

Abstract
The interplay between G protein–coupled receptors (GPCRs) is critical for controlling neuronal activity that shapes neuromodulatory outcomes. Recent evidence indicates that the orphan receptor GPR139 influences opioid modulation of key brain circuits by opposing the actions of the µ-opioid receptor (MOR). However, the function of GPR139 and its signaling mechanisms are poorly understood. In this study, we report that GPR139 activates multiple heterotrimeric G proteins, including members of the Gq/11 and Gi/o families. Using a panel of reporter assays in reconstituted HEK293T/17 cells, we found that GPR139 functions via the Gq/11 pathway and thereby distinctly regulates cellular effector systems, including stimulation of cAMP production and inhibition of G protein inward rectifying potassium (GIRK) channels. Electrophysiological recordings from medial habenular neurons revealed that GPR139 signaling via Gq/11 is necessary and sufficient for counteracting MOR-mediated inhibition of neuronal firing. These results uncover a mechanistic interplay between GPCRs involved in controlling opioidergic neuromodulation in the brain.
Keywords: G protein-coupled receptor 139 (GPR139), brain, neuron, GPCR signaling, opioids, orphan receptor, medial habenula, G protein-coupled receptor (GPCR), heterotrimeric G protein, opiate opioid, cell signaling, cellular regulation, adenylate cyclase (adenylyl cyclase), GIRK channel
The mammalian brain features a staggering number of neuromodulatory systems that often converge on the same neurons and must be integrated and balanced through a system of opposing signaling and feedback mechanisms (1). Most neuromodulators act on G protein–coupled receptors (GPCRs), an extensive protein family that triggers responses by engaging second messenger systems and ion channels. GPCR signals are relayed through an array of heterotrimeric G proteins comprised of 16 Gα subunits (2). The G protein(s) activated by a GPCR dictates the nature of the signal by engaging specific downstream targets. Ultimately, neuronal responses are built from cross-talk between GPCRs engaged by neuromodulatory inputs (1, 3). Understanding the logic and mechanisms of this GPCR signaling coordination is critical yet poorly explored.
Recently, we uncovered a novel regulatory GPCR system that revolves around GPR139 antagonizing the μ-opioid receptor (MOR) (4). MOR couples to the Gi/o class of heterotrimeric G proteins to inhibit production of second messenger cAMP and to activate G protein inwardly rectifying potassium (GIRK) channels (5). It has been heavily targeted therapeutically for its desirable analgesic effects (6–10). However, MOR activation also generates untoward euphoria that leads to dependence (5, 11, 12). Our initial examination of the intersection between MOR and GPR139 revealed that multiple aspects of MOR signaling were impinged by GPR139, which could be aided by a physical interaction between the receptors (4). For instance, GPR139 enhanced β-arrestin recruitment to activated MOR and moderated its coupling to GIRK channels (4). However, the exact mechanism(s) of how GPR139 exerts an inhibitory influence on MOR and its relevance to the regulation of neuronal responses remains unknown.
The biology and signaling mechanisms of GPR139 are also poorly understood. It is an orphan GPCR expressed in select circuits in the brain, most notably in the areas implicated in motivation and reward (13–15). Whereas the endogenous ligand for GPR139 is a lingering question, it has been shown to be activated by the amino acids l-Phe and l-Trp and the neuropeptide α-MSH (13, 16, 17). Additionally, several synthetic ligands can activate GPR139 with high efficiency (18–20). Studies have also shown that GPR139 engages a variety of second messenger pathways when studied in transfected cells (14, 15, 21). However, its coupling to individual G proteins and their role in propagating downstream signals has not been holistically examined, which limits our understanding of the MOR-GPR139 interplay.
Here, we present the results of a systematic investigation of G protein–coupling selectivity of GPR139 and its role in signaling to downstream effectors. We also explore how GPR139 regulates MOR effects in a native neuronal setting. We establish that the key effects of GPR139 are mediated by Gq/11 signaling, which opposes the transmission of MOR signals at several intersecting points. Based on our findings, we propose that GPR139 “tunes” cellular responses to modulate opioid effects.
Results
GPR139 preferentially couples to Gα11/q and Gαi/o
To determine the signaling properties of GPR139, we first examined its complete G protein–coupling profile. We used a bioluminescence resonance energy transfer (BRET) GPCR “fingerprinting” approach that permits the comprehensive study of unmodified receptors and unmodified Gα proteins to examine their coupling selectivity (22). In this assay, Venus-tagged Gβ1γ2 was paired with the nanoluciferase-based sensor mas-GRK3CT. In the presence of a chosen Gα subunit, GPCR activation triggers dissociation of the G protein heterotrimer, generating the BRET signal (Fig. 1A). Using this strategy, we surveyed GPR139 activity on 15 Gα proteins (Fig. 1, B–F) and recorded both the maximum amplitude and initial activation kinetics for each G protein (Fig. 1, G and H).
Figure 1.
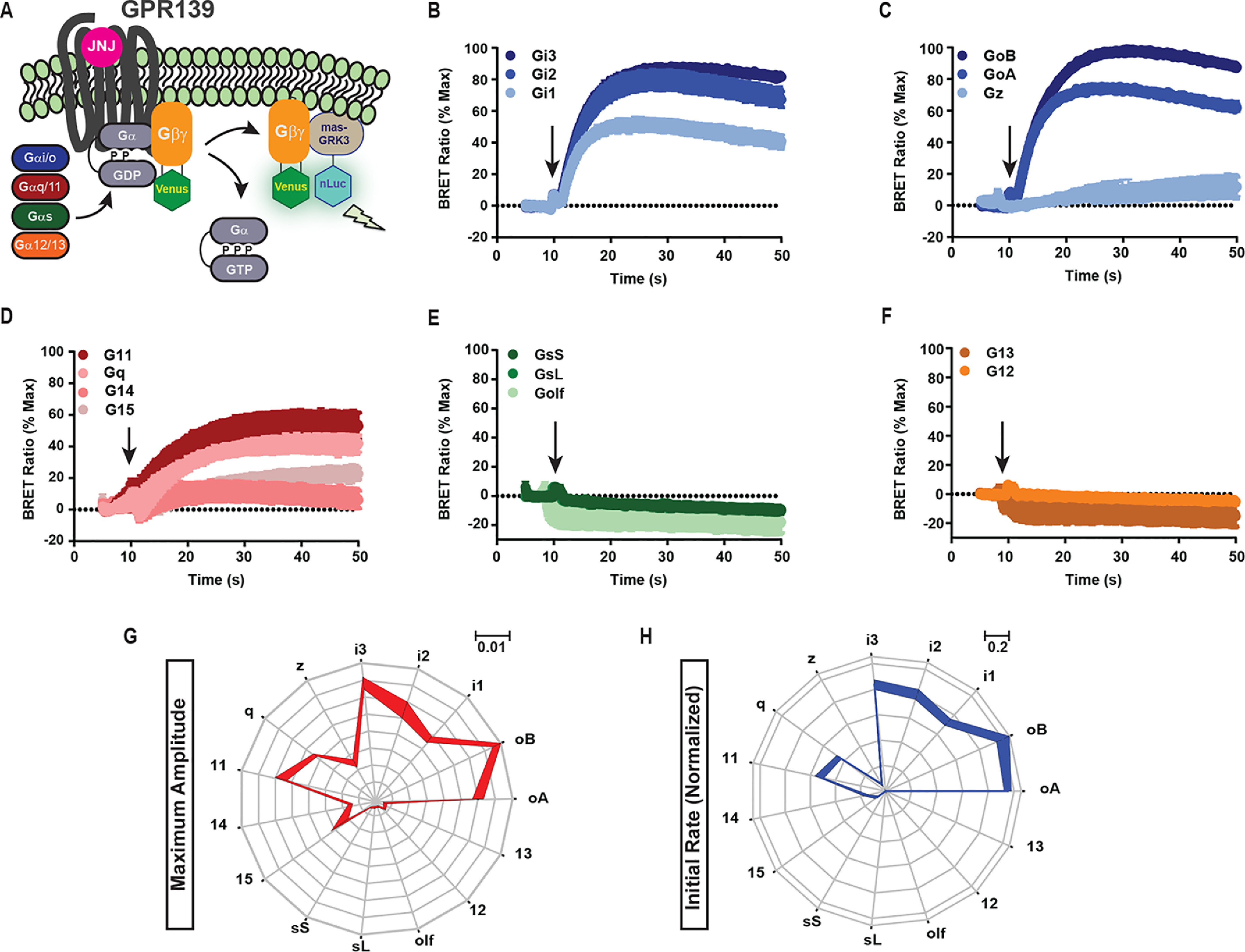
GPR139 G protein–coupling profile. A, schematic representation of the G protein BRET fingerprinting assay in HEK293 cells. B–F, GPR139 activity on the Gi/o class (B and C), Gq/11 class (D), Gs class (E), or G12/13 class (F) of G proteins. Amplitude and rate of activation were measured upon application of 10 μm JNJ-63533054, indicated by an arrow. Shown are the maximum amplitude (G) and the initial activation rate (H) for each G protein by GPR139. A–F, data are mean ± S.E. (error bars) of three independent experiments. G and H, mean of representative data ± S.D.
Interestingly, GPR139 could activate multiple G proteins with varying efficacies and kinetics. Within the Gi/o class, there were no strong preferences for individual subtypes (Fig. 1, B and C). Within the Gq/11 class, only Gq and G11 showed appreciable activation (Fig. 1D). Dose-response studies with representative members further confirmed similar potencies of GPR139 coupling to G11 and Go (Fig. S1). Gs and G12/13 classes were not activated by GPR139 (Fig. 1, E and F). Side-by-side comparison of maximum amplitudes (Fig. 1G) and the activation rates (Fig. 1H) across all classes of G proteins revealed a unique fingerprint-like profile of GPR139 coupling preferences. Together, these results indicate that GPR139 is a dual-specificity receptor capable of activating G proteins of the Gi/o and Gq/11 classes.
GPR139 selectively utilizes Gq/11 but not Gi/o for signaling to downstream effectors
Because GPR139 can activate multiple G protein classes, which can lead to different cellular responses, we examined the effectors engaged by GPR139. We used a panel of sensors to assay the effect of GPR139 activation on Ca2+ mobilization, ion channel regulation, and cAMP generation (Fig. 2A).
Figure 2.

GPR139 signaling to downstream effectors. A, schematic of downstream signaling pathways examined. B, calcium mobilization was measured using the CalFluxVTN calcium biosensor in response to treating HEK293 cells with 10 μm JNJ-63533054. C, the maximal amplitude of the Ca2+ signal across concentrations of JNJ-63533054. D, thallium flux through GIRK1/2 channels in HEK293 cells upon treatment with 10 μm JNJ-63533054. E, the initial activation rate of thallium flux calculated across doses of JNJ-63533054. F, time course of GPR139 influence on cAMP. G, dose dependence of GPR139 activation on cAMP modulation. Data are mean ± S.E. (error bars) of 3–4 independent experiments.
When calcium levels were assayed with the BRET-based sensor CalFluxVTN (23), we observed that GPR139 activation triggered a rapid and transient increase in BRET signal (Fig. 2B) suggestive of Ca2+ release from intracellular stores and consistent with previous reports (14, 15). The application of the Gαq family–selective inhibitor YM-254890 completely blocked the response, indicating that the signal was mediated by Gαq-type proteins (Fig. 2B). Dose-response studies confirmed these observations and established an EC50 of 75 ± 29 nm (Fig. 2C), which is consistent with the EC50 for G11 activation in the G protein BRET assay (119 ± 32 nm) (Fig. S1).
Next, we tested the influence of GPR139 on GIRK channels, which are activated by Gβγ released preferentially by Gi/o proteins (24–26) (Fig. 2, D and E). We monitored GIRK channel opening kinetically by measuring thallium influx with an indicator dye (27). The simultaneous addition of thallium and the GPR139 agonist JNJ-63533054 did not increase the rate of thallium influx compared with the slow, passive entry observed in cells without receptor (Fig. 2D). Dose-response studies quantifying changes in the slope of fluorescence showed no JNJ-63533054–induced changes at any concentration tested, confirming that GPR139 does not activate GIRK channels (Fig. 2E). Positive control experiments with MOR-transfected cells treated with morphine significantly increased the rate of GIRK channel activation, indicating adequate assay sensitivity (Fig. S2, A and B).
Finally, we measured the effect of GPR139 on cAMP generation with a biosensor (Fig. 2F). Unexpectedly, we found that GPR139 stimulation dynamically increased cAMP levels. The increase occurred in a dose-dependent manner with an EC50 of 41 ± 20 nm (Fig. 2G). GPR139 expression without agonist stimulation also increased the baseline cAMP levels, suggesting a high level of constitutive activity (Fig. S3). In summary, these results indicate that GPR139 coupling to Gq/11 propagates to engage the canonical Ca2+ release pathway. However, despite the efficient coupling of GPR139 to Gi/o and no coupling to Gs/olf, we observed no GIRK channel activation and paradoxical increase in cAMP production.
GPR139 signals to secondary effectors adenylyl cyclase and GIRK channels via Gαq/11
The seemingly contradictory results between G protein coupling and downstream modulation of cAMP warranted further investigation. First, we tested whether GPR139 can also alter cAMP dynamics after treatment with the AC activator forskolin (FSK). Preincubation with JNJ-63533054 had no effect on cAMP dynamics in control cells (Fig. S4), but in GPR139-transfected cells, we detected a substantial enhancement of cAMP generation in both the amplitude and the initial activation rate (Fig. 3, A, C, and D).
Figure 3.
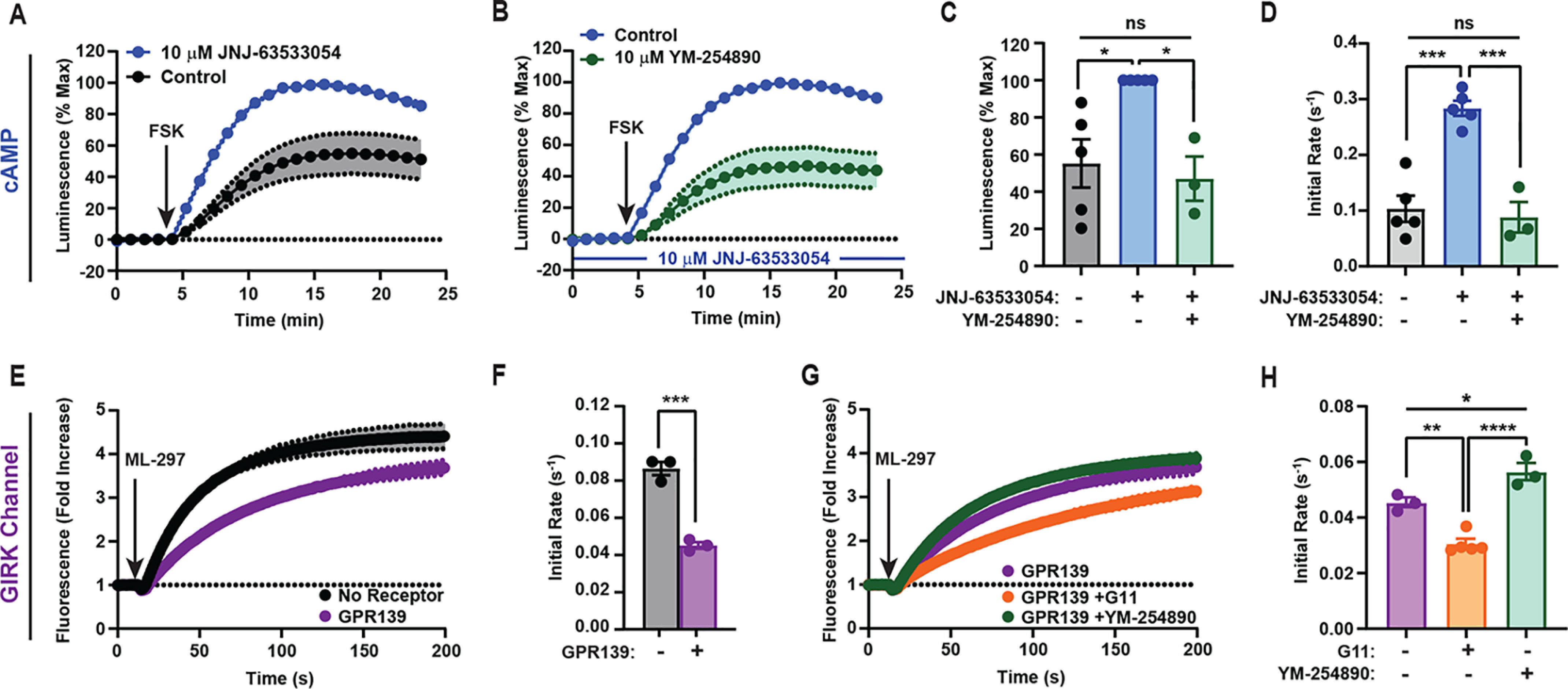
GPR139 signals selectively through Gq/11 to secondary effectors in cultured cells. Shown are changes in forskolin-stimulated cAMP (A–D) or ML-297-mediated GIRK1/2 activation (E–H) in response to GPR139 activation. A, effect of JNJ-63533054 on FSK-stimulated cAMP response. B, effect of pretreatment with Gq inhibitor YM-254890 on GPR139-mediated cAMP modulation. C and D, quantification of the maximum amplitude (C) and the initial activation rates (D) from A and B. E, effect of GPR139 on thallium flux through GIRK1/2 channels activated by ML-297. F, quantification of the initial activation rates from E. Student's t test was used: ***, p < 0.001. G, thallium flux in GPR139-expressing cells either additionally expressing G11 or pretreated with 10 μm YM-254890. H, quantification of the initial activation rates from G. Data are mean ± S.E. (error bars) of 3–5 independent experiments. C, D, and H, one-way analysis of variance with Tukey's post hoc test: ns, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Shaded areas in A, B, E, and G are S.E.
Intriguingly, several AC isoforms known to be expressed in HEK293T can be regulated by calcium generated downstream of Gq/11-coupled GPCRs (28–30). To test the hypothesis that the GPR139-mediated changes in cAMP were driven by Gq/11 signaling, we blocked Gαq/11 with YM-254890 and found that it prevented GPR139-mediated augmentation of cAMP generation (Fig. 3B). The maximal response elicited by co-application of YM-254890 with JNJ-63533054 was decreased compared with cells treated with agonist alone and restored back to the control-treated condition (Fig. 3C). Moreover, YM-254890 also abolished the effect of JNJ-63533054 on the acceleration of cAMP production rate (Fig. 3D).
We next addressed the inability to detect GIRK channel activation by GPR139 despite its coupling to Gi/o (25, 26, 31). Interestingly, GIRK channels have also been shown to be impacted by Gq signaling acting via PIP2 and protein kinase C to inhibit channel activity (31–33). Thus, we hypothesized that GPR139 could negatively regulate GIRK channels via Gq to prevent channel opening in response to Gi/o activation. To test this hypothesis, we activated GIRK directly by agonist ML-297 (34), which requires PIP2 as a cofactor, making it sensitive to Gq-mediated modulation (34, 35). Indeed, expression of GPR139 inhibited GIRK channel opening to 52.3 ± 1.6% of control (Fig. 3, E and F). Overexpression of G11 enhanced this inhibition (Fig. 3, G and H). In contrast, blockade of Gq/11 by YM-254890 diminished the inhibitory effects of GPR139 (Fig. 3, G and H). These effects were selective for GPR139 as MOR had no impact on ML-297–mediated GIRK activation either upon overexpression of G11 or blockade of Gq/11 by YM-254890 (Fig. S5, A and B). We thus conclude that GPR139 negatively regulates GIRK channel opening by engaging the Gq/11 pathway, which predominates over stimulatory Gi/o engagement.
GPR139 signaling via Gq/11 opposes MOR-mediated suppression of neuronal firing
We next sought to determine the relevance of GPR139-mediated Gq/11 signaling to counteracting MOR in the endogenous setting by examining spontaneous neuron firing in the medial habenula (mHb), where MOR and GPR139 are co-expressed (4, 36). Application of the MOR agonist DAMGO significantly dampened neuronal firing and reversed upon DAMGO washout (Fig. 4, A, B, and D). Pretreatment with JNJ-63533054 did not change the baseline firing rate (Fig. 4A) but completely blocked DAMGO's effects on firing (Fig. 4, A, C, and D). This effect of JNJ-63533054 was specific as it did not block DAMGO influence on firing in GPR139−/− mice (Fig. S6, A and B). Importantly, blockade of Gαq/11 with YM-254890 reversed the effects of the JNJ-63533054 and restored inhibition of firing in response to DAMGO (Fig. 4, A, C, and D). Taken together, these data indicate that GPR139-initiated Gq/11 signaling is sufficient to counteract MOR-mediated effects on mHb neuron firing.
Figure 4.

GPR139 signals via Gq/11 to oppose opioid-mediated inhibition of neuronal firing in medial habenula. A, representative firing traces recorded from medial habenular neurons in brain slices. B and C, change in firing frequency upon treatment with MOR agonist DAMGO. Slices were pretreated with vehicle (B) or 10 μm JNJ-63533054 (blue trace) (C) or with 10 μm JNJ-63533054 and 10 μm YM-254890 (green trace). D, maximum change in firing was quantified for (B and C). Data are mean ± S.E. of 8–10 neurons/trace; one-way analysis of variance with Tukey's post hoc test: ns, not significant; *, p < 0.05; **, p < 0.01.
Discussion
Orphan GPCRs comprise ∼100 receptors with unknown ligands and often unknown physiological roles and signaling (37) yet critical involvement in many neuronal functions (38). Thus, elucidating their functional roles presents a compelling research frontier. GPR139 is an orphan GPCR with poorly understood biology and signaling mechanisms. It is an intriguing receptor for its selective expression pattern across brain circuits and involvement in movement (13) and the effects of alcohol and opioids (4, 39). We recently found that GPR139 is molecularly intertwined with MOR and opposes its signaling, offering the first glimpse of a possible physiological role for GPR139 in regulating reward (4). In this report, we provide critical insight into unanswered questions regarding GPR139 signaling mechanisms. We show that GPR139 primarily engages the Gq/11 pathway to activate adenylyl cyclase and inhibit the GIRK channel, key effector molecules that are regulated in an opposite fashion by MOR, thereby providing a mechanistic basis for the signaling cross-talk (Fig. 5). We further show that this Gq/11-mediated signaling is essential for GPR139's ability to inhibit opioid effects in mHb neurons. The Gq/11 mechanism identified in this study may complement the direct inhibitory effects of GPR139 on MOR mediated by heterologous β-arrestin recruitment and heteromerization reported previously (4).
Figure 5.
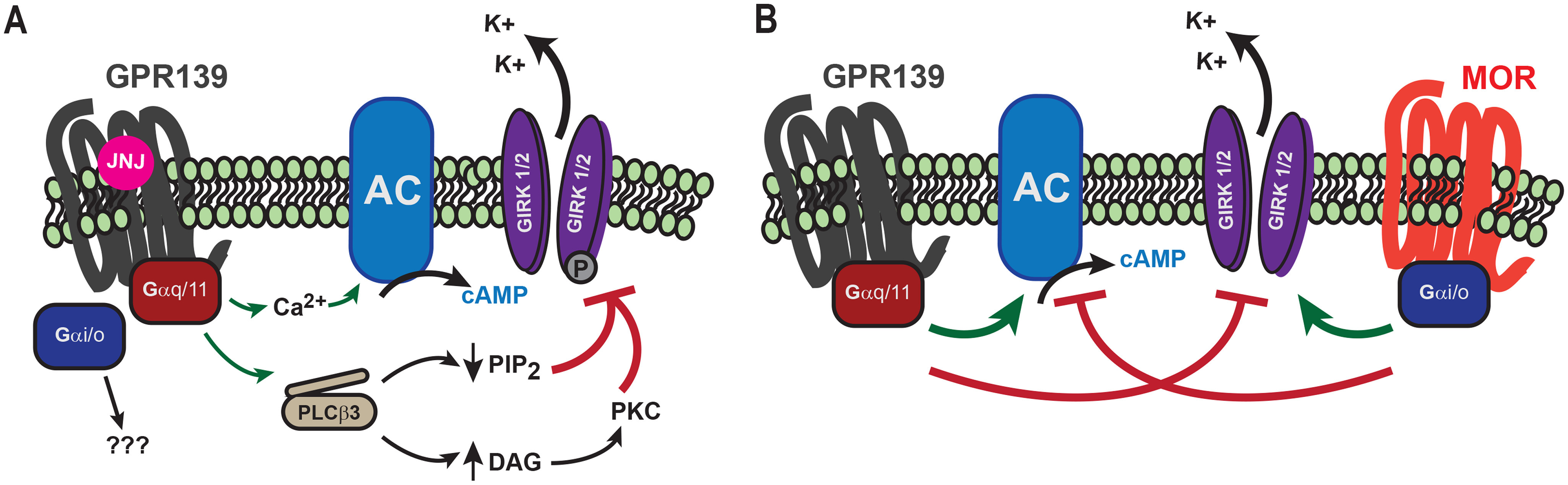
GPR139 signaling mechanisms. A, GPR139 activates both Gq/11 and Gi/o classes of G proteins, but signaling to secondary effectors is mediated by Gq/11. B, GPR139 counteracts MOR signaling by opposing regulation of the downstream effectors AC and GIRK channels.
Our observations that GPR139 effectively utilizes Gq/11 to suppress MOR signaling mediated by Gi/o provides a clear illustration of the signaling cross-talk that occurs at the level of G proteins competing to regulate common effectors. The interplay between Gq/11 and Gi/o has been noted for ion channels and adenylyl cyclase (40, 41). In particular, opposing signaling between Gαq and Gαo pathways has been well-documented in Caenorhabditis elegans (42–44). On the other hand, Gi/o-coupled receptors may act synergistically with Gq/11-coupled receptors to potentiate Gq/11-mediated signaling (45–47). Thus, the cellular context likely affects whether Gq/11 and Gi/o display an inhibitory or synergistic relationship. Therefore, the full extent of MOR-GPR139 cross-talk is likely more nuanced and context-dependent, making it possible that regulation of effector systems may vary in different neuronal populations. We envision that Gq/11 signaling by GPR139 may also similarly interfere with the Gi/o signals generated by other GPCRs co-expressed with GPR139. Such a scenario and the possible range of additional Gi/o-coupled GPCR targets impacted by GPR139 remains to be established. From this perspective, it is intriguing that a recent study reported a possible functional interaction between GPR139 and the Gi/o-coupled dopamine 2 receptor (48). The interplay of GPR139 with other GPCRs may differ from what we observe for MOR and may be shaped by heteromerization, compartment localization, and cellular environment.
Our exhaustive profiling of G protein coupling revealed that GPR139 can activate multiple G proteins. Whereas this study establishes the role of the Gq/11 branch in mediating GPR139 effects on opioid signaling, the relevance and roles of other signaling modalities remain to be determined. It seems possible that other Gα species may fine-tune GPR139 effects or allow it to access different signaling modalities. We are intrigued by the robust coupling of GPR139 to the Gi/o class. This coupling does not result in the engagement of typical Gi/o effectors, likely due to antagonism from Gαq that overpowers it, yet Gi/o is expected to be concurrently activated. We envision two potential mechanisms that could exploit this dual coupling. First, concurrent activation of Gi/o may serve to constrain the Gq/11 signals endowing GPR139 with a self-pacing mechanism. Second, Gi/o signaling might be preferentially realized in some neuronal populations where the Gq/11 branch is weak. This would enable bidirectional control of cellular signaling by GPR139, depending on the G protein contingent in a given neuron. In any event, the complete map of G protein selectiveness presented in this study should enable exploration of GPR139 signaling modalities in future studies.
One key frontier in GPR139 research is related to its activation mechanism. We observed GPR139 activity in the absence of agonist in every assay performed, which likely reflects high constitutive activity. GPR139 could use this constitutive inhibitory influence to dampen or enhance the response of MOR to endogenous opioid peptides. In addition, GPR139 could also be modulated by ligands. The amino acids l-Phe and l-Trp have been reported to activate GPR139 with an EC50 in the 200–300 μm range (16). These amino acids are present in the culture media at a ∼20–70 μm range and thus may enhance GPR139 activity. GPR139 could also be activated by several peptides, most prominently α-MSH, although there are conflicting reports (17, 49). Whether these peptides are involved in mediating GPR139 modulation of opioid signaling in endogenous neural circuits remains to be established.
Our observations suggest a strategy for increasing the therapeutic window of opioids. We predict that GPR139 antagonists and/or inverse agonists would be useful to enhance the behavioral sensitivity to opioids. Several groups have performed high-throughput screens to identify potential antagonists, but most compounds identified exhibit low potency and poor specificity and are therefore of limited utility for in vivo applications (21, 50). Discrete distribution of GPR139 in the brain, its robust anti-opioid activity, and the demonstration of its signaling mechanisms presented in this paper makes this orphan receptor an attractive therapeutic target.
Experimental procedures
Reagents and constructs
Dulbecco's modified Eagle's medium, Opti-MEM, sodium pyruvate, Lipofectamine 3000, nonessential amino acids, and Hanks' balanced saline solution with Ca2+ and Mg2+ were purchased from Invitrogen (Carlsbad, CA). Matrigel was from Corning Life Sciences. NanoGlo Luciferase Reagent, GloSensor cAMP reagent, and the pGloSensor −22F cAMP plasmid were purchased from Promega Corp. (Madison, WI). (S)-JNJ-63533054 was synthesized as described (18). YM-254890 was purchased from Wako Chemicals USA, Inc. (Richmond, VA). The GPR139 cDNA used for all experiments (HASP-HA-GPR139) contained an N-terminal HA signal peptide (HASP), followed by an HA tag and human GPR139 (NM_001002911.4).
Cell culture and transfection
HEK293T/17 cells were maintained in Dulbecco's modified Eagle's medium + 10% (v/v) fetal bovine serum, nonessential amino acids, and 1 mm sodium pyruvate. Transfections were performed as described (51).
G protein BRET assay
HEK293T/17 cells were transfected with HASP-HA-GPR139 pcDNA3.1 and 0.84 µg of Gαo pcMV5, 0.84 µg of Gα11 pcDNA3.1, or 0.84 µg of the indicated Gα protein. Additionally, 0.42 µg of Venus(1–155)-Gγ2 pcDNA3.1, 0.42 µg of Venus(156–299)-Gβ1 pcDNA3.1, and 0.42 µg of mas-GRK3-NanoLuc pcDNA3.1 were transfected, and DNAs were balanced to 10 µg using pcDNA3.1 and assays were performed as described (22).
CalFluxVTN calcium assay
HEK293T/17 cells were transfected with 0.84 µg of HASP-HA-GPR139 pcDNA3.1 and 0.84 μg of CalFluxVTN pcDNA3.1 and incubated overnight, and the assay was performed as described (52), measuring luminescence every 0.06 s using the PHERAstar FSX plate reader.
−22F pGLO assay
HEK293T cells were transfected using 0.42 µg of HASP-HA-GPR139 pcDNA3.1, 0.21 µg of MOR-FLAG, and 0.42 µg of −22F pGloSensor™ cAMP plasmid. The next day, cells were reseeded in a 96-well plate in culture medium at 1 × 105 cells/well for 2 h at 37 °C and 5% CO2. The overlaying medium was then replaced with 40 µl of 1× GloSensor™ cAMP reagent and measured as described (53).
Thallium flux
GIRK channel function was assessed using the Molecular Devices FLIPR® potassium assay kit. The following cDNAs were transfected: 0.84 µg of MOR-FLAG pcDNA3.1, 0.21 µg of HASP-HA-GPR139 pcDNA3.1, 1.26 µg of GIRK-1-AU5 pCMV5, 1.26 µg of mGIRK-2a pCMV5, 0.84 µg of rat Gαo pCMV, 0.42 µg of Gβ1 pcDNA3.1, 0.42 µg of Gγ2 pcDNA3.1, and for the indicated assay conditions, 0.42 µg of hGα11 pcDNA3.1. Cells were incubated for 18–24 h at 37 °C and 5% CO2. The next day, cells were reseeded at 20,000 cells/well of a black 384-well Corning CellBIND® plate and treated with or without 10 μm YM-254890. Cells were incubated for an additional 18–24 h. On the day of the experiment, 20 μl of loading buffer was added to 20 μl of cell culture per the manufacturer's instructions. Plates were incubated in the dark at room temperature for 1 h.
Baseline signaling was assessed for 10 reads at 1 s/read using the FLIPR Tetra®. Data were normalized as -fold over baseline, and the change in activation over time was compared by fitting a straight line to the data within the initial linear range of activation (15–25 s) using GraphPad Prism 8.
Slice electrophysiology
All procedures involving mice were approved by the institutional animal care and use committee at Scripps Research and were carried out in strict compliance with National Institutes of Health guidelines. Mice of either sex aged 2–4 months were anesthetized with isoflurane and decapitated. Brain slices were prepared, and loose-seal, cell-attached recordings from habenular neurons were performed as described (4). Changes in firing frequency following pharmacological treatments were quantified as the mean value of the last 3 min of the recordings in the presence of the drug, normalized with respect to the last 3 min of baseline preceding the drug application. Current clamp signals were high pass–filtered at 300 Hz.
Data availability
Any data not in the article can be shared upon request.
Supplementary Material
Acknowledgments
We thank Natalia Maremyanova for mouse husbandry and Nickolas K. Skamangas for technical support.
This article contains supporting information.
Author contributions—H. M. S. and K. A. M. conceptualization; H. M. S. data curation; H. M. S., S. Z., and I. M. formal analysis; H. M. S., S. Z., and I. M. investigation; H. M. S. and K. A. M. writing-original draft; H. M. S., S. Z., I. M., B. G., and K. A. M. writing-review and editing; B. G. resources; B. G. and K. A. M. funding acquisition; K. A. M. supervision; K. A. M. project administration.
Funding and additional information—This work was supported by National Institutes of Health Grants DA036596 (to K. A. M.), DA048036 (to B. G. and K. A. M.), DA047771 (to H. M. S.), and DA048579 (to S. Z.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—B. G. and K. A. M. have filed a patent on the utility of GPR139 as a drug target.
- GPCR
- G protein–coupled receptor
- AC
- adenylyl cyclase
- MOR
- µ-opioid receptor
- GIRK
- G protein inward rectifying potassium
- BRET
- bioluminescence resonance energy transfer
- FSK
- forskolin
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- mHb
- medial habenula
- HA
- hemagglutinin
- HASP
- HA signal peptide.
References
- 1. Huang Y., and Thathiah A. (2015) Regulation of neuronal communication by G protein-coupled receptors. FEBS Lett. 589, 1607–1619 10.1016/j.febslet.2015.05.007 [DOI] [PubMed] [Google Scholar]
- 2. Milligan G., and Kostenis E. (2006) Heterotrimeric G-proteins: a short history. Br. J. Pharmacol. 147, S46–S55 10.1038/sj.bjp.0706405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Greengard P. (2001) The neurobiology of slow synaptic transmission. Science 294, 1024–1030 10.1126/science.294.5544.1024 [DOI] [PubMed] [Google Scholar]
- 4. Wang D., Stoveken H. M., Zucca S., Dao M., Orlandi C., Song C., Masuho I., Johnston C., Opperman K. J., Giles A. C., Gill M. S., Lundquist E. A., Grill B., and Martemyanov K. A. (2019) Genetic behavioral screen identifies an orphan anti-opioid system. Science 365, 1267–1273 10.1126/science.aau2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Al-Hasani R., and Bruchas M. R. (2011) Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 115, 1363–1381 10.1097/ALN.0b013e318238bba6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen Y., Mestek A., Liu J., Hurley J. A., and Yu L. (1993) Molecular cloning and functional expression of a μ-opioid receptor from rat brain. Mol. Pharmacol. 44, 8–12 [PubMed] [Google Scholar]
- 7. Matthes H. W., Maldonado R., Simonin F., Valverde O., Slowe S., Kitchen I., Befort K., Dierich A., Le Meur M., Dollé P., Tzavara E., Hanoune J., Roques B. P., and Kieffer B. L. (1996) Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the μ-opioid-receptor gene. Nature 383, 819–823 10.1038/383819a0 [DOI] [PubMed] [Google Scholar]
- 8. Pert C. B., and Snyder S. H. (1973) Opiate receptor: demonstration in nervous tissue. Science 179, 1011–1014 10.1126/science.179.4077.1011 [DOI] [PubMed] [Google Scholar]
- 9. Clark M. J., Furman C. A., Gilson T. D., and Traynor J. R. (2006) Comparison of the relative efficacy and potency of μ-opioid agonists to activate Gαi/o proteins containing a pertussis toxin-insensitive mutation. J. Pharmacol. Exp. Ther. 317, 858–864 10.1124/jpet.105.096818 [DOI] [PubMed] [Google Scholar]
- 10. Laugwitz K. L., Offermanns S., Spicher K., and Schultz G. (1993) μ and δ opioid receptors differentially couple to G protein subtypes in membranes of human neuroblastoma SH-SY5Y cells. Neuron 10, 233–242 10.1016/0896-6273(93)90314-H [DOI] [PubMed] [Google Scholar]
- 11. Darcq E., and Kieffer B. L. (2018) Opioid receptors: drivers to addiction? Nat. Rev. Neurosci. 19, 499–514 10.1038/s41583-018-0028-x [DOI] [PubMed] [Google Scholar]
- 12. Navratilova E., Atcherley C. W., and Porreca F. (2015) Brain circuits encoding reward from pain relief. Trends Neurosci. 38, 741–750 10.1016/j.tins.2015.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu C., Bonaventure P., Lee G., Nepomuceno D., Kuei C., Wu J., Li Q., Joseph V., Sutton S. W., Eckert W., Yao X., Yieh L., Dvorak C., Carruthers N., Coate H., et al. (2015) GPR139, an orphan receptor highly enriched in the habenula and septum, is activated by the essential amino acids l-tryptophan and l-phenylalanine. Mol. Pharmacol. 88, 911–925 10.1124/mol.115.100412 [DOI] [PubMed] [Google Scholar]
- 14. Matsuo A., Matsumoto S., Nagano M., Masumoto K. H., Takasaki J., Matsumoto M., Kobori M., Katoh M., and Shigeyoshi Y. (2005) Molecular cloning and characterization of a novel Gq-coupled orphan receptor GPRg1 exclusively expressed in the central nervous system. Biochem. Biophys. Res. Commun. 331, 363–369 10.1016/j.bbrc.2005.03.174 [DOI] [PubMed] [Google Scholar]
- 15. Süsens U., Hermans-Borgmeyer I., Urny J., and Schaller H. C. (2006) Characterisation and differential expression of two very closely related G-protein-coupled receptors, GPR139 and GPR142, in mouse tissue and during mouse development. Neuropharmacology 50, 512–520 10.1016/j.neuropharm.2005.11.003 [DOI] [PubMed] [Google Scholar]
- 16. Isberg V., Andersen K. B., Bisig C., Dietz G. P., Bräuner-Osborne H., and Gloriam D. E. (2014) Computer-aided discovery of aromatic l-α-amino acids as agonists of the orphan G protein-coupled receptor GPR139. J. Chem. Inf. Model. 54, 1553–1557 10.1021/ci500197a [DOI] [PubMed] [Google Scholar]
- 17. Nøhr A. C., Shehata M. A., Hauser A. S., Isberg V., Mokrosinski J., Andersen K. B., Farooqi I. S., Pedersen D. S., Gloriam D. E., and Bräuner-Osborne H. (2017) The orphan G protein-coupled receptor GPR139 is activated by the peptides: adrenocorticotropic hormone (ACTH), α-, and β-melanocyte stimulating hormone (α-MSH, and β-MSH), and the conserved core motif HFRW. Neurochem. Int. 102, 105–113 10.1016/j.neuint.2016.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dvorak C. A., Coate H., Nepomuceno D., Wennerholm M., Kuei C., Lord B., Woody D., Bonaventure P., Liu C., Lovenberg T., and Carruthers N. I. (2015) Identification and SAR of glycine benzamides as potent agonists for the GPR139 receptor. ACS Med. Chem. Lett. 6, 1015–1018 10.1021/acsmedchemlett.5b00247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nøhr A. C., Shehata M. A., Palmer D., Pokhrel R., Vallianou M., Foster S. R., Gentry P. R., Gloriam D. E., and Bräuner-Osborne H. (2019) Identification of a novel scaffold for a small molecule GPR139 receptor agonist. Sci. Rep. 9, 3802 10.1038/s41598-019-40085-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shi F., Shen J. K., Chen D., Fog K., Thirstrup K., Hentzer M., Karlsson J. J., Menon V., Jones K. A., Smith K. E., and Smith G. (2011) Discovery and SAR of a series of agonists at orphan G protein-coupled receptor 139. ACS Med. Chem. Lett. 2, 303–306 10.1021/ml100293q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu L. A., Tang P. M., Eslahi N. K., Zhou T., Barbosa J., and Liu Q. (2009) Identification of surrogate agonists and antagonists for orphan G-protein-coupled receptor GPR139. J. Biomol. Screen. 14, 789–797 10.1177/1087057109335744 [DOI] [PubMed] [Google Scholar]
- 22. Masuho I., Ostrovskaya O., Kramer G. M., Jones C. D., Xie K., and Martemyanov K. A. (2015) Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Sci. Signal. 8, ra123 10.1126/scisignal.aab4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang J., Cumberbatch D., Centanni S., Shi S. Q., Winder D., Webb D., and Johnson C. H. (2016) Coupling optogenetic stimulation with NanoLuc-based luminescence (BRET) Ca2+ sensing. Nat. Commun. 7, 13268 10.1038/ncomms13268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Breitwieser G. E., and Szabo G. (1985) Uncoupling of cardiac muscarinic and beta-adrenergic receptors from ion channels by a guanine nucleotide analogue. Nature 317, 538–540 10.1038/317538a0 [DOI] [PubMed] [Google Scholar]
- 25. Huang C. L., Slesinger P. A., Casey P. J., Jan Y. N., and Jan L. Y. (1995) Evidence that direct binding of Gβγ to the GIRK1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron 15, 1133–1143 10.1016/0896-6273(95)90101-9 [DOI] [PubMed] [Google Scholar]
- 26. Pfaffinger P. J., Martin J. M., Hunter D. D., Nathanson N. M., and Hille B. (1985) GTP-binding proteins couple cardiac muscarinic receptors to a K channel. Nature 317, 536–538 10.1038/317536a0 [DOI] [PubMed] [Google Scholar]
- 27. Weaver C. D., Harden D., Dworetzky S. I., Robertson B., and Knox R. J. (2004) A thallium-sensitive, fluorescence-based assay for detecting and characterizing potassium channel modulators in mammalian cells. J. Biomol. Screen. 9, 671–677 10.1177/1087057104268749 [DOI] [PubMed] [Google Scholar]
- 28. Atwood B. K., Lopez J., Wager-Miller J., Mackie K., and Straiker A. (2011) Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics 12, 14 10.1186/1471-2164-12-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Beazely M. A., and Watts V. J. (2005) Gαq-coupled receptor signaling enhances adenylate cyclase type 6 activation. Biochem. Pharmacol. 70, 113–120 10.1016/j.bcp.2005.04.007 [DOI] [PubMed] [Google Scholar]
- 30. Sadana R., and Dessauer C. W. (2009) Physiological roles for G protein-regulated adenylyl cyclase isoforms: insights from knockout and overexpression studies. Neurosignals 17, 5–22 10.1159/000166277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang C. L., Feng S., and Hilgemann D. W. (1998) Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature 391, 803–806 10.1038/35882 [DOI] [PubMed] [Google Scholar]
- 32. Keselman I., Fribourg M., Felsenfeld D. P., and Logothetis D. E. (2007) Mechanism of PLC-mediated Kir3 current inhibition. Channels (Austin) 1, 113–123 10.4161/chan.4321 [DOI] [PubMed] [Google Scholar]
- 33. Lei Q., Talley E. M., and Bayliss D. A. (2001) Receptor-mediated inhibition of G protein-coupled inwardly rectifying potassium channels involves Gαq family subunits, phospholipase C, and a readily diffusible messenger. J. Biol. Chem. 276, 16720–16730 10.1074/jbc.M100207200 [DOI] [PubMed] [Google Scholar]
- 34. Kaufmann K., Romaine I., Days E., Pascual C., Malik A., Yang L., Zou B., Du Y., Sliwoski G., Morrison R. D., Denton J., Niswender C. M., Daniels J. S., Sulikowski G. A., Xie X. S., et al. (2013) ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem. Neurosci. 4, 1278–1286 10.1021/cn400062a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wydeven N., Marron Fernandez de Velasco E., Du Y., Benneyworth M. A., Hearing M. C., Fischer R. A., Thomas M. J., Weaver C. D., and Wickman K. (2014) Mechanisms underlying the activation of G-protein-gated inwardly rectifying K+ (GIRK) channels by the novel anxiolytic drug, ML297. Proc. Natl. Acad. Sci. U. S. A. 111, 10755–10760 10.1073/pnas.1405190111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boulos L. J., Darcq E., and Kieffer B. L. (2017) Translating the habenula: from rodents to humans. Biol. Psychiatry 81, 296–305 10.1016/j.biopsych.2016.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sriram K., and Insel P. A. (2018) G protein-coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol. Pharmacol. 93, 251–258 10.1124/mol.117.111062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alavi M. S., Shamsizadeh A., Azhdari-Zarmehri H., and Roohbakhsh A. (2018) Orphan G protein-coupled receptors: the role in CNS disorders. Biomed. Pharmacother. 98, 222–232 10.1016/j.biopha.2017.12.056 [DOI] [PubMed] [Google Scholar]
- 39. Kononoff J., Kallupi M., Kimbrough A., Conlisk D., de Guglielmo G., and George O. (2018) Systemic and intra-habenular activation of the orphan G protein-coupled receptor GPR139 decreases compulsive-like alcohol drinking and hyperalgesia in alcohol-dependent rats. eNeuro 5, ENEURO.0153-18.2018 10.1523/ENEURO.0153-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Halls M. L., and Cooper D. M. F. (2017) Adenylyl cyclase signalling complexes: pharmacological challenges and opportunities. Pharmacol. Ther. 172, 171–180 10.1016/j.pharmthera.2017.01.001 [DOI] [PubMed] [Google Scholar]
- 41. Inanobe A., and Kurachi Y. (2014) Membrane channels as integrators of G-protein-mediated signaling. Biochim. Biophys. Acta 1838, 521–531 10.1016/j.bbamem.2013.08.018 [DOI] [PubMed] [Google Scholar]
- 42. Bastiani C., and Mendel J. (2006) Heterotrimeric G proteins in C. elegans. WormBook 1–25 10.1895/wormbook.1.75.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hajdu-Cronin Y. M., Chen W. J., Patikoglou G., Koelle M. R., and Sternberg P. W. (1999) Antagonism between Goα and Gqα in Caenorhabditis elegans: the RGS protein EAT-16 is necessary for Goα signaling and regulates Gqα activity. Genes Dev. 13, 1780–1793 10.1101/gad.13.14.1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miller K. G., Emerson M. D., and Rand J. B. (1999) Goα and diacylglycerol kinase negatively regulate the Gqα pathway in C. elegans. Neuron 24, 323–333 10.1016/S0896-6273(00)80847-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gerwins P., and Fredholm B. B. (1992) Stimulation of adenosine A1 receptors and bradykinin receptors, which act via different G proteins, synergistically raises inositol 1,4,5-trisphosphate and intracellular free calcium in DDT1 MF-2 smooth muscle cells. Proc. Natl. Acad. Sci. U. S. A. 89, 7330–7334 10.1073/pnas.89.16.7330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Seyedi N., Win T., Lander H. M., and Levi R. (1997) Bradykinin B2-receptor activation augments norepinephrine exocytosis from cardiac sympathetic nerve endings: mediation by autocrine/paracrine mechanisms. Circ. Res. 81, 774–784 10.1161/01.res.81.5.774 [DOI] [PubMed] [Google Scholar]
- 47. Shah B. H., Siddiqui A., Qureshi K. A., Khan M., Rafi S., Ujan V. A., Yakoob M. Y., Yaqub Y., Rasheed H., and Saeed S. A. (1999) Co-activation of Gi and Gq proteins exerts synergistic effect on human platelet aggregation through activation of phospholipase C and Ca2+ signalling pathways. Exp. Mol. Med. 31, 42–46 10.1038/emm.1999.7 [DOI] [PubMed] [Google Scholar]
- 48. Wang L., Lee G., Kuei C., Yao X., Harrington A., Bonaventure P., Lovenberg T. W., and Liu C. (2019) GPR139 and dopamine D2 receptor co-express in the same cells of the brain and may functionally interact. Front. Neurosci. 13, 281 10.3389/fnins.2019.00281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nepomuceno D., Kuei C., Dvorak C., Lovenberg T., Liu C., and Bonaventure P. (2018) Re-evaluation of adrenocorticotropic hormone and melanocyte stimulating hormone activation of GPR139 in vitro. Front. Pharmacol. 9, 157 10.3389/fphar.2018.00157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang J., Zhu L. Y., Liu Q., Hentzer M., Smith G. P., and Wang M. W. (2015) High-throughput screening of antagonists for the orphan G-protein coupled receptor GPR139. Acta Pharmacol. Sin. 36, 874–878 10.1038/aps.2015.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Masuho I., Martemyanov K. A., and Lambert N. A. (2015) Monitoring G protein activation in cells with BRET. Methods Mol. Biol. 1335, 107–113 10.1007/978-1-4939-2914-6_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Masuho I., Chavali S., Muntean B. S., Skamangas N. K., Simonyan K., Patil D. N., Kramer G. M., Ozelius L., Babu M. M., and Martemyanov K. A. (2018) Molecular deconvolution platform to establish disease mechanisms by surveying GPCR signaling. Cell Rep. 24, 557–568.e5 10.1016/j.celrep.2018.06.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dunn H. A., Patil D. N., Cao Y., Orlandi C., and Martemyanov K. A. (2018) Synaptic adhesion protein ELFN1 is a selective allosteric modulator of group III metabotropic glutamate receptors in trans. Proc. Natl. Acad. Sci. U. S. A. 115, 5022–5027 10.1073/pnas.1722498115 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Any data not in the article can be shared upon request.