

Abstract
The treatment of cancer patients with α‐particle‐emitting therapeutics continues to gain in importance and relevance. The range of radiopharmaceutically relevant α‐emitters is limited to a few radionuclides, as stable chelators or carrier systems for safe transport of the radioactive cargo are often lacking. Encapsulation of α‐emitters into solid inorganic systems can help to diversify the portfolio of candidate radionuclides, provided, that these nanomaterials effectively retain both the parent and the recoil daughters. We therefore focus on designing stable and defined nanocarrier‐based systems for various clinically relevant radionuclides, including the promising α‐emitting radionuclide 224Ra. Hence, sub‐10 nm barium sulfate nanocontainers were prepared and different radiometals like 89Zr, 111In, 131Ba, 177Lu or 224Ra were incorporated. Our system shows stabilities of >90 % regarding the radiometal release from the BaSO4 matrix. Furthermore, we confirm the presence of surface‐exposed amine functionalities as well as the formation of a biomolecular corona.
Keywords: radiolabeling, radium, nanoencapsulation, radiooncology, barium, therapeutic applications
Well packed behind bars: a range of clinically relevant radionuclides including the α‐emitter radium‐224 is incorporated into the crystal matrix of robust sub‐10 nm barium sulfate nanocontainers.
1. Introduction
Targeted alpha therapy (TAT) is one of the most intensively discussed therapeutic approaches in nuclear medicine and oncology in recent years.1 The concept of this promising treatment option involves the combination of α‐emitters and specific cancer targeting moieties, such as small molecules, peptides, peptidomimetics or biomacromolecules including antibody‐based entities via stable carrier systems.2 The oncotropic vector molecules are then responsible for the selective deposition of the cytotoxic α‐payloads and for the delivery of a tumoricidal radiation dose, leading to tumor cell death without inducing collateral damages to normal surrounding healthy tissues.1a, 3 Due to the high linear energy transfers (LET) of up to 220 keV/μm and their short track path length in biological tissues in the micrometer range,4 α‐particles show an extremely high ionization density, leading to severe cellular radiation damage via DNA double strand breaks, thereby yielding a high relative biological effectiveness (RBE) for cell killing.5 Furthermore, as α‐emitters generate clustered radiation damage independent of oxygenation mostly by direct ionization of target molecules, effective cell killing can be expected, even in hypoxic areas of the tumor. That particular feature renders α‐emitters proficient for taking tumoricidal effects on both the well‐oxygenated and hypoxic cell populations, thus overcoming the limitations of β‐particle emitters.6 However, owing to decay properties and availability,7 only a couple of α‐emitting radionuclides are considered clinically relevant for cancer treatment, including astatine‐2116b, 8 and actinium‐225,1b, 9 of which actinium‐225 has already been successfully applied for prostate tumor targeting in humans.10 In addition to these radionuclides, bismuth‐213, thorium‐227, terbium‐149 as well as the radium isotopes radium‐223 and radium‐224 represent suitable candidates for TAT of cancer.1d, 2, 7, 11 Importantly, the continuous development and optimization of TAT towards clinical translation needs to be complemented by resolute efforts to engineer appropriate tools for pre‐therapeutic dosimetry planning as well as treatment monitoring. The simplest way of achieving this is the replacement of the therapeutic by a diagnostic isotope of the same chemical element (matched pair) within the respective radiopharmaceutical, e. g. yttrium‐90/‐86 or iodine‐131/‐124.12 As no suitable diagnostic isotopes exist for the above‐mentioned clinically relevant α‐emitters except for terbium‐149,13 diagnostic radionuclides with similar charge and physical properties are commonly used in combination with appropriate carrier systems, e. g. actinium‐225/gallium‐68 complexed by the bifunctional chelator DOTA (1,4,7,10‐tetraazacyclododecane‐1,4,7,10‐tetraacetic acid).14 Due to their similar effective ionic radii, analogous chemical behavior as well as attractive decay properties (γ‐photon emission), the radionuclides barium‐131 (t1/2=11.5 d; Eγ=124; 216 keV) and barium‐135m (t1/2=28.7 h; Eγ=268 keV) represent eligible diagnostic alkaline earth metal counterparts to radium‐223/‐224.15 Using these barium isotopes instead of or in combination with the α‐emitting radium isotopes would allow to investigate the pharmacologic properties of the respective radiopharmaceutical in vivo, enable therapy monitoring and facilitate image‐based dose calculations for different tissues and organs. However, robust and sufficiently stable carrier systems for barium and radium ions have not been reported so far by other groups. Macrocyclic chelators based on calixarenes for stable complexation of radium isotopes have been investigated in our group,16 but respective ligands with adequate kinetic stability are yet to be discovered. As an alternative strategy to organic ligands, we and several other groups made substantial efforts to incorporate radium into nanocarrier‐based systems, including polyoxopalladates,17 hydroxyapatite,18 iron oxide,19 barium ferrite, or nanozeolites.20 Recently, we reported on the development of a straightforward carrier system for radium based on barium sulfate nanoparticles (NPs).21 By following the facile approach of barium/radium co‐precipitation, we obtained radiolabeled barium sulfate NPs with an average particle diameter of more than 100 nm and a broad size distribution ((140±50) nm), and with reasonable stability regarding activity release (less than 5 %) in aqueous environment. Noteworthy, incorporation of alendronate into the NP matrix resulted in the introduction of multiple reactive access points available for further functionalization with (bio)molecules. With this in mind, our present work aims on the one hand at reducing the NP diameter to less than 10 nm with an acceptable size distribution striving for a favorable pharmacokinetic profile and distinct clearance behavior. This is of special relevance to decrease normal tissue radiation exposure and toxicity, thereby ensuring reasonable dose deposition at the tumor site as a prerequisite for high therapeutic efficacy and clinical effectiveness. On the other hand and even more important, we examine these smaller NPs for their suitability as feasible transporters for other relevant radionuclides beyond the alkaline earth metals radium and barium. Consequently, we describe herein the synthesis and characterization of stable, radiolabeled, alendronate‐containing, sub‐10 nm barium sulfate NPs with a functionalized surface architecture.
2. Results and Discussion
2.1. Synthesis and Characterization of Alendronate‐Containing BaSO4 NPs
Recently, we have published a novel approach to follow the TAT concept based on the incorporation of barium and radium radionuclides into BaSO4 NPs using a simple and efficient single‐step precipitation method.21 To achieve biological requirements, such as body clearance, we optimized the synthesis procedure to further decrease the mean particle diameter and sharpen the NP size distribution. Thus, we initially precipitated barium alendronate by the addition of ethanol in the first reaction step (Figure 1).
Figure 1.

Representative bright‐field TEM image of ethanol‐stabilized barium alendronate precipitates obtained after the first reaction step.
Using ethanol for stabilizing purposes in NP syntheses has been tested before, but without a distinct template.22 During the second reaction step, the ethanol‐stabilized barium alendronate precipitates form nanotemplates for subsequent growth of the alendronate‐containing BaSO4 NPs.
For synthesis purpose, a 0.1 m Ba(NO3)2 solution (200 μmol, 1 eq.) and sodium alendronate (80 μmol, 0.4 eq) were rapidly mixed with 4 mL of water/ethanol 1/1. Subsequently, a 0.05 m (NH4)2SO4 solution (100 μmol, 0.5 eq.) was added to the barium alendronate dispersion via a syringe pump under a constant volume stream of 8 mL/h. The resulting particles were then washed three times with deionized water, characterized by transmission electron microscopy (TEM), and the elemental distribution was evaluated by energy‐dispersive X‐ray spectroscopy (EDXS). Comparative TEM images of our previous first‐generation as well as second‐generation alendronate‐containing BaSO4 NPs obtained after optimizing the synthesis procedure are shown in Figure 2.
Figure 2.

Representative bright‐field TEM images of first‐generation (a) and second‐generation (b) alendronate‐containing BaSO4 NPs.
Compared to the NPs in Figure 2a, which show various shapes and sizes, the second‐generation NPs using the modified re‐precipitation method (Figure 2b) are more spherically‐like and substantially smaller with a narrower size distribution (9.1±1.9 nm). Selected area electron diffraction (SAED) analysis confirmed the orthorhombic barite structure (a=8.8842(12) Å, b=5.4559(8) Å, c=7.1569(9) Å, space group Pnma (62)).23 The massive particle size reduction concomitant with a much sharper size distribution observed by TEM analyses was further supported by dynamic light scattering (DLS) measurements (Figure 3).
Figure 3.
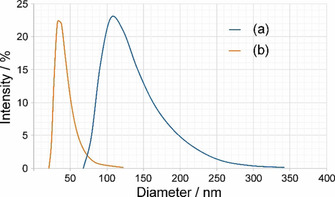
Size distribution of first‐generation (a) and second‐generation (b) alendronate‐containing BaSO4 NPs obtained by dynamic light scattering measurements.
It is clearly shown in Figure 3 that the synthesized second‐generation NPs (Figure 3b) possess a substantially smaller size distribution compared to the first‐generation NPs (Figure 3a). Moreover, the mean particle size (curve maximum point) has been decreased from approximately 110 nm to 35 nm. Not surprisingly, the hydration sphere diameter determined by DLS differs from the primary particle size of the NPs obtained by TEM due to the fundamental differences in the underlying measurement principles.24 Whereas for TEM a precise particle diameter is obtained, DLS measurements are influenced by the NPs’ hydrodynamic properties in the respective solvent. Moreover, as DLS is a low‐resolution batch method, a small number of large particles often masks the smaller ones, if the samples are not ideally monodisperse. In contrast to that, TEM is a direct counting method, which measures a size value for each particle selected for the analysis, thus providing a number‐based particle size distribution. Furthermore, TEM allows to visualize polydisperse samples over a wide size range, thus providing direct shape information. Unlike TEM, measurement of NP sizes from DLS data is an indirect method, based on the determination of the frequency of movement, and modelling of the size from this data.
EDXS data of the synthesized second‐generation BaSO4 NPs show an equal overall distribution of the elements barium, sulfur and oxygen as well as phosphorous. The former three elements originate from the existing BaSO4 crystals, while the latter element is detectable due to the alendronate moieties inside the BaSO4 lattice (Figure 4).
Figure 4.
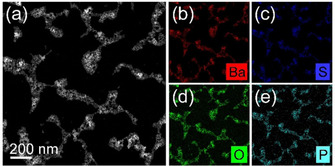
HAADF‐STEM image of second‐generation NP aggregates (a) together with the corresponding element distributions obtained by energy‐dispersive X‐ray spectroscopy analysis for barium (b), sulfur (c), oxygen (d), and phosphorous (e).
In similarity to our first‐generation NPs, we proved the reactivity of the amine groups by the reaction with ninhydrin (data not shown). Moreover, we successfully performed a respective coupling reaction with the fluorescent dye fluorescein isothiocyanate (FITC) leading to fluorescent NPs in aqueous media. To verify our work‐up procedure after the functionalization and to examine the stability of the FITC‐labeled BaSO4 NPs, a comparative electrophoresis experiment analyzing the migration behavior of pure FITC, FITC‐functionalized alendronate and FITC‐labeled, alendronate‐containing BaSO4 NPs in an agarose gel was performed. The resulting image is presented in Figure 5.
Figure 5.
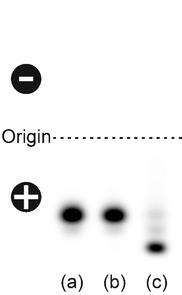
Comparative agarose gel electrophoresis with (a) fluorescein isothiocyanate (FITC), (b) FITC‐labeled alendronate and (c) 25 μg of alendronate‐containing FITC‐labeled BaSO4 NPs. The samples were separated by electrophoresis on a 1 % TAE‐agarose gel (pH 8). Fluorescence signals were documented using a Typhoon fluorescence scanner.
FITC (Figure 5a) as well as FITC‐alendronate (Figure 5b) both cover the same distance from the origin towards the anode, while the dye‐labeled BaSO4 NPs (Figure 5c) migrate even further to the positive pole due to their overall negative charge under these experimental conditions. Particularly worthy to mention here is the almost complete absence of impurities or starting material (FITC) in the BaSO4 NPs preparation. Based on this observation, we conclude that our applied work‐up procedure is adequate and that the dye‐labeled BaSO4 NPs are sufficiently pure and stable under the applied conditions.
During our studies, we noticed a pH‐dependent aggregation behavior of the second‐generation BaSO4 NPs (data not shown), which led us to the investigation of the surface ζ‐potential at slightly acidic and slightly basic pH values (Table 1).
Table 1.
Comparison of the surface ζ‐potentials of different BaSO4 NPs at pH 6 and pH 8
|
Sample |
Surface ζ‐potential |
|
|---|---|---|
|
pH 6 |
pH 8 |
|
|
Unlabeled BaSO4 NPs without alendronate |
18.6 mV |
−15.2 mV |
|
Unlabeled alendronate‐ containing BaSO4 NPs (free amines) |
19.5 mV |
−14.7 mV |
|
FITC‐labeled alendronate‐ containing BaSO4 NPs |
11.2 mV |
−15.1 mV |
The ζ‐potential values indicate that the surface charge of the NPs is strongly influenced by protonation and deprotonation. Since there are furthermore no substantial differences in ζ‐potential regarding dye‐modified and unmodified NPs as well as NPs with vs. without alendronate containment, the charge is mainly influenced by the BaSO4 crystal matrix itself. Free barium and sulfate ions on the outer surface may interact with either hydroxy groups or protons in a pH‐dependent manner and therefore being responsible for the NPs’ surface charge after protonation and deprotonation processes. A strongly charged NP surface certainly represents one of the main reasons for their undesired aggregation at cell membranes as well as for the formation of a biomolecular corona and an unfavorable pharmacokinetic profile in vivo. However, this drawback is at the present research stage of secondary importance as the current focus is predominantly on stable incorporation of radionuclides.
2.2. One‐Pot Radiolabeling of Alendronate‐Containing BaSO4 NPs
Besides the size reduction, the main objective of this study was to investigate the applicability of the BaSO4 NPs as feasible carrier systems not only for barium and radium, but also for other clinically relevant diagnostic and therapeutic radionuclides. Respective radionuclides were added to the first reaction step of the NP synthesis procedure mentioned above. In addition to the priority radionuclides barium‐131/‐133 and radium‐224, the incorporation of fluorine‐18, copper‐64, zirconium‐89, indium‐111, and lutetium‐177 was investigated. After the synthesis, the respective radionuclide‐doped NPs were centrifuged and washed three times with deionized water. Radiochemical yields (RCY) are expressed as the ratio between the amount of activity in the NP pellet and the starting activity (Table 2).
Table 2.
Overview of the used radionuclides, their chemical forms, applied amounts of activity, and the resulting radiochemical yields (RCY) after NP precipitation.
|
Radionuclide |
Counter ion |
Starting activity (MBq) |
RCY (%) |
|---|---|---|---|
|
18F |
H+ |
14.6 |
26[a] |
|
64Cu |
Cl− |
80.0 |
<5[a] |
|
89Zr |
C2O4 2− |
30.0 |
75 |
|
111In |
Cl− |
9.0 |
88 |
|
131Ba |
NO3 − |
7.9 |
40 |
|
133Ba |
Cl− |
0.05 |
41 |
|
177Lu |
Cl− |
48.1 |
69 |
|
224Ra |
NO3 − |
1.1 |
31 |
[a] Decomposition/release during washing steps directly after the synthesis: no sufficient radiolabeling
Radiolabeling reactions were performed using a broad range of clinically relevant radionuclides. Labeling approaches using zirconium‐89, indium‐111 and lutetium‐177 resulted in acceptable RCY of more than two‐thirds of incorporated starting activity. By incorporating barium‐131/‐133 and radium‐224 as the radionuclides of our highest interest, RCY ranged from 30 % to 40 %. These yields meet our expectations, caused by the twofold excess of barium over sulfate. Due to this present stoichiometry, which is required from the methodical point of view, a maximum RCY of 50 % is achievable. Still, we consider our method as highly efficient, as the unincorporated radiometal ions can be recovered from the NP washing solutions by ion exchange chromatography very easily and efficiently. Referring to the β+‐emitter copper‐64, sufficient labeling could not be achieved. This was most likely because of the affinity of the alendronate as a complexing agent for copper ions in the first reaction step. Consequently, the BaSO4 crystal matrix was no longer accessible for 64Cu‐incorporation that further led to a complete wash out (alongside the alendronate excess) continuously during the NP work‐up.
2.3. Stability of Radiolabeled Alendronate‐Containing BaSO4 NPs
After the synthesis and work‐up of the radiolabeled NPs had been accomplished, the dispersions were stored in deionized water and the activity release was monitored after 1, 3 and 7 d after radiolabeling. Especially for therapeutic applications with short‐lived radionuclides (days to weeks) such as α‐emitters, the long‐term stability of the NPs is crucial and highly desirable. The activity release was determined by sonicating the NPs followed by centrifugation of the pellet after a respective period of time. Afterwards, the release was determined as the amount of activity in the supernatant compared to the amount of activity in the particle pellet. The data for the NP stability values after the labeling experiments is presented in Table 3.
Table 3.
Long‐term stability of radiolabeled alendronate‐containing BaSO4 NPs.
|
|
Remaining activity in the NP pellet after the indicated period of time (%) |
||
|---|---|---|---|
|
Radionuclide |
1 d |
3 d |
7 d |
|
89Zr |
99 |
99 |
99 |
|
111In |
95 |
94 |
93 |
|
131Ba |
97 |
97 |
93 |
|
133Ba |
97 |
95 |
93 |
|
177Lu |
96 |
97 |
97 |
|
224Ra |
99[a]/82[b] |
95[a]/78[b] |
96[a]/82[b] |
[a] Regarding 224Ra. [b] Regarding 212Pb.
Overall, the radiolabeled BaSO4 NPs do not show substantial release of activity over the period of investigation and the stability is consistently >90 %, thereby providing a sufficient stability for in vivo application. This leads to the assumption that even the defect ions (apart from barium and radium ions) are stably trapped in the inner particle matrix and do not only bind via surface interactions. An additional TLC analysis with a competing ligand (50 mm EDTA as mobile phase) after one week of storage confirmed that significant amounts of activity were not released (data not shown).
Worth mentioning are the two relative stability values given in Table 3 for radium‐224. The percentage of direct radium‐224 release (<5% after 7 d) is on the one hand very low and the NPs seem to be stable at first sight. Apart from that, the overall γ‐measurement referring to lead‐212 indicates a slightly lower stability value of approx. 80 %. This difference originates from the release of activity during the cascade of decays from radium‐224 to lead‐212 with three consecutive α‐decays and the corresponding recoil energy. Moreover, the chemical characteristics of the first daughter nuclide radon‐220 ‐ being a noble gas ‐ are in general unfavorable for any stable binding and may allow the escape of surface‐located atoms from the BaSO4 crystal matrix by passive diffusion. The recoil effect of α‐emitting radiopharmaceuticals has also been investigated by different research groups strongly advising to consider this observation.25 Therefore, not only the NP distribution, but also the distribution of the radium's daughter nuclides is of high importance for the future planning of in vivo experiments and for the clinical perspectives of radionuclides that decay via a cascade, especially those including α‐decays.
The decayed NP samples were analyzed by TEM to examine potentially occurring decomposition events or other side effects caused by the decay energy. TEM images of the different radiolabeled BaSO4 NPs after a minimum decay time of ten half‐lives are presented in Figure 6.
Figure 6.

Representative bright‐field TEM images of the second‐generation alendronate‐containing BaSO4 NPs (a) after radiolabeling with 64Cu (b), 89Zr (c), 111In (d), 131Ba (e), 177Lu (f) or 224Ra (g).
By comparing the properties of the second‐generation BaSO4 NPs before (see Figure 2b and 6a for comparison) and after the radiolabeling procedures, substantial morphological differences were not noticed, independent of the applied radiation entity (α‐, β+‐, β−‐ or γ‐emitters). The particles show similar size and shape characteristics as well as no signs for any destructive processes caused by the long storage time during the total decay or directly by the radiation energy of the radionuclides.
2.4. Interaction of Alendronate‐Containing BaSO4 NPs with Serum Components
The unspecific adsorption of various biomolecules, such as proteins and lipids, to the NP surface often results in the formation of a biomolecular corona upon exposure to complex biological fluids.26 As a consequence, the corona interacts with biological systems and not the bare NP surface, thus giving these particles a biological identity distinct from that of the originally synthesized material. This dynamic interface, which can significantly affect the cellular uptake and pharmacokinetics of the NPs, depends on various factors like size, charge, shape, and hydrophobicity.27 To investigate the bio‐nano‐interactions of the alendronate‐containing BaSO4 NPs with serum proteins and other biomolecules, we applied an electrophoretic mobility shift assay (EMSA). This technique relies on an altered migration behavior of biomolecule‐NP‐complexes compared to bare NPs in an agarose gel as size and charge can substantially differ upon the corona formation.28 The bare alendronate‐containing BaSO4 NPs migrate through the agarose gel towards the positive pole due to their negative ζ‐potential under these experimental conditions as already observed earlier (Figure 5c and 7).
Figure 7.
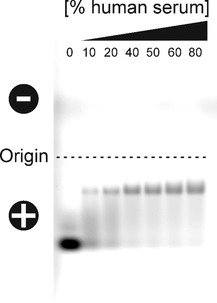
Effect of human serum on the electrophoretic mobility of FITC‐labeled, alendronate‐containing BaSO4 NPs. Increasing concentrations of human serum were incubated with NP concentrations of 0.1 μg/μL for 1 h at 37 °C. Afterwards, the biomolecule‐NP‐complexes were separated by electrophoresis on a 1 % TAE‐agarose gel (pH 8). Fluorescence signals originating from FITC‐labeled NPs were finally documented using a Typhoon fluorescence scanner. The percentage denotes the concentration of human serum used for incubation.
Noteworthy, the appearance of these NPs in the gel as a distinct fluorescent band indicates their high uniformity and homogeneity. Their incubation with increasing concentrations of human serum for 1 h at 37 °C causes a substantial change in their electrophoretic mobility, pointing to the formation of biomolecule‐NP‐complexes. Due to the adsorption of biomolecules, serum‐incubated NPs show a significant shift on the gel, as the corona components increase their hydrodynamic diameter and mask their negative surface charge.
In order to investigate the composition of the biomolecular corona in more detail, the serum‐incubated NP samples were analyzed using sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE). In this experiment, the NP‐associated biomolecules were separated from the supernatant serum by centrifugation, followed by extensive washing with phosphate buffered saline (PBS) solution in order to remove serum components with low affinity for the NP surface (“soft” corona). Finally, biomolecules retained on the surface of the alendronate‐containing BaSO4 NPs (“hard” corona) were detached and denatured by incubating for 5 min at 95 °C in reducing Laemmli sample buffer and subjected to 1D SDS‐PAGE analysis, followed by visualization of protein bands by staining with a colloidal Coomassie G‐250 dye (Figure 8).
Figure 8.
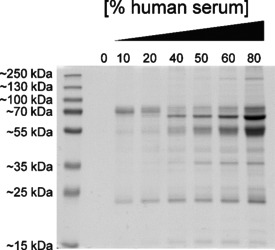
SDS‐PAGE analysis of the serum protein absorption on alendronate‐containing BaSO4 NPs upon incubation with different serum concentrations for 1 h at 37 °C.
As expected from the previous experiment, a protein corona was formed on the alendronate‐containing BaSO4 NPs. Several distinct bands at ∼22, ∼38, ∼54, ∼65, and ∼70 kDa corresponding to surface‐associated serum proteins were observed, and with increasing serum concentration, the total protein amount clearly increases. In particular, the intensity of the recurring band in the size range of ∼65 kDa strongly increases in moving from 40 to 80 % serum.
Although the surface of the synthesized BaSO4 NPs is covered with the hydrophilic alendronate coating, the interaction with biomolecules is not (completely) prevented, most likely due to the surface charge of the NPs. However, the unwanted effect of corona formation has nowadays been transformed into an advantage under certain circumstances and seen as the opportunity to confer extrinsic properties of the cognate corona to the intrinsic functions of the NPs. In other words, the biomolecular corona can be exploited by providing additional features that render nanotherapeutics more effective and precise. For example, a functional protein corona endowing the NPs with both, targeting and immune escape elements, could increase their stability as well as improve their biocompatibility, bioavailability and molecular recognition functions.29
3. Conclusions
In summary, we have presented a facile approach to obtain stable, defined and water‐dispersible alendronate‐containing BaSO4 NPs with a diameter of less than 10 nm as determined by TEM by following a two‐step precipitation procedure using ethanol‐stabilized barium alendronate nanotemplates as precursors. Importantly, this precipitation method provides a straightforward way to dope the BaSO4 crystal matrix with a variety of diagnostic and therapeutic radiometal ions including radium, barium, lutetium, indium, and zirconium. These radiolabeled nanocarriers exhibit >90 % radionuclide retention even after a week in dispersion. Indications of decomposition or destruction due to radioactive decay and recoil processes were not found in the comparative analysis of TEM images. Additionally, the radiation‐tolerant crystal structure of the BaSO4 NPs consequently facilitates a future theranostic approach by doping the NP matrix with diagnostic and therapeutic radionuclides simultaneously. For instance, zirconium‐89 and indium‐111 applied on the here presented class of BaSO4 NPs represent conceivable diagnostic counterparts to radium‐224 and lutetium‐177 for pre‐therapeutic dosimetry planning as well as treatment monitoring by positron emission tomography and single‐photon emission computed tomography, respectively. Doping of the BaSO4 crystal structure with two or more radionuclides emitting γ‐rays of different energies will be pursued as one of the next steps to shed light on the in vivo stability of the nanocarriers. As mentioned in detail above, the alendronate‐containing BaSO4 NPs show some degree of biomolecular corona formation when exposed to high serum concentrations. On the one hand, this could result in rapid recognition and elimination by the mononuclear phagocyte system in vivo as well as further off‐target biodistribution and accumulation. On the other hand, the protein corona could be engineered and exploited with regard to improving molecular targeting and cellular uptake. With that in mind, we are currently focusing on both, the design of ultrasmall polymer‐coated corona‐resistant “stealth” BaSO4 NPs and in‐depth understanding of the corona composition, which will enable its tailoring and engineering. Finally, we strive for conducting in vitro and in vivo studies to examine the diagnostic and therapeutic properties of targeted BaSO4 NPs necessitating the attachment of specific cancer targeting moieties including small molecules, peptides, peptidomimetics, and antibody‐based entities.
Experimental Section
General
All chemicals were purchased from commercial suppliers and used without further purification. Chromatographic analyses were carried out on Merck Silica Gel 60 F254 TLC plates. Dynamic light scattering to determine particle size, particle size distribution and ζ‐potential measurement was performed on a Zetasizer Nano ZS (Malvern Panalytical) using disposable plastic cuvettes. Gel documentation as well as fluorescent TLC plate imaging were performed using an Amersham Typhoon 5 Biomolecular Imager (GE Healthcare). Bright‐field TEM imaging was performed with an image‐Cs‐corrected Titan 80–300 microscope (FEI) operated at an accelerating voltage of 300 kV. To determine their size, 100 particles were analyzed for each sample. The respective particle sizes are given with their standard deviation. To analyze the crystal structure of the synthesized particles, SAED was performed. High‐angle annular dark‐field scanning transmission electron microscopy (HAADF‐STEM) imaging and spectrum imaging analysis based on energy‐dispersive X‐ray spectroscopy (EDXS) were done at 200 kV with a Talos F200X microscope equipped with a Super‐X EDXS detector system (FEI). Prior to TEM analysis, the specimen mounted in a high‐visibility low‐background holder was placed for 2 s into a Model 1020 Plasma Cleaner (Fischione) to remove possible contaminations. TEM specimens were prepared by dispersing the particles in deionized water, followed by ultrasonication, dropping the particle solution onto carbon‐coated copper grids and final drying at ambient temperature.
Radionuclides and Radioanalytics
Solutions of [18F]F−, [64Cu]CuCl2 and [131Ba]Ba(NO3)2 were produced in‐house by proton bombardment of the respective liquid or solid target at the TR‐FLEX Cyclotron from ASCI.30 All cyclotron‐based radionuclides are non‐carrier‐added products. [224Ra]Ra(NO3)2 was separated from a 228Th‐source by ion exchange chromatography and was obtained as non‐carrier‐added solution. [177Lu]LuCl3 was purchased by ITG and [111In]InCl3 by Mallinckrodt. [89Zr]Zr(C2O4)2 was purchased by Perkin Elmer and [133Ba]BaCl2 from Polatom. The specific activity from the manufacturers’ certificate was 50 MBq/mg Ba. Activity count rates were measured using the ISOMED 2160 (MED) sodium iodide detector and the ISOMED 2100 dose calibrator (MED). High‐precision measurements were performed using a HPGe detector by Canberra.
Synthesis of Alendronate‐Containing BaSO4 NPs
A 0.1 m Ba(NO3)2 solution (200 μmol, 1 eq.) and sodium alendronate (80 μmol, 0.4 eq.) were vigorously stirred for 5 min to dissolve the salt. Afterwards, 2 mL of ethanol and 2 mL of water were added to precipitate the barium alendronate template particles. Then, a 0.05 m (NH4)2SO4 solution (100 μmol, 0.5 eq.) was added via a syringe pump (PHD 2000, Harvard Apparatus) over 15 min. The precipitated, alendronate‐containing BaSO4 NPs were centrifuged at 10,000 g for 15 min and washed with deionized water. The washing cycle was repeated two more times. Ready‐to‐use NPs (approx. 10 mg) were redispersed in deionized stored as dispersion.
Synthesis of Radiolabeled BaSO4 NPs
For all radiolabeling experiments, the above‐mentioned method was used as basis. Before the precipitation of the radiolabeled alendronate template particles, the respective radionuclide was added to the first reaction step in aqueous solution in a small volume (approx. 50 μL). The work‐up was proceeded in analogy to the non‐radioactive method and the finalized particles were redispersed in deionized water and stored as dispersion.
Surface Modification of Alendronate‐Containing BaSO4 NPs
To verify the further reactivity of the outer amine sites of the alendronate‐containing NPs, approx. 5 mg of NPs were dispersed in 5 mL (EtOH/H2O 1/1 pH 8 adjusted by Na2CO3). 1 mg of fluorescein isothiocyanate (FITC) was added to the NP dispersion and the reaction was stirred for 2 h at rt. The functionalized particles were centrifuged at 10,000 g for 15 min and the excess of FITC was removed by washing with ethanol/water 1/1 three times. The particles were redispersed in deionized water.
Electrophoretic Mobility Shift Assay
The samples for the mobility shift assay were prepared as described recently with slight modifications.28 A total amount of 100 μg of FITC‐labeled, alendronate‐containing BaSO4 NPs (final concentration 0.1 μg/μL) were incubated with different concentrations of ‘off the clot’ human serum (Biochrom AG) diluted with PBS for 1 h at 37 °C in protein LoBind vials (Eppendorf). After the incubation time, the protein‐NP‐mixtures were loaded on a 1 % TAE (40 mm Tris, 20 mm acetic acid, 1 mm EDTA, pH 8) agarose gel and electrophoresis was carried out at 80 V for 30 min. Gel images were acquired with an Amersham Typhoon 5 Biomolecular Imager (GE Healthcare) using the specific settings for Cy2 (Laser LD488/Filter 525BP20).
Isolation and Characterization of NP‐Associated Proteins
Biomolecular corona formed on alendronate‐containing BaSO4 NPs was isolated as described recently, but with slight modifications.31 Briefly, samples containing a total amount of 100 μg of alendronate‐containing BaSO4 NPs (final concentration 0.1 μg/μL) were incubated with different concentrations of human serum, diluted with PBS, for 1 h at 37 °C in protein LoBind vials (Eppendorf). After incubation in serum, samples were centrifuged for 5 min at 10,000 g and 4 °C to pellet the NP‐corona complexes. The pellets were washed three times with ice‐cold PBS and centrifuged again for 5 min at 10,000 g and 4 °C. Finally, the NP‐corona pellets were air‐dried after the last centrifugation step to evaporate remaining liquid, resuspended in Laemmli buffer containing 355 mm β‐mercaptoethanol and incubated for 5 min at 95 °C. After cooling to rt, the samples were loaded on a 12 % SDS‐polyacrylamide gel and subjected to electrophoresis until the bromophenol blue dye of the sample buffer reached the end of the gel. After electrophoretic separation, the gels were stained with colloidal Coomassie G‐250 (PageBlue Protein Staining Solution, Thermo Fisher Scientific) according to the manufacturer's instructions.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
We thank Dr. Martin Walther and Dr. Martin Kreller as well as the cyclotron operation team for providing the cyclotron‐produced radionuclides. The use of HZDR Ion Beam Center TEM facilities and the funding of TEM Talos by the German Federal Ministry of Education of Research (BMBF), Grant No. 03SF0451, in the framework of HEMCP are gratefully acknowledged.
F. Reissig, K. Zarschler, R. Hübner, H.-J. Pietzsch, K. Kopka, C. Mamat, ChemistryOpen 2020, 9, 797.
Contributor Information
Dr. Kristof Zarschler, Email: k.zarschler@hzdr.de.
Dr. Constantin Mamat, Email: c.mamat@hzdr.de.
References
- 1.
- 1a. McDevitt M. R., Sgouros G., Sofou S., Annu. Rev. Biomed. Eng. 2018, 20, 73–93; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Miederer M., Scheinberg D. A., McDevitt M. R., Adv. Drug Delivery Rev. 2008, 60, 1371–1382; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Boll R. A., Malkemus D., Mirzadeh S., Appl. Radiat. Isot. 2005, 62, 667–679; [DOI] [PubMed] [Google Scholar]
- 1d. Marques I. A., Neves A. R., Abrantes A. M., Pires A. S., Tavares-da-Silva E., Figueiredo A., Botelho M. F., Cancer Treat. Rev. 2018, 68, 47–54. [DOI] [PubMed] [Google Scholar]
- 2. Makvandi M., Dupis E., Engle J. W., Nortier F. M., Fassbender M. E., Simon S., Birnbaum E. R., Atcher R. W., John K. D., Rixe O., Norenberg J. P., Target Oncol. 2018, 13, 189–203. [DOI] [PubMed] [Google Scholar]
- 3. Martins C. D., Kramer-Marek G., Oyen W. J. G., Expert Opin. Drug Delivery 2018, 15, 185–196. [DOI] [PubMed] [Google Scholar]
- 4. Song H., Senthamizhchelvan S., Hobbs R. F., Sgouros G., Antibodies 2012, 1, 124–148. [Google Scholar]
- 5. Pouget J. P., Navarro-Teulon I., Bardies M., Chouin N., Cartron G., Pelegrin A., Azria D., Nat. Rev. Clin. Oncol. 2011, 8, 720–734. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Wild D., Frischknecht M., Zhang H., Morgenstern A., Bruchertseifer F., Boisclair J., Provencher-Bolliger A., Reubi J. C., Maecke H. R., Cancer Res. 2011, 71, 1009–1018; [DOI] [PubMed] [Google Scholar]
- 6b. Gott M., Steinbach J., Mamat C., Open Chem. 2016, 14, 118–129. [Google Scholar]
- 7.
- 7a. Poty S., Francesconi L. C., McDevitt M. R., Morris M. J., Lewis J. S., J. Nucl. Med. 2018, 59, 878–884; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Poty S., Francesconi L. C., McDevitt M. R., Morris M. J., Lewis J. S., J. Nucl. Med. 2018, 59, 1020–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Zalutsky M. R., Pruszynski M., Curr. Radiopharm. 2011, 4, 177–185; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Choi J., Vaidyanathan G., Koumarianou E., Kang C. M., Zalutsky M. R., Nucl. Med. Biol. 2018, 56, 10–20; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Vaidyanathan G., Zalutsky M. R., Curr. Radiopharm. 2011, 4, 283–294; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d. Dziawer L., Majkowska-Pilip A., Gawel D., Godlewska M., Pruszynski M., Jastrzebski J., Was B., Bilewicz A., Nanomaterials 2019, 9, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Robertson A. K. H., Ramogida C. F., Schaffer P., Radchenko V., Curr. Radiopharm. 2018, 11, 156–172; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Thiele N. A., Wilson J. J., Cancer Biother. Radiopharm. 2018, 33, 336–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kratochwil C., Bruchertseifer F., Giesel F. L., Weis M., Verburg F. A., Mottaghy F., Kopka K., Apostolidis C., Haberkorn U., Morgenstern A., J. Nucl. Med. 2016, 57, 1941–1944. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Frantellizzi V., Cosma L., Brunotti G., Pani A., Spanu A., Nuvoli S., De Cristofaro F., Civitelli L., De Vincentis G., Cancer Biother. Radiopharm. 2020; [DOI] [PubMed] [Google Scholar]
- 11b. Müller C., Vermeulen C., Köster U., Johnston K., Türler A., Schibli R., van der Meulen N. P., EJNMMI Radiopharm. Chem. 2017, 1, 5; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Müller C., Reber J., Haller S., Dorrer H., Köster U., Johnston K., Zhernosekov K., Türler A., Schibli R., Pharmaceuticals 2014, 7, 353–365; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11d. Hagemann U. B., Wickstroem K., Hammer S., Bjerke R. M., Zitzmann-Kolbe S., Ryan O. B., Karlsson J., Scholz A., Hennekes H., Mumberg D., Cuthbertson A. S., Cancer Biother. Radiopharm. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Park J. A., Kim J. Y., Curr. Top. Med. Chem. 2013, 13, 458–469; [DOI] [PubMed] [Google Scholar]
- 12b. Bockisch A., Eur. J. Nucl. Med. Mol. Imaging 2011, 38 Suppl 1, S1–3. [DOI] [PubMed] [Google Scholar]
- 13. Müller C., Zhernosekov K., Köster U., Johnston K., Dorrer H., Hohn A., van der Walt N. T., Türler A., Schibli R., J. Nucl. Med. 2012, 53, 1951–1959. [DOI] [PubMed] [Google Scholar]
- 14. Price E. W., Orvig C., Chem. Soc. Rev. 2014, 43, 260–290. [DOI] [PubMed] [Google Scholar]
- 15. Spencer R. P., Lange R. C., Treves S., J. Nucl. Med. 1971, 12, 216-&. [PubMed] [Google Scholar]
- 16.
- 16a. Bauer D., Gott M., Steinbach J., Mamat C., Spectrochim. Acta Part A 2018, 199, 50–56; [DOI] [PubMed] [Google Scholar]
- 16b. Steinberg J., Bauer D., Reissig F., Köckerling M., Pietzsch H. J., Mamat C., ChemistryOpen 2018, 7, 432–438; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16c. Bauer D., Blumberg M., Köckerling M., Mamat C., RSC Adv. 2019, 9, 32357–32366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gott M., Yang P., Kortz U., Stephan H., Pietzsch H. J., Mamat C., Chem. Commun. (Camb.) 2019, 55, 7631–7634. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Vasiliev A. N., Severin A., Lapshina E., Chernykh E., Ermolaev S., Kalmykov S., J. Radioanal. Nucl. Chem. 2016, 311, 1503–1509; [Google Scholar]
- 18b. Uskokovic V., Uskokovic D. P., J. Biomed. Mater. Res. Part B 2011, 96, 152–191. [DOI] [PubMed] [Google Scholar]
- 19. Mokhodoeva O., Vlk M., Málková E., Kukleva E., Mičolová P., Štamberg K., Šlouf M., Dzhenloda R., Kozempel J., J. Nanopart. Res. 2016, 18, 301–312. [Google Scholar]
- 20.
- 20a. Piotrowska A., Meczynska-Wielgosz S., Majkowska-Pilip A., Kozminski P., Wojciuk G., Cedrowska E., Bruchertseifer F., Morgenstern A., Kruszewski M., Bilewicz A., Nucl. Med. Biol. 2017, 47, 10–18; [DOI] [PubMed] [Google Scholar]
- 20b. Piotrowska A., Leszczuk E., Bruchertseifer F., Morgenstern A., Bilewicz A., J. Nanopart. Res. 2013, 15, 2082; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20c. Tsotsalas M. M., Kopka K., Luppi G., Wagner S., Law M. P., Schäfers M., De Cola L., ACS Nano 2010, 4, 342–348. [DOI] [PubMed] [Google Scholar]
- 21. Reissig F., Hübner R., Steinbach J., Pietzsch H. J., Mamat C., Inorg. Chem. Front. 2019, 6, 1341–1349. [Google Scholar]
- 22. Bala H., Fu W. Y., Zhao J. Z., Ding X. F., Hang Y. Q., Yu K. F., Wang Z. C., Colloids Surf. A 2005, 252, 129–134. [Google Scholar]
- 23. Hill R. J., Can. Mineral. 1977, 15, 522–526. [Google Scholar]
- 24.
- 24a. Souza T. G. F., Ciminelli V. S. T., Mohallem N. D. S., J. Phys. Conf. Ser. 2016, 733, 012039; [Google Scholar]
- 24b. Eaton P., Quaresma P., Soares C., Neves C., de Almeida M. P., Pereira E., West P., Ultramicroscopy 2017, 182, 179–190. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Kozempel J., Mokhodoeva O., Vlk M., Molecules 2018, 23, 1–18; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. de Kruijff R. M., Wolterbeek H. T., Denkova A. G., Pharmaceuticals 2015, 8, 321–336; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25c. Wang G., de Kruijff R. M., Rol A., Thijssen L., Mendes E., Morgenstern A., Bruchertseifer F., Stuart M. C., Wolterbeek H. T., Denkova A. G., Appl. Radiat. Isot. 2014, 85, 45–53; [DOI] [PubMed] [Google Scholar]
- 25d. Arazi L., Cooks T., Schmidt M., Keisari Y., Kelson I., Phys. Med. Biol. 2007, 52, 5025–5042. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Cai R., Chen C., Adv. Mater. 2019, 31, e1805740; [DOI] [PubMed] [Google Scholar]
- 26b. Ke P. C., Lin S., Parak W. J., Davis T. P., Caruso F., ACS Nano 2017, 11, 11773–11776; [DOI] [PubMed] [Google Scholar]
- 26c. Docter D., Westmeier D., Markiewicz M., Stolte S., Knauer S. K., Stauber R. H., Chem. Soc. Rev. 2015, 44, 6094–6121. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Wang H., Lin Y., Nienhaus K., Nienhaus G. U., Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 10, e1500; [DOI] [PubMed] [Google Scholar]
- 27b. Schöttler S., Landfester K., Mailänder V., Angew. Chem. Int. Ed. Engl. 2016, 55, 8806–8815; [DOI] [PubMed] [Google Scholar]
- 27c. Feliu N., Docter D., Heine M., Del Pino P., Ashraf S., Kolosnjaj-Tabi J., Macchiarini P., Nielsen P., Alloyeau D., Gazeau F., Stauber R. H., Parak W. J., Chem. Soc. Rev. 2016, 45, 2440–2457; [DOI] [PubMed] [Google Scholar]
- 27d. Zarschler K., Rocks L., Licciardello N., Boselli L., Polo E., Garcia K. P., De Cola L., Stephan H., Dawson K. A., Nanomedicine 2016, 12, 1663–1701. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Pant K., Pufe J., Zarschler K., Bergmann R., Steinbach J., Reimann S., Haag R., Pietzsch J., Stephan H., Nanoscale 2017, 9, 8723–8739; [DOI] [PubMed] [Google Scholar]
- 28b. Pombo-García K., Zarschler K., Barreto J. A., Hesse J., Spiccia L., Graham B., Stephan H., RSC Adv. 2013, 3, 22443–22454. [Google Scholar]
- 29.
- 29a. Maiolo D., Del Pino P., Metrangolo P., Parak W. J., Baldelli Bombelli F., Nanomedicine (Lond) 2015, 10, 3231–3247; [DOI] [PubMed] [Google Scholar]
- 29b. Hamad-Schifferli K., Nanomedicine (Lond) 2013, 8, 1–3; [DOI] [PubMed] [Google Scholar]
- 29c. Nguyen V. H., Lee B. J., Int. J. Nanomed. 2017, 12, 3137–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.
- 30a. Kreller M., Pietzsch H.-J., Walther M., Tietze H., Kaever P., Knieß T., Füchtner F., Steinbach J., Preusche S., Instruments 2019, 3; [Google Scholar]
- 30b. Hoberück S., Wunderlich G., Michler E., Hölscher T., Walther M., Seppelt D., Platzek I., Zöphel K., Kotzerke J., J. Labelled Compd. Radiopharm. 2019, 62, 523–532. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Nsubuga A., Zarschler K., Sgarzi M., Graham B., Stephan H., Joshi T., Angew. Chem. Int. Ed. Engl. 2018, 57, 16036–16040; [DOI] [PubMed] [Google Scholar]
- 31b. Nsubuga A., Sgarzi M., Zarschler K., Kubeil M., Hübner R., Steudtner R., Graham B., Joshi T., Stephan H., Dalton Trans. 2018, 47, 8595–8604. [DOI] [PubMed] [Google Scholar]