

Abstract
Objectives:
The WNT pathway is an important oncologic driver of epithelial ovarian cancer (EOC). The first-in-class recombinant fusion protein ipafricept (IPA) blocks Wnt signaling through binding of Wnt ligands. This phase Ib trial was designed to determine the maximum tolerated dose (MTD) and recommended phase 2 dose (RPh2) for IPA in combination with taxane and platinum therapy (C/P)
Methods:
Dose escalation started with a standard 3+3 design for IPA/C/P with q3w intravenous IPA on Day 1, in cycles 1 to 6 with C (AUC = 5 mg/mL · min) and P (175 mg/m2). For enhanced bone safety the trial was revised to 6-patient cohorts with a q3w regimen of IPA on Day 1 and C/P on Day 3 (IPA→C/P).
Results:
37 patients have been treated; 30 of whom were treated following protocol revision to q3w IPA(D1)→C/P(D3) (2 & 4 mg/kg). IPA-related TEAEs that occurred in ≥15% included: fatigue (40%); nausea (35%); diarrhea and decreased appetite (22%) each; dysgeusia (19%); and vomiting (16.2%). 22% reported ≥1 IPA related TEAE Grade ≥3 the most common of which was neutropenia at 16%. There were no DLTs; the MTD was not reached. The maximum administered dose based on bone safety was 6mg/kg. The overall response rate (ORR) was 75.7%. Median PFS was 10.3 months (95% CI 8.5–14.2) and OS 33 months (95% CI 23.4- NR).
Conclusions:
IPA is well tolerated in combination with sequential C/P. ORR, PFS and OS are comparable to historical data but bone toxicity at efficacy doses of this particular Wnt inhibitor limit further development in EOC.
Introduction:
Epithelial ovarian cancer (EOC) remains the most lethal of the gynecologic malignancies in the developed world. In 2018, there were an estimated 22,240 new cases of EOC and 14,070 deaths in the United States. [1] Despite aggressive research into novel therapies and novel delivery of cytotoxic agents, the 5 year relative survival rates remain unacceptably low at 47% for all subtypes and stages and 30% for advanced stage disease.[2] Ideally, interventions will be developed that move more patients into a cured state following front line chemotherapy. However, until that is a reality, efforts towards developing more effective therapies for use at the time of recurrence may improve overall survival (OS) for patients.
Once recurred, subsequent therapy is decided based on the treatment-free interval from the conclusion of the prior platinum. (TFIp). If this interval is ≥ 6 months, the current paradigm is re-treatment with platinum based therapy whereas patients with a TFIp of < 6 months are moved to non-platinum based agents.[3, 4] For patients who are deemed appropriate for platinum re-treatment, chemotherapy backbones include carboplatin alone, or carboplatin doublets with paclitaxel[5], gemcitabine[6] or pegylated liposomal doxorubicin (PLD)[7]. Median progression free survival (PFS), inclusive of the time during chemotherapy administration ranges from 8.6 to 12 months.
Although response to therapy appears high, the durability of responses following platinum re-treatment is short. This loss of durability brought the concept of maintenance therapy to the forefront. Use of bevacizumab concurrent with chemotherapy and as maintenance following 6–8 cycles of cytotoxic therapy for platinum sensitive disease has been evaluated both with paclitaxel and carboplatin (Gynecologic Oncology Group (GOG) Study 213)[8] and carboplatin and gemcitabine (OCEANS)[9] with positive results. Median PFS is 13.8 and 12.4 months in the bevacizumab included arms for each study, respectively. Overall survival (OS) in GOG 213 was also improved with bevacizumab (42.6 vs. 37.3 months; Hazard ratio (HR) 0.823 (0.680–0.996)).[8] Based on this data, the US FDA approved bevacizumab for concurrent use with either carboplatin and paclitaxel or gemcitabine as well as to follow as a maintenance agent until toxicity or progression[10].
Poly (ADP-ribose polymerase) (PARP) inhibitors have also gained approval as switch maintenance agents to follow any platinum-based regimen used for recurrent platinum sensitive disease as long as the patient has a very robust partial response (PR) or CR to at least 4 cycles of chemotherapy. Among patients with BRCA mutations (either germline or somatic), the PFS (excluding the time during cytotoxic chemotherapy) varies from 16.6 to 21 months as compared to 5.4 to 5.5 months for placebo[11–13]. Among patients with evidence for homologous recombination repair deficiency (HRD) but whom are BRCA wild type (wt) the range was 9.3 versus 3.7 months and among patients with no evidence of HRD the difference was 6.9 vs. 3.8 months.[11] These data led to the approval of olaparib, niraparib and rucaparib as switch maintenance following platinum-based therapy for patients in PR or CR.[12, 14, 15]
Although these advancements are welcomed, there are still limitations. There is no predictive biomarker yet determined for bevacizumab and the PFS and OS gains are modest. PARP inhibitors work best in patients who harbor a germline or somatic BRCA mutation and moderately well in those who are BRCA wt but HRD +. Benefit in HRD- patients exists but is modest.
Many current treatments for metastatic cancers can produce an initial reduction in tumor burden but do not result in long-term benefit. A possible explanation for this observation is the continued presence of cancer stem cells (CSCs) that represent a small percentage of the tumor but are the most tumorigenic and treatment-resistant cell population. CSCs have been most extensively studied in hematologic malignancies, but have now also been identified in many solid tumors.[16, 17] CSCs drive growth and metastasis and are more resistant to radiotherapy and chemotherapy than the remaining bulk of the tumor.[18, 19] Novel approaches that target CSCs are currently being developed. Wnt signaling occurs through both β-catenin dependent and β-catenin independent pathways. The two pathways do not exist in isolation nor in opposition to one another as was previously believed. The two pathways are interconnected in complicated ways and the activation of the pathways involved in Wnt signaling is based on the Wnt pathway ligand that binds as well as binding of co-receptors and can result in activation of pathways that are involved in cellular growth, differentiation and apoptosis as well as those involved in epithelial mesenchymal transition (EMT). [20]
Wnt signaling in epithelial ovarian cancer appears to be a promising therapeutic target. On the side of Wnt dependent signaling, the Wnt ligands Wnt7a and Wnt7b are increased in EOC and increase MMP7, cyclin 1 and proliferation. In addition, inhibitors of Wnt signaling, SFRP4 and SFRP5 are both downregulated in EOC which allows increased EMT, cell migration, growth and invasion.[20–22] On the side of Wnt independent signaling, the co-receptors ROR1 and ROR2 which bind with the ligand Wnt5a activate pathways associated with ovarian cancer progression and poor prognosis.[20, 23, 24]
Ipafricept (IPA), a recombinant protein that inhibits Wnt signaling, was found to have activity against EOC in a panel of patient-derived xenograft models. IPA combined with a taxane showed superior anti-tumor activity to that of either agent alone in the majority of these EOC models. In several tumor types, including EOC, IPA activity has been associated with a profound decrease in cancer cells that can reconstitute tumors in mice, suggesting that cells with CSC-like properties are significantly reduced by the combination of IPA with paclitaxel.[25]
Furthermore, results in an EOC xenograft model suggest a sequential dosing regimen of a Wnt inhibitor prior to a taxane-containing chemotherapeutic regimen might offer optimal efficacy. [25]The current study evaluated two dosing schedules of IPA in combination with the chemotherapeutic agents carboplatin (C) and paclitaxel (P): same day dosing (in Cohorts 1 and 2) and sequential dosing, with IPA administered two days prior to these combination therapies (in Cohorts ≥3).
Patients and Methods:
This phase I, dose-escalation study (NCT02092363) was conducted at five clinical centers in the United States. The primary objectives were to determine the safety and tolerability of IPA (OMP-54F28) in combination with C/P in patients with recurrent platinum-sensitive EOC as well as to identify dose-limiting toxicities (DLT), maximum tolerated dose (MTD), and recommended phase 2 dose (RP2D) for IPA in combination with C/P. Secondary objectives included characterization of the pharmacokinetics (PK), immunogenicity, and antitumor activity of IPA in combination with C/P. Enrollment was sequential in a “3+3” design.
Eligible patients were ≥18 years, with histologically documented EOC, recurrent disease deemed appropriate for retreatment with platinum (> 6 months from last platinum therapy), Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, evaluable or measurable disease per RECIST 1.1, and adequate organ function. Archival tumor was required for all patients and fresh tumor biopsy during study was optional during dose escalation but mandatory during dose expansion.
Osteoporosis based on the total femur and L1–L4 T scores (≤−2.5) on screening bone density scan was exclusionary. T scores had to be obtained for either the right or left hip or right or left femoral neck and lumbar spine (L1–L4). History of bone metastases with a prior history of a pathologic fracture, or with a lytic lesion requiring an orthopedic intervention, or not receiving a bisphosphonate or denosumab as per institutional guidelines were exclusionary. Patients were excluded if they were on glucocorticoid therapy at the equivalent of ≥7.5 mg/day of oral prednisone for ≥4 weeks within the last 8 weeks; fasting β-C-terminal telopeptide (β-CTX) > 1,000 pg/mL; metabolic bone disease; or history of a symptomatic vertebral fragility fracture or any fragility fracture of hip, pelvis, wrist or other location; or a history of moderate morphometric fractures, for a 10 year fracture risk of >20% for any bone or >3% for the hip as determined by the US FRAX calculation tool.[26]
All patients provided written informed consent, and the study was approved by local Institution Review Boards and was conducted in accordance with the declaration of Helsinki, the International Conference on Harmonization Good Clinical Practice guidelines, and all applicable local regulatory requirements and laws.
Four dose cohorts (Cohorts 1, 2, 3, and 4) were planned during the dose escalation stage of the study. In all cohorts, IPA was administered IV on Day 1 of each 21-day cycle. The dose levels of IPA for Cohorts 1 and 2 were 5 and 10 mg/kg q3w, respectively. Due to the fragility fractures observed in the Phase 1 programs of IPA, and more so vantictumab (an antibody that inhibits the Wnt pathway by binding Frizzled [FZD] receptors FZD1, FZD2, FZD5, FZD7, and FZD8), IPA was discontinued for all patients in Cohorts 1 and 2[27]. Subsequently, patients of Cohorts 3, 4 and 5 were dosed at 2, 4 and 6 mg/kg q3w, respectively. No dose escalation of IPA was allowed within a dose cohort. The expansion cohort was dosed at 6mg/kg q3w.
In all cohorts, the combination therapies C/P were administered intravenously at AUC=5 mg/ml•min (C) and 175 mg/m2(P). In Cohorts 1 and 2, C/P were administered on Day 1 of each 21-day cycle. Nonclinical data indicated that administration of a Wnt inhibitor (IPA or vantictumab) 2 or 3 days prior to a taxane resulted in superior efficacy. Therefore, in Cohorts 3, 4, and 5, a staggered administration of IPA on Day 1, and C/P on Day 3, of each cycle was evaluated. A total of 6 cycles of C/P were given. Additional cycles were permitted as per institutional standard of care after discussion with the Medical Monitor. Treatment with IPA continued as maintenance after completion of treatment with C/P until progression or toxicity.
DLTs were defined during the first 21 days (cohorts 1 and 2) or 28 (cohorts 3+) days of treatment, as any possible treatment emergent grade ≥3 toxicities (except for grade 3 infusion reactions that resolve within 24 hours) as assessed using the CTCAE version 4.02 of the NCI, or any AE that results in the subject being discontinued from study that occurs any time from study days 0 to 28, unless the event could be clearly attributed to another cause. Study treatment continued until disease progression, unacceptable toxicity (including DLT criteria above), or withdrawal of consent.
For Cohorts 3, 4, and 5, the same dose escalation rules were followed, except a minimum of 6 patients were enrolled in each cohort. In addition, rules for a bone safety review were included.
The MTD based on bone safety was defined as the highest dose level at which <2 of 6 patients experienced Grade ≥1 fragility fracture. If the highest dose level did not exceed the MTD based on bone safety, that dose level was declared the maximum administered dose (MAD).
Once the MTD or MAD of IPA was determined, up to 20 patients were to be enrolled in the cohort expansion phase to better characterize the safety and tolerability and to preliminarily evaluate the anti-tumor activity of IPA in combination with C/P. Safety and PK assessments, as well as the rules for continued dosing in the cohort expansion stage, were identical to those in the dose escalation stage. If frequency of Grade ≥3 toxicities or other unacceptable chronic toxicities, in particular fragility fractures, in the cohort expansion stage suggested that the MTD was exceeded at that dose level, any remaining accrual at that dose level was to be halted. Consideration would then be given to enrolling additional patients into an expansion cohort at a lower dose level based on bone safety.
Study assessments
Safety assessments were conducted weekly throughout the study and for 30 days after treatment. Adverse events (AE) were graded using Common Terminology Criteria for Adverse Events (CTCAE), version 4.02. In addition to β-isomerized C-terminal telopeptides (β-CTX) and N-terminal propeptide of type 1 collagen (P1NP) at screening, every cycle, and at treatment termination, a bone safety assessment by DEXA was performed at screening, every 2 cycles (cohorts 1 and 2) and every 3 cycles (cohorts 3+), and at treatment termination. Tumor response was assessed after cycle 2 and every other cycle thereafter (cohorts 1 and 2) or at the end of cycle 3 and every third cycle thereafter (in cohorts 3+), according to RECIST version1.1. Tumor markers were followed prospectively.
Pharmacokinetic and Pharmacodynamic Analyses
For all cohorts, blood samples to evaluate pharmacokinetics (PK) were collected on days 1, 8 and 15 of cycle 1 and 3 at predose and 30 min (+/− 10 min) after the end of infusion. On day 1 of cycle 2, 4 and 5 and then every other cycle, PK was collected predose only. PK was also collected at study termination. For PK analysis, only a by patient listing of IPA concentrations was generated.
For all dose cohorts, blood samples for determination of anti-drug antibodies (ADAs) were collected on Day 1 of Cycle 1 and every other cycle thereafter (Cycles 3, 5, 7, 9, etc.), and at treatment termination visit.
Statistical Methods
The general analytical approach for evaluating all endpoints was descriptive in nature. No formal statistical hypothesis testing was conducted. No p-values were presented due to the small sample size of this study. Summary tables presented the data for each dose cohort and overall (all patients regardless of dose).
For continuous variables, descriptive statistics included the number of non-missing values, mean, standard deviation, median, 25th and 75th percentile, minimum, maximum, and 95% confidence intervals (CIs) as appropriate.
For categorical variables, descriptive statistics included counts and percentages per category. Statistics describing time-to-event variables utilized the Kaplan-Meier method. Individual patient data obtained were presented by patient in data listings. The dose group for each patient as based on the dose and dosing frequency assigned at enrollment.
Results:
Patients:
Between February 2014 and June 2017, 37 patients were enrolled and all 37 received IPA, C/P and qualified for inclusion in the safety, IIT and PK populations. Patients’ baseline demographic and tumor characteristics are summarized in Table 1.
Table 1:
Baseline demographics and cancer treatment history for included patients (n=37)
| Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 | Expansion Cohort | Overall n (%) | |
|---|---|---|---|---|---|---|---|
| Dose of Ipafricept (Q3W) |
5 mg/kg n (%) | 10 mg/kg n (%) | 2 mg/kg n (%) | 4 mg/kg n (%) | 6 mg/kg n (%) | 6 mg/kg n (%) | |
| Patients Enrolled | 3 | 4 | 8 | 9 | 6 | 7 | 37 |
| 3 (100.0) | 4 (100.0) | 8 (100.0) | 9 (100.0) | 6 (100.0) | 7 (100.0) | 37 (100.0) | |
| Demographics | |||||||
| Age | |||||||
| Median | 56 | 55.5 | 56 | 59 | 60.5 | 63 | 59 |
| Ethnicity | |||||||
| Not Hispanic or Latino | 3(100%) | 4(100%) | 8(100%) | 9(100%) | 6(100%) | 6(85.7%) | 36(97.3%) |
| Race | |||||||
| White | 3(100%) | 3(75%) | 7(87.5%) | 8(88.9%) | 6(100%) | 6(85.7%) | 33(39.2%) |
| BMI (kg/m2) mean (SD) | 26.7(5.7) | 32.1(8.7) | 29.1(6.8) | 30.6(4.6) | 29.7(4.0) | 26.1(3.3) | 29.1(5.4) |
| Cancer History | |||||||
| Time since diagnosis (mos) | |||||||
| Median | 23.2 | 33.5 | 30.8 | 19 | 27 | 19.8 | 26 |
| Histology | |||||||
| Other | 2(22.2%) | 1(16.7%) | 1(14.3%) | 4(10.8%) | |||
| Prior Lines of Therapy | |||||||
| 2 Regimens | 2(25%) | 1(11.1%) | 3(50%) | 1(14.3%) | 7(18.9%) | ||
| >3 Regimens | 1(12.5%) | 1(11.1%) | 2(28.6%) | 4(10.8%) |
SD= standard deviation, NOS = not otherwise specified; mos = months
Dose Escalation:
Dose escalation, the number of patients treated on each dose cohort, IPA exposure, duration of treatment and their disposition is presented in Table 2. For IPA, the median [25th, 75th%] number of IPA infusions administered per patient in the study was 7 [4,12]. The median number of paclitaxel and carboplatin infusions administered per patient in the study was 6[5,6].
Table 2:
Description of dose escalation and patient disposition
| Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 | Expansion Cohort | Overall n (%) | |
|---|---|---|---|---|---|---|---|
| Dose of Ipafricept (Q3W) |
5 mg/kg n (%) | 10 mg/kg n (%) | 2 mg/kg n (%) | 4 mg/kg n (%) | 6 mg/kg n (%) | 6 mg/kg n (%) | |
| Patients Enrolled | 3 | 4 | 8 | 9 | 6 | 7 | 37 |
| 3 (100.0) | 4 (100.0) | 8 (100.0) | 9 (100.0) | 6 (100.0) | 7 (100.0) | 37 (100.0) | |
| Overall Reason for Ending Study Treatmentd | |||||||
| Lost to Follow-Up | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Withdrawal of Consent | 1(33.3) | 0 | 0 | 0 | 0 | 1(14.3) | 2(5.4) |
| Death | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Adverse Event | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Disease Progression | 0 | 0 | 5 (62.5) | 5 (55.6) | 2 (33.3) | 1 (14.3) | 13 (35.1) |
| Investigator Decision Based on Patient’s Best Interest | 1(33.3) | 0 | 0 | 1(11.1) | 0 | 0 | 2(5.4) |
| Patient completed planned treatment cycles | 1 (33.3) | 4 (100.0) | 1(12.5) | 0 | 0 | 0 | 6(16.2) |
| Clinical/symptomatic progression | 0 | 0 | 1 (12.5) | 0 | 1(16.7) | 0 | 2(5.4) |
| Study terminated by Sponsor | 0 | 0 | 1 (12.5) | 3(33.3) | 3(50.0) | 5(71.4) | 12(32.4) |
| Total Number of Ipafricept Infusion Administered per Patient | |||||||
| Mean (SD) | 4.3 (0.58) | 2.0 (0.82) | 14.4 (8.90) | 14.0 (9.60) | 9.8 (2.32) | 4.7 (1.80) | 9.6 (7.78) |
| Median | 4.0 | 2.0 | 15.0 | 12.0 | 10.5 | 4.0 | 7.0 |
| Total Number of Paclitaxel Infusions Administered per Patient | |||||||
| Mean (SD) | 5.3 (0.58) | 6.0 (0.00) | 6.0 (0.00) | 5.7 (1.80) | 5.2 (2.04) | 4.7 (1.80) | 5.5 (1.45) |
| Median | 5.0 | 6.0 | 6.0 | 6.0 | 6.0 | 4.0 | 6.0 |
| Total Number of Carboplatin Infusions Administered per Patient | |||||||
| Mean (SD) | 5.3 (0.58) | 8.0 (4.00) | 5.5 (1.41) | 7.0 (3.97) | 7.5 (3.67) | 6.4 (6.08) | 6.6 (3.75) |
| Median | 5.0 | 6.0 | 6.0 | 6.0 | 6.0 | 4.0 | 6.0 |
SD= standard deviation
Dose-Limiting Toxicities and Maximum Tolerated Dose
There were no DLTs on this study. The MAD investigated in this study was 10 mg/kg every 3 weeks in Cohort 2, but this dose was not considered safe and development of IPA was ultimately terminated based on the incidence of fragility fractures observed in the ipafricept (6%) and vantictumab (12%) programs.[27] The MAD after the implementation of the revised bone safety plan was 6mg/kg every 3 weeks, at which dose level no patient experienced a treatment emergent adverse event (TEAE) that qualified as a DLT or fragility fractures, thus the MTD for IPA was not determined.
Treatment Emergent Adverse Events (TEAE)
All 37 patients (100.0%) reported ≥1 TEAE during the study. Overall, 32 patients (86.5%) reported at ≥1 IPA -related TEAE and 37 patients (100.0%) reported ≥1 treatment (IPA -, C-, or P-) related TEAE. These are summarized in supplementary table 1.
Overall, 32 patients (86.5%) reported ≥1 IPA related TEAE during the study; 29 patients reported ≥1 IPA related with an incidence of ≥10% in any dose cohort. The most frequently reported IPA related TEAEs were fatigue (15 patients [40.%]); nausea (13 patients [35.1%]); diarrhea and decreased appetite (8 patients [21.6%] each); dysgeusia (7 patients [18.9%]); vomiting (6 patients [16.2%]); rash and neutrophil count decreased (5 patients [13.5%] each); and stomatitis, alopecia, pruritus, and neutropenia (4 patients [10.8%] each). Overall, 8 patients (21.6%) reported ≥1 IPA-related TEAE with a CTCAE Grade ≥3. The most frequently reported IPA-related TEAEs with CTCAE Grade ≥3 were neutropenia 16.2%. All other IPA-related TEAEs with CTCAE Grade ≥3 (i.e., diarrhea, hypophosphatemia, and peripheral neuropathy) occurred in only 1 patient (2.7%) overall. (Table 3)
Table 3:
IPA-Related Treatment-Emergent Adverse Events with CTCAE Grade ≥3 by System Organ Class
| Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 | Expansion Cohort | Overall N=37 n (%) |
|
|---|---|---|---|---|---|---|---|
| Dose of Ipafricept (Q3W) System Organ Class/ |
5 mg/kg N=3 n (%) |
10 mg/kg N=4 n (%) |
2 mg/kg N=8 n (%) |
4 mg/kg N=9 n (%) |
6 mg/kg N=6 n (%) |
6 mg/kg N=7 n (%) |
|
| Patients reporting ≥1 IPA related TEAE with CTCAE≥ Grade 3 | 3 (100.0) | 0 | 1(12.5) | 0 | 1(16.7) | 3(42.9) | 8(21.6) |
| Investigations | 3 (100) | 0 | 0 | 0 | 0 | 3(42.9) | 6(16.2) |
| Neutropenia | 3 (100) | 0 | 0 | 0 | 0 | 3(42.9) | 5(13.5) |
| Metabolism and nutrition disorders | 0 | 0 | 1(12.5) | 0 | 0 | 0 | 1(2.7) |
| Hypophosphatemia | 0 | 0 | 1(12.5) | 0 | 0 | 0 | 1(2.7) |
| Gastrointestinal disorders | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 1(2.7) |
| Diarrhea | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 1(2.7) |
| Nervous system disorders | 0 | 0 | 0 | 0 | 0 | 1(14.3) | 1(2.7) |
| Neuropathy peripheral | 0 | 0 | 0 | 0 | 0 | 1(14.3) | 1(2.7) |
One patient in Cohort 1 (5mg/kg q3w) experienced a pelvic fracture that was considered by both the Investigator and the Sponsor to be a fragility fracture related to IPA. This non-serious event was of CTCAE severity Grade 1.
Bone Turnover Markers and bone safety
Overall, all serum bone turnover markers decreased post-baseline. A positive correlation was found between β-CTX and P1NP (correlation coefficient=0.488), BSAP (correlation coefficient=0.521), and β-CTX and osteocalcin (correlation coefficient=0.499). The maximum increase in β-CTX and concurrent changes in P1NP, BSAP, and osteocalcin are displayed in box plots in Figure 1.
Figure 1:
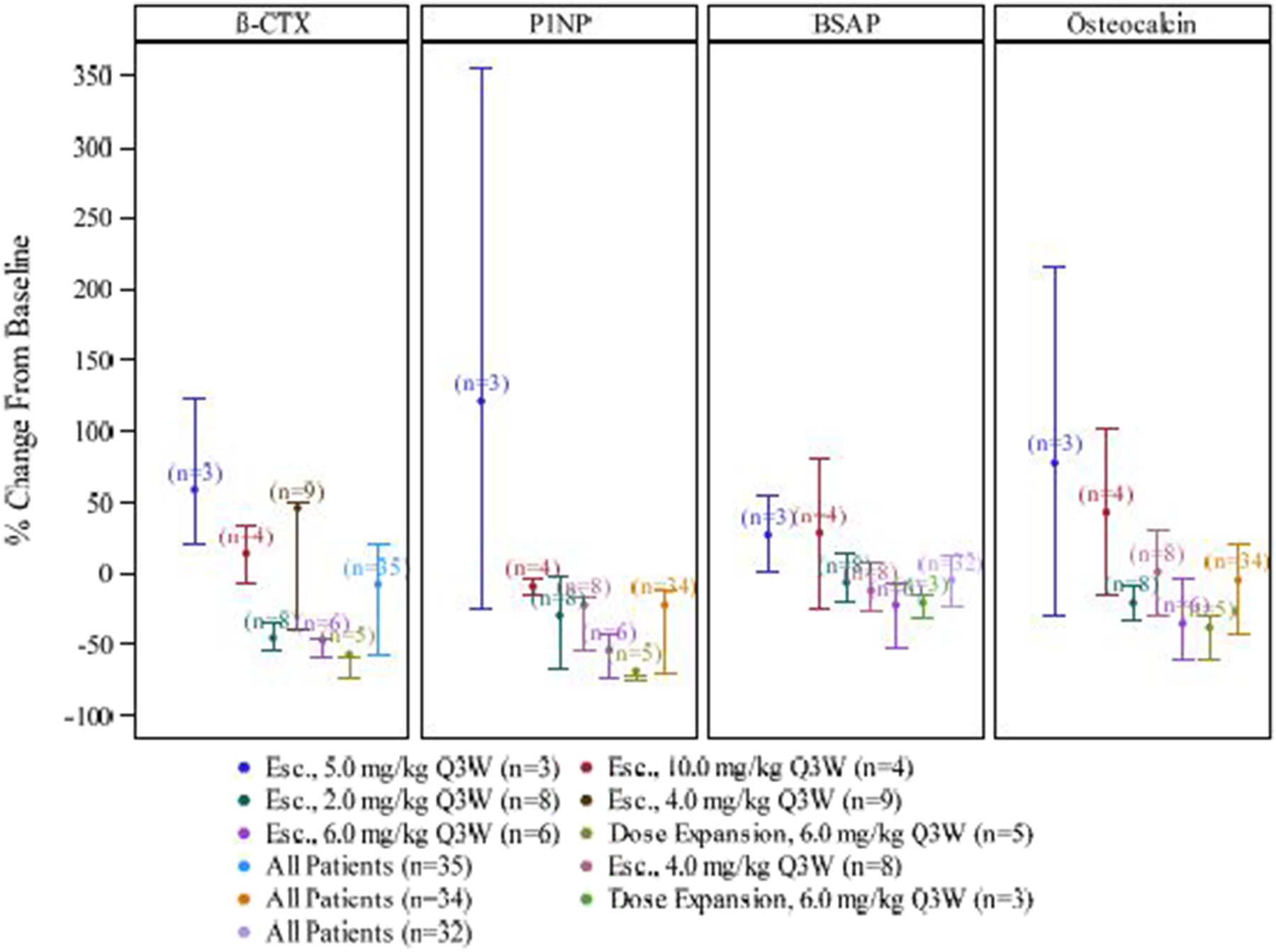
Plots of Maximum Increase in β-CTX and Concurrent Changes in P1NP, BSAP, and Osteocalcin
The ‘dots’ represent the means. The ‘whiskers’ represent the 25th and 75th percentiles. Baseline is defined as the last non-missing value prior to first dose of study drug.
β-CTX = β-C-terminal telopeptide; P1NP= procollagen type 1 amino-terminal propeptide; BSAP= bone-specific alkaline phosphatase
The overall mean (SD) DEXA bone-density T-scores at baseline were: −0.36 (1.19) for lumbar spine [L1–L4], −1.09 (0.81) for any total hip or femoral neck location [with the lowest T-score at baseline], −0.99 (0.87) for left femoral neck, −1.32 (0.68) for right femoral neck, −0.54 (0.96) for left total hip, and −0.84 (1.02) for right total hip. The percent change from baseline to the minimum post-baseline value (SD) was 5.17 (69.68)% for lumbar spine, 4.48 (21.38)% for any total hip or femoral location with lowest T-score at baseline, 5.89 (23.10)% for left femoral neck, 13.11 (12.93)% for right femoral neck, 10.58 (51.80)% for left total hip, 8.76 (32.39)% and for right total hip.
Most patients (31 patients [83.8%] overall) received bone protective therapy (zoledronic acid or denosumab) during the study. The 6 patients who did not receive bone protective therapy were in Cohort 1 (2 patients) and Cohort 2 (4 patients). Bone protective therapy administered in Cohorts 1 and 2 adhered to criteria in the original protocol. The requirements for bone protective therapy were updated in Protocol Amendment 1, making it mandatory for postmenopausal females to receive zoledronic acid in cohorts 3+.
The reasons patients received zoledronic acid were: Increase of β-CTX 2-fold over baseline (Cohorts 1 and 2) (1 patient), bone metastases and no prior use of bisphosphonate/denosumab (1 patient), postmenopausal and no bone metastases at baseline (Cohorts 3+) (29 patients).Due to fragility fractures observed in the IPA, and more so vantictumab, clinical programs, the Sponsor decided to discontinue IPA treatment for all patients in Cohorts 1 and 2 (5 and 10 mg/kg q3w, respectively). Subsequently, patients in Cohorts 3 and 4 were dosed at 2 and 4 mg/kg q3w, respectively. No DLTs or fragility fractures were reported in these cohorts, so enrollment of patients into a higher dose cohort (Cohort 5, 6 mg/kg q3w) proceeded. No DLTs or fragility fractures were reported in Cohort 5, so cohort expansion occurred with an additional 7 patients (Expansion Cohort, 6 mg/kg q3w). Thus, a total of 13 patients were treated with 6 mg/kg q3w IPA. The study was discontinued prematurely and the development of IPA was ultimately stopped based on the incidence of fragility fractures observed in the IPA (6%) and vantictumab (12%) programs.[27]
Efficacy
Overall, 28 patients (75.7% (95% CI 29.5 – 63.1) of the ITT population) had a complete or partial response (unconfirmed). Complete responses were reported in 29.7% (95% CI 15.9, 47.0). A total of 35 patients (94.6% of the ITT population) had a clinical benefit, defined as complete/partial response or stable disease. (Figure 2)
Figure 2:
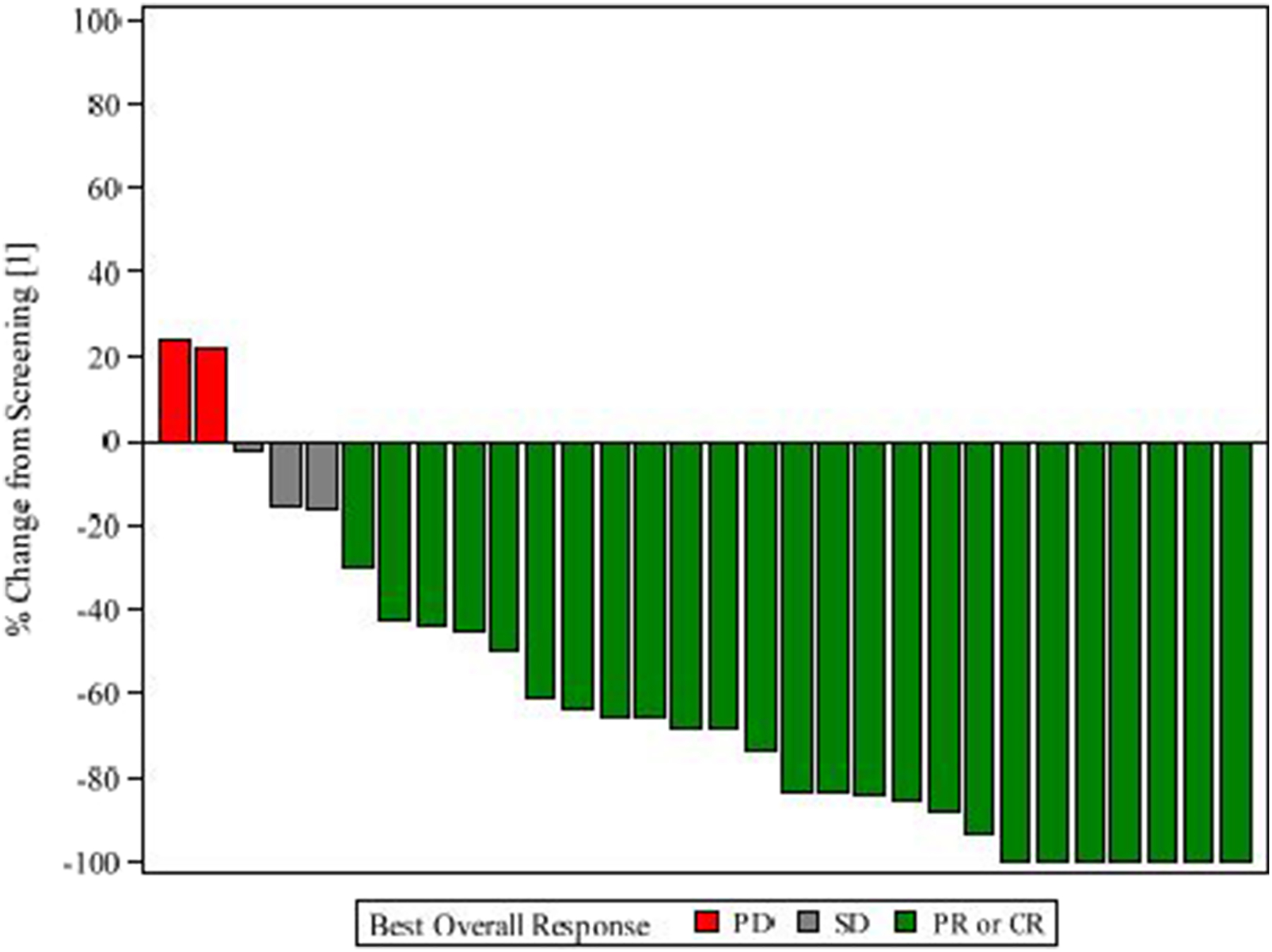
Waterfall Plot of Maximum Change in the Sum of Target Lesion Diameters
CR = Complete Response; PR = Partial Response; SD = Stable Disease; PD = Progressive Disease.
Median PFS was 10.3 months (95% CI 8.5–14.2) and OS 33 months (95% CI 23.4- NR). (not shown).
Pharmacokinetic and Immunogenicity Analysis
IPA serum concentrations in all patients (i.e., drug exposure) were within the expected drug exposure levels, given the dose levels and dosing frequencies studied. Formation of anti-drug antibodies (ADAs) was of very low titer and transient in nature, with an overall immunogenicity incidence of 5.4%. There was no evidence that formation of ADAs affected drug exposure.
Discussion
IPA is a recombinant fusion protein (immunoadhesin) consisting of a combination of human Frizzled (FZD) 8 receptor containing the extracellular ligand binding domain and human IgG1 Fc fragment that acts as a decoy receptor to inhibit Wnt signaling.[25]
IPA exhibited broad spectrum anti-tumor activity in a range of patient-derived xenografts including pancreatic, liver, breast, colon, ovarian, and melanoma. IPA was tested in a panel of 5 patient-derived EOC models in combination with paclitaxel and was found to be active in 3 of these ovarian tumor xenografts in combination with paclitaxel, producing increased inhibition of tumor growth relative to paclitaxel alone (data on file at OncoMed). In addition, a study in a patient-derived EOC xenograft evaluated the efficacy of IPA in combination with either paclitaxel, nab-paclitaxel, carboplatin, or the combination of carboplatin and paclitaxel. The combination of IPA and each of the taxane-containing regimens led to tumor regression whereas the combination with carboplatin alone was less active. The synergy of IPA with taxanes could be explained by the dual effects these drugs have on cell reproduction, blockade of Wnt signaling by IPA and disruption of microtubule formation essential for cell division by taxanes.[28–30]
Due to fragility fractures observed in the IPA, and more so vantictumab, clinical programs, the protocol was revised to decrease this risk and no further fragility fractures were seen. The MAD after implementation of the revised bone safety plan was 6mg/kg q3w, at which dose level no patient experienced a TEAE that qualified as a DLT or a fragility fracture. Thus, the MTD for IPA was not determined.
When assessing the efficacy of this combination, the lack of a control group limits definitive statements. However, if compared to the overall response rates seen among measureable patients who received doublet chemotherapy and bevacizumab on OCEANS (57%) and GOG 213 (56%) the 75.5% overall response rate demonstrated in this study compares favorably. In a similar comparison, the median PFS (inclusive of chemotherapy) for OCEANs and GOG 213 was 8.4 (95% 8.3, 9.7) and 10.4 (95% CI 9.7–11) months, respectively.[8, 9] The median PFS on this current study is 10.3 (95% CI 8.5 – NR) months – again, comparable to expected outcomes using standard of care agents. While outcomes were comparable to those seen with standard of care regimens, the occurrence of fragility fractures at doses associated with efficacy are of concern and limit further development of this particular Wnt targeting combination therapy in ovarian cancer.
Supplementary Material
Supplemental Table 1: Study Treatment Emergent Adverse Events by Preferred Term with Overall Incidence of ≥10%
Figure 3:
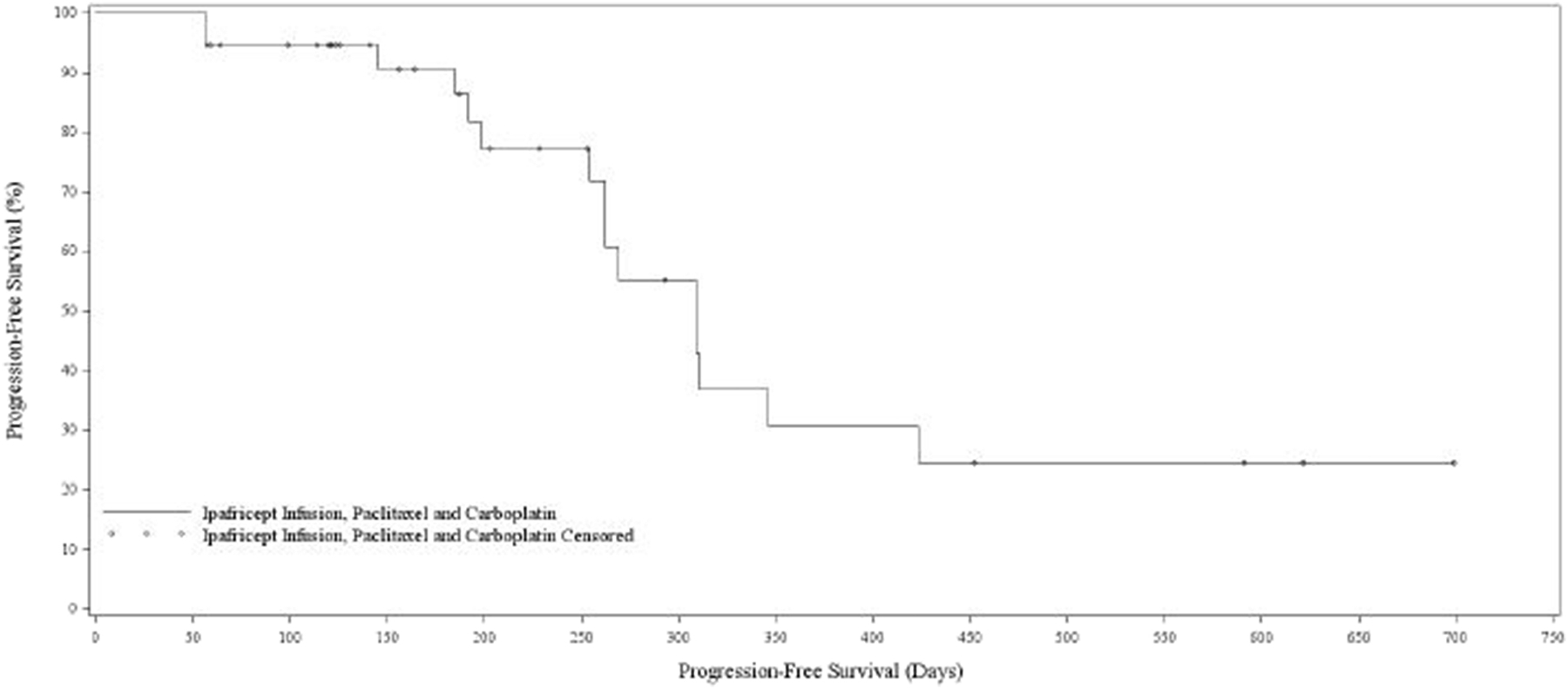
Progression free survival
Highlights.
Wnt signaling in epithelial ovarian cancer is a promising target
Ipafricept is a recombinant protein that inhibits Wnt signaling
Ipafricept, added to paclitaxel and carboplatin was feasible but associated with bone toxicity
Acknowledgments
This study was funded in part through the NIH/NCI Support Grant P30 CA008748 (Dr. O’Cearbhaill, Dr. Sabbatini).
Footnotes
Author Disclosures: KNM reports advisory board participation and reimbursement from Astra Zeneca, Immunogen, Clovis, Genentech/Roche, Tesaro, Janssen, Merck, Pfizer, Aravive, Onco-Med, VBL Therapeutics and Samumed. She also serves on steering committees for Tesaro, Astra Zeneca, VBL Therapeutics, Aravive and Genentech/Roche.
RAB reports consulting fees from Amgen, Astra Zeneca, Tesaro, Clovis, Genentech-Roche Invitae, Merck and VBL Therapeutics
REO reports advisory board participation and reimbursement from Tesaro and Clovis.
AK, RKB and RS were employees at OncoMed at the time that this study was conducted.
GMS and MAM have no disclosures.
References
- 1.FDA label for Pembrolizumab, revised 12/2018; https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125514s014lbl.pdf accessed March 2019.
- 2.SEER 18 Registries, N.C.I.
- 3.Markman M, et al. , Second-line platinum therapy in patients with ovarian cancer previously treated with cisplatin. J Clin Oncol, 1991. 9(3): p. 389–93. [DOI] [PubMed] [Google Scholar]
- 4.Gore ME, et al. , Cisplatin/carboplatin cross-resistance in ovarian cancer. Br J Cancer, 1989. 60(5): p. 767–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parmar MK, et al. , Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON4/AGO-OVAR-2.2 trial. Lancet, 2003. 361(9375): p. 2099–106. [DOI] [PubMed] [Google Scholar]
- 6.Pfisterer J, et al. , Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: an intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J Clin Oncol, 2006. 24(29): p. 4699–707. [DOI] [PubMed] [Google Scholar]
- 7.Pujade-Lauraine E, et al. , Pegylated liposomal Doxorubicin and Carboplatin compared with Paclitaxel and Carboplatin for patients with platinum-sensitive ovarian cancer in late relapse. J Clin Oncol, 2010. 28(20): p. 3323–9. [DOI] [PubMed] [Google Scholar]
- 8.Coleman RL, et al. , Bevacizumab and paclitaxel-carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol, 2017. 18(6): p. 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aghajanian C, et al. , OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol, 2012. 30(17): p. 2039–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.FDA label for bevacizumab; https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125085s305lbl.pdf accessed August 14, 2017.
- 11.Mirza MR, et al. , Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med, 2016. 375(22): p. 2154–2164. [DOI] [PubMed] [Google Scholar]
- 12.Coleman RL, et al. , Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet, 2017. 390(10106): p. 1949–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pujade-Lauraine E, et al. , Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol, 2017. 18(9): p. 1274–1284. [DOI] [PubMed] [Google Scholar]
- 14.FDA label for niraparib: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208447lbl.pdf.
- 15.FDA label for Olaparib https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208558s000lbl.pdf.
- 16.Clarke MF and Fuller M, Stem cells and cancer: two faces of eve. Cell, 2006. 124(6): p. 1111–5. [DOI] [PubMed] [Google Scholar]
- 17.Wicha MS, Liu S, and Dontu G, Cancer stem cells: an old idea--a paradigm shift. Cancer Res, 2006. 66(4): p. 1883–90; discussion 1895–6. [DOI] [PubMed] [Google Scholar]
- 18.Bao S, et al. , Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature, 2006. 444(7120): p. 756–60. [DOI] [PubMed] [Google Scholar]
- 19.Dylla SJ, et al. , Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One, 2008. 3(6): p. e2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ford CE, et al. , Wnt signalling in gynaecological cancers: A future target for personalised medicine? Gynecol Oncol, 2016. 140(2): p. 345–51. [DOI] [PubMed] [Google Scholar]
- 21.Ford CE, et al. , The Wnt gatekeeper SFRP4 modulates EMT, cell migration and downstream Wnt signalling in serous ovarian cancer cells. PLoS One, 2013. 8(1): p. e54362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacob F, et al. , Loss of secreted frizzled-related protein 4 correlates with an aggressive phenotype and predicts poor outcome in ovarian cancer patients. PLoS One, 2012. 7(2): p. e31885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ford CE, et al. , The non-canonical Wnt ligand, Wnt5a, is upregulated and associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Gynecol Oncol, 2014. 134(2): p. 338–45. [DOI] [PubMed] [Google Scholar]
- 24.Peng C, et al. , Wnt5a as a predictor in poor clinical outcome of patients and a mediator in chemoresistance of ovarian cancer. Int J Gynecol Cancer, 2011. 21(2): p. 280–8. [DOI] [PubMed] [Google Scholar]
- 25.Fischer MM, et al. , WNT antagonists exhibit unique combinatorial antitumor activity with taxanes by potentiating mitotic cell death. Sci Adv, 2017. 3(6): p. e1700090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.US FRAX Calculation Tool: https://www.sheffield.ac.uk/FRAX/tool.aspx.
- 27.Mita MM,BC, Richards DA, Mita AC, Shagisultanova E, Osborne CR, O’Shaughnessy J, Zhang C, Henner R, Kapoun AM, Xu L, Dupont J, Brachmann RK, Farooki A, Diamond JR, Phase 1b study of WNT inhibitor vantictumab (VAN, human monoclonal antibody) with paclitaxel (P) in patients (pts) with 1st- to 3rd-line metastatic HER2-negative breast cancer (BC). J Clin Oncol 2016. 34(no.15_suppl(May 2016)): p. 2516.27269942 [Google Scholar]
- 28.Bhalla KN, Microtubule-targeted anticancer agents and apoptosis. Oncogene, 2003. 22(56): p. 9075–86. [DOI] [PubMed] [Google Scholar]
- 29.Acebron SP, et al. , Mitotic wnt signaling promotes protein stabilization and regulates cell size. Mol Cell, 2014. 54(4): p. 663–74. [DOI] [PubMed] [Google Scholar]
- 30.Niehrs C and Acebron SP, Mitotic and mitogenic Wnt signalling. EMBO J, 2012. 31(12): p. 2705–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: Study Treatment Emergent Adverse Events by Preferred Term with Overall Incidence of ≥10%