

Abstract
Background
Cancer-associated fibroblasts (CAFs) are a major component of hepatocellular carcinoma (HCC) stroma that are critically involved in HCC cancer chemoresistance, but the mechanism has not been elucidated. Previous studies have reported CD73 exerted an immunosuppressive function in cancer. Here, we explored the mechanism by which CAFs regulates CD73+ HCC cells and clarified whether CAFs promote chemoresistance of CD73+ cells.
Methods
We used the co-culture method to study the relationship between CAFs and HCC cells. Immunohistochemistry was applied to evaluate the correlation between α-smooth-muscle actin (α-SMA) and CD73. CD73 mRNA and protein were determined by real-time polymerase chain reaction (RT-PCR) and western blotting, and hepatocyte growth factor (HGF) was assayed by enzyme-linked immunosorbent assay (ELISA). Western blotting was used to explore the regulated pathway of CD73+ HCC. We then knocked down CD73 in cells, and then assessed the effect of CD73 on the apoptosis by flow cytometry. Finally, a sphere formation assay was applied to investigate the stemness of cancer cells, and xenograft tumors in nude mice were built to investigate the tumorigenicity.
Results
We found that the proportion of CAFs was positively correlated with CD73 expression in HCC cells. Mechanistically, c-Met and the MEK-ERK1/2 pathway were activated by HGF from CAFs which upregulated CD73 expression in HCC cells. Also, we found that CD73 promote sorafenib and cisplatin resistance in HCC, and CD73+ HCC cells indicated the higher capability of tumorigenicity compared to CD73− HCC cells in vivo. Furthermore, HGF further enhanced the chemoresistant characteristics of CD73+ tumor cells.
Conclusions
Our findings collectively suggest that CD73 is a vital HCC-chemoresistance force controlled by cross-talking between CAFs and HCC cells, thereby establishing CD73 as a potential new therapeutic target for HCC.
Keywords: Hepatocellular carcinoma (HCC), chemoresistance, cancer-associated fibroblast (CAF), CD73
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer worldwide, and is the world’s second leading cause of cancer death (1). There have been several treatments for HCC developed, including surgical resection, liver transplantation, and radiofrequency ablation and chemotherapy; these unfortunately cannot prevent HCC recurrence and metastasis (2,3). Therefore, a further understanding of the mechanism of tumorigenesis is still crucial for the treatment of tumors.
The tumor microenvironment (TME) is the environment around a tumor. It is composed of stromal cells, immune cells, extracellular matrix, secreted cytokines, and growth factors, and is closely related to tumorigenesis. Cancer-associated fibroblasts (CAFs) are a significant part of tumor stromal cells (4). CAFs may secrete various growth factors and cytokines, such as stromal cell-derived factor 1 (SDF1), hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), etc., all which contribute to tumor growth, angiogenesis, and drug resistance (5).
CD73 (ecto-5'-nucleotidase, Ecto-5'-NTase) was found to be capable of dephosphorylating adenosine monophosphate (AMP) into adenosine. Previous studies have mainly focused on its immunosuppressive function in immune response (6). A preliminary study reported that CD73 could promote HCC progression and metastasis (7). However, whether CD73 is responsible for resistance to chemotherapeutic drugs in HCC treatment is unknown.
Sorafenib, an oral multikinase inhibitor, has been shown to suppress proliferation and angiogenesis by inhibiting serine/threonine kinases, and receptor tyrosine kinases, including BRAF, Raf-1, Flt3, VEGFR-2/3, and PDGFR-β (8). Sorafenib is thus far the only choice for systemic therapy, but previous studies have shown its efficiency to be only around 30% in HCC patients, with acquired resistance often developing within 6 months (9). Cisplatin, a first-generation platinum chemotherapeutic drug, is one of the chemotherapeutic agents most commonly used to treat advanced HCC (10). Mechanisms involved in drug resistance to HCC are complicated, and specific tumor-stromal interactions can affect tumor cells and their sensitivity to anti-cancer drugs (11).
The present study investigated the role of CAFs in HCC drug resistance and focused on the regulation of HGF secreted by CAFs of CD73 expression in HCC cells. Here we discovered that the CAF-derived HGF could induce CD73 expression in HCC cells via the Met-ERK1/2 pathway. We further found that HGF could enhance the chemoresistance and tumor formation of CD73+ HCC cells. Our results collectively provide a better understanding of the molecular background of CD73-positive HCCs and may provide a potential therapeutic target.
We present the following article in accordance with the MDAR reporting checklist (available at http://dx.doi.org/10.21037/atm-20-1038).
Methods
The trial was conducted in accordance with the Declaration of Helsinki (as revised in 2013). The studies involving human HCC tissue samples were approved by the ethics committee of the Department of Hepatobiliary Surgery, the First Affiliated Hospital of Nanjing Medical University (China) (No. 2019-SR-127). Informed consent from the patients was obtained in all cases. The protocols for the animal experiment were approved by the Nanjing Medical University (NJMU) Institutional Animal Care and Use Committee (No. IACUC-1912015).
Patients and clinical samples
The 25 HCC tissues were collected from patients who underwent resection between 2017 and 2019 at the Department of Hepatobiliary Surgery, Nanjing Medical University’s First Affiliated Hospital (China), with the patients’ permission. None of these patients received radiotherapy or preoperative chemotherapy. Both samples were examined histologically by pathological analysis with HCC. The detailed clinicopathologic characteristics of patients are summarized in Table S1.
Table S1. Clinicopathological information of patients.
| Hospital number | Sex | Age | Tumor size | Metastasis | Pathologic type | Hepatitis | T stage |
|---|---|---|---|---|---|---|---|
| 0991860 | Male | 56 | 7.3×7.0 | None | HCC | Positive | T3 |
| 0991231 | Male | 56 | 3.8×3.9×3.3 | None | HCC | Positive | T2–3 |
| 0991833 | Male | 75 | 6.5×6×4.8 | None | HCC | Positive | T2 |
| 0992311 | Male | 44 | 5×5×5×5 | None | HCC | Positive | T2 |
| 0992502 | Male | 42 | 1.7×1.1×1 | None | HCC | Positive | T2 |
| 0992822 | Male | 78 | 3.3×3×3 | None | HCC | Negative | T2 |
| 0992228 | Male | 37 | 6×5×5 | None | HCC | Positive | T2–3 |
| 0992981 | Male | 73 | 3.5×2.2×2.2 | None | HCC | Positive | T1–2 |
| 0975542 | Male | 66 | 3.8×3.5×2.7 | None | HCC | Positive | T2 |
| 0993396 | Female | 69 | 14×9.5×9 | None | HCC | Negative | T2 |
| 0992674 | Male | 74 | 4×3.5×3.5 | None | HCC | Negative | T1 |
| 0993691 | Female | 69 | 3×2.5×2 | None | HCC | Positive | T2 |
| 0995868 | Male | 61 | 1.9×1.91 | None | HCC | Positive | T3 |
| 0995832 | Female | 56 | 1.5×1×1 | None | HCC | Positive | T1–2 |
| 0998904 | Female | 79 | 5.5×5×3 | None | HCC | Negative | T1–2 |
| 1000954 | Male | 63 | 10×8×6 | None | HCC | Positive | T2–3 |
| 1014382 | Male | 46 | 11×7.5×7 | None | HCC | Positive | T3 |
| 0490397 | Male | 64 | 12×12×7 | None | HCC | Positive | T2 |
| 1014306 | Male | 64 | 5×4.5×3 | None | HCC | Positive | T2 |
| 0996441 | Female | 64 | 5×4×4 | None | HCC | Positive | T2 |
| 0996252 | Male | 67 | 5×4.8×3.4 | None | HCC | Positive | T2 |
| 1015925 | Female | 41 | 5×4.2×5.5 | None | HCC | Positive | T2 |
| 1016239 | Male | 54 | 3.5×3×2.5 | None | HCC | Positive | T2 |
| 1015609 | Male | 61 | 8×7×5.5 | None | HCC | Positive | T3 |
| 1022016 | Male | 59 | 17×15×9 | None | HCC | Positive | T2–3 |
| 0999159 | Male | 44 | 4×3×2.5 | None | Liver hemangioma | Negative | – |
Patient no. 0999159 was used to extract NFs, no. 0998904 was used to extract CAFs. HCC, hepatocellular carcinoma; CAF, cancer-associated fibroblast.
Isolation of CAFs and paired normal fibroblasts (NFs)
Fresh HCC samples from patients after surgery were cut into pieces of approximately 2×2 mm and put in Petri dishes and cultured in Dulbecco’s Modified Eagle Medium (DMEM) added with 1 mg/mL collagenase I and 100 U/mL hyaluronidase at 37 °C for 2 h. They were then washed twice with phosphate-buffered saline (PBS) and centrifuged at 450 g for 8 min on each occasion, after which time the supernatants were discarded. The samples were finally re-suspended in DMEM supplemented with 10% fetal bovine serum (FBS), 100 IU/mL penicillin, 100 mg/mL streptomycin, and then cultured at 37 °C in a humidified 5% CO2 environment. Media was renewed every 3 days. All experiments were performed with fibroblasts until passage 10 to avoid CAFs senescence.
Cell lines and cell culture
The human HCC cell lines Hep3B, HepG2, MHCC-LM3, Huh7, MHCC-97H were obtained from the Shanghai Institutes for Biological Sciences (China) Cell Center. In a humidified incubator containing 5% CO2 at 37 °C, cells were cultivated in DMEM supplemented with 10% FBS with 100 IU/mL penicillin and 100 µg/mL streptomycin.
RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR)
According to the manufacturer’s instructions, full RNA was extracted from cultured cell lines using TRIzol reagent (Invitrogen, USA). Table S2 lists all the first sequences for qRT-PCR. For relative gene expression in cells, the rates were first normalized to the expression of GAPDH as αCt, then applied to one of the cells and converted to the change of fold (2–ΔΔCt).
Table S2. Primer sequences for qRT-PCR.
| Primer | Forward sequence (5' to 3') | Reverse sequence (5' to 3') |
|---|---|---|
| VEGF | AGGGCAGAATCATCACGAAGT | AGGGTCTCGATTGGATGGCA |
| HGF | GCTATCGGGGTAAAGACCTACA | CGTAGCGTACCTCTGGATTGC |
| bFGF | AGAAGAGCGACCCTCACATCA | CGGTTAGCACACACTCCTTTG |
| PDGF-A | TCGATGAGATGGAGGGTCG | ACCCGGACAGAAATCCAGTCT |
| TGF-β1 | CAATTCCTGGCGATACCTCAG | GCACAACTCCGGTGACATCAA |
| IGF-1 | GCTCTTCAGTTCGTGTGTGGA | GCCTCCTTAGATCACAGCTCC |
| IL-6 | ACTCACCTCTTCAGAACGAATTG | CCATCTTTGGAAGGTTCAGGTTG |
| CCL18 | GAGTGCTGCCTCGTCTATACCT | ACCGGTGACGAGGATGACACCT |
| TNF-α | CCTCTCTCTAATCAGCCCTCTG | GAGGACCTGGGAGTAGATGAG |
| CCL2 | CAGCCAGATGCAATCAATGCC | TGGAATCCTGAACCCACTTCT |
| CCL5 | CCAGCAGTCGTCTTTGTCAC | CTCTGGGTTGGCACACACTT |
| CCL7 | CCACACAGAAGTGGGTCCAG | ACCACTCTGAGAAAGGACAGG |
| CXCL16 | CCCGCCATCGGTTCAGTTC | CCCCGAGTAAGCATGTCCAC |
| SDF-1a | ATTCTCAACACTCCAAACTGTGC | ACTTTAGCTTCGGGTCAATGC |
| CD73 | CCAGTACCAGGGCACTATCTG | TGGCTCGATCAGTCCTTCCA |
| GAPDH | GGAGCGAGATCCCTCCAAAAT | GGCTGTTGTCATACTTCTCATGG |
qRT-PCR, quantitative real-time polymerase chain reaction.
Western blot
Cells were lysed in RIPA lysis buffer (Beyotime, China) containing phenylmethylsulfonyl fluoride (PMSF) (Beyotime, China). The supernatants were collected after centrifugation at 13,000 ×g at 4 °C for 10 min. Protein concentration was determined using a bicinchoninic acid (BCA) assay protein kit (Beyotime, China), and whole lysates were mixed with 5× sodium dodecyl sulfate (SDS) loading buffer. Proteins were isolated from 10% polyacrylamide gels of SDS and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, CA, USA) by electroblotting. The PVDF membranes were tested with primary antibodies and incubated with an anti-rabbit or anti-mouse antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA)-conjugated goat horseradish peroxidase (HRP). Signals were observed with ImmobilonTM Western Chemiluminescent HRP (Millipore) substrate. Primary antibodies used in Western blot were as follows: p-ERK1/2 (Cell Signaling Technology, CST, Cat. No. 4370T), ERK1/2 (Cell Signaling Technology, CST, Cat. No. 4695T), CD73 (abcam, Cat. No. ab91086), vimentin (Cell Signaling Technology, CST, Cat. No. 5741T), GAPDH (Cell Signaling Technology, CST, Cat. No. 5174T).
Collection of conditioned media (CM)
CAFs and NFs were seeded at 105 cells-per-well density on 6-well plates. After cell seeding, the culture medium was removed for 24 h, cells were washed once with PBS, and 1 mL of serum-free medium was applied per well. The CM was collected after 48 h of incubation at 37 °C and passed through a 0.2 µm membrane syringe filter to eliminate any cells and cell debris.
CM was pre-incubated with 10 µg/mL of human HGF antibody (R&D Systems, Cat. No. AF-294-NA) for 1 h at room temperature for neutralization of HGF in the CM of CAFs before it was subjected to experimental use.
Enzyme-linked immunosorbent assay (ELISA)
CAFs and matched NFs (105 cells/well) were seeded into 6-well plates and cultured in DMEM without serum for 3 days. Conditioned mediums were collected and detected for HGF using ELISA kits (Excell) according to the manufacturer’s instructions. Each experiment was repeated at least 3 times.
Immunohistochemistry and immunofluorescence
Paraffin-embedded primary HCC samples were immunostained with the primary anti-human α-smooth-muscle actin (α-SMA) rabbit (1:100, Abcam, Cat. No. ab32575), and the anti-human CD73 mouse (1:100, Abcam, Cat. No. ab91086) antibodies. The expression levels of α-SMA and CD73 were scored semi-quantitatively, according to staining intensity and distribution, as per the immunoreactivity score which has been described previously (12). Staining was assessed by two pathologists under double-blind conditions according to the scoring criteria.
Cell transfection and viral infection
For the knockdown of CD73 expression, short-hairpin RNAs (shRNAs) against CD73 were designed and cloned into the lentiviral vector pLKO.1-puro. The sequences of human CD73 shRNAs were 5'-CCGGCCCATTGATGAACGCAACAATCTCGAGATTGTTGCGTTCATCAATGGGTTTTTG-3' (shRNA-1) and 5'-CCGGGCACTGGGAAATCATGAATTTCTCGAGAAATTCATGATTTCCCAGTGCTTTTTG-3' (shRNA-2). Empty vector was used as a negative control. Puromycin (2 µg/mL) (Thermo Fisher Scientific, USA) was used to generate the antibiotic-resistant cells for subsequent assays.
Flow cytometry
Cells were resuspended into sterile 1× PBS buffer and labeled with PE anti-human CD73 (Biolegend, Cat. No. 344003) or isotype Ctrl and PE Mouse lgG1 (Biolegend, Cat. No. 400112) at 4 °C for 15 min. The labeled cells were washed twice with sterile 1× PBS buffer and sorted with a FACSAria Fusion flow cytometer (BD Biosciences, San Jose, CA, USA). For apoptosis detection, cells were stained in 1× Annexin V binding buffer (BD Biosciences) with 5 µL FITC-conjugated Annexin V (Vazyme) and 5 µL propidium iodide (PI) (Vazyme). The cells were subjected to flow analysis using the Beckman Cytoflex.
Spheroid formation assay
Approximately 200 dissociated cells were seeded in each well of Ultra-Low 24-well plates (Corning Integrated Life Sciences, Acton, MA, USA) and grown in DMEM/F12 (Invitrogen) supplemented by 1% methylcellulose (Sigma-Aldrich, St Louis, MO, Cat. No. M7027), 20 ng/mL of epidermal growth factor (Invitrogen, Cat. No. PHG0311), 20 ng/mL of primary fibroblast growth factor (Invitrogen, Cat. No. PHG0266), and 1× B27 (Invitrogen, Cat. No. 12587010) in a humidified atmosphere of 5% CO2 at 37 °C. The spheres greater than 100 µm were counted under a stereomicroscope (Olympus, Tokyo, Japan) after 5–7 days.
Tumorigenicity assay in nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice
Approximately 1×105 CD73+ HCC cells or CD73– HCC cells were suspended in 50 µL serum-free medium mixed with 50 µL Matrigel Matrix (BD Biosciences, Bedford, MA, USA) and subcutaneously transplanted into the armpit of 4-to-6-week-old female NOD/SCID mice (Vital River, Beijing, China) for assessment of tumor formation capability. The development of tumors was monitored regularly. Tumor volume was calculated as follows: V = (L × W2)/2 (V, volume; L, length diameter; W, width diameter). After 3–4 weeks, mice were sacrificed, and the tumors were dissected. The animal care and experimental protocols were approved by the guidelines of Nanjing Medical University (NJMU) Institutional Animal Care and Use Committee.
Statistical analysis
Using GraphPad Prism 7.0 software, all data were analyzed. Differences between each group were measured by t-test unless otherwise stated. Statistically significance was considered a value of P=0.05. Pearson’s correlation analysis was used to determine the associations between the expression α-SMA and CD73.
Results
HGF was upregulated by CAFs in HCC
A prior study had identified CAFs as the primary component of tumor stroma, which itself is a component of the TME (4). The human hepatic CAFs and normal NFs were isolated from fresh HCC clinical tissue and adjacent non-neoplastic hepatic tissue, and we found that CAFs and NFs that grew in vitro had spindle-shaped fibroblastic morphology (Figure 1A). To characterize CAFs and NFs, we analyzed two markers of activated fibroblasts, α-SMA and vimentin, by cell immunofluorescence staining (Figure 1B) and western blot (Figure 1C). Comparing the CAFs from different patients, we detected the expression of α-SMA by tissue immunofluorescence (Figure 1D).
Figure 1.

HGF was up-regulated by CAFs in HCC. (A) Representative images showed the fibroblasts morphology in vitro. The two pictures on the left show freshly isolated fibroblasts from the tissues, while the two images on the right show fibroblasts cultured in vitro for 2 weeks; (B,C) immunofluorescence staining and western blot showing the expression of α-SMA and vimentin in NFs and CAFs; (D) representative immunofluorescence images showing two HCC cases with high α-SMA expression (case 1) and low α-SMA expression (case 2); (E) qRT-PCR indicated mRNA expression differences of soluble factors that CAFs and NFs secreted; (F) CM from CAFs and NFs was collected, and the concentration of HGF was determined using human HGF ELISA. CAFs secreted a significant amount of HGF (9,000, 12,000 pg/mL). Data are presented as the means ± SEM of three independent experiments, the quantitative analysis are done for western blot. ns: not significantly different. **, P<0.01; ***, P<0.001; ****, P<0.0001, t-test. HGF, hepatocyte growth factor; CAF, cancer-associated fibroblast; HCC, hepatocellular carcinoma; α-SMA, α-smooth-muscle actin; NF, normal fibroblast; qRT-PCR, quantitative real-time polymerase chain reaction; CM, conditioned medium; ELISA, enzyme-linked immunosorbent assay.
Then, we compared the mRNA expression of some soluble factors that were secreted by CAFs and NFs. We found that HGF was the most significantly upregulated cytokine that was secreted by CAFs (Figure 1E). Subsequently, we performed ELISA analysis to further identify the secretion of HGF in CAF-CM compared with matched NF-CM (Figure 1F). Together, these data indicate that CAFs secreted more HGF than NFs.
The CD73 expression in HCC was correlated with α-SMA-positive CAFs and depended on HGF
Previously published RNA-seq data indicated that CD73 was one of the most upregulated genes in response to HGF (13). We concentrated on CD73 to investigate whether CD73 expression in HCC is controlled by CAFs in the TME. By using immunohistochemistry in a cohort of HCC patients (n=25), the association between the CD73 expression and α-SMA positive CAFs was confirmed. We observed CD73- positive tumor cells located mainly at the CAFs interface, and the CD73 expression was higher in HCCs with a higher proportion of α-SMA + CAFs (Figure 2A). We semi-quantitatively evaluated the degree of expression of CD73 and the sum of α-SMA + CAFs, and observed a significant correlation (Figure 2B).
Figure 2.

The CD73 expression in HCC was correlated with α-SMA-positive CAFs and depended on HGF. (A,B) In samples that were double immunostained with α-SMA (blue color) and CD73 (brown color), the correlation between the percentage of α-SMA+ CAFs and the percentage of CD73+ tumor cells in HCC samples was identified by tissue IHC; P<0.001, Pearson’s correlation; (C,D) the qRT-PCR and western blot analysis showing the mRNA and protein levels of CD73 in Hep3B and HepG2 cells cultured with CAFs-CM or without (NC: negative control) for 48 h; (E) the qRT-PCR results showing the mRNA expression of CD73 in HepG2 and Hep3B cells in response to 10 ng/mL of HGF treatment for 24 h, (NC: negative control); (F) Western blot indicated that HGF increased the expression of the CD73 protein in HCC cells; (G) the effect of CAF-induced CD73 expression was abolished when pre-incubation with an HGF-neutralizing antibody in Hep3B and HepG2 cells. Data are presented as the means ± SEM of three independent experiments, the quantitative analysis are done for western blot. **, P<0.01; ***, P<0.001; ****, P<0.0001, t-test. HCC, hepatocellular carcinoma; α-SMA, α-smooth-muscle actin; CAF, cancer-associated fibroblast; HGF, hepatocyte growth factor; qRT-PCR, quantitative real-time polymerase chain reaction; CM, conditioned medium.
We further aimed to investigate the underlying molecular mechanism by which CAFs regulate the expression of CD73 in HCC cells. We collected CM from CAFs and co-cultured them with HepG2 or Hep3B cells in vitro. qRT-PCR and western blotting showed that CD73 was upregulated in HepG2 and Hep3B cells (Figure 2C,D). Similarly, the expression of CD73 after induction of human recombinant HGF was also up-regulated, as indicated by qPCR and western blotting (Figure 2E,F). We then demonstrated that the induction of CM on CD73 expression was abolished when the CM of CAFs was pre-incubated with the HGF-neutralizing antibody (Figure 2G). These results suggest that CAFs regulated CD73 expression through secreting HGF in HCCs.
HGF in CM of CAFs regulated CD73 expression via the MEK-ERK1/2 pathway
We subsequently attempted to identify which signaling pathway initiates CD73 expression. Previous studies had shown that c-Met is the ligand of HGF, and Met is known to drive the invasive growth of HCC via MEK-ERK1/2 (14). We confirmed the phosphorylation of ERK upon treatment with CM in HepG2 and Hep3B cells by western blot analysis, and we observed this phosphorylation effect was abrogated after the administration of c-Met inhibitor PHA-665752, and the mRNA and protein levels of CD73 were also inhibited (Figure 3A,B). Similarly, by using MEK inhibitor U0126 to block the ERK1/2 pathway, phosphorylation of ERK1/2 was also decreased, as was the expression of CD73 (Figure 3C,D). To determine whether HGF is indeed responsible for the CD73 upregulation induced by CM via MEK-ERK1/2 pathway, Hep3B and HepG2 cells were pre-treated with U0126 and then treated with 10 ng/mL HGF. The inhibition of MEK abolished HGF-induced phosphorylation of ERK1/2 and the expression upregulation of CD73 (Figure 3E). We also found an increase in CD73 mRNA levels in the presence of HGF, and this effect was attenuated after using the U0126 (Figure 3E). Taken together, our results suggest that HGF paracrine signaling from CAFs increased CD73 expression via MEK-ERK1/2 pathway.
Figure 3.

HGF in CM of CAFs regulated CD73 expression via the MEK-ERK1/2 pathway. (A,B) The effect of CAFs-induced CD73 expression was suppressed after incubation with c-Met inhibitor PHA-665752. CD73 mRNA and protein expression levels were analyzed by qRT-PCR or Western blot. The phosphorylation of ERK1/2 was also inhibited when treated with a c-Met inhibitor; (C,D) Hep3B and HepG2 cells were pre-treated with MEK inhibitor U0126, followed by the incubation of CM of CAFs. Inhibition of MEK abolished CAF-CM-induced CD73 expression and the phosphorylation of ERK1/2; (E) Hep3B and HepG2 cells were pre-treated with U0126 to inhibit MEK, followed by the administration of 10 ng/mL of HGF. The administration of HGF alone promoted CD73 expression in HCC cells. Inhibition of MEK abolished HGF-induced CD73 expression. Data are presented as the means ± SEM of three independent experiments, the quantitative analysis are done for western blot. *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001, t-test. HGF, hepatocyte growth factor; CM, conditioned medium; CAF, cancer-associated fibroblast; qRT-PCR, quantitative real-time polymerase chain reaction; HCC, hepatocellular carcinoma.
CD73 promoted drug resistance ability of HCC cells
Due to the irreplaceable function of chemotherapeutic therapies in anti-cancer treatment, drug resistance of cancer cells has been seen as a significant challenge. In order to determine whether CD73 expressed in HCC cells increased the chemoresistance to conventional chemotherapeutic agents, such as sorafenib or cisplatin, we firstly analyzed the differential expression of CD73 in HCC cell lines (Figure 4A). We then observed that sorafenib or cisplatin treatment upregulated CD73 expression in HepG2 or Hep3B cells through flow cytometry analysis (Figure 4B,C,D). Also, this drug-induced CD73 upregulation was confirmed by western blot (Figure 4E). We next investigated the effect of CD73 gene-silencing on anti-cancer drug activity and successfully knocked down CD73 (shCD73-1 and shCD73-2) in HepG2 cells using a lentivirus-based knockdown approach. Both mRNA and protein levels verified the efficacy of knockdown (Figure 4F). In line with this, shCtl, rather than shCD73-1 or shCD73-2, effectively protected HepG2 and Hep3B cells from chemotherapy-induced apoptosis (Figure 4G,H). These results show that CD73 could promote chemoresistance in HCC cells.
Figure 4.

CD73 promoted the drug resistance ability of HCC cells. (A) The qRT-PCR and western blot analysis showing the mRNA and protein levels of CD73 in different HCC cell lines; (B,C) HepG2 and Hep3B were treated with sorafenib or cisplatin for 24 h. Cell-surface protein CD73 (relative to untreated cells) was then measured by flow cytometry; (D) histograms showing the statistical analysis of HepG2 and Hep3B cells treated with anti-cancer agents; (E) Western blot analysis of the expression of CD73 in HepG2 and Hep3B cells treated with sorafenib or cisplatin for 24 h; (F) the effects of shRNA-mediated knockdown of CD73 in HepG2 cells were analyzed by qRT-PCR and Western blot at both mRNA and protein levels, respectively; (G,H) CD73 was knocked down in HepG2 cells and treated with different doses of sorafenib or cisplatin in vitro and detected the proportions of Annexin V+/PI– (early apoptosis) and Annexin V+/PI+ (late apoptosis) cells. Data are presented as the means ± SEM of 3 independent experiments, the quantitative analysis are done for western blot. ns: not significantly different. **, P<0.01; ***, P<0.001; ****, P<0.0001, t-test. HCC, hepatocellular carcinoma; qRT-PCR, quantitative real-time polymerase chain reaction; shRNA, short-hairpin RNA; PI, propidium iodide.
CD73+ HCC exhibited stemness properties of self-renewal and promoted tumorigenicity
Multiple potential molecular, cellular, and microenvironmental mechanisms underlie the impaired sensitivity to anti-cancer drugs in HCC, including epithelial-mesenchymal transitions (EMTs), cancer stem cells (CSCs), and stroma-induced acquired chemoresistance (15,16). We thus wanted to explore whether CD73 possesses properties of stemness. CSCs are believed to have those stemness properties capable of expressing specific stemness-related genes that are crucial for establishing and maintaining stem cell-like characteristics. We found the knockdown of CD73 in HepG2 cells remarkably downregulated the expression of these stem cell-related genes, including NANOG, OCT4, SOX2 (Figure 5A). To obtain further evidence for CD73 promoting self-renewal ability, we performed a spheroid formation assay. In the vector control (shCtl) group, the diameter of the sphere was significantly larger than that of the group in which CD73 was knocked down (Figure 5B,C). Considering CD73 is a cell-surface marker, we enriched CD73+ and CD73– populations from Hep3B cell lines by fluorescence-activated cell sorting (FACS) (Figure 5D). In order to determine whether CD73+ cells were more tumorigenic than CD73– cells in vivo, we injected CD73+ cells and CD73– cells subcutaneously enriched by Hep3B cells into NOD/SCID mice. The volume and the weight of tumors derived from CD73+ Hep3B cells group were greater than those derived from the CD73– Hep3B cells group (Figure 5E,F,G). These results taken together indicated that CD73+ HCC cells expressed higher level stemness-related genes and exhibited some stemness features of self-renewal, while promoting tumorigenicity.
Figure 5.

CD73+ HCC exhibited stemness properties of self-renewal and promoted tumorigenicity. (A) qPCR analysis of the expression of indicated stem cell-related genes in HepG2 cells infected with CD73 shRNAs or empty vector; (B,C) representative photographs and histograms showing the average diameters of spheres generated by shCtl or CD73 knockdown (shCD73-1 and shCD73-2) in stem cell medium (n=5); (D) representative FACS of CD73+ and CD73– populations, which were isolated from Hep3B cells by FASC sorting; (E) CD73+ Hep3B cells and CD73– Hep3B cells were subcutaneously injected into the flank of NOD/SCID mice. The images of harvested tumors; n=3/group. Scale bar =2 cm; (F) tumor weights are shown as means ±SEM (n=3/group); (G) tumor growth curve showing the points, with bars representing means ± SEM (n=3/group). Data are presented as the means ± SEM of three independent experiments. ns: not significantly different. *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001, t-test. HCC, hepatocellular carcinoma; qPCR, quantitative polymerase chain reaction; shRNA, short-hairpin RNA; FACS, fluorescence-activated cell sorting; NOD/SCID, nonobese diabetic/severe combined immunodeficient.
HGF secreted by CAFs enhanced the chemoresistance and tumorigenicity of CD73+ HCC cells
We further investigated if HGF secreted by CAFs enhance the chemoresistant characteristics of CD73+ HCC cells. CD73+ Hep3B cells showed higher resistance to sorafenib or cisplatin when treated with HGF (Figure 6A,B,C). Besides this, we found a significant increase of NANOG, OCT4, and SOX2 gene levels in CD73+ Hep3B cells after HGF incubation compared with CD73+ Hep3B cells that were cultured in DMEM alone (Figure 6D). Moreover, HGF profoundly enhanced the spheroid formation ability of CD73+ Hep3B cells (Figure 6E). To determine whether HGF promoted the tumorigenesis of CD73+ HCC cells in vivo, we subcutaneously injected CD73+ Hep3B cells alone into NOD/SCID mice and also did so with added with HGF. We observed the CD73+ Hep3B cells added with HGF displayed tumorigenic features faster than those of the CD73+ Hep3B groups injected alone (Figure 6F,G,H). These results show that the CAFs that originated from HCC tissues promoted the chemoresistance of CD73+ HCC cells and tumor formation.
Figure 6.

HGF secreted by CAFs enhanced the chemoresistance and tumorigenicity of CD73+ HCC cells. (A,B) CD73+ Hep3B cells were cultured in the presence of HGF or DMEM alone for 48 h, then cells were treated with different doses of sorafenib or cisplatin for 24 h, and the apoptotic cells were detected by FACS; (C) histograms showing the statistical results; (D) CD73+ cells treated with HGF showed significantly higher expression of stemness-related genes than CD73+ cells treated without HGF; (E) sphere formation assays showed CD73+ Hep3B cells cultured with HGF generated more spheroids than CD73+ Hep3B cells cultured alone. Diameters larger than 100 µm were counted (n=3/group); (F) CD73+ Hep3B cells added with and without 10 ng/mL HGF were subcutaneously injected into the flank of NOD/SCID mice. The images of harvested tumors (n=5/group). Scale bar =2 cm; (G) tumor growth curve. The points and bars represent means ± SEM (n=5/group). (H) tumor weights are shown as means ± SEM (n=5/group). Data are presented as means ± SEM of three independent experiments. ns: not significantly different. *, P<0.05; **, P<0.01; ***, P<0.001; t-test. HGF, hepatocyte growth factor; CAF, cancer-associated fibroblast; HCC, hepatocellular carcinoma; FACS, fluorescence-activated cell sorting; NOD/SCID, nonobese diabetic/severe combined immunodeficient.
Discussion
Previous documents reported that CAFs are major tumor stromal components of TME, which promote tumor progression and chemoresistance. CAFs or NFs are commonly identified by their expression of α-SMA and vimentin by immunostaining (17). We found that CAFs isolated from HCC secreted a significant amount of HGF compared to NFs. It was reported previously that, under cDNA microarray profiling analysis, several genes were significantly up-regulated in Bel-7402 cells in response to HGF treatment, including CD73 (13). We focused on CD73 to find out whether the expression of CD73 in HCC is regulated by CAFs in the TME. In this study, we demonstrated the proportion of α-SMA-positive CAFs was correlated with the proportion of CD73+ HCC cells by immunohistochemistry; meanwhile, we found that high CD73 expression density is mainly located at the interface of CAFs in HCC samples. Also, previous results showed that positive immunoexpression of HGF was associated with a proportion of α-SMA-positive CAFs (18).
Next, we explored the functional connection between CAFs and CD73 expression. We discovered that the CM from CAFs or HGF could up-regulate CD73 expression in HCC cells. The MET tyrosine kinase is a receptor for HGF, and HGF stimulates aberrant Met activation to promote tumor progression (19). We found the phosphorylation of ERK1/2, the downstream molecular of Met, was elevated after incubation of CM of CAFs or HGF, and this effect was abolished when using a c-Met inhibitor, MEK inhibitor, or HGF-neutralizing antibody.
It has been shown that CD73 promotes HCC growth, metastasis (20), and predicts poor prognosis (7). In addition, CD73 has also been demonstrated to exert an anthracycline resistance effect in triple-negative breast cancer (21). However, CD73 expression in HCC cells related to drug resistance has not been reported. Sorafenib, a multikinase inhibitor, is the best choice for the systemic therapy of advanced HCC. However, it has been suggested that only 30% of patients benefit from it, and this may give rise to acquired drug resistance (15). Here, our study observed sorafenib treatment upregulated CD73 expression in liver cancer cell lines in vitro. By gene-silencing CD73, we found that CD73 protected HCCs from sorafenib-induced cell apoptosis. Cisplatin is another traditional chemotherapeutic agent applied to advanced HCC patients (22), and we found that CD73 exerted the same effect on HCCs after treatment with cisplatin. This finding suggests that CD73 in HCC cells confers chemoresistance to anti-cancer drugs. However, the mechanism for either sorafenib or cisplatin to upregulate the CD73 expression in HCC cells needs to be further investigated.
The underlying mechanisms of acquired resistance might be explained by EMTs, CSC hypothesis, and TME. Previous reports have shown that CD73 promotes EMT by activating PI3K/AKT signaling in HCC (7). In terms of CSC study, CSCs can resist chemotherapy and return to prominence after months or years, which may explain the tragic relapses after surgical or chemotherapeutic treatment (23). Recent studies also suggest that CD73 contributes to the stemness function of CSCs in pancreatic neuroendocrine (24), breast cancer (25), and ovarian carcinoma (26). To determine whether the expression of CD73 in HCC cells possessed some properties of stemness in the regulation of liver cancer, we conducted spheroid formation assays. This revealed that the in vitro self-renewal ability of HepG2 cells were decreased when CD73 was knocked down. In addition, in the shCtl group, HepG2 cells expressed higher levels of stem-related genes, and this effect was abolished upon CD73 knockdown. However, more research needs to be conducted to verify whether CD73 is a marker of CSCs in HCC. Consistent with the previous findings (7), we isolated CD73+ Hep3B cells and CD73-Hep3B cells by a cell-sorting approach and observed that CD73+ Hep3B cells showed faster tumorigenicity than their CD73- counterparts in NOD/SCID mice, which demonstrated an aggressive character.
CAFs play a crucial role in modulating neighboring cancer cells via the secretion of various paracrine factors, including the HGF, SDF-1, IL6, TGFβ, EGF, and FGF families (27). Moreover, the magnitude of drug resistance varies, depending on the particular stroma and therapy being evaluated (16). Our study investigated CAF-derived HGF enhanced chemoresistance in CD73+ HCC cells in vitro: the CD73+ HCC cells treated with HGF exhibited stronger features of stemness via sphere formation assays and manifested faster tumorigenicity capability.
In conclusion, our findings suggest that CAFs induce the upregulation of CD73 expression, which mediates chemotherapeutic resistance and promotes tumor growth. Moreover, this regulation depends on HGF secreted by CAFs via the Met-ERK1/2 pathway. Meanwhile, HGF enhances the drug resistance and tumorigenicity of CD73+ HCC cells (Figure 7). Our findings provide a better understanding of how CAFs interact with tumor cells to induce drug resistance in the TME and revealed a potential therapeutic target in HCC.
Figure 7.
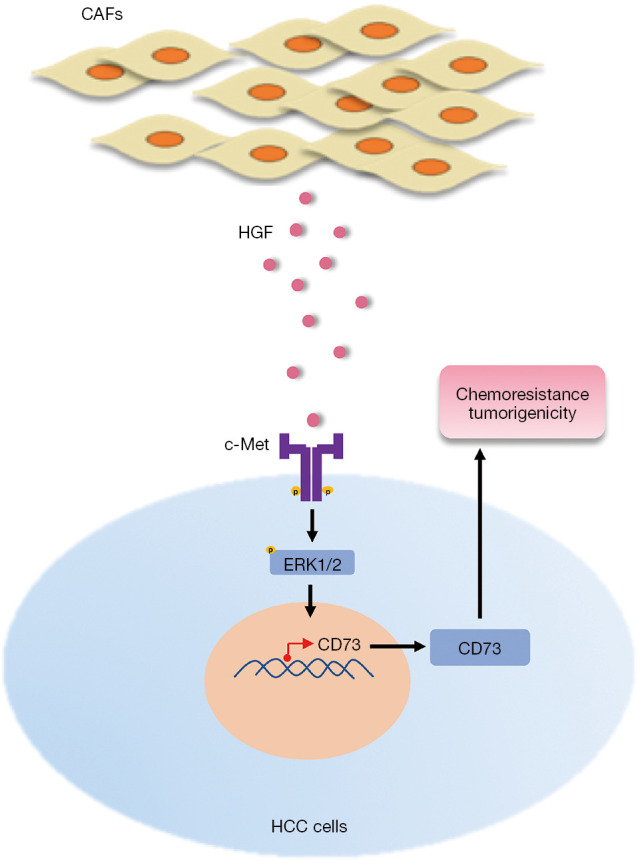
A schematic illustration of the crosstalk between CAFs and HCC cells. CAFs secrete HGF, which phosphorylates ERK1/2 at residues Thy202/Tyr204 in HCC cells through c-Met signaling. Following activation, phosphorylated ERK1/2 translocate to the nucleus, which regulates the expression of CD73 to promote the chemoresistance and tumorigenicity of HCC. CAF, cancer-associated fibroblast; HCC, hepatocellular carcinoma; HGF, hepatocyte growth factor.
Acknowledgments
We would like to thank Dr. Jingjing Chen, Dr. Yuye Yin, and the other lab members of Prof. Xiaoming W for their helpful technical assistance and device support.
Funding: This work was supported by grants from the National Natural Science Foundation of China (81971495, 81571564, and 91442117), the CAMS Innovation Fund for Medical Sciences (No. 2019-I2M-5-035), the National Science Foundation of Jiangsu Province (BRA2017533, BK20191490, and BE2016766), the 863 Young Scientists Special Fund (SS2015AA0209322), and the Foundation of Jiangsu Collaborative Innovation Center of Biomedical Functional Materials.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The trial was conducted in accordance with the Declaration of Helsinki (as revised in 2013). The studies involving human HCC tissue samples were approved by the ethics committee of the Department of Hepatobiliary Surgery, the First Affiliated Hospital of Nanjing Medical University (China) (No. 2019-SR-127). Informed consent from the patients was obtained in all cases. The protocols for the animal experiment were approved by the Nanjing Medical University (NJMU) Institutional Animal Care and Use Committee (No. IACUC-1912015).
Open Access Statement: This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license). See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Footnotes
Reporting Checklist: The authors have completed the MDAR reporting checklist. Available at http://dx.doi.org/10.21037/atm-20-1038
Data Sharing Statement: Available at http://dx.doi.org/10.21037/atm-20-1038
Peer Review File: Available at http://dx.doi.org/10.21037/atm-20-1038
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm-20-1038). LL serves as an unpaid editorial board member of Annals of Translational Medicine from Jun 2019 to May 2024. Xuehao W serves as an unpaid editorial board member of Annals of Translational Medicine from Aug 2019 to Jul 2024. The other authors have no conflicts of interest to declare.
References
- 1.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394-424. 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 2.Holzwanger DJ, Madoff DC. Role of interventional radiology in the management of hepatocellular carcinoma: current status. Chin Clin Oncol 2018;7:49. 10.21037/cco.2018.07.04 [DOI] [PubMed] [Google Scholar]
- 3.Nio K, Yamashita T, Kaneko S. The evolving concept of liver cancer stem cells. Mol Cancer 2017;16:4. 10.1186/s12943-016-0572-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xing F, Saidou J, Watabe K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci (Landmark Ed) 2010;15:166-79. 10.2741/3613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eckstein M, Gupta S. New insights in predictive determinants of the tumor immune microenvironment for immune checkpoint inhibition: a never ending story? Ann Transl Med 2019;7:S135. 10.21037/atm.2019.06.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gourdin N, Bossennec M, Rodriguez C, et al. Autocrine adenosine regulates tumor polyfunctional CD73(+)CD4(+) effector T cells devoid of immune checkpoints. Cancer Res 2018;78:3604-18. [DOI] [PubMed] [Google Scholar]
- 7.Ma XL, Shen MN, Hu B, et al. CD73 promotes hepatocellular carcinoma progression and metastasis via activating PI3K/AKT signaling by inducing Rap1-mediated membrane localization of P110beta and predicts poor prognosis. J Hematol Oncol 2019;12:37. 10.1186/s13045-019-0724-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keating GM, Santoro A. Sorafenib a review of its use in advanced hepatocellular carcinoma. Drugs 2009;69:223-40. 10.2165/00003495-200969020-00006 [DOI] [PubMed] [Google Scholar]
- 9.Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol 2009;10:25-34. 10.1016/S1470-2045(08)70285-7 [DOI] [PubMed] [Google Scholar]
- 10.Wei L, Wang X, Lv L, et al. The emerging role of microRNAs and long noncoding RNAs in drug resistance of hepatocellular carcinoma. Mol Cancer 2019;18:147. 10.1186/s12943-019-1086-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McMillin DW, Negri JM, Mitsiades CS. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discov 2013;12:217-28. 10.1038/nrd3870 [DOI] [PubMed] [Google Scholar]
- 12.Wei L, Ye H, Li G, et al. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis 2018;9:1065. 10.1038/s41419-018-1104-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lau EY, Lo J, Cheng BY, et al. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in hepatocellular carcinoma through c-Met/FRA1/HEY1 signaling. Cell Rep 2016;15:1175-89. 10.1016/j.celrep.2016.04.019 [DOI] [PubMed] [Google Scholar]
- 14.Giordano S, Columbano A. Met as a therapeutic target in HCC: facts and hopes. J Hepatol 2014;60:442-52. 10.1016/j.jhep.2013.09.009 [DOI] [PubMed] [Google Scholar]
- 15.Chen J, Jin R, Zhao J, et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett 2015;367:1-11. 10.1016/j.canlet.2015.06.019 [DOI] [PubMed] [Google Scholar]
- 16.McMillin DW, Delmore J, Weisberg E, et al. Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nat Med 2010;16:483-9. 10.1038/nm.2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eyden B. The myofibroblast: phenotypic characterization as a prerequisite to understanding its functions in translational medicine. J Cell Mol Med 2008;12:22-37. 10.1111/j.1582-4934.2007.00213.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rhee H, Kim HY, Choi JH, et al. Keratin 19 expression in hepatocellular carcinoma is regulated by fibroblast-derived HGF via a MET-ERK1/2-AP1 and SP1 axis. Cancer Res 2018;78:1619-31. 10.1158/0008-5472.CAN-17-0988 [DOI] [PubMed] [Google Scholar]
- 19.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov 2008;7:504-16. 10.1038/nrd2530 [DOI] [PubMed] [Google Scholar]
- 20.Shali S, Yu J, Zhang X, et al. Ecto-5'-nucleotidase (CD73) is a potential target of hepatocellular carcinoma. J Cell Physiol 2019;234:10248-59. 10.1002/jcp.27694 [DOI] [PubMed] [Google Scholar]
- 21.Loi S, Pommey S, Haibe-Kains B, et al. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc Natl Acad Sci U S A 2013;110:11091-6. 10.1073/pnas.1222251110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Llovet JM, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: chemoembolization improves survival. Hepatology 2003;37:429-42. 10.1053/jhep.2003.50047 [DOI] [PubMed] [Google Scholar]
- 23.Lathia JD, Liu H. Overview of cancer stem cells and stemness for community oncologists. Target Oncol 2017;12:387-99. 10.1007/s11523-017-0508-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katsuta E, Tanaka S, Mogushi K, et al. CD73 as a therapeutic target for pancreatic neuroendocrine tumor stem cells. Int J Oncol 2016;48:657-69. 10.3892/ijo.2015.3299 [DOI] [PubMed] [Google Scholar]
- 25.Yu J, Liao X, Li L, et al. A preliminary study of the role of extracellular -5'- nucleotidase in breast cancer stem cells and epithelial-mesenchymal transition. In Vitro Cell Dev Biol Anim 2017;53:132-40. 10.1007/s11626-016-0089-y [DOI] [PubMed] [Google Scholar]
- 26.Lupia M, Angiolini F, Bertalot G, et al. CD73 regulates stemness and epithelial-mesenchymal transition in ovarian cancer-initiating cells. Stem Cell Reports 2018;10:1412-25. 10.1016/j.stemcr.2018.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mazzocca A, Dituri F, Lupo L, et al. Tumor-secreted lysophostatidic acid accelerates hepatocellular carcinoma progression by promoting differentiation of peritumoral fibroblasts in myofibroblasts. Hepatology 2011;54:920-30. 10.1002/hep.24485 [DOI] [PubMed] [Google Scholar]