol pellet removal, the volumes of the control tumors reduced 50% in 1 week, whereas the LMNA-NTRKl-expressing tumors remained stable in size with modest shrinkage in the following days (Figure 2E). Treatment with larotrectinib significantly diminished the LMNA-NTRKl-expressing tumors, indicating the proliferative dependency of these tumor cells on LMNA-NTRK1 fusion protein after estrogen deprivation (Figure 2E).
Functional studies: EML4-ALK fusion promotes hormone-independent growth.
The identification of EML4-ALK fusion in patient P-0013299 prompted us to investigate the functional impact of ALK fusion on BC cells in response to ET. The EML4-ALK fusion consists of C-terminal kinase domain of ALK fused with N-terminal fragment coded by EML4 exons 1–6. Expressing EML4-ALK fusion in T47D cells promoted proliferation under estrogen-depleted culture conditions (Figure 3A). Western blot analysis showed elevated activation of AKT and ERK signaling pathways in the cells expressing EML4-ALK fusion (Figure 3B). Ceritinib, a potent ALK inhibitor, suppressed the AKT and ERK signaling activation (Figure 3B). T47D cells expressing the fusion exhibited great sensitivity to ceritinib treatment, especially with estrogen deprivation (Figure 3C). We also treated these cells with fulvestrant to assess the capability of EML4-ALK fusion in promoting ET resistance. Significant growth advantage was observed in fusion-expressing cells compared with the control cells in response to fulvestrant treatment. Addition of ceritinib totally abolished this growth advantage (Figure 3D).
Figure 3. EML4-ALK fusion promotes hormone-independent growth and confers resistance to fulvestrant.
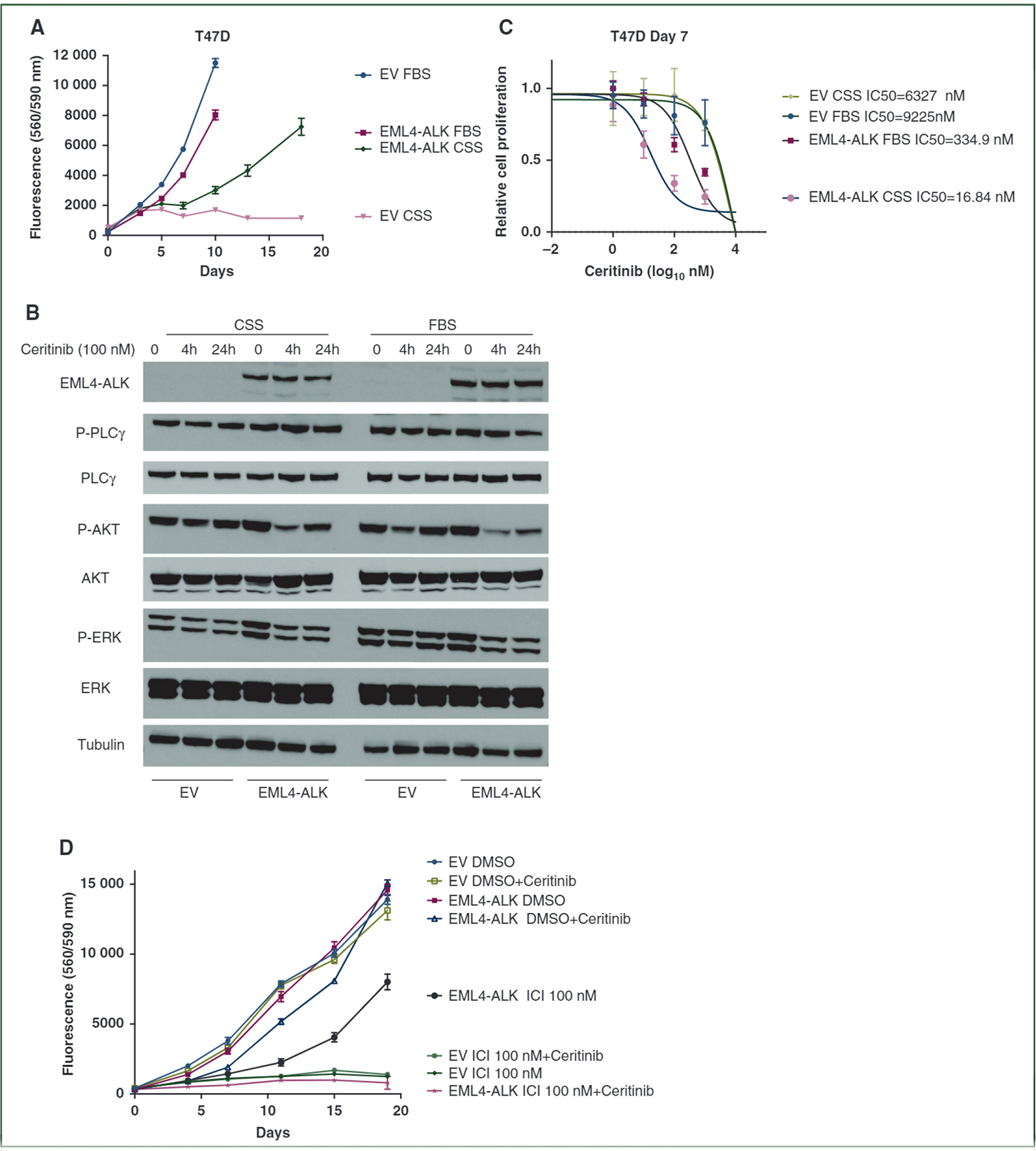
(A) EML4-ALK fusion promoted hormone independent growth of T47D cells. T47D cells with empty vector (EV) or EML4-ALK fusion were seeded into 96-well plates in Dulbecco’s Modified Eagle Medium (DMEM)/F12 medium supplemented with either 10% fetal bovine serum (FBS) or 10% charcoal stripped bovine serum (CSS), and cell proliferation was assayed with resazurin. (B) Western blot analysis of phosphorylated AKT, ERK and PLCγ in T47D cells expressing EML4-ALK fusion that were cultured with either 10% FBS or 10% CSS and treated with 100 nM ceritinib for 4 h or 24 h. (C) Proliferative inhibition of T47D cells expressing EML4-ALK fusion by ceritinib. T47D cells with empty vector (EV) or EML4-ALK fusion were treated with various doses of ceritinib in medium supplemented with 10% FBS or 10% CSS. (D) EML4-ALK fusion confers resistance to fulvestrant. T47D cells with EV or EML4-ALK fusion seeded into 96-well plates were treated with 100 nM fulvestrant (ICI) and/or 100 nM ceritinib.
DISCUSSION
Kinase fusions occur in both ductal and lobular tumors and are not mutually exclusive with common BC mutations such as PIK3CA or AKT1 mutations. While kinase fusions are extremely rare in BC, occurring in only 0.6% of patients, they appear enriched in post-progression specimens after ET. The six cases with acquired kinase fusions confirmed to be present only at progression after ET lacked ESR1 hotspot mutations, and two of those cases showed concomitant loss of ER expression. Kinase fusions represent potential new therapeutic targets in these patients and highlight the importance of fusion testing in the setting of resistance/ progression of disease. The first patient who received a kinase inhibitor targeting her acquired LMNA-NTRK1 fusion demonstrated a complete response of her systemic metastatic disease, while the second patient with this same fusion demonstrated a dramatic biochemical response with a carcinoembryonic antigen (CEA) reduction of approximately 80% within 23 days.
The most frequently involved kinases were the FGFR genes (n = 11). Early stage clinical trials are ongoing for various FGFR inhibitors, yet the majority of trials have focused on intrahepatic cholangiocarcinoma, which often carries a poor prognosis and may result in a different response to treatment compared with BC.15,16 BRAF fusions were the second most frequent kinase fusions in the cohort (n = 5), and all were identified in progression samples after ET. Other identified fusions for which kinase inhibitors are available included NTRK1, ALK, ROS1, and RET.
Paratala et al.17 recently reported a case of a woman in whom an NCOA4-RET fusion was identified in a metastatic ER+/ HER2+ BC sample after 5 years of tamoxifen. The patient was treated with cabozantinib and demonstrated radiologic response in multiple lesions, representing another example of the efficacy of targeted therapy matched to an acquired kinase fusion in this clinical setting. While this trend is novel in BC, acquired kinase fusions have been described in lung cancer,18 and acquired oncogenic BRAF structural variants have been described in colorectal carcinoma after Raf-inhibitor therapy.19 Typically, kinase fusions like other strong MAPK pathway drivers such as KRAS exon 2 and BRAF p.V600E mutations are thought to be present early and clonally in a malignancy as ‘drivers’. The current paper highlights a unique scenario in which kinase fusions develop later in the course of disease, likely under selective pressure during ET.
ET is the most effective treatment of ER-positive primary and metastatic BC; however, acquired resistance remains a major clinical challenge. Loss of ER expression status (ER-positive primary tumor, ER-negative relapse) occurs in approximately 10%–35% of patients treated with ET.20–22 Loss of ER is thought to be due to clonal selection and is associated with an increased risk of local and systemic relapse and shorter survival.22,23 Interestingly, two of the four cases with acquired kinase fusions in this dataset also showed a change in ER status from positive (pre-ET) to negative (post-ET). Overall, half of the patients with kinase fusions had ER-negative tumors at the time of fusion detection. In keeping with these findings, the results of the AURORA trial (ClinicalTrials.gov Identifier: NCT02102165) were recently presented by Aftimos et al. where they identified the presence of subtype switching in metastatic samples along with mutations in ESR1, PTEN, KAT6A, MYC, MDM4, and AKT3, and copy number losses in RBI and ARID1A.24
There are known instances of new primary tumors with a different receptor subtype as opposed to a ‘switching’ of the subtype of the original cancer. It is often very difficult to distinguish between these two entities in clinical practice. As demonstrated in Figure 1, two cases (P-0023994, P-0006601) with post-ET loss of ER showed clonal relatedness by NGS of the pre-ET and post-ET samples. The pre-treatment sample was not available for NGS testing for the remaining three of five cases with loss of ER in the metastasis. Pathologic and radiologic findings formed the clinical basis for treating the new lesions as metastases. Irrespectively of whether the metastasis arose from a new primary (still status post ET) or was an example of subtype switching, in either case a kinase fusion may be a driver which was not previously detected.
In 2018, the enrichment of MAPK activating alterations independent of ESR1 mutations in endocrine-resistant advanced BC was identified.5 The findings in our study are concordant with those of Razavi et al.5 insofar as all fusion-positive BC specimens were ESR1 wild type. Thus, kinase fusions may represent another mechanism of MAPK pathway-mediated resistance separate from direct reactivation of ER activity and a new opportunity for treatment in advanced BC. Congruent with the findings of Razavi et al.,5 we found that the introduction of an LMNA-NTRK1 fusion into ER-positive MCF7 cells showed a growth advantage when treated with antiestrogen therapy. The fusion-positive MCF7 cells demonstrated resulting increased MAPK output, and further, the growth advantage ceased when a Trk inhibitor (larotrectinib) was introduced.
The findings in this study add to our knowledge of the genomic landscape of BC and the list of targetable alterations that can serve as predictive biomarkers. The ESMO Scale for Clinical Actionability of molecular Targets (ESCAT) has been developed to rank the level of evidence of individual recurrent genomic alterations observed in BC and help clinicians to prioritize treatment.25 Regarding the kinase fusions identified in our study cohort, NTRK1–3 fusions are ranked in tier 1C with the clinical implication that access to treatment should be considered standard of care. The other kinase fusions in this study were not ranked at the time of publication.
This study has several limitations. Kinase fusions are extremely rare, and the cohort of fusion-positive BC is in turn small despite the very large number of BC analyzed. Due to the small size of the fusion-positive cohort, statistical associations such as mutual exclusion with ESR1 hotspot mutations may not be uncovered due to low power. We also did not have pre-treatment samples on all fusion-positive BC as our hospital is a large referral center and many patients had their initial treatment elsewhere. Only two patients with a kinase fusion went on to receive targeted therapy as other patients did not meet performance criteria for ongoing trials or did not require further therapy at the time of this manuscript.
In conclusion, kinase fusions in BC are rare, occurring in 0.6% of samples in the current study. At least 22% (6/27) of the fusions identified in the current study were confirmed to have been acquired at progression after ET. Our findings show that fusion testing of metastatic BC, particularly those progressing after ET, may benefit select patients who harbor targetable kinase fusions.
Supplementary Material
{kind=link}
FUNDING
This work was supported by the National Institutes of Health/National Cancer Institute (NIH/NCI) Cancer Center Support [grant number P30 CA008748] and the Marie-Josee and Henry R Kravis Center for Molecular Oncology, the Cancer Couch Foundation (to SC), and the Breast Cancer Research Foundation (to SC and MS). MS is also funded by the NIH [grant number R01CA190642–01] and by a kind gift from Mrs. Barbara Smith. This study was approved by the institutional review board at Memorial Sloan Kettering Cancer Center.
Footnotes
DISCLOSURE
PR has received honoraria for consulting/advisory board for Novartis, AstraZeneca, Foundation Medicine and institutional research support from GRAIL, Inc. MS has received research funds from Puma Biotechnology, AstraZeneca, Daiichi Sankyo, Immunomedics, Targimmune, and Menarini Ricerche. He is in the scientific advisory board (SAB) of Menarini Ricerche and Bioscience Institute and is a cofounder of Medendi.org. ML has received honoraria for ad hoc advisory board participation from Merck, AstraZeneca, Bristol Myers Squibb, Takeda, Lilly Oncology and Bayer, and research support from LOXO Oncology, Merus, and Helsinn Therapeutics. DMH has received personal fees from Chugai Pharma, Boehringer Ingelheim, AstraZeneca, Pfizer, Bayer, Debiopharm Group, and Genentech/Roche, as well as grants from Bayer, AstraZeneca, Puma Biotechnology, and Loxo Oncology, and employment and equity in Eli Lilly. All personal fees and grants are for work outside the submitted work. AD has honoraria from Medscape, OncLive, PeerVoice, Physician Education Resources, Tyra Biosciences, Targeted Oncology, MORE Health, Research to Practice, Foundation Medicine, PeerView, AstraZeneca, Ignyta/ Genentech/Roche, Bayer; consulting roles at Ignyta, Loxo/ Lilly, TP Therapeutics, AstraZeneca, Pfizer, Blueprint Medicines, Genentech/Roche, Takeda, Helsinn Therapeutics, BeiGene, Hengrui Therapeutics, Exelixis, 14ner/Elevation Oncology, GlaxoSmithKline, Exelixis, Teva, Taiho, Merck, Puma, Merus, Boeringer Ingelheim, PharmaMar, and Bayer. RB has received a grant from Archer, honoraria for advisory board participation from Loxo oncology and speaking fees from lllumina. SC has received honoraria for ad hoc consulting for Eli Lilly, Sermonix, BMS, Paige Al, Context Therapeutics, Revolution Medicine, and Novartis; research support to the institution from Daiichi Sankyo, Novartis, Eli Lilly, Genentech, and Sanofi. JFH has received honoraria from Medscape, Cor2Ed, ClearView Healthcare Partners, lllumina, and Axiom Healthcare Strategies, as well as research funding from Bayer, Eli Lilly, and Boehringer Ingelheim. All remaining authors have declared no conflicts of interest.
REFERENCES
- 1.Toy W, Shen Y, Won H, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45(12):1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robinson DR, Wu YM, Vats P, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45(12): 1446–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toy W, Weir H, Razavi P, et al. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov. 2017;7(3):277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartmaier RJ, Trabucco SE, Priedigkeit N, et al. Recurrent hyperactive ESR1 fusion proteins in endocrine therapy-resistant breast cancer. Ann Oncol. 2018;29(4):8728–8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Razavi P, Chang MT, Xu G, et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell. 2018;34(3):427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matissek KJ, Onozato ML, Sun S, et al. Expressed gene fusions as frequent drivers of poor outcomes in hormone receptor-positive breast cancer. Cancer Discov. 2018;8(3):336–353. [DOI] [PubMed] [Google Scholar]
- 7.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17(3):251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu G, Benayed R, Ho C, et al. Diagnosis of known sarcoma fusions and novel fusion partners by targeted RNA sequencing with identification of a recurrent ACTB-FOSB fusion in pseudomyogenic hemangioendothelioma. Mod Pathol. 2019;32(5):609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20(12):1479–1484. [DOI] [PubMed] [Google Scholar]
- 10.Cocco E, Benhamida J, Middha S, et al. Colorectal carcinomas containing hypermethylated MLH1 promoter and wild-type BRAF/KRAS are enriched for targetable kinase fusions. Cancer Res. 2019;79(6): 1047–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tognon C, Knezevich SR, Huntsman D, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2(5):367–376. [DOI] [PubMed] [Google Scholar]
- 12.Hammond ME, Hayes DF, Dowsett M, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28(16):2784–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolff AC, Hammond MEH, Allison KH, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline focused update. J Clin Oncol. 2018;36(20):2105–2122. [DOI] [PubMed] [Google Scholar]
- 14.Hechtman JF, Benayed R, Hyman DM, et al. Pan-Trk immunohistochemistry is an efficient and reliable screen for the detection of NTRK fusions. Am J Surg Pathol. 2017;41(11):1547–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Javle M, Lowery M, Shroff RT, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol. 2018;36(3):276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goyal L, Saha SK, Liu LY, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 2017;7(3):252–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paratala BS, Chung JH, Williams CB, et al. RET rearrangements are actionable alterations in breast cancer. Nat Commun. 2018;9(1):4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu HA, Suzawa K, Jordan E, et al. Concurrent alterations in EGFR-mutant lung cancers associated with resistance to EGFR kinase inhibitors and characterization of MTOR as a mediator of resistance. Clin Cancer Res. 2018;24(13):3108–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Geel RMJM, Tabernero J, Elez E, et al. A phase lb dose-escalation study of encorafenib and cetuximab with or without alpelisib in metastatic BRFA-mutant colorectal cancer. Cancer Discov. 2017;7(6): 610–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindstrom LS, Karlsson E, Wilking UM, et al. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression. J Clin Oncol. 2012;30(21):2601–2608. [DOI] [PubMed] [Google Scholar]
- 21.Broom RJ, Tang PA, Simmons C, et al. Changes in estrogen receptor, progesterone receptor and Her-2/neu status with time: discordance rates between primary and metastatic breast cancer. Anticancer Res. 2009;29(5):1557–1562. [PubMed] [Google Scholar]
- 22.Lower EE, Glass EL, Bradley DA, et al. Impact of metastatic estrogen receptor and progesterone receptor status on survival. Breast Cancer Res Treat. 2005;90(1):65–70. [DOI] [PubMed] [Google Scholar]
- 23.Van Poznak C, Somerfield MR, Bast RC, et al. Use of biomarkers to guide decisions on systemic therapy for women with metastatic breast cancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2015;33(24):2695–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aftimos PG, Antunes De Melo e Oliveira AM, Hilbers F, et al. First report of AURORA, the breast international group (BIG) molecular screening initiative for metastatic breast cancer (mbc) patients (pts). Ann Oncol. 2019;30(suppl_3):iii47–iii64. [Google Scholar]
- 25.Condorelli R, Mosele F, Verret B, et al. Genomic alterations in breast cancer: level of evidence for actionability according to ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann Oncol. 2019;30(3):365–373 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.