

Summary
Some Listeria monocytogenes (Lm) strains harbor a prophage within the comK gene, which renders it inactive. During Lm infection of macrophage cells, the prophage turns into a molecular switch, promoting comK gene expression and therefore Lm intracellular growth. During this process, the prophage does not produce infective phages or cause bacterial lysis, suggesting it has acquired an adaptive behavior suited to the pathogenic lifestyle of its host. In this study, we demonstrate that this non-classical phage behavior, named active lysogeny, relies on a transcriptional response that is specific to the intracellular niche. While the prophage undergoes lytic induction, the process is arrested midway, preventing the transcription of the late genes. Further, we demonstrate key phage factors, such as LlgA transcription regulator and a DNA replicase, that support the phage adaptive behavior. This study provides molecular insights into the adaptation of phages to their pathogenic hosts, uncovering unusual cooperative interactions.
Keywords: listeria, phage, mammalian infection, active lysogeny, phage excision, phage transcriptional response, phage adaptaion, bacterial pathogen
Graphical Abstract
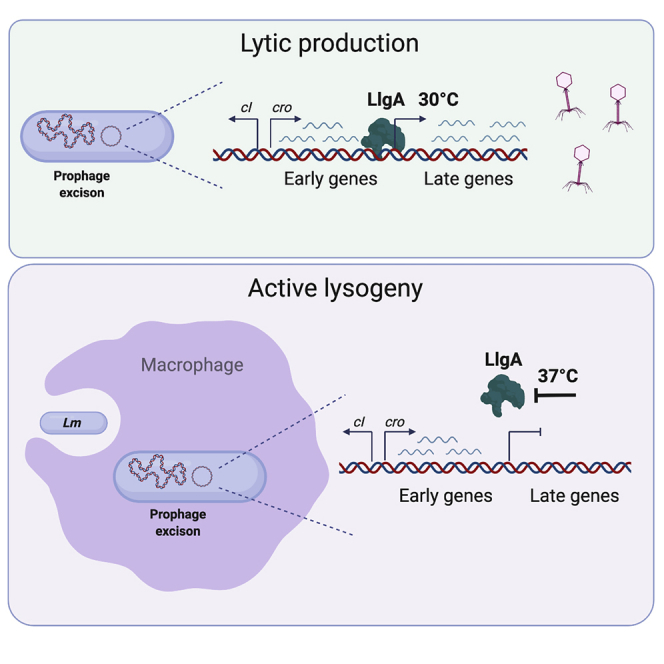
Highlights
-
•
L. monocytogenes strain 10403S harbors a prophage in its comK gene
-
•
During infection of macrophage cells, the prophage lytic pathway is induced
-
•
The phage lytic response is arrested, preventing the expression of the late genes
-
•
LlgA, the late lytic gene activator is specifically inhibited at 37°C
Pasechnek at al. describe a phage transcriptional response that supports the pathogenic lifestyle of its host.
Introduction
Bacteriophages (or phages) are obligatory parasites that exploit bacterial cells for propagation and play a critical role in bacterial evolution. Phages are classified as lytic or lysogenic, based on their infection life cycle (Oppenheim et al., 2005). Upon infection, lytic phages enter a productive cycle, generating infective virions that are liberated via bacterial lysis. In contrast, lysogenic phages utilize various strategies to propagate without activating the lytic cycle. For example, some phages integrate their genome into the bacterial chromosome, turning into prophages that persist in what is considered a phage “dormant state” (Ptashne, 2004). These prophages replicate together with their host chromosome but can switch into lytic production upon exposure to stressful conditions (e.g., DNA damage)—a process termed prophage induction. Even though most bacterial pathogens carry prophages within their genome (Asadulghani et al., 2009; Burns et al., 2015; Figueroa-Bossi et al., 2001; Matos et al., 2013; Ohnishi et al., 2001; Wang et al., 2010), the mechanisms by which they are controlled under the stresses imposed within the mammalian niche remain unclear.
Listeria monocytogenes (Lm) is the causative agent of listeriosis disease in humans (Swaminathan and Gerner-Smidt, 2007). It is a Gram-positive, facultative intracellular pathogen that invades a wide array of mammalian cells (Lecuit, 2005). Upon invasion, it resides within a vacuole (or a phagosome), from which it escapes into the host cell cytosol in order to replicate (Barry et al., 1992; Hamon et al., 2006). Most Lm strains carry prophages in their genome (e.g., A118, A500, A006, and PSA-like phages) (Dorscht et al., 2009; Klumpp and Loessner, 2013; Zink and Loessner, 1992), yet the impact of these prophages on the pathogenesis of Lm is not understood. We previously described an unusual interaction between Lm strain 10403S and its prophage φ10403S, in which the prophage promotes the virulence of its host via an adaptive behavior (Rabinovich et al., 2012). φ10403S is a ∼38-kb-long phage of the Siphoviridae family that is integrated within the comK gene (similar to A118) (Rabinovich et al., 2012). Many Lm strains and Listeria species carry a prophage within the comK gene. To date, over ∼8300 comK-associated prophages have been sequenced together with their listerial hosts. Owing to the prophage insertion, the listerial comK gene was considered non-functional. In Bacillus subtilis, ComK functions as the master transcription activator of the competence system (Com), which is known to facilitate the uptake of exogenous DNA (Dubnau, 1999). Noticeably, transcriptome studies of Lm 10403S indicated that the com genes are highly activated during macrophage infection. Further investigation demonstrated that two major components of the Com system—the Com translocation channel (encoded by comEC) and the cell-wall-crossing pseudopilus (encoded by the comG operon)—play a role in the escape of Lm from the macrophages’ phagosomes to the cytosol (Rabinovich et al., 2012). Expression of the com genes during Lm intracellular growth was found to require the formation of an intact comK gene via precise excision of φ10403S-prophage. Prophage excision was found to be highly induced in bacteria that are located within macrophages’ phagosomes, yet unlike classic prophage excision, this did not lead to the production of progeny virions and bacterial lysis (Rabinovich et al., 2012). These findings indicated that during mammalian cell infection, the prophage functions as a DNA molecular switch that regulates comK gene expression to support Lm intracellular growth. We termed this adaptive phage behavior active lysogeny, representing cases where phages regulate bacterial gene expression via genomic excision without triggering the lytic cycle (Argov et al., 2017a; Feiner et al., 2015). The current study was designed to investigate the molecular mechanisms that uphold φ10403S active-lysogenic behavior in macrophage cells. We analyzed the phage genome and transcriptional response under lysogenic, lytic, and active lysogenic conditions and characterized its regulatory and lytic genes. The results uncovered that φ10403S acquired a non-classical transcriptional response that supports the survival of its host in the intracellular niche. This finding led to the identification of phage-encoded factors that promote active lysogeny (e.g., factors that facilitate phage excision and re-integration) and a transcriptional regulator that plays a critical role in this bacteria-phage cooperative interaction. The findings presented here demonstrate that in nature, phages have evolved to acquire innovative responses that are beyond the classic lysogenic and lytic, which can support bacteria-phage cooperation under certain circumstances, such as within the mammalian niche.
Results
φ10403S-Prophage Switches into Lytic Production in Response to UV Irradiation
To gain a better understanding of the interaction between Lm strain 10403S and its prophage φ10403S, we first investigated the phage response to conditions that induce the lytic cycle (i.e., conditions that cause DNA damage and trigger the SOS response). Bacteria were grown in the rich brain heart infusion (BHI) medium and exposed to ultraviolet (UV) radiation (4 J/cm2), a treatment that is known to trigger phage induction (Lamont et al., 1989). Both bacterial growth and infective virion production were monitored following UV irradiation, the latter by using a plaque forming assay. As shown in Figure 1, the UV treatment inhibited the growth of Lm bacteria compared to the control (i.e., non-treated bacteria) (Figure 1A). Infective phages appeared at 4 h post-UV irradiation (liberated via bacterial lysis), reaching a maximum number of ∼3.5 × 106 plaque-forming units per milliliter of culture (PFUs/ml) (Figure 1B). Of note, mitomycin C, another agent that causes DNA damage, was found to be more potent, yielding 108 to 109 PFUs/ml (Table S1). To evaluate the burst size of φ10403S-phage, a classic one-step growth experiment was performed by infecting Lm bacteria, which are lacking the phage and the comK gene (ΔcomK), with free φ10403S-phage particles. The results indicated the phage latent period to be ∼80 min, the rise period to be ∼40 min, and the phage burst size to be ∼40 virions per cell (Figure 1C). Examining virion production using transmission electron microscopy revealed phage capsids of ∼60 nm inside the bacteria, as well as free phage particles at 4 h post-UV irradiation (Figure 1D). Notably, comparing the lytic production of φ10403S to that of a related comK-phage of Lm strain EGDe revealed that in contrast to φ10403S, the EGDe comK-phage fails to produce infective phages in response to UV irradiation (Figure 1E). Under mitomycin C treatment, the EGDe phage produced ∼10 to 50 PFUs/ml, whereas φ10403S produced >108 PFUs/ml (Table S1). Taken together, these experiments characterize the lytic response of φ10403S upon induction and infection, establishing the basis for further investigation of its response in macrophage cells.
Figure 1.

φ10403S Lytic Production
(A) Growth analysis of Lm 10403S in BHI medium with and without UV irradiation. Cultures were grown for 3 h in BHI to mid-log phase and then irradiated with UV light (at 4 J/cm2) and incubated at 30°C. Optical density (OD600) measurements were taken every hour as indicated. The data represent a mean of three independent experiments. Error bars represent the standard deviation.
(B) A plaque-forming assay detecting the formation of infective phages after UV irradiation (shown as plaque-forming units, PFUs). The data represent a mean of three independent experiments. Error bars represent the standard deviation.
(C) One-step growth analysis of φ10403S lytic infection. Free virion particles of φ10403S were used to infect exponentially grown Lm bacteria lacking the comK gene. Virion production was assayed as PFUs at the indicated time points. The data represent the mean of three independent experiments. Error bars represent the standard deviation.
(D) Transmission electron microscopy of Lm 10403S at 4 h post-UV irradiation. Representative images of three independent biological experiments.
(E) A plaque-forming assay detecting the formation of infective phages upon UV irradiation (shown as PFUs) of Lm 10403S and EGDe strains grown at 30°C. The data represent the mean of three independent experiments. Error bars represent the standard deviation. ND, not detected.
φ10403S Lysogenic and Lytic Transcriptional Responses
To study φ10403S behavior during Lm infection of macrophage cells, we sought to analyze its transcriptional response within the intracellular niche. For this purpose, we first analyzed the phage transcription profile under lysogenic and lytic conditions, using strand-specific RNA sequencing (RNA-seq) analysis. Fifty-two active open reading frames (ORFs) were identified (out of the 54 annotated ORFs) as well as three putative non-coding RNAs (lasRNA, rli140, and rliG) (Toledo-Arana et al., 2009) (Table S2). We could assign a putative function to ∼60% of the phage ORFs, while the rest had no homology to known proteins or functional domains (Table S2). We found the φ10403S genome to be generally organized in five modules: immunity/integration, lysogeny-lysis switch, early genes (mediating gene regulation and DNA replication), late genes (mediating phage DNA packaging, capsid and tail formation, and bacterial lysis), and lastly, “accessory” genes, whose function is unknown (Figure 2A). Notably, the lysogeny-lysis switch was similar to that of λ-phage, harboring two oppositely directed promoters transcribing cI-like and cro-like repressor genes (LMRG_01514 and LMRG_01515, respectively) (Johnson et al., 1981; Little et al., 1999; Ptashne, 2004).
Figure 2.

φ10403S Transcriptional Response under Lysogenic and Lytic Conditions
(A) Schematic representation of the φ10403S-genome including predicted genes and functional modules. Regulatory genes are marked in black, ncRNAs are marked in bright yellow, immunity/integration and accessory genes are marked in green, early genes are marked in orange, late genes are marked in peach, and lysis genes are marked in red.
(B) Strand-specific RNA-seq analysis of φ10403S under lysogenic conditions (i.e., bacteria grown in BHI to exponential phase at 30°C). Transcription levels are indicated as RPKM (scaled reads per kilobase per million reads). Data represent the mean of three independent experiments.
(C) Strand-specific RNA-seq analysis of φ10403S-phage under lytic conditions (4 h post-UV irradiation). Transcription levels are indicated as RPKM. Data represent the mean of three independent experiments.
(D) A plaque-forming assay detecting the formation of infective phages upon UV irradiation of Lm 10403S and indicated phage mutants grown at 30°C, as well as a strain overexpressing CI-like repressor (pPL2-cI-like). Data are presented as a percentage relative to the levels observed in WT Lm. Data represent a mean of three independent experiments. Error bars represent the standard deviation. ND, not detected.
φ10403S transcriptome analysis under lysogeny indicated that most of the phage genes are repressed, except for a few accessory genes (LMRG_01555-01556, LMRG_01558-01559) and two putative non-coding RNAs (lasRNA and rliG) for which the function is unknown (Figure 2B; Table S3). As expected, the phage CI-like repressor and its downstream genes, including the integrase gene (LMRG_01511 or int), were expressed under lysogeny (Figure 2B). Under lytic conditions (i.e., UV irradiation), the phage early and late genes, including the cro-like gene, were activated, whereas the cI-like repressor gene and its downstream genes were concomitantly repressed (Figure 2C; Table S3). Most of the accessory genes were upregulated under UV irradiation, except for LMRG_01557, which was repressed under all tested conditions (Figures 2B and 2C).
To assess the role of the phage genes in the production of infective virions, we generated a library of gene deletion mutants (deleted for one or two genes), which collectively covers the phage genome (32 mutants in total, excluding the phage cI-like repressor and functionally redundant genes, such as capsid and tail genes) (Figure 2D). These mutants were subjected to a plaque-forming assay under UV irradiation, and their capacity to produce infective virions was compared to wild-type (WT) bacteria carrying a WT prophage (WT Lm) (Figure 2D). The results indicated that most of the phage genes are involved in virion production, but they also identified a number of dispensable genes that encode unknown functions (LMRG_02918, 01516, lasRNA, 02984, 01527, 02921, rliG, 01530, and 01555-1556). Taken together, these results establish φ10403S lysogenic and lytic responses and further identify genes that are essential and non-essential to the lytic cycle.
φ10403S Exhibits a Unique Transcriptional Response in Macrophage Cells
Next, we analyzed the transcription profile of φ10403S during Lm infection of macrophage cells. For this purpose, the NanoString technology was employed, which is based on the hybridization of specific probes (Table S4) (Kulkarni, 2011). Bone-marrow-derived macrophage (BMDM) cells were infected with Lm bacteria, and the phage transcription profile was detected at 2, 4, and 6 h post-infection (h.p.i). The dynamics of the phage response during macrophage cell infection was compared to the lytic response under UV irradiation at 1, 2, 3, 4, and 5 h post-treatment. The data revealed a unique phage transcriptional response that is specific to the intracellular niche (Figure 3A; Tables S3 and S5). While there was a gradual activation of the phage early and late genes under UV irradiation, during Lm infection of macrophage cells, only the early genes were activated, whereas the late genes were essentially repressed (the latter starting at the terS gene that mediates phage DNA-packaging and ending at the lysin and holin genes that mediate bacterial lysis) (Figures 3A–3C). Notably, the data further demonstrated that the cro-like repressor gene was strongly activated under UV irradiation and during macrophage cell infection, overall indicating that the phage is effectively induced under both these conditions (Figures 3A and 3D). These observations demonstrate that φ10403S is differentially regulated in the intracellular niche. While it appears that the prophage undergoes lytic induction within macrophages, the lytic pathway is arrested halfway, with no transcription of the late genes and, hence, no production of infective virions and bacterial lysis in the intracellular niche.
Figure 3.

φ10403S Transcriptional Response in Macrophage Cells
(A) φ10403S transcriptional response under UV irradiation and active lysogenic conditions, the latter during Lm infection of BMDM cells. Strand-specific RNA-seq analysis under UV irradiation was performed by Illumina. Transcription analysis during active lysogeny was performed using nCounter analysis. Transcription levels are presented as relative counts, compared to the levels observed in the lysogenic state (i.e., bacteria grown exponentially in BHI medium at 30°C). Data represent the mean of three independent experiments.
(B–D) Transcription profile of φ10403S (B) early genes, (C) late genes, and (D) regulatory genes under lytic and active lysogenic conditions.
Identifying Early Genes That Promote Active Lysogeny
Given the observation that the phage early genes are activated in the intracellular niche, we hypothesized that a number of them may play a role in promoting active lysogeny. Active lysogeny involves the excision and re-integration of φ10403S genome from the comK gene, thereby controlling its expression (Rabinovich et al., 2012). We previously demonstrated that the phage integrase (LMRG_01511 or Int) mediates prophage excision, yet many aspects of this process remain unclear, particularly whether the phage-DNA replicates after excision and which factors, if any, facilitate its re-integration into comK. Since many of the phage early genes encode proteins predicted to be functionally associated with the phage DNA (e.g., recombinases, a replicase, and an endonuclease; Table S2), we speculated that some of these genes may contribute to the ability of φ10403S to function as a DNA-regulatory switch. To address this hypothesis, mutants deleted for each one of the early genes were monitored for phage excision and potential extra-chromosomal replication during Lm infection of macrophage cells. Phage DNA excision and extra-chromosomal replication were evaluated by quantifying the number of attB and attP sites (using qRT-PCR), which are formed only upon phage excision and circularization of its DNA, respectively (Rabinovich et al., 2012). A mutant deleted for the phage integrase gene (Δint) was used as a control, as it fails to excise the chromosome and therefore to replicate as an episome. As expected, in contrast to WT Lm, no attB or attP sites (representing the intact comK gene and the circular phage-DNA, respectively) were detected in the intracellularly grown Δint mutant (Figure 4A). Notably, a similar analysis of the early gene mutants detected genes that are important for prophage excision and replication in the intracellular niche. Specifically, a mutant lacking LMRG_01522 demonstrated a phenotype similar to that of Δint, indicating this gene plays a critical role in φ10403S excision (Figure 4A). In accordance with the finding that φ10403S excision is a prerequisite for comK gene expression and, therefore, the escape of Lm from the phagosome to the cytosol, we found ΔLMRG_01522 mutant to be impaired in phagosomal escape and intracellular growth in macrophage cells (Figures 4B and 4C). Over the course of this study, a homolog of LMRG_01522, Gp44 of A118-phage, was reported to act as a recombination directionality factor (RDF) that activates the phage integrase to promote excision (Mandali et al., 2017). While this finding is in accordance with our observations, it further strengthened the premise that LMRG_01522 plays a critical role in promoting Lm phagosomal escape by triggering the formation of an intact comK gene during Lm infection of macrophages.
Figure 4.

Phage Early Genes That Contribute to Active Lysogeny
(A) qRT-PCR analysis of attB and attP sites in WT Lm and indicated mutants grown in BMDM cells for 6 h. Data are presented as relative quantity (RQ), compared to the levels in WT bacteria. The data represent a mean of three independent experiments. Error bars indicate the 95% confidence interval.
(B) A bacterial phagosomal escape assay. Percentage of bacteria that escaped the macrophage phagosomes as determined by a microscope fluorescence assay. Macrophages were infected with WT Lm and ΔLMRG_01522, as well as with Δint as a control. The data represent two biological repeats. ∗p < 0.05 as calculated by the χ-test.
(C) Intracellular growth of WT Lm and ΔLMRG_01522 mutant in BMDM cells. The data are representative of three independent experiments. Error bars represent the standard deviation of a triplicate.
(D) Analysis of phage loss in ΔLMRG_01526 mutant in comparison to WT Lm grown in macrophage cells for 6 h. The number of phage-cured bacteria isolated from macrophages infected with ΔLMRG_01526 mutant was normalized to that of WT bacteria. The data represent the mean of three independent experiments. Error bars represent the standard deviation. ∗p < 0.05 as calculated by Student’s t test.
(E) Analysis of phage loss in the ΔLMRG_01526 mutant in comparison to WT Lm during in vivo infection of C57BL/6 mice (48 h.p.i.). The number of phage-cured bacteria isolated from the spleens and livers of infected mice was normalized to the total number of bacteria recovered. The data represent 3 to 5 mice per sample. ∗p = 0.05 as calculated by Student’s t test.
Another mutant that exhibited a strong phenotype in the attP/attB analysis was ΔLMRG_01526 (Figure 4A). LMRG_01526 encodes a protein containing a DnaD-like domain, which is a putative replicative DNA-helicase that initiates phage DNA replication. In line with this prediction, intracellularly grown ΔLMRG_01526 bacteria demonstrated a very low level of attP sites in comparison to WT Lm (∼60-fold less), while the attB level was reduced by only 2.5-fold. This finding indicates that φ10403S is indeed replicated extra-chromosomally in the intracellular niche. Since the transcriptome data implied that there is no phage DNA packaging into newly formed capsids in the intracellular niche (functions that are encoded by the late genes), we speculated that the extra-chromosomal replication of the phage DNA might be necessary to increase the occurrence of its re-integration into comK, thus promoting the phage persistence in the Lm chromosome during mammalian infection. To address this hypothesis, we designed a system that monitors the loss of the phage from the Lm chromosome during intracellular growth, which is based on the counter selection pheS∗ gene. This gene encodes a mutated phenylalanyl-tRNA synthetase that is toxic upon growth in medium supplemented with p-chloro-phenylalanine (Argov et al., 2017b). The pheS∗ gene was cloned into the phage genome (in the accessory module) and was used to evaluate the emergence of phage-cured bacteria upon Lm intracellular growth. We surmised that if the phage DNA fails to re-integrate into comK, it will be lost during Lm intracellular growth. Using this system, we found that φ10403S re-integration into comK is a highly efficient process, with a phage-loss rate of 1 in 500,000 bacteria (using WT Lm carrying a WT phage) (Argov et al., 2019). Notably, performing this analysis using a ΔLMRG_01526 mutant, we found it to exhibit a phage-loss rate of 1 in 30,000 bacteria, which is 15-fold greater than WT bacteria (Figure 4D). A similar phenotype was observed upon intravenous mice infections. Monitoring phage-cured bacteria 48 h post-mice infection revealed that the number of phage-cured bacteria recovered from the livers of mice infected with ΔLMRG_01526 was greater than ∼30-fold the number recovered using WT Lm (Figure 4E). These findings indicate that phage DNA replication in the intracellular niche contributes to phage re-integration into comK, hence promoting its persistence within the Lm chromosome during infection of mammalian cells. Altogether, these findings provide an insight into the fate of φ10403S within the mammalian niche and further explain the activation of the early genes in this environment.
Characterizing φ10403S Main Transcription Regulators
Having discovered that the transcription of the late genes is specifically blocked during Lm infection of macrophage cells, we next aimed to identify the phage regulator(s) that control the phage late genes. To this end, we identified four putative regulatory proteins in the φ10403S-genome. Among these are CI and Cro-like repressors and two additional regulators: LMRG_02920 and LMRG_01529, which encode for a putative anti-repressor and an ArpU-like transcription regulator, respectively (Quiles-Puchalt et al., 2013) (Figure 2A). Mutants deleted for each of these regulatory genes (except for the cI-like repressor gene) were severely impaired in the production of infective virions under UV irradiation (Figure 2D). Of note, deletion of the cI-like gene could not be achieved because it results in the immediate induction of the phage and bacterial lysis. To validate the role of the CI-like repressor as the main phage repressor, we overexpressed it using a constitutive promoter from the integrative pPL2 plasmid (pPL2-cI-like) and examined its ability to repress virion production under UV irradiation. As predicted, overexpression of the CI-like repressor blocked the production of infective virions, validating its function as the main phage repressor that maintains lysogeny (shown in Figure 2D; pPL2-cI-like). To examine the function of the other regulators, we analyzed their effect on the transcription of early and late genes using qRT-PCR (Figure 5). Bacterial mutants deleted for each regulatory gene were subjected to UV irradiation, and total RNA was extracted before (time 0) and at 1, 3, and 5 h post-UV irradiation. Performing this analysis confirmed that LMRG_01515 is a classic Cro-like repressor, which represses the transcription of the early genes in the course of the lytic pathway (including LMRG_02920 and LMRG_01529) (Figure 5A). The function of the putative anti-repressor LMRG_02920 was less definitive. A mutant lacking this gene exhibited an increase in the transcription of the early genes and a decrease in the transcription of the late genes at later time points (4 to 5 h post-UV), suggesting it may temporally regulate the transcription of the early and late genes in the course of the lytic pathway (Figure 5B). Analyzing the LMRG_01529 putative regulator, we found that it functions as the main activator of the late genes. A mutant deleted of LMRG_01529 failed to transcribe the late genes, whereas the early and regulatory genes were transcribed at levels similar to those in WT Lm harboring a WT phage (Figure 5C). We therefore named LMRG_01529 LlgA (late-lytic gene activator of φ10403S).
Figure 5.

Characterization of φ10403S Regulatory Proteins
qRT-PCR analysis of selected phage genes representing the early gene module (LMRG_01516 and LMRG_01524-endonuclease), the late gene module (LMRG_01532-terL and LMRG_01553-holin), and the putative regulatory genes (LMRG_01515, LMRG_02920, and LMRG_01529) in WT Lm and regulatory gene mutants as indicated.
(A) Analysis of genes regulated by LMRG_01515.
(B) Analysis of genes regulated by LMRG_02920.
(C) Analysis of genes regulated by LMRG_01529.
RNA was isolated from bacteria grown in BHI medium at 30°C after UV irradiation at the indicated time points. The data are representative of three independent experiments. Transcription levels are presented as a RQ, compared to their levels in WT bacteria prior to UV irradiation, indicated as t = 0. Error bars indicate the 95% confidence interval of the triplicate.
LlgA Protein Activity Is Inhibited in the Intracellular Niche
Given our finding that LlgA is the activator of the late genes, we speculated that it may be specifically inhibited in the intracellular niche. The transcriptome data indicated that llgA is transcribed within the macrophages together with the early genes, and yet the late genes are not expressed (Figure 3A). This observation implies that LlgA is inhibited post-transcriptionally, possibly during translation or even later, at the protein level. To examine these possibilities, a six-histidine-tag was fused to the carboxy-terminus of LlgA (in the prophage genome), and western blot analysis was performed to detect the level of LlgA proteins in intracellularly grown bacteria. No LlgA proteins were detected in intracellular bacteria, yet we cannot conclude that LlgA is not translated, as many listerial proteins are hard to detect in macrophage cells due to their relatively low levels. As an alternative approach, we examined the impact of LlgA overexpression on intracellularly grown bacteria by expressing it from pPL2 plasmid using the promoter of the actA virulence gene, which was shown to be highly activated intracellularly (pPL2-PactA-llgA) (Reniere et al., 2016). Remarkably, LlgA expression resulted in the immediate killing of the bacteria within the macrophage cells. This killing was driven by the phage lysis proteins (holin and lysin), as bacteria that were cured of the phage or deleted of the lysis genes (Δφ-pPL2-PactA-llgA and Δ(lysis)φ-pPL2-PactA-llgA, respectively) grew similarly to WT Lm in the macrophage cells (Figure 6A). This observation supports the premise that LlgA is inhibited in the intracellular niche, in order to prevent the activation of the late lytic genes.
Figure 6.

LlgA Activity Is Thermo-Regulated
(A) Intracellular growth analysis of WT Lm and mutants that are cured of the phage (Δφ) or deleted of lysis genes (Δ(lysis)φ) expressing llgA (pPL2-PactA-llgA) in BMDM cells. Data are representative of three independent experiments. Error bars represent the standard deviation of a triplicate.
(B and C) Growth analysis of WT Lm and bacteria expressing llgA-his from a TetR-dependent promoter (pPL2-PtetR-llgA-his), as well as a mutant deleted of the phage lysis genes (Δ(lysis)φ) harboring pPL2-PtetR-llgA-his with and without the addition of anhydrotetracycline (AT) at (B) 30°C and (C) 37°C. Data represent the mean of three independent experiments. Error bars represent the standard deviation.
(D) qRT-PCR analysis of llgA transcription levels at 30°C and 37°C in WT Lm and in Δ(lysis)φ and Δφ strains harboring pPL2-PtetR-llgA-his. Transcription levels are presented as a RQ, compared to the levels in WT bacteria grown at 30°C. mRNA levels were normalized to the levels of 16S rRNA. The data are representative of three independent experiments. Error bars indicate the 95% confidence interval.
(E) Western blot analysis of LlgA-6His protein obtained from WT Lm grown at 30°C and 37°C and from Δ(lysis)φ strain and a strain cured of the phage Δφ, all harboring pPL2-PtetR-llgA-his. Equal amounts of total protein were separated on a 15% SDS-PAGE that was blotted and probed with anti-6His antibody. The experiment was performed three times, and the figure shows a representative blot. Coomassie staining was used as a loading control (lower panel)
(F) qRT-PCR analysis of selected phage genes, representative of the early and late gene modules in WT Lm and in Δ(lysis)φ mutant harboring pPL2-PtetR-llgA-his grown in BHI at 30°C and 37°C. Transcription levels are presented as a RQ, compared to the levels in WT bacteria grown at 30°C. mRNA levels were normalized to the levels of 16S rRNA. The data are representative of three independent experiments. Error bars indicate the 95% confidence interval.
(G) qRT-PCR analysis of terS and llgA genes expressed from the pPL2 plasmid (pPL2-PtetR-llgA-terS) in Lm bacteria that are cured of the phage (Δφ) grown in BHI at 30°C and 37°C. Transcription levels are presented as a RQ, compared to the levels in bacteria grown at 30°C. mRNA levels were normalized to the levels of 16S rRNA. The data are representative of three independent experiments. Error bars indicate the 95% confidence interval.
(H) qRT-PCR analysis of terS and llgA genes expressed from amyE::Phs-llgA-terS in B. subtillis bacteria grown in LB at 30°C and 37°C with IPTG. Transcription levels are presented as a RQ, compared to the levels in bacteria grown at 30°C. mRNA levels were normalized to the levels of 16S rRNA. The data are representative of three independent experiments. Error bars indicate the 95% confidence interval.
A clue to the mode of regulation of LlgA within the intracellular niche came from in vitro experiments designed to investigate LlgA translation and protein activity. For this purpose, llgA-his-tagged (llgA-his) was cloned into the pPL2 plasmid, this time under the regulation of an inducible tetracycline repressor (TetR) dependent promoter (pPL2-PtetR-llgA-his). Monitoring the growth of LlgA-expressing bacteria (WT Lm-pPL2-PtetR-llgA-his) in BHI medium without UV irradiation unexpectedly revealed that the bacteria failed to grow at 30°C but did grow at 37°C, as compared to WT Lm not carrying the llgA plasmid (this phenotype was observed without the addition of a TetR inducer) (Figures 6B and 6C). Interestingly, similarly to the LlgA-mediated bacterial killing observed in macrophages, the lack of bacterial growth at 30°C was entirely attributed to the triggering of bacterial lysis by the phage lysis proteins. A mutant deleted for the phage lysis genes harboring pPL2-PtetR-llgA-his (Δ(lysis)φ-pPL2-PtetR-llgA-his) grew similarly to WT bacteria at both 30°C and 37°C (Figures 6B and 6C). While these findings demonstrated that ectopic expression of LlgA is sufficient to activate the expression of the late genes even in the absence of UV irradiation, they further revealed that LlgA is preferentially inhibited under 37°C, which is the temperature of the intracellular niche. Of note, increasing the expression of LlgA by the addition of the TetR inducer anhydrotetracycline (AT) resulted in bacterial killing also at 37°C, demonstrating that a high level of LlgA can override its temperature-dependent inhibition (as observed in intracellularly grown bacteria overexpressing LlgA) (Figures 6B and 6C). To further investigate the observed temperature-dependent regulation of LlgA, we compared its mRNA and protein levels in bacteria grown at 30°C and 37°C (without UV irradiation), as well as its ability to activate the transcription of early and late genes. LlgA transcription and translation were assessed in a phage-cured strain or a strain deleted of the phage lysis genes (Δφ or Δ(lysis)φ) in order to avoid unwanted bacterial lysis and therefore the loss of mRNA and proteins. Interestingly, both qRT-PCR and western blot analyses indicated that llgA is transcribed and translated equally well at 30°C and 37°C (Figures 6D and 6E). However, the late genes were preferentially activated only at 30°C and not at 37°C (Figure 6F). As expected, the early genes remained repressed at both temperatures, since the phage lysogeny-lysis switch is not induced in the absence of UV irradiation, and hence the early genes are not activated (Figure 6F). The results of these experiments suggest that the activity of LlgA is thermo-regulated (i.e., active at 30°C and less active at 37°C), which is in line with the phage transcriptional response in the intracellular niche (Figure 3A). To examine whether LlgA thermo-regulation is mediated by another phage-encoded factor, we constructed a reporter system for LlgA activity and tested it in Lm bacteria lacking the prophage (Δφ). To this end, the terS gene, which is positively regulated by LlgA, was cloned with its upstream region (including LlgA putative binding sites) into pPL2 plasmid that carries llgA under TetR regulation (pPL2-PtetR-llgA-terS). The plasmid was then introduced into Δφ bacteria, and, using qRT-PCR, the transcription levels of terS and llgA were evaluated during growth at 30°C and 37°C. The data indicated that LlgA thermo-regulation does not involve other phage factors, as the terS gene was preferentially activated at 30°C and less so at 37°C, even in the absence of the prophage (Figure 6G). Of note, llgA transcription from the pPL2 plasmid was comparable under both temperatures. To examine whether a listerial factor is responsible for the observed LlgA thermo-regulation, we introduced the llgA-terS reporter system to Bacillus subtilis bacteria. The llgA gene was cloned under the regulation of the hyperspank promoter (Phs), and the entire Phs-llgA-terS cassette was integrated into the B. subtillis chromosome (in the amyE gene) using AES777 plasmid (amyE::Phs-llgA-terS). The bacteria were then grown in Luria Bertani (LB) medium at 30°C and 37°C (with the addition of isopropyl β-d-1 thiogalactopyranoside [IPTG]), and the transcription levels of terS and llgA were evaluated. Interestingly, we found the terS gene to be equally transcribed under both temperatures, suggesting that LlgA thermo-regulation is mediated by a listerial specific factor that is absent in B. subtilis (Figure 6H). Although further attempts to identify this presumably existing listerial factor were not successful, the findings presented here indicate that LlgA is a critical factor in the interaction of Lm with its inhabiting prophage in the mammalian environment.
Discussion
Lysogeny was first described in 1925 with the observation that some bacterial strains lyse and produce infective virions (Lwoff, 1953). D’Herelle, who also observed this phenomenon, suggested that these strains are “symbiotic,” maintaining mutualistic interactions with their phages (Lwoff, 1953). Since then, a number of bacteria-phage symbiotic interactions have been documented, describing cases where lysogenic phages provide immunity to their hosts (superinfection exclusion) as well as virulence or metabolic genes (lysogenic conversion) (Bondy-Denomy et al., 2016; Brüssow et al., 2004; Waldor and Friedman, 2005). Since under lysogeny, the bacteria and the phage become a single genetic unit, a unique situation is formed in which their interests are temporarily aligned. Within this context, innovative and complex interactions evolve that balance the bacteria and the phage needs, sometimes leading to cooperative behaviors under certain circumstances. In line with this premise, the results of this study describe an example of a bacteria-phage interaction in which non-classical phage transcriptional regulation and molecular adaptations operate to promote bacteria-phage cooperation in the intracellular environment.
This study relies on our previous observation that the Lm φ10403S-prophage acts as a molecular switch to regulate comK gene expression during Lm infection of macrophage cells. While this finding suggested that the phage had acquired an adaptive behavior aligned to the pathogenic lifestyle of its host, the underlying mechanisms and molecular adaptations that uphold this interaction were unknown. In this study, we comprehensively investigated the φ10403S genome, transcriptional responses, and regulatory proteins, revealing a unique transcription profile and key factors that collectively control the phage active lysogenic behavior in mammalian cells. We characterized the lytic production of φ10403S under UV irradiation and upon phage infection, and we found the phage to be functional, producing dozens of infective virions per cell. Interestingly, we further found that this characteristic is not necessarily shared by all listerial comK-phages: for example, the comK-phage of Lm strain EGDe hardly produced infective virions in response to SOS, suggesting that it has evolved differently.
Characterizing the transcription profile of φ10403S under lysogenic and lytic conditions, we identified 52 active ORFs and 3 putative ncRNAs. The data demonstrated that during lysogeny, most of the phage genes are repressed, yet the data also identified a number of genes that are highly expressed during lysogeny that require further investigation (lasRNA, rliG, LMRG_01555, 01556, 01558, and 01559). Notably, four of these genes are located in the accessory gene module, suggesting that they possess functions that benefit the bacteria and/or the phage under lysogeny. For example, the “accessory” gene module of related Listeria phages has been shown to encode for anti-CRISPR proteins (Acrs) that target the type II CRISPR-Cas system of Lm (Osuna et al., 2019; Rauch et al., 2017). These proteins were shown to function under lysogeny and less upon lytic infection, suggesting they have been evolved to protect the bacteria and the phage under this condition. Under UV irradiation, we found the φ10403S lysogeny-lysis switch to be induced, resulting in the robust activation of the phage early and late lytic genes. Systematic analysis of the phage genome indicated that most of the genes are involved in the production of infective virions, whereas some are completely dispensable. Intriguingly, the dispensable genes were found to be distributed across the whole phage genome. This observation relates to one of the greatest mysteries in phage research that is the remarkably large reservoir of unknown genes found in phage genomes, which implies that bacteria-phage interactions in nature are even more complex than currently understood.
The main observation of this study concerns the transcriptional response of φ10403S during Lm infection of macrophage cells (i.e., active lysogeny). The results indicate that φ10403S is induced upon macrophage infection; however, unlike classic phage induction, the lytic pathway is arrested halfway. These findings establish that active lysogeny is a tightly regulated process that prevents the formation of infective virions and bacterial lysis within the intracellular niche. Further, we found that this unique transcriptional response supports the phage function as a DNA regulatory switch (regulating comK gene), which is designed to facilitate Lm phagosomal escape and intracellular growth. Within the early gene module, we identified genes that mediate prophage excision and re-integration, thus playing an important role in comK regulation. The data support the premise that LMRG_01522 functions as an RDF that activates prophage excision and that LMRG_01526 functions as a replicative factor that mediates the phage DNA replication. We found that upon excision, the phage DNA is replicated in the intracellular niche as an episome, and this replication increases the chances of its re-integration into comK. Bacteria carrying a mutated phage that fails to replicate lost the phage more frequently upon macrophage and mice infections, overall indicating that phage replication plays an important role in the persistence of φ10403S within the Lm chromosome.
The transcription profile of φ10403S during macrophage cell infection revealed that the late genes are specifically repressed in the intracellular niche. This finding corroborated our previous observation that the phage does not produce infective virions or trigger bacterial lysis in the intracellular environment, thereby supporting Lm intracellular growth (Rabinovich et al., 2012). Notwithstanding, we recently demonstrated that φ10403S is not the only lytic phage element that inhabits the Lm chromosome (Argov et al., 2017b). Lm 10403S harbors another cryptic phage element that encodes for tail-like bacteriocins named monocins. We found that this monocin element is also activated under UV irradiation and triggers the production and release of monocins via bacterial lysis, independently of φ10403S (Argov et al., 2017b, 2019). Interestingly, we also found that φ10403S and the monocin element are tightly co-regulated, as they share a common anti-repressor, MpaR, which is a metalloprotease that is encoded by the monocin element. Under SOS stress or infection of mammalian cells, MpaR concomitantly cleaves the CI-like repressors of both elements (φ10403S and monocin), thus synchronizing their lytic induction. Further investigation indicated that the lytic genes of the monocin element (including its lysis genes) are also repressed in the intracellular niche, yet the mechanism of their repression is still not clear (Argov et al., 2019). Of note, the monocin element is evolutionarily more ancient than φ10403S and hence may have acquired a different mechanism to regulate its lytic genes in the intracellular niche. While the data indicate that LlgA is not involved in the regulation of the monocin genes (Figure 6A), the findings reveal that a controlled inhibition of phage-derived lytic genes (particularly lysis genes) in the intracellular environment is a common mechanism that is essential for the adaptation of prophages (infective and cryptic) to the pathogenic lifestyle of their host.
LlgA belongs to the Ltr super-family of late transcriptional regulators (Quiles-Puchalt et al., 2013). These activators were shown to bind upstream the terS gene, which is the first gene of the late gene module. We identified hundreds of LlgA homologs in Listeria genomes, located in various comK and non-comK prophages (e.g., A500 and A006-like phages), many of them exhibiting a remarkable amino acid sequence identity (examples are in Figure S1). Interestingly, overexpression of LlgA immediately triggered a bacterial killing in macrophage cells. This killing was solely dependent on the phage and associated with its lysis proteins, supporting the hypothesis that LlgA is inhibited in the intracellular niche. The phage transcription profile indicated that while llgA is transcribed in the intracellular niche, there is no expression of the downstream-regulated late genes, implying that LlgA regulation may be post-transcriptional. Since we could not investigate LlgA translation and activity in the intracellular environment (the LlgA protein was undetectable), we studied LlgA expression from the pPL2 plasmid. The experiments revealed that LlgA directly activates the late genes and thus triggers bacterial lysis, even in the absence of UV irradiation. In addition, further investigation demonstrated that the LlgA protein is preferentially active at 30°C and less active at 37°C, which is the temperature of the intracellular niche. Despite similar levels of transcription and translation at both temperatures, at 37°C, the LlgA protein failed to activate the transcription of the late genes. We found this thermo-regulation of LlgA to be independent of other phage factors, yet possibly reliant on a listerial specific factor, as it was not observed in B. subtilis. Taken together, the data point out LlgA as a critical factor in the interaction of Lm with its prophage and further suggest that LlgA thermo-regulation has evolved to support the survival of Lm (and the phage) in the mammalian host. Finally, this study strengthens the premise that lysogeny is a highly versatile state, in which bacteria and their inhabiting phages are continuously subjected to evolutionary selection (Bobay et al., 2013; Howard-Varona et al., 2017). We posit that like Lm and its prophage, many other bacterial pathogens also maintain unique lysogenic interactions with their prophages that are designed to support mutual survival in the mammalian host.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-6His tag antibody | Abcam | ab18184; RRID: AB_444306 |
| anti-listeria-FITC antibody | Bio-Rad | 0400-0027; RRID: AB_619120 |
| Bacterial and Virus Strains | ||
| Lm 10403S | Prof. Daniel Portnoy (University of California, Berkley) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| mitomycin C | Sigma | M4287 |
| rhodamine-phalloidin | Biotium | #00027 |
| Brain Heart Infusion Broth | Sigma | 53286-500G |
| Critical Commercial Assays | ||
| Ribo-Zero rRNA Removal Kit (Bacteria) | Illumina | MRZB12424 |
| RNAeasy kit | QIAGEN | 74104 |
| NEBNext® Ultra Directional RNA Library Prep Kit for Illumina | NEB | E7420S |
| Deposited Data | ||
| RNA-Seq data (BHI) | This study | Mendeley Data: https://doi.org/10.17632/kjyh2swhg9.1 |
| RNA-Seq data (UV treated) | This study | Mendeley Data: https://doi.org/10.17632/mz484h884h.2 |
| Source file including raw experimental data | This study | Mendeley Data: https://doi.org/10.17632/9jzf6vk3p7.1 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6JOlaHsd mice, 8-week old | Envigo, Israel | N/A |
| Oligonucleotides | ||
| See Table S4 | This study | N/A |
| Recombinant DNA | ||
| pheS∗ | Argov et al., 2017b | Addgene, 98783 |
| Software and Algorithms | ||
| StepOne V2.1 | Applied Biosystems | N/A |
| nSolver 4.0 software | Kulkarni, 2011 | N/A |
| Other | ||
| nCounter system | NanoString Technologies | N/A |
| Synergy HT | BioTek | N/A |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anat A. Herskovits (anathe@tauex.tau.ac.il).
Materials Availability
This study did not generate new unique reagents. The plasmids and strains used in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and Code Availability
The RNA-Seq data are available in Mendeley Data via these links:
Mendeley Data: https://doi.org/10.17632/kjyh2swhg9.2
Mendeley Data: https://doi.org/10.17632/mz484h884h.3
Source file including raw experimental data
Mendeley Data: https://doi.org/10.17632/9jzf6vk3p7.1
Experimental Model and Subject Details
Bacterial strains
Listeria monocytogenes (Lm) strain 10403S was obtained from Prof. Daniel Portnoy (University of California, Berkley) and used as the WT strain. Lm 10403S strain cured of φ10403S phage (DPL-4056) was generated by Prof. Richard Calendar (University of California, Berkley) by biological curing. E. coli XL-1 Blue (Stratagene) was utilized for vector propagation. E. coli SM-10 was utilized for conjugative plasmid delivery to Lm bacteria. Lm strains were grown in brain heart infusion (BHI) (Merck) medium at 37°C or 30°C as specified, and E. coli strains were grown in Luria-Bertani (LB) (Acumedia) medium at 37°C. B. subtilis strain PY79 was obtained from Prof. Avigdor Eldar (Tel Aviv University). The strains and mutants used in this study are described further in Table S6.
Animals
The use of animals in this study was approved by the Tel Aviv University Animal Care and Use Committee (04-18-028 and 04-20-005) according to the Israel Welfare Law (1994) and the National Research Council guide (Guide for the Care and Use of Laboratory Animals 2010). Animals used were 8-week-old female C57BL/6 mice (Envigo, Israel). Mice were infected via the tail vein or used for bone marrow derived macrophages (BMDM) isolation. BMDMs were cultured in Dulbecco’s Modified Eagle Medium (DMEM)-based media supplemented with 20% fetal bovine serum, sodium pyruvate (1 mM), L-glutamine (2 mM), β- Mercaptoethanol (0.05 mM), and monocyte-colony stimulating factor (M-CSF, L929-conditioned medium); BMDM medium.
Method Details
Generation of gene deletion mutants and overexpression strains
All in-frame deletions generated in this work were constructed using the Lm 10403S strain as the parental strain. Upstream and downstream regions of selected genes were amplified using Phusion DNA polymerase and cloned into pKSV7oriT vector (Smith and Youngman, 1992). Cloned plasmids were sequenced and conjugated to Lm using the E. coli SM-10 strain. Lm conjugants were then grown at 41°C for two days in BHI with chloramphenicol to promote plasmid integration into the bacterial chromosome by homologous recombination. For plasmid curing, bacteria were passed several times in fresh BHI without chloramphenicol at 30°C to allow plasmid excision via the generation of an in-frame deletion. The bacteria were then seeded on BHI plates and chloramphenicol sensitive colonies were picked for validation of gene deletion using PCR. The llgA expressing strains were generated by using the pPL2 integrative plasmid to introduce a copy of the llgA gene in trans under the control of PactA or PtetR promoters (Lauer et al., 2002). For the detection of LlgA activity under 30°C and 37°C, a reporter system was designed that includes the llgA gene under the regulation of a TetR dependent promoter, cloned up-stream to terS gene with its upstream region, including LlgA putative binding sites (according to Quiles-Puchalt et al., 2013). For the expression of this system in B. subtilis strain PY79, the llgA-terS cassette was cloned in the AES777 plasmid under the inducible hyperspank (hs) promoter. The construct was then introduced into B. subtilis chromosome, in the amyE gene, using standard transformation and Spp1 transduction protocols (amyE::Phs-llgA-terS) (Bareia et al., 2018).
Phage induction by UV irradiation
Lm bacteria were grown in BHI medium over-night (O.N.) at 37°C with agitation. To obtain lysogenic bacteria, the culture was diluted by a factor of 100 in fresh BHI and incubated without agitation at 30°C to reach OD600 of 0.7-0.8. For induction of the lytic cycle, O.N. cultures were diluted by a factor of 10 in 10 mL of fresh BHI, and incubated without agitation at 30°C to reach an OD600 of 0.5. The cultures were then irradiated by UV light at 4 J/cm2 (using CL 508S model UV cross-linker oven), were supplemented with 5 mL of fresh BHI medium, and were then incubated without agitation at 30°C. At specified time points, the cultures were either filtered through 0.22 μm filters to isolate virions, or centrifuged to collect bacteria that were snap-frozen in liquid nitrogen for further transcription analysis.
One-step phage growth analysis and plaque assay
For the one-step growth analysis over-night cultures of Lm ΔcomK bacteria were diluted in 10 mL of BHI to an OD600 of 0.1, and incubated with agitation at 30°C to reach an OD600 of 0.5 (∼5∗108 cells/ml). The cultures were then supplemented with 10 mM CaCl2 and infected with 0.5 mL of isolated virions (108 virions/ml, induced by MC) at a multiplicity of infection of ∼100:1 (bacteria:virions) for 10 min at 30°C (the adsorption step). One ml of culture was then centrifuged at 16000 g for 1 min and resuspended in fresh BHI to remove unattached virions. A 0.1 mL aliquot of the sample was then diluted into 10 mL of fresh pre-warmed BHI (to avoid a second infection) and incubated at 30°C without shaking. Samples were collected every 20 min, and plaque forming units (PFUs) were quantified by adding 100 μL of virion containing sample at appropriate dilution, to 300 μL of an O.N. culture of Lm Mack861 bacteria (used as the indicator strain). Three ml of melted LB-0.7% agar medium (at 56°C) supplemented with 10 mM CaCl2, was added, mixed, and then quickly overlaid on BHI-agar plates. Plates were incubated for 2 days at 30°C to allow the formation of visible plaques. To calculate the phage burst size the PFU number at the plateau (∼1000) was divided by the PFU number at the latent period (∼25), yielding ∼40 virions per infected cell.
Transmission electron microscopy
4 h post UV irradiation, bacteria were collected by centrifugation and fixed in 2.5% glutaraldehyde in PBS over night at 4°C. After several washings in PBS, the bacteria were post fixed in 1% OsO4 in PBS for 2 h at 4°C. Dehydration was carried out in graded ethanol followed by embedding in glycidether. Thin sections were mounted on Formvar/Carbon coated grids, stained with uranyl acetate and lead citrate, and examined by a Jeol 1400 – Plus transmission electron microscope (Jeol, Japan). Images were captured using SIS Megaview III and iTEM the TEM imaging platform (Olympus).
RNA-seq analysis
Analysis of the phage transcription profile under lysogenic and lytic conditions was performed using strand-specific RNA-seq analysis by Illumina. Bacteria were grown in BHI medium (with and without UV irradiation) and harvested at 4 h post UV irradiation for RNA-seq analysis. Total bacterial RNA was extracted using the RNAsnap method (Stead et al., 2012) followed by DNase I (QIAGEN) treatment on QIAGEN RNAeasy columns. The RNA integrity number (RIN) was evaluated using a TapeStation instrument (Agilent Technologies) (the RIN values ranged from 7.5 to 9.6, except for one sample that was 7.1), and then the rRNA was depleted using the Ribo- Zero kit (Illumina). Strand specific RNA-seq libraries were prepared using the NEBNext® Ultra Directional RNA Library Prep Kit for Illumina (NEB) and sequenced (50 nt per read) by HiSeq 2500 instrument (Illumina) at the Technion Genome Centre (Haifa, Israel). Mapping of reads followed by upper quartile normalization by gene expression and differential expression analysis by the negative binomial distribution as the statistical model was performed using Rockhopper V2.03 with default parameters (McClure et al., 2013; Tjaden, 2015). The complete results are provided in Table S3. The heatmap was generated using R plotting. The RNA-seq analysis was performed on three independent biological repeats.
NanoString analysis of phage transcripts in intracellularly grown bacteria
To analyze the transcription profile of φ10403S during Lm infection of macrophages the NanoString technology was employed (using the nCounter system), which is based on the hybridization of specific fluorescently labeled probes (Kulkarni, 2011). Sixty phage-specific barcoded probes were designed (∼80-100 nucleotides each) that, based on the Illumina sequencing data, cover all the phage transcripts from both strands. RNA was purified from intracellularly grown bacteria in bone marrow-derived macrophage cells (BMDM) at time points 2, 4 and 6 h post infection as described previously (Sigal et al., 2016). BMDM cells used for infection experiments were isolated from 6–8 week-old female C57BL/6 mice (Envigo, Israel) as described previously (Portnoy et al., 1988) and cultured in Dulbecco’s Modified Eagle Medium (DMEM)-based media supplemented with 20% fetal bovine serum, sodium pyruvate (1 mM), L-glutamine (2 mM), β- Mercaptoethanol (0.05 mM), and monocyte-colony stimulating factor (M-CSF, L929-conditioned medium); BMDM medium. For each time point, three 145 mm dishes were seeded with 2 × 107 BMDMs that were then infected (in parallel) with 2 × 109 of WT Lm bacteria. 30 min post infection, BMDM monolayers were washed twice with PBS to remove unattached bacteria, and fresh medium was added. At 1 h post-infection (h.p.i.), gentamicin (50 μg/ml) was added to limit extracellular bacterial growth. At 2, 4, and 6 h. p. i. the macrophages were lysed with 20 mL cold water, and cell debris and nuclei were removed by centrifugation at 800 g for 3 min at 4°C. Released bacteria were quickly collected on 0.45 μm filter membranes (Millipore) using a vacuum apparatus and snap-frozen in liquid nitrogen. Bacteria were recovered from the filters by vortexing into AE buffer (50 mM sodium acetate pH 5.2, 10 mM EDTA), and bacterial nucleic acids were extracted using RNAsnap followed by ethanol precipitation (Stead et al., 2012). RNeasy Mini Kit DNase on column (QIAGEN) was used for DNase treatment. For nCounter analysis of mRNA transcripts, a multiplexed CodeSet was assembled with two sequence-specific probes for each target gene of interest (Table S4). A 150 ng aliquot of each RNA sample was hybridized with the CodeSet. Transcription levels of phage genes in total RNA samples were measured with specific probes using the NanoString nCounter system, according to the manufacturer’s standard procedures. Images of color-coded reporter probes bound to their complementary mRNA target, image acquisition, and data processing were collected in a reporter code count (RCC) file and the raw counts were normalized to the internal positive controls; rpoD, bglA, and rpoB using the nSolver 4.0 software (Kulkarni, 2011). The data were normalized to the counts detected in samples of lysogenic WT Lm bacteria and are presented as a relative quantity (RQ). The complete results are provided in Table S5. The heatmap was generated with R plotting. The NanoString analysis was performed on three independent biological repeats.
Lm intracellular growth
In order to assess the intracellular growth of Lm bacteria, 2 × 106 BMDM cells were seeded in a 60 mm Petri dish on glass coverslips in 5 mL of BMDM medium and incubated O.N. in a 37°C, 5% CO2 forced-air incubator. Lm bacteria were grown O.N. at 30°C without agitation and 8 × 106 bacteria were used to infect BMDM cells. 30 min post-infection, the macrophage monolayers were washed and fresh medium was added. Gentamicin was supplemented at 1 h.p.i. (50 μg/ml) to limit the growth of extracellular bacteria. At the indicated time points, three coverslips were transferred into 2 mL of sterile water and vortexed to release intracellular bacteria. Appropriate dilutions of the resulting lysate were plated on BHI agar plates and CFUs were counted after 24 h of incubation at 37°C. Each experiment was repeated at least three times.
Phagosomal escape assay
1 × 106 BMDM cells were seeded on 20 mm coverslips and infected as described above. Cells were fixed at 2.5 h.p.i. with 3.7% paraformaldehyde, and permeabilized with 0.05% Triton. Slides were then washed and stained appropriately: bacteria were stained with anti-listeria-FITC antibody (Bio-Rad); actin was stained with rhodamine-phalloidin (Biotium); and DNA was stained with DAPI containing Vectashield® mounting media. Images were collected using a Nikon eclipse Ti-E microscope. For each infection experiment ∼200 bacteria were counted in 4-5 different frames and statistical analysis was performed using χ-test.
Phage loss assays
To measure the loss of φ10403S-prophage from the Lm chromosome, pheS∗ (encoding a mutated phenylalanyl-tRNA synthetase) (Argov et al., 2017b) and kanamycin resistance genes were cloned into the phage genome downstream to the LMRG_01556 gene in WT Lm and ΔLMRG_01526 bacteria. These strains were then used to infect macrophage cells for 6 h as described above (Intracellular growth of Lm). For in vivo experiments, 8-week-old female C57BL/6 mice (Envigo, Israel) were infected via the tail vein with 1.5 × 105 bacteria in 200 μl PBS. Animals were observed daily for any signs of illnesses and were euthanized at 48 h.p.i. Spleens and livers were harvested and homogenized in 0.2% saponin. Bacteria were released from BMDMs or organs and plated on BHI-agar plates to quantify total CFU, and on selective p-chloro-phenylalanine (18 mM) plates for phage-cured bacteria (Argov et al., 2017b). Phage-loss in bacteria growing on p-Cl−phe plates was verified by re-plating on kanamycin-selective plates and by PCR.
qRT-PCR analysis
Total nucleic acids were isolated from bacterial pellets through standard phenol-chloroform extraction methods. An aliquot of 0.04 ng of total nucleic acids was used for qRT-PCR analysis of attB and attP levels with bacterial 16S rRNA gene used as a reference for sample normalization. For transcription analysis, samples were treated with DNaseI, and 1 μg of RNA was reverse transcribed to cDNA using a qScript (Quanta) kit. qRT-qPCR was performed on 10 ng of cDNA. The relative transcription of bacterial genes was determined by comparing the level of transcript with bacterial 16S rRNA or rpoD genes, which served as a reference. All qRT-qPCR analyses were performed using FastStart Universal SYBR Green Master Mix (Roche) on the StepOnePlus RT-PCR system (Applied Biosystems). Statistical analysis was performed using StepOne V2.1 software. Error bars represent the 95% confidence interval.
Growth of LlgA expressing bacteria
Single colonies of Lm strains (WT, Δ(lysis)φ, and Δφ) harboring a C’-His-tagged llgA gene under the regulation of a TetR dependent promoter on the integrative pPL2 plasmid (pPL2-PtetR-llgA-his) were suspended in BHI medium with or without the TetR inducer anhydrotetracycline (AT, 10 ng/ml) and 200 μl were pipetted in triplicates into a 96-well plate. Plates were incubated at 30°C or 37°C using a Synergy HT BioTek plate reader, and OD600 measurements were taken every 15 min, preceding 1 min of shaking. For transcription analysis, Lm bacteria were grown in BHI and B. subtilis bacteria were grown in Luria-Bertani (LB) medium with the addition of 10 μM IPTG, at 30°C and 37°C. The bacteria were collected by centrifugation at exponential phase and snap-frozen in liquid nitrogen for further transcription analysis.
Western blot analysis
Lm Δ(lysis)φ and Δφ strains harboring 6His-tagged llgA under the regulation of a TetR dependent promoter in the integrative pPL2 plasmid (pPL2-PtetR-llgA-his) were grown at 30°C or 37°C in 50 mL BHI to an OD600 of 0.6. The bacteria were then collected by centrifugation and washed with Buffer-A (20mM Tris-HCl pH 8, 0.5M NaCl, and 1 mM EDTA), suspended in 1 mL Buffer-A supplemented with 1 mM PMSF and lysed by ultra-sonication. Total protein content was assayed using a modified Lowry assay and samples with equal amounts of total proteins were separated on 15% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Proteins were probed with mouse anti-6His tag antibody (Abcam ab18184) at 1:1000 dilution, followed by HRP-conjugated goat anti-mouse IgG (Jackson ImmunoResearch, USA) at 1:20,000 dilution. Western blots were developed by a homemade enhanced chemiluminescence reaction (ECL).
Quantification and Statistical Analysis
All data are presented as mean of three biological repeats (n = 3) ± 1 standard deviation, unless indicated otherwise. Statistical significance was calculated using Student’s t test, except for Figure 4B where it was calculated by the χ-test. Replicate values in Figures 4C, 5, and 6A are in triplicates. For each intracellular growth experiment and western blotting analysis a representative experiment is shown, and additional biological repeats can be found in the provided raw data file. For qRT-PCR experiments statistical analysis was performed using StepOne V2.1 software and error bars represent the 95% confidence interval. Details of statistical analysis can be found in the Figure Legends.
Acknowledgments
We thank Tasneem Bareia and Avigdor Eldar for their help with the B. subtillis experiments. This work was funded by European Research Council (ERC) starting and consolidator grants for A.A.H. (Patho-Phage-Host 335400 and Co-Patho-Phage 817842) and an Israel Science Foundation grant for A.A.H. (ISF 1381/18).
Author Contributions
A.P. and A.A.H. designed the study. A.P., L.R., O.S., G.A., J.M., T.A., and N.S. performed the experiments. N.S. validated the results. I.B. performed bioinformatic analysis, and A.A.H. prepared the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: July 28, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107956.
Supplemental Information
References
- Argov T., Azulay G., Pasechnek A., Stadnyuk O., Ran-Sapir S., Borovok I., Sigal N., Herskovits A.A. Temperate bacteriophages as regulators of host behavior. Curr. Opin. Microbiol. 2017;38:81–87. doi: 10.1016/j.mib.2017.05.002. [DOI] [PubMed] [Google Scholar]
- Argov T., Rabinovich L., Sigal N., Herskovits A.A. An Effective Counterselection System for Listeria monocytogenes and Its Use To Characterize the Monocin Genomic Region of Strain 10403S. Appl. Environ. Microbiol. 2017;83:e02927-16. doi: 10.1128/AEM.02927-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argov T., Sapir S.R., Pasechnek A., Azulay G., Stadnyuk O., Rabinovich L., Sigal N., Borovok I., Herskovits A.A. Coordination of cohabiting phage elements supports bacteria-phage cooperation. Nat. Commun. 2019;10:5288. doi: 10.1038/s41467-019-13296-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asadulghani M., Ogura Y., Ooka T., Itoh T., Sawaguchi A., Iguchi A., Nakayama K., Hayashi T. The defective prophage pool of Escherichia coli O157: prophage-prophage interactions potentiate horizontal transfer of virulence determinants. PLoS Pathog. 2009;5:e1000408. doi: 10.1371/journal.ppat.1000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bareia T., Pollak S., Eldar A. Self-sensing in Bacillus subtilis quorum-sensing systems. Nat. Microbiol. 2018;3:83–89. doi: 10.1038/s41564-017-0044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry R.A., Bouwer H.G., Portnoy D.A., Hinrichs D.J. Pathogenicity and immunogenicity of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect. Immun. 1992;60:1625–1632. doi: 10.1128/iai.60.4.1625-1632.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobay L.M., Rocha E.P., Touchon M. The adaptation of temperate bacteriophages to their host genomes. Mol. Biol. Evol. 2013;30:737–751. doi: 10.1093/molbev/mss279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J., Qian J., Westra E.R., Buckling A., Guttman D.S., Davidson A.R., Maxwell K.L. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016;10:2854–2866. doi: 10.1038/ismej.2016.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüssow H., Canchaya C., Hardt W.D. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004;68:560–602. doi: 10.1128/MMBR.68.3.560-602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns N., James C.E., Harrison E. Polylysogeny magnifies competitiveness of a bacterial pathogen in vivo. Evol. Appl. 2015;8:346–351. doi: 10.1111/eva.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorscht J., Klumpp J., Bielmann R., Schmelcher M., Born Y., Zimmer M., Calendar R., Loessner M.J. Comparative genome analysis of Listeria bacteriophages reveals extensive mosaicism, programmed translational frameshifting, and a novel prophage insertion site. J. Bacteriol. 2009;191:7206–7215. doi: 10.1128/JB.01041-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubnau D. DNA uptake in bacteria. Annu. Rev. Microbiol. 1999;53:217–244. doi: 10.1146/annurev.micro.53.1.217. [DOI] [PubMed] [Google Scholar]
- Feiner R., Argov T., Rabinovich L., Sigal N., Borovok I., Herskovits A.A. A new perspective on lysogeny: prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol. 2015;13:641–650. doi: 10.1038/nrmicro3527. [DOI] [PubMed] [Google Scholar]
- Figueroa-Bossi N., Uzzau S., Maloriol D., Bossi L. Variable assortment of prophages provides a transferable repertoire of pathogenic determinants in Salmonella. Mol. Microbiol. 2001;39:260–271. doi: 10.1046/j.1365-2958.2001.02234.x. [DOI] [PubMed] [Google Scholar]
- Hamon M., Bierne H., Cossart P. Listeria monocytogenes: a multifaceted model. Nat. Rev. Microbiol. 2006;4:423–434. doi: 10.1038/nrmicro1413. [DOI] [PubMed] [Google Scholar]
- Howard-Varona C., Hargreaves K.R., Abedon S.T., Sullivan M.B. Lysogeny in nature: mechanisms, impact and ecology of temperate phages. ISME J. 2017;11:1511–1520. doi: 10.1038/ismej.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A.D., Poteete A.R., Lauer G., Sauer R.T., Ackers G.K., Ptashne M. lambda Repressor and cro--components of an efficient molecular switch. Nature. 1981;294:217–223. doi: 10.1038/294217a0. [DOI] [PubMed] [Google Scholar]
- Klumpp J., Loessner M.J. Listeria phages: Genomes, evolution, and application. Bacteriophage. 2013;3:e26861. doi: 10.4161/bact.26861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni M.M. Digital multiplexed gene expression analysis using the NanoString nCounter system. Curr. Protoc. Mol. Biol. 2011;25:25B.10. doi: 10.1002/0471142727.mb25b10s94. [DOI] [PubMed] [Google Scholar]
- Lamont I., Brumby A.M., Egan J.B. UV induction of coliphage 186: prophage induction as an SOS function. Proc. Natl. Acad. Sci. USA. 1989;86:5492–5496. doi: 10.1073/pnas.86.14.5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer P., Chow M.Y., Loessner M.J., Portnoy D.A., Calendar R. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J. Bacteriol. 2002;184:4177–4186. doi: 10.1128/JB.184.15.4177-4186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit M. Understanding how Listeria monocytogenes targets and crosses host barriers. Clin. Microbiol. Infect. 2005;11:430–436. doi: 10.1111/j.1469-0691.2005.01146.x. [DOI] [PubMed] [Google Scholar]
- Little J.W., Shepley D.P., Wert D.W. Robustness of a gene regulatory circuit. EMBO J. 1999;18:4299–4307. doi: 10.1093/emboj/18.15.4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lwoff A. Lysogeny. Bacteriol. Rev. 1953;17:269–337. doi: 10.1128/br.17.4.269-337.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandali S., Gupta K., Dawson A.R., Van Duyne G.D., Johnson R.C. Control of Recombination Directionality by the Listeria Phage A118 Protein Gp44 and the Coiled-Coil Motif of Its Serine Integrase. J. Bacteriol. 2017;199:e00019-17. doi: 10.1128/JB.00019-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos R.C., Lapaque N., Rigottier-Gois L., Debarbieux L., Meylheuc T., Gonzalez-Zorn B., Repoila F., Lopes Mde.F., Serror P. Enterococcus faecalis prophage dynamics and contributions to pathogenic traits. PLoS Genet. 2013;9:e1003539. doi: 10.1371/journal.pgen.1003539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure R., Balasubramanian D., Sun Y., Bobrovskyy M., Sumby P., Genco C.A., Vanderpool C.K., Tjaden B. Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res. 2013;41:e140. doi: 10.1093/nar/gkt444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi M., Kurokawa K., Hayashi T. Diversification of Escherichia coli genomes: are bacteriophages the major contributors? Trends Microbiol. 2001;9:481–485. doi: 10.1016/s0966-842x(01)02173-4. [DOI] [PubMed] [Google Scholar]
- Oppenheim A.B., Kobiler O., Stavans J., Court D.L., Adhya S. Switches in bacteriophage lambda development. Annu. Rev. Genet. 2005;39:409–429. doi: 10.1146/annurev.genet.39.073003.113656. [DOI] [PubMed] [Google Scholar]
- Osuna B.A., Karambelkar S., Mahendra C., Christie K.A., Garcia B., Davidson A.R., Kleinstiver B.P., Kilcher S., Bondy-Denomy J. Listeria phages induce Cas9 degradation to protect lysogenic genomes. bioRxiv. 2019 doi: 10.1101/787200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy D.A., Jacks P.S., Hinrichs D.J. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 1988;167:1459–1471. doi: 10.1084/jem.167.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashne M. Cold Spring Harbor Laboratory Press; 2004. A Genetic Switch–Phage Lambda Revisited. [Google Scholar]
- Quiles-Puchalt N., Tormo-Más M.A., Campoy S., Toledo-Arana A., Monedero V., Lasa I., Novick R.P., Christie G.E., Penadés J.R. A super-family of transcriptional activators regulates bacteriophage packaging and lysis in Gram-positive bacteria. Nucleic Acids Res. 2013;41:7260–7275. doi: 10.1093/nar/gkt508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich L., Sigal N., Borovok I., Nir-Paz R., Herskovits A.A. Prophage excision activates Listeria competence genes that promote phagosomal escape and virulence. Cell. 2012;150:792–802. doi: 10.1016/j.cell.2012.06.036. [DOI] [PubMed] [Google Scholar]
- Rauch B.J., Silvis M.R., Hultquist J.F., Waters C.S., McGregor M.J., Krogan N.J., Bondy-Denomy J. Inhibition of CRISPR-Cas9 with Bacteriophage Proteins. Cell. 2017;168:150–158.e110. doi: 10.1016/j.cell.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reniere M.L., Whiteley A.T., Portnoy D.A. An In Vivo Selection Identifies Listeria monocytogenes Genes Required to Sense the Intracellular Environment and Activate Virulence Factor Expression. PLoS Pathog. 2016;12:e1005741. doi: 10.1371/journal.ppat.1005741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal N., Pasechnek A., Herskovits A.A. RNA Purification from Intracellularly Grown Listeria monocytogenes in Macrophage Cells. J. Vis. Exp. 2016;(112):54044. doi: 10.3791/54044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K., Youngman P. Use of a new integrational vector to investigate compartment-specific expression of the Bacillus subtilis spoIIM gene. Biochimie. 1992;74:705–711. doi: 10.1016/0300-9084(92)90143-3. [DOI] [PubMed] [Google Scholar]
- Stead M.B., Agrawal A., Bowden K.E., Nasir R., Mohanty B.K., Meagher R.B., Kushner S.R. RNAsnap™: a rapid, quantitative and inexpensive, method for isolating total RNA from bacteria. Nucleic Acids Res. 2012;40:e156. doi: 10.1093/nar/gks680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan B., Gerner-Smidt P. The epidemiology of human listeriosis. Microbes Infect. 2007;9:1236–1243. doi: 10.1016/j.micinf.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Tjaden B. De novo assembly of bacterial transcriptomes from RNA-seq data. Genome Biol. 2015;16:1. doi: 10.1186/s13059-014-0572-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo-Arana A., Dussurget O., Nikitas G., Sesto N., Guet-Revillet H., Balestrino D., Loh E., Gripenland J., Tiensuu T., Vaitkevicius K. The Listeria transcriptional landscape from saprophytism to virulence. Nature. 2009;459:950–956. doi: 10.1038/nature08080. [DOI] [PubMed] [Google Scholar]
- Waldor M.K., Friedman D.I. Phage regulatory circuits and virulence gene expression. Curr. Opin. Microbiol. 2005;8:459–465. doi: 10.1016/j.mib.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Wang X., Kim Y., Ma Q., Hong S.H., Pokusaeva K., Sturino J.M., Wood T.K. Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 2010;1:147. doi: 10.1038/ncomms1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink R., Loessner M.J. Classification of virulent and temperate bacteriophages of Listeria spp. on the basis of morphology and protein analysis. Appl. Environ. Microbiol. 1992;58:296–302. doi: 10.1128/aem.58.1.296-302.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-Seq data are available in Mendeley Data via these links:
Mendeley Data: https://doi.org/10.17632/kjyh2swhg9.2
Mendeley Data: https://doi.org/10.17632/mz484h884h.3
Source file including raw experimental data
Mendeley Data: https://doi.org/10.17632/9jzf6vk3p7.1